Synthesis, Molecular Docking and Anticancer Activity of Diflunisal Derivatives as Cyclooxygenase Enzyme Inhibitors

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
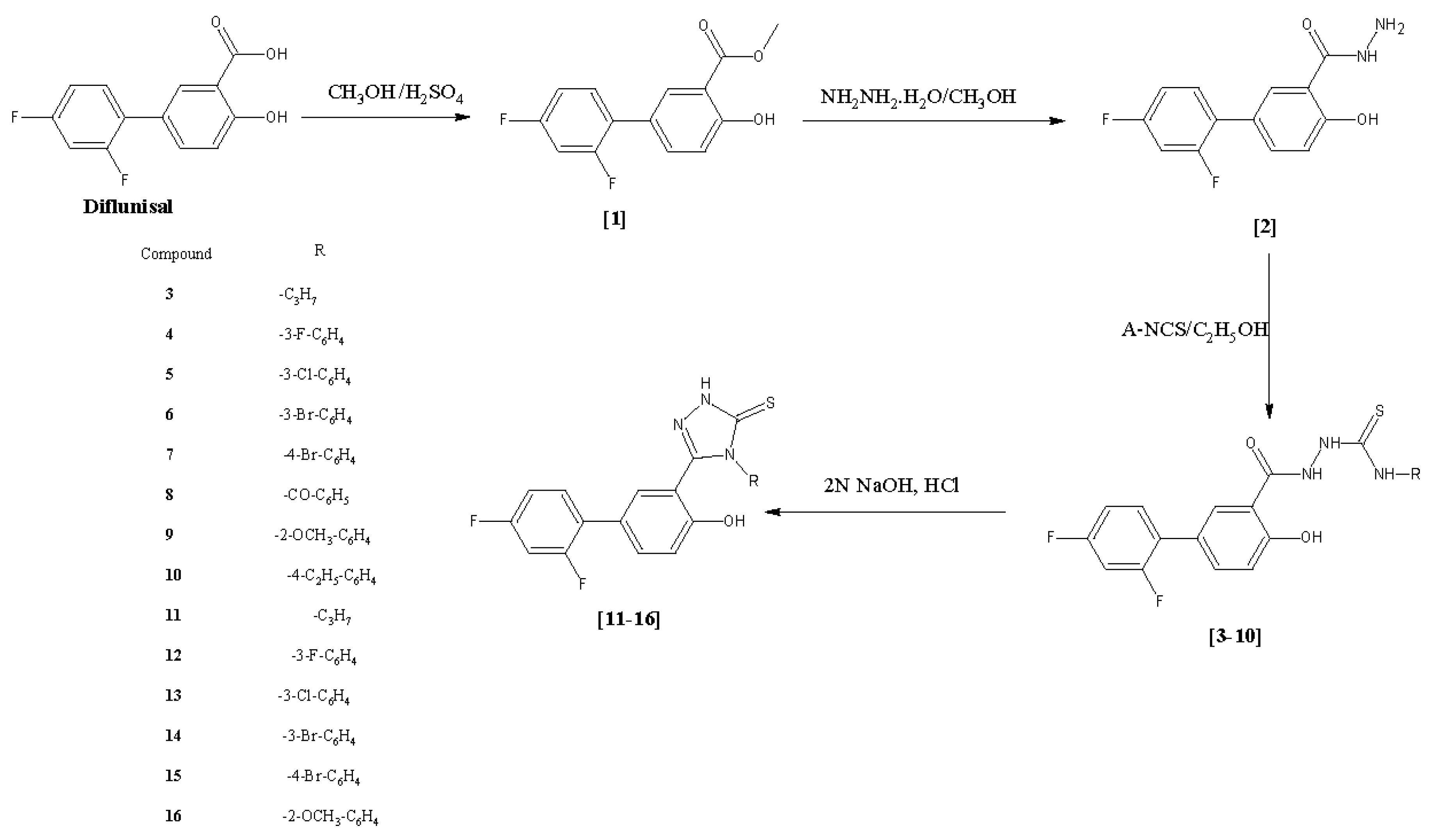
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Anticancer Activity
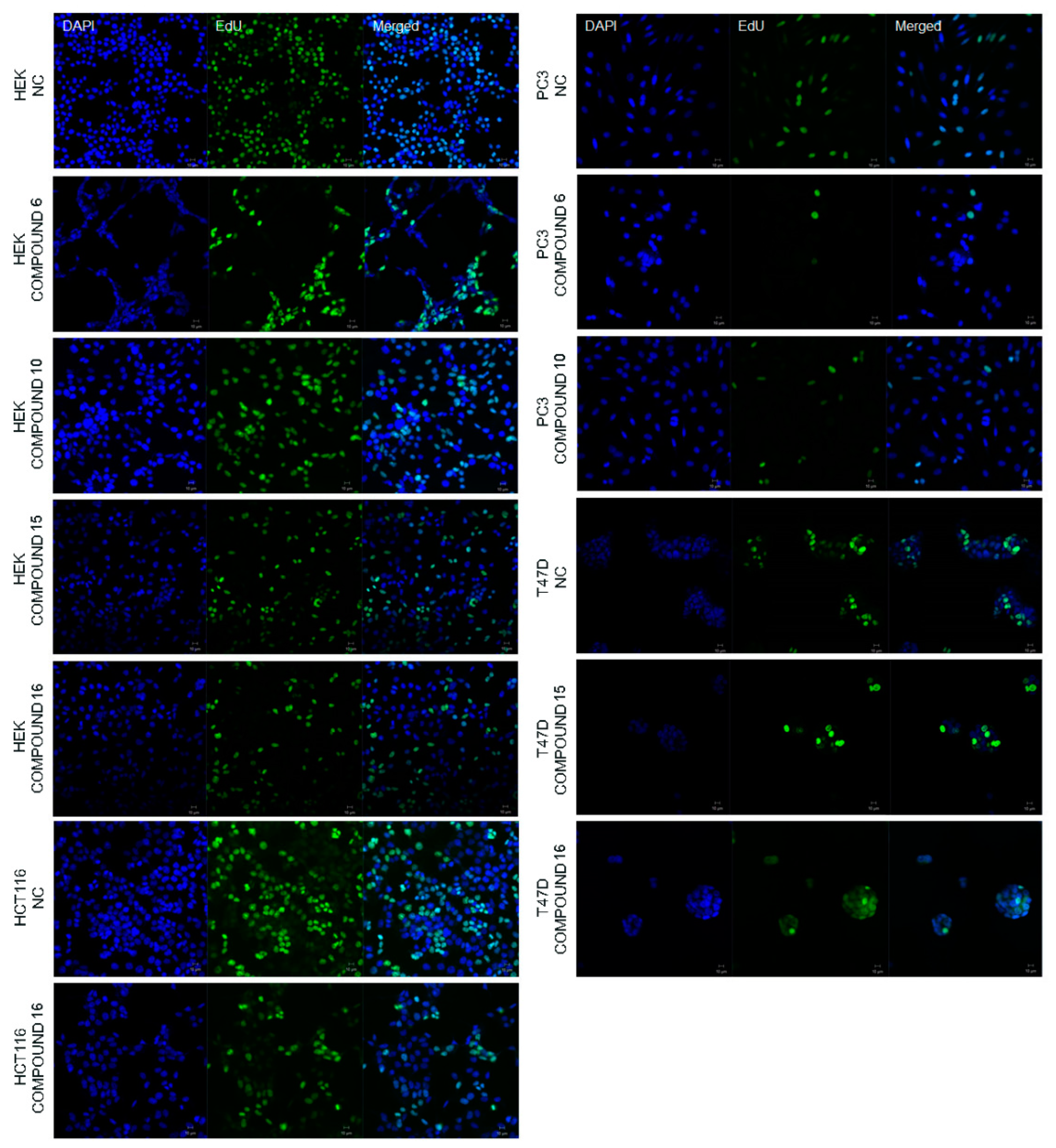
2.2.2. Effect of the Synthesized Compounds on DNA Synthesis
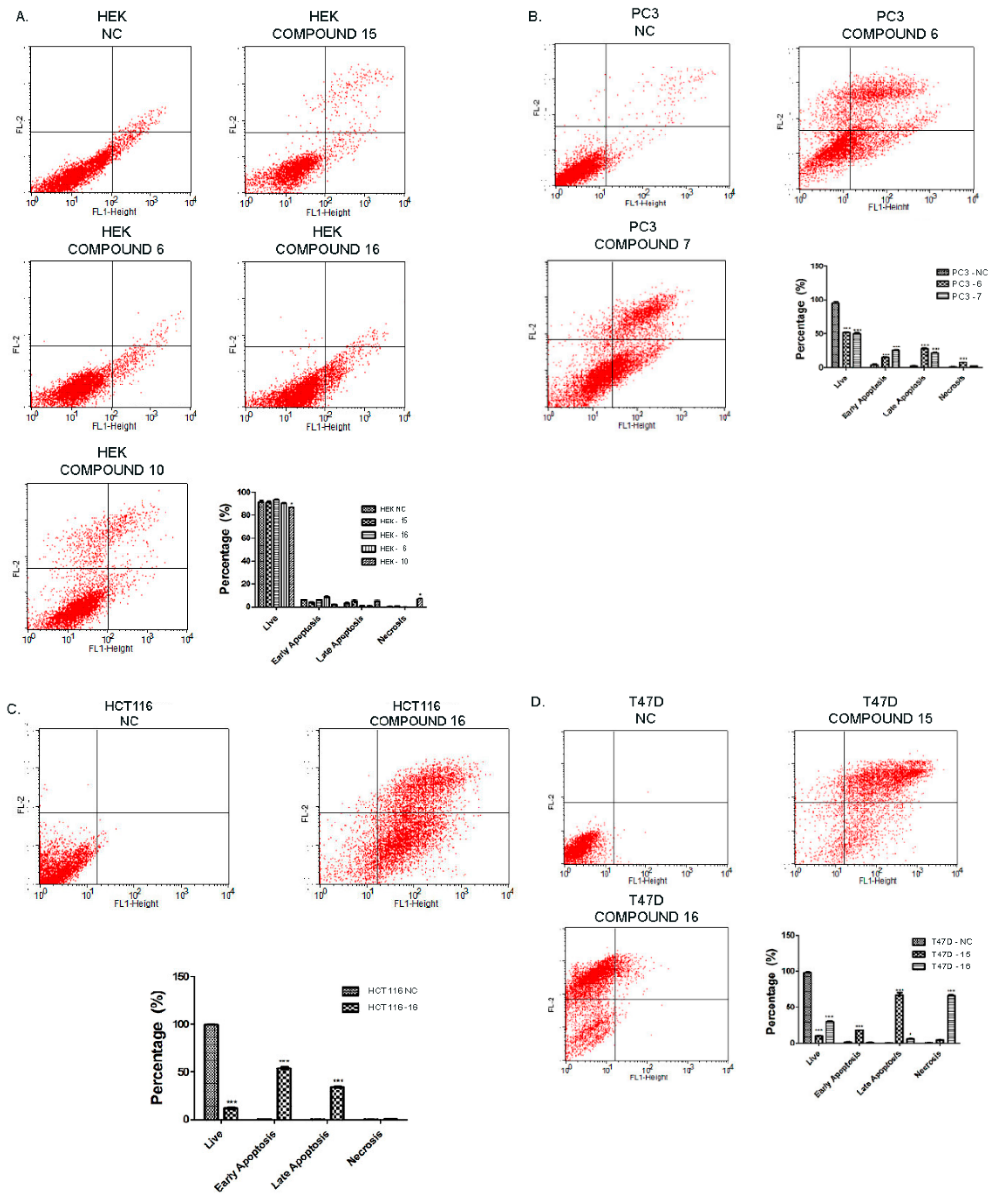
2.2.3. Effect of Synthesized Compounds on Apoptosis
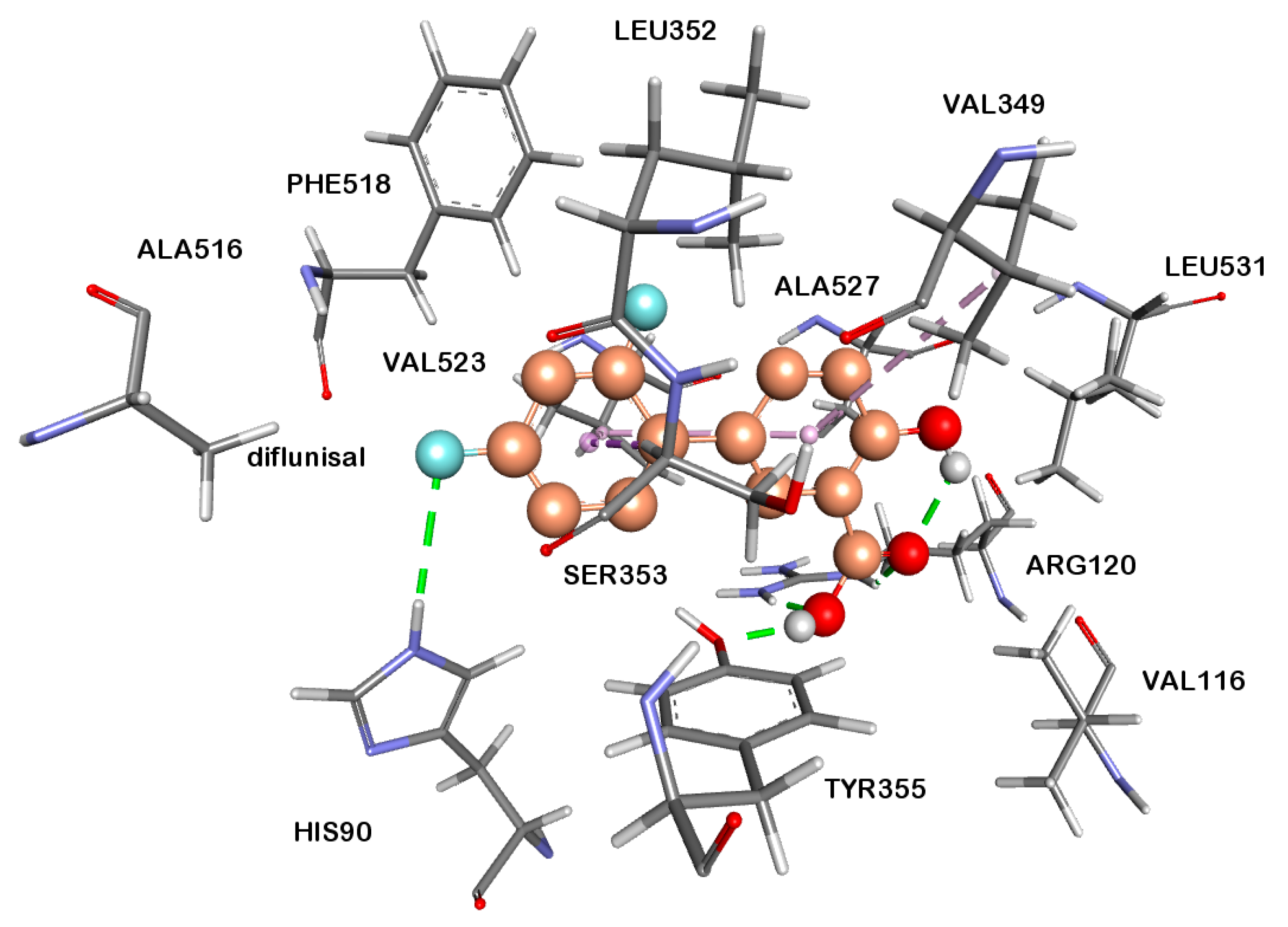
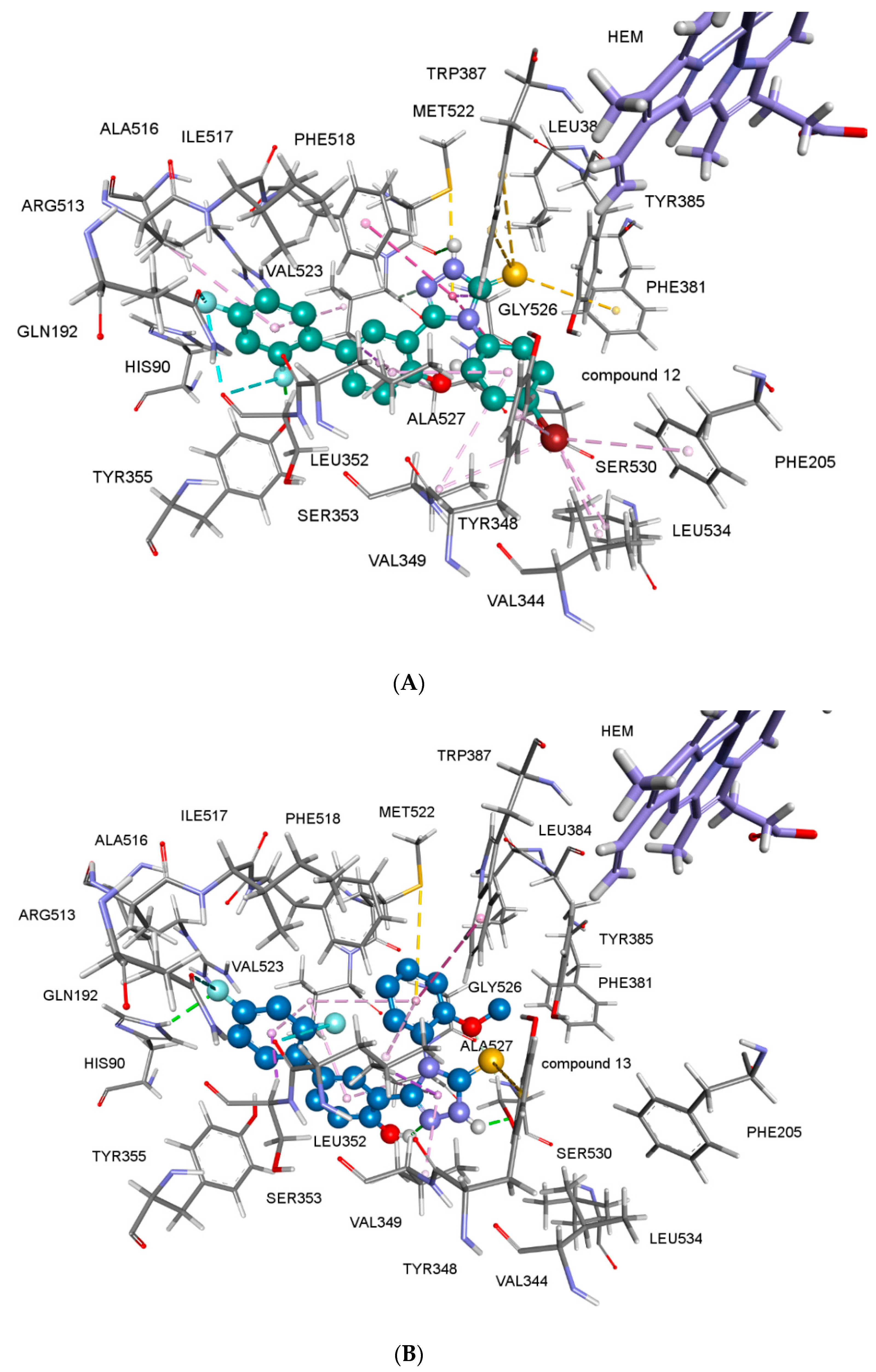
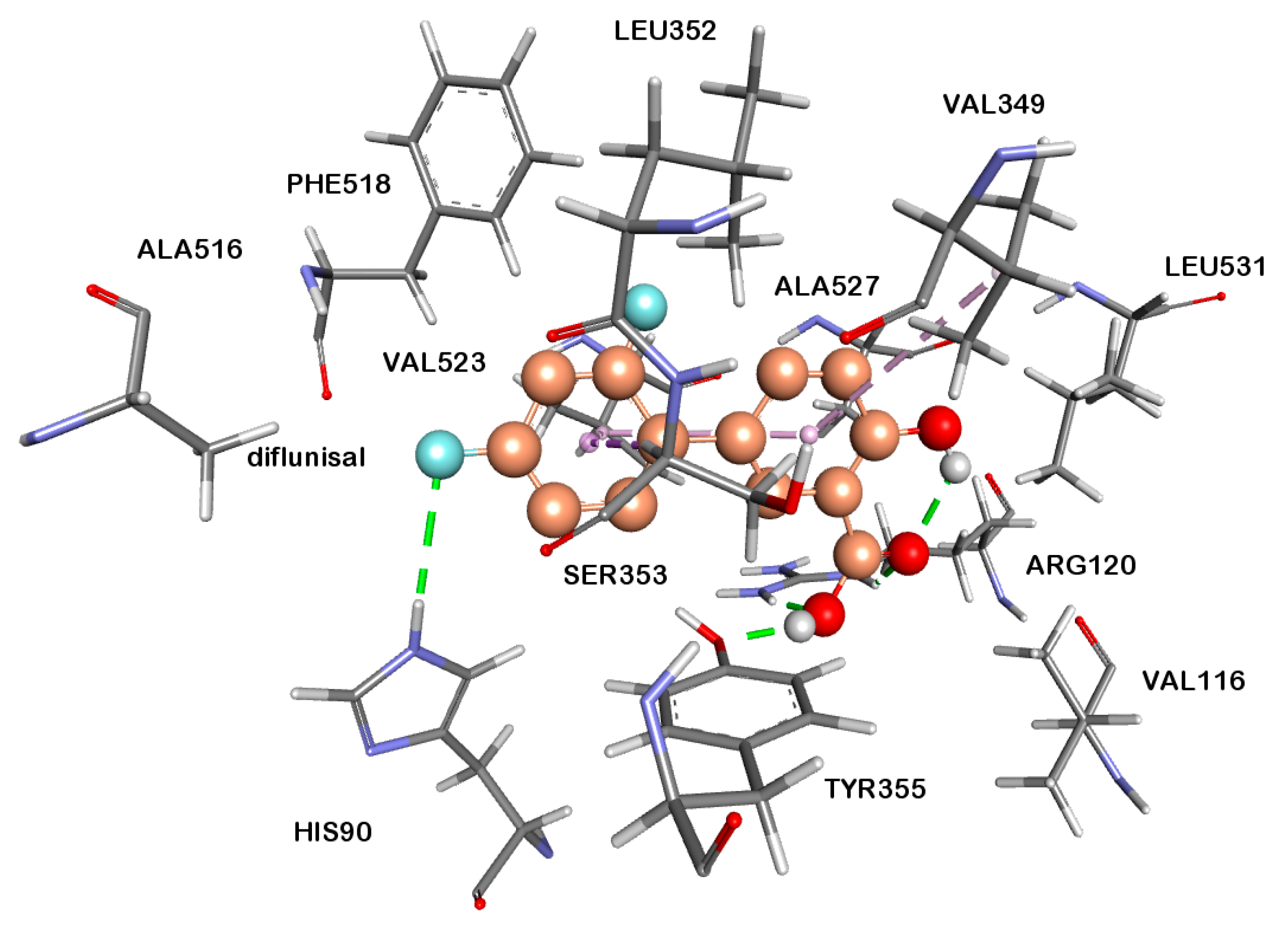
2.2.4. Docking Studies
3. Materials and Methods
3.1. General
3.2. Synthesis of Methyl 2′,4′-Difluoro-4-hydroxybiphenyl-3-carboxylate (Diflunisal Ester, 1, CAS Number: 55544-00-8) and 2′,4′-Difluoro-4-hydroxybiphenyl-3-carbohydrazide (Diflunisal Hydrazide, 2)
3.3. General Procedure for the Synthesis of 2-[(2′,4′-Difluoro-4-hydroxybiphenyl-3-yl)carbonyl]-N-(substituted)hydrazinocarbothioamides 3–10
3.4. General Procedure for the Synthesis of 5-(2′,4′-Difluoro-4-hydroxybiphenyl-3-yl) -4-(substituted)-2,4-dihydro-3H-1,2,4-triazole-3-thiones 11–16
3.5. Cell Culture Conditions
3.5.1. Cytotoxicity Assay and Determination of IC50 Values of Synthesized Compounds
3.5.2. Annexin V
3.5.3. 5-Ethynyl-2′-deoxyuridine (EdU) Staining
3.6. Docking Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shamsudin, K.Y.; Gutiérrez-de-Terán, H.; Boukharta, L.; Aqvist, J. Toward an optimal docking and free energy calculation scheme in ligand design with application to COX-1 inhibitors. J. Chem. Inf. Model. 2014, 54, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, G.; Panella, A.; Perrone, M.G.; Vitale, P.; Di Mauro, G.; Fortuna, C.G.; Armen, R.S.; Ferorelli, S.; Smith, W.L.; Scilimati, A. Structural basis for selective inhibition of Cyclooxygenase-1 (COX-1) by diarylisoxazoles mofezolac and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6). Eur. J. Med. Chem. 2017, 138, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botting, R.; Ayoub, S.S. COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukot. Essent. Fatty Acids 2005, 72, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Wang, P.F.; Li, Z.; Ma, J.T.; Wang, X.M.; Yang, Y.H.; Zhu, H.L. Synthesis of dihydropyrazole sulphonamide derivatives that act as anti-cancer agents through COX-2 inhibition. Pharmacol. Res. 2016, 104, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, M.A.; Bayomi, S.M.; El-Sherbeny, M.A.; Abdel-Aziz, N.I.; ElTahir, K.E.; Shehatou, G.S.; Abdel-Aziz, A.A. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg. Med. Chem. 2016, 24, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Veitonmäki, T.; Murtola, T.J.; Talala, K.; Taari, K.; Tammela, T.; Auvinen, A. Non-steroidal anti-inflammatory drugs and cancer death in the finnish prostate cancer screening trial. PLoS ONE 2016, 11, 0153413. [Google Scholar] [CrossRef] [PubMed]
- Koki, A.T.; Masferrer, J.L. Celecoxib: A specific COX-2 inhibitor with anticancer properties. Cancer Control 2002, 9, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xie, W.; Reed, D.; Bradshaw, W.S.; Simmons, D.L. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 7961–7965. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Mazi, A.; Sahin, F.; Öztürk, S.; Stables, J. Synthesis and biological activities of diflunisal hydrazide-hydrazones. Eur. J. Med. Chem. 2003, 38, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Kocatepe, A.; Clercq, E.D.; Şahin, F.; Güllüce, M. Synthesis and biological activity of 4-thiazolidinones, thiosemicarbazides derived from diflunisal hydrazide. Eur. J. Med. Chem. 2006, 41, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Küçükgüzel, İ.; Tatar, E.; Rollas, S.; Şahin, F.; Güllüce, M.; De Clercq, E.; Kabasakal, L. Synthesis of some novel heterocyclic compounds derived from diflunisal hydrazide as potential anti-infective and anti-inflammatory agents. Eur. J. Med. Chem. 2007, 42, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Şenkardeş, S.; Kaushik-Basu, N.; Durmaz, İ.; Manvar, D.; Basu, A.; Atalay, R.; Küçükgüzel, Ş.G. Synthesis of novel diflunisal hydrazide-hydrazones as anti-hepatitis C virus agents and hepatocellular carcinoma inhibitors. Eur. J. Med. Chem. 2016, 108, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Şenkardeş, S.; Bingöl-Özakpınar, Ö.; Özsavcı, D.; Şener, A.; Çevik, Ö.; Küçükgüzel, Ş.G. Synthesis of diflunisal thiazolidinones as anticancer agents. Anticancer Agents Med. Chem. 2016, 16, 1266–1274. [Google Scholar] [PubMed]
- Kreutz, W. Diflunisal for the Treatment of Cancer. U.S. Patent 2006/0293390, 28 December 2006. [Google Scholar]
- Küçükgüzel, Ş.G.; Coşkun, G.P. Macromolecular Drug Targets in Cancer Treatment and thiosemicarbazides as anticancer agents. Anticancer Agents Med. Chem. 2016, 16, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Süzgün, P. Recent advances bioactive 1,2,4-triazole-3-thiones. Eur. J. Med. Chem. 2015, 97, 830–870. [Google Scholar] [CrossRef] [PubMed]
- Çıkla-Süzgün, P.; Kaushik-Basu, N.; Basu, A.; Arora, P.; Talele, T.T.; Durmaz, İ.; Cetin-Atalay, R.; Kucukguzel, Ş.G. Anti-cancer and anti-hepatitis C virus NS5B polymerase activity of Etodolac 1,2,4-Triazoles. J. Enzym. Inhib. Med. Chem. 2015, 30, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, S.; Shi, L.; Zhang, S.; Ren, J.; Yao, S.; Yun, D.; Huang, L.; Wang, J.; Li, W.; et al. Design, synthesis, and evaluation of asymmetric EF24 analogues as potential anti-cancer agents for Lung Cancer. Eur. J. Med. Chem. 2017, 125, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Aydın, S.; Kaushik-Basu, N.; Özbaş-Turan, S.; Akbuğa, J.; Tiber, P.M.; Orun, O.; Gurukumar, K.R.; Basu, A.; Küçükgüzel, Ş.G. Synthesis of 1-aroyl-3,5-dimethyl-1h-pyrazoles as anti-HCV and anticancer agents. Lett. Drug Des. Discov. 2014, 11, 121–131. [Google Scholar] [CrossRef]
- Küçükgüzel, Ş.G.; Şenkardeş, S. Recent advances in bioactive pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. [Google Scholar] [CrossRef] [PubMed]
- Dadaş, Y.; Coşkun, G.P.; Bingöl-Özakpınar, Ö.; Özsavcı, D.; Küçükgüzel, Ş.G. Synthesis and Anticancer Activity of Some Novel Tolmetin Thiosemicarbazides. Marmara Pharm. J. 2015, 19, 259–267. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Yashima, K.; Morisawa, Y.; Shiota, G.; Kawasaki, H.; Hasegawa, J. Effects of cyclooxygenase-2 inhibitor NS-398 on APC and c-myc expression in rat colon carcinogenesis induced by azoxymethane. J. Gasteroenterol. 2002, 37, 186–193. [Google Scholar] [CrossRef]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. Br. Med. J. 2006, 332, 1302–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coşkun, G.P.; Djikic, T.; Kalaycı, S.; Yelekçi, K.; Şahin, F.; Küçükgüzel, Ş.G. Synthesis, Molecular Modelling and antibacterial activity against Helicobacter pylori of novel diflunisal derivatives as urease enzyme inhibitors. Lett. Drug Des. Discov. 2018, in press. [Google Scholar]
- Bhardwaj, A.; Kaur, J.; Wuest, F.; Knaus, E.E. Fluorophore-labeled cyclooxygenase-2 inhibitors for the imaging of cyclooxygenase-2 overexpression in cancer: Synthesis and biological studies. Chem. Med. Chem. 2014, 9, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Yadagiri, B.; Gurrala, S.; Bantu, R.; Nagarapu, L.; Polepalli, S.; Srujana, G.; Jain, N. Synthesis and evaluation of benzosuberone embedded with 1,3,4-oxadiazole, 1,3,4-331 thiadiazole and 1,2,4-triazole moieties as new potential anti proliferative agents. Bioorg. Med. Chem. Lett. 2014, 25, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Khaled, R.A.A.; Eman, K.A.A.; Mohamed, A.A.; Rasha, R.A.; Rania, B.B. Synthesis and Anticancer Activity of Some New Pyrazolo [3,4-d] pyrimidin-4-one Derivatives. Molecules 2014, 19, 3297–3309. [Google Scholar]
- Sunil, D.; Isloor, A.M.; Shetty, P.; Chandrakantha, B.; Satyamoorthy, K. Synthesis, characterization and in vitro cytotoxic properties of some new Schiff and Mannich bases in Hep G2 cells. Med. Chem. Res. 2011, 20, 1024–1032. [Google Scholar] [CrossRef]
- Karakuş, S.; Çoruh, U.; Barlas-Durgun, B.; Vázquez-López, E.M.; Özbaş-Turan, S.; Akbuğa, J.; Rollas, S. Synthesis and cytotoxic activity of some 1,2,4-triazoline-3-thione and 2,5-disubstituted-1,3,4-thiadiazole derivatives. Marmara Pharm. J. 2010, 14, 84–90. [Google Scholar] [CrossRef]
- Li, Z.; Gu, Z.; Yin, K.; Zhang, R.; Deng, Q.; Xiang, J. Synthesis of substituted-phenyl-1,2,4-triazol-3-thione analogues with modified D-glucopyranosyl residues and their antiproliferative activities. Eur. J. Med. Chem. 2009, 44, 4716–4720. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Pan, F.Y. New 3-[(4-hydroxy-6-methly-2(1H)-pyridinones)-3-yl]-4-substituted-(1H)-1,2,4-triazole-5-thiones: Efficient synthesis, X-ray crystallographic analysis, and antitumor activity. Org. Chem. Lett. 2007, 4, 137–141. [Google Scholar] [CrossRef]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Idelman, G.; Jacobson, E.M.; Tuttle, T.R.; Ben-Jonathan, N. Lactogens and estrogens in breast cancer chemoresistance. Expert Rev. Endocrinol. Metab. 2011, 6, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, L.; Bianchi, P.; Ruff, P.; Arbuthnot, P. Differential modulation by estradiol of P-glycoprotein drug resistance protein expression in cultured MCF7 and T47D breast cancer cells. Anticancer Res. 2002, 22, 2253–2259. [Google Scholar] [PubMed]
- Lee, M.T.; Ho, S.M.; Tarapore, P.; Chung, I.; Leung, Y.K. Estrogen receptor β isoform 5 confers sensitivity of breast cancer cell lines to chemotherapeutic agent-induced apoptosis through interaction with Bcl2L12. Neoplasia 2013, 15, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, S.; Furuse, M.; Yokoyama, N.; Sasazuki, T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993, 260, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Houdt, W.J.; Hoogwater, F.J.H.; Bruijn, M.T.; Emmink, B.L.; Nijkamp, M.W.; Raats, D.A.E.; Groep, P.; Diest, P.; Inne, H.M.; Rinkes, B.; et al. Oncogenic KRAS Desensitizes Colorectal Tumor Cells to Epidermal Growth Factor Receptor Inhibition and Activation. Neoplasia 2010, 12, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Humphrey, L.E.; Yang, H.; Brattain, M.G. Expression of TGFα autocrine activity in human colon carcinoma CBS cells is autoregulated and independent of exogenous epidermal growth factor. J. Cell. Physiol. 1998, 175, 174–183. [Google Scholar] [CrossRef]
- Yuan, S.; Trachtenberg, J.; Mills, G.B.; Brown, T.J.; Xu, F.; Keating, A. Androgen-induced inhibition of cell proliferation in an Androgen-insensitive Prostate Cancer Cell Line (PC-3) Transfected with a Human Androgen Receptor Complementary DNA. Endocrinology 1993, 53, 1304–1311. [Google Scholar]
- Palapattu, G.S.; Wu, C.; Silvers, C.R.; Martin, H.B.; Williams, K.; Salamone, L.; Bushnell, T.; Huang, L.S.; Yang, Q.; Huang, J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 2009, 69, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Tippayamontri, T.; Kotb, R.; Paquette, B.; Sanche, L. Cellular Uptake and Cytoplasm/DNA distribution of cisplatin and oxaliplatin and their liposomal formulation in human colorectal cancer cell HCT116. Investig. New Drugs 2011, 29, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines—An Isobolographic Analysis. PLoS ONE 2015, 10, 0143013. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Karen, H.V. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, M.H.; Youssif, B.G.; Abdelgawad, M.A.; Abdelazeem, A.H.; Ibrahim, H.M.; Moustafa, A.E.; Treamblu, L.; Bukhari, S.N. Synthesis, Biological evaluation, docking study and ulcerogenicity profiling of some novel quinoline-2-carboxamides as dual COXs/LOX inhibitors endowed with anti-inflammatory activity. Eur. J. Med. Chem. 2017, 127, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Selinsky, B.S.; Gupta, K.; Sharkey, C.T.; Loll, P.J. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 2001, 40, 5172–5180. [Google Scholar] [CrossRef] [PubMed]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2010, 285, 34950–34959. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PC-3 | HCT116 | T47D | MCF7 | HEK |
|---|---|---|---|---|---|
| 3 | ND | ND | ND | ND | ND |
| 4 | PCV | PCV | PCV | PCV | 617 + 35 |
| 5 | 67,2 ± 4.5 | PCV | 28,2 ± 2.5 | PCV | 44,4 ± 1,7 |
| 6 | 41.8 ± 3.4 | PCV | 100 ± 2.3 | PCV | 93 ± 3.43 |
| 7 | 11.7 ± 4.2 | PCV | 16.73 ± 2.8 | PCV | 3.17 ± 1.06 |
| 8 | 2.85 ± 3.6 | 3.68 ± 2.1 | 8.07 ± 2.0 | PCV | 5.04 ± 1.5 |
| 9 | 1.575 ± 1.4 | 1.83 ± 0.92 | 3.86 ± 0.7 | 3.92 ± 0.27 | 3.02 ± 1.9 |
| 10 | 11.7 ± 2.6 | PCV | NSC | PCV | 94 ± 3.74 |
| 11 | 174.13 ± 6.3 | NSC | 10.05 ± 1.57 | 22.18 ± 2.3 | 11.52 ± 0.2 |
| 12 | 33.95 ± 2.1 | NSC | 9.24 ± 1.01 | 15.03 ± 1.3 | 10.66 ± 0.96 |
| 13 | 34.09 ± 3.7 | NSC | 5.55 ± 2.7 | 12.883 ± 2.1 | 8.7 ± 1.79 |
| 14 | PCV | PCV | 11.47 ± 1.8 | NSC | 12,8 ± 2.1 |
| 15 | 145 ± 2.8 | PCV | 43.4 ± 2.72 | PCV | 145 ± 2.3 |
| 16 | 168.86 ± 3.9 | 6.2 ± 3.1 | 27.3 ± 2.28 | PCV | 53 ± 2.39 |
| Cisplatin | * 39.9 µM | * 1.25 µM | * 0.836 µM |
| Compound | COX-1 | COX-2 |
|---|---|---|
| 13 | ΔG = −10.07 kcal/mol | ΔG = −11.20 kcal/mol |
| 14 | ΔG = −10.33 kcal/mol | ΔG = −10.66 kcal/mol |
| 15 | ΔG = −10.11 kcal/mol | ΔG = −10.57 kcal/mol |
| 16 | ΔG = −9.41 kcal/mol | ΔG = −9.60 kcal/mol |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coşkun, G.P.; Djikic, T.; Hayal, T.B.; Türkel, N.; Yelekçi, K.; Şahin, F.; Küçükgüzel, Ş.G. Synthesis, Molecular Docking and Anticancer Activity of Diflunisal Derivatives as Cyclooxygenase Enzyme Inhibitors. Molecules 2018, 23, 1969. https://doi.org/10.3390/molecules23081969
Coşkun GP, Djikic T, Hayal TB, Türkel N, Yelekçi K, Şahin F, Küçükgüzel ŞG. Synthesis, Molecular Docking and Anticancer Activity of Diflunisal Derivatives as Cyclooxygenase Enzyme Inhibitors. Molecules. 2018; 23(8):1969. https://doi.org/10.3390/molecules23081969
Chicago/Turabian StyleCoşkun, Göknil Pelin, Teodora Djikic, Taha Bartu Hayal, Nezaket Türkel, Kemal Yelekçi, Fikrettin Şahin, and Ş. Güniz Küçükgüzel. 2018. "Synthesis, Molecular Docking and Anticancer Activity of Diflunisal Derivatives as Cyclooxygenase Enzyme Inhibitors" Molecules 23, no. 8: 1969. https://doi.org/10.3390/molecules23081969