A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives
by
 ,
,
Costel Moldoveanu
1, 
Ionel Mangalagiu
1,
Dragos Lucian Isac
2,
Anton Airinei
2 and
Gheorghita Zbancioc
1,* 1
Chemistry Department, Alexandru Ioan Cuza University of Iasi, 700506 Iasi, Romania
2
Petru Poni Institute of Macromolecular Chemistry, 700487 Iasi, Romania
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(8), 1968; https://doi.org/10.3390/molecules23081968
Submission received: 17 July 2018
/
Revised: 26 July 2018
/
Accepted: 3 August 2018
/
Published: 6 August 2018
(This article belongs to the Section Organic Chemistry)
Abstract
:In this study an efficient and straightforward method for obtaining a new class of blue fluorescent bezofuran derivatives, under microwave irradiation, as well as under conventional thermal heating, is presented. Under conventional TH the reactions occur selectively, and a single type of benzofuran ester derivative was obtained. The synthesis under MW irradiation also led to benzofuran derivatives, but in a time-dependent manner. Irradiation for a short period of time led to a mixture of two types of benzofuran derivatives (3a–c and 4a–c), while MW irradiation for a longer period of time led to a single type of benzofuran (3-methylbenzofuran), the reaction becoming highly selective. Taking into consideration the advantages offered by MW irradiation in terms of a substantial decrease of solvent consumed, a substantial reduction in reaction time (from days to hours), and a consequent diminution in energy consumption, these methods could be considered environmentally friendly. Here, feasible reaction mechanisms for the benzofuran derivatives formation are described. The absorption and fluorescence emission of the obtained benzofuran derivatives were studied, with part of these compounds being intense blue emitters. A certain influence of the benzofuran substituents concerning absorption and fluorescent properties was observed. Only compounds anchored with a carbomethoxy group of furan ring have shown good quantum yields.
1. Introduction
The synthesis of highly fluorescent derivatives with extended π-conjugation has been a topic of considerable research because of their potential applications [1,2,3,4,5,6,7,8,9]. Fused O-heterocyclic rings offer very interesting optical properties. Benzofuran derivatives represent such a class, being a “pure” blue-emitting moiety [1,2,3,4]. In the case of benzofuran derivatives, the literature data shows that they are chromophores with high photoluminescence and good quantum yields [2]. For this reason, benzofuran derivatives have applications in a variety of fields, including sensors and biosensors [5,6], electroluminescent materials [3,4,7], lasers, and other optoelectronic devices.
The synthesis of benzofuran derivatives has attracted special attention [8,9,10,11], thanks to their appearance in a large number of natural products [12] and synthetic pharmaceuticals [13]. Various other classes of benzofurans are attractive to chemists for their biological activities such as antimicrobial [14], antioxidant [15], anti-inflammatory [16], antifungal [17], PPARD (Peroxisome Proliferator Activated Receptor Delta) agonists [18], antifeedant, anti-HIV, anti-tumor and antiplatelet activities [19], and in other fields of chemistry and agriculture [20].
MW irradiation has become a powerful tool in organic chemistry, offering a versatile and facile pathway in a large variety of syntheses. MW-assisted synthesis has numerous advantages compared with classical synthesis: rapid energy transfer, faster reactions and substantial decreases of reaction time, fewer byproducts, improved yields and high purity of the compounds, less energy consumption, lower costs, and simplicity of handling and processing [21,22,23,24].
In previous research works [25,26], we synthesized a series of dihydroxyacetophenone (DHA) derivatives with antimicrobial and anticancer activity. The aim of this work was to find a new method of synthesis for fluorescent benzofurans, starting from DHA, to study the relationship between optical properties and structure (the effect of substituents and conjugation), and to develop a new method for the preparation of these derivatives under MW irradiation.
2. Results and Discussion
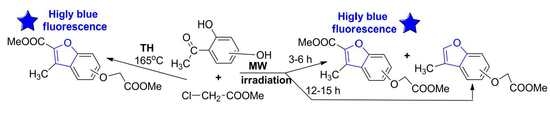
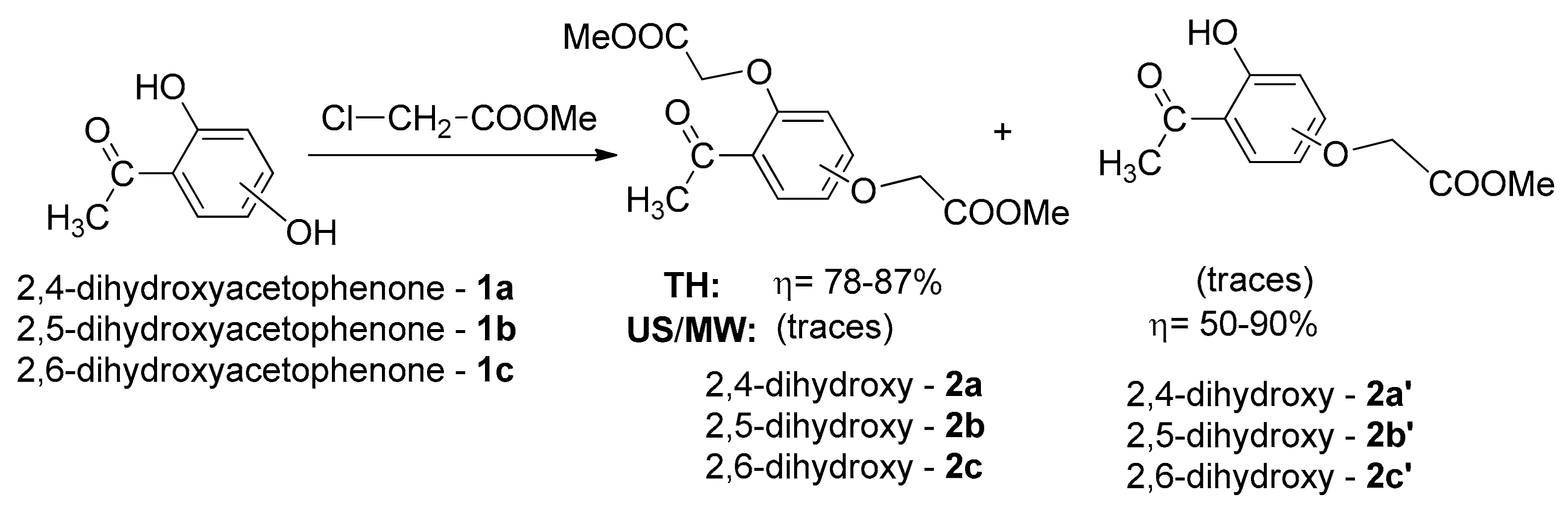
In a continuation of our search for new entities with antimicrobial and/or anticancer activity, in previous research works [25,26] we synthesized a series of new azaheterocycle derivatives with a dihydroxyacetophenone skeleton, with the reactions being studied under conventional thermal heating (TH) and under ultrasound (US) or MW irradiation. The first stage of synthesis involved an O-alkylation reaction of DHA and interesting behavior was noticed in the case of DHA with a hydroxyl group at the second position of the benzene ring: a selective bis-O-alkylation took place under conventional TH while under US or MW irradiation a selective mono-O-alkylation took place (see Scheme 1).
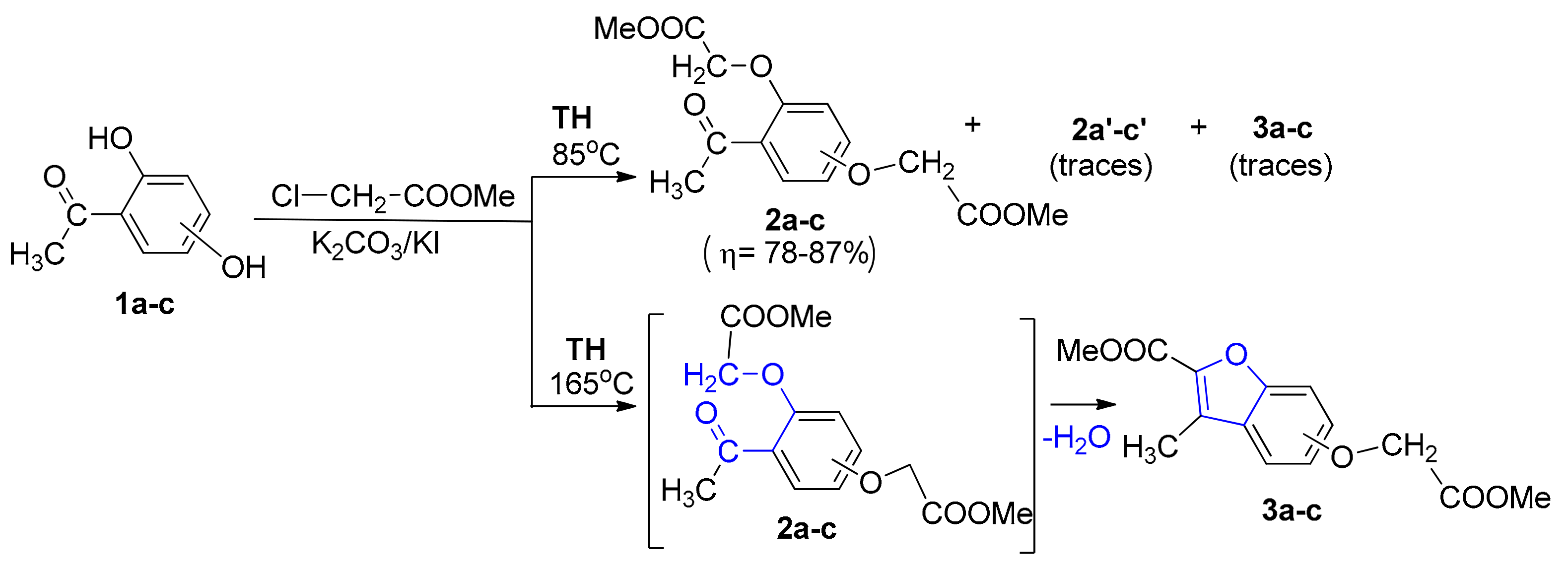
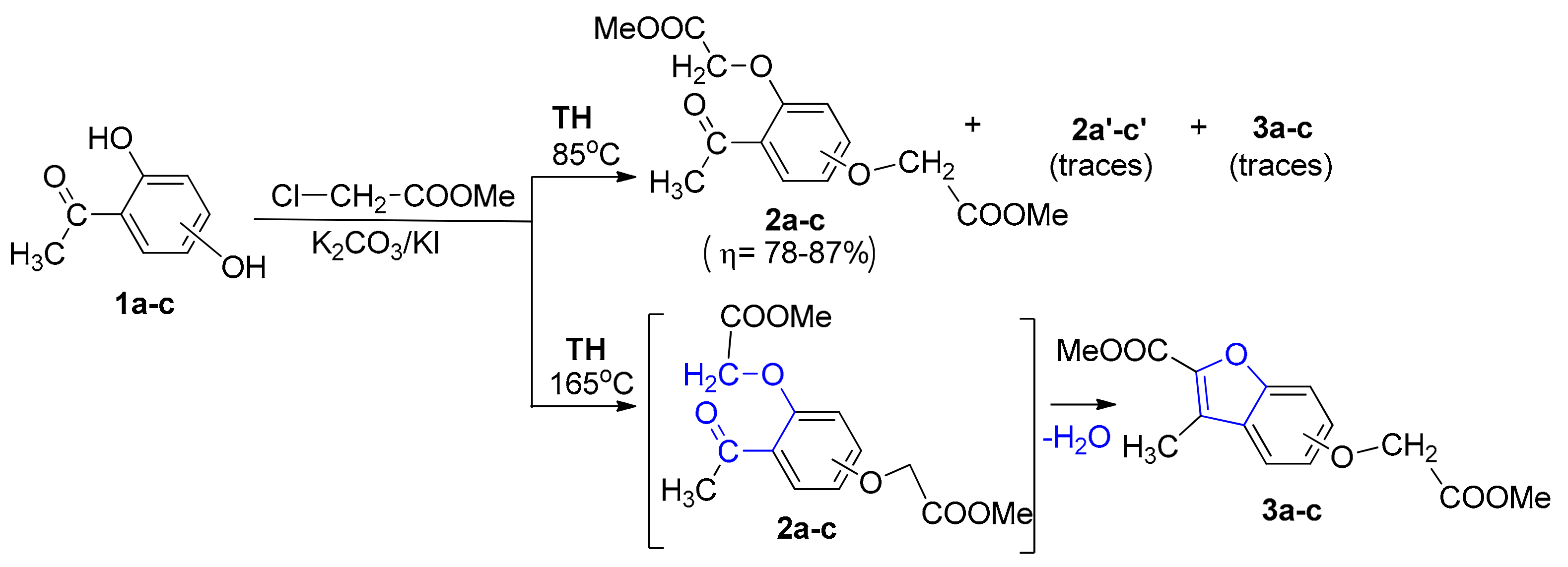
Intrigued by these unusual behaviors, we decided to study these reactions more thoroughly. In the case of TH bis-O-alkylation reactions, traces of a fluorescent compound were observed besides the two types of compounds obtained (bis-akylated DHA 2a–c and mono-akylated compounds 2a′–c′). In order to improve the poor yields of bis-akylated DHA 2a–c, we modified the synthesis procedure by raising the temperature. Unfortunately, the yield in bis-akylated DHA 2a–c decreased, but, at the same time, the yield of fluorescent compound (which proved to be benzofuran ester derivative type 3) increased substantially, and at 165 °C total conversion was observed (see Scheme 2 and Table 1).
As to the reaction mechanism, the formation of benzofuran ester derivatives could be easily explained via an intramolecular cyclocondensation of bis-akylated DHA 2a–c, Scheme 2. This reaction mechanism is in full agreement with that previously reported by Suzuki et al. [8].
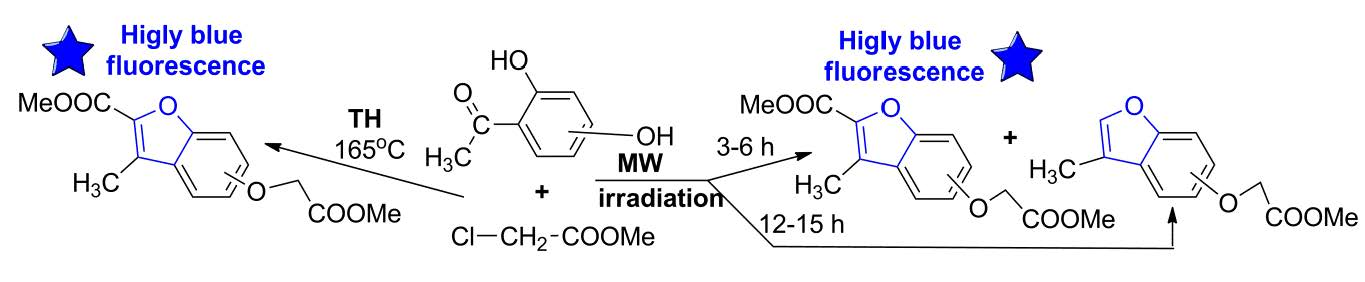
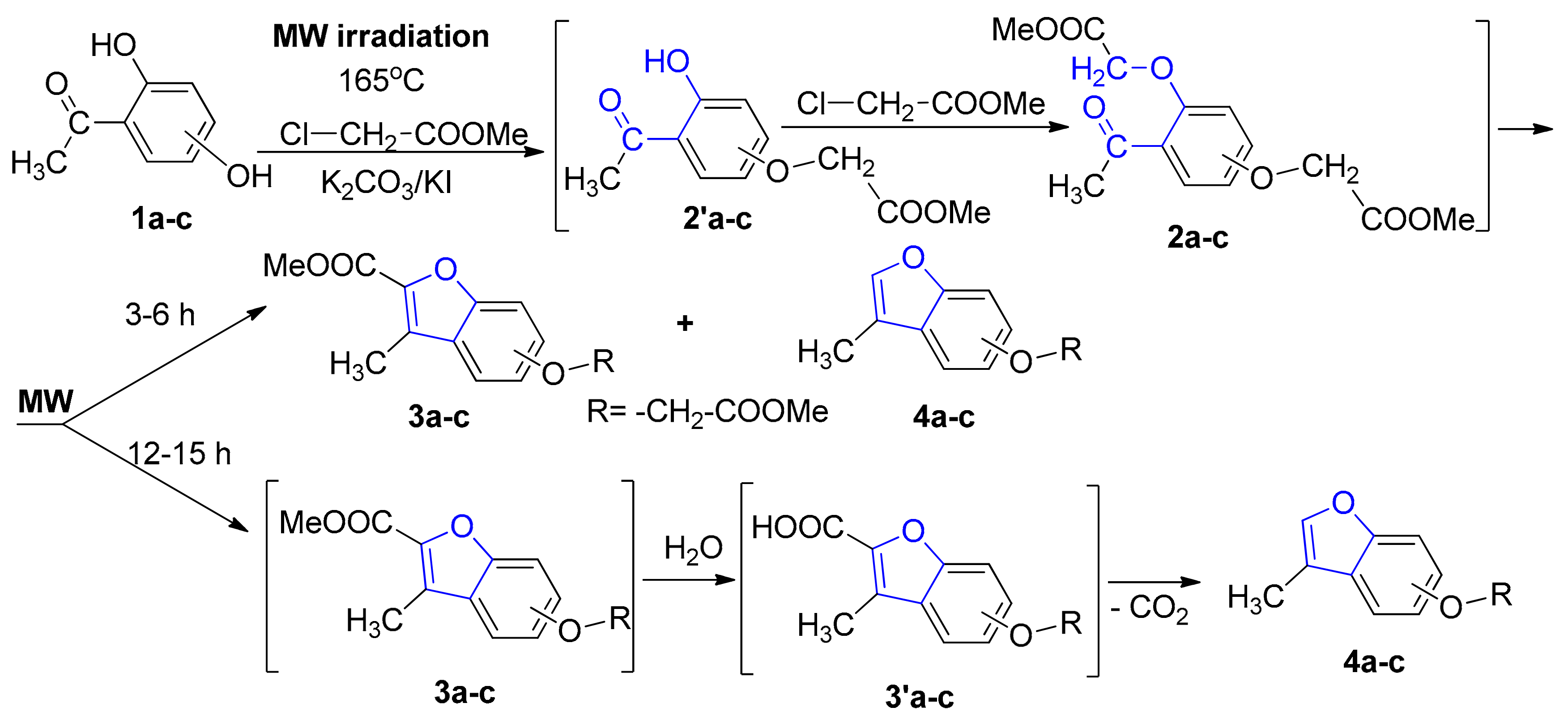
Taking into account our expertise in the field of MW-assisted reactions in organic synthesis, we decided to perform the synthesis of the new benzofuran derivatives under MW irradiation, using a monomode reactor Monowave 300 (Anton Paar, Graz, Austria). This reactor is equipped with a stirring system (0 to 1200 rpm) and can reach up to 300 °C, with temperature control. The reactions take place in a closed vessel, at 30 bars maximum pressure. Under MW irradiation, the optimal reaction conditions were found to be at 165 °C, 17–19 bars, Scheme 3, Table 1. In contrast with conventional TH, when only one type of benzofuran derivative was obtained (3a–c), the MW-assisted reactions led either to a mixture of benzofuran derivatives, 3a–c and 4a–c, or to another type of benzofuran derivative (4a–c), according to the reaction time employed (see Scheme 3).
The formation of the 3-methylbenzofuran derivatives (4a–c) could be explained by the following reaction mechanism: an initial hydrolysis of the ester derivative (3a–c) led to the corresponding acids (3′a–c), which are unstable under these conditions, and undergo a subsequent oxidative decarboxylation to 4a–c (see Scheme 3). This type of compound (3-methylbenzofuran derivatives) was reported to be obtained in the literature, but only as a trace [8], or in multiple steps with low overall yields [9,10,11]. Our synthetic strategy has the major advantages of good yield and easy workup (single-step reaction).
The data listed in Table 1 indicate that, when the reactions were carried out under TH conditions, a selective cyclocondensation reaction occurred, leading to the benzofuran ester derivative type 3a–c, in good yields (74–79%). Under MW irradiation, according to the reaction time employed, the reactions occurred differently. When MW irradiation was performed for a short period of time (3–6 h), a mixture of benzofuran derivatives (3a–c and 4a–c) was obtained in approximately equal proportions, in almost quantitative yields overall (around 95%). When MW irradiation was performed for a longer period of time (12–15 h), only one type of benzofuran derivative (3-methylbenzofurans (4a–c)) was obtained in a similar quantitative yield.
Also, it is worth mentioning that, under MW irradiation, the reaction time decreased substantially (from days to hours), the consumed energy decreased considerably, and the amount of solvent used was at least five times lower (see Section 3) compared with conventional TH. Thus, the cyclocondensation reaction could be considered environmentally friendly.
The structures of all new compounds were proven unambiguously by elemental and spectral analysis (IR, 1H NMR, 13C NMR, two-dimensional experiments 2D-COSY, 2D-HETCOR (HMQC), long-range 2D-HETCOR (HMBC).
2.1. UV-Vis Study
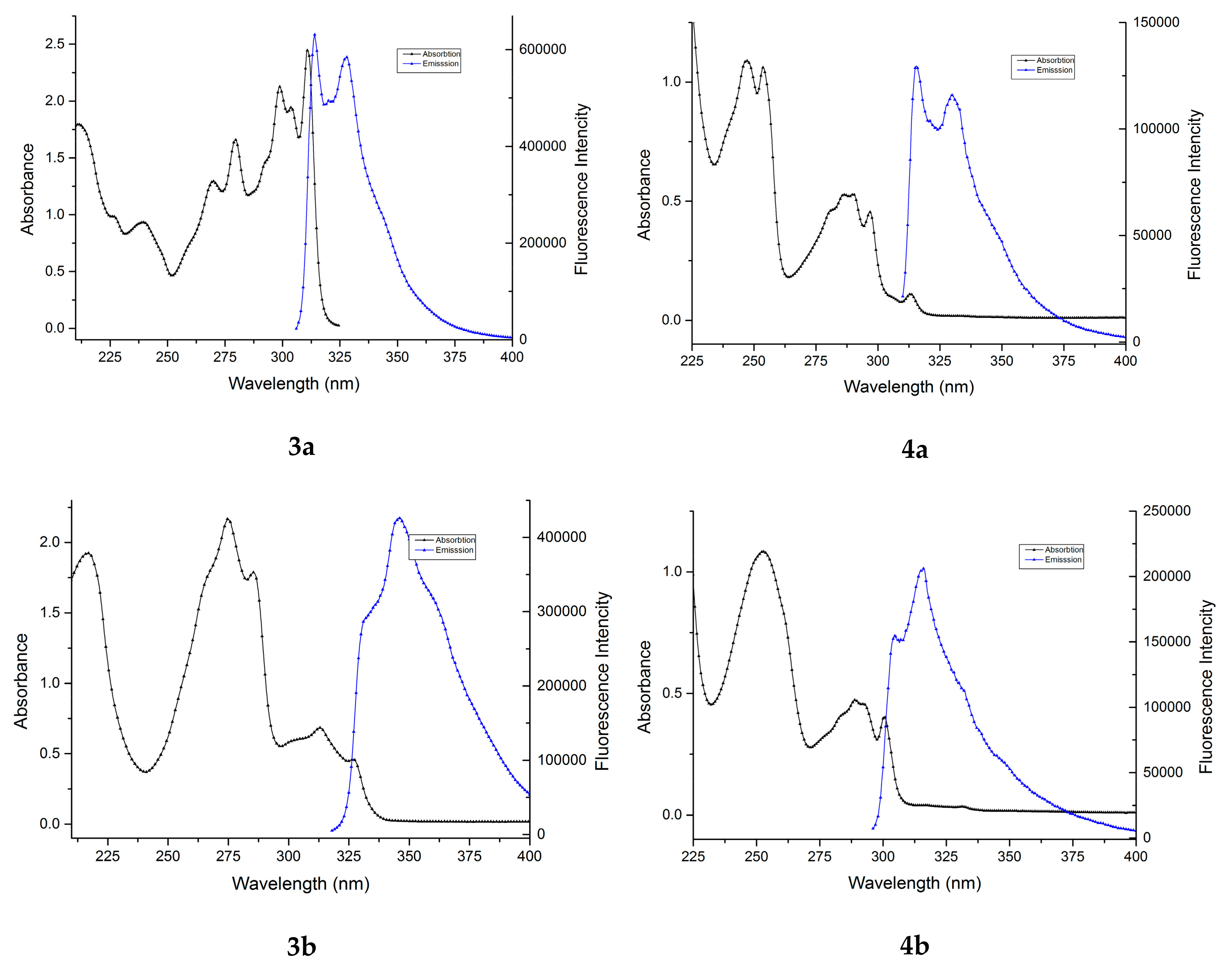
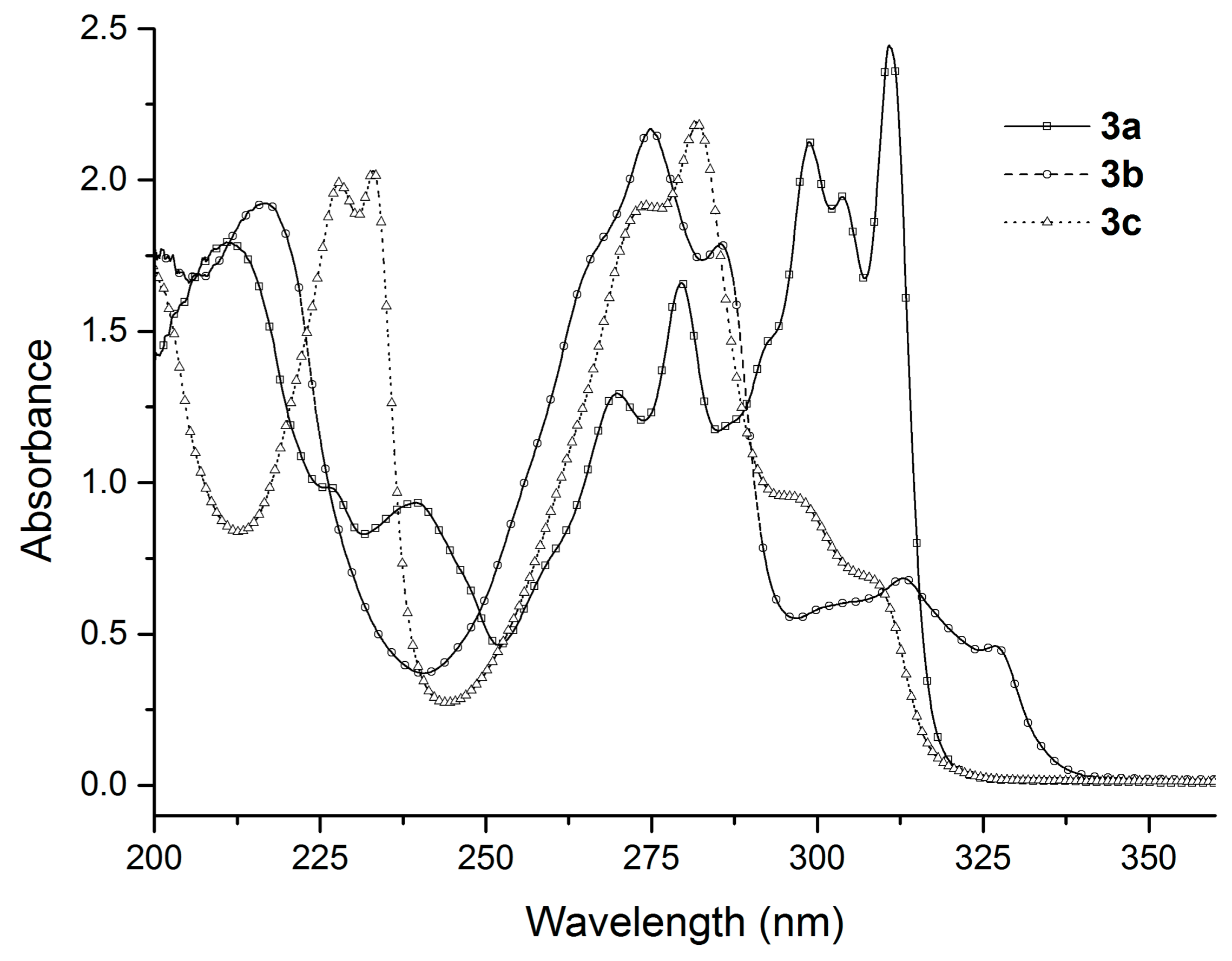
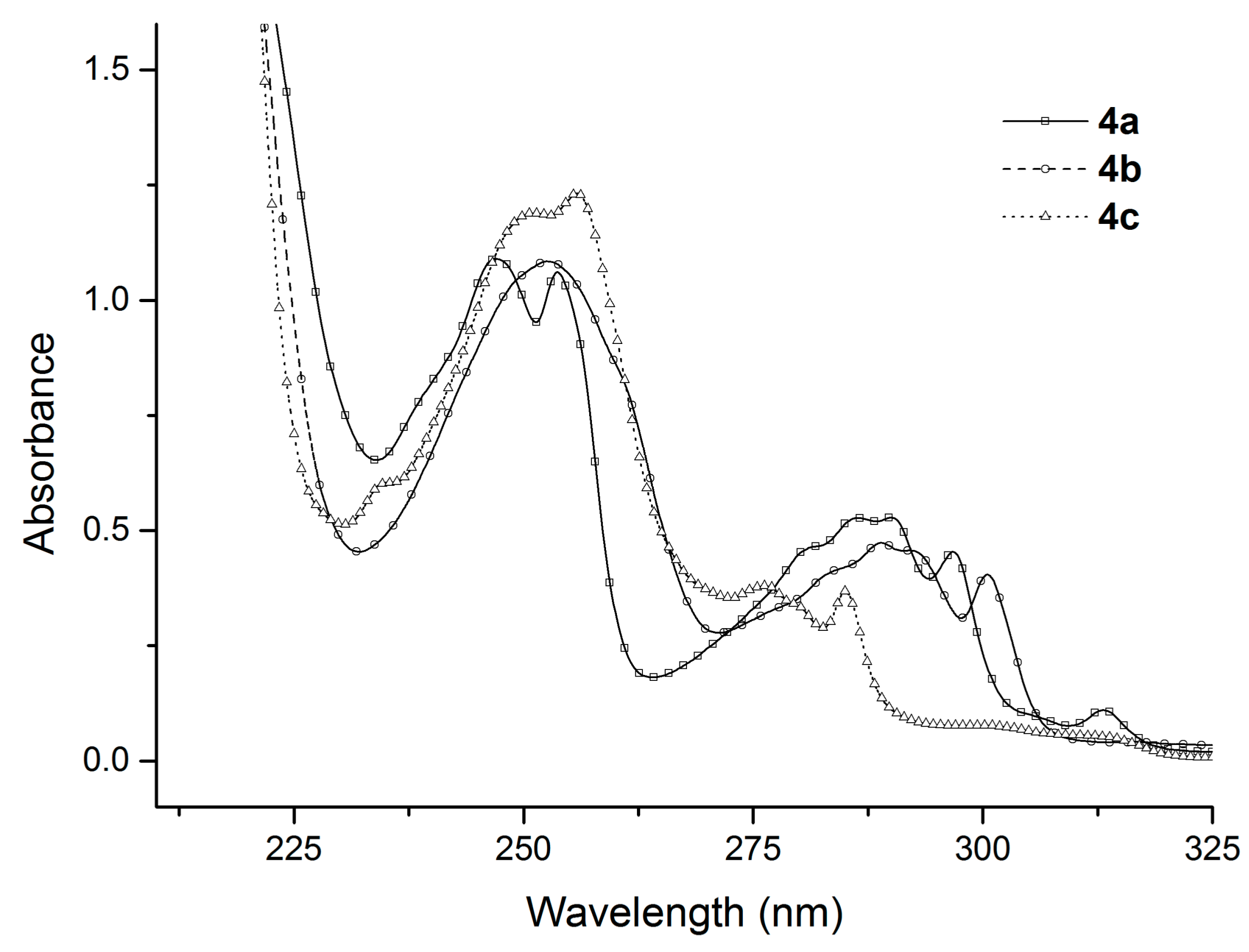
In the next stage of our work, we studied the absorption and emission spectra of the obtained benzofuran derivatives. Although compounds 3a–c and 4a–c have a relatively similar structure, they exhibit clear differences in their experimental UV-Vis absorption spectra, as can be seen from Figure 1 and Figure 2.
The absorption maxima of the six benzofuran derivatives in cyclohexane and the corresponding molar extinction coefficients, ε, are summarized in Table 2.
As we can observe in Table 2 and Figure 1 and Figure 2, the position of the absorption bands and the values of the molar extinction coefficients show the influence of the substituents on the benzofuran ring.
In the case of compounds 4a–c (Figure 2), two well-separated absorption regions are observed (between 230–270 nm and 270–310 nm, respectively). The absorption bands from the first region have a higher (more than double) intensity than the absorption bands from the second region. Also, in the first region the absorption bands have similar intensities and profiles, while in the second region the differences between the absorption bands are higher. The maximum of the absorption of all three compounds 4a–c have close values: 247.0 nm for the compound 4a, 252.4 nm for the compound 4b, and 255.8 nm for the compound 4c.
In the case of compounds 3a–c (Figure 1), the absorption spectral profile is quite different. In this case, two well-separated absorption regions were also observed (between 230–295 nm and 295–335 nm, respectively). For compounds 3b and 3c, as in the previous case, the absorption bands from the first region had a higher intensity (more than double) than the absorption bands from the second region. Regarding the maximum of the absorption of the compounds 3b and 3c, these appear in the first region, but in different bands. Thus, for compound 3b the absorption maximum appeared at 274.8 nm and a secondary band appeared around 285.4 nm, while for compound 3c the situation was reversed: the absorption maximum appeared at 281.8 nm and the secondary band appeared around 274.2 nm. Surprisingly, for compound 3a, the absorption spectrum was totally different. The absorption bands from the first region had a smaller intensity compared to the absorption bands from the second region. Accordingly, a bathochromic shift of the absorption maximum to 310.8 nm was observed.
2.2. Fluorescence Study
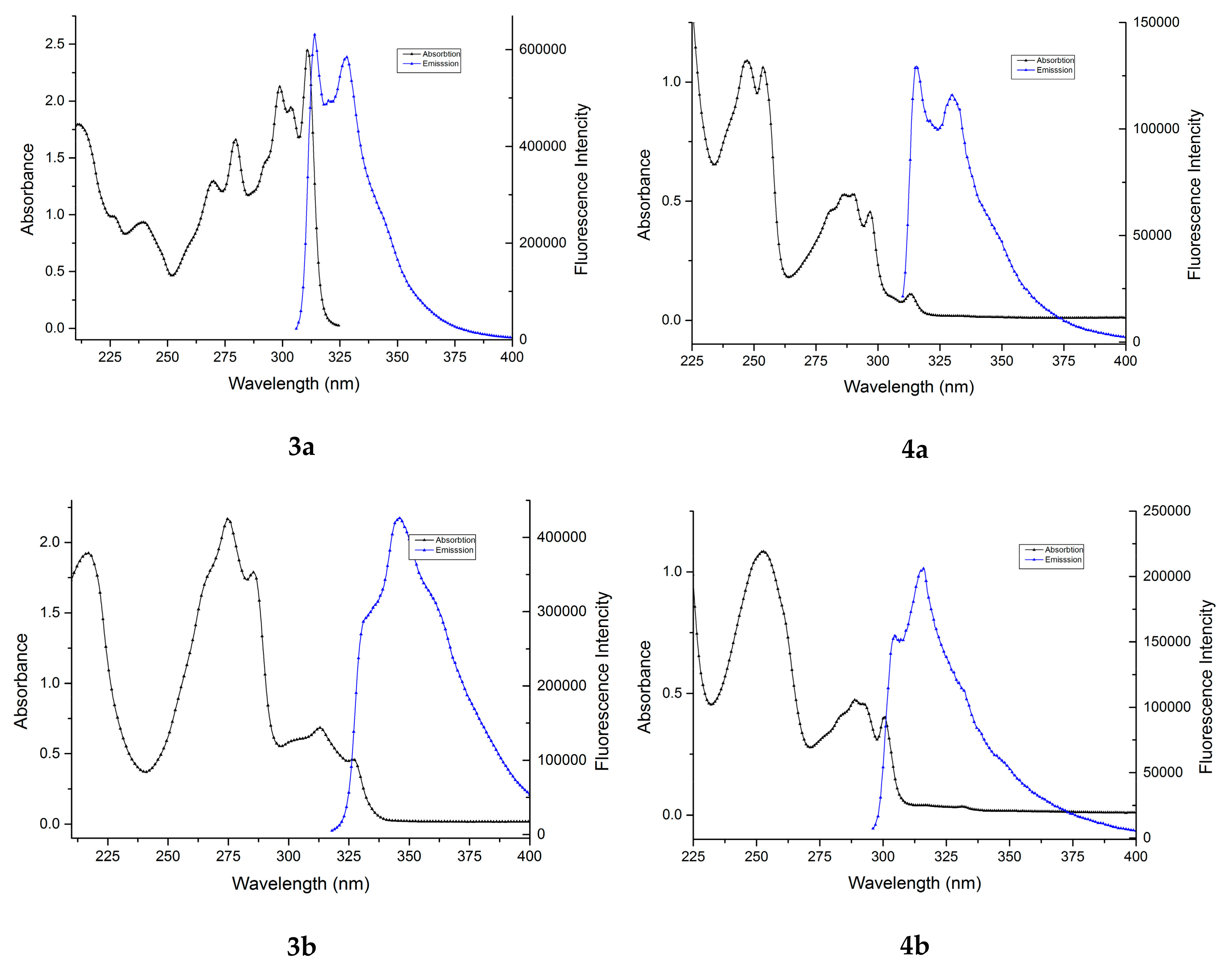
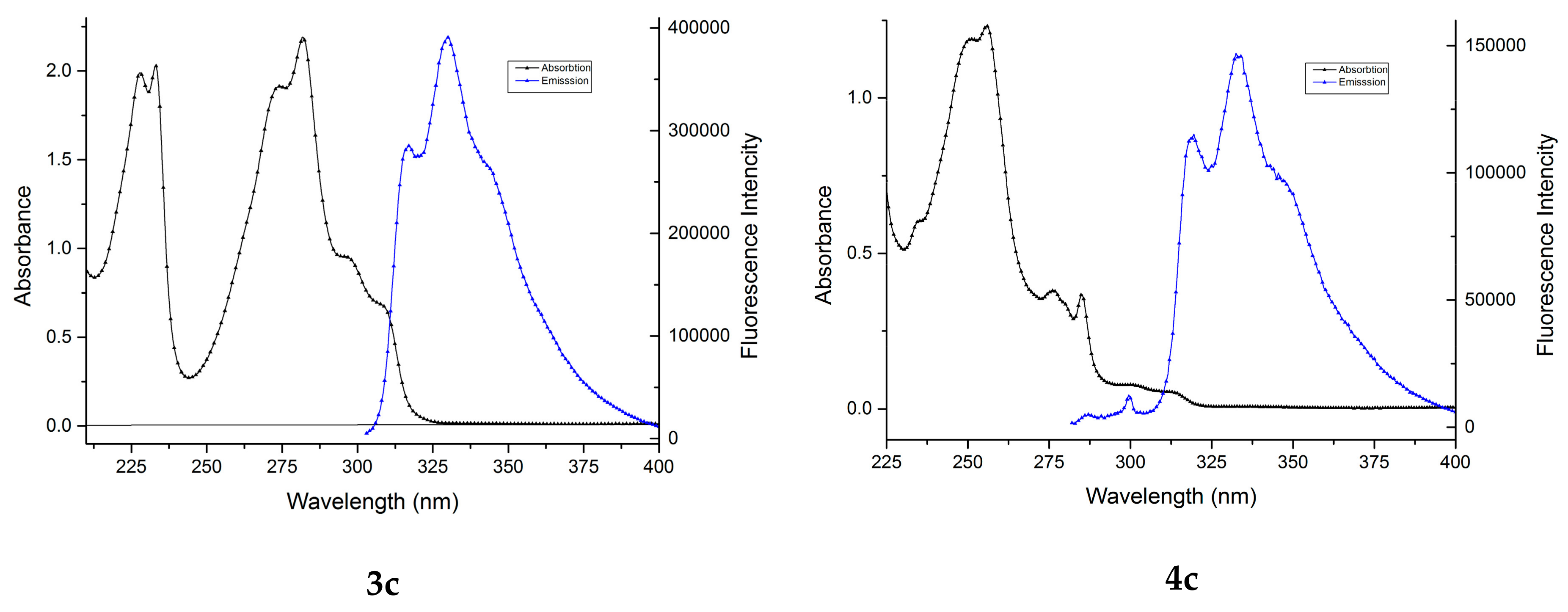
All the studied benzofuran derivatives have emission spectra consisting of one structured band in the 305–370 nm region (Figure 3), indicating a planar structure of the molecules. The position of the band is significantly influenced by the presence of a carbomethoxy group at the 2 position of benzofuran ring. In the first case, a bathochromic shift dropped of Δmax = 3.5 nm (3a compared with 4a and 3c with 4c) and in the second case a hypsochromic shift of Δmax = 29.5 nm (3b compared with 4b) could be observed (Table 3).
The fluorescence quantum yields of benzofuran derivatives are dramatically dependent on the presence of a carbomethoxy group in the furan ring. As seen in Table 3, compounds 3a–c have a high quantum yield (41–55%), while compounds 4a–c have moderate quantum yield values (17–20%). The highest fluorescence absolute quantum yield (54.85%) was observed for compound 3b, thus making it interesting for applications as fluorophores in biolabeling and environmental trace analysis.
Figure 3 presents the absorption and emission spectra of compounds 3a–c and 4a–c in cyclohexane.
3. Experimental Section
3.1. General Procedure
All the reagents and solvents were purchased from commercial sources and used without further purification. Melting points were recorded on a MEL-TEMP II apparatus in open capillary tubes and are uncorrected. Analytical thin-layer chromatography was performed with commercial silica gel plates 60 F254 (Merck Darmstadt, Germany) and visualized with UV light. The NMR spectra were recorded on a (Bruker Vienna, Austria) Avance III 500 MHz or (Bruker Vienna, Austria) Avance DRX 400 MHz spectrometer operating at 500/400 MHz for 1H and 125/100 MHz for 13C. The following abbreviations were used to designate chemical shift multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet. Chemical shifts were reported in delta (δ) units, part per million (ppm) and coupling constants (J) in Hz. Infrared (IR) data were recorded as films on potassium bromide (KBr) pellets on a FT-IR (Shimadzu Kyoto, Japan) Prestige 8400s spectrophotometer. The purity of the compounds was proved by HPLC on a (Thermo Scientific Massachusetts, USA) UHPLC UltiMate 3000 apparatus. For the microwave irradiation we used a monomode reactor Monowave 300 (Anton Paar Graz, Austria). This reactor can reach up to 300 °C and controls the temperature via a built-in IR sensor. Additionally, simultaneous IR and internal temperature monitoring is possible with the so-called Ruby Thermometer. Some additional specifications of Monowave 300: microwave power 850 W, operation limits at 300 °C and 30 bars, reaction vial Borosilicat, operation volume between 0.5–20 mL, pressure control by hydraulic system, agitation with an integrated magnetic stirrer (0 to 1200 rpm) and cooling with compressed air. UV-Vis spectra were recorded on a Shimadzu 1800 PC spectrophotometer in cyclohexane (spectroscopic grade) solution. The fluorescence measurements were made using a (Perkin Elmer Massachusetts, USA) LS55 luminescence spectrometer, in the same solvents as for the UV-Vis spectra, with the excitation wavelength set to the absorption band maximum. For all spectral determinations the solutions were kept in 10 mm path length quartz cells. The fluorescence quantum yield was determined at room temperature with an (Edinburgh Instruments, Edinburgh, UK FLS 980 fluorimeter with an integrating sphere and the excitation wavelength corresponds to the maximum of the absorption band. MS spectra were recorded on a (Shimadzu Kyoto, Japan) GCMS-QP 2010. The instrument was running in chemical ionization (CI) mode. The system was equipped with a 25 m × 0.25 mm × 0.25 μm DB-5ms capillary column. The ion source, quadrupole, and interface temperatures were 330 °C. Helium was used as the carrier gas at constant flow (1.54 mL/min) with an initial pressure of 90.7 kPa, while methane was used as the reagent gas in the mass spectrometer. The electron multiplier voltage was set to 1750 V. Two microliters of diluted solution were injected in cold pulsed splitless mode (initial injector temperature at 250 °C). The temperature of the DB-5ms column was programmed to go from 50 °C, where it stayed for 2 min, to 320 °C at a rate of 15 °C/min and finally stayed for 5 min at 320 °C.
3.1.1. General Procedure for Synthesis of Benzofuran Derivatives 3a–c under Conventional TH Conditions
To a mixture of dihydroyacetophenone derivatives (5 mmol, 0.761 g), potassium carbonate (10 mmol, 1.382 g) and potassium iodide (a spatula tip) in 100 mL acetonitrile, methylchloroacetate were added (10 mmol, 0.88 mL) at room temperature. Under conventional TH, the solution was then refluxed at 165 °C for four days on an oil bath. After the completion of the reaction (TLC), the obtained solution was cooled at room temperature; water was added and then the compounds were extracted with chloroform (3 × 50 mL), washed with a solution of sodium hydroxide (10%), dried and evaporated, with precipitate formation. The purification of the crude product was done by column chromatography on silica gel (eluted with dichloromethane).
3.1.2. General Procedure for Syntheses of Benzofuran Derivatives 3a–c and 4a–c under Microwave Irradiation
Under MW irradiation, the mixture of reagents were dissolved in 40 mL acetonitrile, placed in the reaction vessel (borosilicate), and exposed to microwave irradiation (for 3–6 h; see Table 1). Using MW irradiation, the best results were obtained using a “temperature control” method. The “temperature control” method ensures a constant temperature (in this case 165 °C) and the power and pressure are kept constant. This method takes place in three stages. In the first step the temperature is raised as quickly as possible (within less than 1 min) by applying the maximum power. In the second step the reaction mixture is kept at a constant temperature with the control of the magnetron power. In the last step the reaction tube is cooled to 55 °C by stopping the irradiation and blowing the reaction vial with compressed air. Once the heating cycle is completed, the reaction vial is removed from the reactor, and processed as indicated for TH.
Methyl 6-(2-methoxy-2-oxoethoxy)-3-methylbenzofuran-2-carboxylate (3a). (1.06 g, 76% (under classical heating) and 0.63 g, 45% (under microwaves)) as a yellowish crystals, m.p. 134–135 °C); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.61; IR (cm−1): 3096, 3073, 3030 (C-H arom.), 2955, 2920 (C-H aliph.), 1768, 1718 (C=O, ester), 1618, 1595, 1576, 1443, 1425, 1368 (aromatic and heteroaromatic ring), 1271, 1219, 1169, 1148, 1128, 1097, 1078 (C–O–C, ester); 1H NMR (400 MHz, CDCl3): δ 7.51 (1H, d, J = 8.8 Hz, H-4), 7.00 (2H, m, overlapped peaks, H-5, H-7), 4.69 (2H, CH2 of methyl acetate group from 6 position, s), 3.96 (3H, CH3 of methoxycarbonyl group from 2 position, s), 3.82 (3H, CH3 of methyl acetate group from 6 position, s), 2.55 (3H, CH3 group from 3 position, s); 13C NMR (100 MHz, CDCl3): δ 168.8 (CO keto ester from 6 position), 160.7 (CO keto ester from 2 position), 158.7 (C-7a), 155.3 (C-6), 140.4 (C-2), 126.2 (C-3), 123.3 (C-3a), 121.7 (C-4), 113.4 (C-5), 96.7 (C-7), 65.6 (CH2 of methyl acetate group from 6 position), 52.3 (CH3 of methyl acetate group from 6 position), 51.8 (CH3 of methoxycarbonyl group from 2 position), 9.3 (CH3 group from 3 position); MS (CI, m/z): 280 (M + 2; 7.5%), 279 (M + 1; 45.3%), 278 (M+, base peak, 100%), 247 (M–OCH3; 18.9%), 219 (M–COOCH3; 28.3%), 205 (M–CH2COOCH3; 26.4%).
Methyl 5-(2-methoxy-2-oxoethoxy)-3-methylbenzofuran-2-carboxylate (3b). (1.10 g, 79% (under classical heating) and 0.68 g, 49% (under microwaves)) as a yellowish crystals, m.p. 124–125 °C); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.57; IR (cm−1): 3105, 3084, 3010 (C-H arom.), 2957, 2920 (C-H aliph.), 1759, 1707 (C=O, ester), 1615, 1581, 1473, 1437, 1369 (aromatic and heteroaromatic ring), 1300, 1223, 1207, 1155, 1082 (C–O–C, ester); 1H NMR (400 MHz, CDCl3): δ 7.45 (1H, d, J = 9.2 Hz, H-7), 7.13 (1H, dd, J = 9.2, 2.4 Hz, H-6), 7.01 (1H, d, J = 2.4 Hz, H-4), 4.70 (2H, CH2 of methyl acetate group from 5 position, s), 3.98 (3H, CH3 of methoxycarbonyl group from 2 position, s), 3.83 (3H, CH3 of methyl acetate group from 5 position, s), 2.55 (3H, CH3 group from 3 position, s); 13C NMR (100 MHz, CDCl3): δ 169.5 (CO keto ester from 5 position), 160.9 (CO keto ester from 2 position), 154.6 (C-5), 150.1 (C-7a), 141.8 (C-2), 129.6 (C-3a), 125.9 (C-3), 118.0 (C-6), 113.2 (C-7), 104.4 (C-4), 66.4 (CH2 of methyl acetate group from 5 position), 52.4 (CH3 of methyl acetate group from 5 position), 52.1 (CH3 of methoxycarbonyl group from 2 position), 9.5 (CH3 group from 3 position); MS (CI, m/z): 280 (M + 2; 5.7%), 279 (M + 1; 47.2%), 278 (M+, base peak, 100%), 247 (M–OCH3; 17.0%), 219 (M–COOCH3; 26.4%), 205 (M–CH2COOCH3; 17.0%).
Methyl 4-(2-methoxy-2-oxoethoxy)-3-methylbenzofuran-2-carboxylate (3c). (1.03 g, 74% (under classical heating) and 0.64 g, 46% (under microwaves)) as a yellowish crystals, m.p. 108–109 °C); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.68; IR (cm−1): 3080, 3051, 3011 (C-H arom.), 2957, 2918 (C-H aliph.), 1769, 1707 (C=O, ester), 1611, 1589, 1502, 1435, 1360 (aromatic and heteroaromatic ring), 1277, 1217, 1147, 1111 (C–O–C, ester); 1H NMR (400 MHz, CDCl3): δ 7.30 (1H, t, J = 8.4, 8.0 Hz, H-6), 7.15 (1H, d, J = 8.4 Hz, H-7), 6.50 (1H, d, J = 8.0 Hz, H-5), 4.74 (2H, CH2 of methyl acetate group from 4 position, s), 3.97 (3H, CH3 of methoxycarbonyl group from 2 position, s), 3.82 (3H, CH3 of methyl acetate group from 4 position, s), 2.79 (3H, CH3 group from 3 position, s); 13C NMR (100 MHz, CDCl3): δ 168.9 (CO keto ester from 4 position), 160.9 (CO keto ester from 2 position), 155.8 (C-7a), 154.43 (C-4), 139.9 (C-2), 128.7 (C-6), 127.00 (C-3), 119.2 (C-3a), 106.2 (C-7), 104.1 (C-5), 65.4 (CH2 of methyl acetate group from 4 position), 52.4 (CH3 of methyl acetate group from 4 position), 52.0 (CH3 of methoxycarbonyl group from 2 position), 11.3 (CH3 group from 3 position); MS (CI, m/z): 280 (M + 2; 3.8%), 279 (M + 1; 26.4%), 278 (M+, base peak, 100%), 247 (M–OCH3; 11.3%), 219 (M–COOCH3; 13.2%), 205 (M–CH2COOCH3; 18.9%).
Methyl 2-(3-methylbenzofuran-6-yloxy)acetate (4a). (0.00 g, 0% (under classical heating) and 0.54 g, 49% (under microwaves)) as a yellowish crystals, m.p. 73–74 °C); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.84; IR (cm−1): 3117, 3080, 3009 (C-H arom.), 2951, 2922 (C-H aliph.), 1753 (C=O, ester), 1627, 1591, 1491, 1437, 1394 (aromatic and heteroaromatic ring), 1267, 1219, 1152, 1086, 1057 (C–O–C, ester); 1H NMR (500 MHz, CDCl3): δ 7.39 (1H, d, J = 8.0 Hz, H-4), 7.33 (1H, d, J = 1.0 Hz, H-2), 6.97 (1H, d, J = 2.0 Hz, H-7), 6.92 (1H, dd, J = 8.0, 2.0 Hz, H-5), 4.67 (2H, CH2 of methyl acetate group from 6 position, s), 3.81 (3H, CH3 of methyl acetate group from 6 position, s), 2.20 (3H, CH3 group from 3 position, d, J = 1.0 Hz); 13C NMR (125 MHz, CDCl3): δ 169.5 (CO keto ester from 6 position), 156.1 (C-7a), 155.9 (C-6), 141.0 (C-2), 123.6 (C-3), 119.8 (C-4), 115.6 (C-3a), 111.6 (C-5), 97.4 (C-7), 66.1 (CH2 of methyl acetate group from 6 position), 52.3 (CH3 of methyl acetate group from 6 position), 7.9 (CH3 group from 3 position); MS (CI, m/z): 221 (M+1; 13.7%), 220 (M+, base peak, 100%), 161 (M–COOCH3; 11.8%), 147 (M–CH2COOCH3; 47.1%).
Methyl 2-(3-methylbenzofuran-5-yloxy)acetate (4b). (0.00 g, 0% (under classical heating) and 0.51 g, 46% (under microwaves)) as a yellowish liquid); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.82; IR (cm−1): 3117, 3073, 3010 (C-H arom.), 2957, 2926 (C-H aliph.), 1751 (C=O, ester), 1604, 1479, 1452, 1440 (aromatic and heteroaromatic ring), 1319, 1287, 1263, 1207, 1180, 1089 (C–O–C, ester); 1H NMR (500 MHz, CDCl3): δ 7.38 (1H, d, J = 1.5 Hz, H-2), 7.35 (1H, d, J = 9.0 Hz, H-7), 6.98 (1H, d, J = 2.5 Hz, H-4), 6.94 (1H, dd, J = 9.0, 2.5 Hz, H-6), 4.69 (2H, CH2 of methyl acetate group from 5 position, s), 3.82 (3H, CH3 of methyl acetate group from 5 position, s), 2.20 (3H, CH3 group from 3 position, d, J = 1.5 Hz); 13C NMR (125 MHz, CDCl3): δ 169.8 (CO keto ester from 5 position), 154.1 (C-5), 150.9 (C-7a), 142.7 (C-2), 129.7 (C-3a), 115.8 (C-3), 113.3 (C-6), 112.0 (C-7), 103.9 (C-4), 66.6 (CH2 of methyl acetate group from 5 position), 52.3 (CH3 of methyl acetate group from 5 position), 8.0 (CH3 group from 3 position); MS (CI, m/z): 221 (M + 1; 13.7%), 220 (M+, base peak, 100%), 161 (M–COOCH3; 19.6%), 147 (M–CH2COOCH3; 35.3%).
Methyl 2-(3-methylbenzofuran-4-yloxy)acetate (4c). (0.00 g, 0% (under classical heating) and 0.50 g, 45% (under microwaves)) as a yellowish liquid); Rf (99.9/0.1 CH2Cl2/CH3OH) 0.88; IR (cm−1): 3117, 3078, 3036 (C-H arom.), 2955, 2930 (C-H aliph.), 1763 (C=O, ester), 1609, 1495, 1437, 1360 (aromatic and heteroaromatic ring), 1292, 1259, 1213, 1171, 1115, 1092 (C–O–C, ester); 1H NMR (500 MHz, CDCl3): δ 7.24 (1H, s, H-2), 7.09 (2H, m, overlapped peaks, H-6, H-7), 6.43 (1H, d, J = 7.0 Hz, H-5), 4.66 (2H, CH2 of methyl acetate group from 4 position, s), 3.76 (3H, CH3 of methyl acetate group from 4 position, s), 2.39 (3H, CH3 group from 3 position, s); 13C NMR (125 MHz, CDCl3): δ 169.2 (CO keto ester from 4 position), 156.9 (C-7a), 153.2 (C-4), 140.3 (C-2), 124.6 (C-6), 118.7 (C-3), 115.9 (C-3a), 105.6 (C-7), 103.7 (C-5), 65.4 (CH2 of methyl acetate group from 4 position), 52.1 (CH3 of methyl acetate group from 4 position), 9.8 (CH3 group from 3 position); MS (CI, m/z): 221 (M + 1; 13.7%), 220 (M+, base peak, 100%), 185 (M–OCH3; 9.8%),161 (M–COOCH3; 35.3%), 147 (M–CH2COOCH3; 58.8%).
4. Conclusions
In conclusion, we report herein an efficient and straightforward pathway for obtaining a new class of blue fluorescent bezofuran derivatives, under microwave irradiation as well as under conventional thermal heating. Under conventional TH the reactions occur selectively, and a single type of benzofuran ester derivatives (3a–c) is obtained. Under MW irradiation, the reactions occur in different manners. When MW irradiation was performed for a short period of time, a mixture of benzofuran derivatives (3a–c and 4a–c) were obtained, while when MW irradiation was performed for a longer period of time, the reaction became highly selective, and only one type of benzofuran derivative corresponding to 3-methylbenzofuran (4a–c) was obtained. Taking into consideration the advantages offered by MW irradiation in terms of yield, easy workup of the reaction, substantial decrease in solvent consumed (several-fold less compared with conventional TH), substantial reduction in reaction time (from days to hours) and consequent diminution in energy consumption, these methods could be considered environmentally friendly. Feasible reaction mechanisms for the benzofuran derivatives formation are described fully in this study and are in agreement with the literature. The absorption and fluorescence emission of the obtained benzofuran derivatives were studied, with part of these compounds being intense blue emitters. The fluorescence quantum yields of benzofuran derivatives are dramatically dependent on their structure, with only compounds anchored by a carbomethoxy group of furan ring having good quantum yields.
Supplementary Materials
Details of the IR spectra, NMR spectra (1H NMR and 13C NMR) and HPLC chromatograms of the synthesized compounds can be found in the Supporting Information. Supplementary material is available online at https://www.mdpi.com/1420-3049/23/8/1968/s1, on the publisher’s web site.
Author Contributions
Design, synthesis, experimental work, and writing were performed by I.M., G.Z. and C.M. The evaluation of fluorescent properties was performed by D.L.I., A.A., G.Z. and C.M., I.M., G.Z. and C.M. did the revision of the first draft of paper. All authors reviewed and approved the final version.
Funding
This research was funded by UEFISCDI Bucharest, Romania grant number PN-III-P1-1.1-TE-2016-1205.
Acknowledgments
The authors are grateful to UEFISCDI Bucharest, Romania, project PN-III-P1-1.1-TE-2016-1205 for financial support and the POSCCE-O 2.2.1, SMIS-CSNR 13984-901, No. 257/28.09.2010 Project, CERNESIM for NMR experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bodke, Y.D.; Shankerrao, S.; Harishkumar, H.N. Synthesis of 2-(1-Benzofuran-2-yl)-4-(1,3-benzoxazol-2-yl/1,3-benzothiazol-2-yl) Quinolines as Blue Green Fluorescent Probes. J. Chem. 2013, 2013, 794810. [Google Scholar] [CrossRef]
- Berlman, B. Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, NY, USA, 1971; ISBN 9780323161671. [Google Scholar]
- Anderson, S.; Taylor, P.N.; Verschoor, G.L.B. Benzofuran Trimers for Organic Electroluminescence. Eur. J. Chem. 2004, 10, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Hwu, J.R.; Chuang, K.S.; Chuang, S.H.; Tsay, S.C. New benzo[b]furans as electroluminescent materials for emitting blue light. Org. Lett. 2005, 7, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Kandaz, M.; Güney, O.; Senkal, F.B. Fluorescent chemosensor for Ag (I) based on amplified fluorescence quenching of a new phthalocyanine bearing derivative of benzofuran. Polyhedron 2009, 28, 3110–3114. [Google Scholar] [CrossRef]
- Oter, O.; Ertekin, K.; Kirilmis, C.; Koca, M.; Ahmedzade, M. Characterization of a newly synthesized fluorescent benzofuran derivative and usage as a selective fiber optic sensor for Fe(III). Sens. Actuators B Chem. 2007, 122, 450–456. [Google Scholar] [CrossRef]
- Leung, M.K.; Chang, C.C.; Wu, M.H.; Chuang, K.H.; Lee, J.H.; Shieh, S.J.; Lin, S.C.; Chiu, C.F. 6-N,N-diphenylaminobenzofuran-derived pyran containing fluorescent dyes: A new class of high-brightness red-light-emitting dopants for OLED. Org. Lett. 2006, 8, 2623–2626. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanemura, K.; Horaguchi, T.; Shimizu, T.; Sakakibara, T. Benzofuran derivatives. Part 4, synthesis of benzofurans and 2,3,4,5-tetrahydro-1-benzoxepin-3,5-diones. J. Het. Chem. 1992, 29, 423–429. [Google Scholar] [CrossRef]
- Podea, P.V.; Tosca, M.I.; Paizs, C.; Irimie, F.D. Chemoenzymatic preparation of enantiopure l-benzofuranyl- and l-benzo[b]thiophenyl alanines. Tetrahedron Asymmetry 2008, 19, 500–511. [Google Scholar] [CrossRef]
- Paul, N.M.; Taylor, M.; Kumar, R.; Deschamps, J.R.; Luedtke, R.R.; Newman, A.H. Structure-Activity Relationships for a Novel Series of Dopamine D2-like Receptor Ligands Based on N-Substituted 3-Aryl-8-azabicyclo[3.2.1]octan-3-ol. J. Med. Chem. 2008, 51, 6095–6109. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, T.; Sato, S.; Shibuya, M.; Kanoh, N.; Taniguchi, T.; Monde, K.; Iwabuchi, Y. Enantioselective Intramolecular Aza-Spiroannulation onto Benzofurans Using Chiral Rhodium Catalysis. Heterocycles 2014, 89, 631–639. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium π-Olefin and π-Alkyne Chemistry. Chem. Rev. 2004, 104, 2285–2310. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Yokoyama, T.; Inamura, K.; Katakura, S.; Komoriya, S.; Yamaguchi, H.; Hara, T.; Iwamoto, M. Dibasic (Amidinoaryl)propanoic Acid Derivatives as Novel Blood Coagulation Factor Xa Inhibitors. J. Med. Chem. 1994, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, F.; Jiang, X.; Zhang, W.; Liu, J.; Liu, W.; Fu, L. Synthesis and antimicrobial evaluation of 3-methanone-6-substituted-benzofuran derivatives. Eur. J. Med. Chem. 2012, 54, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, J.; Kumar, H.V.; Harini, S.T.; Naik, N. Synthesis of benzofuran based 1,3,5-substituted pyrazole derivatives: As a new class of potent antioxidants and antimicrobials--a novel accost to amend biocompatibility. Bioorg. Med. Chem. Lett. 2012, 22, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Singh, P.; Tewari, A.K. Design, synthesis, docking and anti-inflammatory evaluation of novel series of benzofuran based prodrugs. Bioorg. Med. Chem. Lett. 2014, 24, 2251–2255. [Google Scholar] [CrossRef] [PubMed]
- Telvekar, V.N.; Belubbi, A.; Bairwa, V.K.; Satardekar, K. Novel N’-benzylidene benzofuran-3-carbohydrazide derivatives as antitubercular and antifungal agents. Bioorg. Med. Chem. Lett. 2012, 22, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Filzen, G.F.; Bratton, L.; Cheng, X.M.; Erasga, N.; Geyer, A.; Lee, C.; Lu, G.; Pulaski, J.; Sorenson, R.J.; Unangst, P.C.; et al. Synthesis and SAR of selective benzothiophene, benzofuran, and indole-based peroxisome proliferator-activated receptor delta agonists. Bioorg. Med. Chem. Lett. 2007, 13, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Yang, X.D.; Zeng, X.H.; Xu, X.L.; Zhang, G.L.; Zhang, H.B. Synthesis and cytotoxic activities of novel hybrid compounds of imidazole scaffold-based 2-substituted benzofurans. RSC Adv. 2012, 2, 4612–4615. [Google Scholar] [CrossRef]
- Habermann, J.; Ley, S.V.; Smits, R. Three-step synthesis of an array of substituted benzofurans using polymer-supported reagents. J. Chem. Soc. Perkin Trans. 1999, 1, 2421–2423. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. Nonthermal Effects of Microwaves in Organic Synthesis. In Microwaves in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 134–218. ISBN 9783527314522. [Google Scholar]
- Rodriquez, M.; Taddei, M. Synthesis of Heterocycles via Microwave-Assisted Cycloadditions and Cyclocondensations. In Microwave-Assisted Synthesis of Heterocycle; Van der Eycken, E., Kappe, C.O., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; pp. 213–266. ISBN 9783540309833. [Google Scholar]
- Perreux, L.; Loupy, A.; Petit, A. Nonthermal Effects of Microwaves in Organic Synthesis. In Microwaves in Organic Synthesis, 3rd ed.; de la Hoz, A., Loupy, A., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 127–207. ISBN 9783527331161. [Google Scholar]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbancioc, G.; Zbancioc, A.M.; Mangalagiu, I. Ultrasound and microwave assisted synthesis of dihydroxyacetophenone derivatives with or without 1,2-diazine skeleton. Ultrason. Sonochem. 2014, 21, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Zbancioc, A.M.; Miron, A.; Tuchilus, C.; Rotinberg, P.; Mihai, C.T.; Mangalagiu, I.; Zbancioc, G. Synthesis and in vitro analysis of novel dihydroxyacetophenone derivatives with antimicrobial and antitumor activities. Med. Chem. 2014, 10, 476–483. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
Scheme 1.
O-alkylation reactions of 2,n(4,5,6)-dihydroxyacetophenone derivatives.

Scheme 2.
Reaction pathway for the preparation of benzofuran derivatives under thermal heating conditions.
Scheme 2.
Reaction pathway for the preparation of benzofuran derivatives under thermal heating conditions.
Scheme 3.
Reaction pathway for benzofuran derivatives preparation under MW irradiation.

Figure 1.
The absorption spectra of the benzofuran derivatives 3a–c recorded in 10−4 M cyclohexane solution.
Figure 1.
The absorption spectra of the benzofuran derivatives 3a–c recorded in 10−4 M cyclohexane solution.

Figure 2.
The absorption spectra of the benzofuran derivatives 4a–c recorded in 10−4 M cyclohexane solution.
Figure 2.
The absorption spectra of the benzofuran derivatives 4a–c recorded in 10−4 M cyclohexane solution.

Figure 3.
The absorption and emission spectra of benzofuran derivatives 3a–c and 4a–c in cyclohexane. Excitation wavelengths were: 300 nm for 3a, 312 nm for 3b, 297 nm for 3c, 304 nm for 4a, 290 nm for 4b, and 276 nm for 4c.
Figure 3.
The absorption and emission spectra of benzofuran derivatives 3a–c and 4a–c in cyclohexane. Excitation wavelengths were: 300 nm for 3a, 312 nm for 3b, 297 nm for 3c, 304 nm for 4a, 290 nm for 4b, and 276 nm for 4c.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The optimized conditions for synthesis of benzofuran derivatives (3a–c or/and 4a–c), under MW and TH conditions.
Table 1.
The optimized conditions for synthesis of benzofuran derivatives (3a–c or/and 4a–c), under MW and TH conditions.
| Compound | Microwaves | Conventional TH | ||||
|---|---|---|---|---|---|---|
| Reaction Time, (Hours) | Temperature, °C | Yield, % | Reaction Time, (Hours) | Temperature, °C | Yield, % | |
| 3a | 5 | 165 | 45 | 96 | 165 | 76 |
| 3b | 6 | 165 | 49 | 96 | 165 | 79 |
| 3c | 3 | 165 | 46 | 96 | 165 | 74 |
| 4a | 5 | 165 | 49 | 96 | 165 | 0 |
| 13 | 88 | |||||
| 4b | 6 | 165 | 46 | 96 | 165 | 0 |
| 15 | 85 | |||||
| 4c | 3 | 165 | 45 | 96 | 165 | 0 |
| 12 | 80 | |||||
Table 2.
Absorption (λabs and ε) for the benzofuran derivatives 3a–c and 4a–c in cyclohexane.
| Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 4a | 4b | 4c | ||||||
| λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 |
| 211.6 | 17.93 | 216.6 | 19.23 | 227.8 | 19.90 | 247.0 | 10.89 | 252.4 | 10.84 | 235.2 | 6.04 |
| 226.0 | 9.84 | 274.8 | 21.66 | 233.0 | 20.31 | 252.8 | 10.27 | 288.8 | 4.73 | 251.2 | 11.89 |
| 239.4 | 9.33 | 285.4 | 17.87 | 274.2 | 19.15 | 286.4 | 5.27 | 292.0 | 4.57 | 255.8 | 12.32 |
| 269.8 | 12.95 | 313.0 | 6.83 | 281.8 | 21.91 | 290.0 | 5.29 | 300.4 | 4.04 | 276.2 | 3.80 |
| 279.4 | 16.60 | 326.8 | 4.60 | 295.6 | 9.55 | 296.6 | 4.54 | 285.0 | 3.68 | ||
| 298.8 | 21.26 | 313.0 | 1.10 | ||||||||
| 303.8 | 19.44 | ||||||||||
| 310.8 | 24.46 | ||||||||||
λabs (nm); ε × 103 (L mol−1 cm−1).
Table 3.
Fluorescence parameters: emission maximum wavelength, λmem and fluorescence quantum yield, of compounds 3a–c and 4a–c in cyclohexane.
Table 3.
Fluorescence parameters: emission maximum wavelength, λmem and fluorescence quantum yield, of compounds 3a–c and 4a–c in cyclohexane.
| Compound | λmem (nm) | Quantum Yields (%) |
|---|---|---|
| 3a | 314.0 | 49.61 |
| 3b | 345.5 | 54.85 |
| 3c | 330.0 | 41.58 |
| 4a | 315.5 | 18.98 |
| 4b | 316.0 | 20.22 |
| 4c | 333.5 | 17.18 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Moldoveanu, C.; Mangalagiu, I.; Isac, D.L.; Airinei, A.; Zbancioc, G. A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules 2018, 23, 1968. https://doi.org/10.3390/molecules23081968
AMA Style
Moldoveanu C, Mangalagiu I, Isac DL, Airinei A, Zbancioc G. A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules. 2018; 23(8):1968. https://doi.org/10.3390/molecules23081968
Chicago/Turabian StyleMoldoveanu, Costel, Ionel Mangalagiu, Dragos Lucian Isac, Anton Airinei, and Gheorghita Zbancioc. 2018. "A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives" Molecules 23, no. 8: 1968. https://doi.org/10.3390/molecules23081968