Theoretical Study of the C2H5 + HO2 Reaction: Mechanism and Kinetics


1
Key Laboratory of Integrated Exploitation of Bayan Obo Multi-Metal Resources, Inner Mongolia University of Science &Technology, Baotou 014010, China
2
School of Science, Inner Mongolia University of Science and Technology, Baotou 014010, China
3
College of Physics, Jilin University, Changchun 130012, China
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(8), 1919; https://doi.org/10.3390/molecules23081919
Submission received: 24 May 2018
/
Revised: 26 July 2018
/
Accepted: 27 July 2018
/
Published: 1 August 2018
(This article belongs to the Special Issue Theoretical Investigations of Reaction Mechanisms)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The mechanism and kinetics for the reaction of the HO2 radical with the ethyl (C2H5) radical have been investigated theoretically. The electronic structure information of the potential energy surface (PES) is obtained at the MP2/6-311++G(d,p) level of theory, and the single-point energies are refined by the CCSD(T)/6-311+G(3df,2p) level of theory. The kinetics of the reaction with multiple channels have been studied by applying variational transition-state theory (VTST) and Rice–Ramsperger–Kassel–Marcus (RRKM) theory over wide temperature and pressure ranges (T = 220–3000 K; P = 1 × 10−4–100 bar). The calculated results show that the HO2 radical can attack C2H5 via a barrierless addition mechanism to form the energy-rich intermediate IM1 C2H5OOH (68.7 kcal/mol) on the singlet PES. The collisional stabilization intermediate IM1 is the predominant product of the reaction at high pressures and low temperatures, while the bimolecular product P1 C2H5O + OH becomes the primary product at lower pressures or higher temperatures. At the experimentally measured temperature 293 K and in the whole pressure range, the reaction yields P1 as major product, which is in good agreement with experiment results, and the branching ratios are predicted to change from 0.96 at 1 × 10−4 bar to 0.66 at 100 bar. Moreover, the direct H-abstraction product P16 C2H6 + 3O2 on the triplet PES is the secondary feasible product with a yield of 0.04 at the collisional limit of 293 K. The present results will be useful to gain deeper insight into the understanding of the kinetics of the C2H5 + HO2 reaction under atmospheric and practical combustion conditions.
1. Introduction
With low molecular weight, small polarity, and high volatility, alkanes can easily enter the atmosphere and are one of the main components of urban atmospheric pollutants. They are mainly released into the atmosphere through abstraction, distillation, refining, and combustion of fossil fuel, as well as combustion and natural decomposition of organics. Alkanes mainly make oxidization reactions with free radical –OH to produce an active alkane radical (R∙). R∙ is also the initial product of pyrolysis, oxidization, combustion, or a photochemical reaction of saturated hydrocarbons. Besides, it can make a quick addition reaction with O2, an important step to generate an alkane peroxy radical (RO2∙). RO2∙, the simplest organic peroxy radical, is an important intermediate of combustion and oxidization of hydrocarbons. It is the key to low temperature combustion, flame propagation, and spontaneous combustion of fuels. Considering the actual application values of R∙ in combustion, atmospheric chemistry, and biological process [1,2,3,4,5,6,7,8], many experimental and theoretical research studies on their reactions with H, O and O2, as well as their own reactions have been reported. It is common knowledge that the hydrogen peroxide radical (HOO∙) is an important transient intermediate of hydrocarbon fuel combustion, atmospheric photolysis circulation, and biochemical process, and has a considerable high concentration in the troposphere [9]. Hence, R∙+HOO∙ reactions play an important role in the combustion chemistry of hydrocarbons and degradation of alkanes. Additionally, they will affect the balance of R∙ + O2 ↔ RO2∙significantly and generate a potential deep chain source.
The ethyl radical C2H5 has the simplest structure and could show many oxidation characteristics of big R∙, including those generated by olefins [2]. Therefore, studying the C2H5 + HOO reaction is important to establish a model of the R∙+ HOO∙ reaction.
The bimolecular C2H5 + HOO reaction can occur indirectly by generating an intermediate with rich energy:
or through hydrogen abstraction:
where the * represents internal excitation.
C2H5 + HOO→CH3CH2OOH * (+M)
C2H5 + HOO→CH3CH2O + OH
C2H5 + HOO→CH3CHO + H2O
C2H5 + HOO→CH3CH2OOH
C2H5 + HOO→CH3CH3 + 3O2
C2H5 + HOO→CH3CH3 + 1O2
C2H5 + HOO→CH2=CH2 + H2O2
Three experimental groups have studied the C2H5 + HOO reaction. Tsang and Hampson [10] first studied the kinetics of the C2H5 + HOO reaction over the temperature range 300–2500 K. They suggested that the generation channel of C2H5O + OH is the most feasible reaction channel, and estimated the reaction rate constant of this channel k1a = 4.98 × 10−11 cm3·molecule−1·s−1. They also proposed another two products: C2H4 + H2O2 and C2H6 + O2, whose reaction rate constants were estimated as k1f = k1e = 5.00 × 10−13 cm3·molecule−1·s−1. In 1993, Dobis and Benson discovered that the reaction rate constant of C2H4 + H2O2 is k1f = 2.97 × 10−12 cm3·molecule−1·s−1 over the temperature range of 243–368 K [11], but they did not detect the expected major product C2H5O + OH. Recently, Temps and his partners studied the C2H5 + HOO reaction by combing time-dependent mass spectrum and laser photolysis/recirculation reactor [3]. They found that the overall reaction rate constant at 293 K is 5.15 (±1.66) × 10−11 cm3·molecule−1·s−1. They detected C2H5O and pointed out that the generation channel of C2H5O + OH is the major reaction channel. However, no clear experimental result on the mechanism of this complicated reaction with multiple potential wells and multiple channels, pressure and temperature dependence of its products within a wider measuring range, as well as product distribution has been reached yet. As far as we know, there has been no theoretical research on this reaction. Considering the significance of the C2H5 + HOO reaction in combustion and atmospheric chemistry, it is necessary to make a comprehensive theoretical study on its mechanism and kinetics. In this paper, potential energy surfaces (PES) of the C2H5 + HOO reaction were explored through quantum chemistry and its kinetics were computed using the Rice–Ramsperger–Kassel–Marcus (RRKM) unimolecular reaction rate theory of microcanonical ensemble [12]. Data of potential energy surfaces are theoretical data. Variations of reaction rate constants and branching ratios of many dissociation products and intermediates with temperature and pressure were discussed.
2. Calculation Methods
The geometries of all of the reactants, products, intermediates, and transition states involved in the C2H5 + HO2 reaction were optimized at the second-order Møller-Plesset perturbation MP2 [13] method in conjunction with the 6-311++G(d,p) basis set. Frequency analysis was performed at the same level to check whether the obtained species is a local minima (with all real frequencies) or a transition state (with only one imaginary frequency). The minimum reaction path (MEP) was calculated by intrinsic reaction coordinate (IRC) [14,15,16,17] to confirm that the transition states connect the designated intermediates. To obtain more reliable energetics, the single-point energies were refined at the CCSD(T)/6-311+G(3df,2p) level, which has been proved to provide accurate energies in most cases. Unless noted, the CCSD(T) energies with inclusion of MP2 zero-point vibrational energy (ZPE) are used throughout. All calculations were carried out using the Gaussian 03 program packages [18].
According to the variational transition-state theory (VTST) and microcanonical RRKM [19] theory, the kinetic calculations for this multi-channel and multi-well reaction were carried out using the Multiwell 2011 program [20,21] on the basis of the PES obtained above in order to obtain the rate constants and the branching ratios for the key product channels.
3. Results and Discussion
3.1. Potential Energy Surface and Reaction Mechanism
The optimized geometries of the reactants, products, intermediates, and transition states for the C2H5 + HO2 reaction are shown in Figure 1a,b along with the available experimental data from the literature. It is seen that when comparison is available, the agreement between theoretical and experimental results is good, with the largest discrepancy within a factor of 2.0%. The schematic profiles of the singlet and triplet potential energy surfaces of the title reaction are depicted in Figure 2a,b. The total energy of the reactant R (C2H5 + HO2) is set to be zero for reference, and the values in parentheses are relative energies in kcal/mol with reference to R.
3.1.1. Association and Decomposition Channels
The reaction of HO2 with the C2H5 radical may proceed barrierlessly via the addition of the O-atom of HO2 to the C-atom of C2H5, leading to the energy-rich entrance intermediate IM1 C2H5OOH (−68.7 kcal/mol) (see Figure 2a). The process is a characteristic feature of the typical radical–radical reaction mechanism. This process makes the intermediate IM1 highly activated so that further isomerization or dissociation from it is possible.
Firstly, IM1 can decompose by direct bond-breaking to form different products P1 C2H5O + OH (−28.4 kcal/mol), P10 CH2CH2OOH + H (30.5 kcal/mol) and P11 CH3CHOOH + H (25.1 kcal/mol) with the breaking bonds of O–O, Cα–H and Cβ–H, respectively. These processes occur via loose, variational transition states without any barriers. Since P10 and P11 have much less thermodynamic stability, the pathways of P10 and P11 formation are energetically unfeasible and could be neglected in the kinetic calculations. Product P1 may have taken place in a secondary dissociation reaction leading to P7 CH2CHOH + H2O (−117.4 kcal/mol) via a H-abstraction transition state TS13 (−18.3 kcal/mol), P8 CH3 + CH2O + OH (−18.9 kcal/mol) via direct C–C bond rupture, and P9 H + CH3CHO + OH (−14.8 kcal/mol) via direct H-extrusion. However, due to much higher energies and more steps required in the formations of P7, P8, and P9, these three secondary reaction channels do not need to be considered in the kinetic calculations.
Secondly, starting from IM1, six kinds of other fragmentation pathways through tight transition states are also identified: (1) 1,3 H2O-elimination through a four-membered ring transition state TS1 (−23.6 kcal/mol) to form P2 CH3CHO + H2O (−129.2 kcal/mol), (2) 1,3-OH migration through transition state TS8 (5.9 kcal/mol) to yield P5 CH3OH + CH2O (−107.1 kcal/mol), (3) concerted 1,3 H-shift and OH-extrusion through four-membered ring transition state TS2 (−11.4 kcal/mol) to produce P12 CH2CH2OH + OH (−29.7 kcal/mol), (4) 1,3 H2-elimination to yield P13 CH3CHOO + H2 (−19.8 kcal/mol) via a five-membered ring transition state TS3 (−3.6 kcal/mol), (5) concerted 1,4-H-shift and C–C bond rupture process leading to P14 CH4 + CH2OO (−23.5 kcal/mol) via TS4 (4.0 kcal/mol), and (6) concerted 1,4 H-migration and C-O bond fission process to yield P15 C2H4 + H2OO (−3.5 kcal/mol) via TS5 (−4.5 kcal/mol). It is easily seen that among them, the most feasible channel is the formation of P2, while the other channels are negligible because the energies of TS8, TS2, TS3, TS4, and TS5 in channels (2)–(6) are much higher than TS1 in channel (1).
In addition, IM1 can also isomerize to intermediate IM2 CH3CH2O(H)O (−24.9 kcal/mol) by 1,2 H-shift transition state TS7 (−18.3 kcal/mol) or a three-membered ring transition state TS6 (11.0 kcal/mol). Once isomer IM2 is formed, four possible reaction pathways could take place: (a) a concerted 1,4 H-migration and C–O bond fission process to yield P4 C2H4 + H2O2 (−50.1 kcal/mol) via TS9 (−10.8 kcal/mol), (b) dissociation to P3 C2H6 + 1O2 (−22.4 kcal/mol) via a 1,2 H-shift transition state TS10 (8.6 kcal/mol), (c) O-extrusion of the O–O bond to form P6 C2H5OH + O (20.8 kcal/mol) with no distinct barriers, and (d) O–H bond formation accompanied by C–H and O–O bond rupture to form IM3 CH2(OH)CH2OH (−123.2 kcal/mol) via TS11 after surmounting a high energy barrier of 42.3 kcal/mol, then IM3 can undergo a concerted 1,3 H-migration and C–C bond fission process to yield P5 via TS12 (−29.3 kcal/mol). Clearly, these dissociation paths that start from IM2 would make negligible contribution to the reaction, because of their much larger barriers.
3.1.2. H-Abstraction Channels
As shown in Figure 2b, the HO2 can directly abstract the H atom from C2H5 via TS15 to form P4 C2H4 + H2O2 (−50.1 kcal/mol), or C2H5 directly abstracts the H atom from HO2 via TS14 to produce P3 C2H6 + 1O2 (−22.4 kcal/mol) on the singlet PES. The reaction barriers are 15.3 and 11.7 kcal/mol, respectively. Furthermore, the H-abstraction reaction can take place on a triplet PES. A van der Waals hydrogen-bonded complex, 3IM1 (−3.8 kcal/mol), is formed first at the entrance channel with the O-H bond distance of 2.05 Å, which is then followed by a hydrogen abstraction that fragments readily via 3TS1 to give P16 C2H6 + 3O2 (−22.4 kcal/mol). This process needs to overcome a small barrier of just 0.7 kcal/mol. The reaction channels that form P3 and P4 are infeasible in the kinetics due to their high energy barriers.
In summary, on the singlet PES, the formations of one intermediate IM1 as well as two primary product fragments P1, P2 and one direct H-abstraction product P16 through the triplet PES are most likely accessible in energy for the reaction of C2H5 + HO2 (see Figure 1 and Figure 2). Because the competition between the variety products under different temperatures and pressures cannot be determined solely on the PESs, VTST and RRKM calculations are performed to obtain the rate constants and branching ratios of all these competitive channels in the following section.
3.2. Kinetic Calculations
Product distributions for the key products are calculated based on the PES obtained above for the C2H5 + HO2 reaction using the MultiWell 2011 program [20,21] in the temperature range of 220 to 3000 K and in the pressure range of 1 × 10−4 to 100 bar. For the barrierless channels, such as the entrance channel for the formation of the chemically activated IM1 C2H5OOH and the dissociation channel from IM1 to P1 C2H5O + OH, VTST [29,30] was used to locate the kinetic bottleneck. For this purpose, we carried out constrained optimizations at fixed C–O bond length or fixed O–O bond length in C2H5OOH at the multi-reference self-consistent field theory CASSCF(8,6)/aug-cc-pvdz level of theory. The total energies along the reaction coordinate were refined by single-point energy calculations using the CASPT2(8,6)/aug-cc-pvdz level. CASPT2//CAS calculations were done with the MOLPRO 2006 program [31,32]. The energetic and molecular parameters (reaction barriers, moment of inertia, and vibrational frequencies) of the reactants, intermediates and transition states from the ab initio calculations were used in kinetic calculations. Rate constants for direct H abstraction reactions, including the formation channels of P3, P4, and P16, were also obtained using the variation transition-state theory (CVT) with the small curvature tunneling (SCT) correction by means of the POLYRATE 9.7 program [33,34]. The total rate constant (ktot) is obtained as the sum of the individual rate constants associated with the corresponding channels. The calculated rate constant value, 6.88 × 10−11 cm3·molecule−1·s−1 at 293 K, is in good agreement with the experimental values, 4.98 × 10−11 and (5.15 ± 1.66) × 10−11 cm3·molecule−1·s−1.
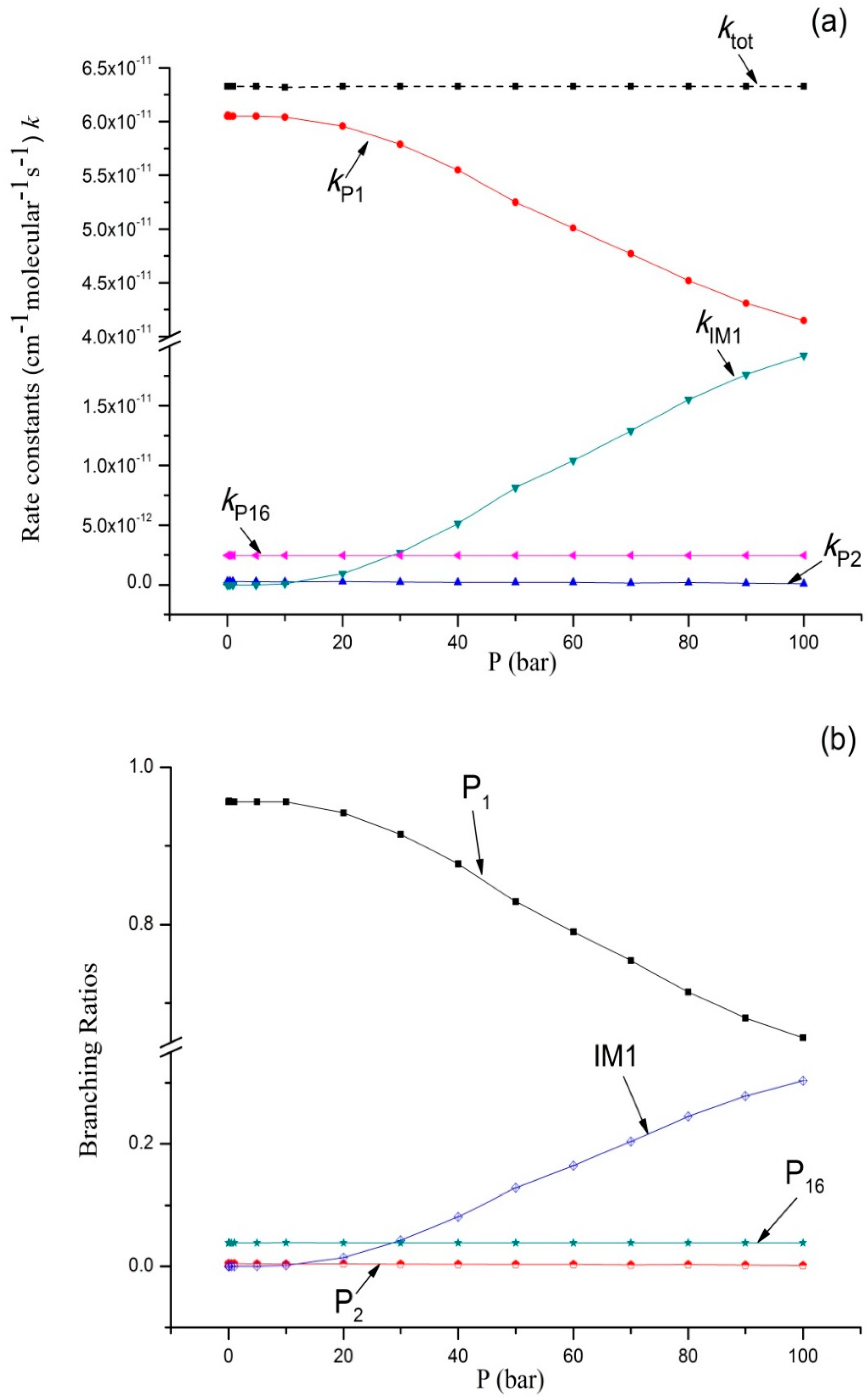
Variations of the reaction rate constants and branching ratios of the reaction channels at 293K when pressure increases from 1 × 10−4 bar–100 bar are shown in Figure 3a,b. ktot seemed independent from pressure. The dissociation bimolecular product P1 C2H5O + OH was the major product, which is negatively correlated with pressure. The output of P1 decreased from 0.96 at 1 × 10−4 bar to 0.66 at 100 bar. It is interesting that kP16 and kP2 were predicted independently from pressure, and kP16 > kP2. The branching ratio of kP16 reached 0.04 throughout the whole pressure range. The contribution of P2 generation to the total reaction rate can be ignored. kIM1 increases with an increase in pressure (>10 bar), and the output of IM1 C2H5OOH increased continuously and reached 0.31 at 100 bar. These computational results were compared with the available experimental data; our findings showed that P1 C2H5O + OH was the major product. According to the theoretical computation, bimolecular products P2 CH3CHO + H2O and P16 C2H6 + 3O2, as well as unimolecular product IM1 C2H5OOH, had small outputs. This is why they were difficult to detect in this experiment.
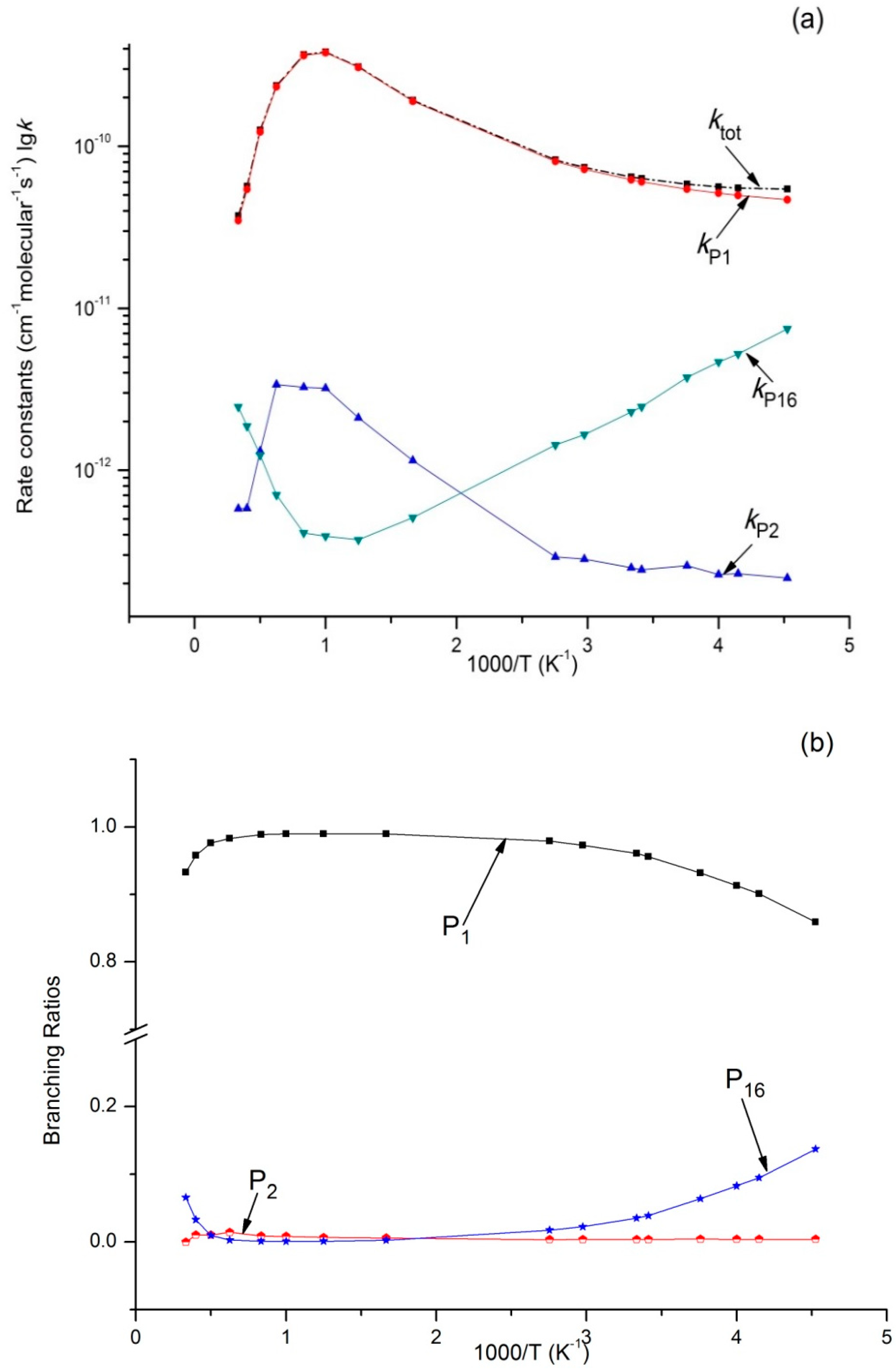
Variations of ktot as well as reaction rate constants and branching ratios of reaction channels at 1 × 10−4 bar, 1 bar, and 100 bar when the temperature increases from 220–3000 K are presented in Figure 4a,b, Figure 5a,b, and Figure 6a,b. At the very beginning, ktot increased from 5.45 × 10−11 cm3·molecule−1·s−1 at 220 K to 3.87 × 10−10 cm3·molecule−1·s−1 at 1000 K, but then decreased to 3.73 × 10−11 cm3·molecule−1·s−1 at 3000 K. Outputs of bimolecular products P1 and P2 showed similar temperature dependence with ktot. However, the output of P16 showed the opposite, firstly decreasing to 3.72 × 10−13 cm3·molecule−1·s−1 at 800 K and then increasing to 2.46 × 10−12 cm3·molecule−1·s−1 at 3000 K. Meanwhile, kIM1 was negatively correlated with temperature under high pressure at 100 bar. According to Figure 4b and Figure 5b, the reaction mainly produced dissociation products under 1 × 10−4 bar and 1 bar, while the impact stabilization effect of the intermediates was neglected. The bimolecular product P1 was the main product. Its output increased from 0.86 at 220 K to 0.99 at 1000 K and then decreased to 0.93 (1 × 10−4 bar) and 0.84 (1 bar) at 3000 K. Under 1 × 10−4 bar and 1 bar, the maximum branching ratio of secondary product P16 at 220 K was 0.14 and reached the peak (0.07) at 300 K. Under high pressure (100 bar) (Figure 6b), output of P1 increased quickly with the temperature rise and reached the peak (0.99) at 1000 K. P1 became the major product after the temperature reached 227 K. However, the intermediate stabilized by impact (IM1) was the major product under low temperature, while other products like P2 and P16 had a very small branching ratio within the whole studying temperature range (≤0.1). According to our kinetic calculation of the C2H5 + HOO reaction, the effect of pressure on product distribution declined after temperatures exceeded 293 K and the dissociation product P1 C2H5O + OH became the major product. In other words, under low temperature, relative output showed strong pressure dependence, and the stabilization effect of IM1 C2H5OOH became increasingly important as pressure increased.
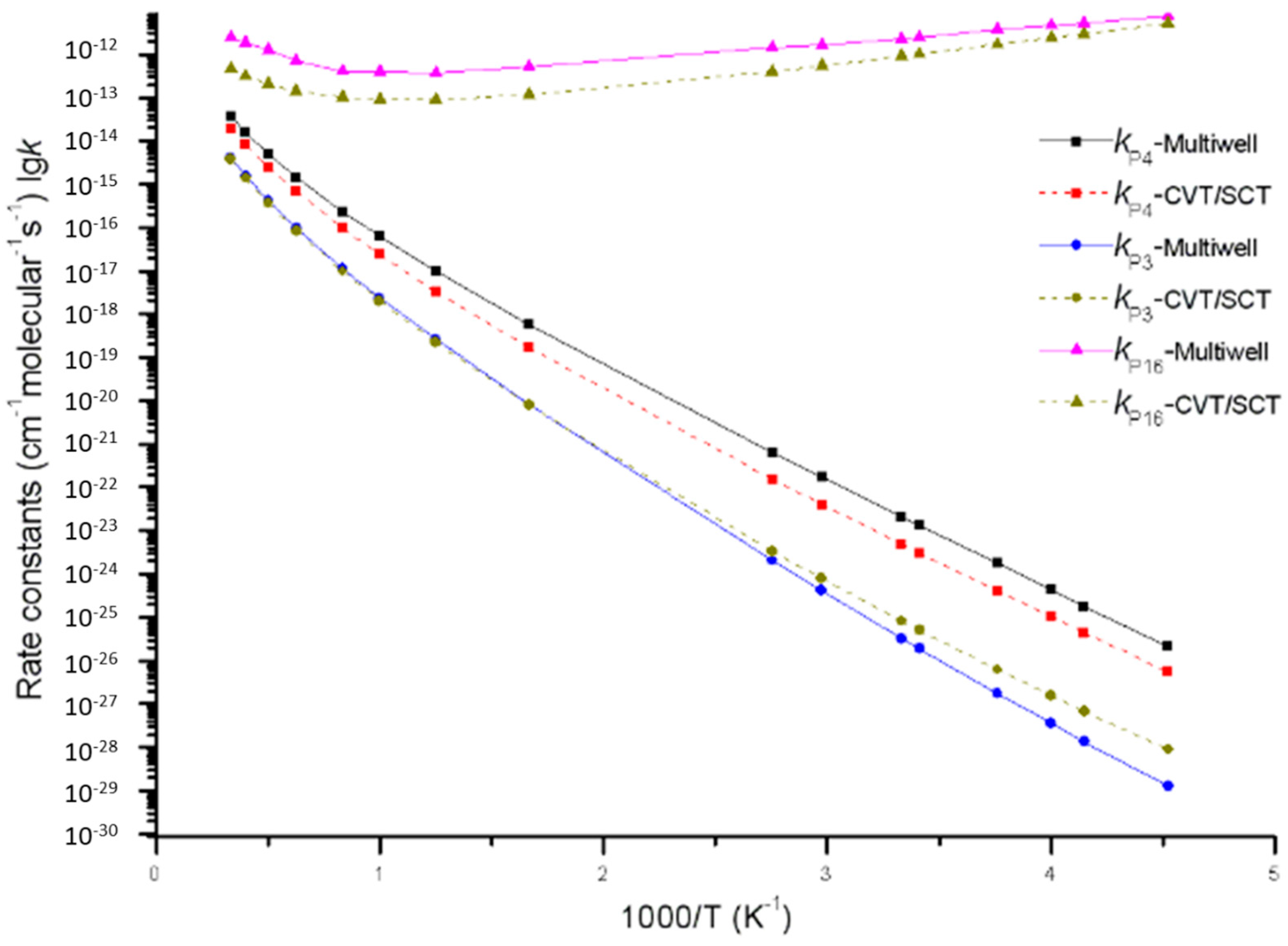
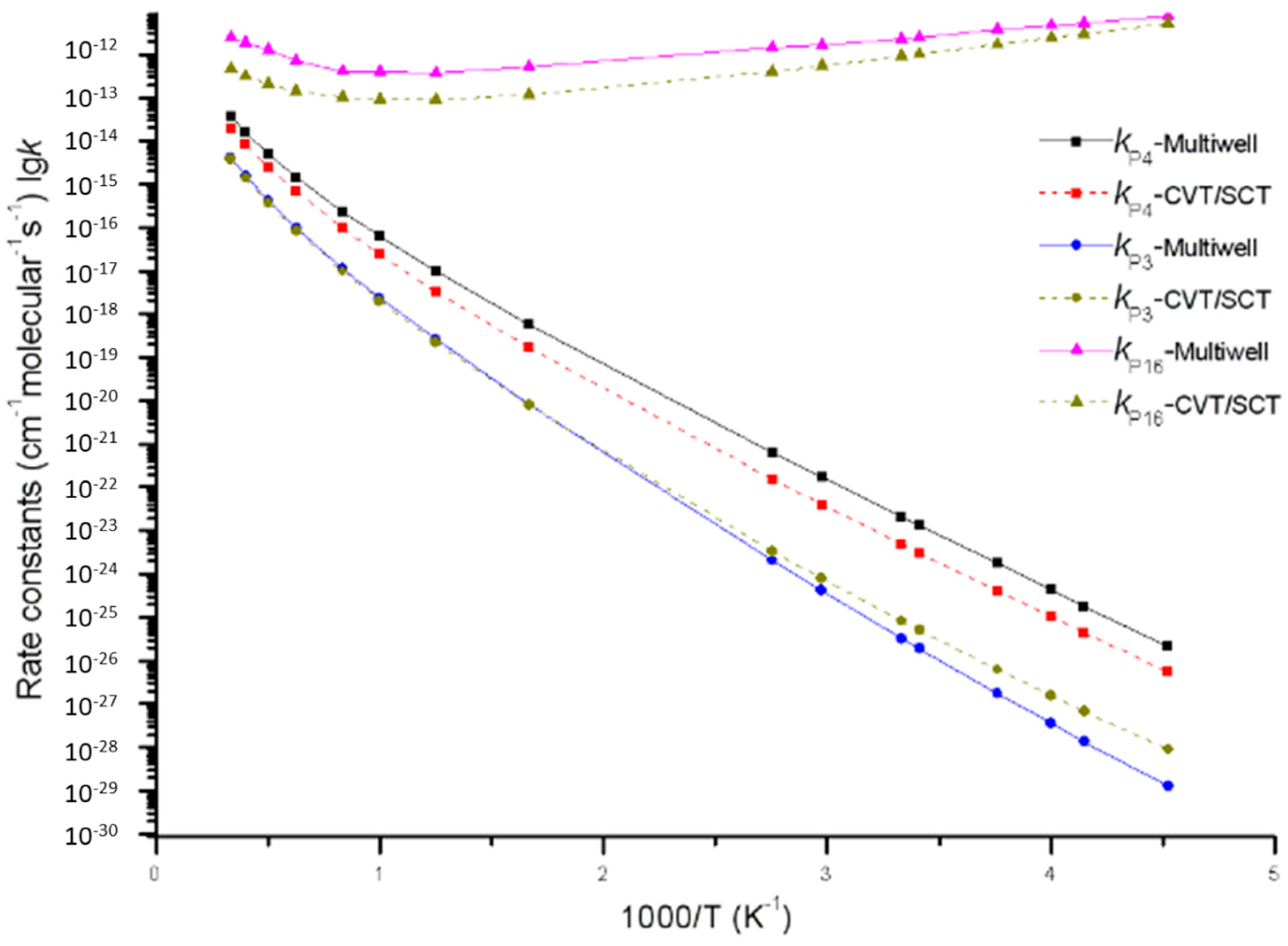
Furthermore, it is very important to compare the MultiWell program results of direct H-abstraction reaction channels (including generation channels of P3 and P4 on a singlet potential energy surface and generation channel of P16 on a triplet potential energy surface) with the Polyrate predicted results. Reaction rate constants calculated by the MultiWell program and the Polyrate program are shown in Figure 7. Under low temperature, the MultiWell and Polyrate programs report similar rate constants of triplet product P16 C2H6 + 3O2. Under high temperature, they conformed to each other more in terms of singlet product P3 C2H6 + 1O2 and P4 C2H4 + H2O2. At 220 K, kP3, kP4, and kP16 calculated by the MultiWell program were 0.14, 3.3, and 1.4 times higher than those calculated by the CVT/SCT model. At 3000 K, kP3, kP4, and kP16 calculated by the MultiWell program were 1.1, 2, and 5 times more satisfactory than those calculated by the CVT/SCT model. This paper concludes that under low energy barrier and low temperature range (or high energy barrier and high temperature range), reaction rate constants calculated by both the MultiWell and Polyrate programs have high accuracy.
4. Conclusions
In this paper, singlet and triplet potential energy surfaces of the C2H5 + HOO reaction were computed on the CCSD(T)/6-311+G(3df,2p)//MP2/6-311++G(d,p) theoretical level. Rate constants and branching ratios of major reaction channels under the temperature range 220 K–3000 K and pressure range 1 × 10−4 bar–100 bar were predicted. According to the computational results, the C2H5 + HOO reaction occurred through addition and abstraction on the singlet potential energy surface. In other words, the O atom in HOO attacked the C atom in C2H5 to form an entrance intermediate with rich energy IM1 C2H5OOH through an energy barrier-free addition reaction. Starting from IM1, direct energy barrier-free O–O bond breakage generated the major product P1 C2H5O + OH, and P2 CH3CHO + H2O was the secondary product. Alternatively, HOO can directly abstract a H atom in C2H5 to produce P4 C2H4 + H2O2, and C2H5 can also directly abstract a H atom in HOO to produce P3 C2H6 + 1O2. These two H-abstraction reaction channels have to overcome high energy barriers and are unfeasible in kinetics. On the triplet potential energy surface, generation of P16 C2H6 + 3O2 through a H-abstraction reaction has to pass through a loose van der Waals complex, 3IM1 3C2H5HOO, and overcome a small energy barrier. The kinetic calculation demonstrates that the impact-stabilized intermediate IM1 is the major product under high pressure and low temperature, but the bimolecular product P1 is the major product under low pressure or high temperature. Under 293 K, P1 was the major product within the studied pressure range, which conformed well to experimental prediction. The branching ratio of P1 output was inversely proportional to pressure, decreasing from 0.96 at 1 × 10−4 bar to 0.66 at 100 bar. IM1 became the secondary product after pressure exceeded 30 bar (the maximum output is 0.31). Research results in this paper are conducive to deepening our understanding on the mechanisms and kinetics of the C2H5 + HOO reaction and determining its possible products.
Author Contributions
N.-N.W. designed the research, performed the experimental work and wrote the manuscript. N.-N.W. and S.-L.O.-Y. provided direction and contributed to the revisions of the manuscript. M.-Z.Z. and L.L. read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21363013, 11364027, 11564031), Natural Science Foundation of Inner Mongolia Autonomous Region (Grant No. 2018LH02004), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region, China (No. NJYT-17-B10).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- Naik, C.V.; Dean, A.M. Detailed kinetic modeling of ethane oxidation. Combust. Flame 2006, 145, 16–37. [Google Scholar] [CrossRef]
- Kaiser, E.W. Mechanism of the reaction C2H5 + O2 from 298 To 680 K. J. Phys. Chem. A. 2002, 106, 1256–1265. [Google Scholar] [CrossRef]
- Ludwig, W.; Brandt, B.; Friedrichs, G.; Temps, F. Kinetics of the reaction C2H5 + HO2 by time-resolved mass spectrometry. J. Phys. Chem. A 2006, 110, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S. Addition of peroxyl radicals to alkenes and the reaction of oxygen with alkyl radicals. J. Am. Chem. Soc. 2000, 122, 4162–4170. [Google Scholar] [CrossRef]
- Platt, U.; Lebras, G.; Poulet, G.; Burrows, J.P.; Moortgat, G. Peroxy radicals from night-time reaction of NO3 with organic compounds. Nature 1990, 348, 147–149. [Google Scholar] [CrossRef]
- Lightfoot, P.D.; Cox, R.A.; Crowley, J.N.; Destriau, M.; Hayman, G.D.; Jenkin, M.E.; Moortgat, G.K.; Zabe, F. Organic peroxy radicals: Kinetics, spectroscopy and tropospheric chemistry. Atmos. Environ. Part A 1992, 26, 1805–1961. [Google Scholar] [CrossRef]
- Meloni, G.; Zou, P.; Klippenstein, S.J.; Ahmed, M.; Leone, S.R.; Taatjes, C.A.; Osborn, D.L. Energy-resolved photoionization of alkylperoxy radicals and the stability of their cations. J. Am. Chem. Soc. 2006, 128, 13559–13567. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; OuYang, S.L.; Li, L. Theoretical study of ClOO + NO reaction: Mechanism and kinetics. Molecules 2017, 22, 2121. [Google Scholar] [CrossRef] [PubMed]
- Hrušák, J.; Friedrichs, H.; Schwarz, H.; Razafinjanahary, H.; Chermette, H. Electron affinity of hydrogen peroxide and the [H2, O2]•− potential energy surface. A comparative DFT and ab Initio study. J. Phys. Chem. 1996, 100, 100–110. [Google Scholar] [CrossRef]
- Tsang, W.; Hampson, R.F. Chemical kinetic data base for combustion chemistry. Part I. Methane and related compounds. J. Phys. Chem. Ref. Data 2009, 15, 1087–1279. [Google Scholar] [CrossRef]
- Dobis, O.; Benson, S. Reaction of the ethyl radical with oxygen at millitorr pressures at 243–368 K and a study of the Cl + HO2, ethyl + HO2, and HO2 + HO2 reactions. J. Am. Chem. Soc. 1993, 115, 8798–8809. [Google Scholar] [CrossRef]
- Holbrook, K.A.; Pilling, M.J.; Robertson, S.H. Unimolecular Reactions, 2nd ed.; John Wiley & Sons: Chichester, UK, 1996. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.R.; Pople, J. AB INITIO Molecular Orbital Theory; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1986; Volume 9, pp. 399–406. [Google Scholar]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 471–476. [Google Scholar] [CrossRef]
- Charles, D., Jr.; James, W.M., Jr.; Michael, P. Singlet biradicals as intermediates. Canonical variational transition-state theory results for trimethylene. J. Chem. Phys. 1988, 92, 4367–4371. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Frisch, M.J.; Truck, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.J.; Stratmann, R.E.; Burant, J.C.; et al. GAUSSIAN 03, Revision A1; Guassian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Holbrook, K.A.; Pilling, M.J.; Robertson, S.H. Unimolecular Reactions; Wiley: Chichester, UK, 1972; Volume 15, pp. 357–362. [Google Scholar]
- Barker, J.R. Multiple-Well, multiple-path unimolecular reaction systems. I. MultiWell computer program suite. Int. J. Chem. Kinet. 2001, 33, 232–245. [Google Scholar] [CrossRef]
- Barker, J.R.; Ortiz, N.F.; Preses, J.M.; Lohr, L.L.; Maranzana, A.; Stimac, P.J. MultiWell-2011 Software; University of Michigan: Ann Arbor, MI, USA, 2007; Available online: http://aoss.engin.umich.edu/multiwell/ (accessed on January 2011).
- Lubic, K.G.; Amano, T.; Uehara, H.; Kawaguchi, K.; Hirota, E. The V1 band of the DO2 radical by difference frequency laser and diode laser spectroscopy: The equilibrium sturcture of the hydroperoxyl radical. J. Chem. Phys. 1984, 81, 4826–4831. [Google Scholar] [CrossRef]
- Hellwege, K.H.; Hellwege, A.M. Structure data of free polyatomic molecules. In Landolt-Bornstein: Group II: Atomic and Molecular Physics; Springer Science & Business Media: Berlin, Germany, 1976; Volume 7. [Google Scholar]
- Hollenstien, H.; Gunthard, H.H. Solid State and gas infrared spectra and normal coordinate analysis of 5 isotopic species of acetaldehyde. Spec. Act. Part A 1971, 27A, 2027–2060. [Google Scholar] [CrossRef]
- Redington, R.L.; Olson, W.B.; Cross, P.C. Studies of hydrogen peroxide: The infrared spectrum and the internal rotation problem. J. Chem. Phys. 1962, 36, 1311–1326. [Google Scholar] [CrossRef]
- Venkateswarlu, P.; Gordy, W. Methyl alcohol II. molecular structure. J. Chem. Phys. 1955, 23, 1200–1202. [Google Scholar] [CrossRef]
- Gurvich, L.V.; Veyts, I.V.; Alcock, C.B. Thermodynamic Properties of Individual Substances, 4th ed.; Hemisphere Pub. Co.: New York, NY, USA, 1989. [Google Scholar]
- Hoy, A.R.; Bunker, P.R. A precise solution of the rotation beninding schrodinger equation for a triatomic molecule with application to the water molecule. J. Mol. Spectrosc. 1979, 74, 1–8. [Google Scholar] [CrossRef]
- Klippenstein, S.J. An efficient procedure for evaluating the number of available states within a variably defined reaction coordinate framework. J. Phys. Chem. 1994, 98, 11459–11464. [Google Scholar] [CrossRef]
- Klippenstein, S.J. A bond length reaction coordinate for unimolecular reactions. II. Microcanonical and canonical implementations with application to the dissociation of NCNO. J. Chem. Phys. 1991, 94, 6469–6482. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J. A second order multiconfiguration SCF procedure with optimum convergence. J. Chem. Phys. 1985, 82, 5053–5063. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. 2006 MOLPRO Quantum Chemistry Software, version 2006.1 a Package of ab Initio Programs. Available online: http://www.molpro.net (accessed on January 2011).
- Garrett, B.C.; Truhlar, D.G. Generalized transition state theory. Bond energy-bond order method for canonical variational calculations with application to hydrogen atom transfer reactions. J. Am. Chem. Soc. 1979, 101, 4534–4548. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G.; Grev, R.S. Improved treatment of threshold contributions in variational transition-state theory. J. Phys. Chem. 1980, 84, 1730–1748. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
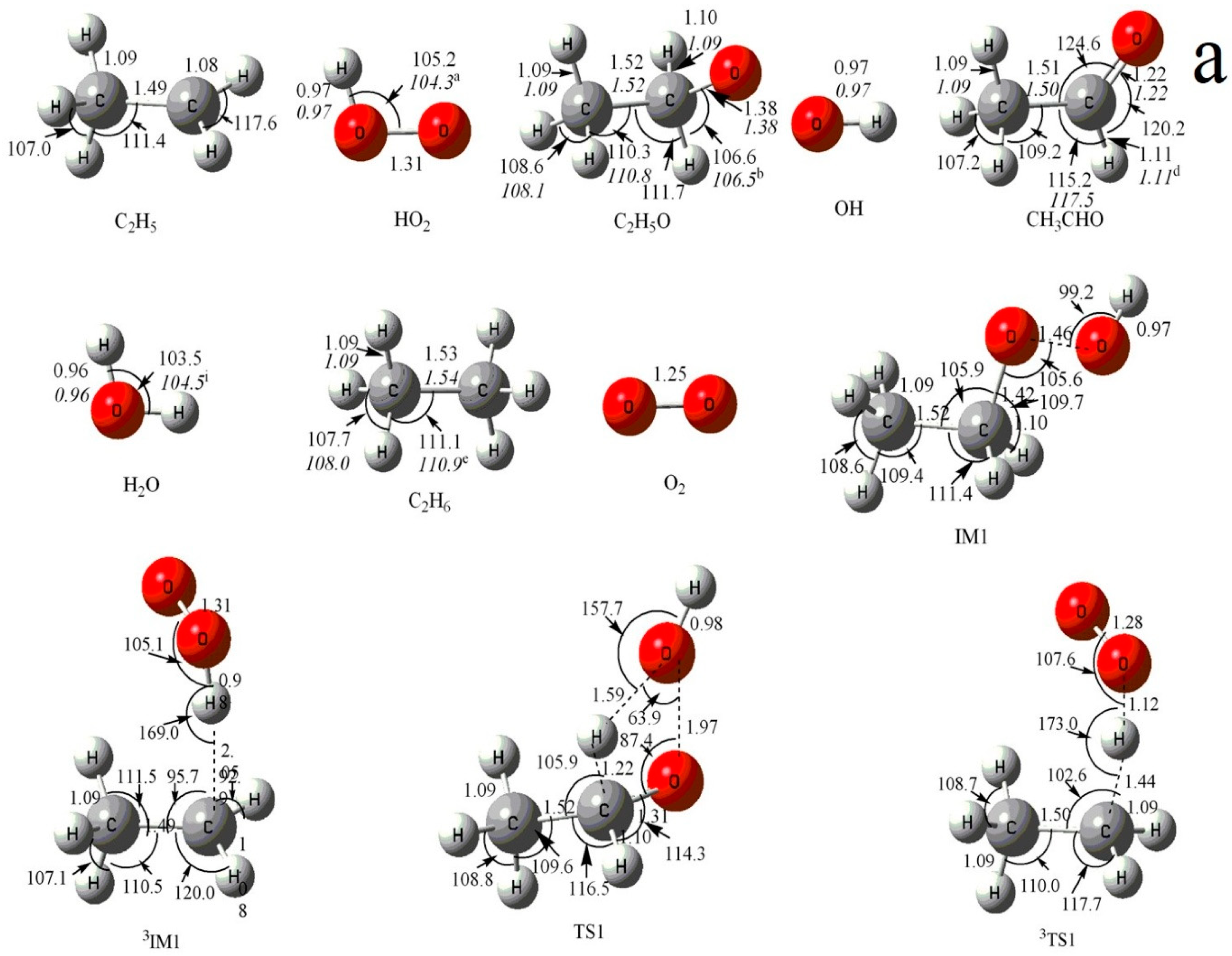
Figure 1.
(a) MP2/6-311++G(d,p) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of the primary product channels of the C2H5 + HO2 reaction. (b) MP2/6-311++G(d,p) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of the secondary product channels of the C2H5 + HO2 reaction. The values in parentheses are the pertinent experimental data from the literature, and a–i represent refs. [17,18,22,23,24,25,26,27,28], respectively. Bond lengths are in Å, and bond angles are in degree.
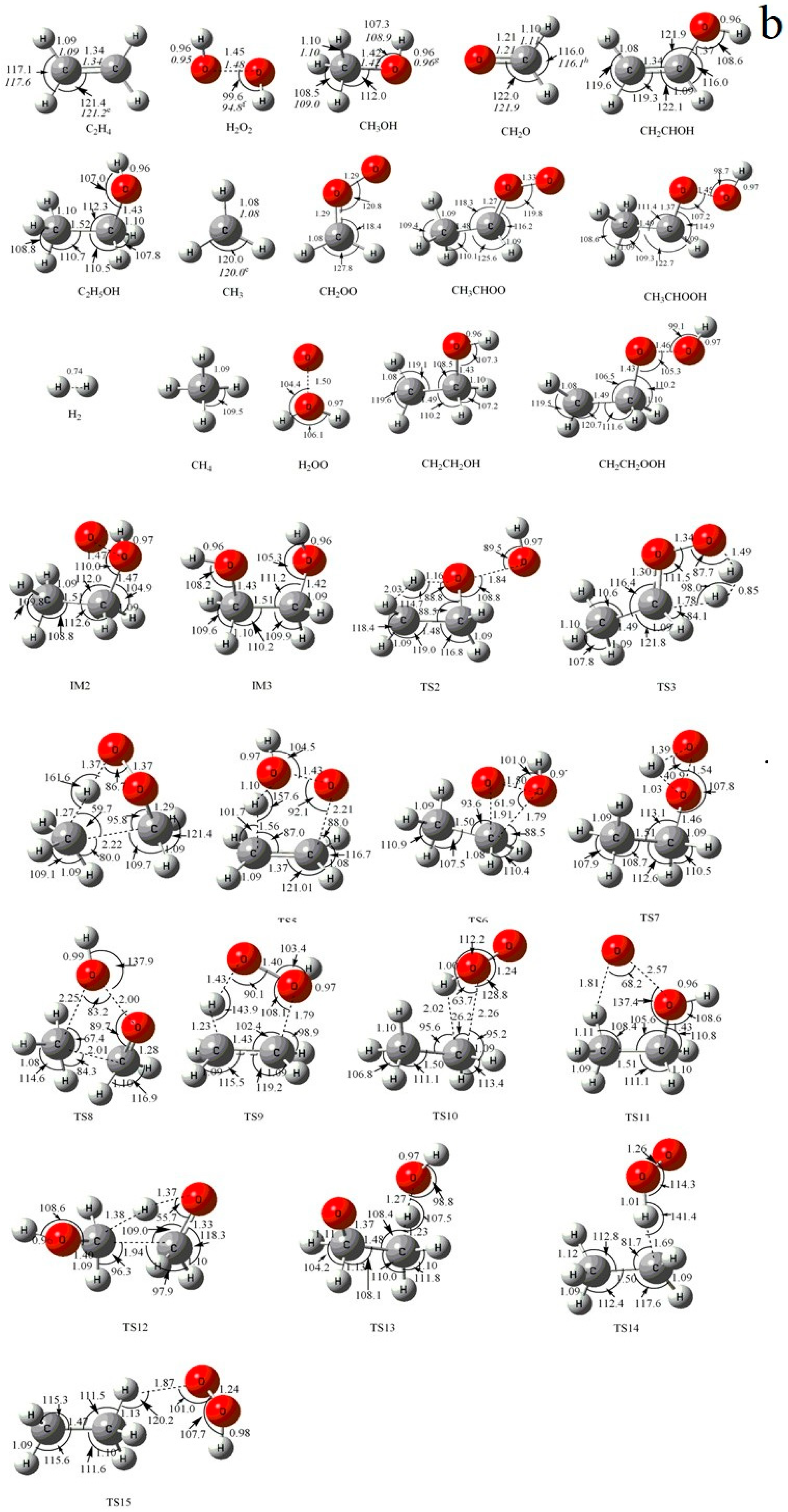
Figure 1.
(a) MP2/6-311++G(d,p) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of the primary product channels of the C2H5 + HO2 reaction. (b) MP2/6-311++G(d,p) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of the secondary product channels of the C2H5 + HO2 reaction. The values in parentheses are the pertinent experimental data from the literature, and a–i represent refs. [17,18,22,23,24,25,26,27,28], respectively. Bond lengths are in Å, and bond angles are in degree.

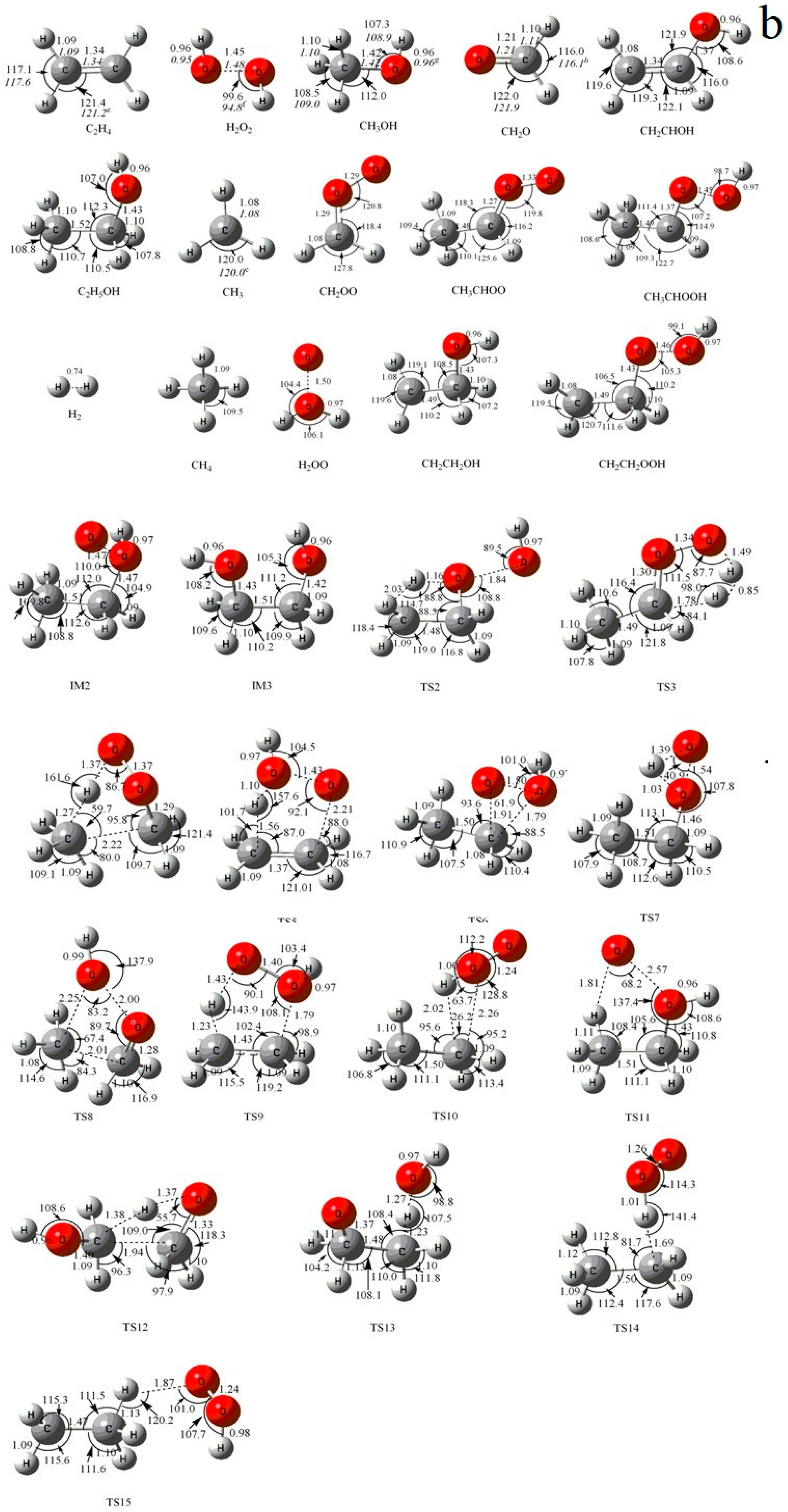
Figure 2.
(a) Schematic singlet potential energy surface (PES) of the addition and dissociation reaction channels for the C2H5 + HO2 reaction at the CCSD(T)/6-311+G(3df,2p)//MP2/6-311++G(d,p)+ZPE level. (b) Schematic singlet and triplet PESs of the abstraction reaction channels for the C2H5 + HO2 reaction at the CCSD(T)/6-311+G(3df,2p)//MP2/6-311++G(d,p)+ZPE level.
Figure 2.
(a) Schematic singlet potential energy surface (PES) of the addition and dissociation reaction channels for the C2H5 + HO2 reaction at the CCSD(T)/6-311+G(3df,2p)//MP2/6-311++G(d,p)+ZPE level. (b) Schematic singlet and triplet PESs of the abstraction reaction channels for the C2H5 + HO2 reaction at the CCSD(T)/6-311+G(3df,2p)//MP2/6-311++G(d,p)+ZPE level.
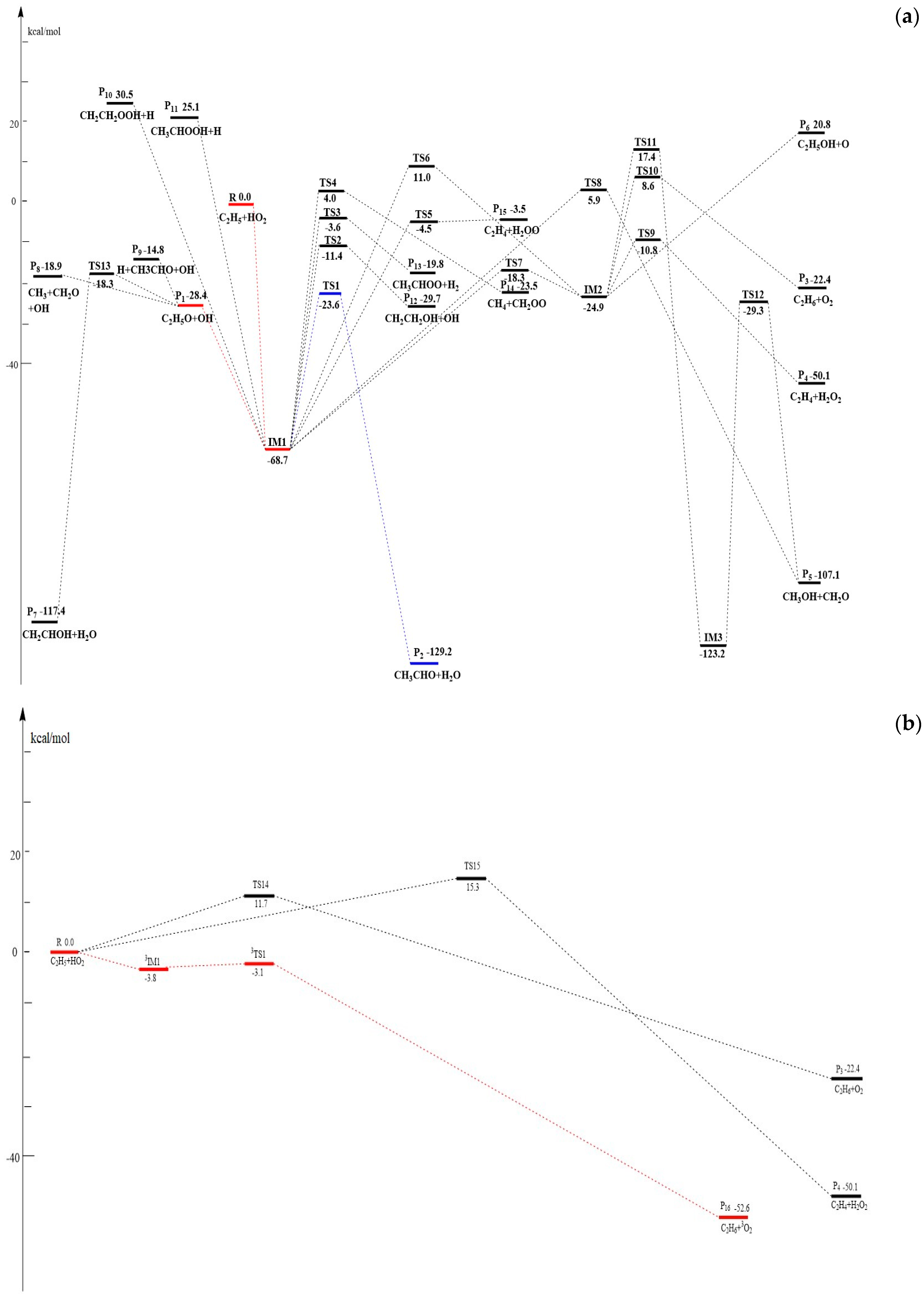
Figure 3.
(a) Rate constants of total and various reaction channels at 293 K in a pressure range from 1 × 10−4 bar to 100 bar; (b) Branching ratios of various reaction channels at 293 K in a pressure range from 1 × 10−4 bar to 100 bar.
Figure 3.
(a) Rate constants of total and various reaction channels at 293 K in a pressure range from 1 × 10−4 bar to 100 bar; (b) Branching ratios of various reaction channels at 293 K in a pressure range from 1 × 10−4 bar to 100 bar.
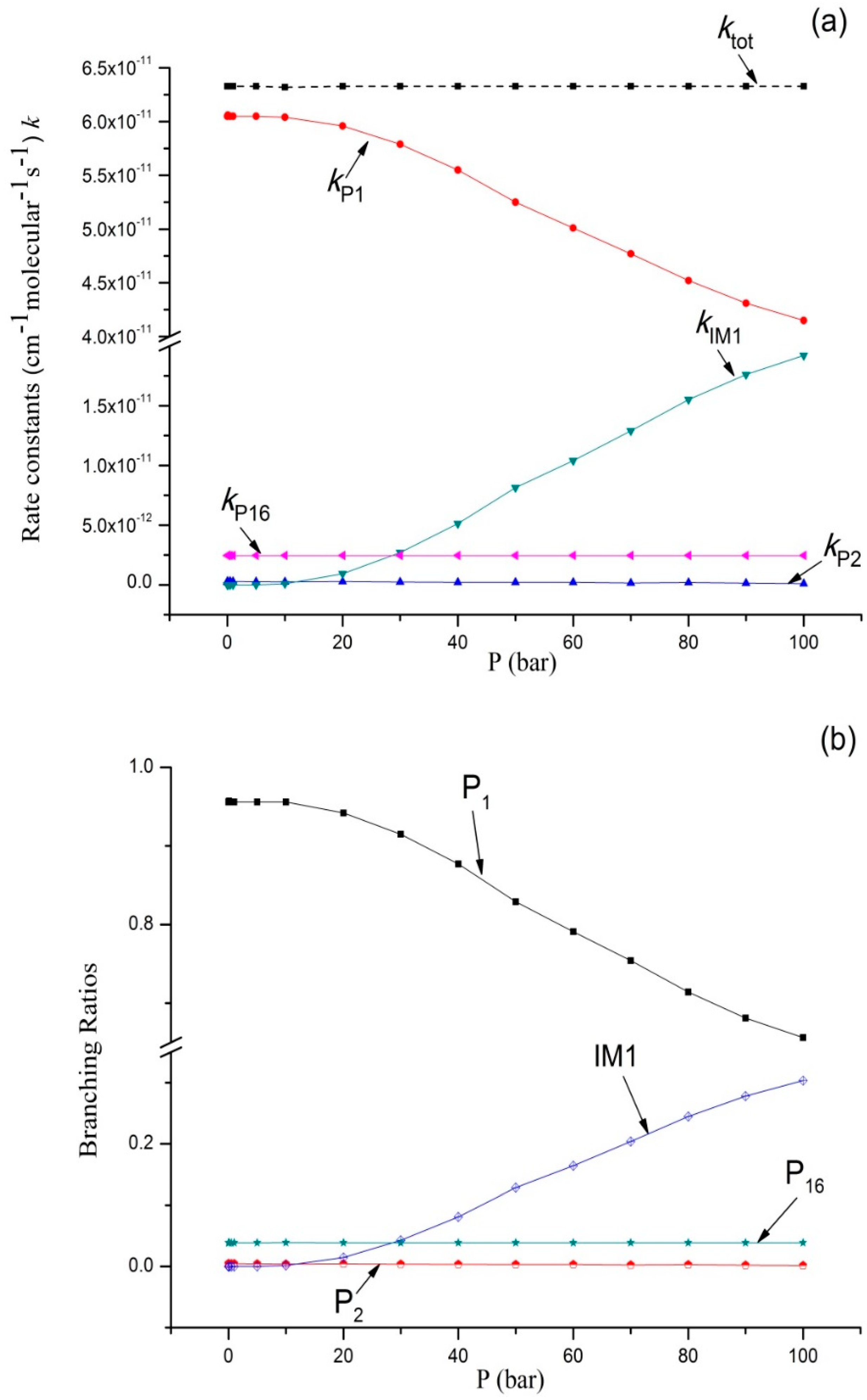
Figure 4.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 1 × 10−4 Bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 1 × 10−4 Bar in a temperature range of 220 K to 3000 K.
Figure 4.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 1 × 10−4 Bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 1 × 10−4 Bar in a temperature range of 220 K to 3000 K.
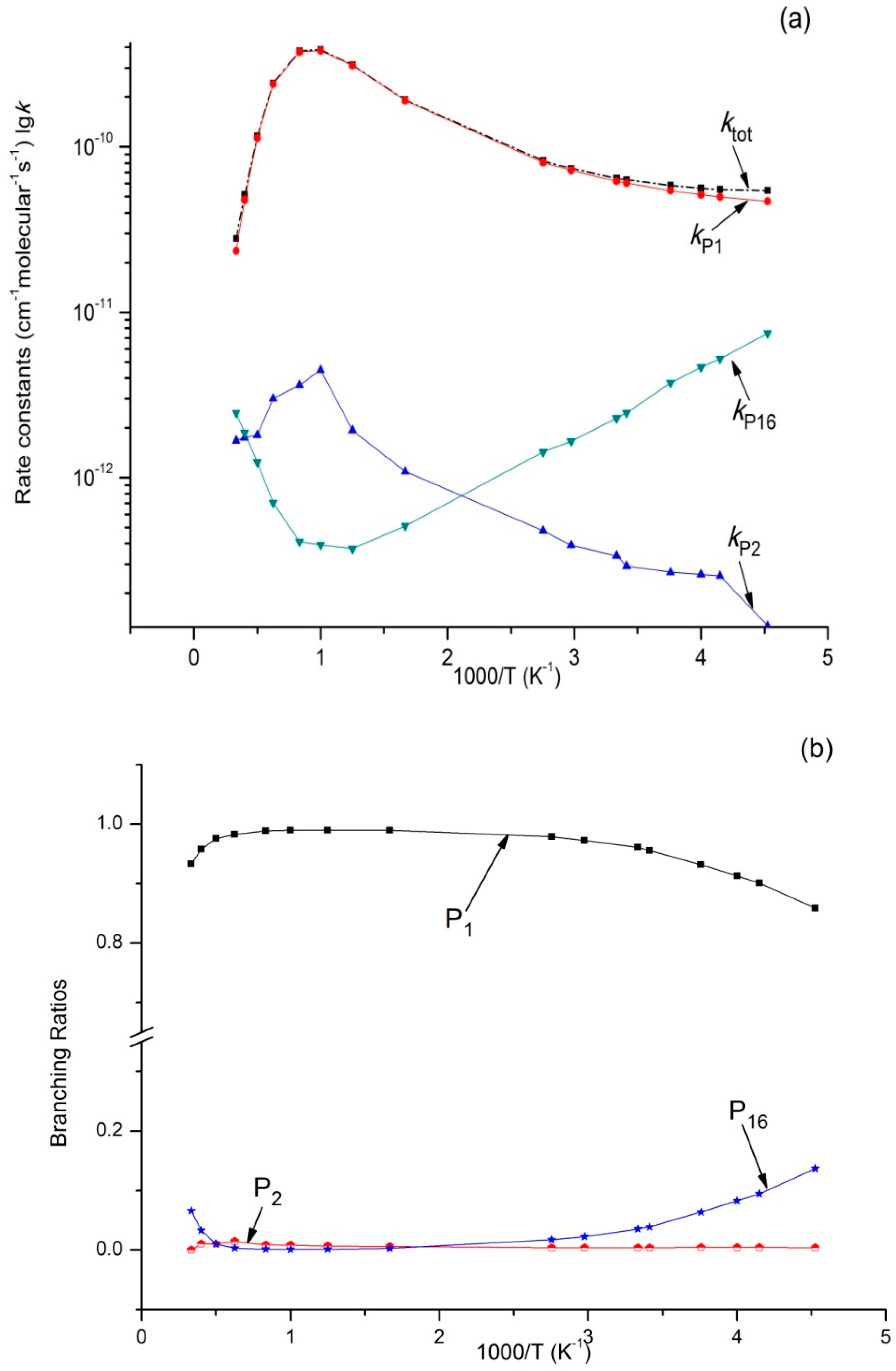
Figure 5.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 1 bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 1 bar in a temperature range of 220 K to 3000 K.
Figure 5.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 1 bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 1 bar in a temperature range of 220 K to 3000 K.
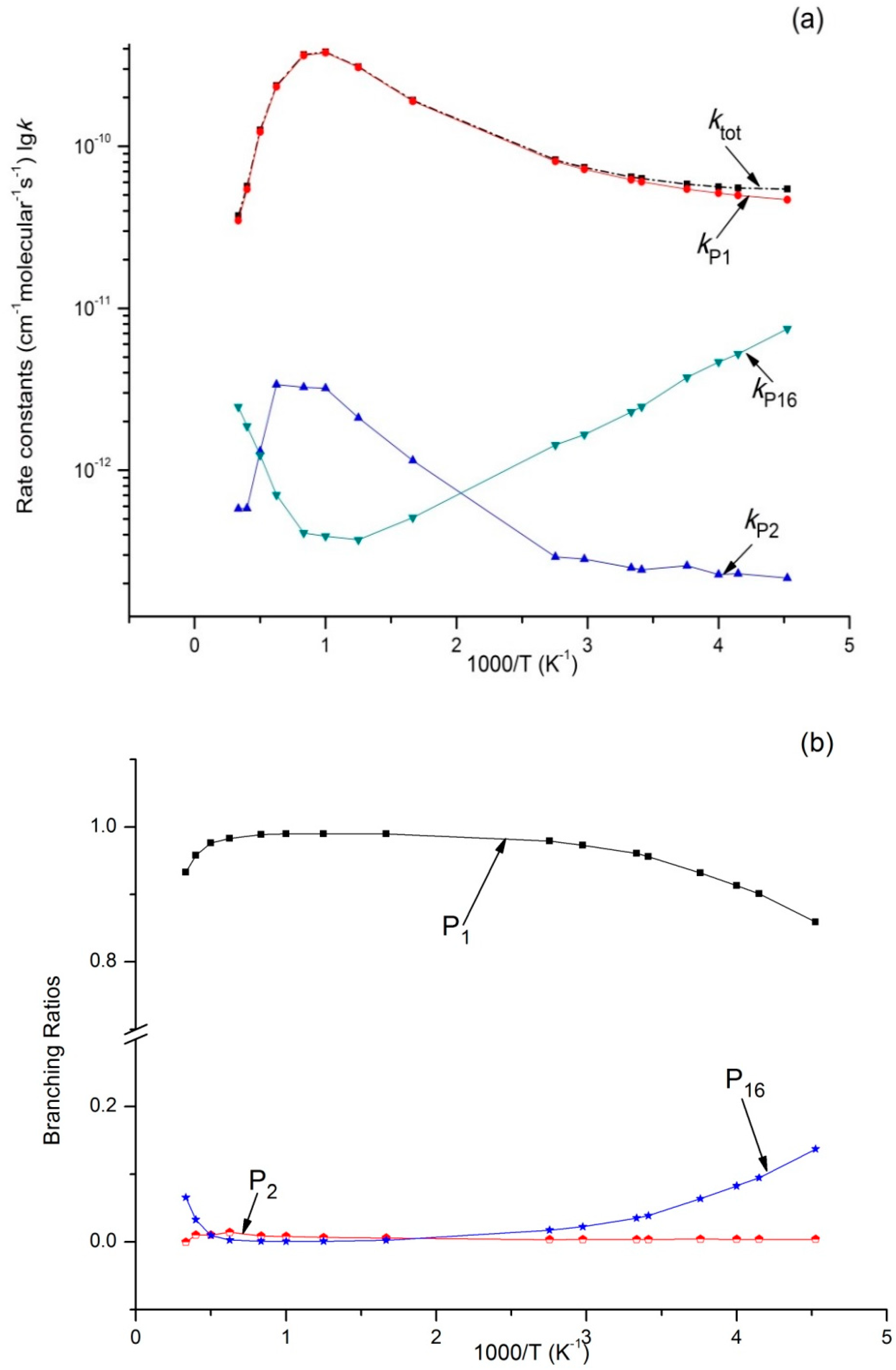
Figure 6.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 100 bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 100 bar in a temperature range of 220 K to 3000 K.
Figure 6.
(a) Rate constants of total (ktot) and various reaction channels (kP1, kP2, kP16, and kIM1) at 100 bar in a temperature range of 220 K to 3000 K; (b) Branching ratios of various reaction channels at 100 bar in a temperature range of 220 K to 3000 K.
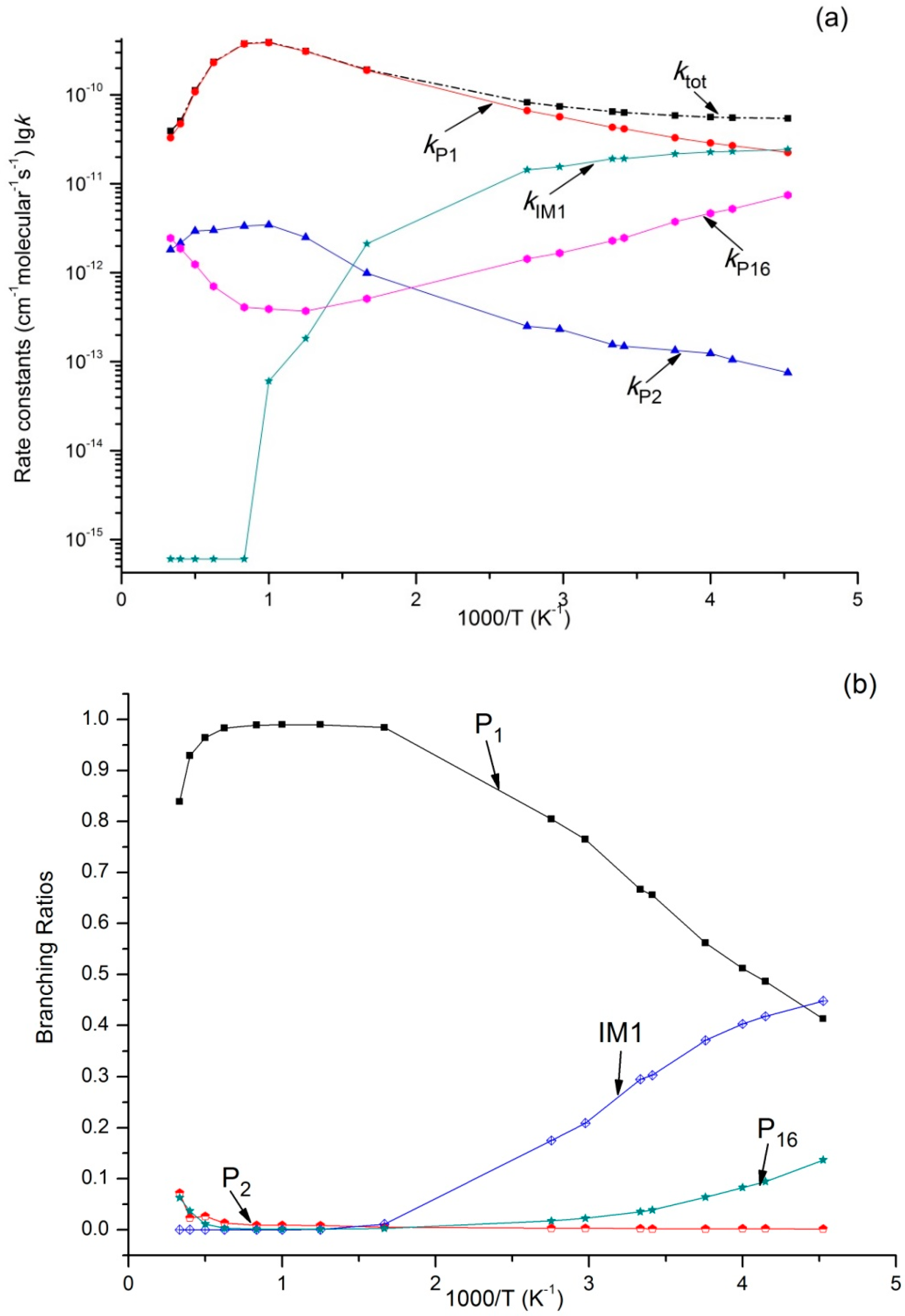
Figure 7.
Rate constants of direct H-abstraction reaction channels (including generation channels of P3, P4 and P16) calculated by using the MultiWell program and the Polyrate program, respectively.
Figure 7.
Rate constants of direct H-abstraction reaction channels (including generation channels of P3, P4 and P16) calculated by using the MultiWell program and the Polyrate program, respectively.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, N.-N.; Zhang, M.-Z.; Ou-Yang, S.-L.; Li, L. Theoretical Study of the C2H5 + HO2 Reaction: Mechanism and Kinetics. Molecules 2018, 23, 1919. https://doi.org/10.3390/molecules23081919
AMA Style
Wu N-N, Zhang M-Z, Ou-Yang S-L, Li L. Theoretical Study of the C2H5 + HO2 Reaction: Mechanism and Kinetics. Molecules. 2018; 23(8):1919. https://doi.org/10.3390/molecules23081919
Chicago/Turabian StyleWu, Nan-Nan, Ming-Zhe Zhang, Shun-Li Ou-Yang, and Liang Li. 2018. "Theoretical Study of the C2H5 + HO2 Reaction: Mechanism and Kinetics" Molecules 23, no. 8: 1919. https://doi.org/10.3390/molecules23081919