
Comparative Study of Carborane- and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay


Abstract
:
1. Introduction
2. Results and Discussion
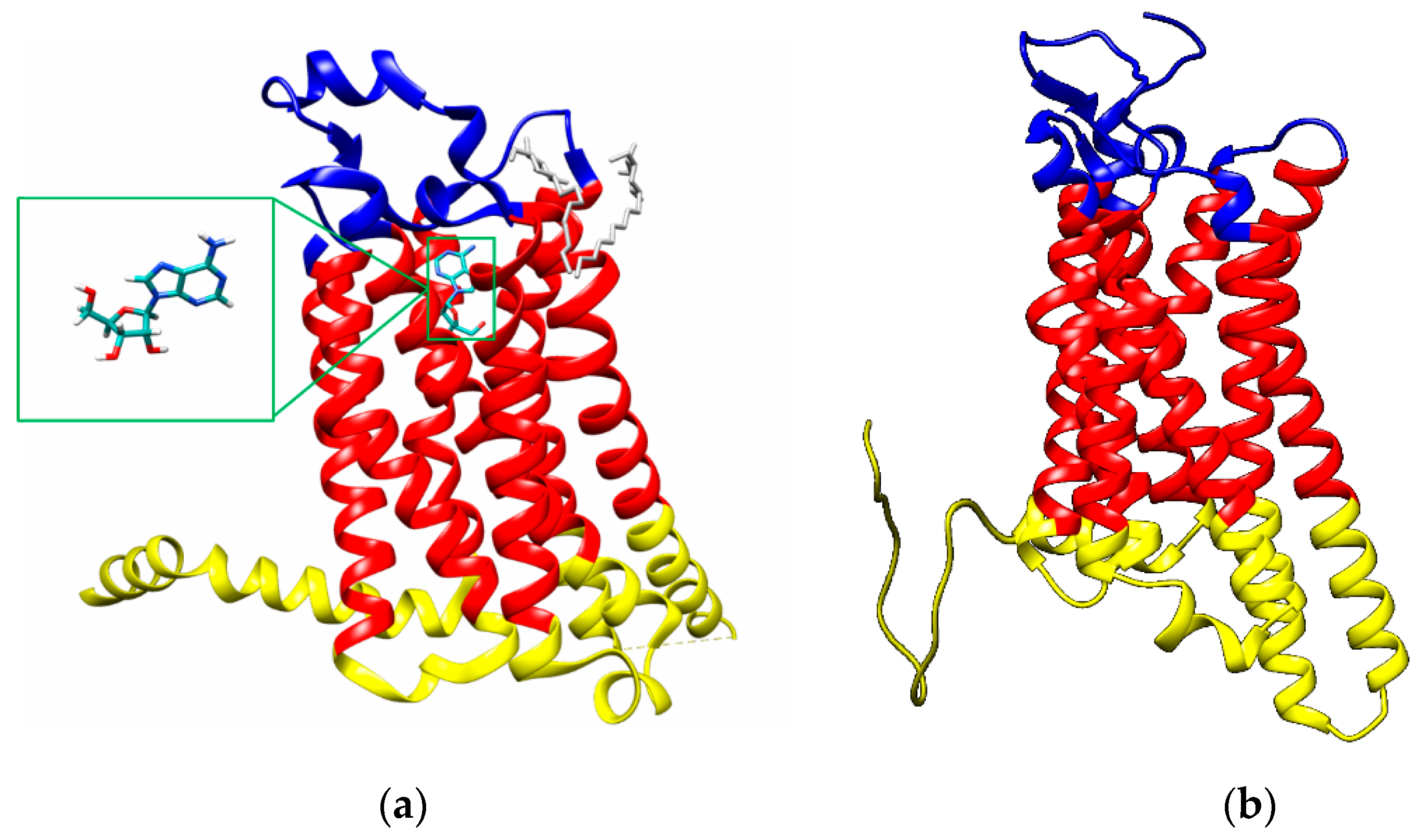
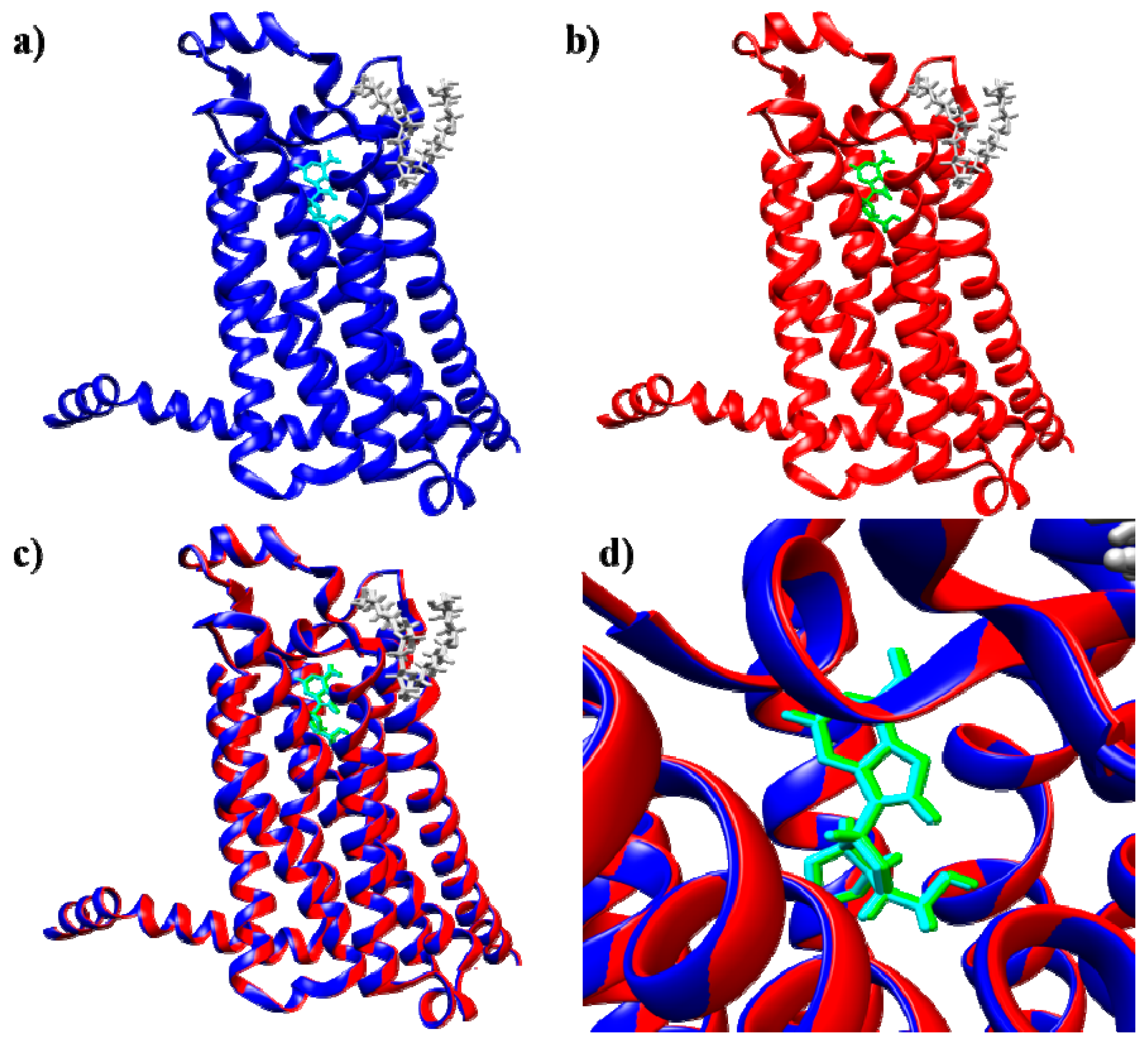
2.1. Protein Structure Selection and Modeling
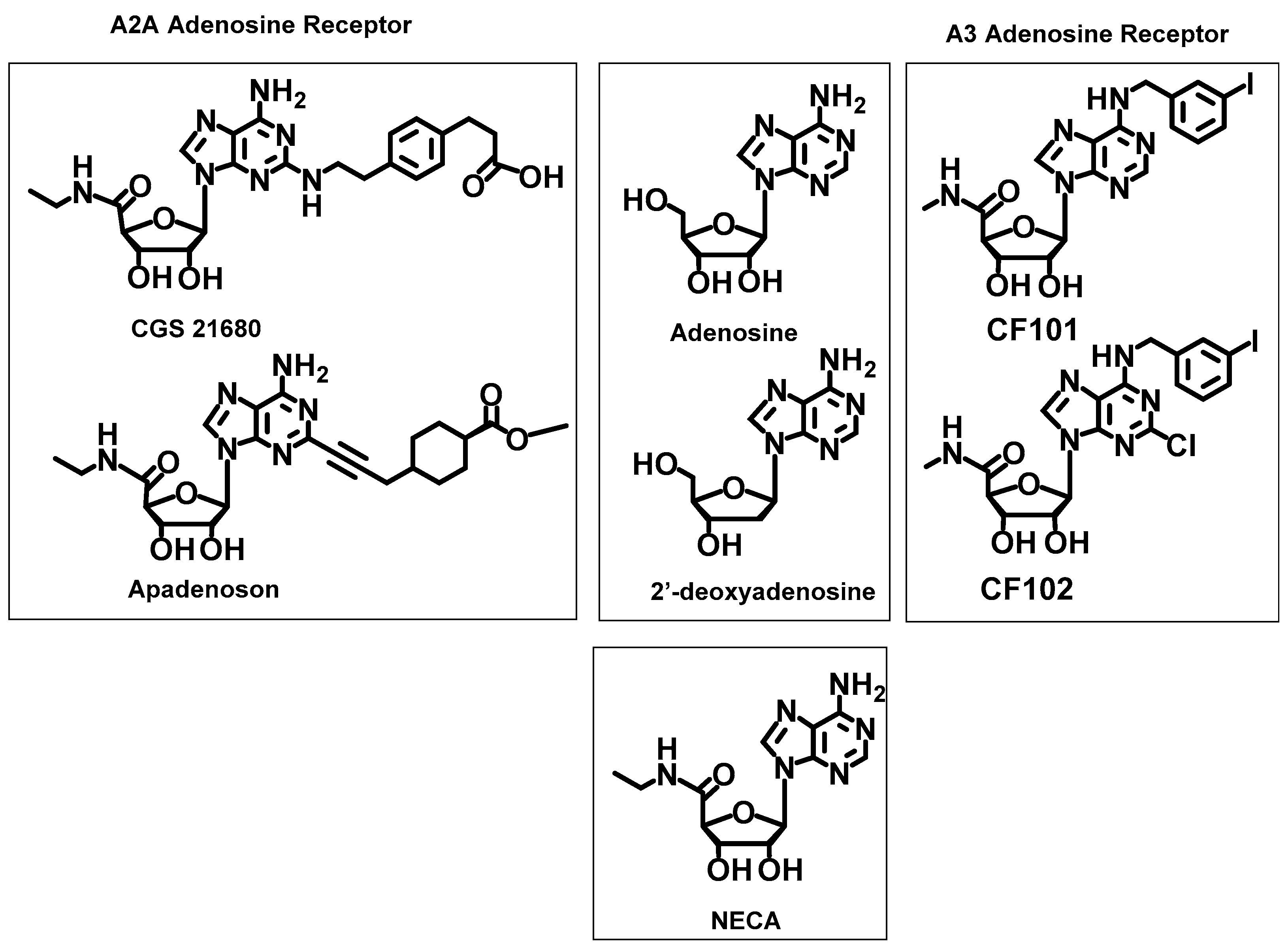
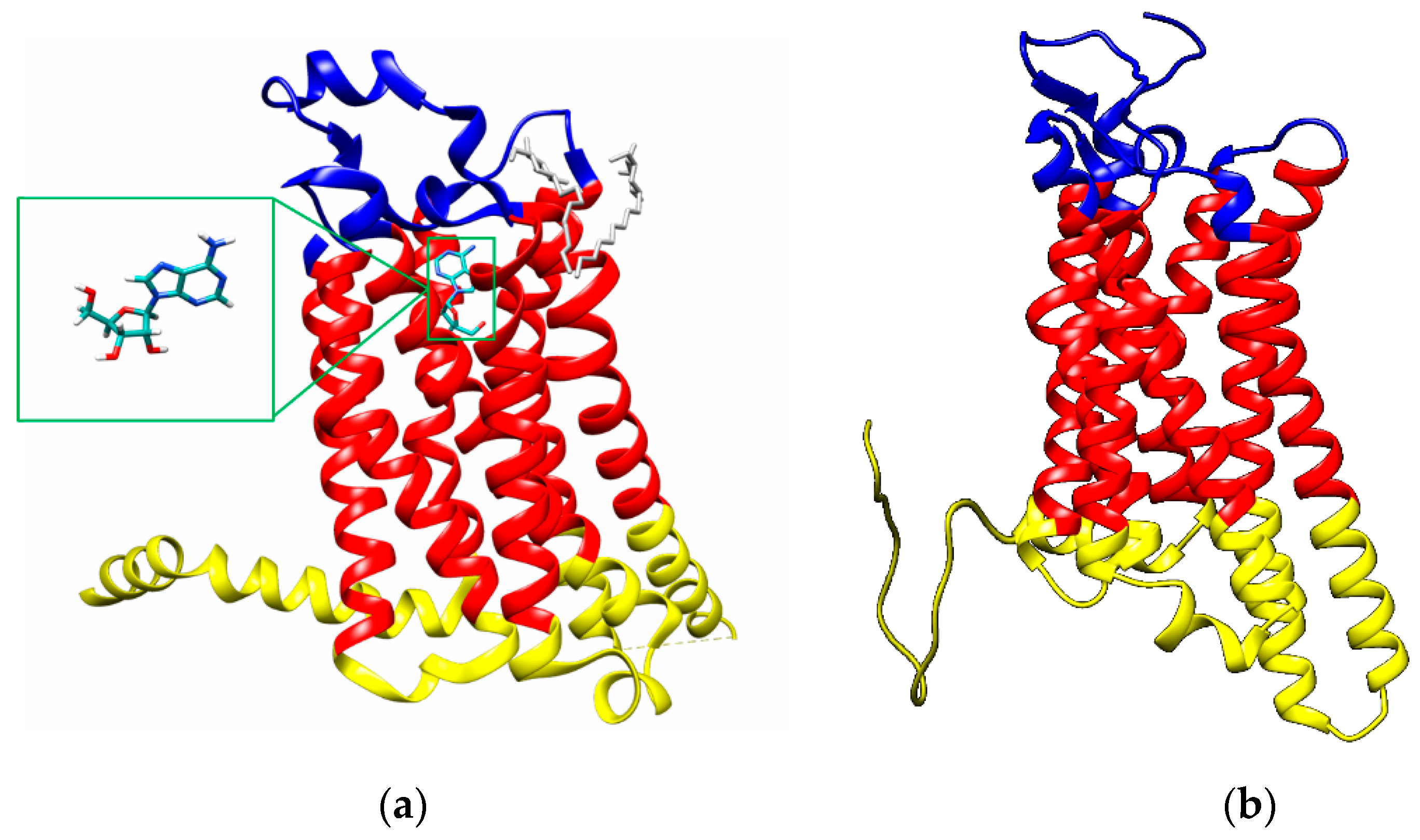
2.1.1. A2A Adenosine Receptor
2.1.2. A3 Adenosine Receptor
2.2. Docking Method Validation and Optimization
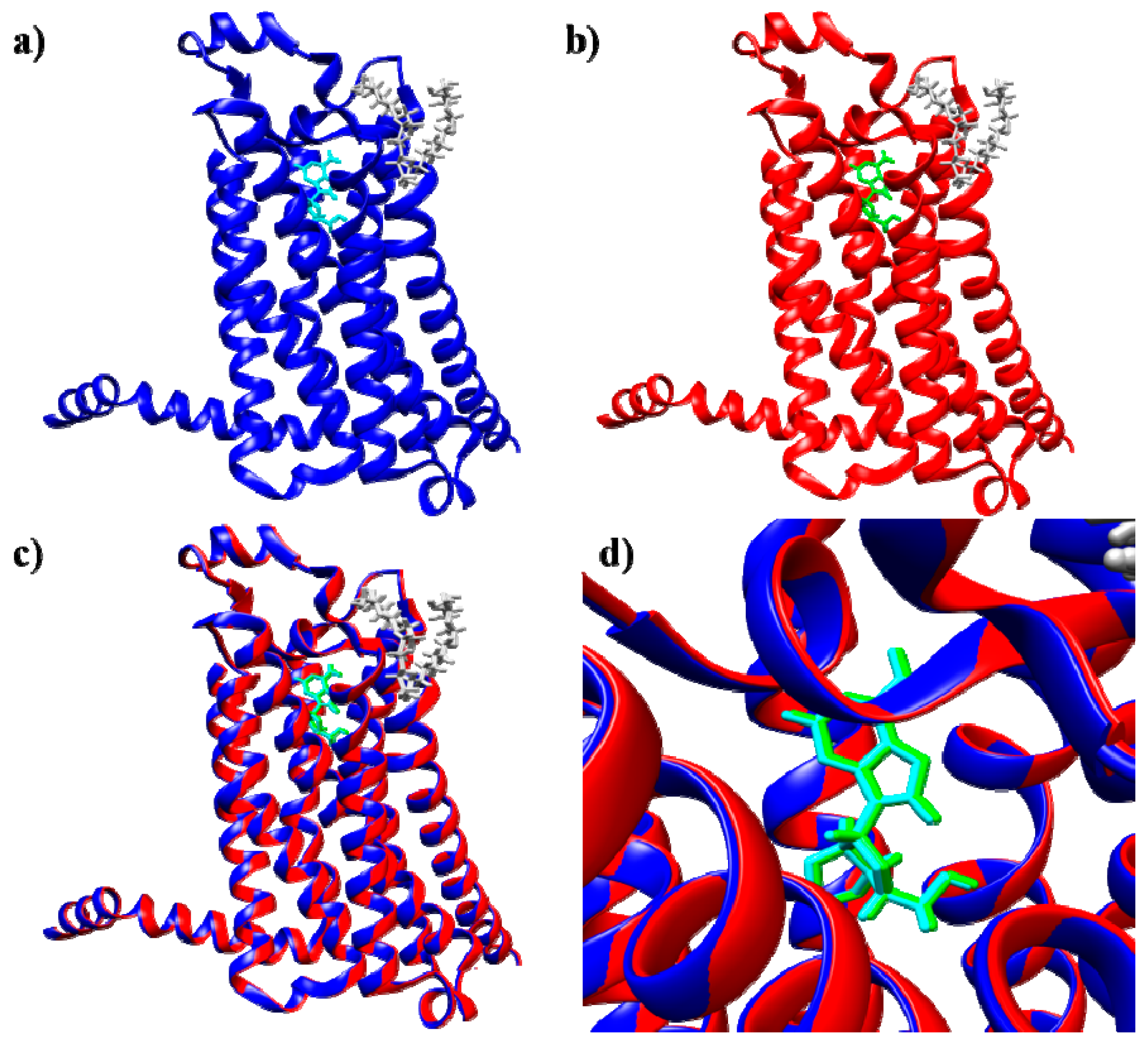
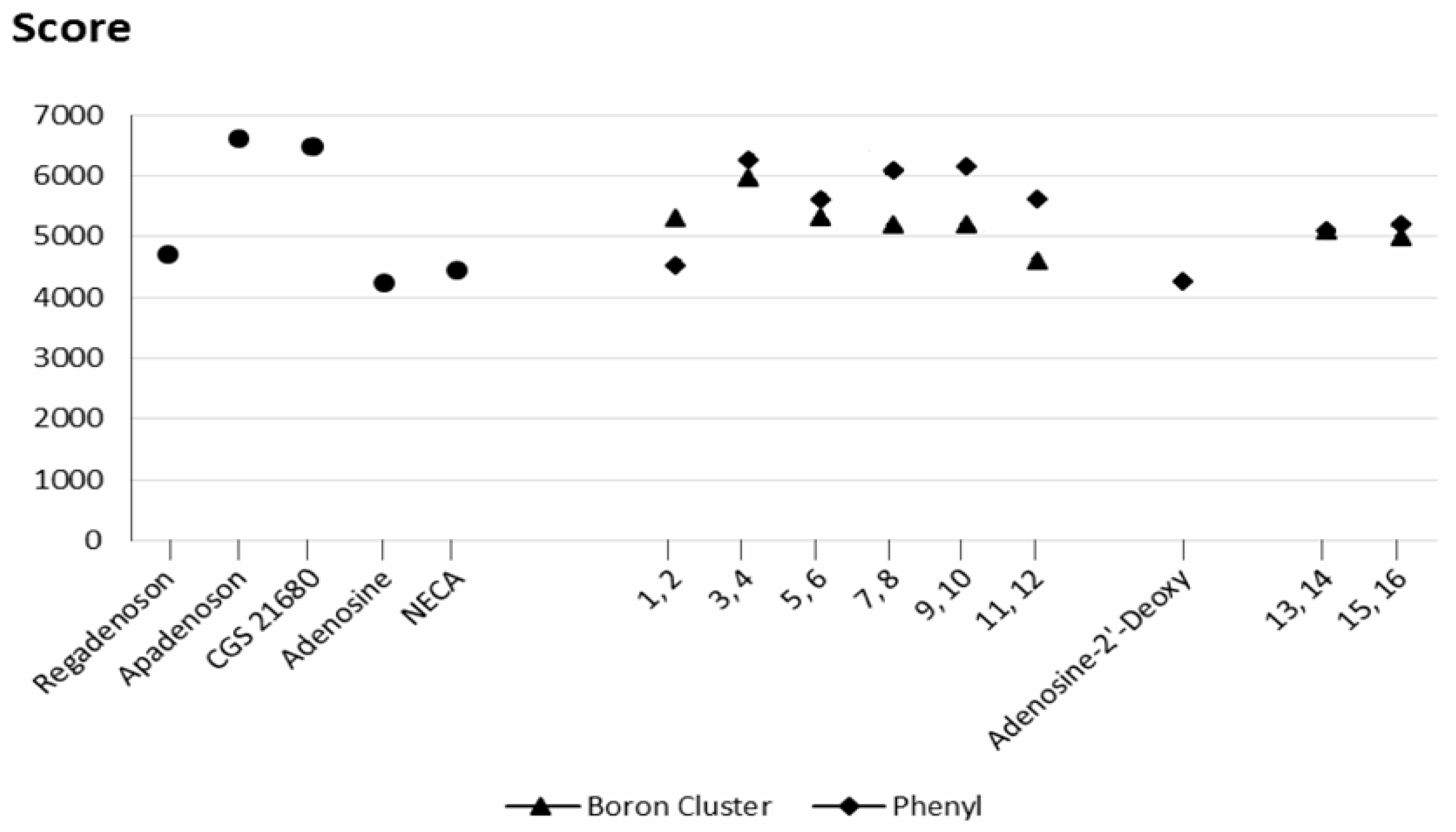
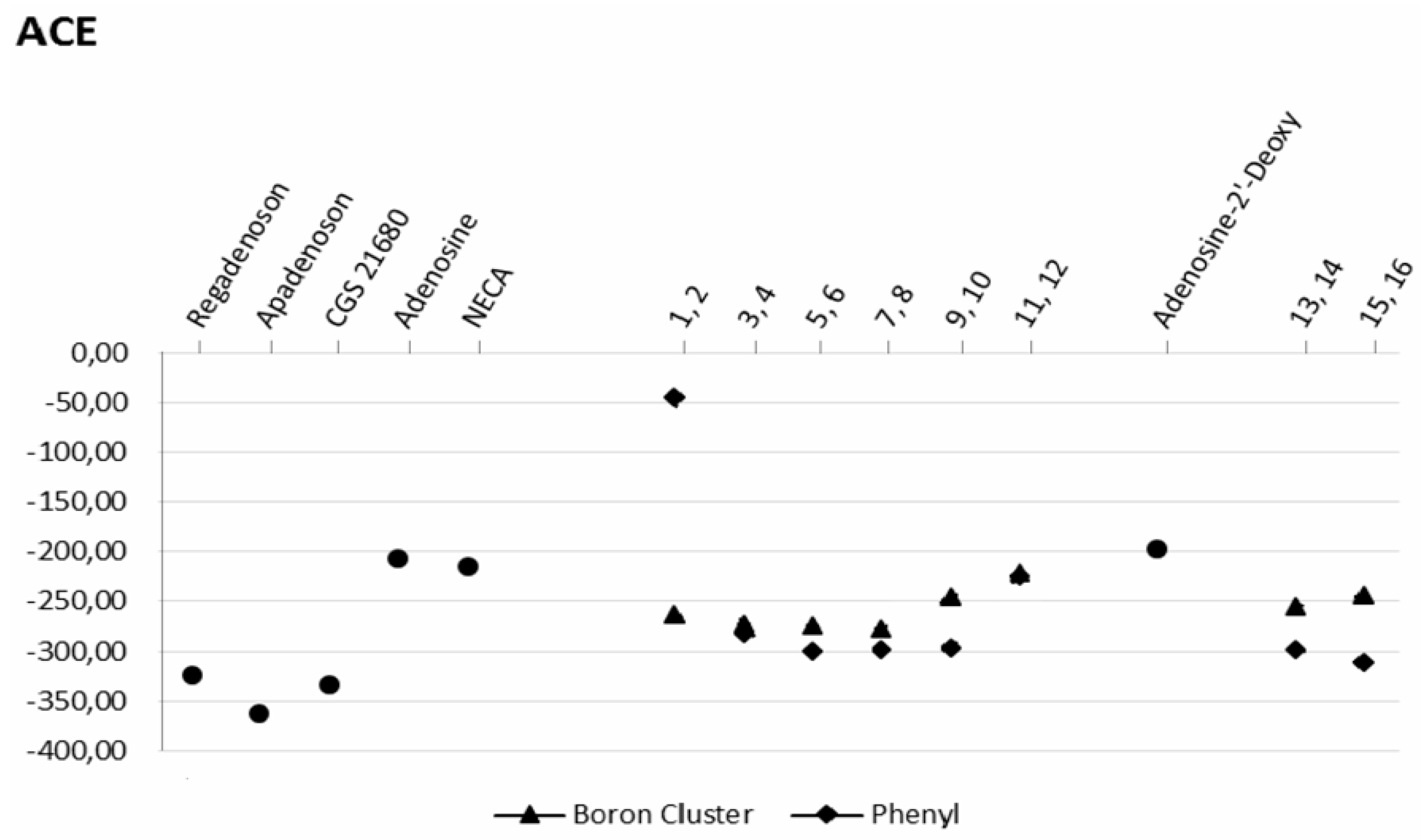
2.2.1. A2A Adenosine Receptor
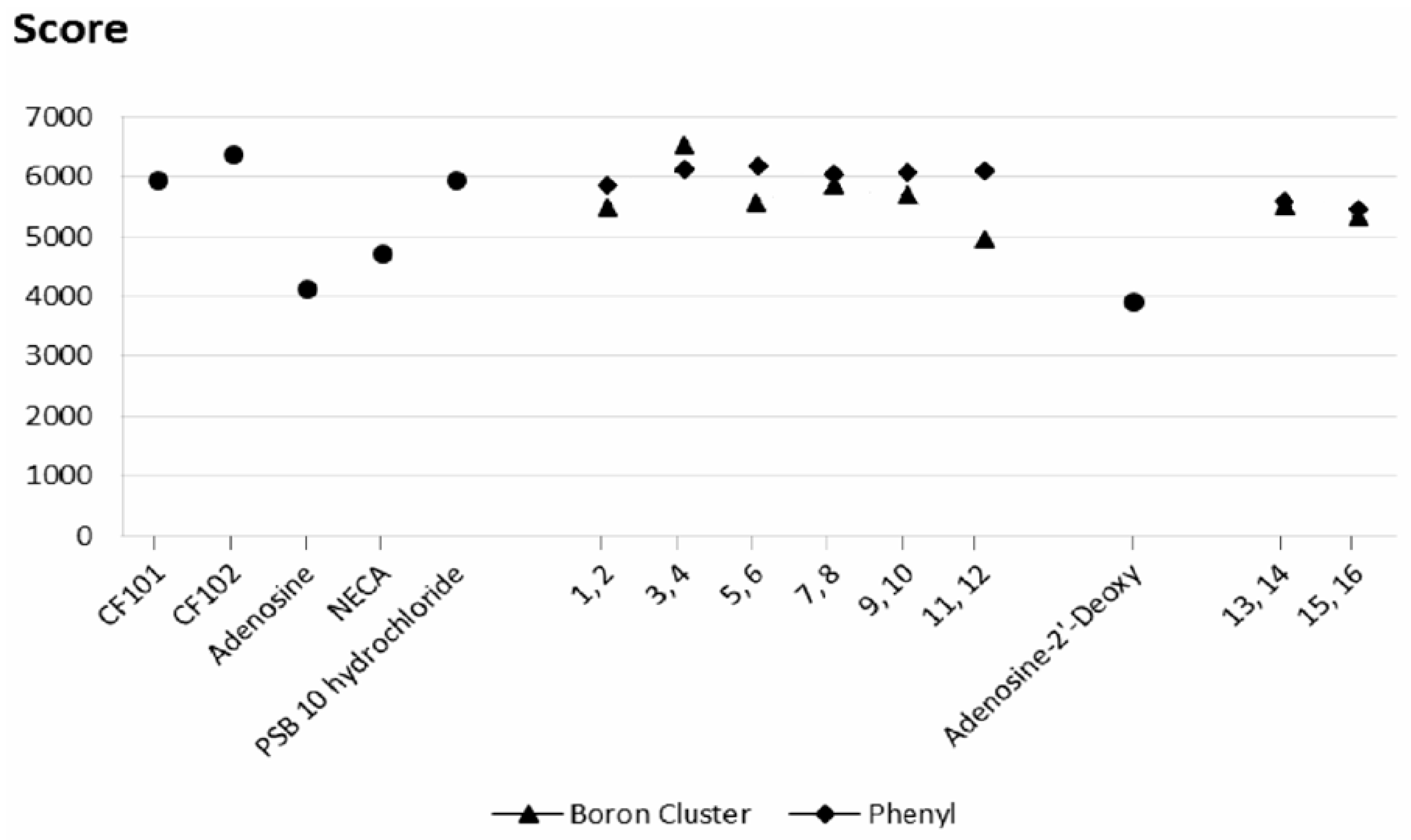
2.2.2. A3 Adenosine Receptor
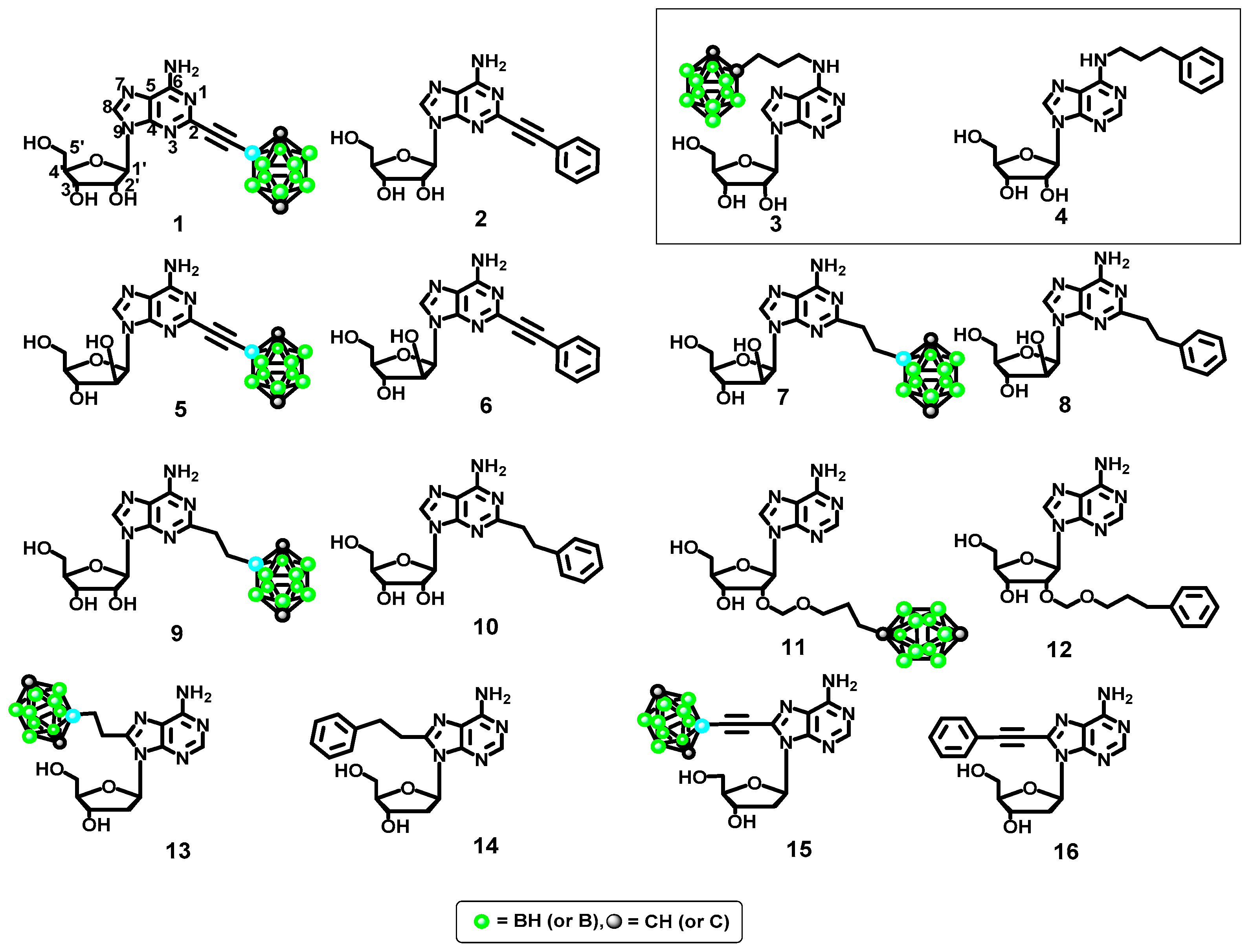
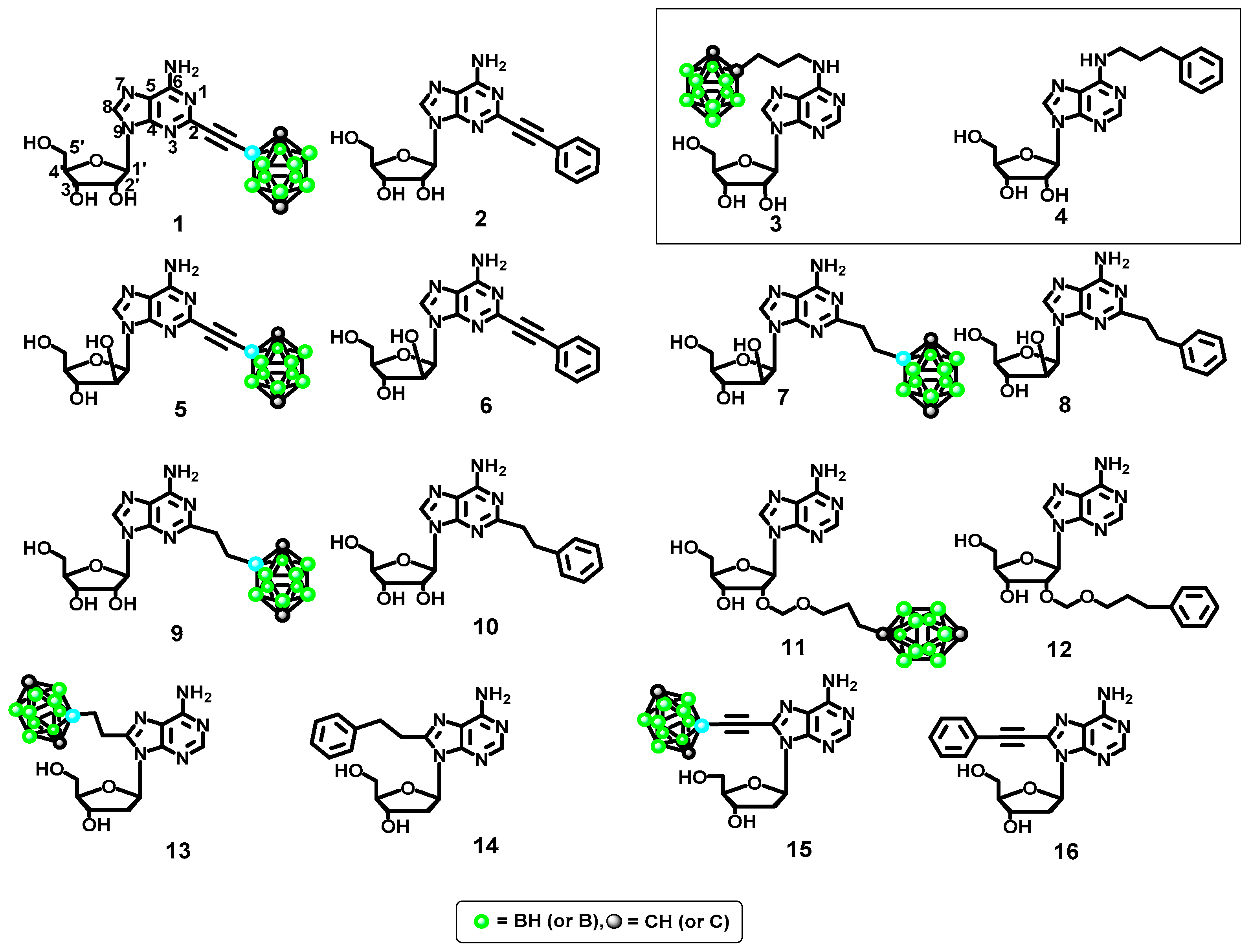
2.2.3. Ligand Modeling
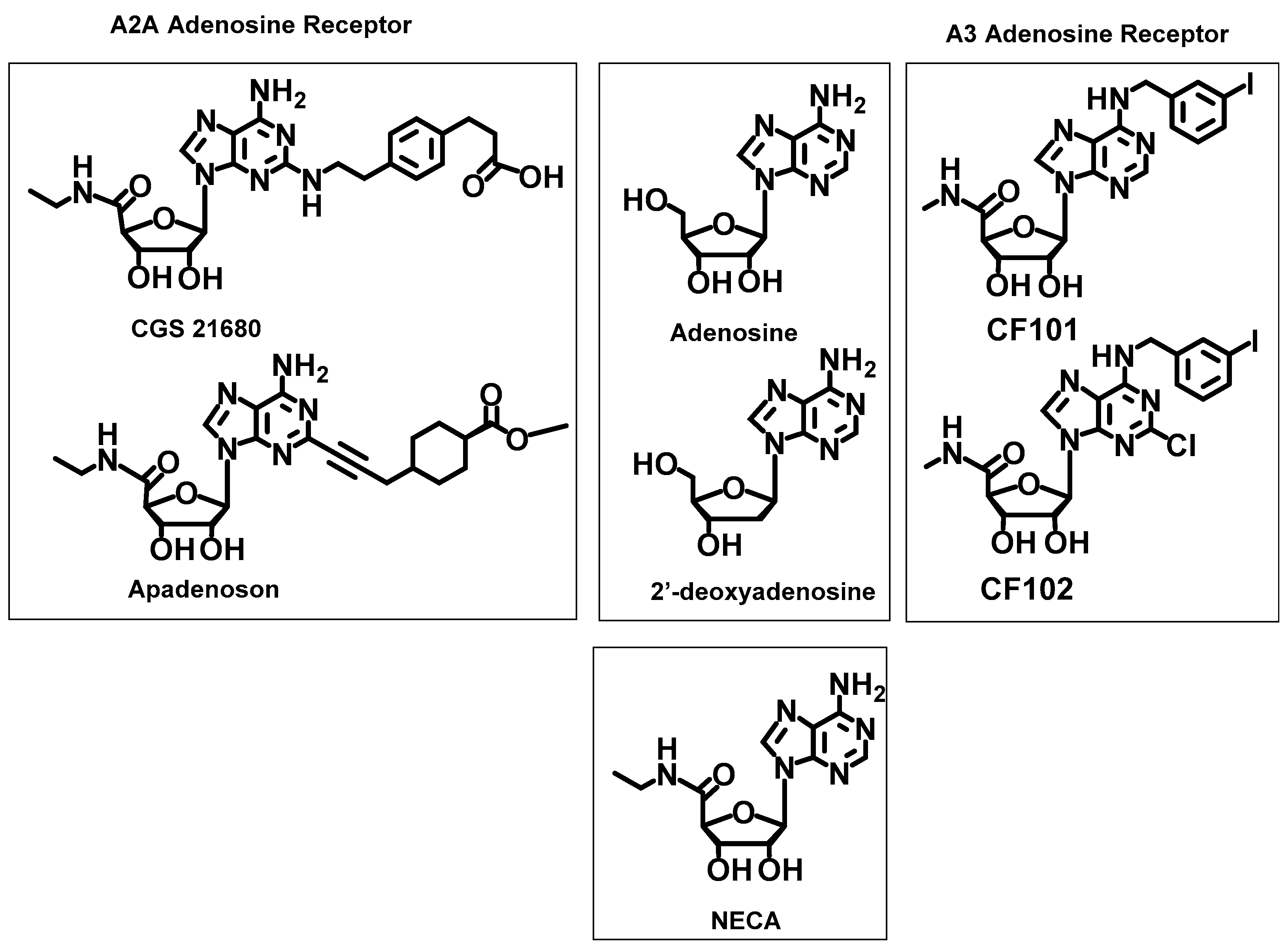
2.2.4. Reference Ligands
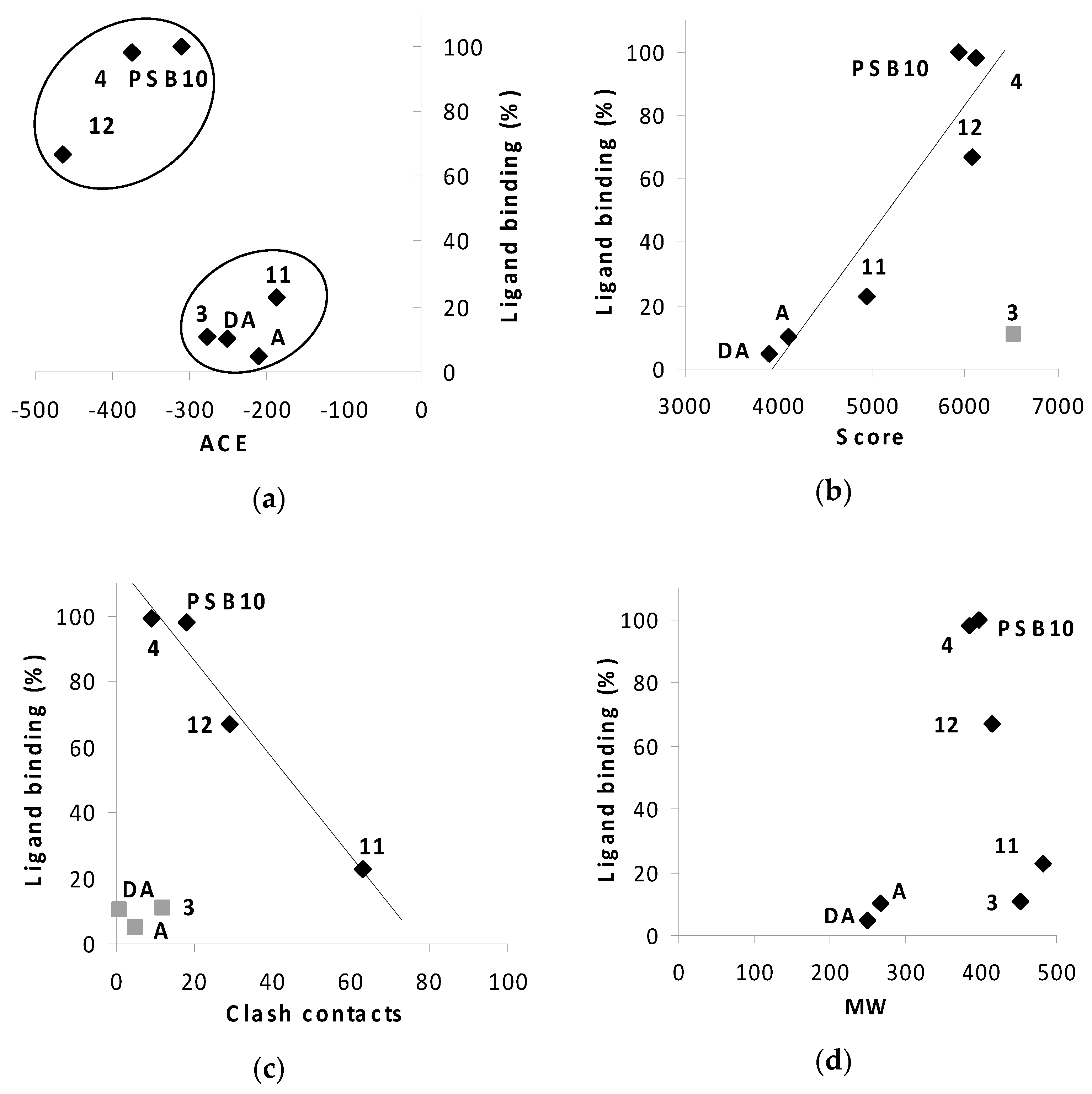
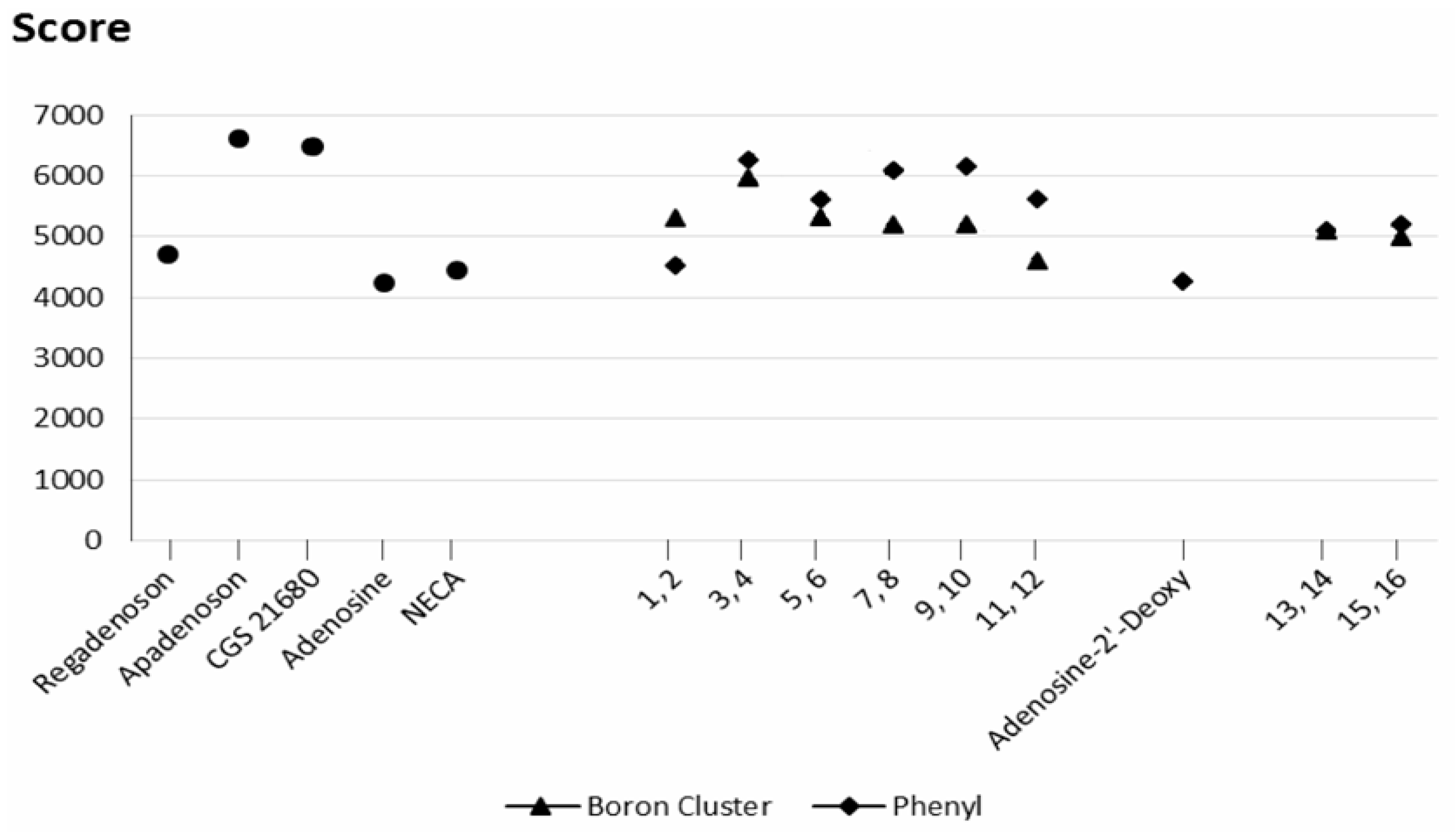
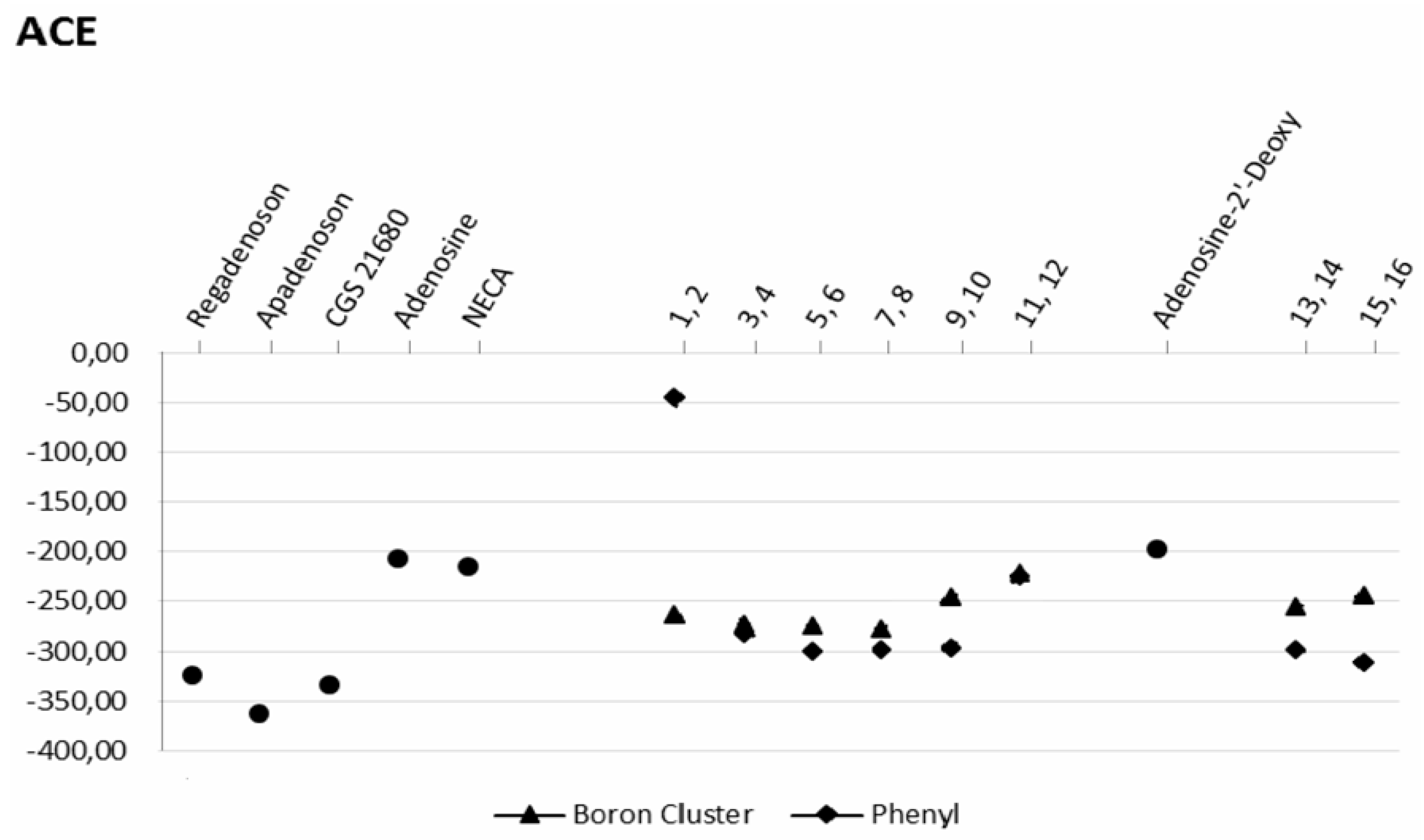
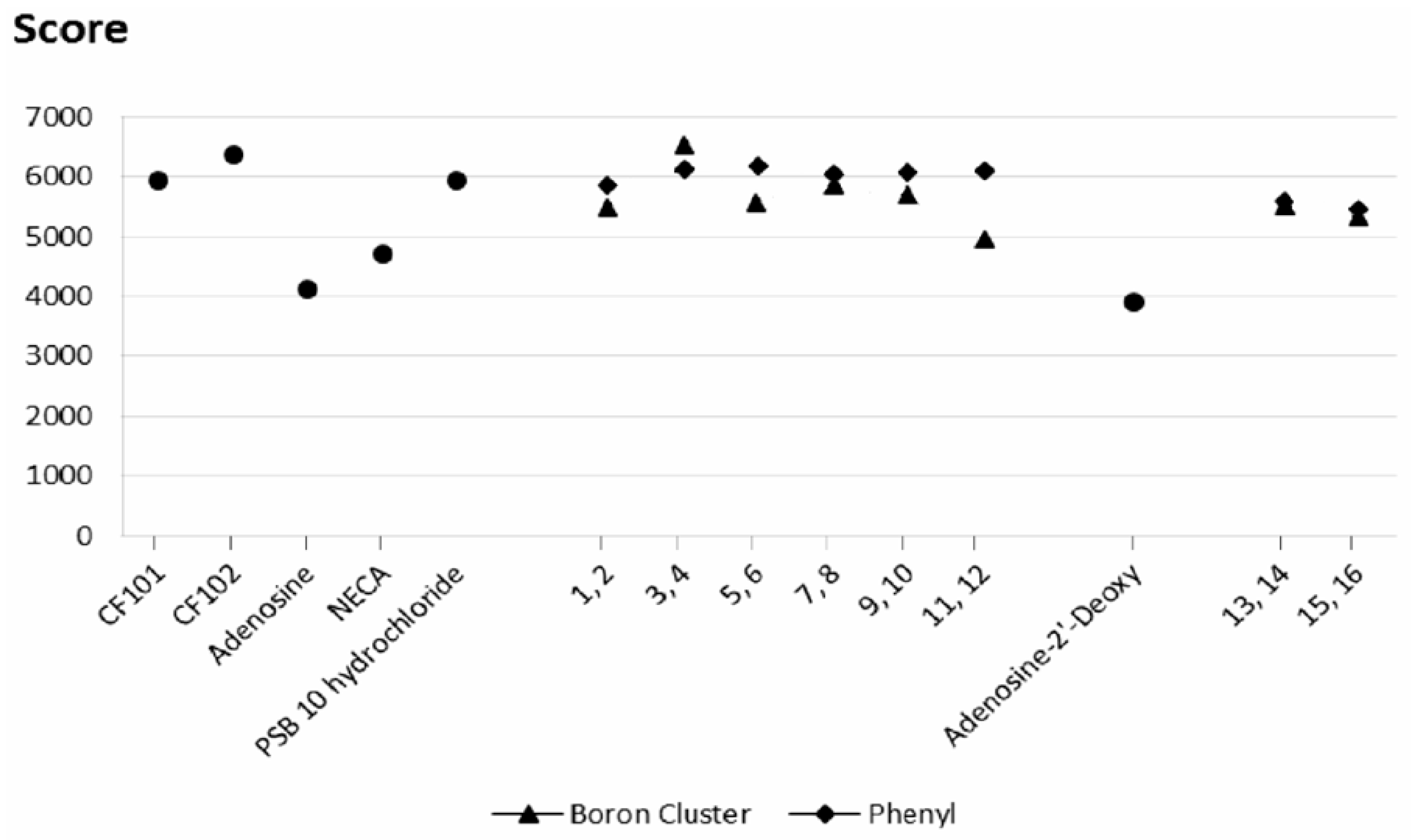
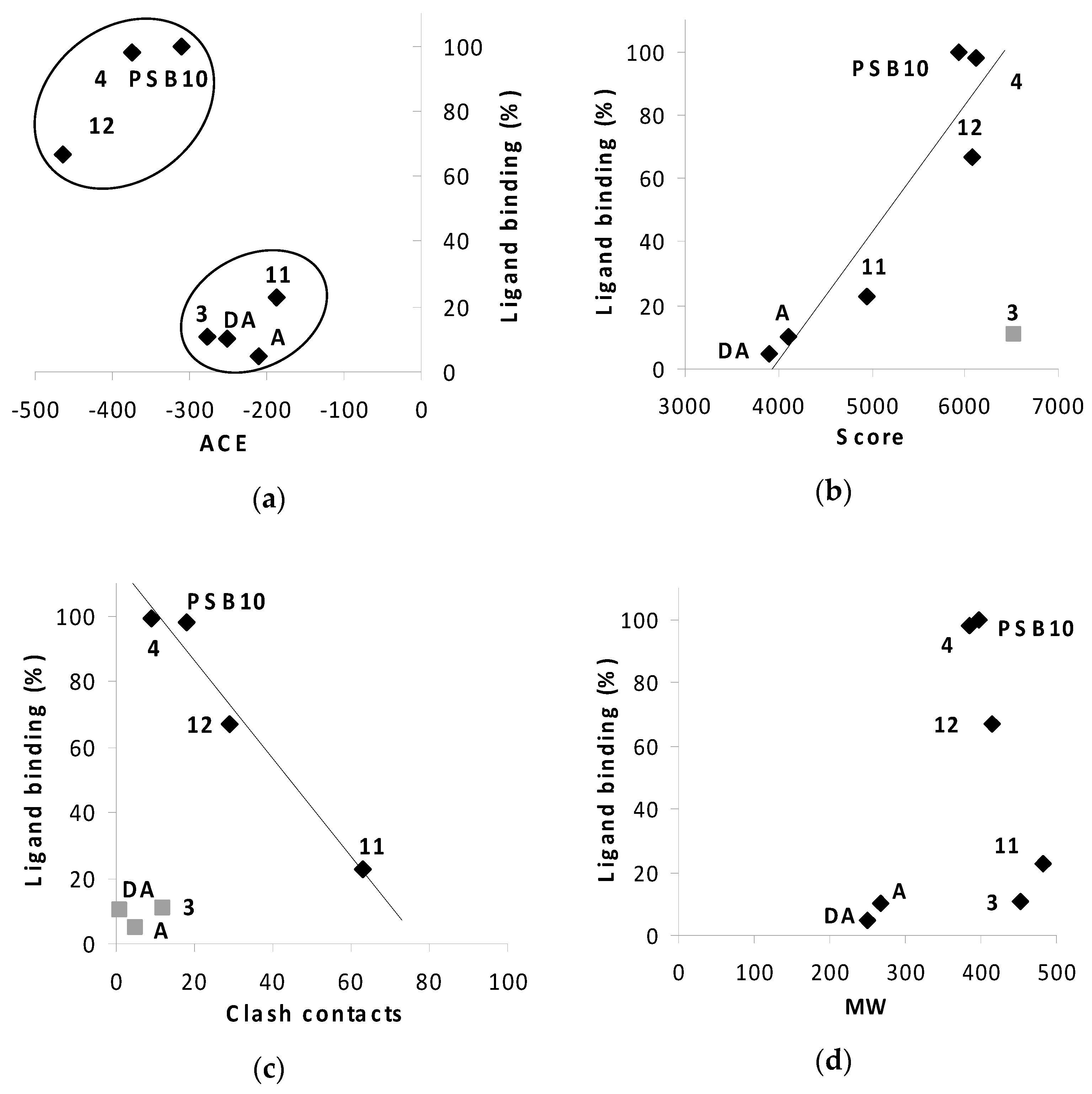
2.3. Docking Results
2.3.1. Phenyl Ring vs. Boron Cluster
2.3.2. Sugar Configuration
2.3.3. Spacer Flexibility
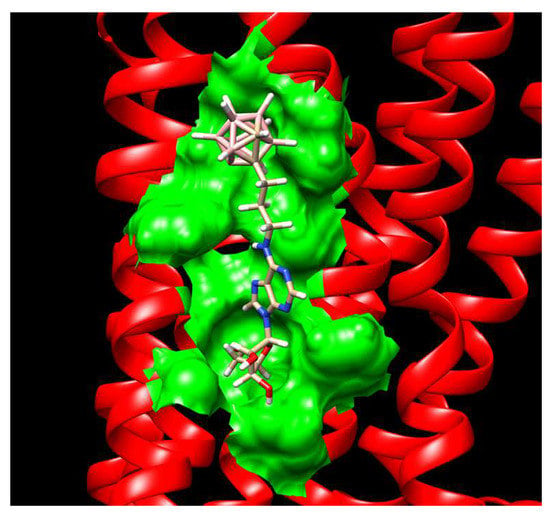
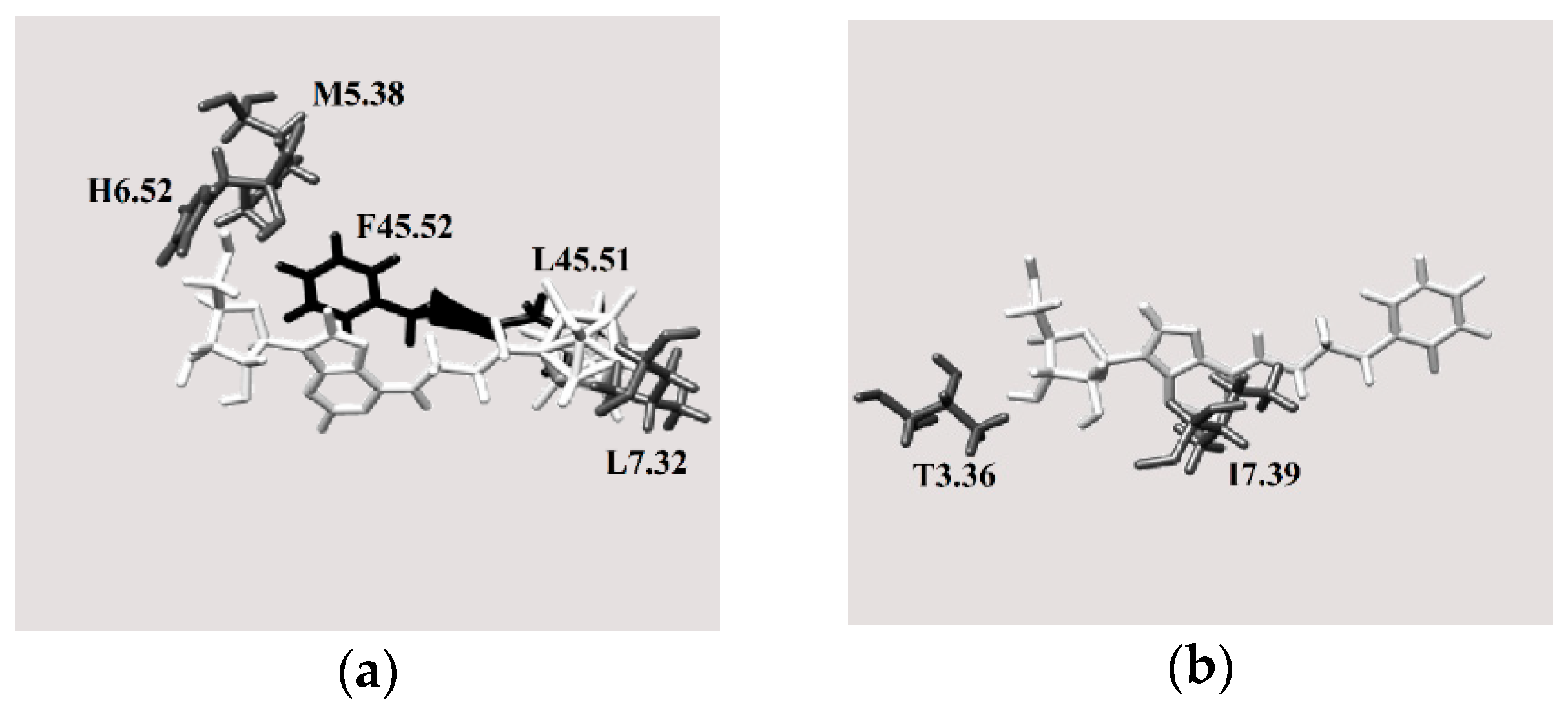
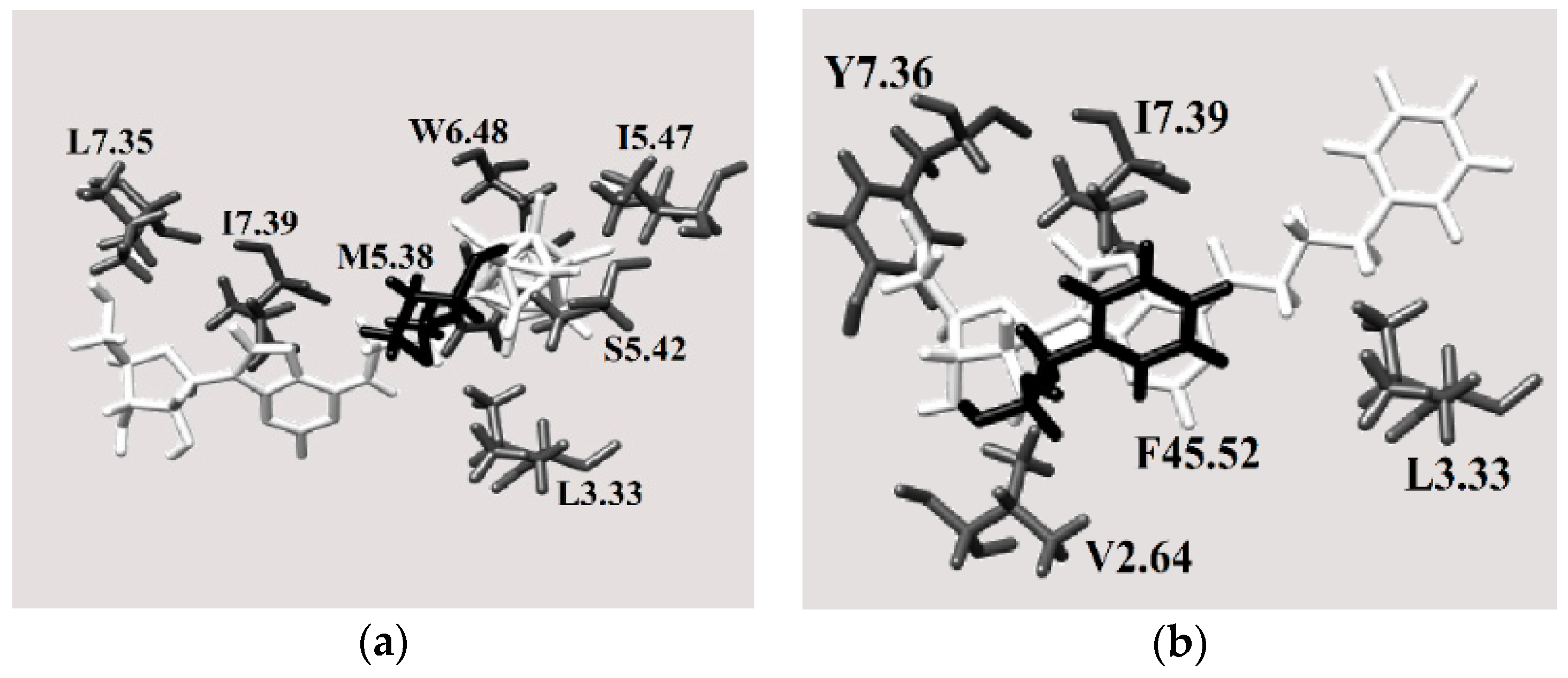
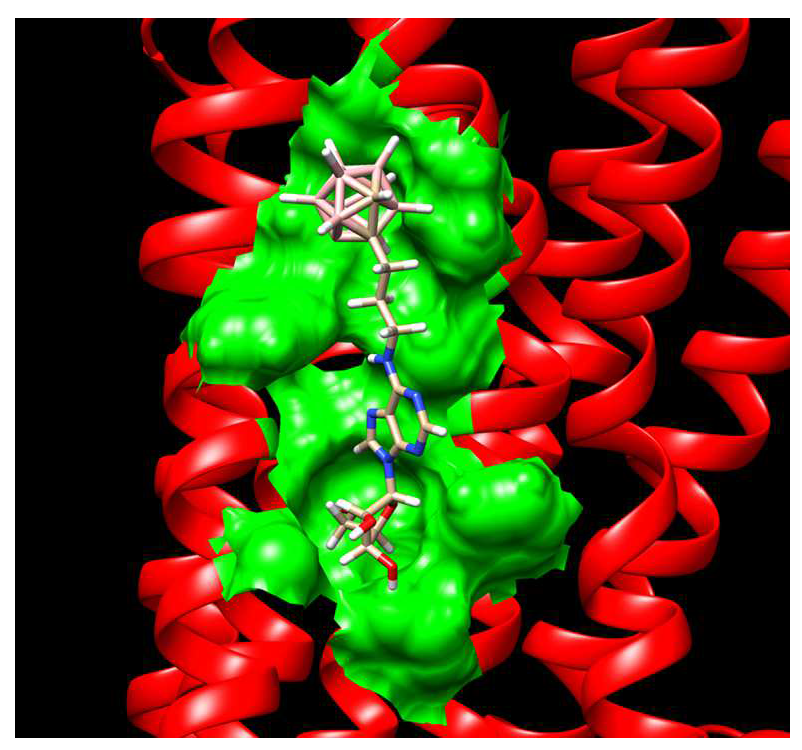
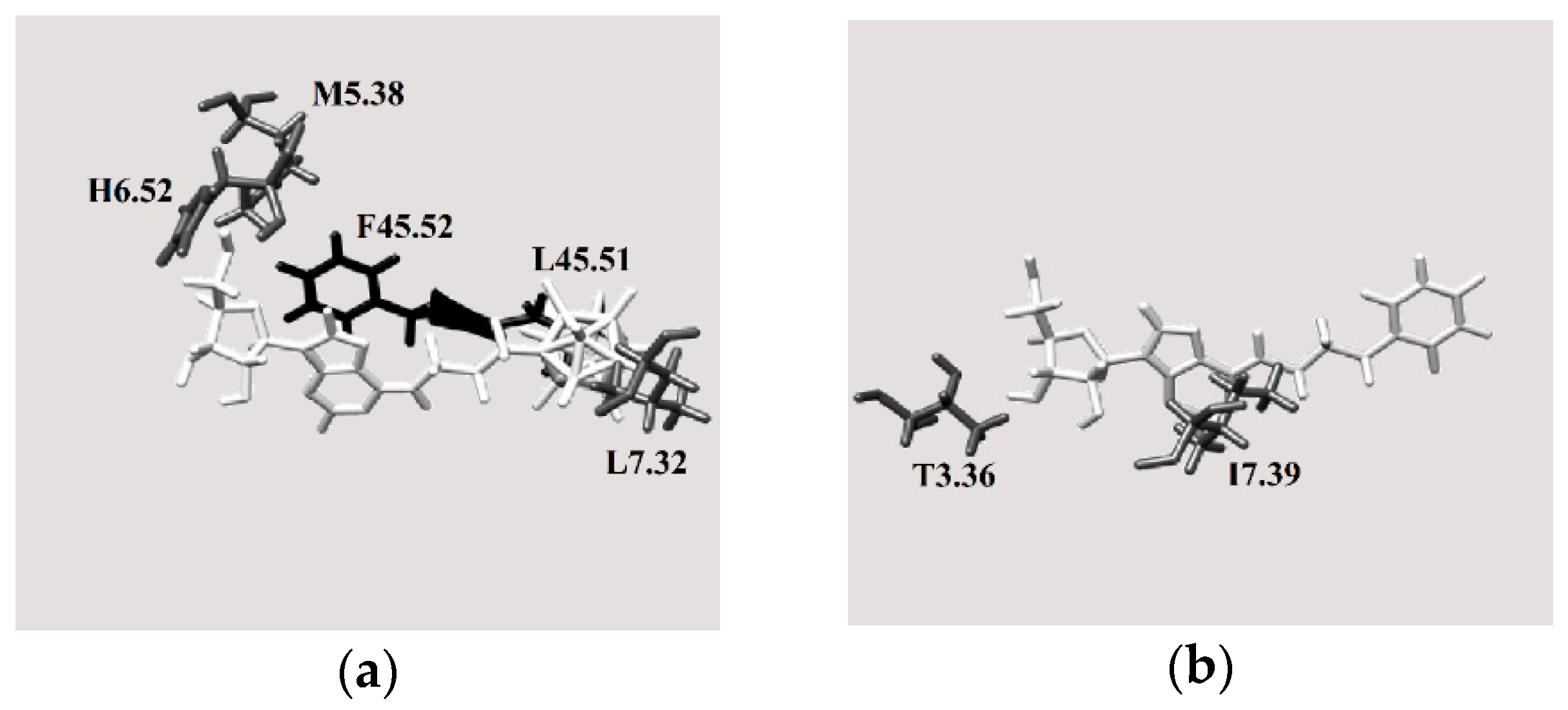
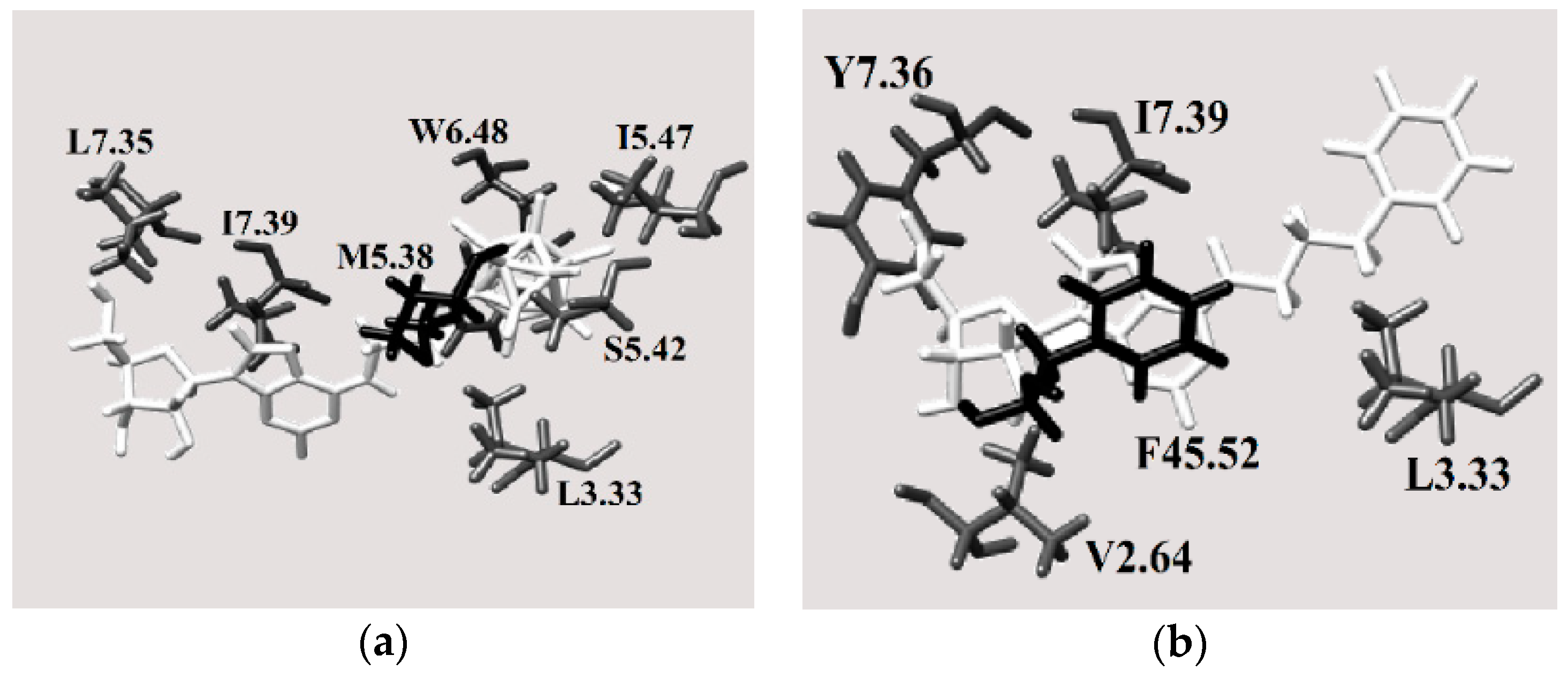
2.3.4. Binding Pocket
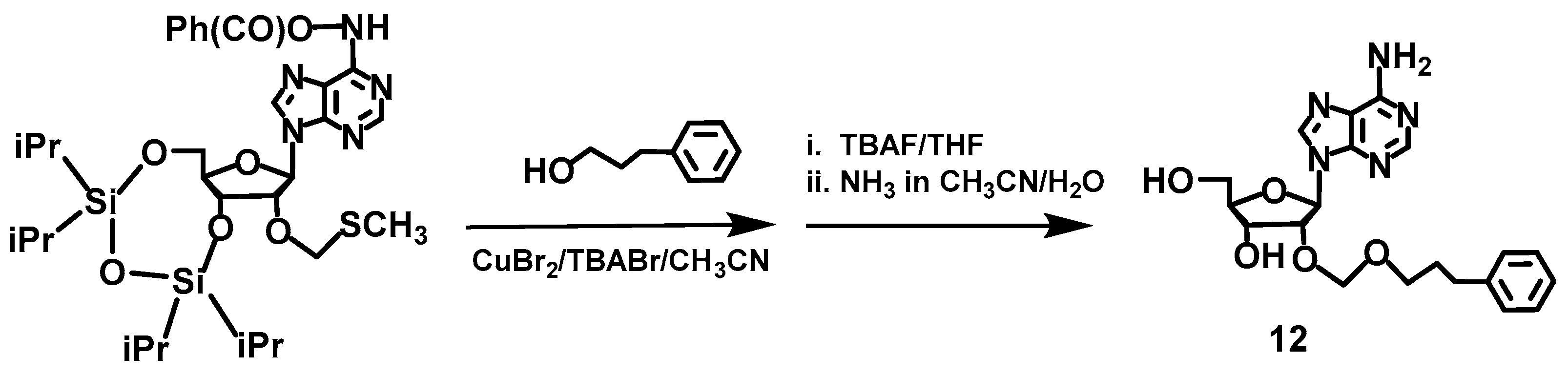
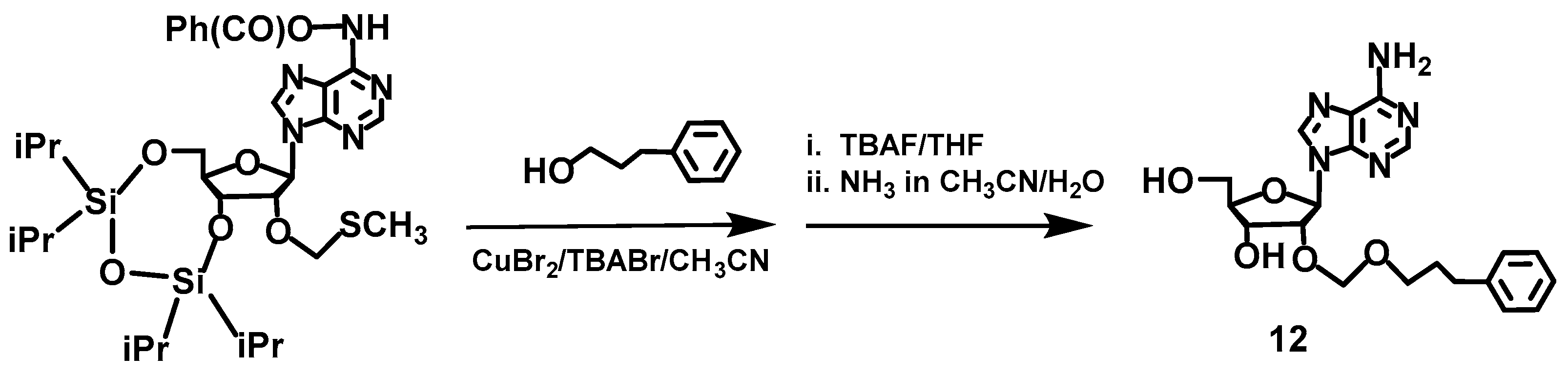
2.4. Synthesis of Compounds 3,4 and 11,12, Which Contain a Boron Cluster or Phenyl Group
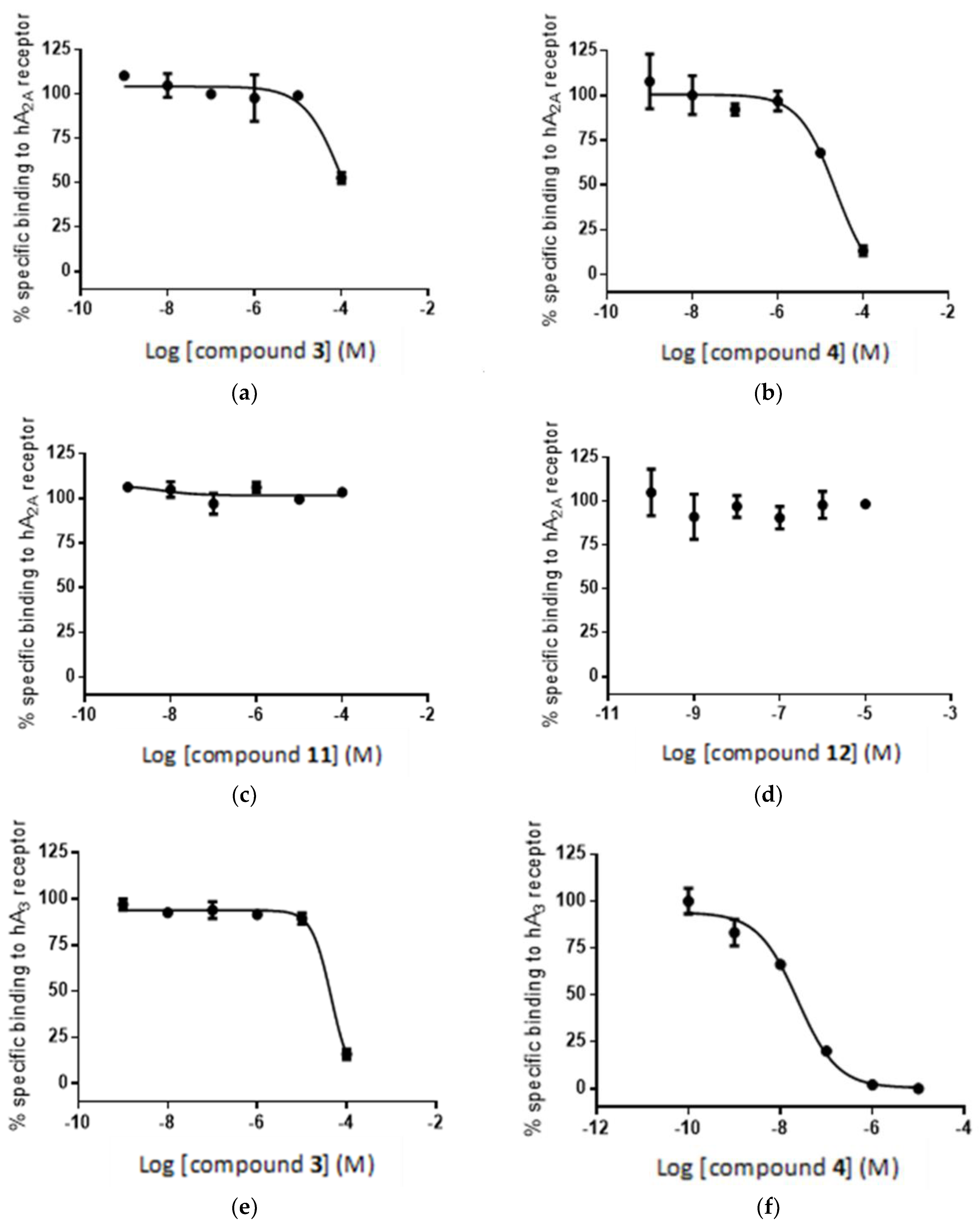
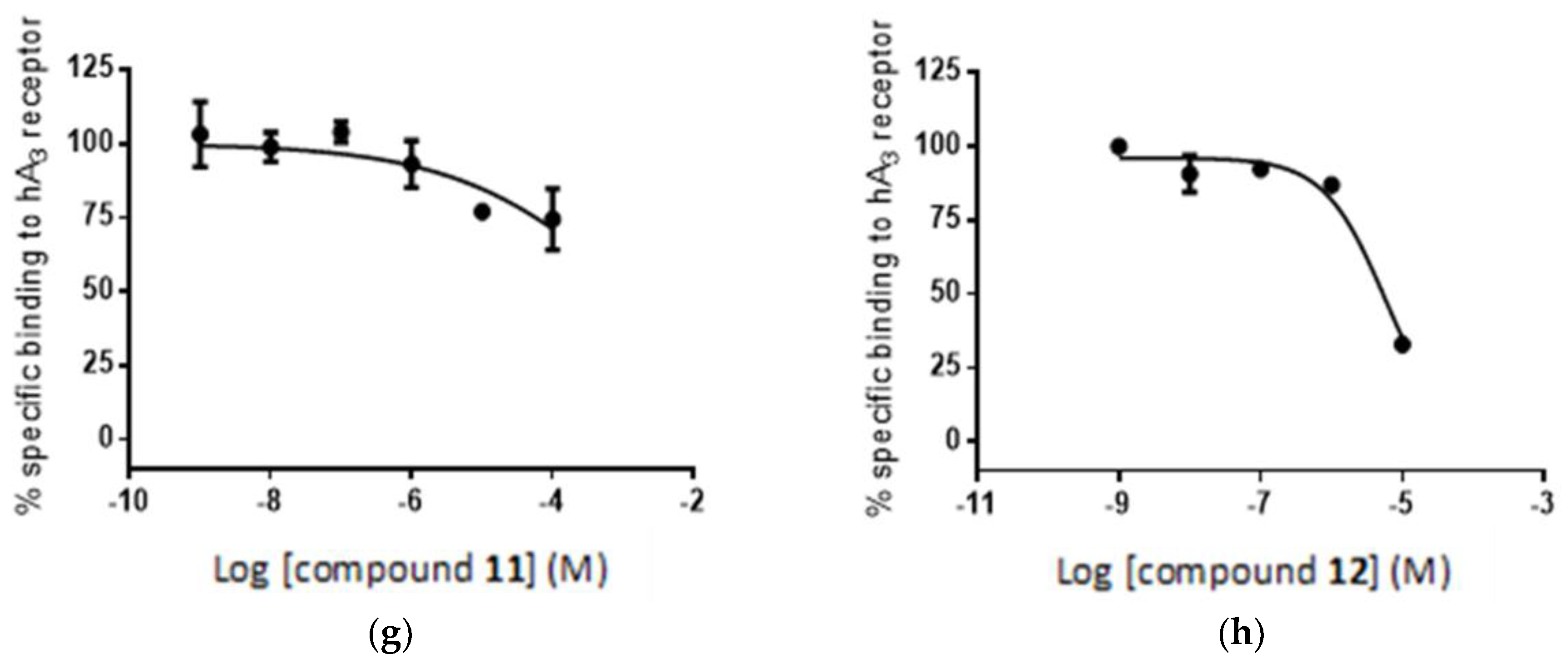
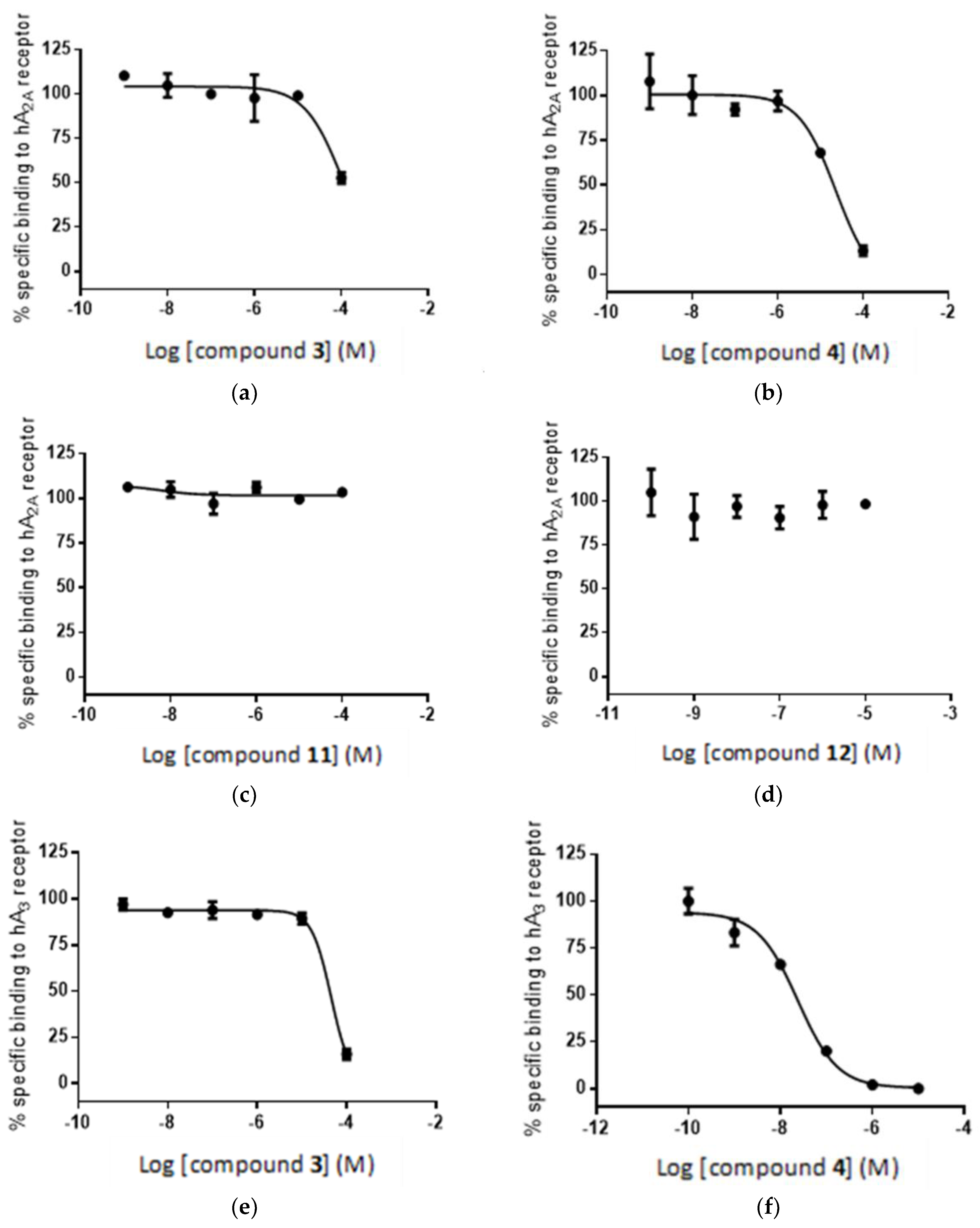
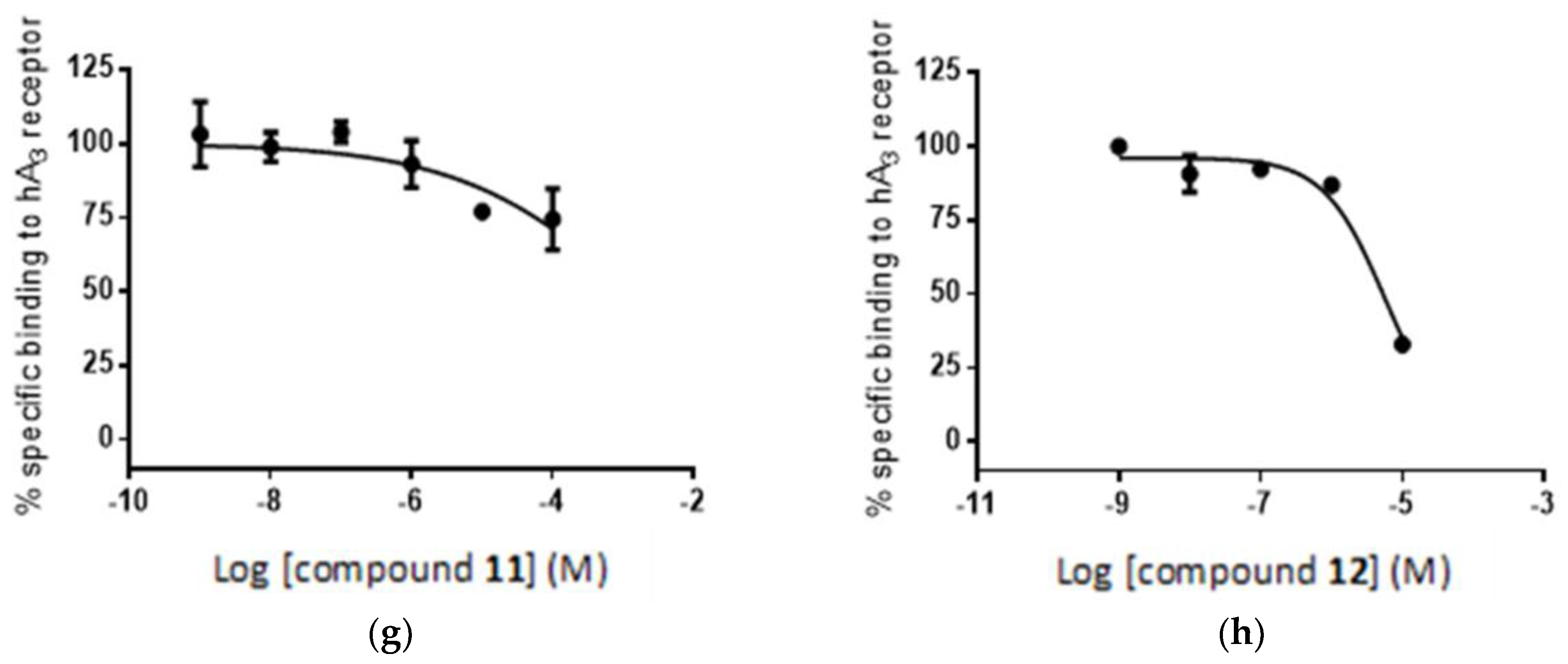
2.5. Radioligand Assay
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2′-O-(3-Phenylpropyleneoxymethyl)-adenosine (12)
3.3. Docking Analysis
3.4. Radioligand Replacement Assay for Human Adenosine Receptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Trincavelli, M.L.; Daniele, S.; Martini, C. Adenosine receptors what we know and what we are learning. Curr. Top. Med. Chem. 2010, 10, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.G.; Ijzerman, A.P. Allosteric modulation of purine and pyrimidine receptors. Adv. Pharmacol. 2011, 61, 187–220. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Daly, J.W.; Manganiello, V.F. Purines in Cellular Signaling—Targets for New Drugs; Springer-Verlag: New York, NY, USA, 1990; ISBN 978-0-387-97244-2. [Google Scholar]
- Tosh, D.K.; Paoletta, S.; Chen, Z.; Moss, S.M.; Gao, Z.G.; Salvemini, D.; Jacobson, K.A. Extended N6 substitution of rigid C2-arylethynyl nucleosides for exploring the role of extracellular loops in ligand recognition at the A3 adenosine receptor. Bioorg. Med. Chem. Lett. 2014, 15, 3302–3306. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Phan, K.; Gao, Z.G.; Gakh, A.A.; Xu, F.; Deflorian, F.; Abagyan, R.; Stevens, R.C.; Jacobson, K.A.; Katritch, V. Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. J. Med. Chem. 2012, 55, 4297–4308. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores—Properties, synthesis, and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef] [PubMed]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Exprt Opin. Drug Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Challenges and opportunities for the application of boron clusters in drug design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.F.; Valiant, J.F. The bioinorganic and medicinal chemistry of carboranes from new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 38, 4240–4251. [Google Scholar] [CrossRef] [PubMed]
- Lesnikowski, Z.J. New opportunities in boron chemistry for medical applications. In Boron Sciences. New Technologies and Applications, 1st ed.; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 3–19. ISBN 9781439826621. [Google Scholar]
- Lesnikowski, Z.J.; Shi, J.; Schinazi, R. Nucleic acids and nucleosides containing carboranes. J. Organomet. Chem. 1999, 581, 156–169. [Google Scholar] [CrossRef]
- Druzina, A.A.; Bregadze, V.I.; Mironov, A.F.; Semioshkin, A.A. Synthesis of conjugates of polyhedral boron hydrides with nucleosides. Russ. Chem. Rev. 2016, 85, 1229–1254. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J.; Olejniczak, A.B.; Schinazi, R.F. Frontiers in Nucleic Acids, 1st ed.; Schinazi, R.F., Liotta, D.C., Eds.; IHL Press: Tucker, GA, USA, 2004; ISBN 0975418807. [Google Scholar]
- Lesnikowski, Z.J. Boron Clusters—A New Entity for DNA-Oligonucleotide Modification. Eur. J. Org. Chem. 2003, 23, 4489–4500. [Google Scholar] [CrossRef]
- Baxter, A.; Bent, J.; Bowers, K.; Braddock, M.; Brough, S.; Fagura, M.; Lawson, M.; McInally, T.; Mortimore, M.; Robertson, M.; et al. Hit-to-Lead studies: The discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 13, 4047–4050. [Google Scholar] [CrossRef] [PubMed]
- Bednarska, K.; Olejniczak, A.B.; Wojtczak, B.; Sułowska, Z.; Leśnikowski, Z.J. Adenosine and 2′-deoxyadenosine modified with boron cluster pharmacophores as new classes of human blood platelet function modulators. Chem. Med. Chem. 2010, 5, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Bednarska, K.; Olejniczak, A.B.; Piskała, A.; Klink, M.; Sułowska, Z.; Leśnikowski, Z.J. Effect of adenosine modified with a boron cluster pharmacophore on reactive oxygen species production by human neutrophils. Bioorg. Med. Chem. 2012, 20, 6621–6629. [Google Scholar] [CrossRef] [PubMed]
- Białek-Pietras, M.; Olejniczak, A.B.; Paradowska, E.; Studzińska, M.; Suski, P.; Jabłońska, A.; Leśnikowski, Z.J. Synthesis and in vitro antiviral activity of lipophilic pyrimidine nucleoside/carborane conjugates. J. Organomet. Chem. 2015, 798, 99–105. [Google Scholar] [CrossRef]
- Żołnierczyk, J.D.; Olejniczak, A.B.; Mieczkowski, A.J.; Błoński, J.Z.; Kiliańska, Z.M.; Robak, T.; Leśnikowski, Z.J. In vitro antileukemic activity of novel adenosine derivatives bearing boron cluster modification. Bioorg. Med. Chem. 2016, 24, 5076–5087. [Google Scholar] [CrossRef] [PubMed]
- Bours, M.J.L.; Swennen, E.L.R.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Levin, R.I.; Belanoff, J.; Weissmann, G.; Hirschhorn, R. Adenosine: An endogenous inhibitor of neutrophil-mediated injury to endothelial cells. J. Clin. Investig. 1986, 78, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Pacher, P.J. A2A receptors in inflammation and injury: Lessons learned from transgenic animals. J. Leukoc. Biol. 2008, 83, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Areias, F.M.; Brea, J.; Gregori-Puigjané, E.; Zaki, M.E.A.; Carvalho, M.A.; Domínguez, E.; Gutiérrez-de-Terán, H.; Proença, M.F.; Loza, M.I.; Mestres, J. In silico directed chemical probing of the adenosine receptor family. Bioorg. Med. Chem. 2010, 18, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.; Hemmings, J.L.; Winfield, I.; Leuenberger, M.; Frattini, E.; Frenguelli, B.G.; Dowell, S.J.; Lochner, M.; Ladds, G. Discovery of novel adenosine receptor agonists that exhibit subtype selectivity. J. Med. Chem. 2016, 59, 947–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirci, F.; Goracci, L.; Rodriguez, D.; van Muijlwijk-Koezen, J.; Gutierrez-de-teran, H.; Mannhold, R. Ligand-, structure- and pharmacophore-based molecular fingerprints: A case study on adenosine A1, A 2A, A2B and A3 receptor antagonists. J. Comput. Aided Mol. Des. 2012, 26, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Almerico, A.M.; Tutone, M.; Pantano, L.; Lauria, A. A3 adenosine receptor: Homology modeling and 3D-QSAR studies. J. Mol. Graph. Model. 2013, 42, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucl. Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucl. Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; Jzerman, A.P.; Marshall, F.H.; et al. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucl. Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Sevryugina, Y.V.; Khan, A.; Ye, S.Q. Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase. J. Med. Chem. 2012, 55, 7290–7294. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.M.; Gunosewoyo, H.; Barron, M.L.; Boucher, A.; McDonnell, M.; Turner, P.; Morrison, D.E.; Bennett, M.R.; Mc Gregor, I.S.; Rendina, L.M.; et al. The first CNS-active carborane: A novel P2X7 receptor antagonist with antidepressant activity. ACS Chem. Neurosci. 2014, 5, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kracke, G.R.; VanGordon, M.R.; Sevryugina, Y.V.; Kueffer, P.J.; Kabytaev, K.; Jalisatgi, S.S.; Hawthorne, M.F. Carborane-derived local anesthetics are isomer dependent. Chem. Med. Chem. 2015, 10, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Mahasenan, K.; Pavlovicz, R.; Li, C.; Tjarks, W.J. Carborane clusters in computational drug design: A comparative docking evaluation using AutoDock, FlexX, Glide and Surflex. J. Chem. Inf. Model. 2009, 49, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.; Thirumamagal, B.T.; Yang, W.; Eriksson, S.; Barth, R.F.; Tjarks, W. Preparation and biological evaluation of 10B-enriched 3-[5-{2-(2,3-dihydroxyprop-1-yl)-o-carboran-1-yl}pentan-1-yl]thymidine (N5-2OH), a new boron delivery agent for boron neutron capture therapy of brain tumors. J. Med. Chem. 2006, 18, 5513–5523. [Google Scholar] [CrossRef] [PubMed]
- Johnsamuel, J.; Byun, Y.; Jones, T.P.; Endo, Y.; Tjarks, W. A new strategy for molecular modeling and receptor-based design of carborane containing compounds. J. Organomet. Chem. 2003, 680, 223–231. [Google Scholar] [CrossRef]
- Narayanasamy, S.; Thirumamagal, B.T.; Johnsamuel, J.; Byun, Y.; Al-Madhoun, A.S.; Usova, E.; Cosquer, G.Y.; Yan, J.; Bandyopadhyaya, A.K.; Tiwari, R.; et al. Hydrophilically enhanced 3-carboranyl thymidine analogues (3CTAs), for boron neutron capture therapy BNCT, of cancer. Bioorg. Med. Chem. 2006, 14, 6886–6899. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, R.; Fanfrlik, J.; Pecina, A.; Hnyk, D.; Hobza, P.; Lepšík, M. Noncovalent Interactions of Heteroboranes. In Boron: The Fifth Element (Challenges and Advances in Computational Chemistry and Physics), 1st ed.; Hnyk, D., McKee, M.L., Eds.; Springer International Publishing: Basel, Switzerland, 2015; Volume 20, pp. 219–239. ISBN 978-3-319-22281-3. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Rork, T.H.; Wallace, K.L.; Kennedy, D.P.; Marshall, M.A.; Lankford, A.R.; Linden, J. Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am. J. Physiol. Heart Circ. Physiol. 2008, 5, H1825–H1833. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- SCH 58261 Product Datasheet. Available online: https//www.tocris.com/products/sch-58261_2270; https//documents.tocris.com/pdfs/tocris_coa/2270_9_coa.pdf (accessed on 21 August 2017).
- Nishikawa, S.; Kumazawa, Z.; Kashimura, N.; Niskimi, Y.; Uemura, S. Alternating Dependency of Cytokinin Activity on the Number of Methylene Units in ω-Phenylalkyl Derivatives of Some Purine Cytokinins and 4-Substituted Pyrido[3,4-d]pyrimidme. Agric. Biol. Chem. 1986, 50, 2243–2249. [Google Scholar] [CrossRef]
- Wojtczak, B.; Semenyuk, A.; Olejniczak, A.B.; Kwiatkowski, M.; Lesnikowski, Z.J. General method for the synthesis of 2′-O-carboranyl-nucleosides. Tetrahedron Lett. 2005, 46, 3969–3972. [Google Scholar] [CrossRef]
- Sproat, B.S. Synthesis of 2′-O-Alkyloligoribonucleotides. In Protocols for Oligonucleotides and Analogs. Synthesis and Properties, 1st ed.; Agrawal, S., Ed.; Humana Press Inc.: Totowa, NJ, USA, 1993; pp. 115–141. ISBN 978-0-89603-281-1. [Google Scholar]
- Hovinen, J.; Azhayeva, E.; Azhayev, A.; Guzaev, A.; Lönberg, H. Synthesis of 3′-O-(ω-aminoalkoxymethyl,thymidine 5′-triphosphates, terminators of DNA synthesis that enable 3′-labelling. J. Chem. Soc. Perkin Trans. 1994, 2, 211–217. [Google Scholar] [CrossRef]
- Volpini, R.; Costanzi, S.; Lambertucci, C.; Taffi, S.; Vittori, S.; Klotz, K.-N.; Cristalli, G. N(6,-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3, receptor and a starting point for searching A2B, ligands. J. Med. Chem. 2002, 45, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Volpini, R.; Cristalli, G. The importance of alkynyl chain presence for the activity of adenine nucleosides/nucleotides on purinergic receptors. Curr. Med. Chem. 2011, 18, 1444–1463. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3, 4, 11 and 12 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Inhib. 10 μM A2A | % Inhib. 10 μM A3 | Ki (nM) A3 |
|---|---|---|---|
| Adenosine 2′-deoxyadenosine 3 4 11 12 | 2 ± 1 | 10 ± 1 | n.d. |
| 3 ± 1 | 5 ± 1 | n.d. | |
| 2 ± 2 | 11 ± 4 | n.d. | |
| 32 ± 1 | 98 ± 1 | 7.5 | |
| 1 ± 1 | 23 ± 3 | n.d. | |
| 2 ± 1 | 67 ± 1 | 2208 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincenzi, M.; Bednarska, K.; Leśnikowski, Z.J. Comparative Study of Carborane- and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay. Molecules 2018, 23, 1846. https://doi.org/10.3390/molecules23081846
Vincenzi M, Bednarska K, Leśnikowski ZJ. Comparative Study of Carborane- and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay. Molecules. 2018; 23(8):1846. https://doi.org/10.3390/molecules23081846
Chicago/Turabian StyleVincenzi, Marian, Katarzyna Bednarska, and Zbigniew J. Leśnikowski. 2018. "Comparative Study of Carborane- and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay" Molecules 23, no. 8: 1846. https://doi.org/10.3390/molecules23081846