
Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors

Department of Biology, Geology and Physical Sciences, Sul Ross State University, Alpine, TX 79832, USA
Molecules 2018, 23(7), 1753; https://doi.org/10.3390/molecules23071753
Submission received: 20 June 2018
/
Revised: 13 July 2018
/
Accepted: 15 July 2018
/
Published: 17 July 2018
(This article belongs to the Special Issue Chemical Biology of Sterols, Triterpenoids and Other Natural Products: A Themed Issue in Honor of Professor W. David Nes on the Occasion of His 65th Birthday)
Abstract
:Sterol 14α-demethylase (SDM) is essential for sterol biosynthesis and is the primary molecular target for clinical and agricultural antifungals. SDM has been demonstrated to be a valid drug target for antiprotozoal therapies, and much research has been focused on using SDM inhibitors to treat neglected tropical diseases such as human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis. Sterol C24-methyltransferase (24-SMT) introduces the C24-methyl group of ergosterol and is an enzyme found in pathogenic fungi and protozoa but is absent from animals. This difference in sterol metabolism has the potential to be exploited in the development of selective drugs that specifically target 24-SMT of invasive fungi or protozoa without adversely affecting the human or animal host. The synthesis and biological activity of SDM and 24-SMT inhibitors are reviewed herein.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
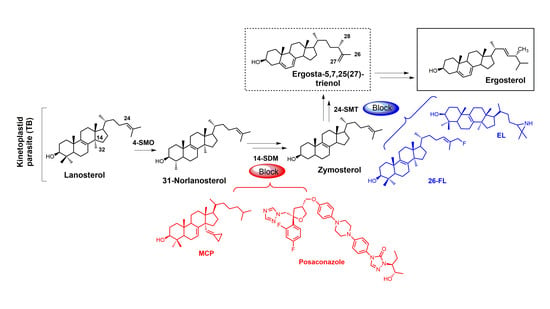
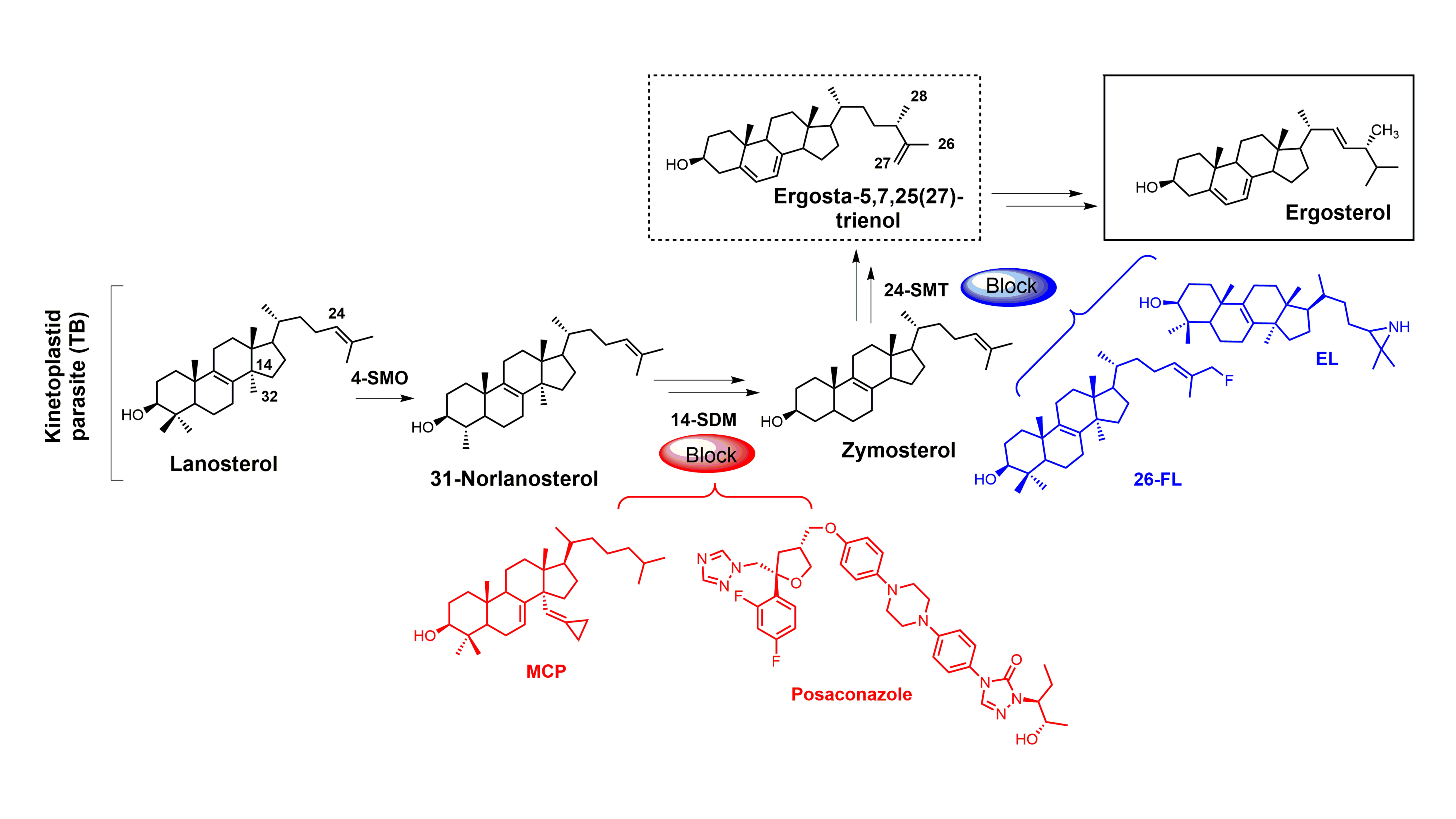
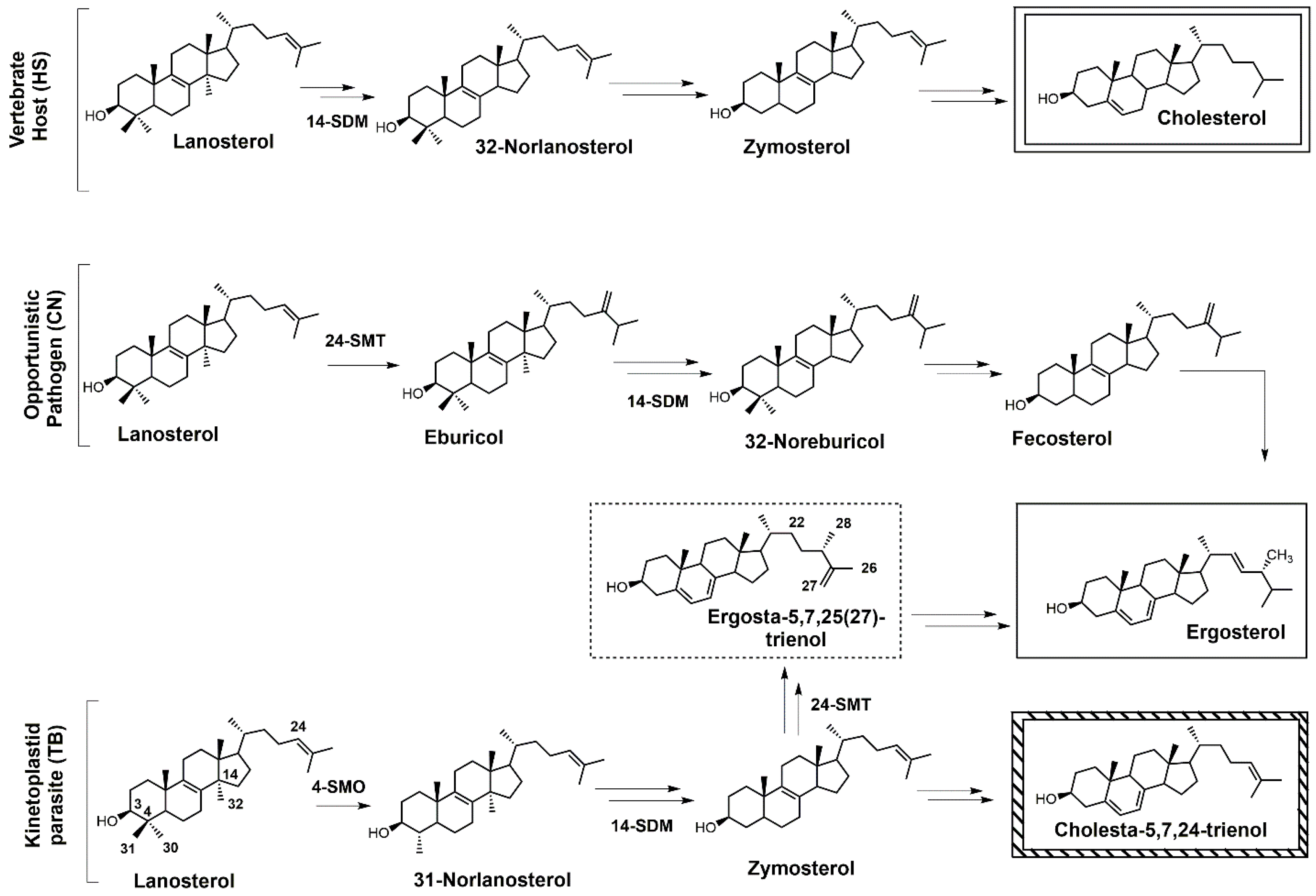
Sterols such as ergosterol and cholesterol are essential lipid molecules, and they perform numerous cellular roles associated with membrane (bulk) and signal (sparking) functions [1,2,3]. Cholesterol is biosynthesized in humans, while ergosterol or other 24-alkylated sterols are biosynthesized in opportunistic fungi and parasitic protozoa (Figure 1) [2,3,4]. This difference in sterol production can be exploited in the development of drugs that are designed to selectively block ergosterol biosynthesis in invasive fungi or protozoan parasites without harming the human host [3,4,5,6,7]. Infectious diseases caused by parasitic protozoa take a heavy toll on human health, are widespread, and are increasing in resistance to current chemotherapies [8]. Leishmaniasis is threatening around 350 million people in more than 98 countries [9], while the World Health Organization (WHO) has estimated that 16–18 million people are infected with Chagas disease [10], and it has been suggested that 70 million people in Africa are at risk of human African trypanosomiasis (HAT; sleeping sickness) [11]. Leishmaniasis is caused by various species of Leishmania, while the causative pathogens for Chagas and HAT are Trypanosoma cruzi and Trypanosoma brucei, respectively. It is well known that these diseases are life-threatening, and without proper treatment they are often fatal. Leishmania spp., T. cruzi, and T. brucei all require ergosterol for growth, and inhibiting the ergosterol biosynthesis pathway in these parasitic protozoa is an ideal approach to treat these infections without harming the human host. It should be noted that fungal infections caused by Cryptococcus neoformans have become the leading cause of morbidity and mortality in acquired immune deficiency syndrome (AIDS) patients and other immunocompromised patients, and it is reported that 5–10% of AIDS patients in the United States suffer from these life-threatening infections [12,13].
Selective inhibition of fungal SDM is one of the most common ways to treat fungal infections, and the majority of drugs that target fungal sterol 14α-demethylase (SDM) possess an azole side chain [12,13,14,15,16,17]. For these azole drugs to be efficacious, they need to have greater affinity for fungal SDM versus mammalian SDM [15,16]. Sterol C24-methyltransferase (24-SMT), unlike SDM, is not found in humans but is present in both fungi and protozoa (Figure 1), offering a selective way to inhibit ergosterol biosynthesis [15]. It is important to note that antifungal azoles are heavily used in agriculture [17,18,19], and azole resistance is becoming a major problem [20]. One mechanism of resistance to these antifungal azoles is the activation of efflux pumps that transport azoles out of fungal cells [21]. There is a huge medical need to develop new fungicides and medicinal drugs that specifically block ergosterol biosynthesis and are not likely to develop resistance.
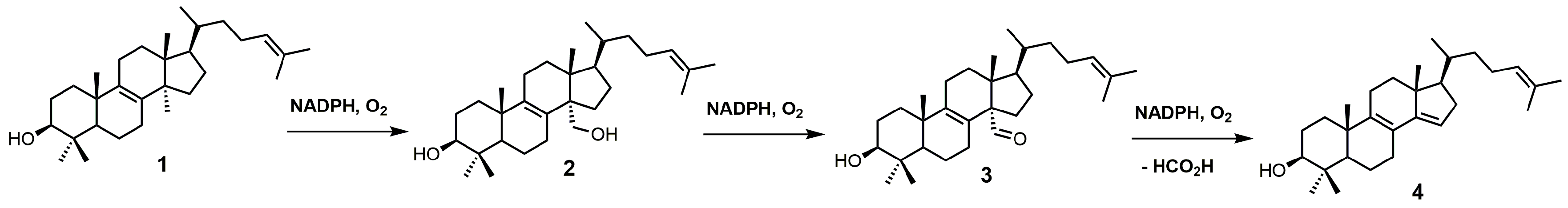
SDM, also known as P45014DM or CYP51, catalyzes the removal of the C-32 methyl group of lanosterol (1) via a repetitive three-step process that uses reduced nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen (Figure 2) to ultimately produce 4,4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol (4) [22,23]. SDM transforms the C-32 methyl group of lanosterol into an alcohol (2), an aldehyde (3), and then formic acid with the insertion of the Δ14–15 double bond (Figure 2) [24]. Each P450 catalytic cycle involves the reduction of heme ferric iron to the ferrous state, the binding of molecular oxygen, and subsequent protonation to form a ferric hydroperoxo intermediate [24]. Further protonation of the distal oxygen atom of the ferric hydroperoxo intermediate causes heterolytic scission of the O–O bond, resulting in the loss of water and the formation of an Fe4+ oxo porphyrin cation radical, which is the catalytically active species [24]. Once the substrate is oxygenated, the iron returns to its ferric state ready for another catalytic cycle [24].
2. Sterol SDM Inhibitors
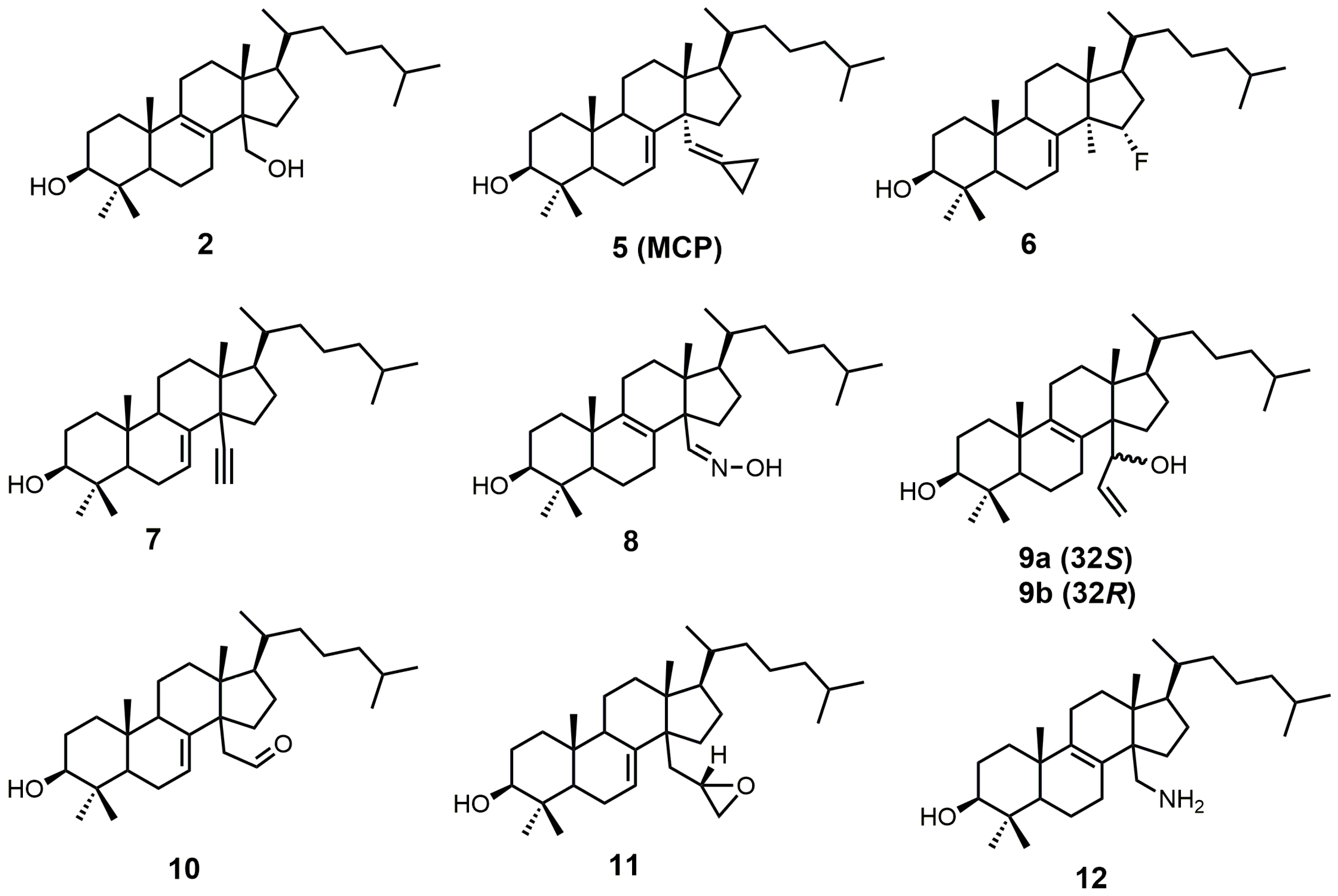
Sterol-based SDM inhibitors have been reported in the literature [22,23,24,25,26,27,28,29,30] (Figure 3); however, they are not as commonly reported as azoles [12,17,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. This is likely due to the limited number of functional groups on lanosterol that can be synthetically modified [47], in addition to the difficult and time-consuming syntheses involved with sterol functional-group manipulation. The preferred substrate of SDM is compound 2, which is the most potent natural inhibitor of SDM [25]. Compound 2 has a half-maximal inhibitory concentration (IC50) value of 7.8 μM against SDM, and it can be made synthetically in nine steps starting from lanosterol [25,48].
14α-Methylenecyclopropyl-Δ7-24,25-dihydrolanosterol (5 or MCP) was observed to be a competitive inhibitor of F105-containing T. brucei CYP51 (TbCYP51)and Lesihmania infantum orthologos, while for T. cruzi, MCP was presumed to act as a mechanism-based inhibitor (suicide substrate) [24]. The cyclopropyl ring of MCP is presumably opened as MCP binds to T. cruzi CYP51 (TcCYP51), forming a covalent bond with the prosthetic heme group [24]. The crystal structure of TbCYP51 covalently bound with MCP has been reported [24]. Despite MCP having the same Kd values of 0.5 μM for both TbCYP51 and TcCYP51, the observed half-maximal effective concentration (EC50) of MCP against T. brucei was >50 μM, while T. cruzi cell growth was inhibited by 50% at a MCP concentration of 3 μM [49]. MCP inhibits TcCYP51 more than TbCYP51, which is likely due to MCP acting as a suicide substrate for TcCYP51 and as a competitive inhibitor for TbCYP51 [24].
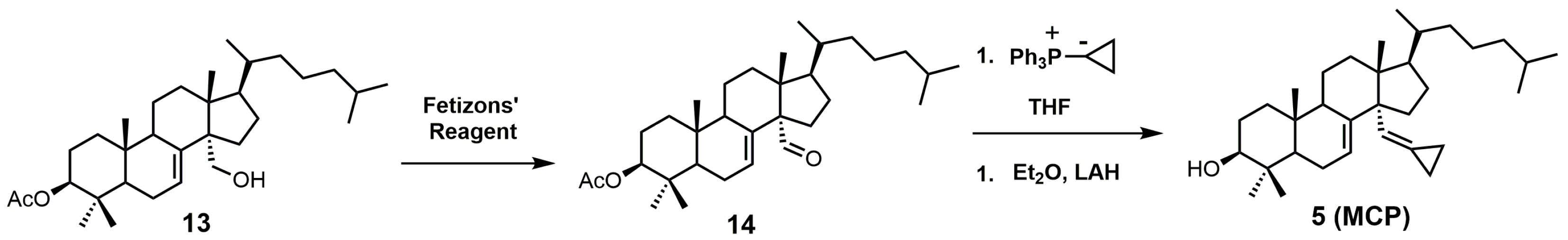
MCP can be synthesized in three steps starting with 3β-acetyloxylanost-7-en-30-ol (13) (Figure 4) [24]. Compound 13 can be synthesized directly from lanosterol in 12 steps [29,48]. Oxidation of compound 13 with Fetizon’s reagent produced 3β-acetyloxylanost-7-en-30-al (14), which was then converted into MCP (5) via a Wittig reaction using a cyclopropyltriphenylphosphium ylide, followed by acetyl deprotection with lithium aluminum hydride (LAH) [24].
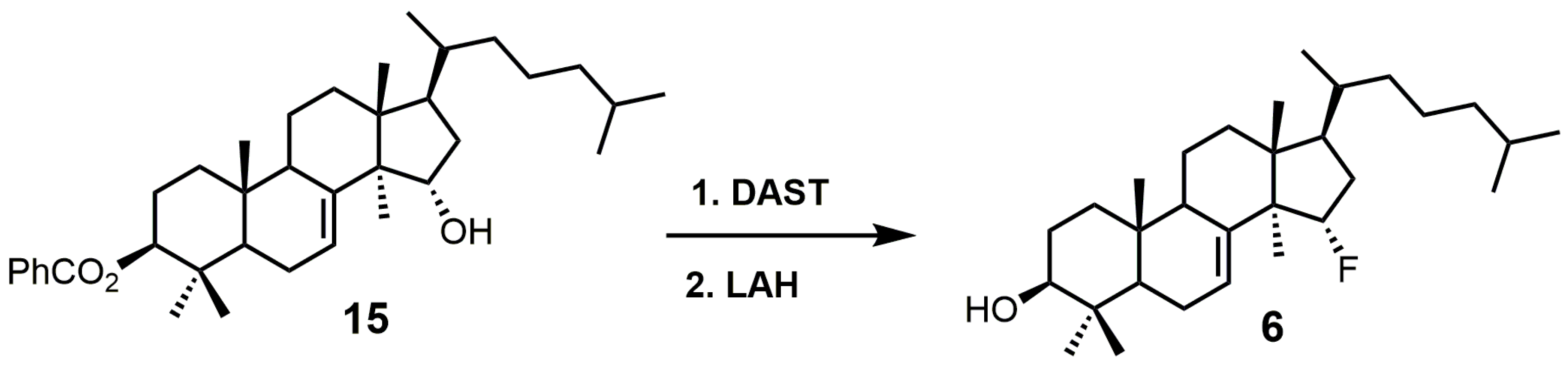
15α-Fluorolanost-7-en-3β-ol (6) was noted to be a weak competitive inhibitor of SDM with a Ki value of 315 μM [28]. Metabolic studies have indicated that compound 6 is converted to 15α-fluoro-3β-hydroxylanost-7-en-32-al by hepatic microsomal SDM and that the 15α-fluoro substitution blocks further metabolic conversion into other cholesterol biosynthetic intermediates [28]. The starting material used to synthesize compound 6 was 3β-benzoyloxy-lanost-7-en-15α-ol (15) (Figure 5) [50,51]. Compound 15 was reacted with diethylaminosulfur trifluoride (DAST) to install the fluorine at C-15, and the benozyl protecting group was removed by LAH [28].
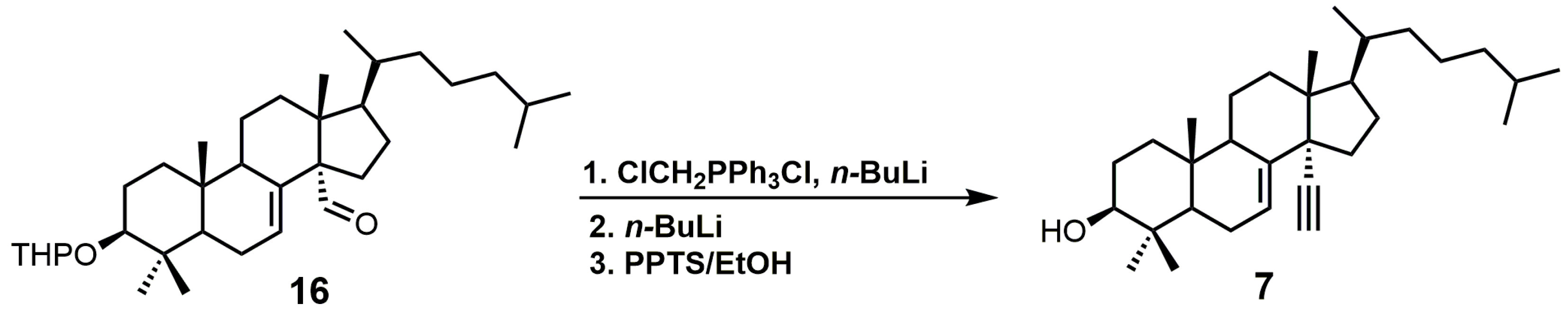
4,4-Dimethyl-14α-ethynylcholest-7-en-30-ol (7) was observed to have Kd values of 1.2 μM against TbCYP51 and 1.3 μM against TcCYP51 [49]. Compound 7 had a weaker affinity for both TbCYP51 and TcCYP51 in comparison to MCP. Compound 16 (Figure 6) is the starting material required to synthesize compound 7, and compound 16 can be synthesized in 11 steps starting from lanosterol [23]. Aldehyde 16 was reacted with the ylide of chloromethyltriphenylphosphonium chloride, followed by the addition of n-butyllithium to introduce the desired alkyne functionality (Figure 6) [29]. The tetrahydropyran (THP) protecting group was removed by the use of pyridinium p-toluenesulfonate (PPTS) in ethanol to yield compound 7 [29].
Lanost-8-en-32-alkoxime-3β-ol (8) was reported as having an IC50 value of 1.1 μM against SDM [25]. Compound 8 was readily prepared from 3β-benzoyloxy-lanost-8-en-32-al (17) in two steps (Figure 7) [52]. The aldehyde functional group of compound 17 was converted into an oxime by the use of hydroxylamine hydrochloride in pyridine [52]. The benzoyl protecting group was removed using potassium hydroxide in ethanol to yield compound 8 [52].
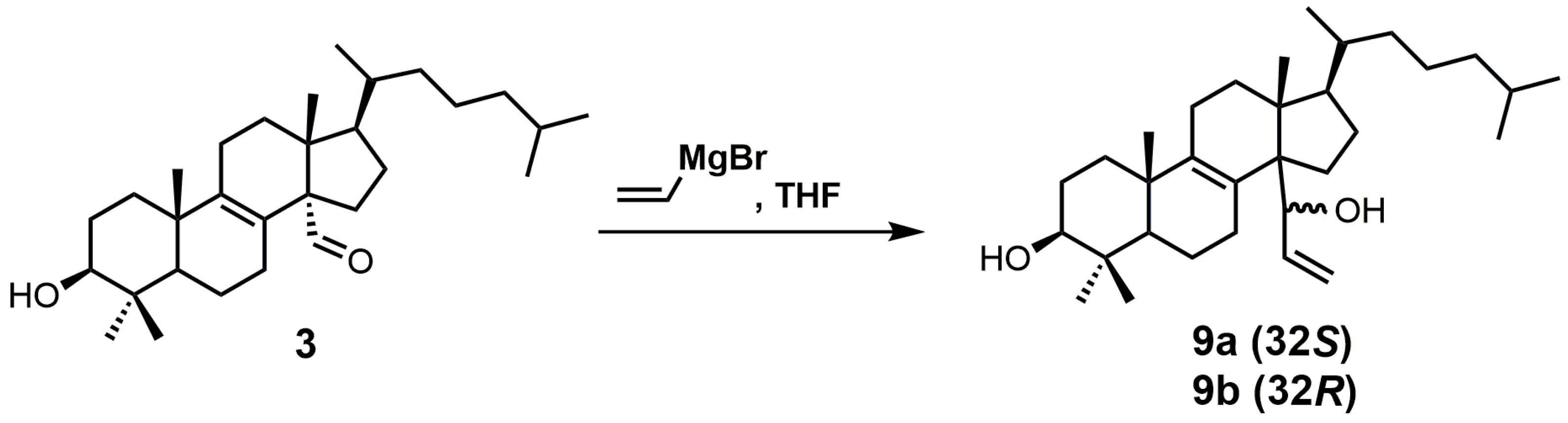
A stereochemical preference of the 32-vinyl alcohols (compounds 9a and 9b; Figure 8) was observed against SDM [25]. The 32S-isomer (compound 9a) was observed to have an IC50 value of 0.75 μM against SDM in comparison to the 32R-isomer (compound 9b), which has a reported IC50 value of 3.20 μM [25]. This result illustrates the importance of having optimized stereochemistry in SDM inhibitors. Compounds 9a and 9b were readily prepared from lanost-8-en-32-al-3β-ol (3) via a Grignard reaction using vinyl magnesium bromide in THF [52]. The Grignard reaction was compatible without needing a protecting group for the 3β alcohol. The two diastereoisomers 9a and 9b were successfully separated by medium pressure liquid chromatography (MPLC) [52].
Aldehyde 10 and epoxide 11 (Figure 9) were both observed to inhibit total cholesterol and lathosterol biosynthesis by >89% at an inhibitor concentration of 10 μM, and these compounds were believed to inhibit SDM [27]. Compounds 10 and 11 have Ki values of 3 and 0.61 μM, respectively, while the 32R-oxiranyllanost-7-en-3β-ol isomer has a Ki value of 2 μM [27]. The synthesis of 32S-oxiranyllanost-7-en-3β-ol (11) and the 32R isomer started with a Wittig reaction between aldehyde 14 and the ylide of (methoxymethyl)triphenylphosphonium chloride to yield compound 18 (Figure 9) [22]. Cleavage of the methyl enol ether of compound 18 was achieved by the use of perchloric acid to yield aldehyde 10 [22]. Corey–Chaykovsky reaction conditions were then used to transform compound 10 into compound 11 [22]. A 6:1 diastereomeric mixture of 32S-oxiranyllanost-7-en-3β-ol (11) and 32R-oxiranyllanost-7-en-3β-ol was obtained, and this mixture was successfully purified by high-performance liquid chromatography (HPLC) [22].
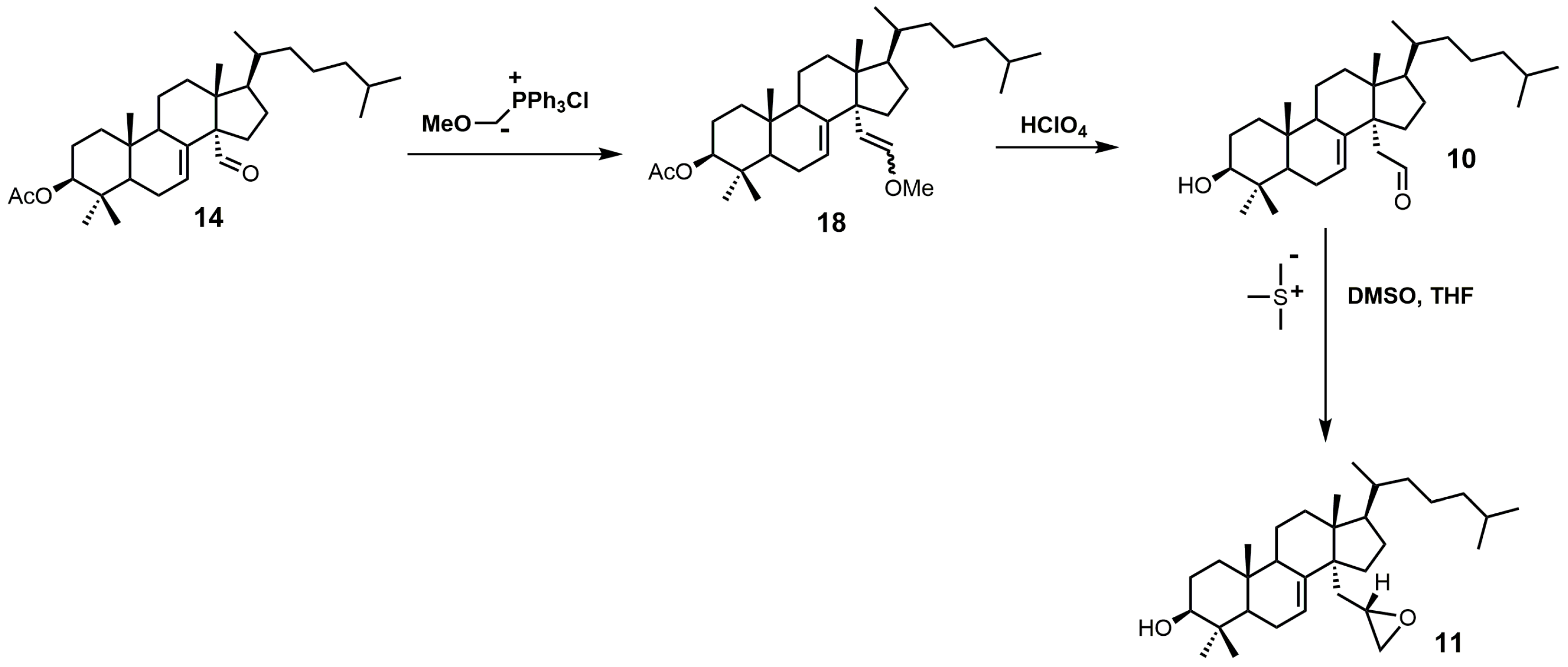
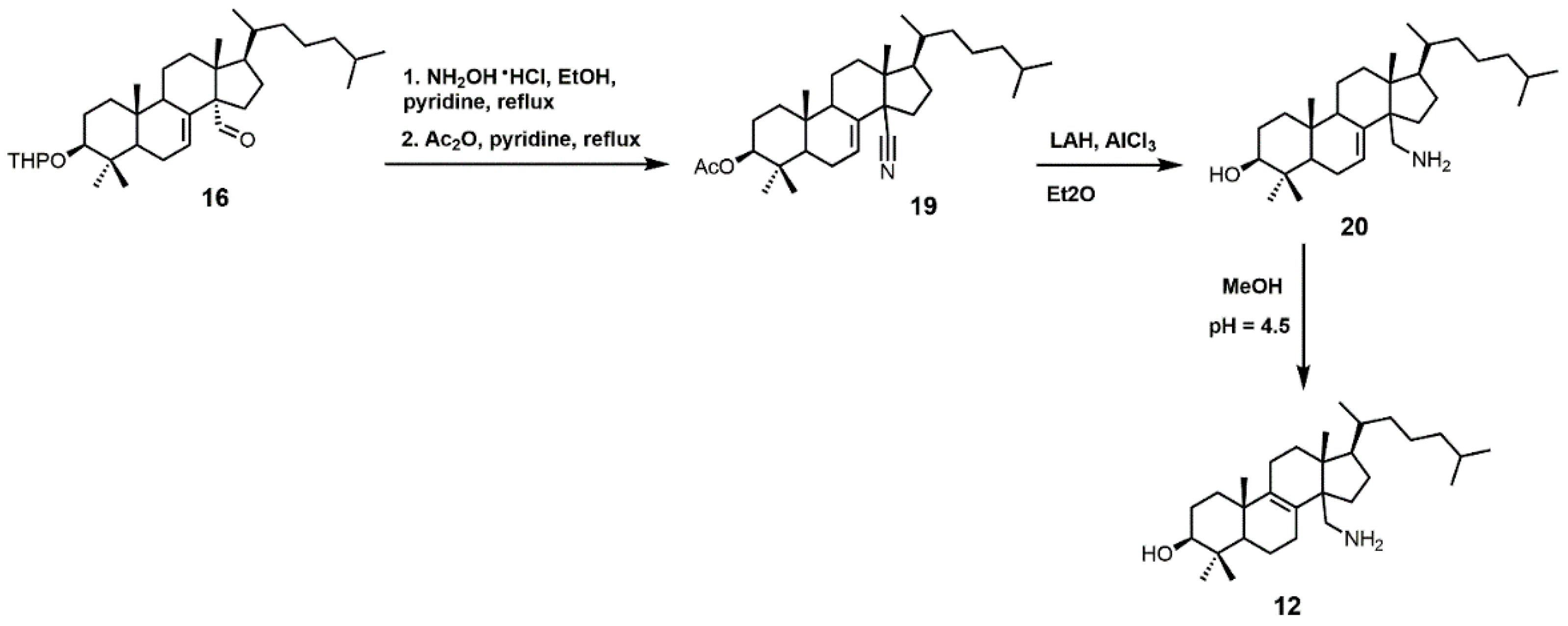
4,4-Dimethyl-14α-aminomethyl-cholest-8-en-3β-ol (12) and 4,4-dimethyl-14α-aminomethyl-cholest-7-en-3β-ol (20) were observed to be active against T. cruzi SDM with apparent Kd values of 5.1 and 1.3 μM, respectively [30]. These two amino derivatives have around a 3-fold stronger inhibitory effect on Candida albicans SDM in comparison to T. cruzi SDM and produce IC50 values of around 4 μM against C. albicans growth [30]. 4,4-Dimethyl-14α-aminomethyl-cholest-8-en-3β-ol (12) can be synthesized starting with compound 16 (Figure 10) [30]. The aldehyde functional group of compound 16 was converted into an oxime with hydroxylamine hydrochloride, which in turn was transformed into nitrile 19 with acetic anhydride and pyridine [30]. Nitrile 19 was then reduced to a primary amine with lithium aluminum hydride and aluminum trichloride to yield compound 20, which was easily isomerized into compound 12 with acidic methanol [30].
3. Azole SDM Inhibitors
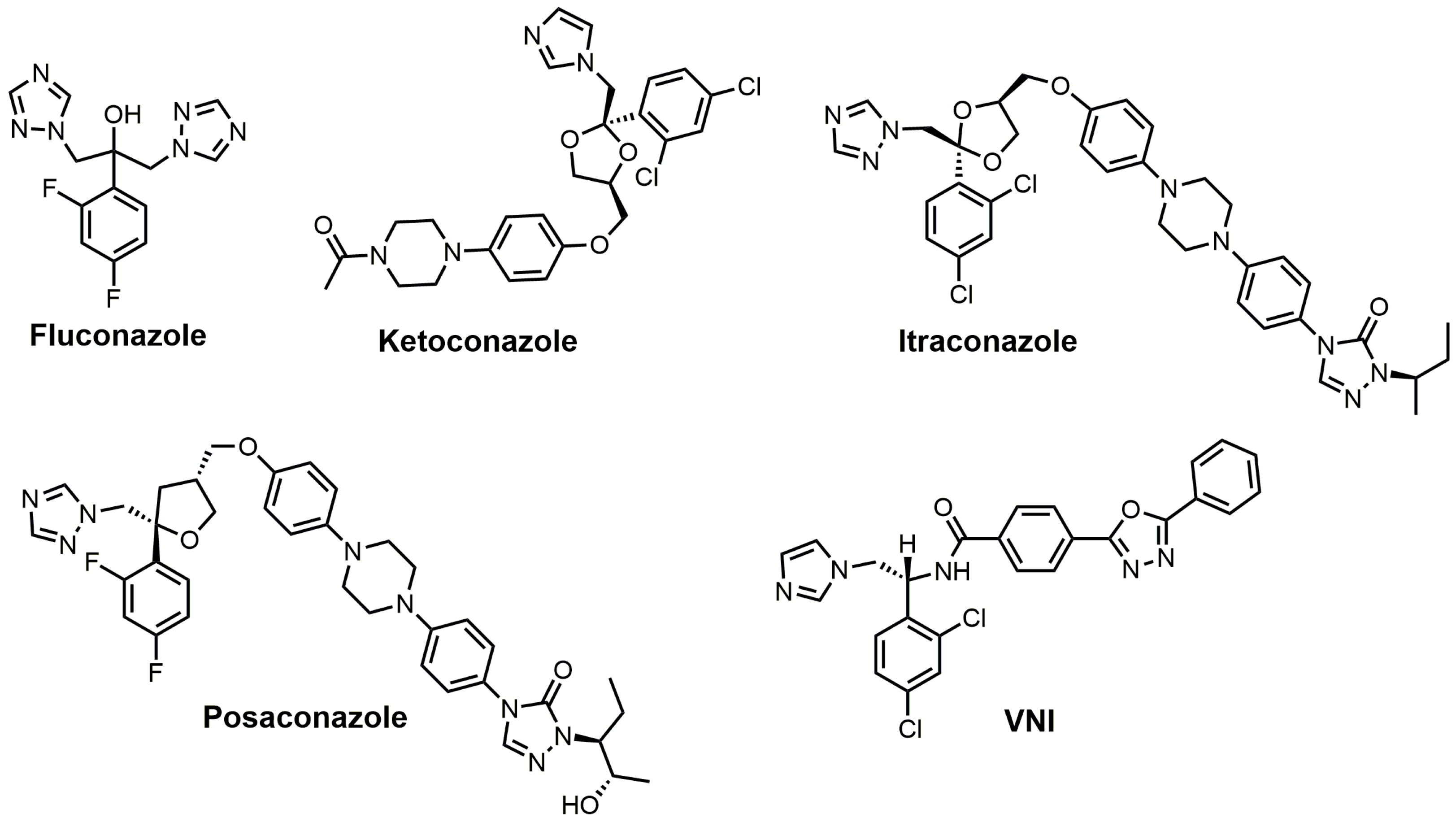
Azoles are the largest class of SDM inhibitors, and this group of inhibitors is continuously expanding with the creation of new drugs or molecules with drug-like properties. 1,2,4-Triazole fungicides such as difenoconazole (Score® (Syngenta, Basel, Switzerland)), epoxiconazole (Opal® (TRC, North York, ON, Canada)), flusilazole (Punch® (DuPunt, Wilmington, DE, USA)), and so forth are well-known SDM inhibitors used against agricultural relevant fungal diseases, including powdery mildews, rusts, and leaf-spotting fungi from Ascomycetes and Basidiomycetes [17]. Human fungal infections have been treated with antifungal azoles for a long period of time; chlormidazole was the first azole drug, introduced in 1958 for the treatment of topical mycosis [53]. The older antifungal azoles that were predominately discovered in the 1950–1960s have undergone numerous structural modifications to yield the next generation of antifungal azole drugs. In addition, many of these older antifungal azole drugs have reemerged or undergone structural modifications to be used as potential anti-trypanosomiasis drugs. The renaissance of using old antifungal agents for treating or attempting to treat trypanosomiasis was largely driven by large pharmaceutical companies not prepared to invest heavily in neglected diseases that are prevalent in developing countries where there would be no chance of cost recovery [53,54]. Some of the classic azoles used as standards for fungal SDM inhibition include ketoconazole, fluconazole, itraconazole, and posaconazole, and structures of these drugs are shown in Figure 11. Fluconazole (Diflucan®) was released by Pfizer Canada Inc, Kirkland, QC, Canada and was active against Candida spp., while itraconazole (Sporanox®) was released from Janssen Pharmaceutica in the early 1990s and was active against both Candida spp. and Aspergillus spp. [17]. Itraconazole from Janssen Pharmaceutica, Beerse, Belgium was approved for use in 1992, while posaconazole (Noxafil®) from the Schering-Plough Research Institute, Kenilworth, New Jersey, USA was approved by the food and drug administration (FDA) in 2006 [53]. Posaconazole was noted to be more effective than amphotericin B in the treatment of Aspergillus spp. infections, and it is structurally similar to itraconazole (Figure 11) [53]. (R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadi-azol-2-yl)-benzamide (VNI) (Figure 11), a new-generation imidazole, has been shown to exert a curative effect for both acute and chronic Chagas disease in a murine model with 100% survival and no observable side effects [14,55]. VNI is available at a low cost (<0.10/mg) and has good oral bioavailability, low toxicity, and favorable pharmacokinetics, which makes this compound an attractive candidate for clinical trials to treat patients with Chagas disease [55].
Fluconazole is a water-soluble, well-tolerated, and cheap first-generation antifungal drug that penetrates the blood–brain barrier [21]. Fluconazole has reported minimum inhibitory concentrations (MIC90) required to inhibit the growth of various Candida spp. isolates by 90% ranging from 2 to 64 mg/mL [56]. Fluconazole was observed to maintain an in vivo effective dose for 50% (ED50) of the murine population with <1.0 mg/kg for 5 days when orally administered in a murine candidiasis model, making it 50 times more potent than ketoconazole [57]. The plasma half-life for fluconazole was 6.1 h, and 75% of the drug was excreted in urine with no changes to its structure [12]. The 2,4-difluorophenyl side was chosen because it was the only phenyl analog that was water-soluble (8 mg/mL), which was needed to enable it to be formulated for intravenous administration. Fluconazole went through safety studies and passed evaluation in humans, where it was shown to very successful in the treatment of C. albicans and C. neoformans infections but was not as effective against Aspergillus infections when compared with itraconazole [12]. Fluconazole has a reported Kd value of 0.23 μM against T. cruzi SDM and an EC50 value of approximately 40 μM against T. cruzi, while fluconazole has a Kd value of 0.34 μM for T. brucei SDM [21,58]. Fluconazole has an in vitro IC50 value of 8 μM against T. cruzi, and a crystal structure of T. cruzi SDM cocrystallized with fluconazole has been reported [21,59,60,61]. A 6 week oral course of fluconazole was shown to be safe and useful for treating leishmaniasis caused by Leishmania major [62].
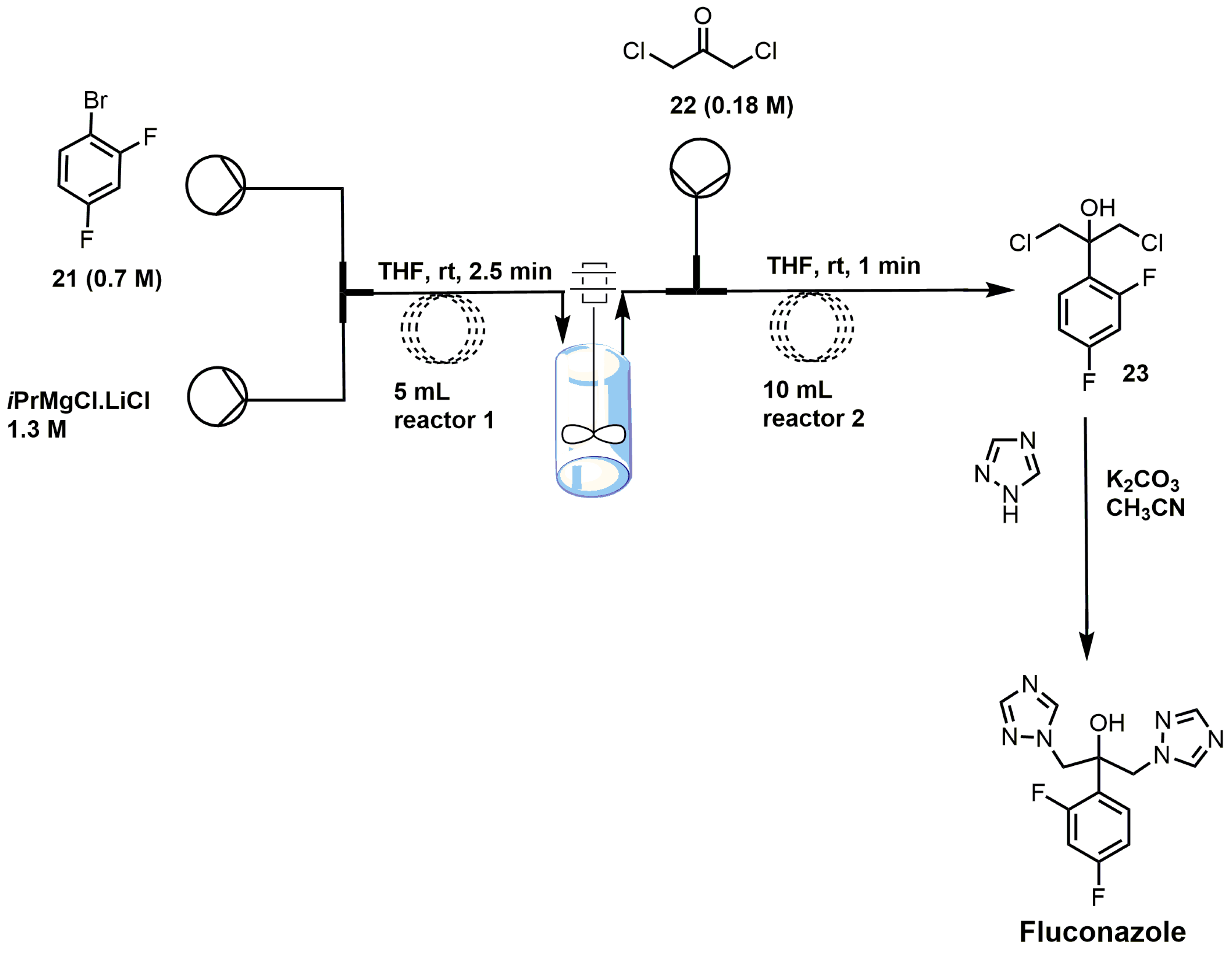
There are several different ways to synthesize fluconazole. One of the popular routes is the oxirane ring opening of 1-[[2-(2,4-difluorophenyl)oxiranyl]methyl]-1H-1,2,4-triazole with 1,2,4-triazole and potassium carbonate [17]. One of the more recent syntheses of fluconazole uses a semi-continuous flow method (Figure 12) [39]. 2,4-Difluorobromobenzene (21) was reacted with isopropyl magnesium chloride in THF to form a Grignard reagent that was reacted under flow conditions with 1,3-dichloroacetone (22) to yield 2-(2,4-difluorophenyl)-1,3-dichloro-2-propanol (23) [39]. The synthesis of fluconazole was completed with the nucleophilic attack of intermediate 23 with two 1,2,4-triazoles without using flow chemistry [39].
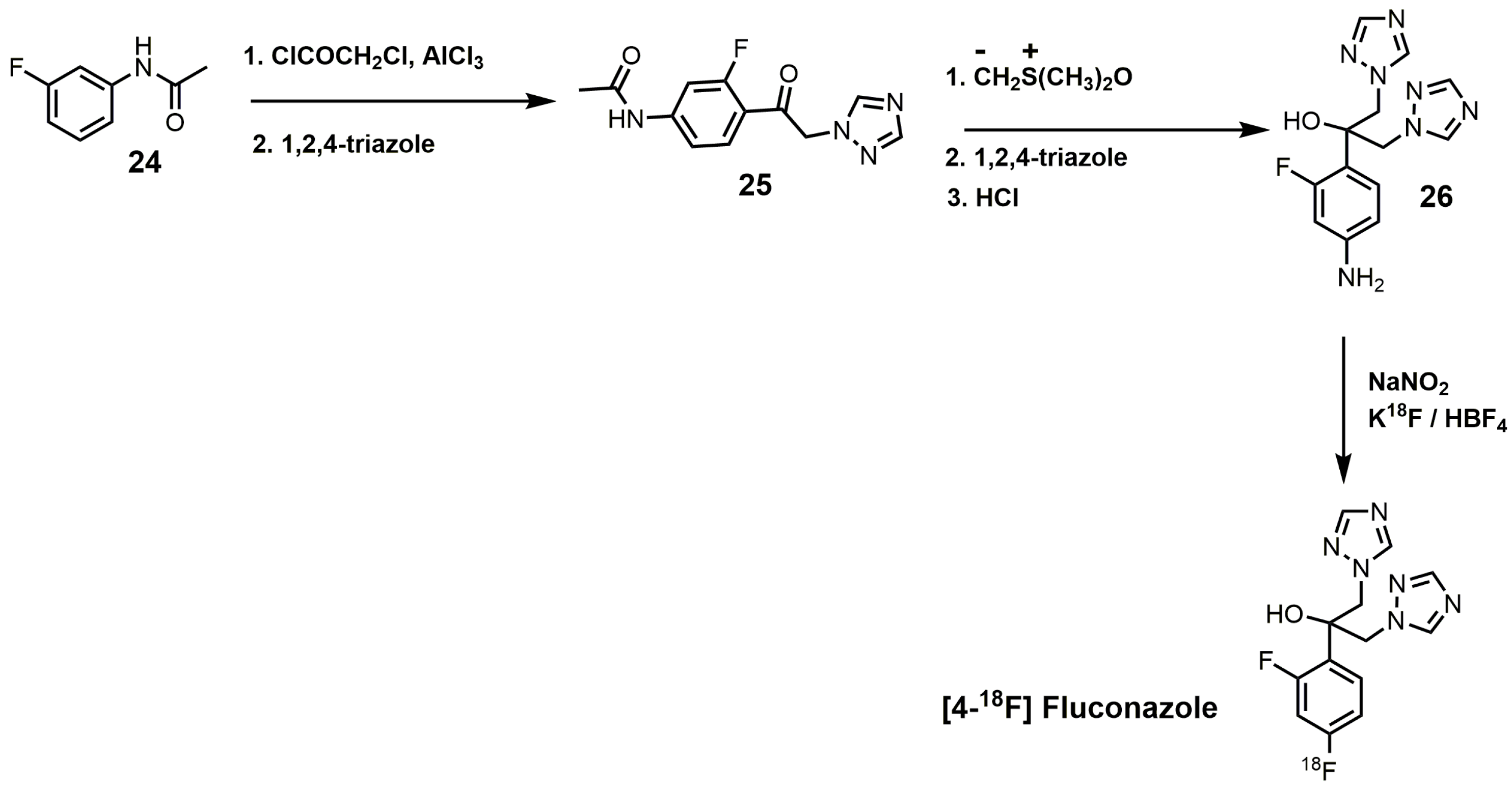
An interesting synthesis of fluconazole is the 18F-labeled synthesis of [4-18F]fluconazole (Figure 13) [38]. Under Friedyl–Crafts acylation conditions, m-fluoroacetanilide (24) was reacted with chloroacetylchloride to yield N-[4-(2-chloroacetyl)-3-fluorophenyl]acetamide, which was reacted with 1,2,4-triazole to produce compound 25 (Figure 13) [38]. Corey–Chaykovsky conditions were used to convert intermediate 25 into an epoxide that was ring-opened by 1,2,4-triazole, and the N-acetyl group was removed by acid hydrolysis to form compound 26. [4-18F]Fluconazole was obtained by a modified Schiemann reaction using 25% tetrafluoroboric acid, sodium nitrite, and potassium [18F]-fluoride [38]. [4-18F]Fluconazole was synthesized to measure the pharmacokinetics of fluconazole in rats by radioactive measurements of excised tissues and in rabbits by positron emission tomography (PET) [38]. A uniform distribution of radioactivity in most organs was observed in both species upon equilibration of [4-18F]fluconazole [38].
Ketoconazole developed by Janssen Pharmaceutica was approved by the FDA in 1981 and was the first broad-spectrum antifungal agent with oral activity; its reported MIC90 values range from 0.06 to 4 μg/mL against various Candida spp. [12,53]. Ketoconazole is highly active against Trichophyton mentagrophytes at 0.1 μg/mL, while Trichophyton rubrum and C. neoformans are inhibited at 1 μg/mL [12]. Ketoconazole has been shown to prolong the survival of mice infected with T. cruzi; however, curing effects were not observed [63,64]. Ketoconazole has a reported Kd value of 4.4 μM against cloned TbCYP51 [65]. Ketoconazole at a concentration of 0.1 μM in conjunction with 24(R,S)-25-epiminolanosterol (EL) at a concentration of 0.3 μM exerts strong antiproliferative effects on T. cruzi epimastigotes, leading to >80% inhibition of growth [66].
The synthesis of ketoconazole (Figure 14) started with the ketalization of 3′,5′-dichloroacetophenone (27) with glycerine, and the crude product was brominated [31]. Benzylation of the resulting primary alcohol produced a mixture of cis/trans isomers, and the desired cis isomer (28) was obtained by crystallization from ethanol [31]. After the bromine atom was displaced by nucleophilic attack by imidazole, the benzoyl ester was cleaved by sodium hydroxide to produce an alcohol that was activated as a mesylate (29). Mesylate 29 was reacted with the sodium salt of 1 acetyl-4-(4-hydroxyphenyl)piperazine to produce ketoconazole [31].
Itraconazole has MIC90 values ranging from 0.03 to 64 μg/mL against various Candida spp. and a MIC90 value of 2.7 μg/mL against Malassezia furfur [12]. Itraconazole was noted to have potent in vitro activity against C. neoformans [67]. Itraconazole was observed to successfully achieve irreversible damage against various fungi, including C. albicans, C. neoformans, T. rubrum, Paracoccidiodes brasiliensis, and Aspergillus fumigatus [12,68]. Itraconazole absorption is enhanced by food intake, and oral solutions provide higher serum concentrations in comparison to capsules [12,69,70]. Itraconazole is very active in vitro against Microsporum canis and other dermatophytes associated with scalp ringworm infections [12]. The selectively index of itraconazole for fungal SDM versus human SDM was reported to be 25, which was much better than the selectively index of ketoconazole [53,71]. Itraconazole was observed to have an ED50 value of 1 μM against T. brucei parasites, and when combined with 25-azalanosterol (AZAL), parasite death resulted [2]. Itraconazole exhibited in vitro activity against Leishmania mexicana mexicana amastigotes in macrophages, and 50% inhibition of ergosterol synthesis was observed at 0.15 μM, which was more potent than that observed for ketoconazole (0.21 μM) [72]. Itraconazole has been reported to successfully treat cutaneous leishmaniasis; however, it was not as effective against Leishmania braziliensis [53,73,74,75]. It is noted that itraconazole has not been able to completely eradicate T. cruzi from experimentally infected animals or human patients in studies investigating it as a possible treatment for Chagas disease [76].
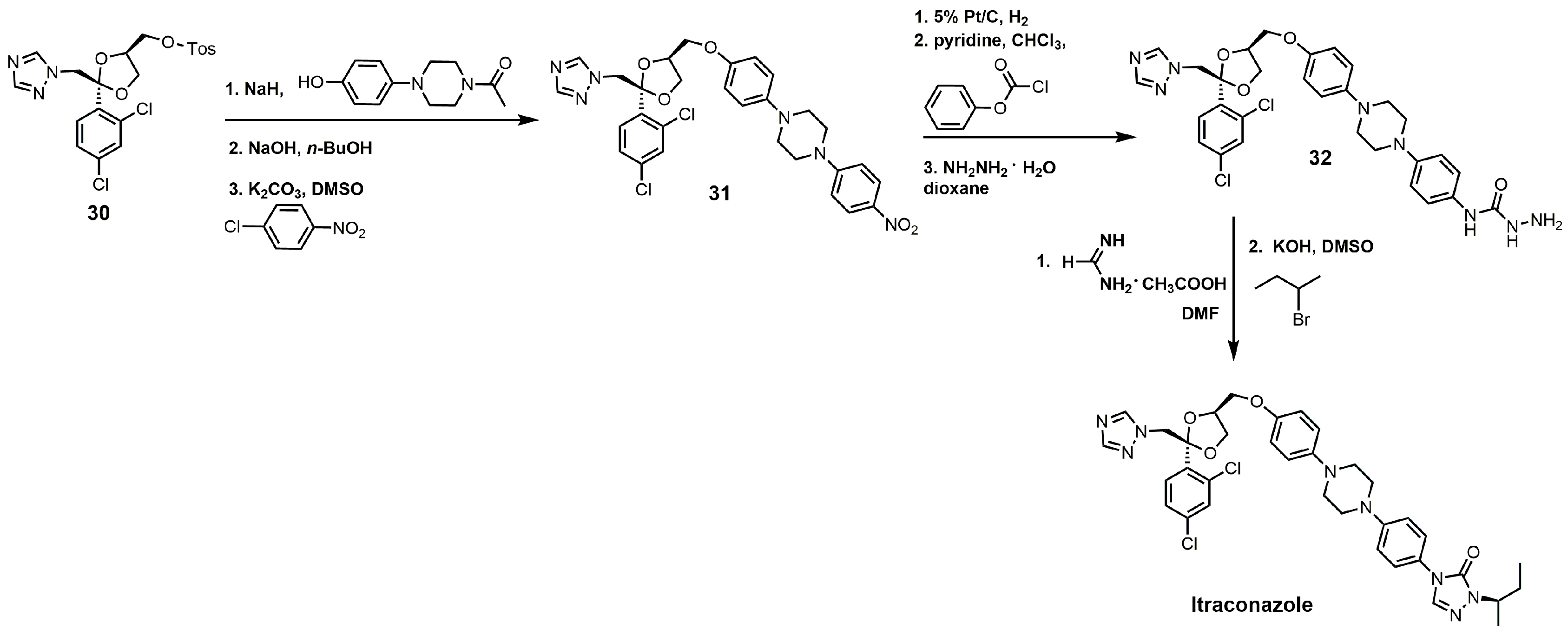
The synthesis of itraconazole can be started by reacting compound 30 with sodium hydride and 1 acetyl-4-(4-hydroxyphenyl)piperazine to yield cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy] phenyl]-piperazine (Figure 15) [34]. The N-acetyl group can be removed by sodium hydroxide, and the unprotected piperazine nitrogen under basic conditions with potassium carbonate can undergo a nucleophilic aromatic substitution with p-chloronitrobenzene to yield compound 31. The aromatic nitro group was reduced to an aniline functional group with hydrogen gas and 5% platinum on carbon, and the aniline derivative was converted into a carbamate with phenyl chloroformate [34]. Reacting the carbamate with hydrazine produced the hydrazine carboxamide 32. Compound 32 was initially treated with formamidine acetate and then with 2-bromobutane in the presence of potassium hydroxide to yield itraconazole (Figure 15) [34].
Posaconazole (Figure 11) has already been registered in the European Union (2005), Australia (2005), and the United States of America (2006) as a prophylactic for invasive fungal infections in addition to azole-resistant candidiasis [77]. Posaconazole was observed to have a MIC value of 20 nM and an IC50 value of 14 nM against the epimastigote form of T. cruzi and a MIC value of 3 nM and an IC50 value of 0.25 nM against the clinically relevant intracellular amastigote form in vitro [78]. Posaconazole demonstrated potent in vivo anti-parasitic activity in a murine model of acute Chagas disease, which was most likely due to its high binding affinity for TcCYP51, good pharmacokinetic properties, and long terminal half-life [77,78]. The crystal structure of posaconazole bound to SDM of T. brucei has been solved and provides valuable insights into how posaconazole binds to its target [60]. Posaconazole is active against nitrofuran- and nitroimidazole-resistant T. cruzi strains and is able to induce radical parasitological cures in both chronic and acute experimental Chagas disease [76]. An Argentinean woman in Spain who suffered from chronic Chagas disease was successfully treated with posaconazole, although requiring concurrent immunosuppressive therapy [79]. The woman was initially treated with benznidazole, which only resulted in a reduction of T. cruzi levels and not an eradication as was achieved with the use of posaconazole [79]. The major drawback of posanconazole is that it is very expensive to produce (>$1000/patient) as it is very low yielding (<1% overall), which will limit its widespread use in developing countries even if it is successful in obtaining approval for treating Chagas disease [55,77,79].
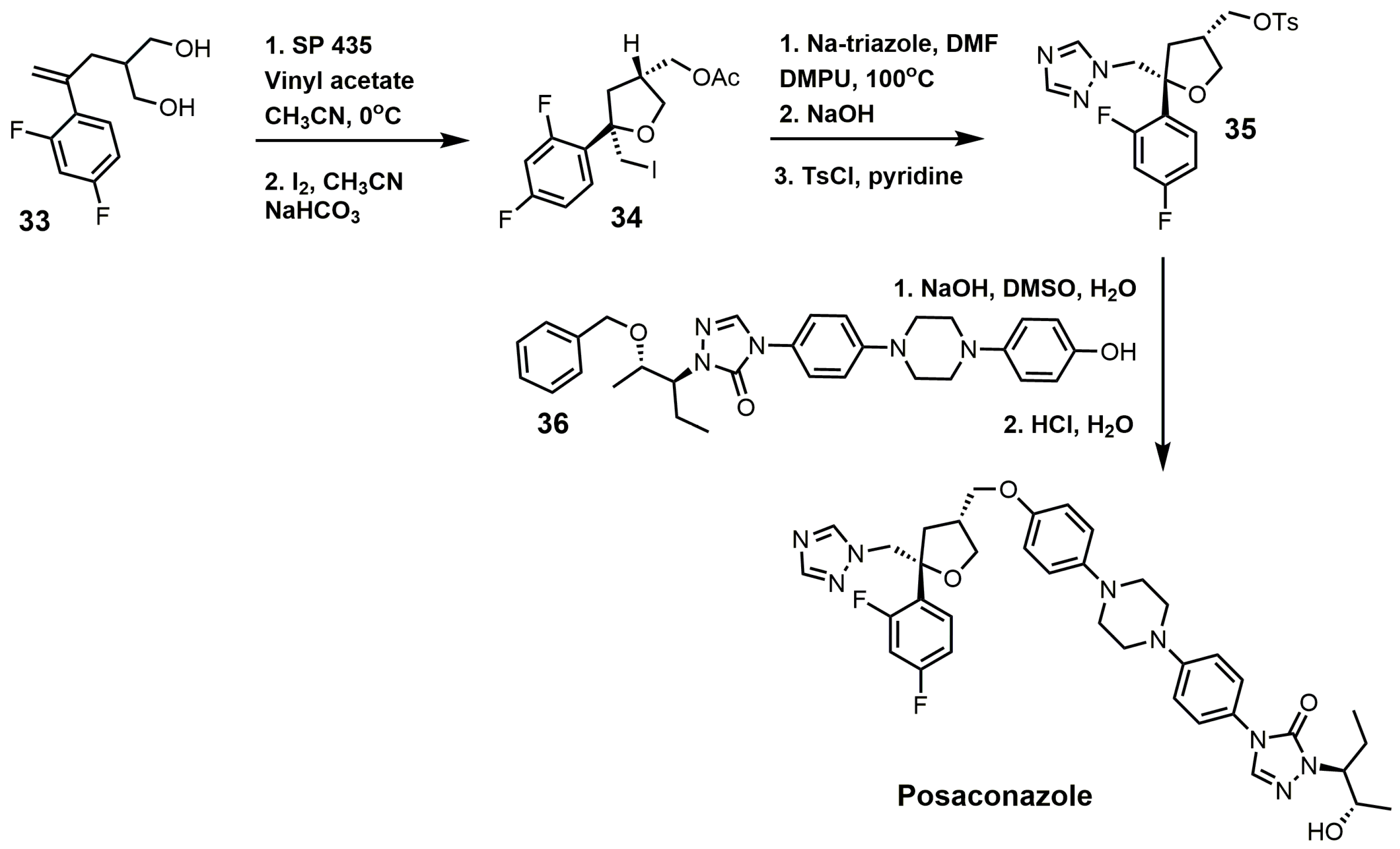
One of the well-established synthetic routes for the synthesis of posaconazole starts with the enzymatic desymmetrization of the homochiral diol (33) with hydrolase Novo SP 435 and vinyl acetate, followed by iodocyclization to produce compound 34 (Figure 16) [37,80]. It should be noted that 169 hydrolases were screened in order to make the first step a practical synthetic route [37]. A 1,2,4-triazole displaced the iodine in compound 34, and the acetate group was replaced with a tosylate to yield compound 35. Compound 35 was then reacted with compound 36 under basic conditions, and the benzyl group was removed under acidic conditions to yield posaconazole [81,82].
VNI (Figure 11) is a potent inhibitor of T. cruzi SDM and is nontoxic and highly selective [55]. Mice infected with an acute or chronic T. cruzi infection were treated orally with VNI at 25 mg/kg for 30 days, which resulted in 100% survival of the infected mice [55]. VNI cocrystallized with both T. cruzi and T. brucei SDM has been reported [21,83]. The Kd value for VNI against T. brucei SDM is 0.37 μM, and it is 0.09 μM against T. cruzi SDM [14,83]. VNI exhibits low general cytotoxicity and a potent cellular effect on T. cruzi amastigotes, with an EC50 value of 1.2 nM, in addition to an excellent selectivity index (human/T. cruzi of >50,000) [14]. An advantage of VNI, unlike posaconazole or fluconazole, is that it does not induce T. cruzi gene expression, which means it is not as likely to cause drug resistance [14]. As mentioned earlier, VNI is available at a low cost (<$0.10/mg), which makes this compound an attractive candidate for clinical trials [55]. A 2-fluoro-4-(2,2,2-trifluoroethoxy)phenyl derivative of VNI was recently reported to inhibit C. albicans and A. fumigatus SDM by 89% and 65%, respectively [46]. This 2-fluoro-4-(2,2,2-trifluoroethoxy)phenyl derivative of VNI was observed to have a Kd value of 10 nM against C. albicans SDM and of 20 nM against A. fumigatus, while posaconazole had a Kd value of 81 nM against C. albicans SDM and of 131 nM against A. fumigatus [46]. A crystal structure of A. fumigataus SDM cocrystallized with this VNI derivative was obtained [46].
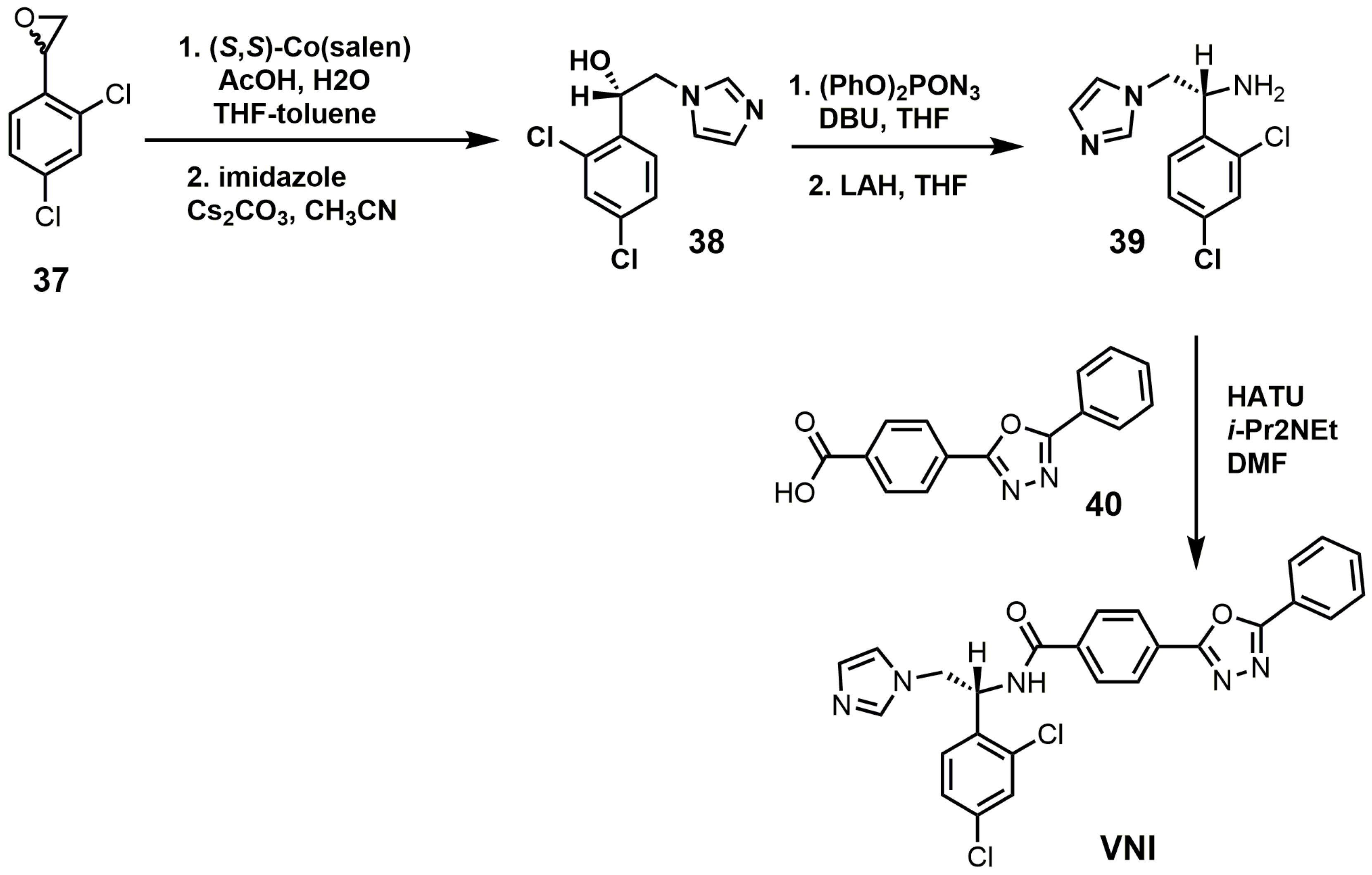
VNI can be synthesized in five steps that are scalable to multi-gram quantities with >98% purity as needed for in vivo animal studies (Figure 17) [84]. The racemic epoxide (37) was resolved with (S,S)-Co(salen) and then opened with imidazole under basic conditions to yield compound 38 [84]. Compound 38 was reacted with diphenylphosphoryl azide to replace the secondary alcohol with an azide group and was subsequently reduced with lithium aluminum hydride to yield amine 39 [84]. VNI was obtained by reacting compounds 39 and 40 under amide bond coupling conditions with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluoro-phosphate (HATU) [84].
4. 24-SMT Inhibitors
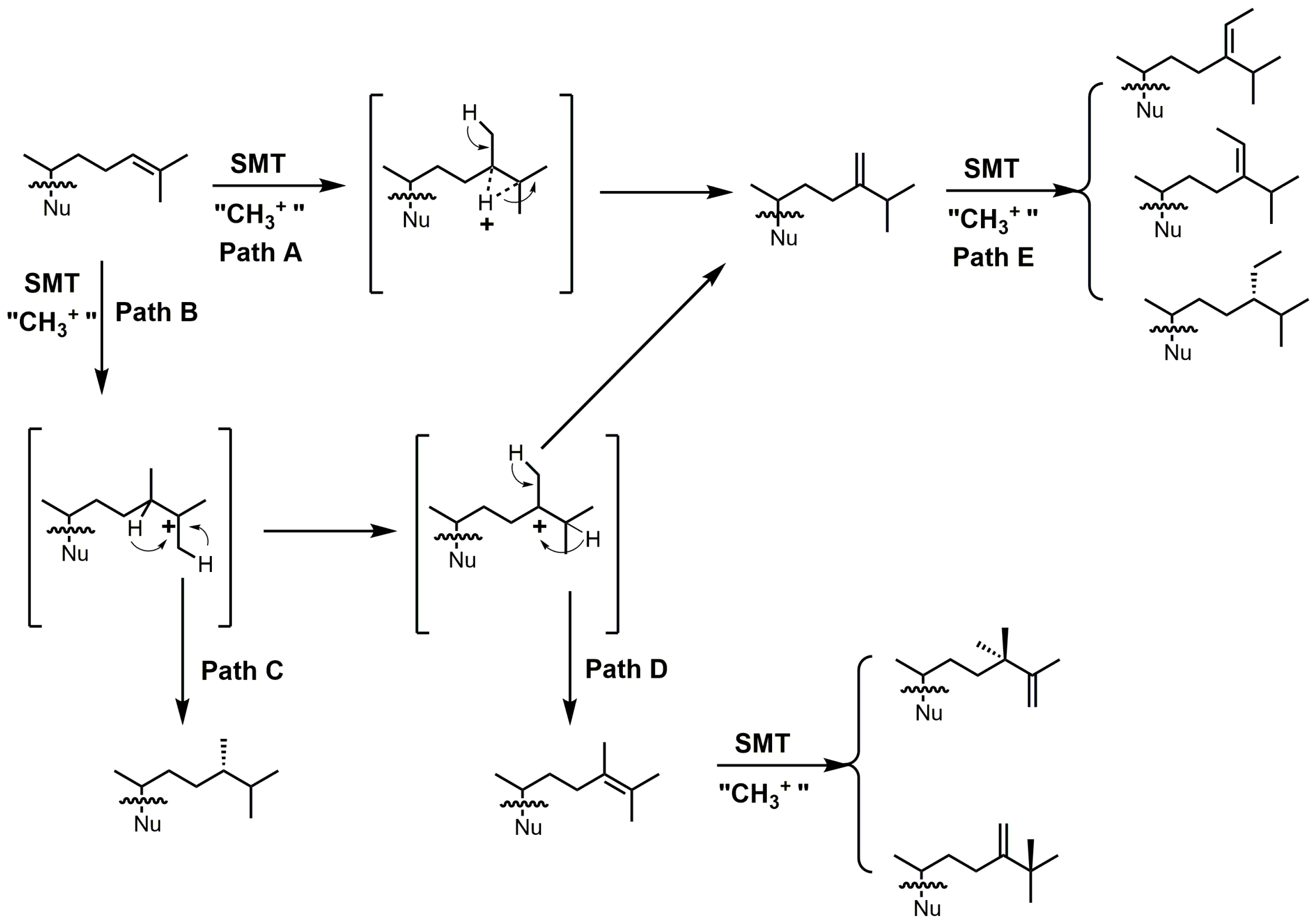
24-SMT catalyzes a methylation–deprotonation reaction that involves electrophilic alkylations of a double bond at C-24 by a methyl cation originating from S-adenosyl methionine (SAM) (Figure 18) [4]. The majority of characterized 24-SMTs are multifunctional and possess a very high substrate specificity, often yielding only a single C-24 methylated olefin product; however, there are a few atypical 24-SMTs that convert substrates to a variety of 24-alkyl(idene) products including T. brucei SMT1 and Glycine max SMT1 [4]. The various pathways possible with 24-SMT catalysis are outlined in Figure 18.
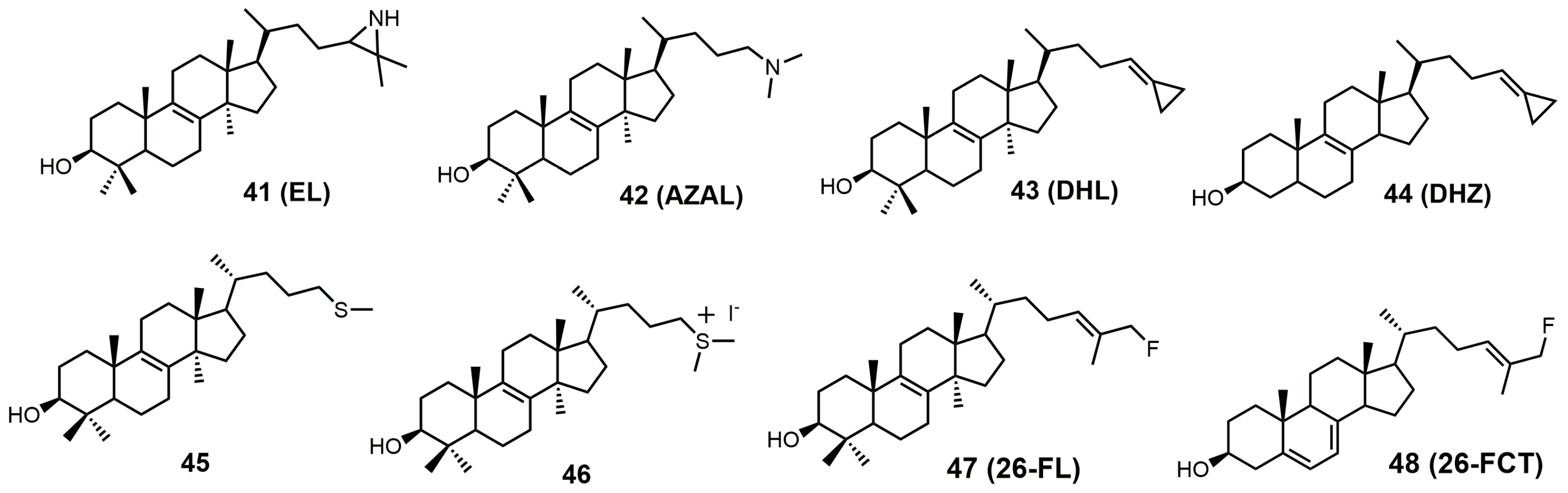
The structures of 24-SMT inhibitors typically have a modified lanosterol side chain (Figure 19).
Most of the inhibitors outlined in Figure 19 are suicide/irreversible inhibitors that have an electron withdrawing group strategically placed at or near position 25 on the sterol side. The cyclopropylidine derivatives (43–44) are spring-loaded electrophiles that are opened irreversibly with nucleophilic attack from 24-SMT (Figure 20).
Having a strong electron withdrawing group near the Δ24–25 bond, such as fluorine, affects the intermediate cation formation and timing of the C-24 methylation reaction, promoting partitioning towards irreversible covalent modification over turnover products (Figure 21) [5].
24(R,S)-25-Epiminolanosterol (EL; 41) has a reported IC50 value of around 0.3 μM against C. neoformans and has comparable activity to itraconazole [6]. EL was observed to be a potent non-competitive inhibitor of 24-SMT from sunflower embryos with a Ki value of 3.0 nM, while sitosterol and 24(28)-methylene cycloartenol were observed to be competitive inhibitors, with Ki values of 26 and 14 μM, respectively [85]. EL as 2-tritio-24(R,S)-25-epiminolanosterol was reported to inhibit 24-SMT of Gibberella fujikuroi and was metabolized to 25-aminolanosterol and lanosterol [86]. EL is a potent inhibitor against C. albicans 24-SMT with a Ki value of 11 nM and an IC50 value of 5 μM [3]. EL added to cultures of rat hepatoma cells (H4) interrupted the conversion of lanosterol to cholesterol [87]. EL caused the accumulation of zymosterol at 45 μM and at 4.5 μM caused the accumulation of desmosterol [87]. H4 rat hepatoma cells seeded into either full growth or lipid-depleted medium containing 22.5 μM EL would not grow unless the media was supplemented with low-density lipoproteins (60 μg/mL) [87].
EL was observed to have a Ki value of 49 nM against T. brucei 24-SMT [88]. An amastigote form of T. cruzi proliferating in a liver infusion tryptose medium was treated with EL, and growth was completely arrested; lysis occurred at an EL concentration of 6 μM [89]. EL was recently reported as an inhibitor of Acanthamoeba spp. trophozoite growth with an IC50 value of 7 nM, and in this study, EL yielded 20-fold higher inhibition compared to the reference drug voriconazole [90]. EL exhibited tight biding against both 24- and 28-SMT with Ki values of around 9 nM [90]. EL (41) can readily be synthesized directly from unprotected lanosterol with an excess of iodine azide and the addition of LAH (Figure 22) [91].
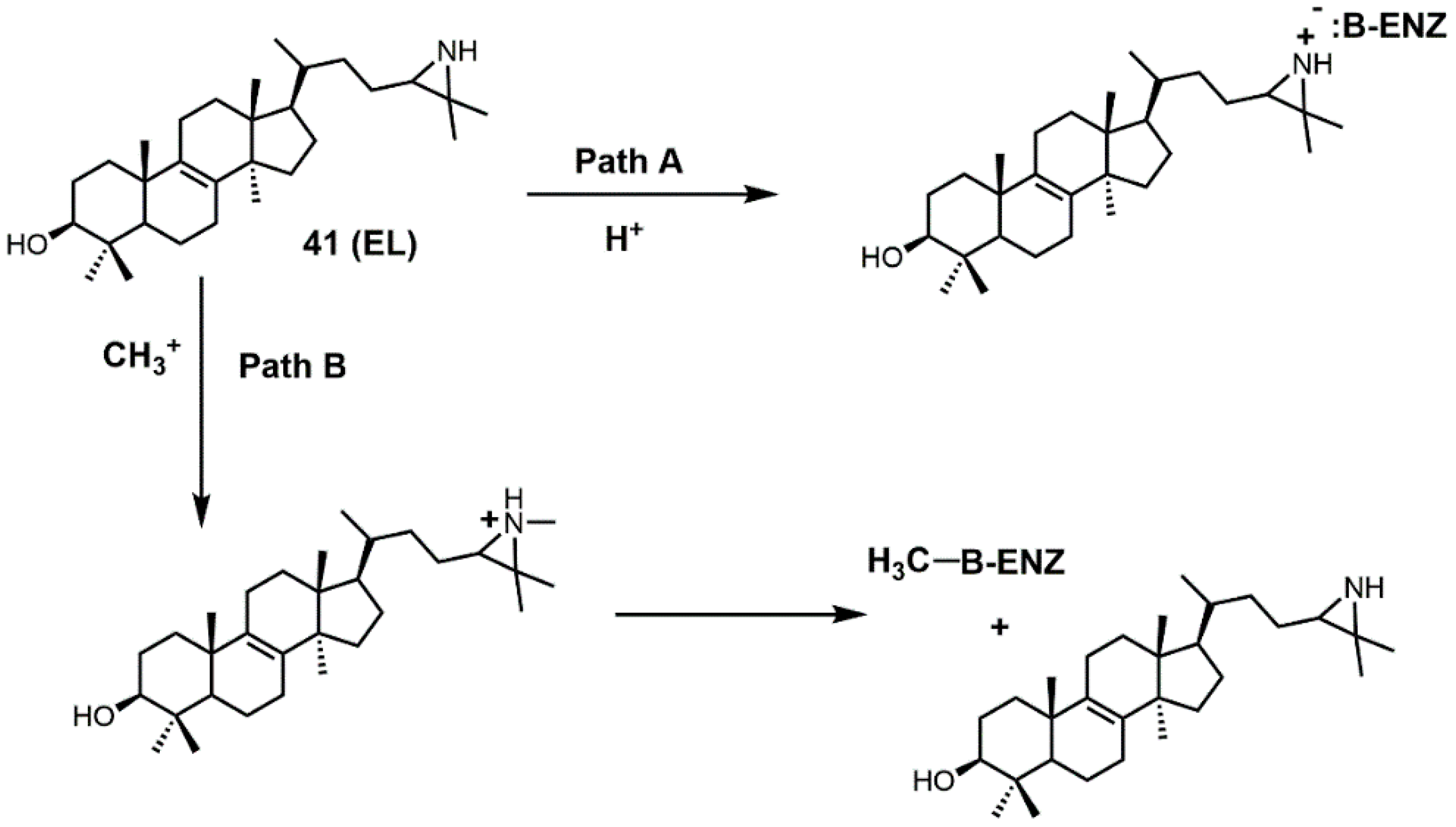
There are several different ways EL is thought to inhibit 24-SMT (Figure 23). EL could have its aziridine nitrogen atom protonated (path A) and then form an ammonium salt that can electrostatically interact with a polar amino acid in the active site of 24-SMT (such as a carboxylate group) [92]. Alternatively, EL could be methylated by 24-SMT and donate its N-methyl group to the active-site residue of 24-SMT, thereby inactivating it [92].
AZAL was reported to have a Ki value of 54 nM and an IC50 value of 3 μM against 24-SMT of C. albicans [3], while it had a Ki value of 30 nM against 24-SMT from sunflower embryos [3,85]. AZAL was noted to be a potent inhibitor of the ascomycetous fungus P. brasiliensis (Pb) with an IC50 value of 30 nM and a Ki value of 14 nM against Pb 24-SMT [93]. AZAL was observed to inhibit P. brasiliensis growth much more than for the related yeasts Saccharomyces cerevisiae and C. albicans [93]. AZAL has a Ki value of 39 nM against T. brucei 24-SMT and an ED50 value of 1 μM against T. brucei parasites [2,88]. AZAL failed to inhibit cultured human epithelial cells (HEK) with an ED50 value of >50 μM and a therapeutic index of 25 [2]. AZAL has a reported IC50 value of approximately 1 μM against both the procylic and bloodstream forms of T. brucei, and the bloodstream form of T. brucei was not rescued with cholesterol absorption from the host, highlighting the importance of ergosterol in cell proliferation of the parasite [94]. Combing AZAL and itraconazole at ED50 concentrations to the bloodstream form of T. brucei in lipid-depleted medium resulted in cell death and significant growth inhibition when grown in full growth medium [2].
AZAL can readily be synthesized via three different synthetic routes, which are outlined in Figure 24 [4,95,96]. All three routes involve converting the Δ24–25 double bond of lanosterol into an activated carbonyl group (aldehyde, carboxylic acid, or ester) followed by nucleophilic attack with dimethylamine. Each synthetic route was completed with protecting group removal by LAH.
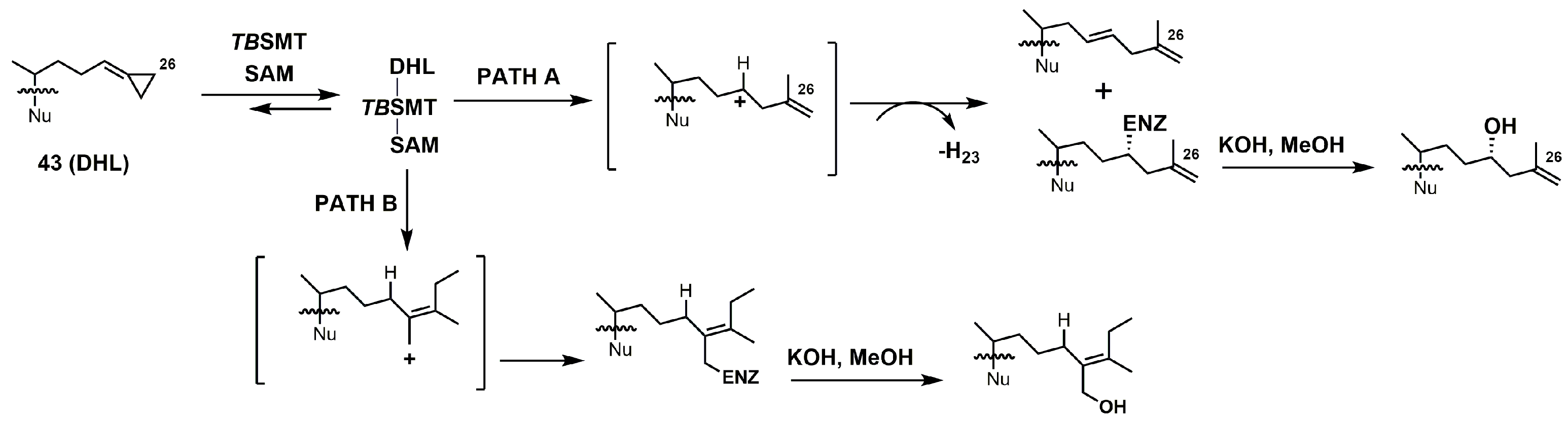
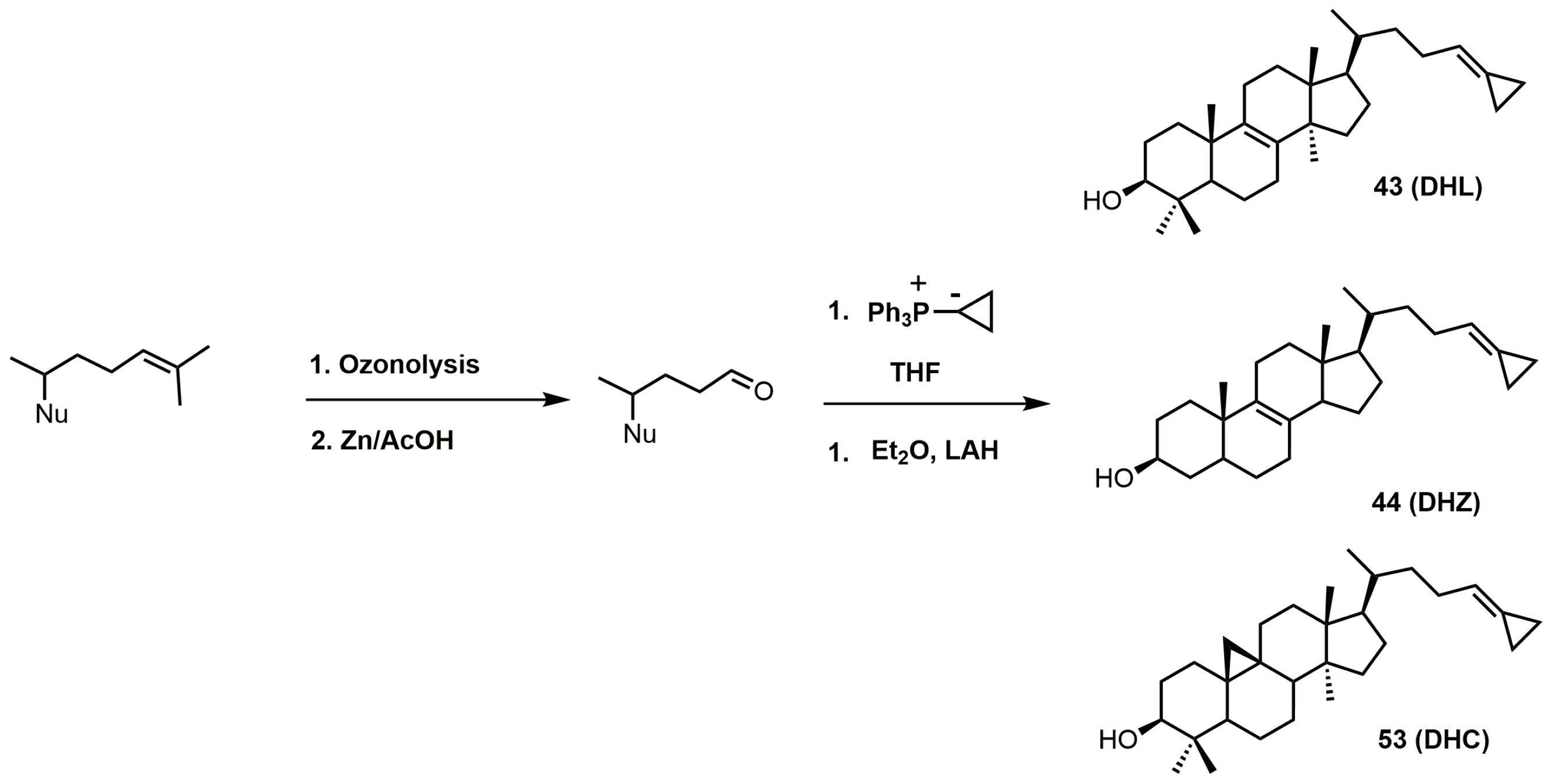
26,27-Dehydrolanosterol (DHL; 43) was reported as an inhibitor of Acanthamoeba spp. trophozoite growth, with an IC50 value of 6 μM making it a much weaker binder in comparison to EL (IC50 of 7 nM) [90]. DHL is metabolized to a favorable substrate, which irreversibly inhibits Tb-24SMT, and the ED50 values of DHL incubated with procyclic or bloodstream forms of T. brucei are 10 and 20 μM, respectively [97]. 26,27-Dehydrozymosterol (DHZ) was reported to inhibit 24-SMT from C. albicans with a Ki value of 9 μM and a reported kinact value of 0.03 min−1 [3]. [3-3H]26,27-Dehydrozymosterol was noted to inhibit 24-SMT from S. cerevisiae with an apparent Ki value of 1.1 μM [98]. DHZ also inhibits SMT2-2 of Glycine max (soybean) with a Ki value of 9.3 μM and a Kinact value of 0.023 min−1 [99], while DHZ irreversibly inhibits Arabidopsis SMT2 with a Ki value of 49 μM and a kinact value of 0.009 s−1 [100]. T. brucei 24-SMT was inhibited by DHZ with a Ki value of 29 μM and a kinact value of 0.26 min−1. Against T. brucei 24-SMT, DHZ was noted to be a weaker inhibitor in comparison to EI and AZAL [88]. DHL cannot inhibit T. brucei 24-SMT or yeast 24-SMT because “uncharged” 4,4-dimethylsterols cannot bind productively to these types of SMT [88]. 26,27-Dehydrocycloartenol (DHC; 53) was observed to be a substrate for soybean SMT with Km and kcat values of 10 mM and 0.018, respectively [101]. There are two main pathways along which DHL can irreversibly inhibit TbSMT (Figure 20) [97]. Path A has a carbocation intermediate on carbon 24, while path A has the intermediate on carbon 27, and both carbocations are reactive with SMT. Either biosynthetic route results in the metabolized DHL to irreversibly bind to TbSMT, and the resulting prosthetic group was hydrolyzed with potassium hydroxide in methanol to yield the C-24 or C-27 alcohol (Figure 23) that was characterized by gas chromatography–mass spectrometry (GCMS).
The 26,27-dehydrosterols can be prepared in a few steps starting with the 3-acetylated sterol (Figure 25). The 24–25 double bond of the corresponding sterol is transformed into an aldehyde by ozonolysis with a reductive workup [102]. The 26,27-cyclopropylidine moiety is installed via a Wittig reaction using the desired sterol aldehyde and the phosphorus ylide formed from cyclopropyltriphenylphosphonium bromide and butyllithium [102]. The acetate group was easily removed with LAH.
Another class of 24-SMT inhibitors comprises sulfur-containing sterols such as 25-thialanosterol (45) and 25-thialanosterol iodide (46) (Figure 19) [103]. 24-SMT from C. albicans was irreversibly inhibited with 25-thialanosterol with a Ki value of 5 μM and an apparent kinact value of 0.013 min−1, while the corresponding sulfonium salt was a reversible transition-state inhibitor with a Ki value of 20 nM [103]. 24-Thialanosterol was observed to inhibit C. albicans 24-SMT with an IC50 value of 20 μM [3]. 25-Thialanosterol iodide has a Ki value of 86 nM against T. brucei 24-SMT and an ED50 value of 2 μM against both the procyclic and bloodstream forms of the T. brucei parasite [2]. 24-SMT can methylate the 25-sulfur atom of 25-thialanosterol to form a sulfonium cation that can act as a methyl donor to irreversibly methylate SMT (Figure 26) [4].
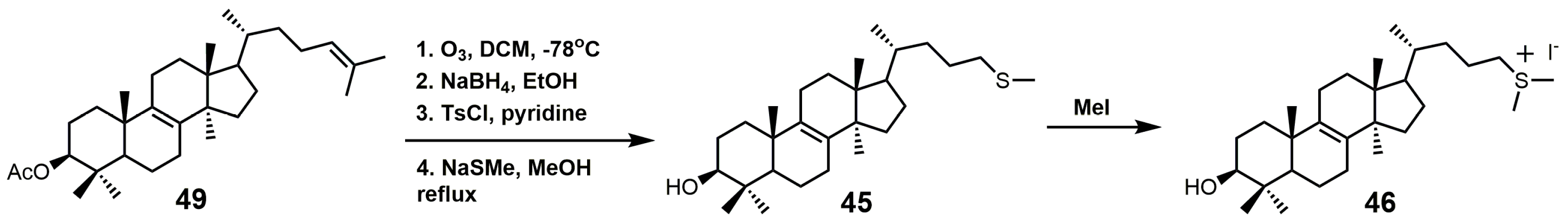
The synthesis of 25-thialanosterol (45) and 25-thialanosterol iodide (46) is outlined in Figure 27 [103]. The synthesis started with ozonolysis of acetylated lanosterol (49) to yield 3-acetoxylanosta-8,24-dienol-26-al. 3-Acetoxylanosta-8,24-dienol-26-al was then reduced with sodium borohydride to yield an alcohol that was activated as a tosylate. Methanethiolate then displaced the tosylate group to yield 25-thialanosterol (45). Treatment of 25-thialansterol with methyl iodide yielded 25-thialanosterol iodide (46).
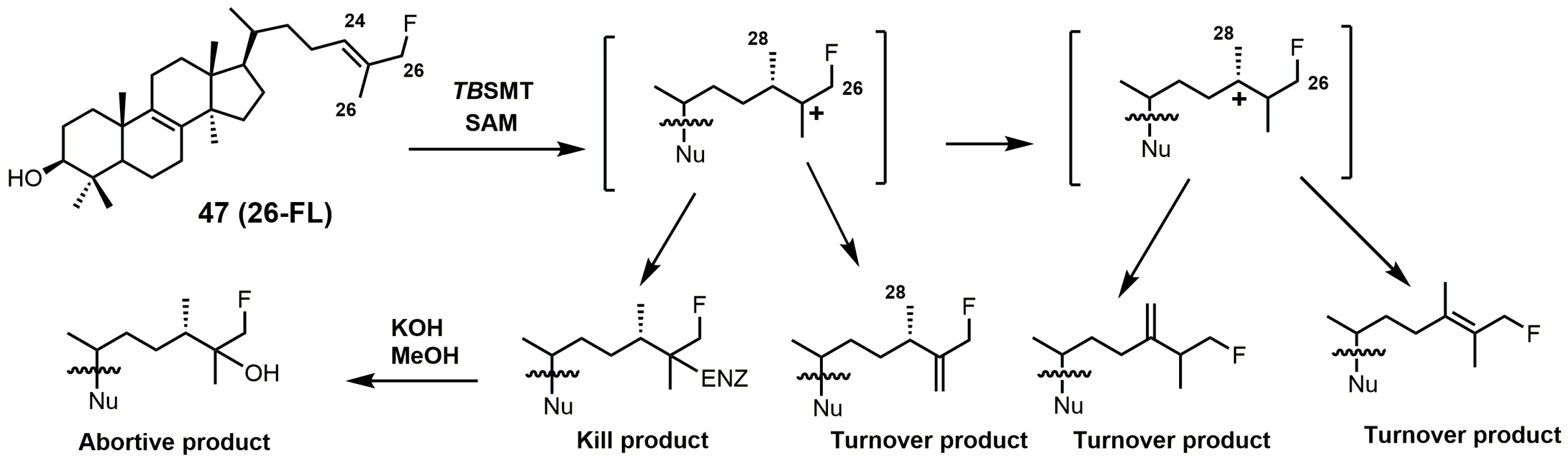
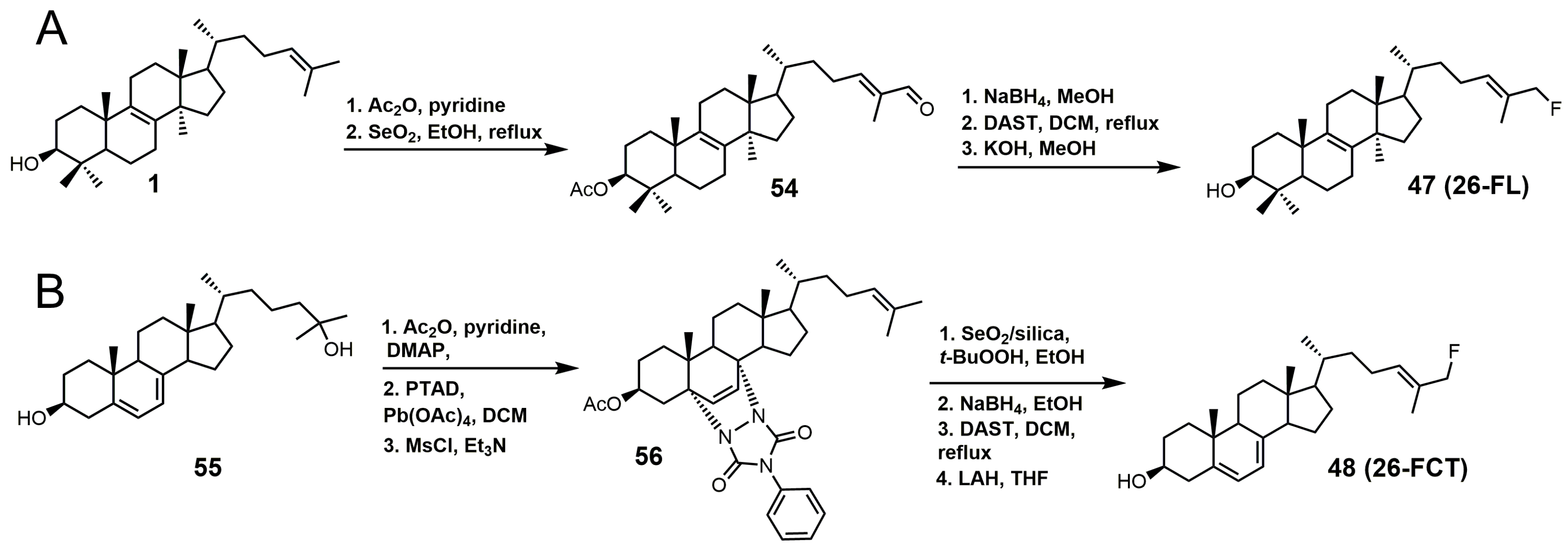
26-Fluorolanosterol (26-FL; 47) is metabolically converted by T. brucei into a fluorinated substrate (26-fluorozymosterol) that irreversibly binds to 24-SMT and inhibits ergosterol biosynthesis and growth of both the procyclic and bloodstream forms of T. brucei [5]. T. brucei cell-based studies were conducted with 26-FL, and IC50 values for the procyclic and bloodstream forms were 3 and 16 μM, respectively, while 26-FL had no effect on HEK cell growth at up to 100 μM [5]. In order to further investigate the preferred substrate for TbSMT, 26-fluorocholesta-5,7,24-trienol (26-FCT) was synthesized (Figure 28). 26-FCT was synthesized instead of 26-fluorozymosterol because of the difficulty in obtaining sufficient amounts of zymosterol to enable synthetic manipulation. 26-FCT was observed to be a competitive inhibitor of 24-SMT with respect to zymosterol and exhibited a Ki value of 75 μM [5]. 26-FCT was confirmed to be a metabolite of 26-FL when metabolized by the procyclic form of T. brucei by GCMS with an authentic sample of 26-FCT [5]. 26-FCT has an excellent partition ration of 1.08, comparing favorably with eflornithine, which has a partition ratio of 3.3 against L-ornithine decarboxylase [5]. Both 26-FL and 26-FCT exhibit desirable drug characteristics with good specificity and low toxicity, and they might be useful in treating sleeping sickness or other protozoan infections [5]. When 26-FL is methylated by TbSMT, two carbocations can form as short-lived intermediates that can be transformed into various turnover products (Figure 27) [5]. The kill product is where the 24-SMT enzyme has a prosthetic group attached, whereby SMT is irreversibly inhibited.
26-FL was synthesized by the method used to synthesize 26-fluorocycloarentol (Figure 28A) [104]. The 3-hydroxy group of lanosterol was acetylated, and then the 26 methyl group was oxidized to an aldehyde (54) with selenium dioxide. The aldehyde was then reduced to the alcohol with sodium borohydride, and then the fluorine atom was installed via DAST. The acetate group was removed by potassium hydroxide in methanol to yield 26-FL.
The synthesis of 26-FCT started with compound 55, which can be synthesized in eight steps starting with ergosterol (Figure 28B) [105]. Compound 55 was then acetylated, the Δ5 and Δ7 double bonds were protected with 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD), and the Δ24–25 double bond was successfully installed with the use of mesyl chloride and triethylamine to yield compound 56. The last four steps in the synthesis of 25-FCT were similar to the last four steps in the synthesis of 26-FL, except that global deprotection in the last step was accomplished with LAH.
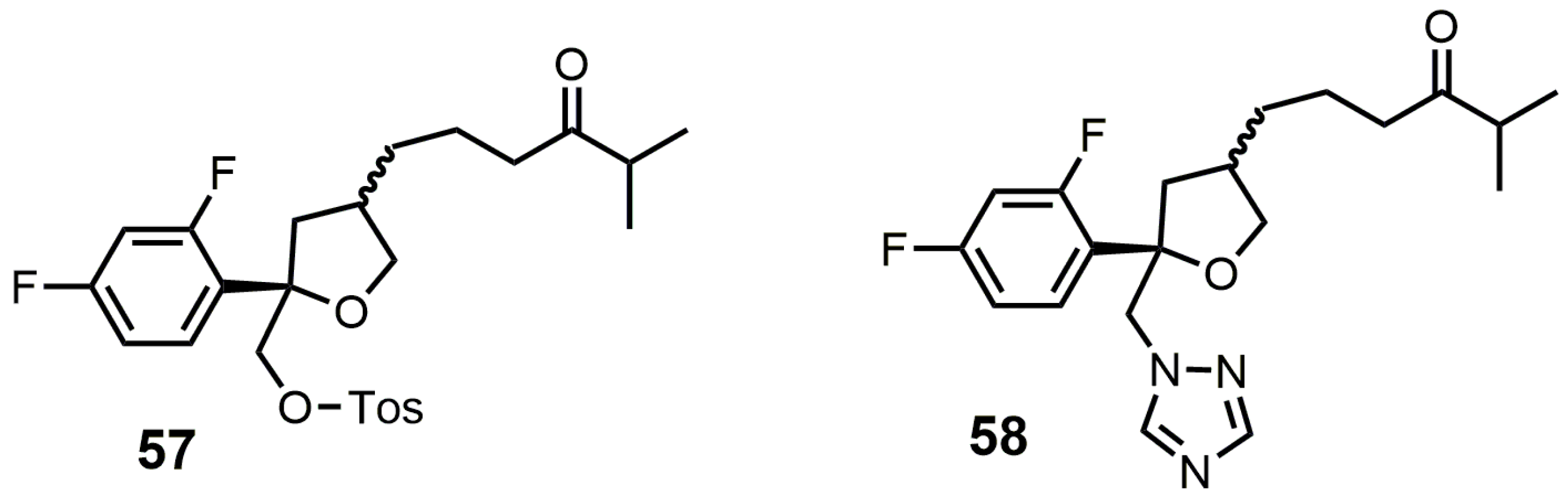
5. Bifunctional 24-SMT and SDM Inhibitors
Compounds 57 and 58 (Figure 29) were designed to be dual functional SDM and 24-SMT inhibitors [106]. These compounds completely inhibited SDM from rat liver microsomes at 10 μM and showed reasonable in vitro potencies against C. albicans, C. neoformans, A. fumigatus, T. mentagrophytes, Candida. pseudotropicalis, and Candida. krusei with MIC values ranging from <0.1 to 50 mg/mL [106]. Compounds 57 and 58 were then tested in vivo against a murine candidiasis antifungal model, and unfortunately both compounds were ineffective against the induced infection. No explanation was provided as to why these compounds were ineffective in the in vivo model.
6. Conclusions
The inhibition of ergosterol biosynthesis in fungi and parasitic protozoa via the inhibition of SDM or 24-SMT with small molecules has been shown to be effective. Azole antifungals that target SDM have already been approved for the treatment of various fungal infections; however, they have not been officially approved to treat protozoan infections, despite various azoles advancing to clinical trials. Posaconazole was investigated in two phase II clinical trials as a possible agent to treat Chagas disease, and on the basis of the results provided thus far, it is unlikely that posaconazole will progress to phase III clinical trials [107,108,109]. The inhibition of fungal and protozoan 24-SMT with rationally designed molecules that are specific, selective, and non-toxic to humans have the potential to be used as the next generation of drugs to treat fungal infections or neglected tropical diseases that are demonstrating resistance against current therapies. It is anticipated that in the near future, both fungal and protozoan infections will be treated with a combination therapy that utilizes the cocurrent administration of a SDM and a 24-SMT inhibitor.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef] [PubMed]
- Haubrich, B.A.; Singha, U.K.; Miller, M.B.; Nes, C.R.; Anyatonwu, H.; Lecordier, L.; Patkar, P.; Leaver, D.J.; Villalta, F.; Vanhollebeke, B.; et al. Discovery of an ergosterol-signaling factor that regulates Trypanosoma brucei growth. J. Lipid Res. 2015, 56, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, K.; Kanagasabai, R.; Nguyen, T.T.M.; Nes, W.D. Purification, characterization and inhibition of sterol C24-methyltransferase from Candida albicans. Arch. Biochem. Biophys. 2011, 505, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nes, W.D. Steroidal triterpenes: Design of substrate-based inhibitors of ergosterol and sitosterol synthesis. Molecules 2009, 14, 4690–4706. [Google Scholar] [CrossRef] [PubMed]
- Leaver, D.J.; Patkar, P.; Singha, U.K.; Miller, M.B.; Haubrich, B.A.; Chaudhuri, M.; Nes, W.D. Fluorinated sterols are suicide inhibitors of ergosterol biosynthesis and growth in Trypanosoma brucei. Chem. Biol. 2015, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Zhou, W.; Ganapathy, K.; Liu, J.; Vatsyayan, R.; Chamala, S.; Hernandez, K.; Miranda, M. Sterol 24-C-methyltransferase: An enzymatic target for the disruption of ergosterol biosynthesis and homeostasis in Cryptococcus neoformans. Arch. Biochem. Biophys. 2009, 481, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Rachakonda, G.; Wawrzak, Z.; Pomel, S.; Cojean, S.; Nde, P.N.; Nes, W.D.; Locuson, C.W.; Calcutt, M.W.; et al. VFV as a new effective CYP51 structure-derived drug candidate for chagas disease and visceral leishmaniasis. J. Infect. Dis. 2015, 212, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Scutera, S.; Savoia, D. New chemotherapeutic strategies against malaria, leishmaniasis and trypanosomiases. Curr. Med. Chem. 2013, 20, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Tavangar, P.; Keighobadi, M. An overview of azoles targeting sterol 14α-demethylase for antileishmanial therapy. Eur. J. Med. Chem. 2017, 135, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Duschank, V.G.; Couto, A.S. Targets and patented drugs for chemotherapy of Chagas disease. In Frontiers in Anti-Infective Drug Discovery; Choudhary, M.I., Ed.; Bentham Science Publishers: Oak Park, IL, USA, 2010; Volume 1, pp. 323–408. [Google Scholar]
- Simarro, P.P.; Cecchi, G.; Franco, J.R.; Paone, M.; Diarra, A.; Ruiz-Postigo, J.A.; Fѐvre, E.M.; Mattioli, R.C.; Jannin, J.G. Estimating and mapping the population at risk of sleeping sickness. PLoS Negl. Trop. Dis. 2012, 6, e1859. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Meerpoel, L.; Lewi, P. Conazoles. Molecules 2010, 15, 4129–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, H.L.; Ernst, E.J.; Klepser, M.E. Novel triazole antifungal agents. Exp. Opin. Investig. Drugs 2000, 9, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Kim, K.; Soeiro, M.N.C.; da Silva, C.F.; Batista, D.G.J.; Batista, M.M.; Yazlovitskaya, E.M.; Waterman, M.R.; Sulikowski, G.A.; Lepesheva, G.I. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Song, Z.; Kanagasabai, R.; Liu, J.; Jayasimha, P.; Sinha, A.; Veeramachanemi, P.; Miller, M.B.; Nes, W.D. Mechanism-based enzyme inactivators of phytosterol biosynthesis. Molecules 2004, 9, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [PubMed]
- Worthington, P. Sterol biosynthesis inhibiting triazole fungicides. In Bioactive Heterocyclic Compound Classes: Agrochemicals; Lamberth, C., Dinges, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 129–145. [Google Scholar]
- Parker, J.E.; Warrilow, A.G.S.; Cools, H.J.; Martel, C.M.; Nes, W.D.; Fraaije, B.A.; Lucas, J.A.; Kelly, D.E.; Kelly, S.L. Mechanism of binding of prothioconazole to Mycosphaerella graminicola CYP51 differs from that of other azole antifungals. Appl. Environ. Microbiol. 2011, 77, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Price, C.L.; Warrilow, A.G.S.; Parker, J.E.; Mullins, J.G.L.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Novel substrate specificity and temperature-sensitive activity of Mycosphaerella graminicola CYP51 supported by the native NADPH cytochrome P450 reductase. Appl. Environ. Microbiol. 2015, 81, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Price, C.L.; Parker, J.E.; Warrilow, A.G.S.; Kelly, D.E.; Kelly, S.L. Azole fungicides–understanding resistance mechanisms in agricultural fungal pathogens. Pest Manag. Sci. 2015, 71, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Anderson, S.; Kleshchenko, Y.; Furtak, V.; Wawrzak, Z.; Villalta, F.; Waterman, M.R. Structural insights into inhibition of sterol 14α-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 2010, 285, 25582–25590. [Google Scholar] [CrossRef] [PubMed]
- Tuck, S.F.; Robinson, C.H.; Silverton, J.V. Assessment of the active-site requirements of lanosterol 14α-demethylase: Evaluation of novel substrate analogues as competitive inhibitors. J. Org. Chem. 1991, 56, 1260–1266. [Google Scholar] [CrossRef]
- Frye, L.L.; Cusack, K.P.; Leonard, D.A. 32-Methyl-32-oxylanosterols: Dual-action inhibitors of cholesterol biosynthesis. J. Med. Chem. 1993, 36, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-7-24,25-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Trzaskos, J.M.; Ko, S.S.; Magolda, R.L.; Favata, M.F.; Fischer, R.T.; Stam, S.H.; Johnson, P.R.; Gaylor, J.L. Substrate-based inhibitors of lanosterol 14α-methyl demethylase: I. Assessment of inhibitor structure-activity relationship and cholesterol biosynthesis inhibition properties. Biochemistry 1995, 34, 9670–9676. [Google Scholar] [CrossRef] [PubMed]
- Trzaskos, J.M.; Fischer, R.T.; Ko, S.S.; Magolda, R.L.; Stam, S.H.; Johnson, P.R.; Gaylor, J.L. Substrate-based inhibitors of lanosterol 14α-methyl demethylase: II. Time-dependent enzyme inactivation by selected oxylanosterol analogs. Biochemistry 1995, 34, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Tuck, S.F.; Patel, H.; Safi, E.; Robinson, C.H. Lanosterol 14α-demethylase (P45014DM): Effects of P45014DM inhibitors on sterol biosynthesis downstream of lanosterol. J. Lipid Res. 1991, 32, 893–902. [Google Scholar] [PubMed]
- Trzaskos, J.M.; Magolda, R.L.; Favata, M.F.; Fischer, R.T.; Johnson, P.R.; Chen, H.W.; Ko, S.S.; Leonard, D.A.; Gaylor, J.L. Modulation of 3-hydroxy-3-methylglutaryl-CoA reductase by 14α-flurolanost-7-en-3-ol. J. Biol. Chem. 1993, 268, 22591–22599. [Google Scholar] [PubMed]
- Frye, L.L.; Robinson, C.H. Synthesis of potential mechanism-based inactivators of lanosterol 14α-methyl demethylase. J. Org. Chem. 1990, 55, 1579–1584. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Zaitseva, N.G.; Nes, W.D.; Zhou, W.; Arase, M.; Liu, J.; Hill, G.C.; Waterman, M.R. CYP51 from Trypanosoma cruzi: A phyla-specific residue in the B’ helix defines substrate preferences of sterol 14α-demethylase. J. Biol. Chem. 2006, 281, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Backx, L.J.J.; Mostmans, J.H.; Van Cutsem, J. Antimycotic imidazoles. Part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. J. Med. Chem. 1979, 22, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Van Cutsem, J. Antimycotic Imidazoles. 5. Synthesis and antimycotic properties of 1-[[2-aryl-4-(arylalkyl)-1,3-dioxolan-3-yl]methyl]-1H-imidazoles. J. Med. Chem. 1981, 24, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Hendrickx, R.; Van Cutsem, J. Antimycotic azoles. 6. Synthesis and antifungal properties of terconazole, a novel triazole ketal. J. Med. Chem. 1983, 26, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Backx, L.J.J.; Van Cutsem, J. Antimycotic azoles. 7. Synthesis and antifungal properties of a series of novel triazol-3-ones. J. Med. Chem. 1984, 27, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.; Cooper, K.; Marriott, M.S.; Tarbit, M.H.; Troke, P.F.; Whittle, P.J. Discovery of fluconazole, a novel antifungal agent. Rev. Infect. Dis. 1990, 12, S267–S271. [Google Scholar] [CrossRef] [PubMed]
- Saksena, A.K.; Girijavallabhan, V.M.; Wang, H.; Liu, Y.T.; Pike, R.E.; Ganguly, A.K. Concise asymmetric routes to 2,2,4-trisubstituted tetrahydrofurans via chiral titanium imide enolates: Key intermediates towards synthesis of highly active azole antifungals SCH 51048 and SCH 56592. Tetrahedron Lett. 1996, 37, 5657–5660. [Google Scholar] [CrossRef]
- Saksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.T.; Pinto, P.; Bennett, F.; Jao, E.; Patel, N.; et al. Advances in the chemistry of novel broad-spectrum orally active azole antifungals: Recent studies leading to the discovery of SCH 56592. Spec. Publ. R. Soc. Chem. 1997, 198, 180–199. [Google Scholar] [CrossRef]
- Livni, E.; Fischman, A.J.; Ray, S.I.; Elmaleh, D.R.; Alpert, N.M.; Weiss, S.; Correia, J.A.; Webb, D.; Dahl, R.; Robeson, W.; et al. Synthesis of 18F-labeled fluconazole and positron emission tomography studies in rabbits. Nucl. Med. Biol. 1992, 19, 191–199. [Google Scholar] [CrossRef]
- Korwar, S.; Amir, S.; Tosso, P.N.; Desai, B.K.; Kong, C.J.; Fadnis, S.; Telgang, N.S.; Ahmad, S.; Roper, T.D.; Gupton, B.F. The application of a continuous Grignard reaction in the preparation of fluconazole. Eur. J. Org. Chem. 2017, 6495–6498. [Google Scholar] [CrossRef]
- Aher, N.G.; Pore, V.S.; Mishra, N.N.; Kumar, A.; Shukla, P.K.; Sharma, A.; Bhat, M.K. Synthesis and antifungal activity of 1,2,3-triazole containing fluconazole analogues. Bioorg. Med. Chem. Lett. 2009, 19, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Garvey, E.P.; Shaver, S.R.; Schotzinger, R.J.; Hoekstra, W.J. Design and optimization of highly-selective, broad spectrum fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Wiederhold, N.P.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. The tetrazole VT-1161 is a potent inhibitor of Trichophyton rubrum through its inhibition of T. rubrum CYP51. Antimicrob. Agents Chemother. 2017, 61, e00333-17/1–e00333-12/11. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Hull, C.M.; Rolley, N.J.; Parker, J.E.; Nes, W.D.; Smith, S.N.; Kelly, D.E.; Kelly, S.L. Clotrimazole as a potent agent for treating the oomycete fish pathogen Saprolegnia parasitica through inhibition of sterol 14α-demethylase (CYP51). Appl. Environ. Microbiol. 2014, 80, 6154–6166. [Google Scholar] [CrossRef] [PubMed]
- Gagnepain, J.; Maity, P.; Lamberth, C.; Cederbaum, F.; Rajan, R.; Jacob, O.; Blum, M.; Bieri, S. Synthesis and fungicidal activity of novel imidazole-based ketene dithioacetals. Bioorg. Med. Chem. 2018, 26, 2009–2016. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Blobaum, A.L.; Rachakonda, G.; Lindsley, C.W.; Villalta, F.; Nes, W.D.; Botta, M.; Guengerich, F.P.; et al. Sterol 14α-demethylase structure-based design of VNI ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide)) derivatives to target fungal infections: Synthesis, biological evaluation, and crystallographic analysis. J. Med. Chem. 2018, 61, 5679–5691. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Podust, L.M.; Roush, W.R. Drug strategies targeting CYP51 in neglected tropical diseases. Chem. Rev. 2014, 114, 11242–11271. [Google Scholar] [CrossRef] [PubMed]
- Parish, E.J.; Schroepfer, G.J. Sterol synthesis. A simplified method for the synthesis of 32-oxygenated derivatives of 24,25-dihydrolanosterol. J. Lipid Res. 1981, 22, 859–868. [Google Scholar] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.N.; Quiocho, F.A.; Sass, R.L.; Werness, P.; Emery, H.; Knapp, F.F.; Schroepfer, G.J. Sterol biosynthesis: Establishment of the structure of 3-p-bromobenzoyloxy-5α-cholest-8(14)-en-15-ol. Bioorg. Chem. 1976, 5, 1–10. [Google Scholar] [CrossRef]
- Gibbons, G.F.; Ramanada, K. Synthesis and configuration at C-15 of the epimeric 5-lanost-8-en-3,15-diols. J. Chem. Soc. Chem. Commun. 1975, 6, 213–214. [Google Scholar] [CrossRef]
- Gaylor, J.L.; Johnson, P.R.; Ko, S.S.; Magolda, R.L.; Stam, S.H.; Trzaskos, J.M. Steroid Derivatives Useful as Hypocholesterolemics. U.S. Patent 5034548, 20 August 1991. [Google Scholar]
- Musiol, R.; Kowalczyk, W. Azole antimycotics—A highway to new drugs or a dead end? Curr. Med. Chem. 2012, 19, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Villalta, F.; Dobish, M.C.; Nde, P.N.; Kleshchenko, Y.Y.; Hargrove, T.Y.; Johnson, C.A.; Waterman, M.R.; Johnston, J.N.; Lepesheva, G.I. VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis. 2013, 208, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.; Cuenca-Estrella, M.; Monzon, A.; Rodriguez-Tudela, J.L. In-vitro comparative activity of UR-9825, itraconazole and fluconazole against clinical isolates of Candida spp. J. Antimicrob. Chemother. 1999, 44, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.; Brammer, K.W.; Marriott, M.S.; Troke, P.F. Activity of UK-49,858, a bis triazole derivative, against experimental infections with Candida albicans and Trichophyton mentagrophytes. Antimicrob. Agents Chemother. 1985, 27, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Ott, R.D.; Hargrove, T.Y.; Kleshchenko, Y.Y.; Schuster, I.; Nes, W.D.; Hill, G.C.; Villalta, F.; Waterman, M.R. Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: Enzyme inhibition and parasite cell growth. Chem. Biol. 2007, 14, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, E.; Samanovic, M.; Murkin, A.S.; Raper, J.; Buckner, F.; Rodriguez, A. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl. Trop. Dis. 2009, 3, e384. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Leung, S.S.F.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Villalta, F.; Waterman, M.R. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv. Parasitol. 2011, 75, 65–87. [Google Scholar] [PubMed]
- Alrajhi, A.A.; Ibrahim, E.A.; De Vol, E.B.; Khairat, M.; Faris, R.M.; Maguire, J.H. Fluconazole for the treatment of cutaneous leishmaniasis caused by Lesihmania major. N. Engl. J. Med. 2002, 346, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Payares, G.; Molina, J.; Sanoja, C.; Liendo, A.; Lazzardi, K.; Piras, M.M.; Piras, R.; Perez, N.; Wincker, P.; et al. Cure of short- and long term experimental Chagas’ disease using D0870. Science 1996, 273, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Payares, G.; Sanoja, C.; Lira, R.; Romanha, A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents 2003, 21, 27–38. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Nes, W.D.; Zhou, W.; Hill, G.C.; Waterman, M.R. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 2004, 43, 10789–10799. [Google Scholar] [CrossRef] [PubMed]
- Vivas, J.; Urbina, J.A.; de Souza, W. Ultrastructural alterations in Tyrpanosoma (Schizotrypanum) cruzi induced by Δ24(25) sterol methyl transferase inhibitors and their combinations with ketoconazole. Int. J. Antimicrob. Agents 1996, 7, 235–240. [Google Scholar] [CrossRef]
- Espinel-Ingroff, A.; Shadomy, S.; Gebhart, R.J. In vitro studies with R 51,211 (Itraconazole). Antimicrob. Agents Chemother. 1984, 26, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Borgers, M.; Van de Ven, M.A. Degenerative changes in fungi after itraconazole treatment. Rev. Infect. Dis. 1987, 9, 33–44. [Google Scholar] [CrossRef]
- Lipp, H.-P. Antifungal agents-clinical pharmacokinetics and drug interactions. Mycoses 2008, 51, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Gubbins, P.O. Mould-active azoles: Pharmacokinetics, drug interactions in neutropenic patients. Curr. Opin. Infect. Dis. 2007, 20, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Kelly, D.E.; Baldwin, B.C.; Kelly, S.L. Differential inhibition of human CYP3A4 and Candida albicans CYP51 with azole antifungal agents. Chem. Biol. Interact. 2000, 125, 165–175. [Google Scholar] [CrossRef]
- Hart, D.T.; Lauwers, W.J.; Willemsens, G.; Bossche, H.V.; Opperdoes, F.R. Perturbation of sterol biosynthesis by itraconazole and ketoconazole in Leishmania mexicana mexicana infected macrophages. Mol. Biochem. Parasitol. 1989, 33, 123–134. [Google Scholar] [CrossRef]
- Momeni, A.Z.; Jalayer, T.; Emamjomeh, M.; Bashardost, N.; Ghassemi, R.L.; Meghdadi, M.; Javadi, A.; Aminjavaheri, M. Treatment of cutaneous leishmaniasis with itraconazole. Randomized double-blind study. Arch. Dermatol. 1996, 132, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Baroni, A.; Aiello, F.S.; Vozza, A.; Vozza, G.; Faccenda, F.; Brasiello, M.; Ruocco, E. Cutaneous leishmaniasis treated with itraconazole. Dermatol. Ther. 2009, 22 (Suppl. 1), S27–S29. [Google Scholar] [CrossRef] [PubMed]
- Minodier, P.; Parola, P. Cutaneous leishmaniasis treatment. Travel Med. Infect. Dis. 2007, 5, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Chemotherapy of Chagas disease. Curr. Pharm. Des. 2002, 8, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Ergosterol biosynthesis for the specific treatment of Chagas disease: From basic science to clinical trials. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery, 1st ed.; Jäger, T., Koch, O., Flohé, L., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 489–514. [Google Scholar]
- Benaim, G.; Sanders, J.M.; Garcia-Marchán, Y.; Colina, C.; Lira, R.; Caldera, A.R.; Payares, G.; Sanoja, C.; Burgos, J.M.; Leo-Rossell, A.; et al. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J. Med. Chem. 2006, 49, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Pinazo, M.; Espinosa, G.; Gállego, M.; López-Chejade, P.L.; Urbina, J.A.; Gascón, J. Case report: Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am. J. Trop. Med. Hyg. 2010, 82, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Saksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Ganguly, A.K.; Morgan, B.; Zaks, A.; Puar, M.S. Highly stereoselective access to novel 2,2,4-trisubstituted tetrahydrofurans by halocyclization: Practical chemoenzymatic synthesis of SCH 51048, a broad-spectrum orally active antifungal agent. Tetrahedron Lett. 1995, 36, 1787–1790. [Google Scholar] [CrossRef]
- Chidambaram, V.S.; Miryala, A.K.; Wadhwa, L. Process for Preparing Posaconazole and Intermediates Thereof. PCT International Application WO2009141837A2, 26 November 2009. [Google Scholar]
- Charyulu, P.V.R.; Gowda, D.J.C.; Rajmahendra, S.; Raman, M. Crystalline Forms of Posaconazole Intermediate and Process for the Preparation of Amorphous Posaconazole. U.S. PCT International Application WO2017051342A1, 30 March 2017. [Google Scholar]
- Lepesheva, G.I.; Park, H.; Hargrove, T.Y.; Vanhollebeke, B.; Wawrzak, Z.; Harp, J.M.; Sundaramoorthy, M.; Nes, W.D.; Pays, E.; Chaudhuri, M.; et al. Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010, 285, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.; Christov, P.; Sulikowski, G.A.; Kim, K. A convergent, scalable and stereoselective synthesis of azole CYP51 inhibitors. Tetrahedron Lett. 2017, 58, 4248–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nes, W.D.; Janssen, G.G.; Norton, R.A.; Kalinowska, M.; Crumley, F.G.; Tal, B.; Bergenstrahle, A.; Jonsson, L. Regulation of sterol biosynthesis in sunflower by 24(R,S)25-epiminolanosterol, a novel C-24 methyl transferase inhibitor. Biochem. Biophys. Res. Commun. 1991, 177, 566–574. [Google Scholar] [CrossRef]
- Nes, W.D.; Xu, S.; Parish, E.J. Metabolism of 24(R,S),25-epiminolanosterol to 25-aminolanosterol and lanosterol by Gibberella fujikuroi. Arch. Biochem. Biophys. 1989, 272, 323–331. [Google Scholar] [CrossRef]
- Popjak, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of cholesterol synthesis and cell growth by 24(R,S),25-iminolanosterol and triparanol in cultured rat hepatoma cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [PubMed]
- Zhou, W.; Lepesheva, G.I.; Waterman, M.R.; Nes, W.D. Mechanistic analysis of a multiple product sterol methyltransferase implicated in ergosterol biosynthesis in Trypanosoma brucei. J. Biol. Chem. 2006, 281, 6290–6296. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Vivas, J.; Lazardi, K.; Molina, J.; Payares, G.; Piras, M.M.; Piras, R. Antiproliferative effects of Δ24(25) sterol methyl transferase inhibitors on Trypanosoma (Schizotrypanum) cruzi: In vitro and in vivo studies. Chemotherapy 1996, 42, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Kidane, M.E.; Vanderloop, B.H.; Zhou, W.; Thomas, C.D.; Ramos, E.; Singha, U.; Chaudhuri, M.; Nes, W.D. Sterol methyltransferase a target for anti-amoebatherapy: Towards transition state analog and suicide substrate drug design. J. Lipid Res. 2017, 58, 2310–2323. [Google Scholar] [CrossRef] [PubMed]
- Parish, E.J.; Nes, W.D. Synthesis of new epiminoisopentenoids. Synth. Commun. 1998, 18, 221–226. [Google Scholar] [CrossRef]
- Nes, W.D. Sterol methyl transferase: Enzymology and inhibition. Biochim. Biophys. Acta 2000, 1529, 63–88. [Google Scholar] [CrossRef]
- Pereira, M.; Song, Z.; Santos-Silva, L.K.; Richards, M.H.; Nguyen, T.T.M.; Liu, J.; Soares, C.M.A.; Cruz, A.H.S.; Ganapathy, K.; Nes, W.D. Cloning, mechanistic and functional analysis of a fungal sterol C24-methyltransferase implicated in brassicasterol biosynthesis. Biochim. Biophys. Acta 2010, 1801, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cross, G.A.M.; Nes, W.D. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J. Lipid Res. 2007, 48, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Kohen, F.; Counsell, R.E. Hypocholesterolemic agents. 8. Synthesis of 25-azadihydrolanosterol and derivatives. J. Med. Chem. 1971, 14, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Oehlschlager, A.C.; Angus, R.H.; Pierce, A.M.; Pierce, H.D.; Srinivasan, R.J. Azasterol inhibition of 24-sterol methyltransferase in Saccharomyces cerevisiae. Biochemistry 1984, 23, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Patkar, P.; Singha, U.K.; Chaudhuri, M.; Nes, W.D. 24-Methylenecyclopropane steroidal inhibitors: A Trojan horse in ergosterol biosynthesis that prevents growth of Trypanosoma brucei. Biochim. Biophys. Acta 2017, 1862, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Nes, W.D. Isolation and characterization of an active-site peptide from a sterol methyl transferase with a mechanism-based inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 1533–1536. [Google Scholar] [CrossRef]
- Neelakandan, A.K.; Song, Z.; Wang, J.; Richards, M.H.; Wu, X.; Valliyodan, B.; Nguyen, H.T.; Nes, W.D. Cloning, functional expression and phylogenetic analysis of plant sterol 24C-methyltransferases involved in sitosterol biosynthesis. Phytochemistry 2009, 70, 1982–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Nes, W.D. Sterol methyltransferase2: Purification, properties, and inhibition. Arch. Biochem. Biophys. 2003, 420, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Zhou, W.; Liu, J.; Nes, W.D. Mechanism-based active site modification of the soybean sterol methyltransferase by 26,27-dehydrocycloartenol. Bioorg. Med. Chem. Lett. 2004, 14, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhou, W.; Guo, D.; Nes, W.D. Synthesis of rationally designed mechanism-based inactivators of the (S)-adenosyl-L-methionine: Δ24(25)-sterol methyl transferase. Synth. Commun. 1996, 26, 3841–3848. [Google Scholar] [CrossRef]
- Kanagasabai, R.; Zhou, W.; Liu, J.; Nguyen, T.T.M.; Veeramachaneni, P.; Nes, W.D. Disruption of ergosterol biosynthesis, growth, and the morphological transition in Candida albicans by sterol methyltransferase inhibitors containing sulfur at C-25 in the sterol side chain. Lipids 2004, 39, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Patkar, P.; Haubrich, B.A.; Qi, M.; Nguyen, T.T.M.; Thomas, C.D.; Nes, W.D. C24-Methylation of 26-fluorocycloartenols by recombinant sterol C24-methyltransferase from soybean: Evidence for channel switching and its phylogenetic implications. Biochem. J. 2013, 456, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Fuse, S.; Mifune, Y.; Tanabe, N.; Takahashi, T. Continuous-flow synthesis of activated vitamin D3 and its analogues. Org. Biomol. Chem. 2012, 10, 5205–5211. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-K.; Lee, K.-W.; Kang, H.I.; Yamashita, C.; Kudo, M.; Yoshida, Y. Design and synthesis of potential inhibitors of the ergosterol biosynthesis as antifungal agents. Bioorg. Med. Chem. 2000, 8, 2475–2486. [Google Scholar] [CrossRef]
- Yang, G.; Lee, N.; Ioset, J.-R.; No, J.H. Evaluation of parameters impacting drug susceptibility in intracellular Trypanosoma cruzi assay protocols. SLAS Discov. 2017, 22, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Prat, J.G.; Salvador, F.; Trevino, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; del Carmen Bangher, M.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. Benznidazole and posaconazole in eliminating parasites in asymptomatic T. cruzi carriers: The stop-Chagas trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Comparative sterol biosynthesis pathways across kingdoms (adapted from [2]). HS: Homo sapiens; CN: Cryptococcus neoformans; TB: Trypanosoma brucei; 4-SMO: sterol C4-methyl oxidase; 14-SDM: sterol 14α-demethylase; 24-SMT: sterol C24-methyltransferase.
Figure 1.
Comparative sterol biosynthesis pathways across kingdoms (adapted from [2]). HS: Homo sapiens; CN: Cryptococcus neoformans; TB: Trypanosoma brucei; 4-SMO: sterol C4-methyl oxidase; 14-SDM: sterol 14α-demethylase; 24-SMT: sterol C24-methyltransferase.
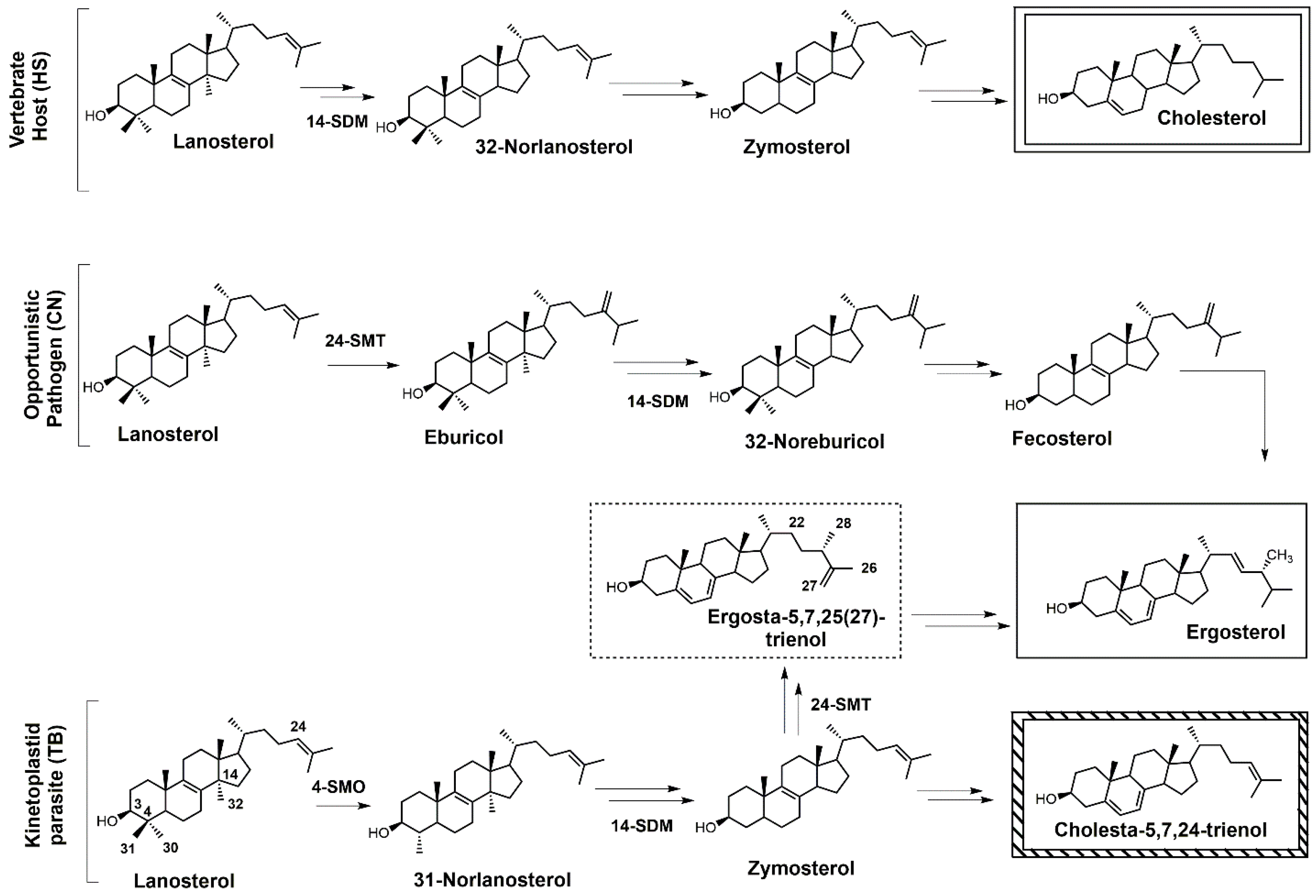
Figure 2.
Conversion of lanosterol (1) into 4,4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol (4) by sterol 14α-demethylase (SDM).
Figure 2.
Conversion of lanosterol (1) into 4,4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol (4) by sterol 14α-demethylase (SDM).
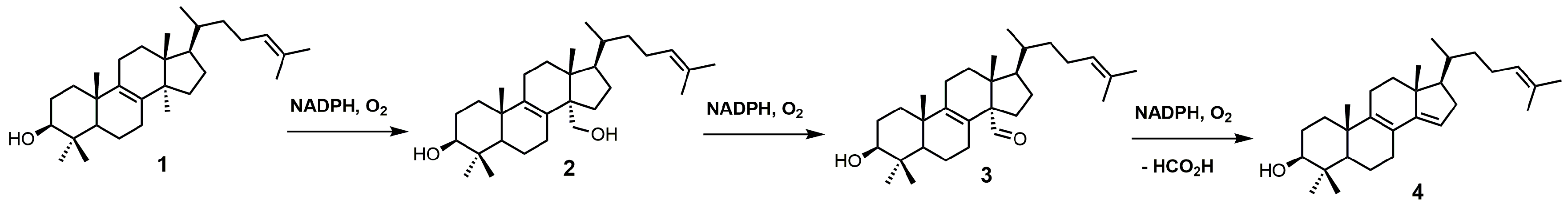
Figure 3.
Sterol-based inhibitors of sterol 14α-demethylase (SDM).

Figure 4.
The synthesis of 14α-methylenecyclopropyl-Δ7-24,25-dihydrolanosterol (MCP) (5).

Figure 5.
The synthesis of 15α-fluoroIanost-7-en-3β-ol (6).
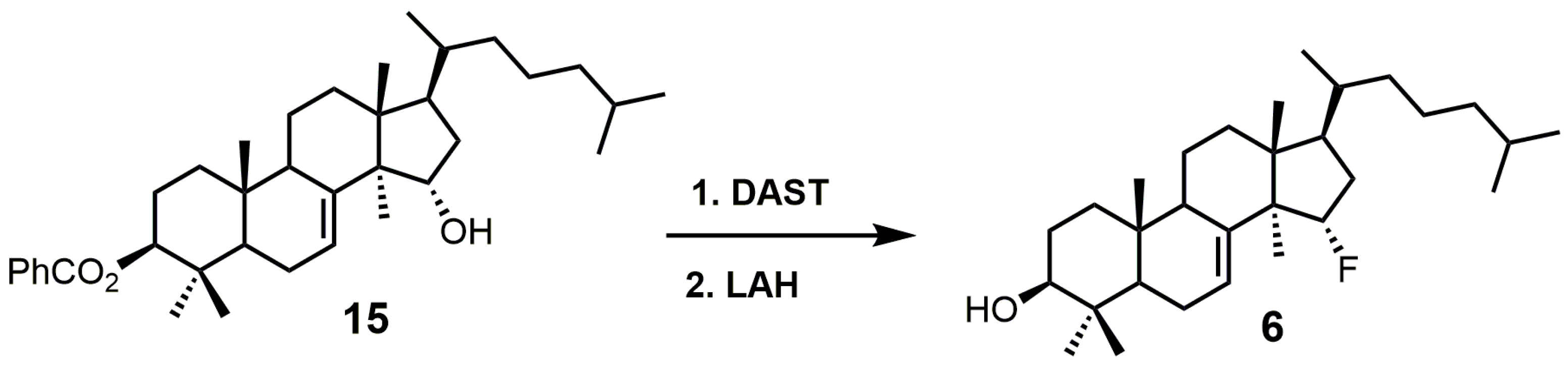
Figure 6.
The synthesis of 4,4-dimethyl-14α-ethynylcholest-7-en-30-ol (7).

Figure 7.
The synthesis of lanost-8-en-32-alkoxime-3β-ol (8).

Figure 8.
The synthesis of 4,4-dimethyl-14α-(1′hydroxy-2′-vinyl)-5α-cholest-8-en-3β-ols.

Figure 9.
The synthesis of 32S-oxiranyllanost-7-en-3β-ol (11).
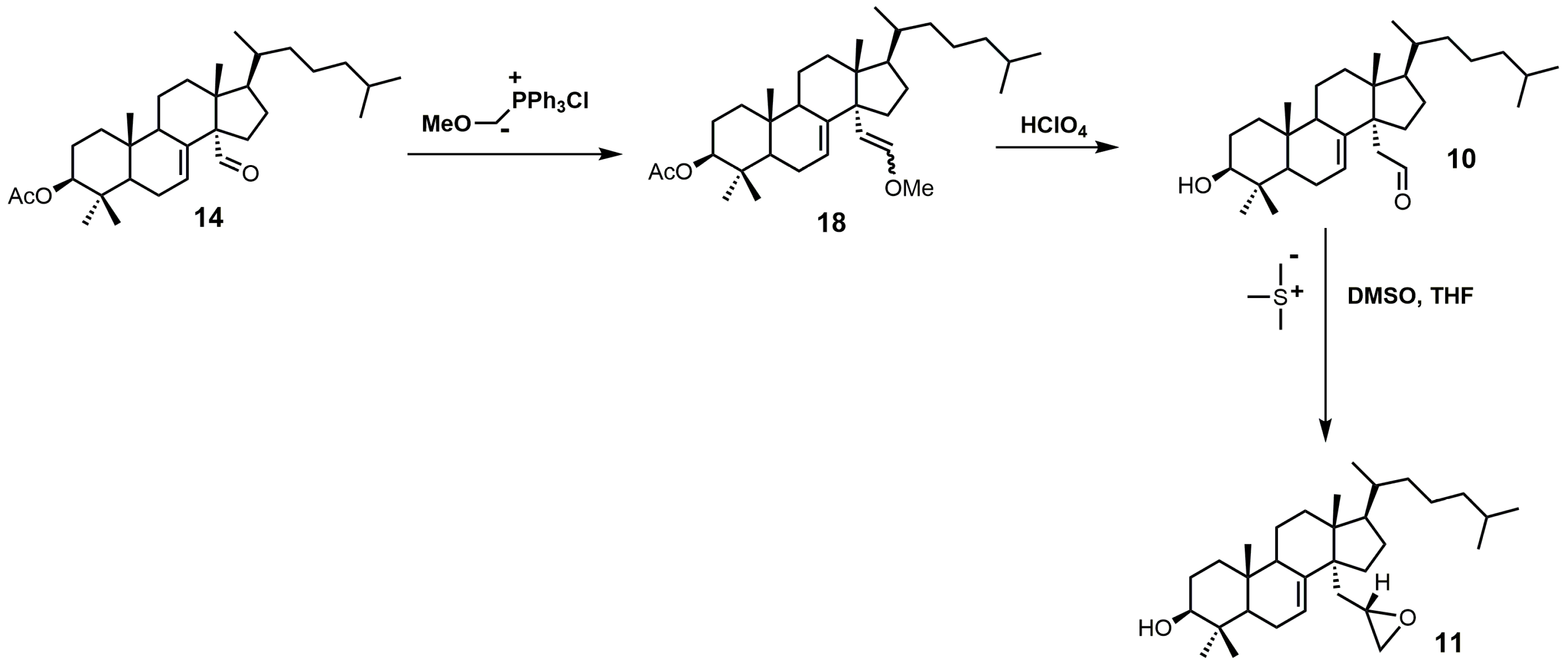
Figure 10.
The synthesis of 4,4-dimethyl-14α-aminomethyl-cholest-8-en-3β-ol (12).
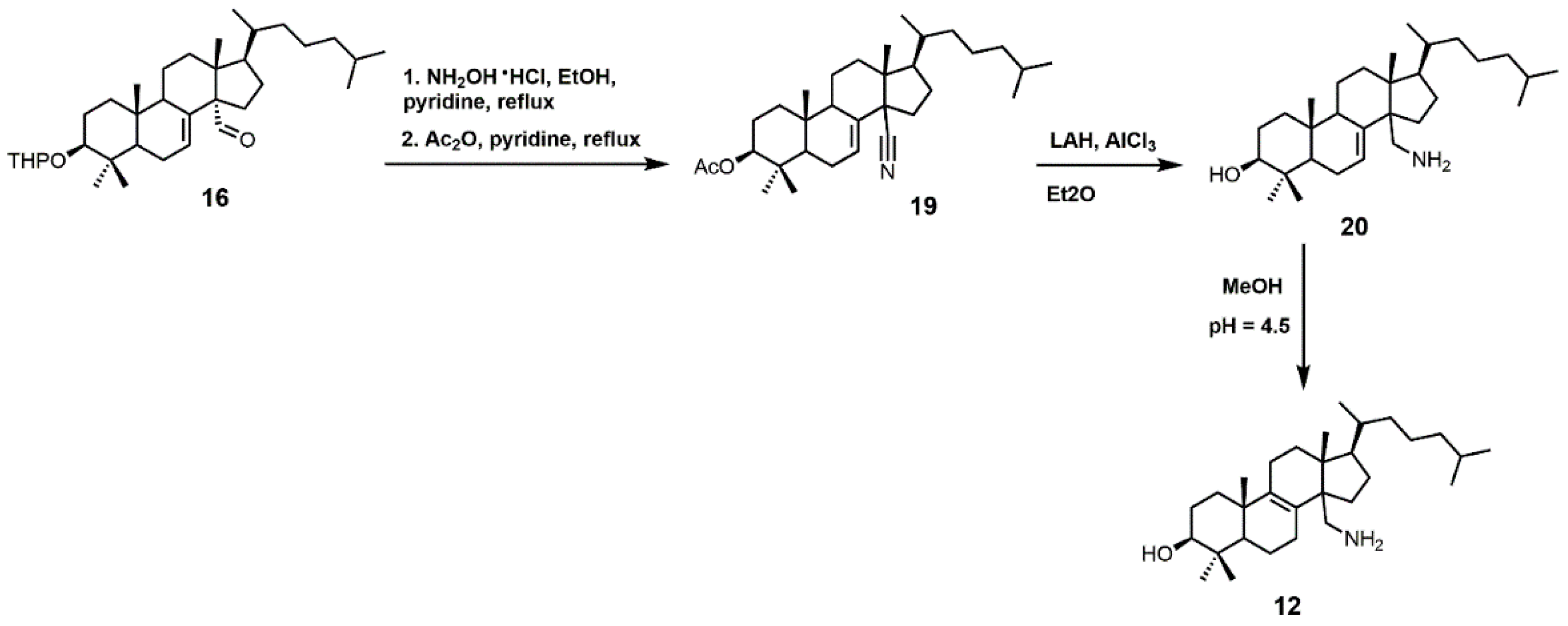
Figure 11.
Azole inhibitors of sterol 14α-demethylase (SDM).

Figure 12.
Synthesis of fluconazole using flow-injection methodology (adapted from [39]).
Figure 12.
Synthesis of fluconazole using flow-injection methodology (adapted from [39]).

Figure 13.
Synthesis of [4-18F]fluconazole.
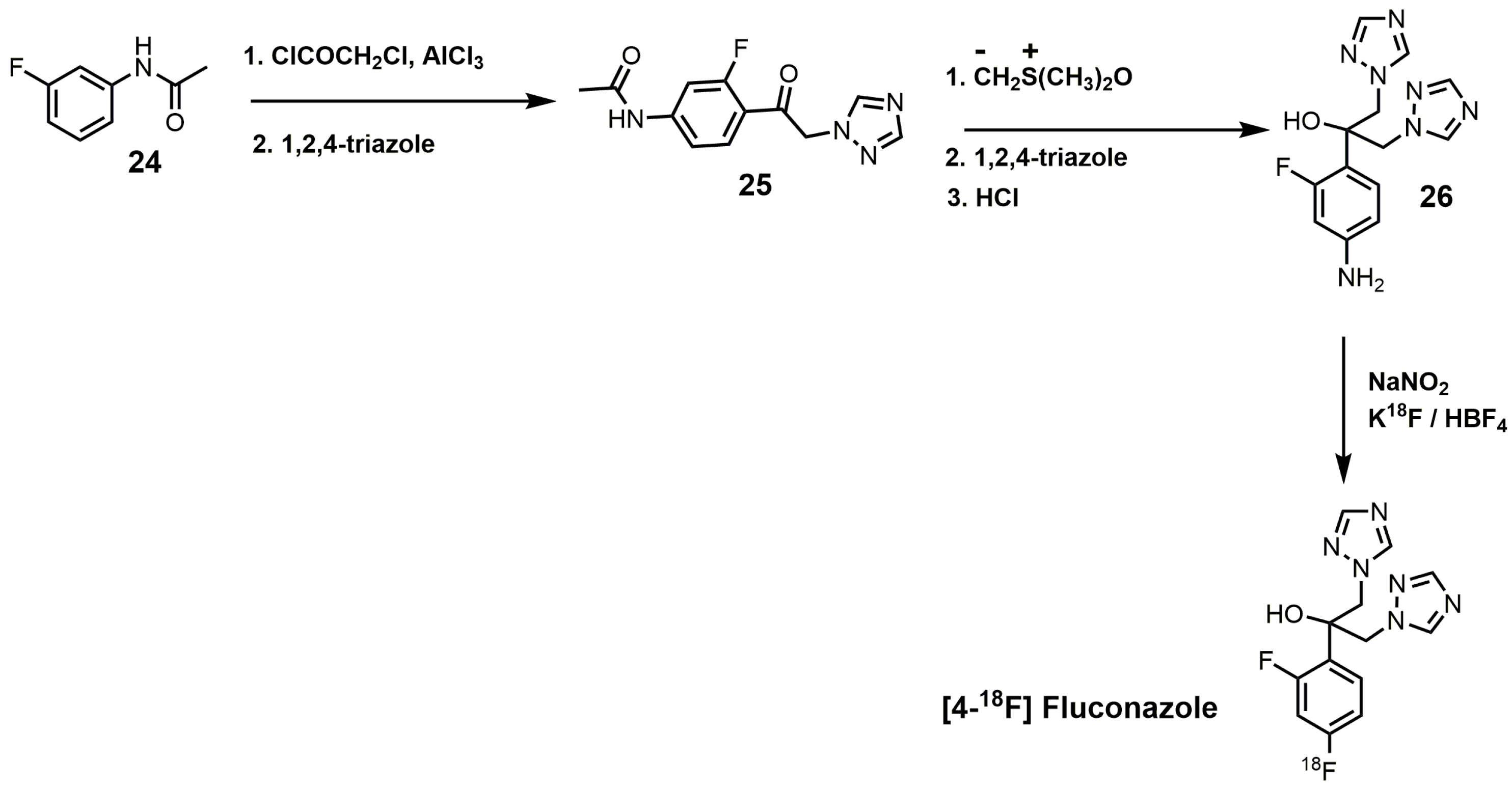
Figure 14.
Synthesis of ketoconazole.

Figure 15.
The synthesis of itraconazole.
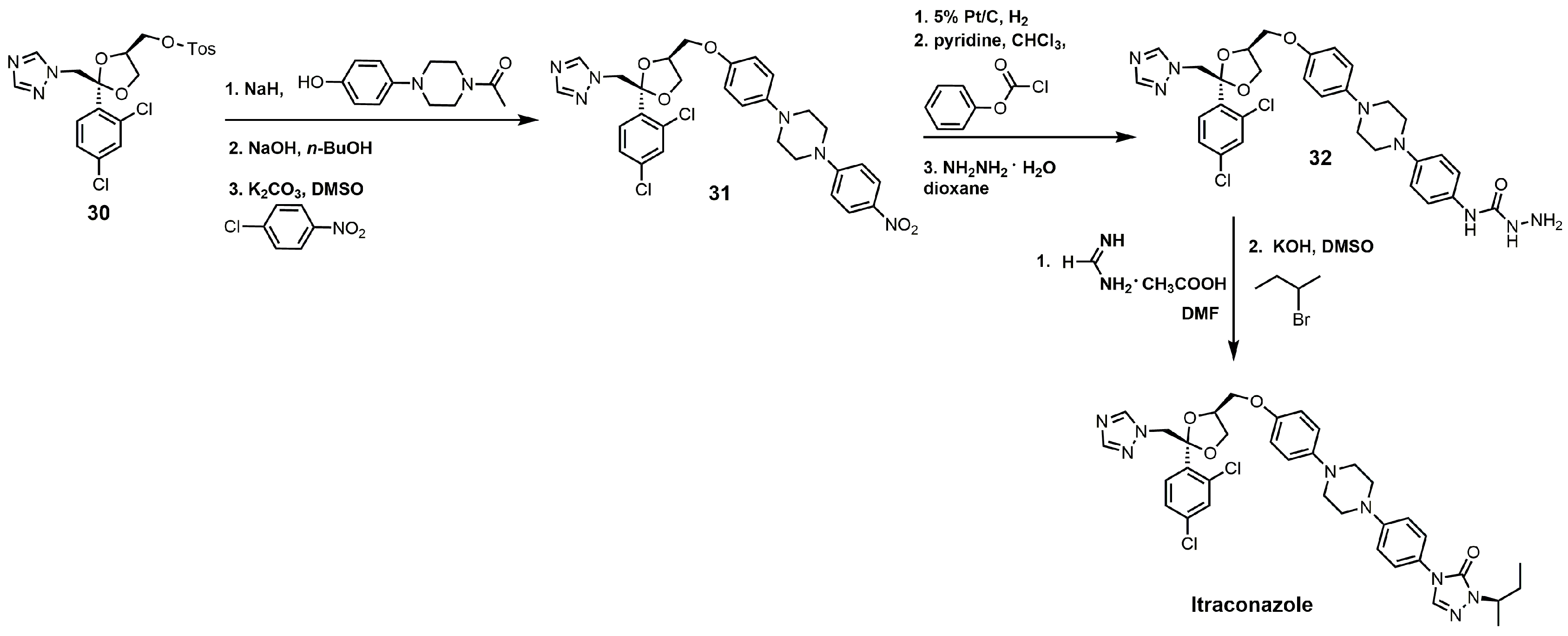
Figure 16.
Synthesis of posaconazole.

Figure 17.
Synthesis of (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadi-azol-2-yl)-benzamide (VNI).
Figure 17.
Synthesis of (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadi-azol-2-yl)-benzamide (VNI).
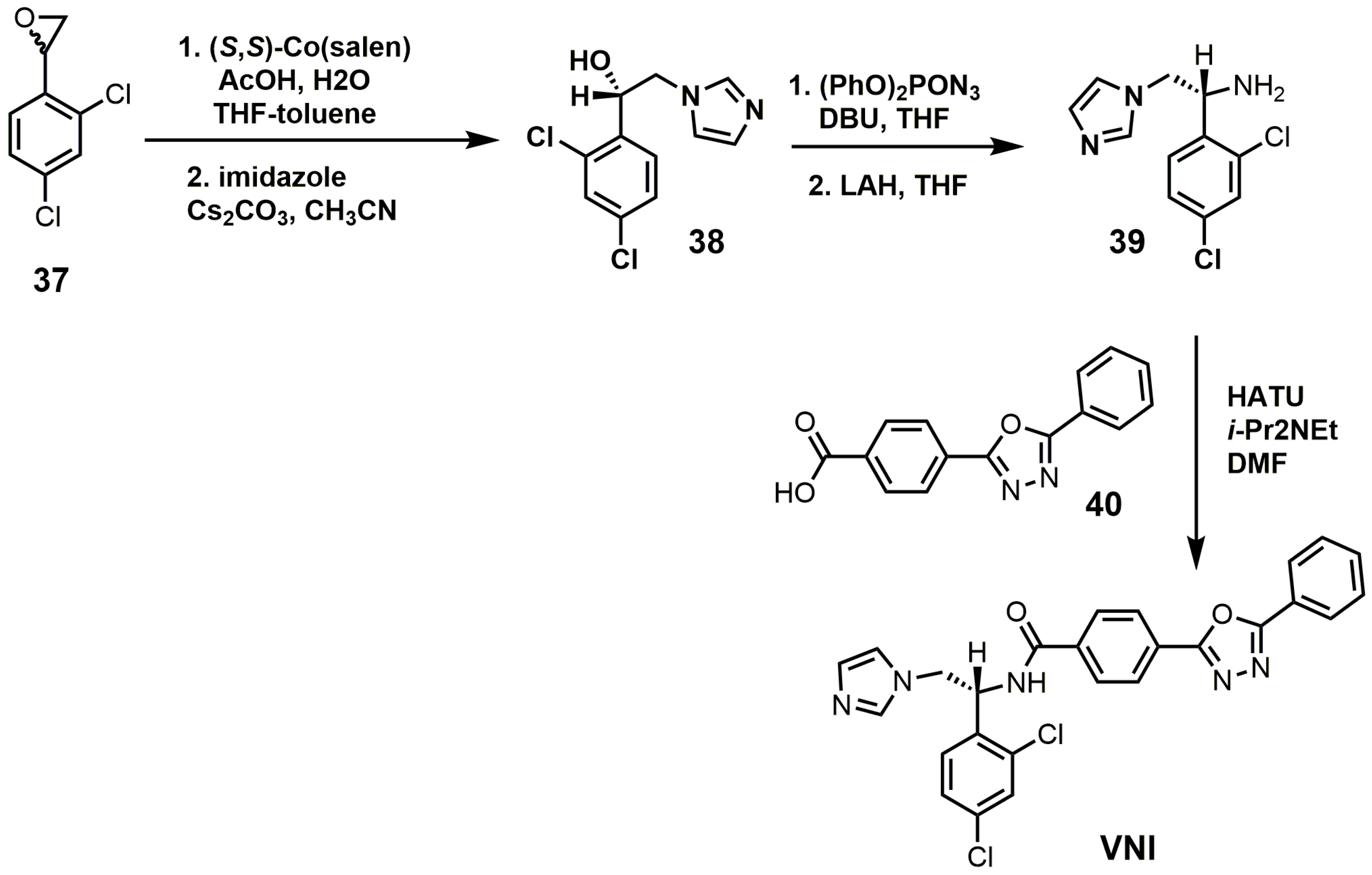
Figure 18.
Different C24-alkylation pathways for sterol C24-methyltransferase (24-SMT) substrates (adapted from [4]). Nu: sterol nucleus.
Figure 18.
Different C24-alkylation pathways for sterol C24-methyltransferase (24-SMT) substrates (adapted from [4]). Nu: sterol nucleus.
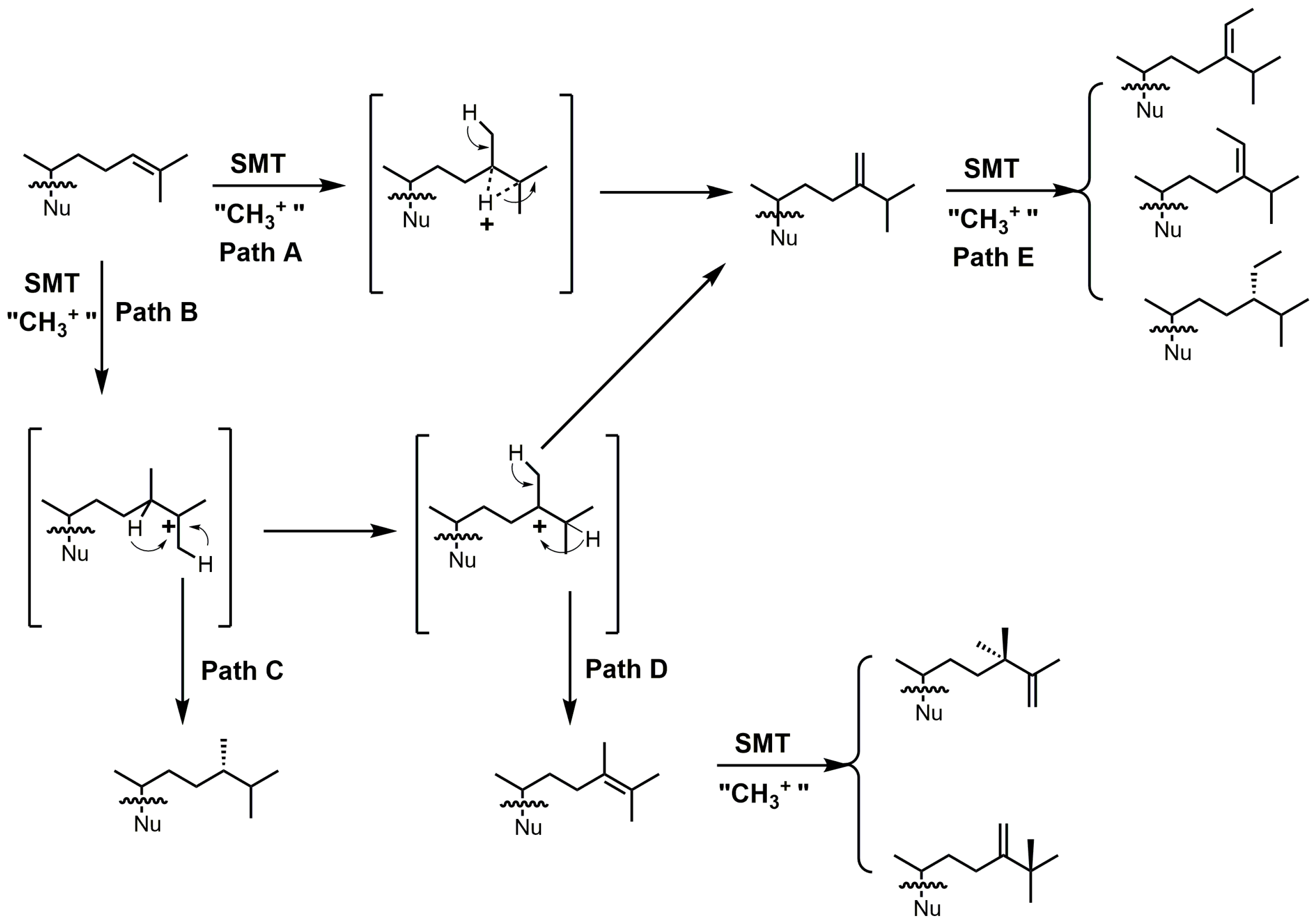
Figure 19.
Structures of sterol C24-methyltransferase (24-SMT) sterol-based inhibitors.

Figure 20.
Proposed inhibitory mechanism of TbSMT with 26,27-dehydrolanosterol (DHL) (adapted from Miller et al., 2017). Nu: lanosterol nucleus.
Figure 20.
Proposed inhibitory mechanism of TbSMT with 26,27-dehydrolanosterol (DHL) (adapted from Miller et al., 2017). Nu: lanosterol nucleus.
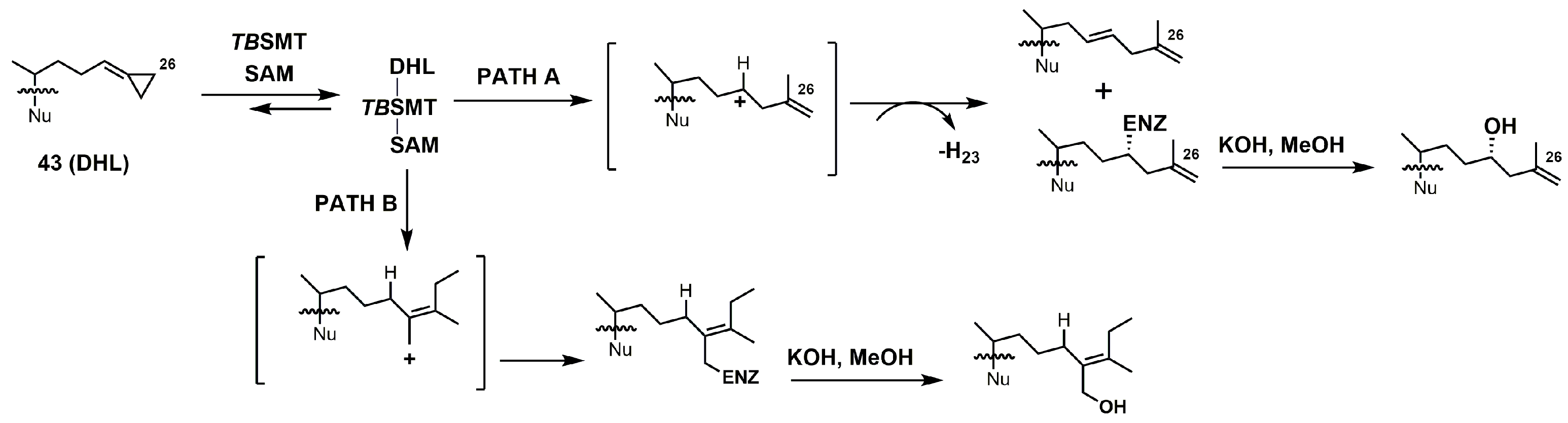
Figure 21.
C24-methylation pathway of 26-fluorolanosterol (26-FL) (47) with TbSMT. Adapted from [5].
Figure 21.
C24-methylation pathway of 26-fluorolanosterol (26-FL) (47) with TbSMT. Adapted from [5].
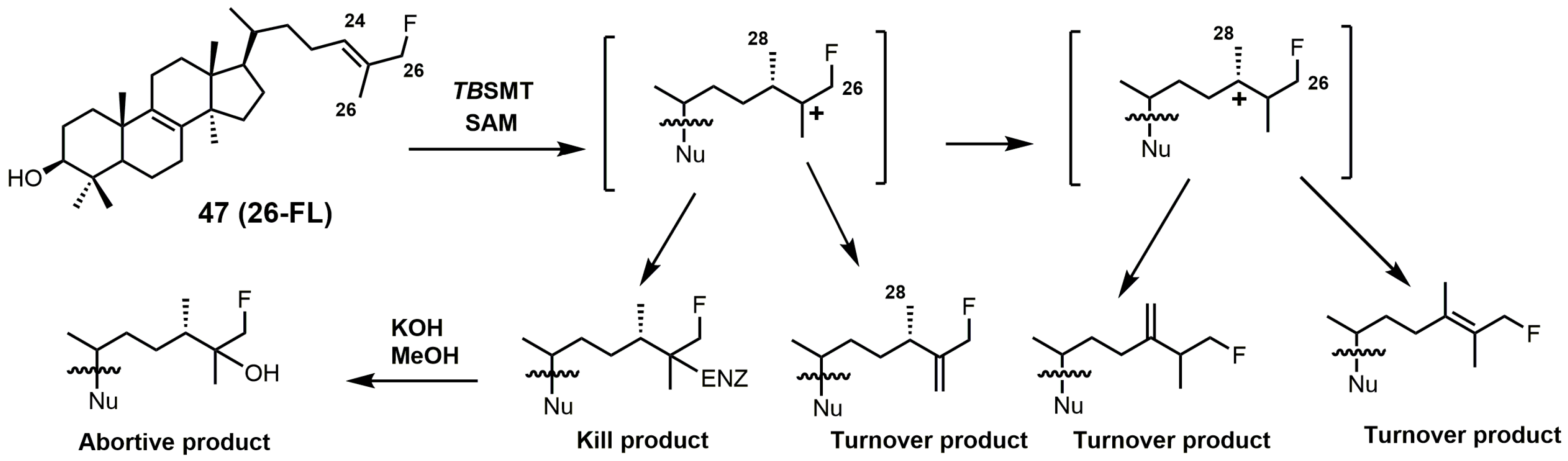
Figure 22.
The synthesis of 24(R,S),25-epiminolanosterol (EL).
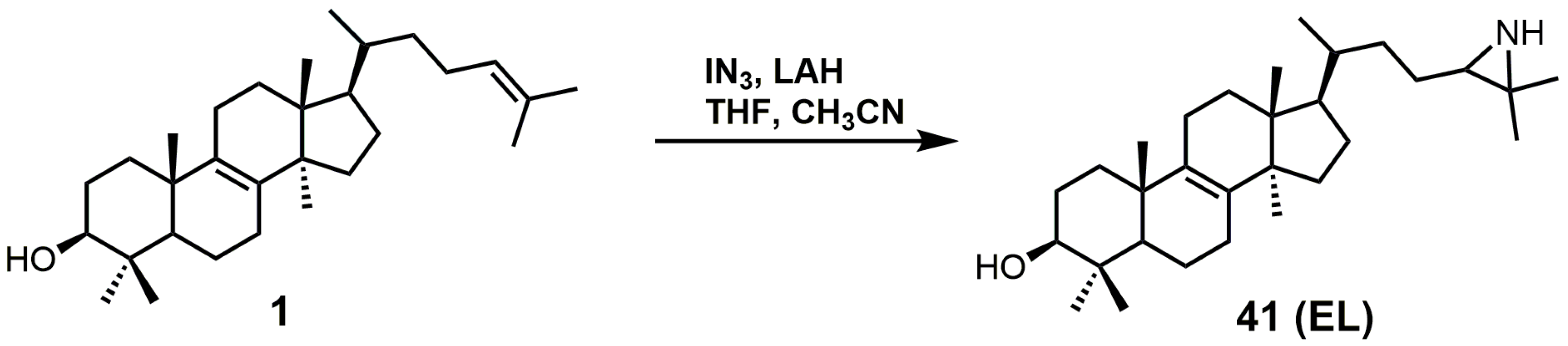
Figure 23.
Proposed pathways for inhibition of sterol C24-methyltransferase (24-SMT) with 24(R,S)-25-epiminolanosterol (EL) (adapted from [92]).
Figure 23.
Proposed pathways for inhibition of sterol C24-methyltransferase (24-SMT) with 24(R,S)-25-epiminolanosterol (EL) (adapted from [92]).

Figure 24.
Three synthetic routes used to prepare 25-azalanosterol (AZAL).
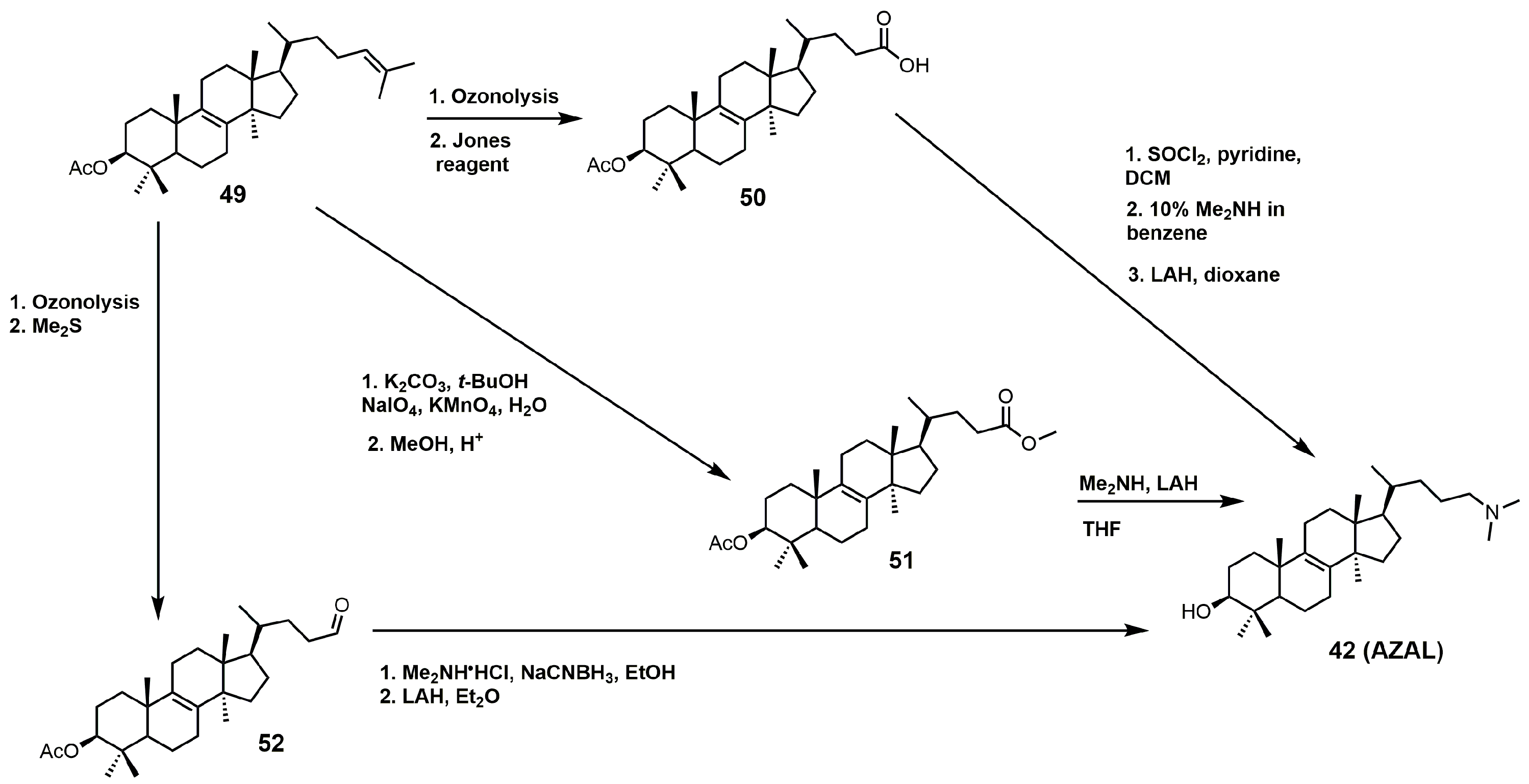
Figure 25.
Synthesis of cyclopropylidine sterol derivatives. Nu: sterol nucleus (cycloartenol, lanosterol, or zymosterol).
Figure 25.
Synthesis of cyclopropylidine sterol derivatives. Nu: sterol nucleus (cycloartenol, lanosterol, or zymosterol).

Figure 26.
Proposed inhibitory mechanism of SMT with 25-thialanosterol (adapted from [4]).
Figure 26.
Proposed inhibitory mechanism of SMT with 25-thialanosterol (adapted from [4]).
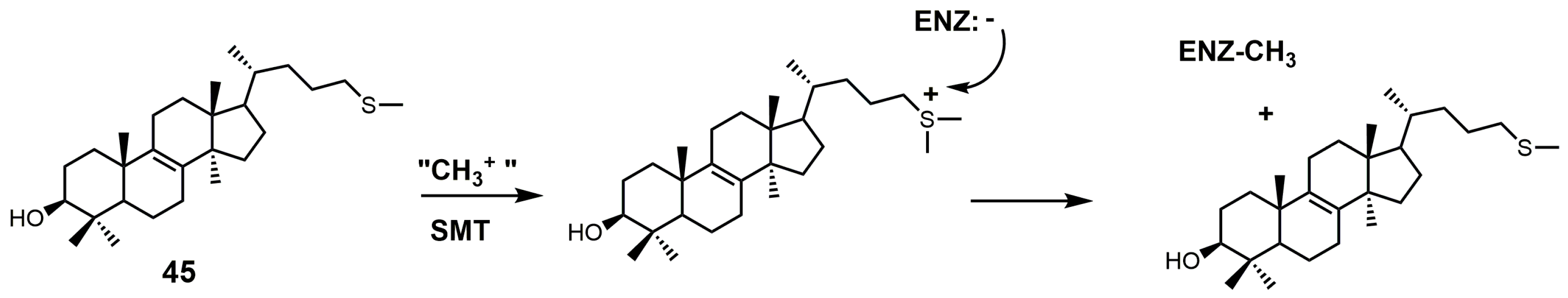
Figure 27.
Synthesis of 25-thialanosterol and 25-thialanosterol salt.
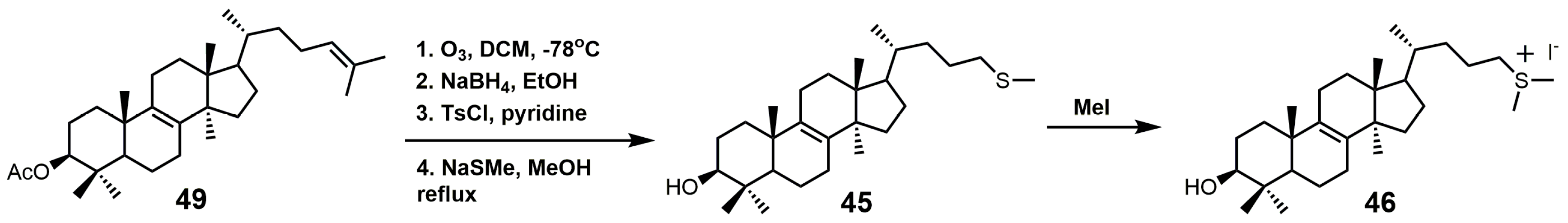
Figure 28.
(A) Synthesis of 26-fluorolanosterol (26-FL) (47); (B) synthesis of 26-fluorocholesta-5,7,24-trienol (26-FCT) (48).
Figure 28.
(A) Synthesis of 26-fluorolanosterol (26-FL) (47); (B) synthesis of 26-fluorocholesta-5,7,24-trienol (26-FCT) (48).
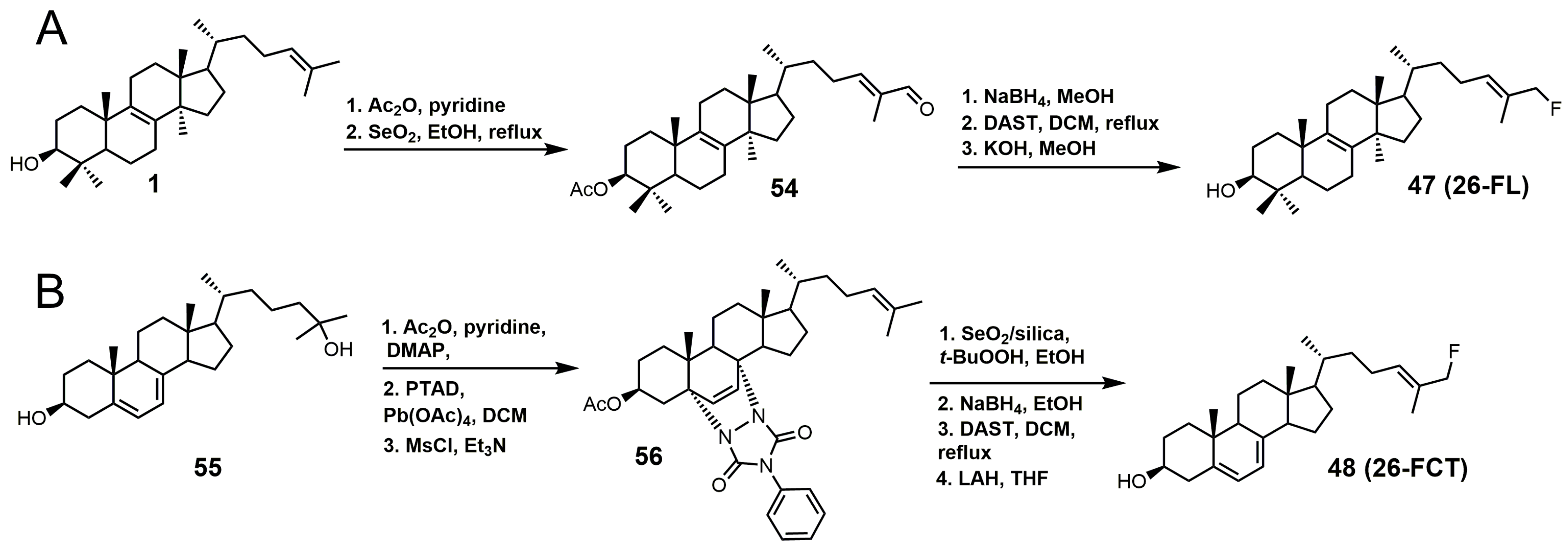
Figure 29.
Structures of compounds designed to be bifunctional SDM and sterol C24-methyltransferase (24-SMT) inhibitors.
Figure 29.
Structures of compounds designed to be bifunctional SDM and sterol C24-methyltransferase (24-SMT) inhibitors.
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Leaver, D.J. Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors. Molecules 2018, 23, 1753. https://doi.org/10.3390/molecules23071753
AMA Style
Leaver DJ. Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors. Molecules. 2018; 23(7):1753. https://doi.org/10.3390/molecules23071753
Chicago/Turabian StyleLeaver, David J. 2018. "Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors" Molecules 23, no. 7: 1753. https://doi.org/10.3390/molecules23071753