Theoretical Studies on Catalysis Mechanisms of Serum Paraoxonase 1 and Phosphotriesterase Diisopropyl Fluorophosphatase Suggest the Alteration of Substrate Preference from Paraoxonase to DFP

 , ,
, ,
Abstract
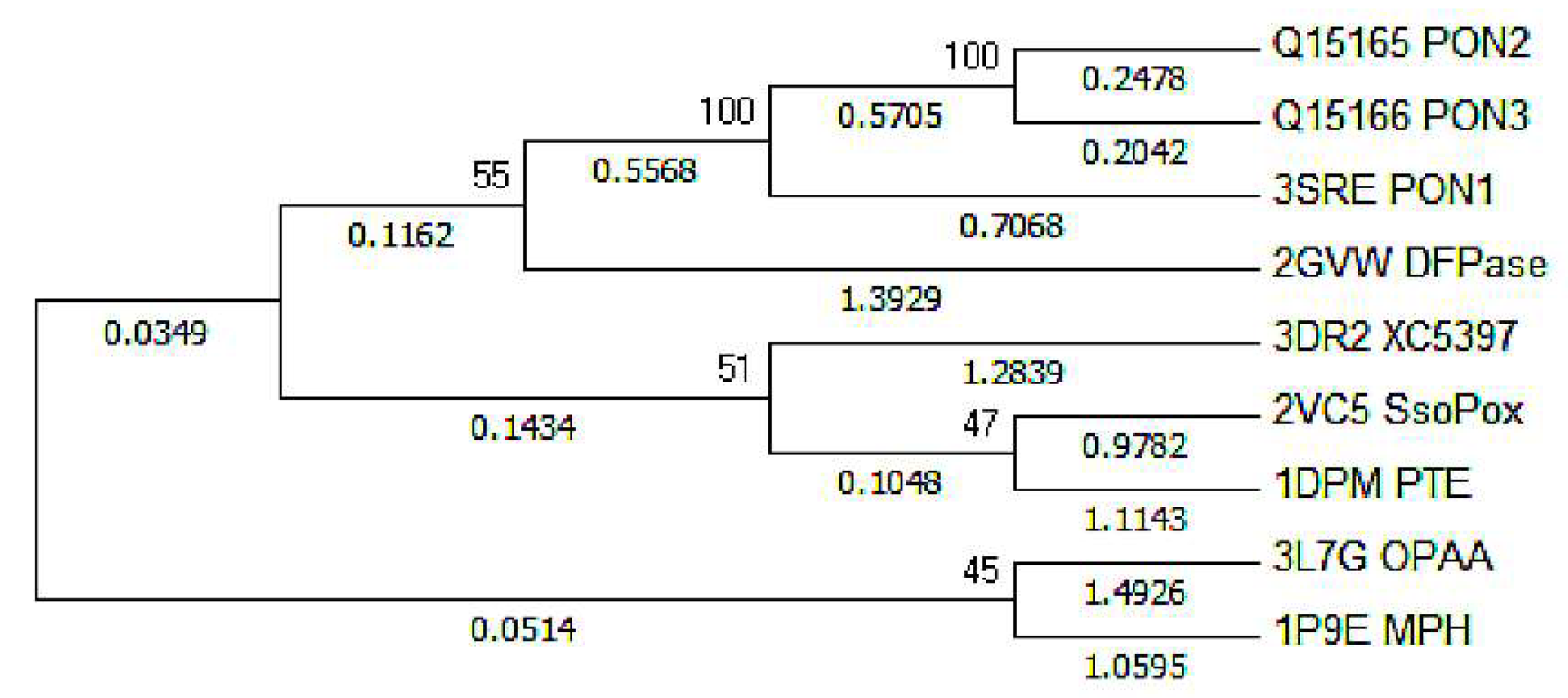
:1. Introduction
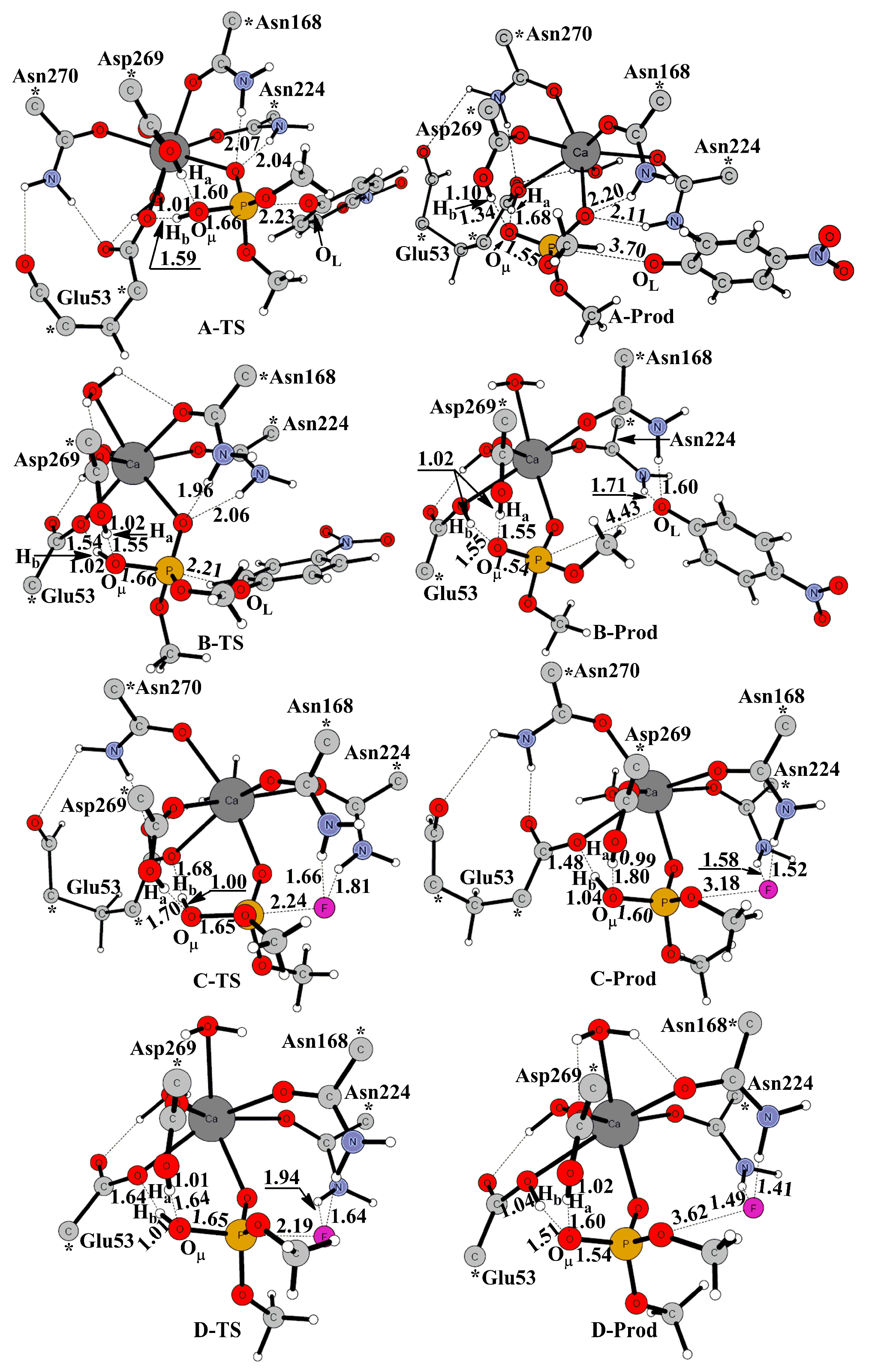
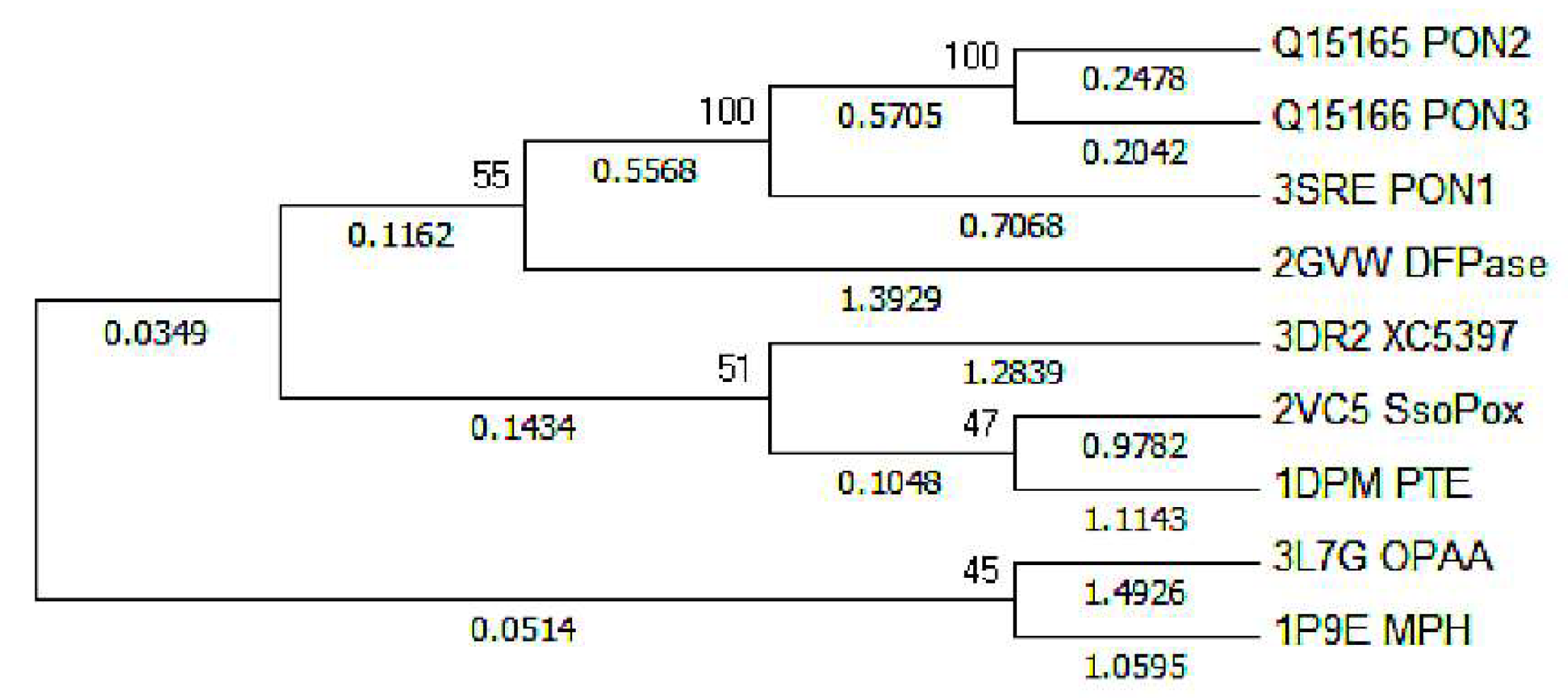
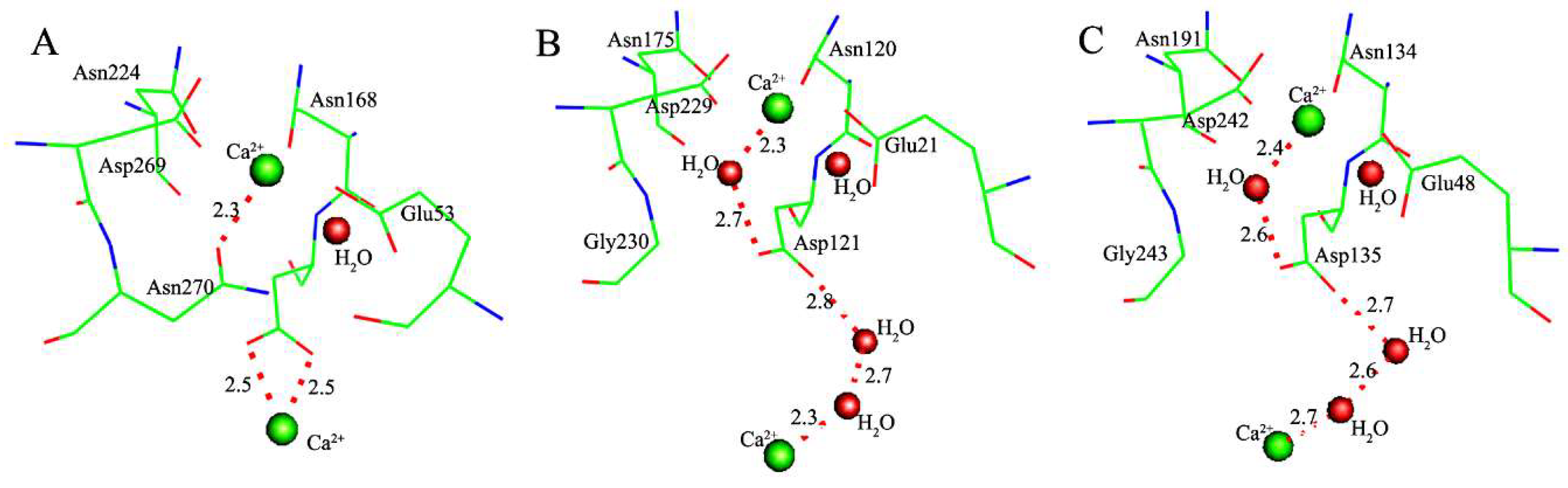
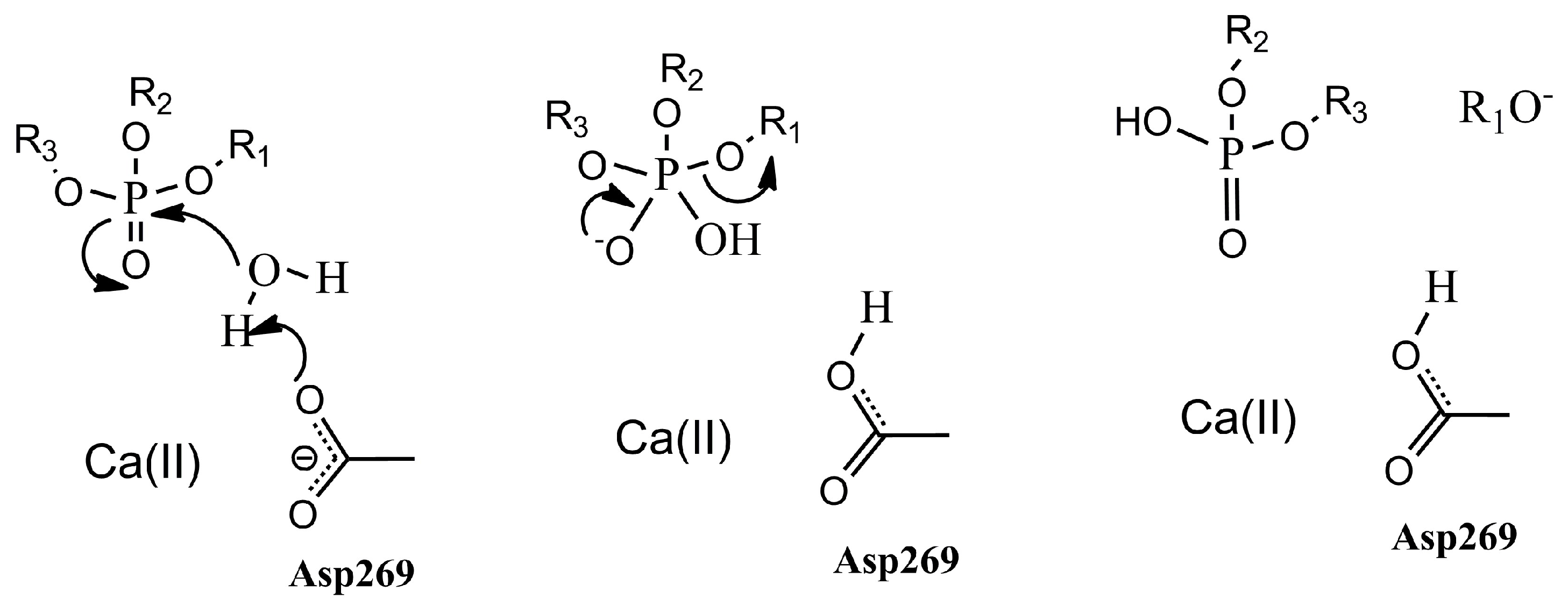
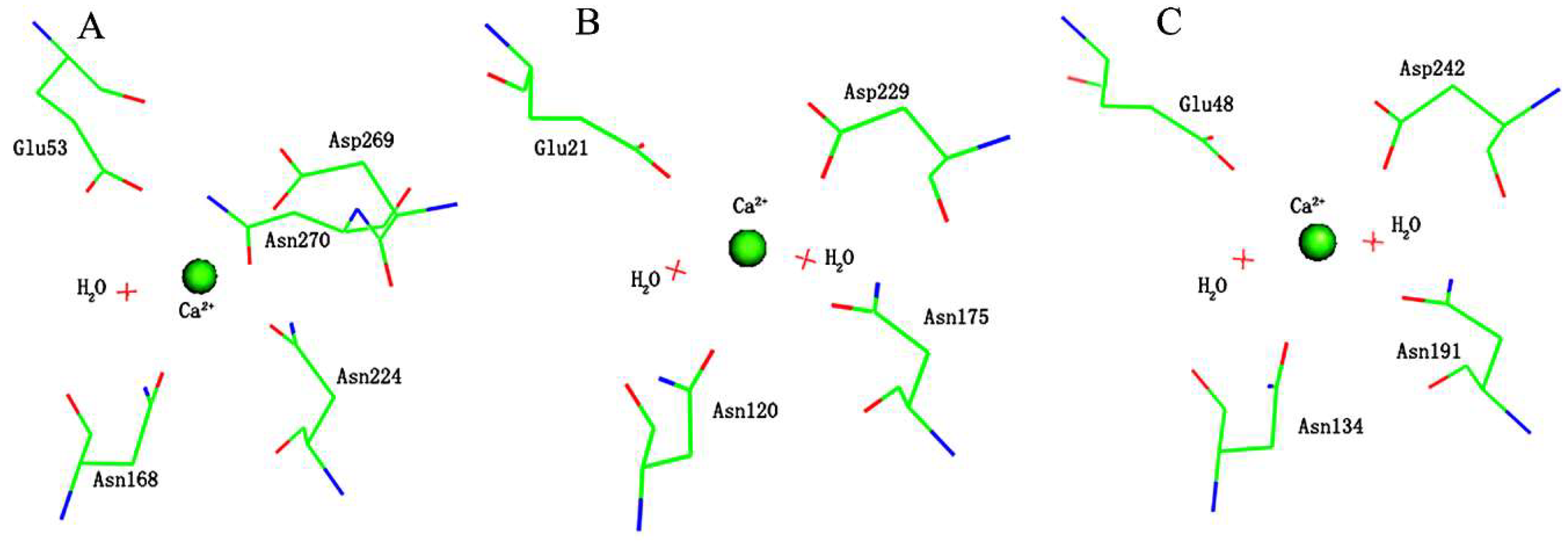
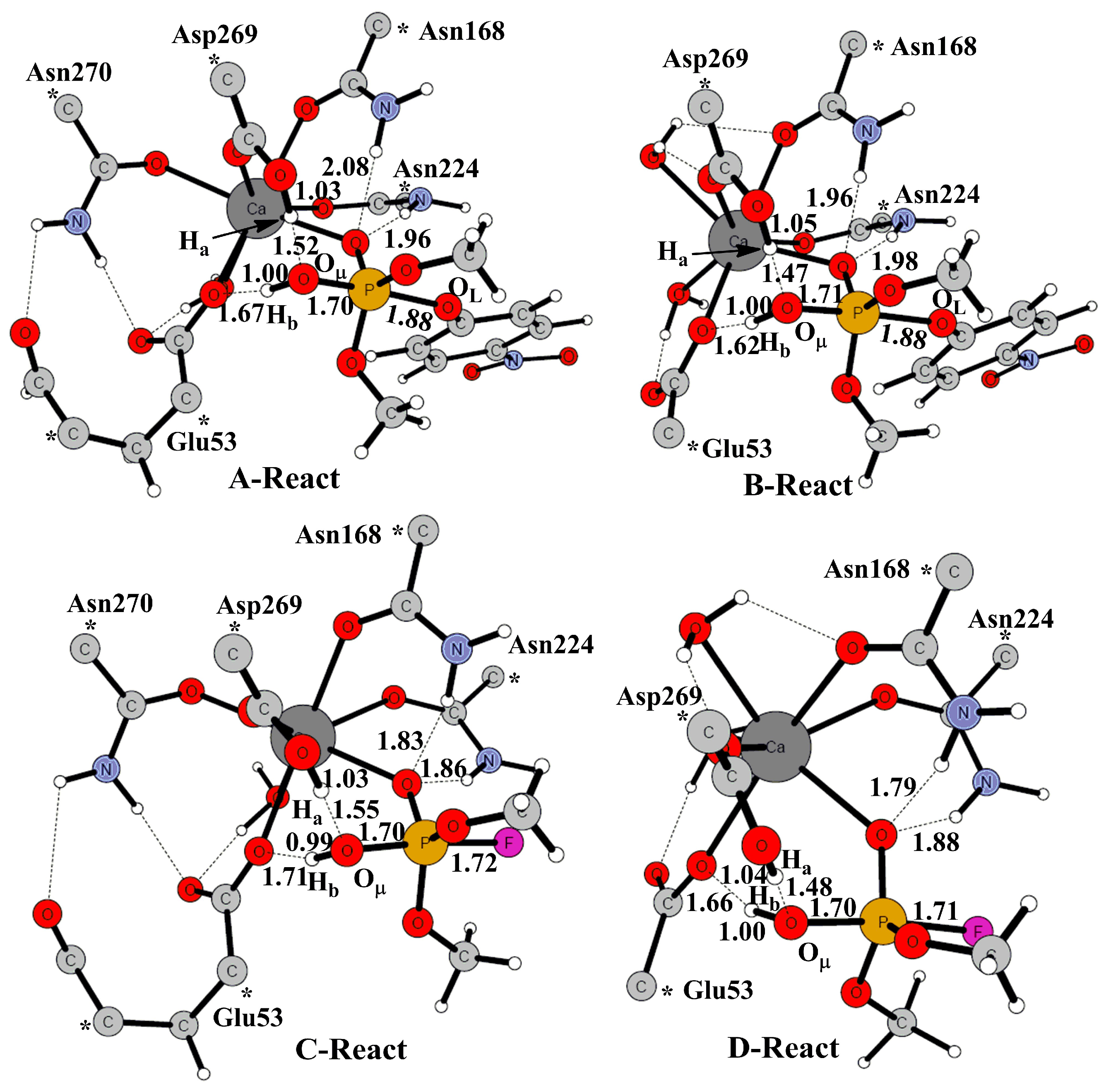
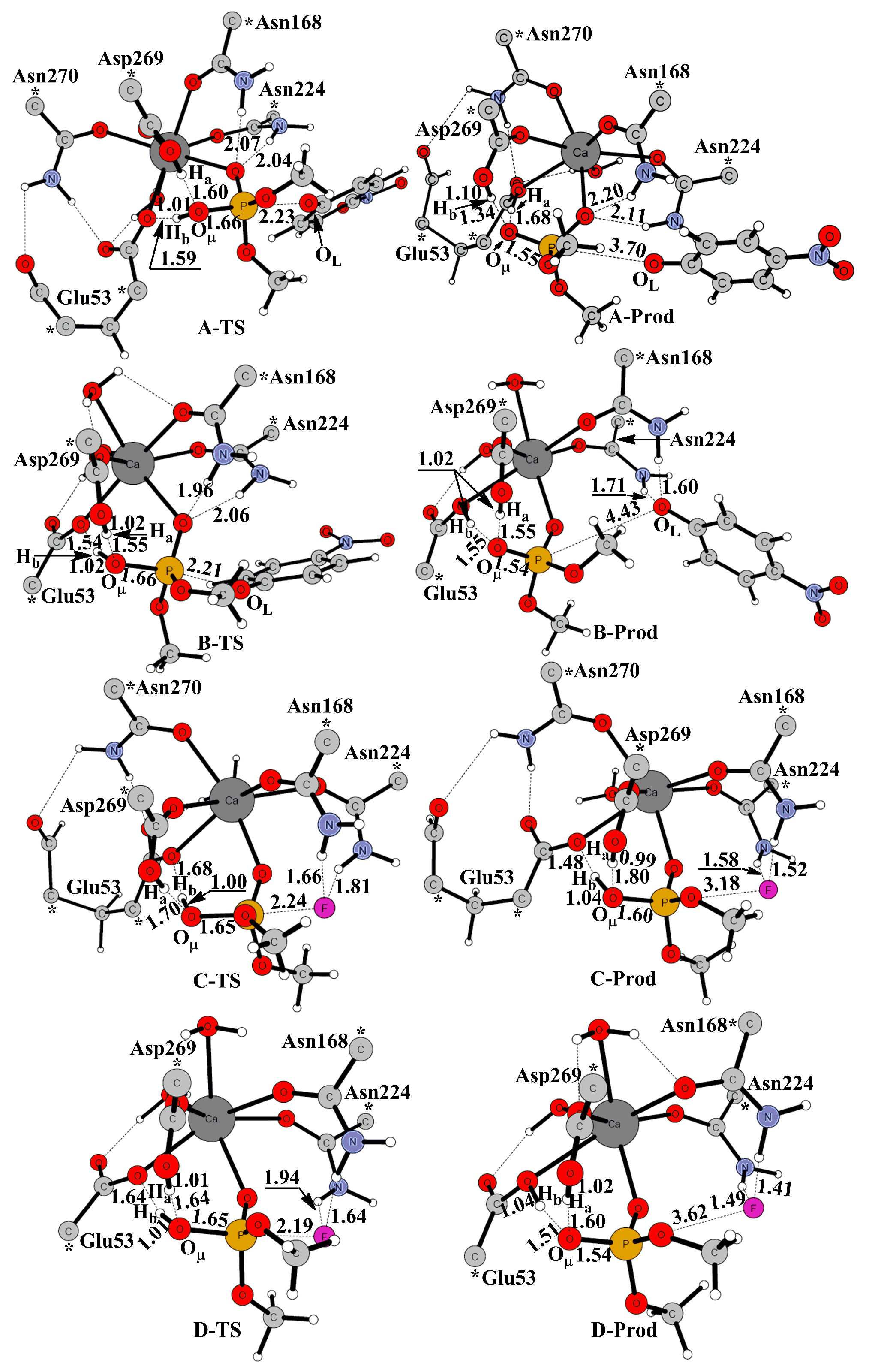
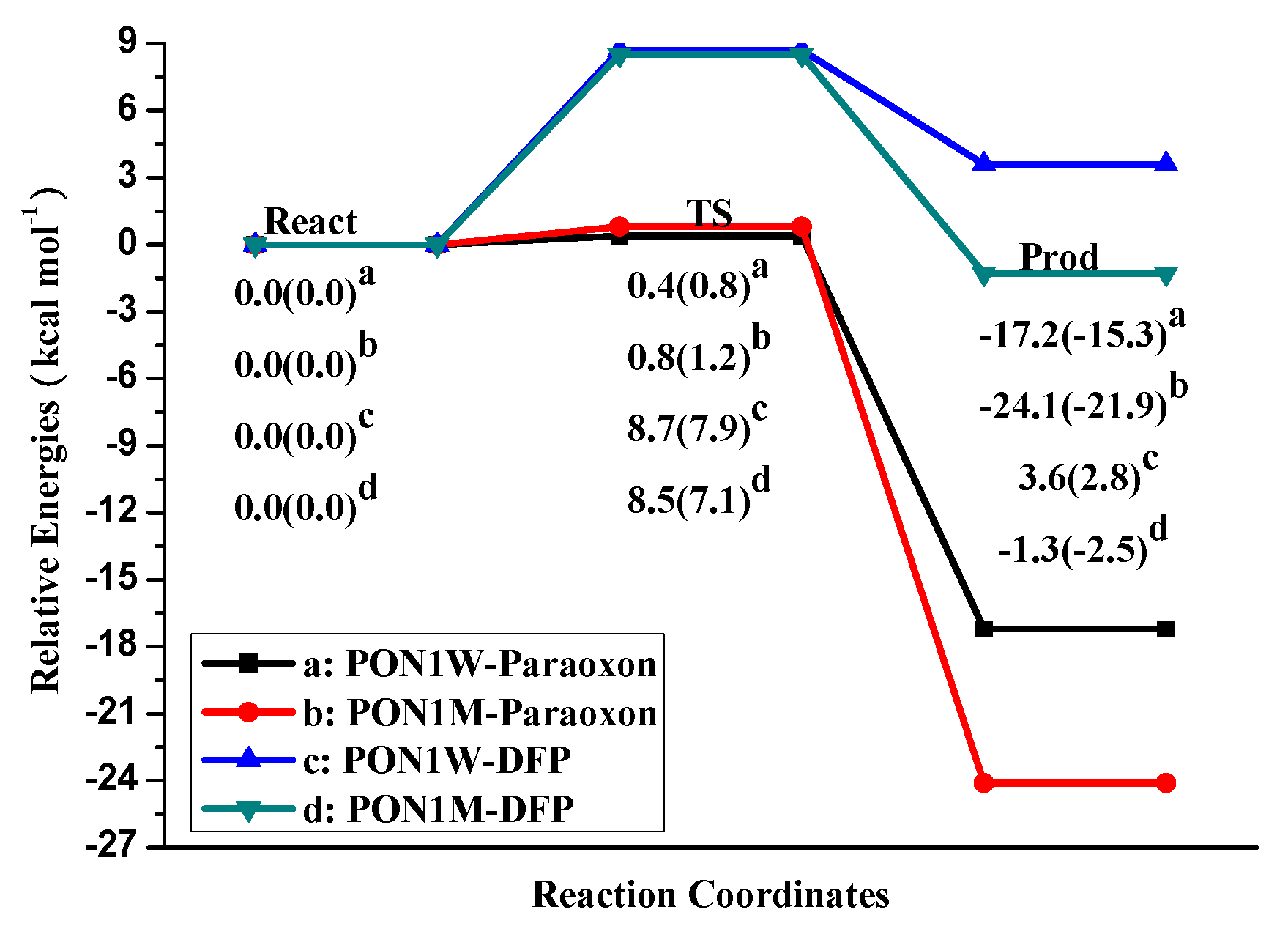
2. Results and Discussion
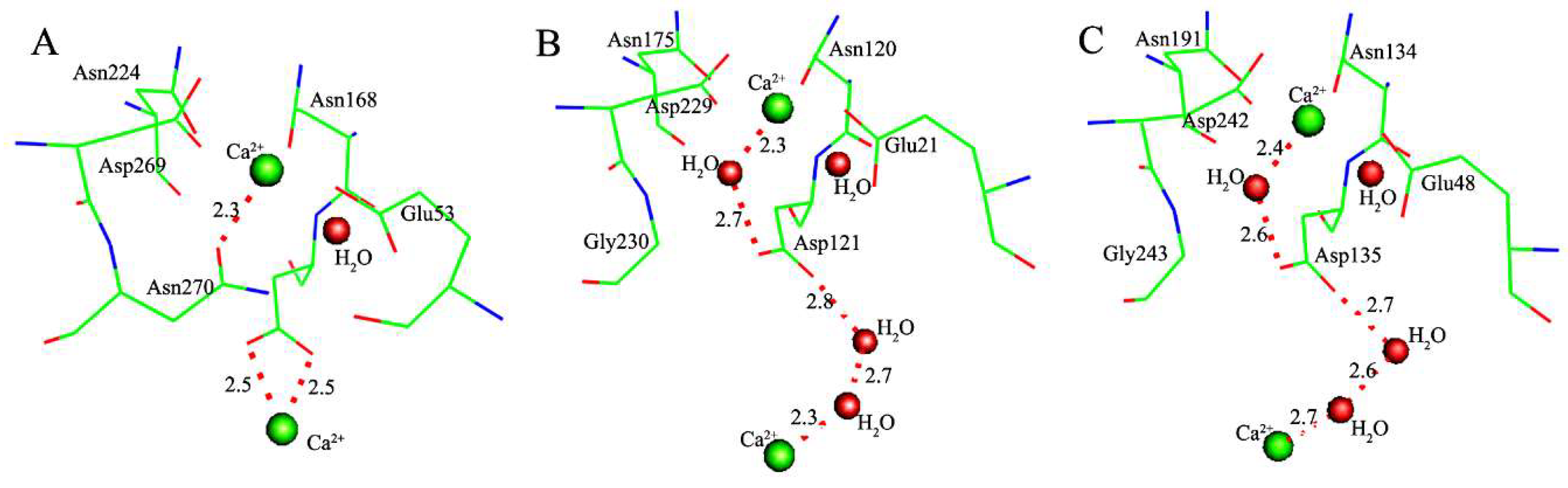
2.1. Effects of Asn270 Mutation on Catalytic Reaction
2.2. Effects of Asn270 Mutation on Enzymatic Structure
3. Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zalatan, J.G.; Fenn, T.D.; Herschlag, D. Comparative enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J. Mol. Biol. 2008, 384, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Patskovsky, Y.; Toro, R.; Farelli, J.D.; Pandya, C.; Almo, S.C.; Allen, K.N.; Dunaway-Mariano, D. Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry 2011, 50, 8937–8949. [Google Scholar] [CrossRef] [PubMed]
- López-Canut, V.; Roca, M.; Bertrán, J.; Moliner, V.; Tuñón, I. Promiscuity in alkaline phosphatase superfamily. Unraveling evolution through molecular simulations. J. Am. Chem. Soc. 2011, 133, 12050–12062. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, M.; Wieczorek, G.; Elias, M.; Silman, I.; Sussman, J.L.; Tawfik, D.S. Catalytic metal ion rearrangements underline promiscuity and evolvability of a metalloenzyme. J. Mol. Biol. 2013, 425, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Babbitt, P.C. New insights about enzyme evolution from large scale studies of sequence and structure relationships. J. Biol. Chem. 2014, 289, 30221–30228. [Google Scholar] [CrossRef] [PubMed]
- Bora, R.P.; Mills, M.J.; Frushicheva, M.P.; Warshel, A. On the challenge of exploring the evolutionary trajectory from phosphotriesterase to arylesterase using computer simulations. J. Phys. Chem. B 2015, 119, 3434–3445. [Google Scholar] [CrossRef] [PubMed]
- Sunden, F.; AlSadhan, I.; Lyubimov, A.Y.; Ressl, S.; Wiersma-Koch, H.; Borland, J.; Brown, C.L.; Johnson, T.A.; Singh, Z.; Herschlag, D. Mechanistic and evolutionary insights from comparative enzymology of phosphomonoesterases and phosphodiesterases across the alkaline phosphatase superfamily. J. Am. Chem. Soc. 2016, 138, 14273–14287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, L.; Yan, L.-F.; Liao, R.-Z.; Tian, W.-Q. Evolution of phosphotriesterase activities of the metallo-β-lactamase family: A theoretical study. J. Inorg. Biochem. 2018, 184, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Aubert, S.D.; Li, Y.; Raushel, F.M. Mechanism for the hydrolysis of organophosphates by the bacterial phosphotriesterase. Biochemistry 2004, 43, 5707–5715. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-Y.; Gao, J. The reaction mechanism of paraoxon hydrolysis by phosphotriesterase from combined QM/MM simulations. Biochemistry 2007, 46, 13352–13369. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fang, W.; Himo, F. Theoretical study of the phosphotriesterase reaction mechanism. J. Phys. Chem. B 2007, 111, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvo, M.C.; Gil, F.; Hernandez, A.F.; Rodrigo, L.; Villanueva, E.; Pla, A. Human liver paraoxonase (PON1): Subcellular distribution and characterization. J. Biochem. Mol. Toxicol. 1998, 12, 61–69. [Google Scholar] [CrossRef]
- Gaidukov, L.; Tawfik, D.S. High affinity, stability, and lactonase activity of serum paraoxonase PON1 Anchored on HDL with ApoA-I. Biochemistry 2005, 44, 11843–11854. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Gaidukov, L.; Khersonsky, O.; Gould, S.M.; Roodveldt, C.; Tawfik, D.S. The ‘evolvability’ of promiscuous protein functions. Nat. Genet. 2005, 37, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, M.; Elias, M.; Filippi, J.-J.; Duñach, E.; Silman, I.; Sussman, J.L.; Tawfik, D.S. Catalytic versatility and backups in enzyme active sites: The case of serum paraoxonase 1. J. Mol. Biol. 2012, 418, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.G.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Scharff, E.I.; Koepke, J.; Fritzsch, G.; Lücke, C.; Rüterjans, H. Crystal structure of diisopropylfluorophosphatase from Loligo vulgaris. Structure 2001, 9, 493–502. [Google Scholar] [CrossRef]
- Blum, M.-M.; Löhr, F.; Richardt, A.; Rüterjans, H.; Chen, J.C.H. Binding of a designed substrate analogue to diisopropyl fluorophosphatase: Implications for the phosphotriesterase mechanism. J. Am. Chem. Soc. 2006, 128, 12750–12757. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.-M.; Chen, J.C.H. Structural characterization of the catalytic calcium-binding site in diisopropyl fluorophosphatase (DFPase)—Comparison with related β-propeller enzymes. Chem. Biol. Interact. 2010, 187, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.-H.; Mustyakimov, M.; Schoenborn, B.P.; Langan, P.; Blum, M.-M. Neutron structure and mechanistic studies of diisopropyl fluorophosphatase (DFPase). Acta Crystallogr. D 2010, 66, 1131–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, M.-M.; Timperley, C.M.; Williams, G.R.; Thiermann, H.; Worek, F. Inhibitory potency against human acetylcholinesterase and enzymatic hydrolysis of fluorogenic nerve agent mimics by human paraoxonase 1 and squid diisopropyl fluorophosphatase. Biochemistry 2008, 47, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Hartleib, J.; Ruterjans, H. High-yield expression, purification, and characterization of the recombinant diisopropylfluorophosphatase from Loligo vulgaris. Protein Expr. Purif. 2001, 21, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Bigley, A.N.; Raushel, F.M. Catalytic mechanisms for phosphotriesterases. BBA Proteins Proteom. 2013, 1834, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khersonsky, O.; Tawfik, D.S. Structure–Reactivity studies of serum paraoxonase PON1 Suggest that Its native activity is Lactonase. Biochemistry 2005, 44, 6371–6382. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. The Histidine 115-Histidine 134 dyad mediates the lactonase activity of mammalian serum paraoxonases. J. Biol. Chem. 2006, 281, 7649–7656. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.-M.; Mustyakimov, M.; Rüterjans, H.; Kehe, K.; Schoenborn, B.P.; Langan, P.; Chen, J.C.-H. Rapid determination of hydrogen positions and protonation states of diisopropyl fluorophosphatase by joint neutron and X-ray diffraction refinement. Proc. Natl. Acad. Sci. USA 2009, 106, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Yang, L.; Yu, J.-G.; Liao, R.-Z. What roles do the residue Asp229 and the coordination variation of calcium play of the reaction mechanism of the diisopropyl-fluorophosphatase? A DFT investigation. Theor. Chem. Acc. 2016, 135, 138. [Google Scholar] [CrossRef]
- Chen, C.-N.; Chin, K.-H.; Wang, A.H.J.; Chou, S.-H. The first crystal structure of gluconolactonase important in the glucose secondary metabolic pathways. J. Mol. Biol. 2008, 384, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Lichtarge, O.; Bourne, H.R.; Cohen, F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Babbitt, P.C. Divergent evolution of enzymatic function: Mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 2001, 70, 209–246. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Herschlag, D. Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol. 1999, 6, R91–R105. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Unraveling the mechanism of the farnesyltransferase enzyme. J. Biol. Inorg. Chem. 2005, 10, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Farnesyltransferase—New insights into the zinc-coordination sphere paradigm: Evidence for a carboxylate-shift mechanism. Biophys. J. 2005, 88, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Farnesyltransferase: Theoretical studies on peptide substrate entrance—Thiol or thiolate coordination? J. Mol. Struct.-THEOCHEM 2005, 729, 125–129. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Theoretical studies on farnesyltransferase: Evidence for thioether product coordination to the active-site zinc sphere. J. Comput. Chem. 2007, 28, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Siegbahn, P.E.M. Quantum chemical studies of radical-containing enzymes. Chem. Rev. 2003, 103, 2421–2456. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M.; Borowski, T. Modeling enzymatic reactions involving transition metals. Acc. Chem. Res. 2006, 39, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M. The effect of backbone constraints: The case of water oxidation by the oxygen-evolving complex in PSII. ChemPhysChem 2011, 12, 3274–3280. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. WIREs Comput. Mol. Sci. 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. Which one among Zn(II), Co.(II), Mn(II), and Fe(II) is the most efficient ion for the methionine aminopeptidase catalyzed reaction? J. Am. Chem. Soc. 2007, 129, 7776–7784. [Google Scholar] [CrossRef] [PubMed]
- Abashkin, Y.G.; Burt, S.K.; Collins, J.R.; Cachau, R.E.; Russo, N.; Erickson, J.W. Metal-Ligand Interactions: Structure and Reactivity; Russo, N., Salahub, D.R., Eds.; Nato Science Series; Kluwer: Dordrecht, The Netherlands, 1996; Volume 474, pp. 1–22. [Google Scholar]
- Marino, T.; Russo, N.; Toscano, M. A comparative study of the catalytic mechanisms of the zinc and cadmium containing carbonic anhydrase. J. Am. Chem. Soc. 2005, 127, 4242–4253. [Google Scholar] [CrossRef] [PubMed]
- Leopoldini, M.; Russo, N.; Toscano, M. Role of the metal ion in formyl-peptide bond hydrolysis by a peptide deformylase active site model. J. Phys. Chem. B 2006, 110, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Allen, K.N.; Dunaway-Mariano, D. Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.E.; Smith, Q.A.; Nechay, M.R.; Alexandrova, A.N. Mysteries of metals in metalloenzymes. Acc. Chem. Res. 2014, 47, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liao, R.-Z.; Ding, W.-J.; Liu, K.; Yu, J.-G.; Liu, R.-Z. Why calcium inhibits magnesium-dependent enzyme phosphoserine phosphatase? A theoretical study. Theor. Chem. Acc. 2012, 131, 1275. [Google Scholar] [CrossRef]
- Salter, E.A.; Honkanen, R.E.; Wierzbicki, A. Modeling the antiferromagnetic MnIIMnII system within the protein phosphatase-5 catalytic site. J. Mol. Model. 2015, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Wymore, T.; Field, M.J.; Langan, P.; Smith, J.C.; Parks, J.M. Hydrolysis of DFP and the Nerve Agent (S)-Sarin by DFPase proceeds along two different reaction pathways: Implications for engineering bioscavengers. J. Phys. Chem. B 2014, 118, 4479–4489. [Google Scholar] [CrossRef] [PubMed]
- Rochu, D.; Chabrière, E.; Masson, P. Human paraoxonase: A promising approach for pre-treatment and therapy of organophosphorus poisoning. Toxicology 2007, 233, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Suzuki, S.M.; Cole, T.B.; Park, S.S.; Richter, R.J.; Furlong, C.E. Engineered recombinant human paraoxonase 1 (rHuPON1) purified from Escherichia coli protects against organophosphate poisoning. Proc. Natl. Acad. Sci. USA 2008, 105, 12780–12784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, M.; Dupuy, J.; Merone, L.; Mandrich, L.; Porzio, E.; Moniot, S.; Rochu, D.; Lecomte, C.; Rossi, M.; Masson, P.; et al. Structural basis for natural lactonase and promiscuous phosphotriesterase activities. J. Mol. Biol. 2008, 379, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Vanhooke, J.L.; Benning, M.M.; Raushel, F.M.; Holden, H.M. Three-Dimensional Structure of the Zinc-Containing Phosphotriesterase with the Bound Substrate Analog Diethyl 4-Methylbenzylphosphonate. Biochemistry 1996, 35, 6020–6025. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.K.; Nickitenko, A.; Rastogi, V.K.; Shah, S.S.; Quiocho, F.A. Structural insights into the dual activities of the nerve agent degrading organophosphate Anhydrolase/Prolidase. Biochemistry 2009, 49, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-J.; Bartlam, M.; Sun, L.; Zhou, Y.-F.; Zhang, Z.-P.; Zhang, C.-G.; Rao, Z.; Zhang, X.-E. Crystal structure of methyl parathion hydrolase from Pseudomonas sp. WBC-3. J. Mol. Biol. 2005, 353, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Heinz, U.; Adolph, H.W. Metallo-β-lactamases: Two binding sites for one catalytic metal ion? Cell. Mol. Life Sci. 2004, 61, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cammi, R.; Mennucci, B.; Tomasi, J. Second-order Møller–Plesset analytical derivatives for the polarizable continuum model using the relaxed density approach. J. Phys. Chem. A 1999, 103, 9100–9108. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds that the optimized geometries along the reaction paths are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordination Bonds | Active-Site-W | Active-Site-M/Δ | PON1W-Paraoxon/Δ | PON1M-Paraoxon/Δ’ | PON1W-DFP/Δ | PON1M-DFP/Δ’ |
|---|---|---|---|---|---|---|
| Ca-OGlu53 | 2.51 | 2.47/−0.04 | 2.45/−0.06 | 2.37/−0.14 | 2.46/−0.05 | 2.39/−0.12 |
| Ca-OAsn168 | 2.73 | 2.67/−0.06 | 2.61/−0.12 | 2.52/−0.21 | 2.64/−0.09 | 2.56/−0.17 |
| Ca-OAsn224 | 2.40 | 2.38/−0.02 | 2.42/0.02 | 2.46/0.06 | 2.43/0.03 | 2.48/0.08 |
| Ca-OAsp269 | 2.49 | 2.51/0.02 | 2.52/0.03 | 2.64/0.15 | 2.51/0.02 | 2.62/0.13 |
| Ca-OAsn270 | 2.41 | / | 2.42/0.01 | / | 2.43/0.02 | / |
| TS Bonds | PON1W-Paraoxon | PON1M-Paraoxon/Δ | PON1W-DFP | PON1M-DFP/Δ |
|---|---|---|---|---|
| Ha-OW1 | 1.60 | 1.55/−0.05 | 1.70 | 1.64/−0.06 |
| Hb-OW1 | 1.01 | 1.02/0.01 | 1.00 | 1.01/0.01 |
| OW1-P | 1.66 | 1.66/0.00 | 1.65 | 1.65/0.00 |
| P-O/F | 2.23 | 2.21/−0.02 | 2.24 | 2.19/−0.05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Yang, L.; Ma, Y.-Y.; Zhu, C.; Lin, S.; Liao, R.-Z. Theoretical Studies on Catalysis Mechanisms of Serum Paraoxonase 1 and Phosphotriesterase Diisopropyl Fluorophosphatase Suggest the Alteration of Substrate Preference from Paraoxonase to DFP. Molecules 2018, 23, 1660. https://doi.org/10.3390/molecules23071660
Zhang H, Yang L, Ma Y-Y, Zhu C, Lin S, Liao R-Z. Theoretical Studies on Catalysis Mechanisms of Serum Paraoxonase 1 and Phosphotriesterase Diisopropyl Fluorophosphatase Suggest the Alteration of Substrate Preference from Paraoxonase to DFP. Molecules. 2018; 23(7):1660. https://doi.org/10.3390/molecules23071660
Chicago/Turabian StyleZhang, Hao, Ling Yang, Ying-Ying Ma, Chaoyuan Zhu, Shenghsien Lin, and Rong-Zhen Liao. 2018. "Theoretical Studies on Catalysis Mechanisms of Serum Paraoxonase 1 and Phosphotriesterase Diisopropyl Fluorophosphatase Suggest the Alteration of Substrate Preference from Paraoxonase to DFP" Molecules 23, no. 7: 1660. https://doi.org/10.3390/molecules23071660