Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase


Abstract
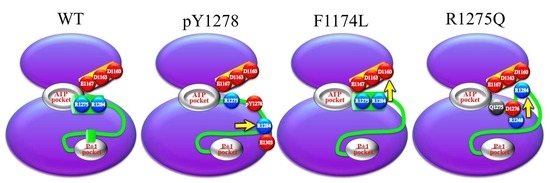
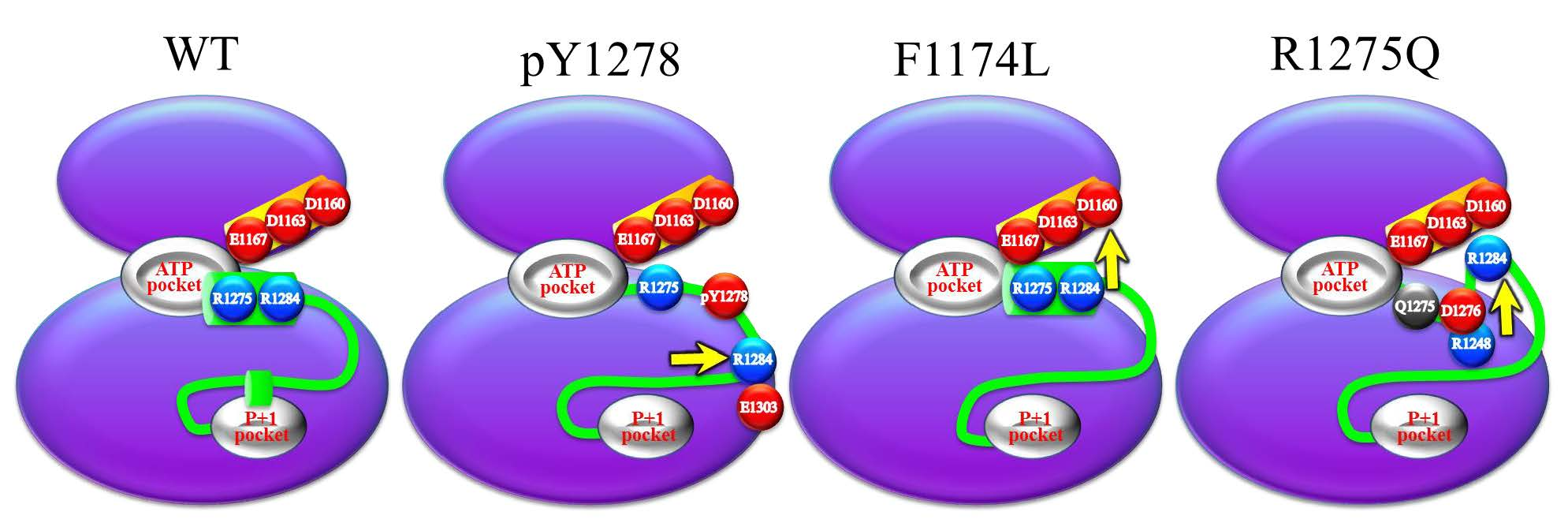
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
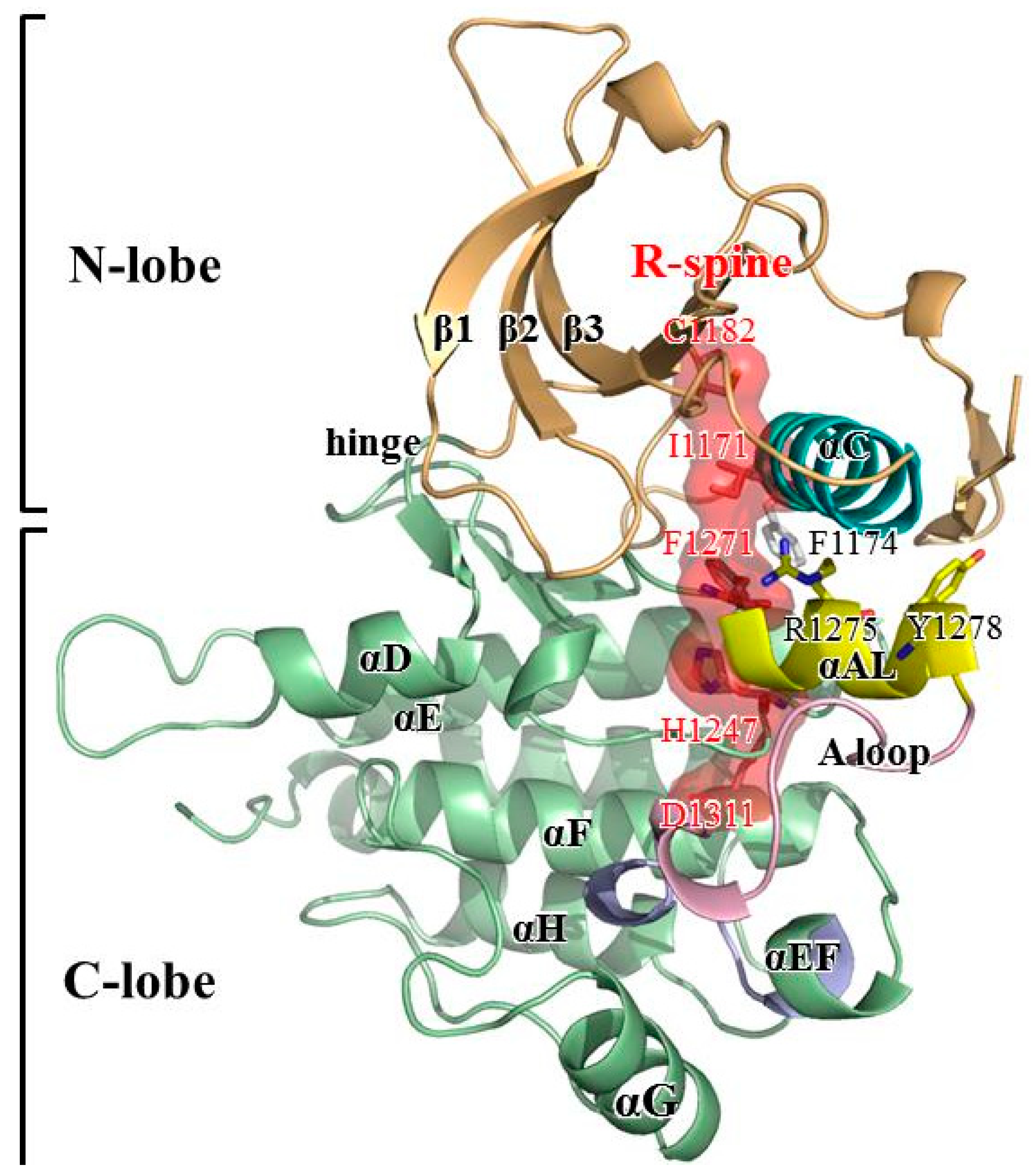
System Setup
3. Results and Discussion
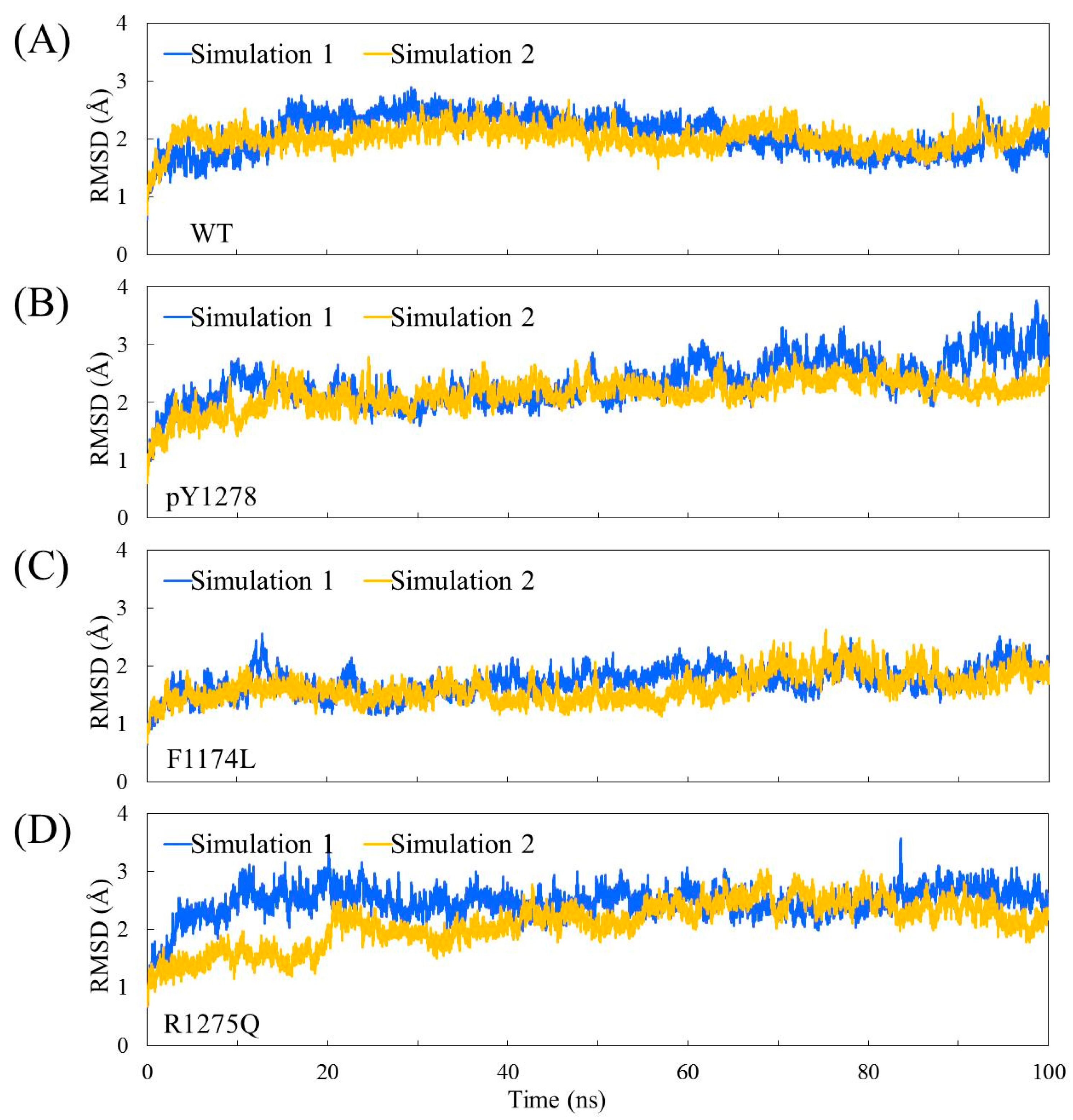
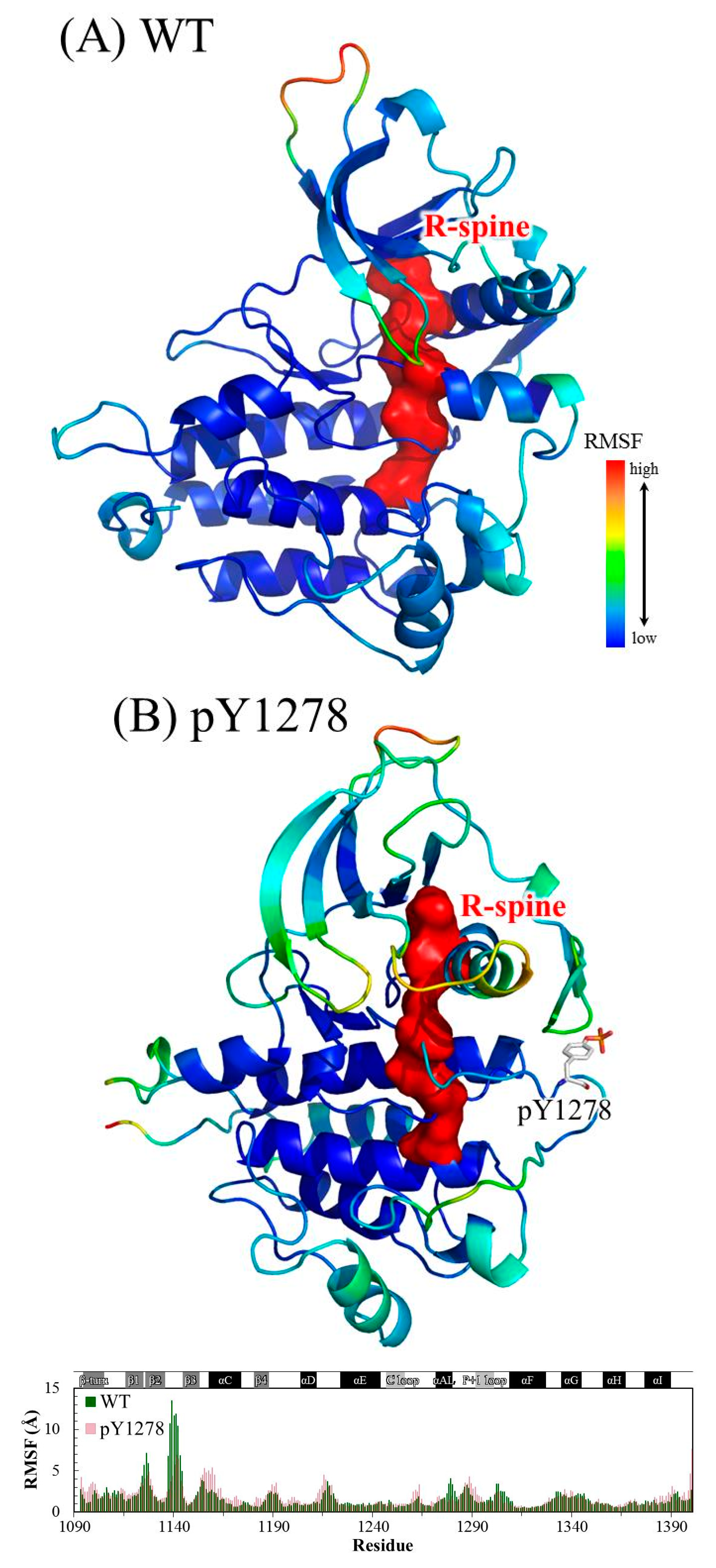
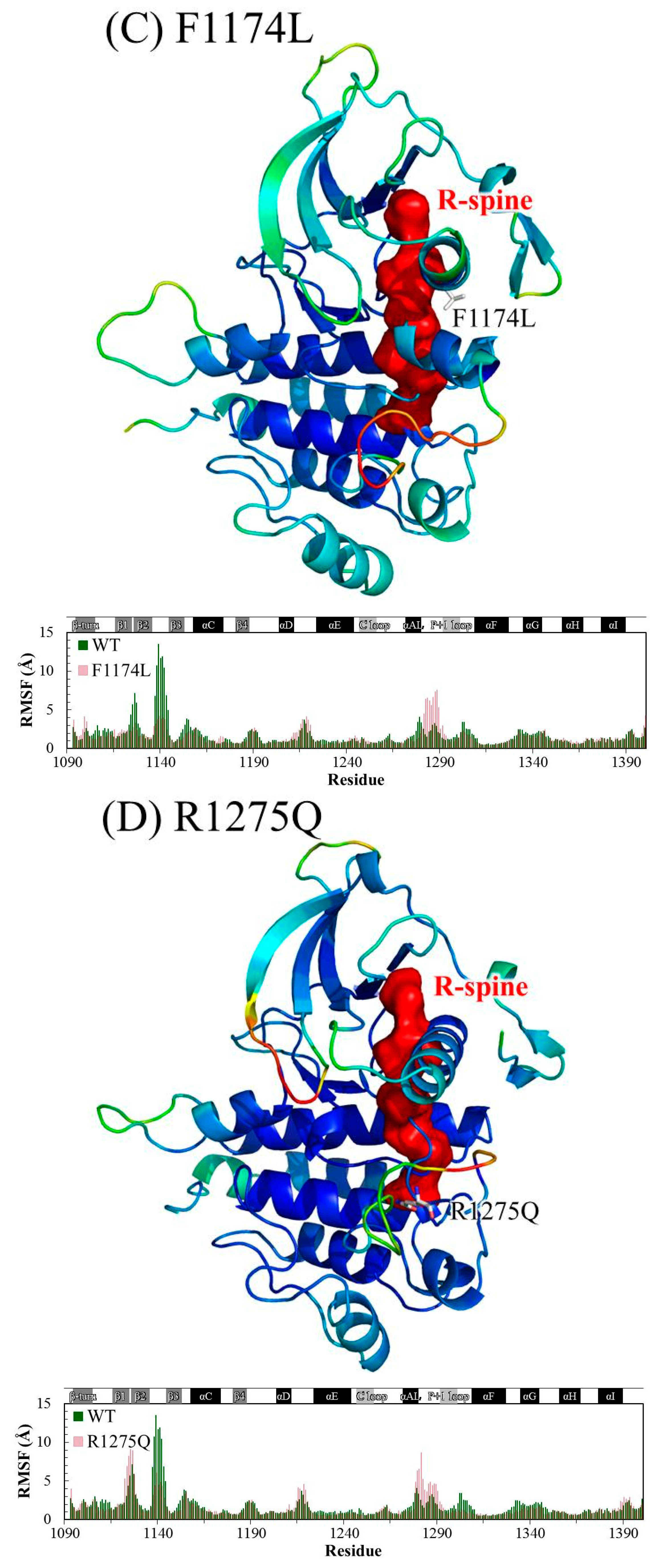
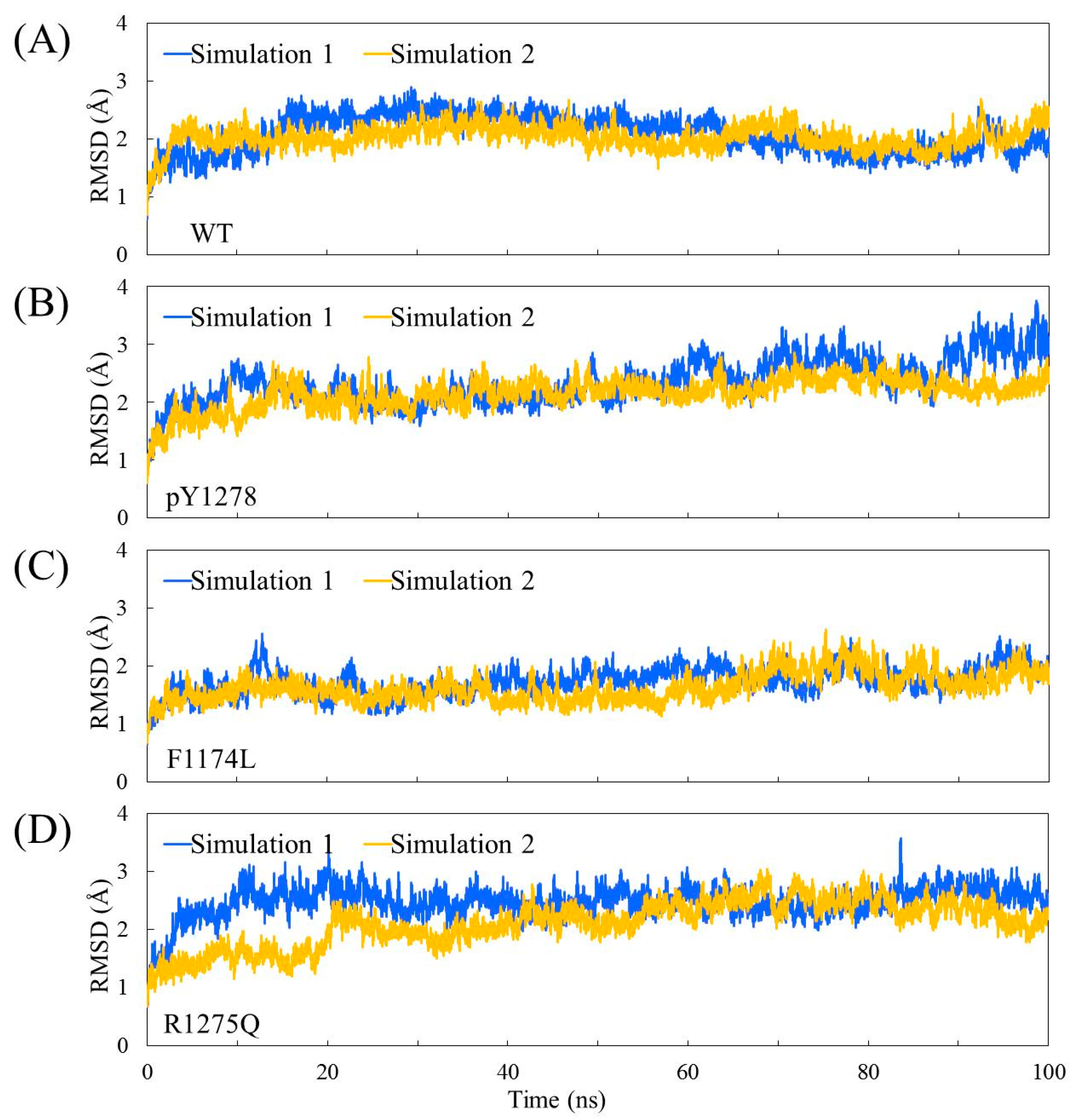
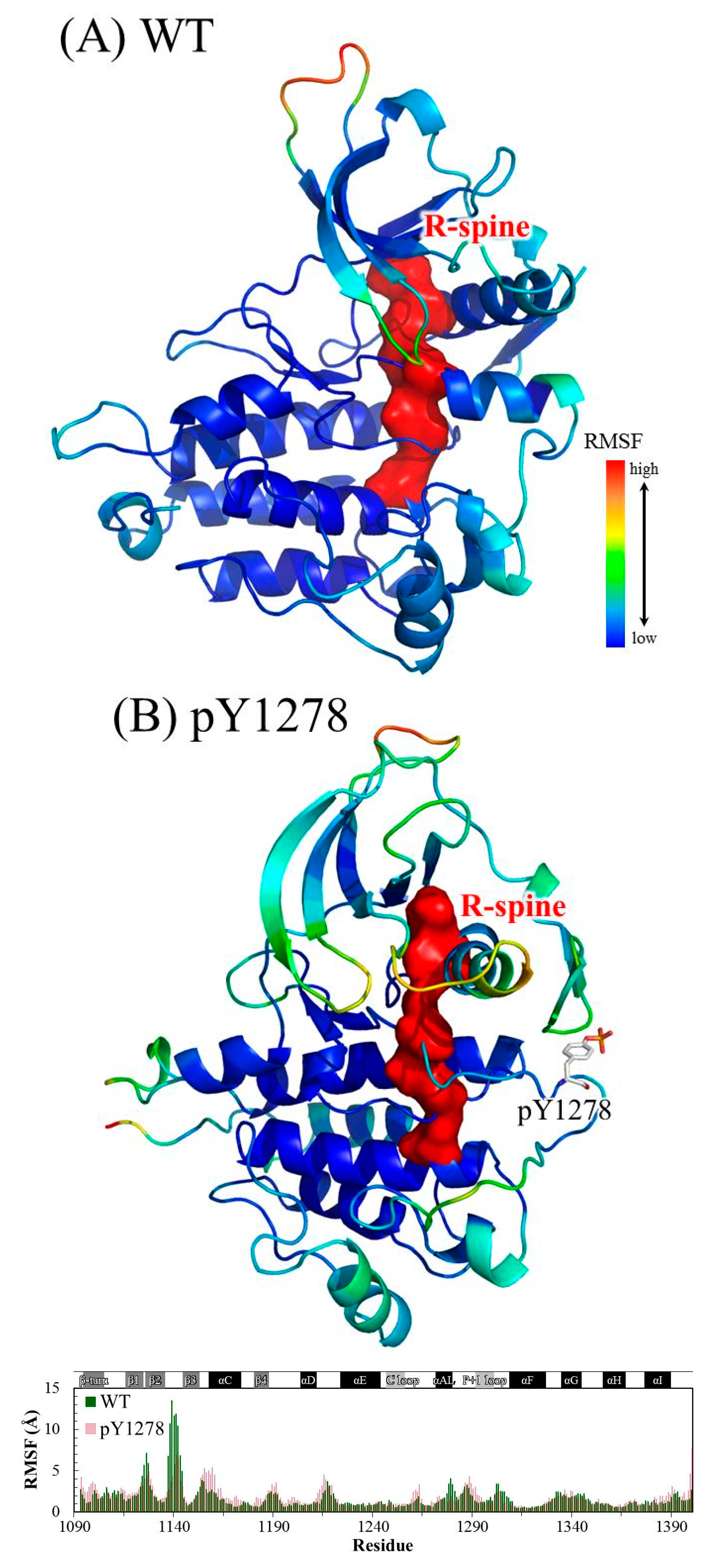
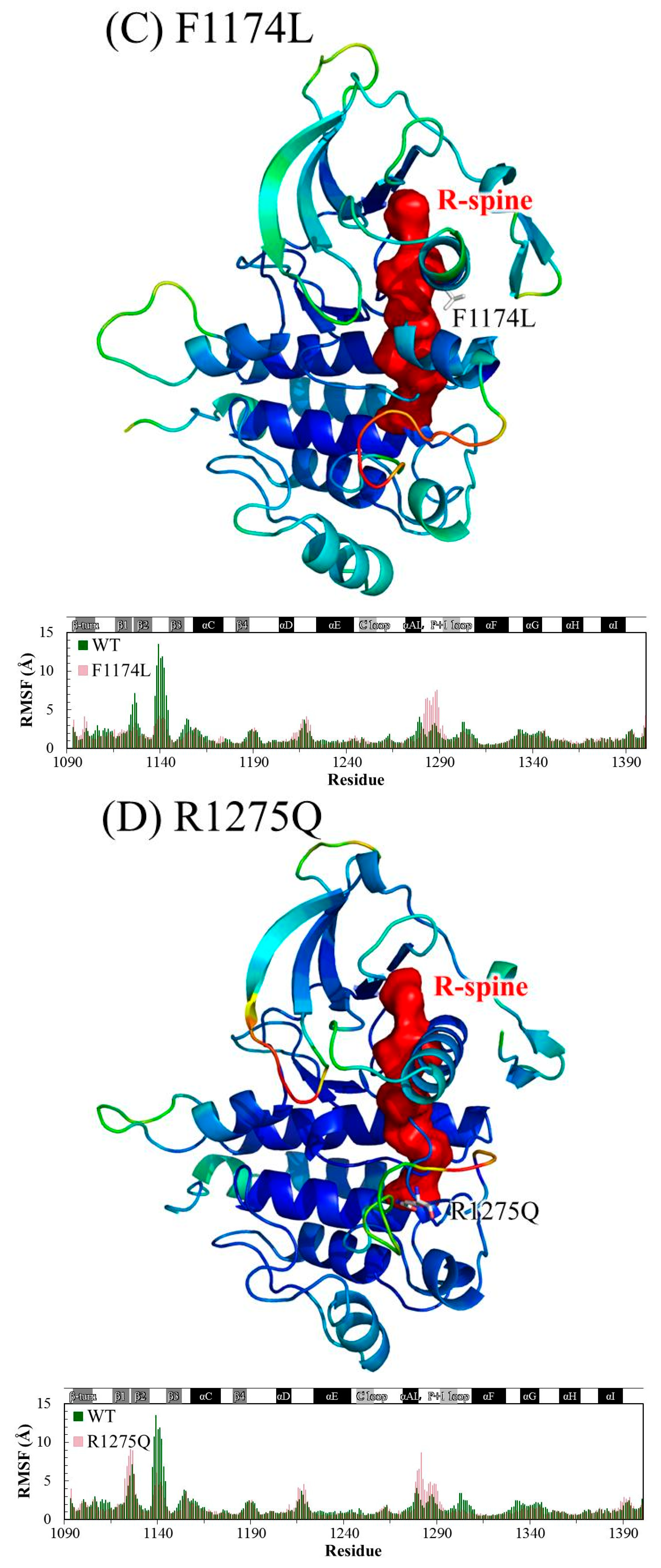
3.1. MD Stability
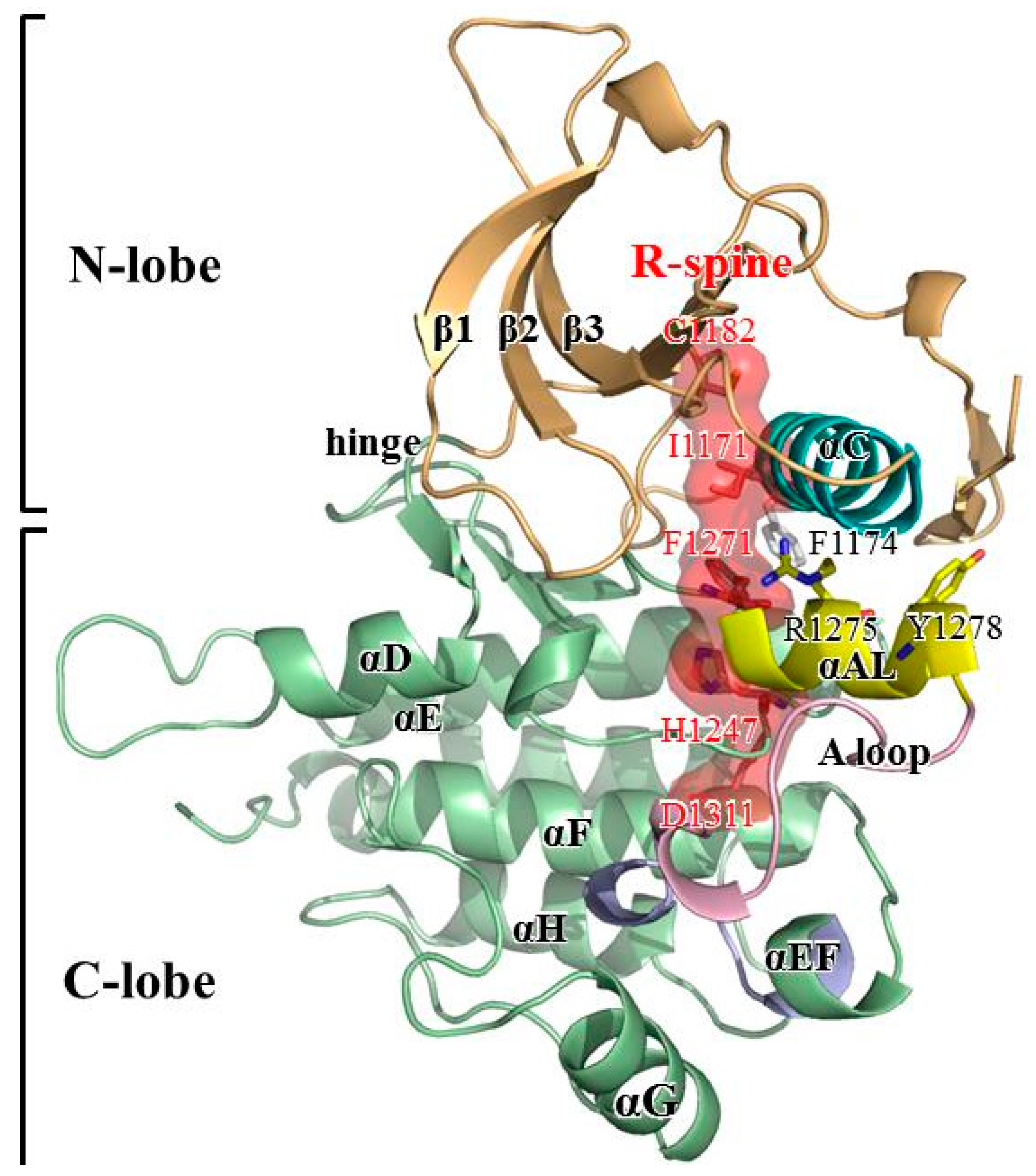
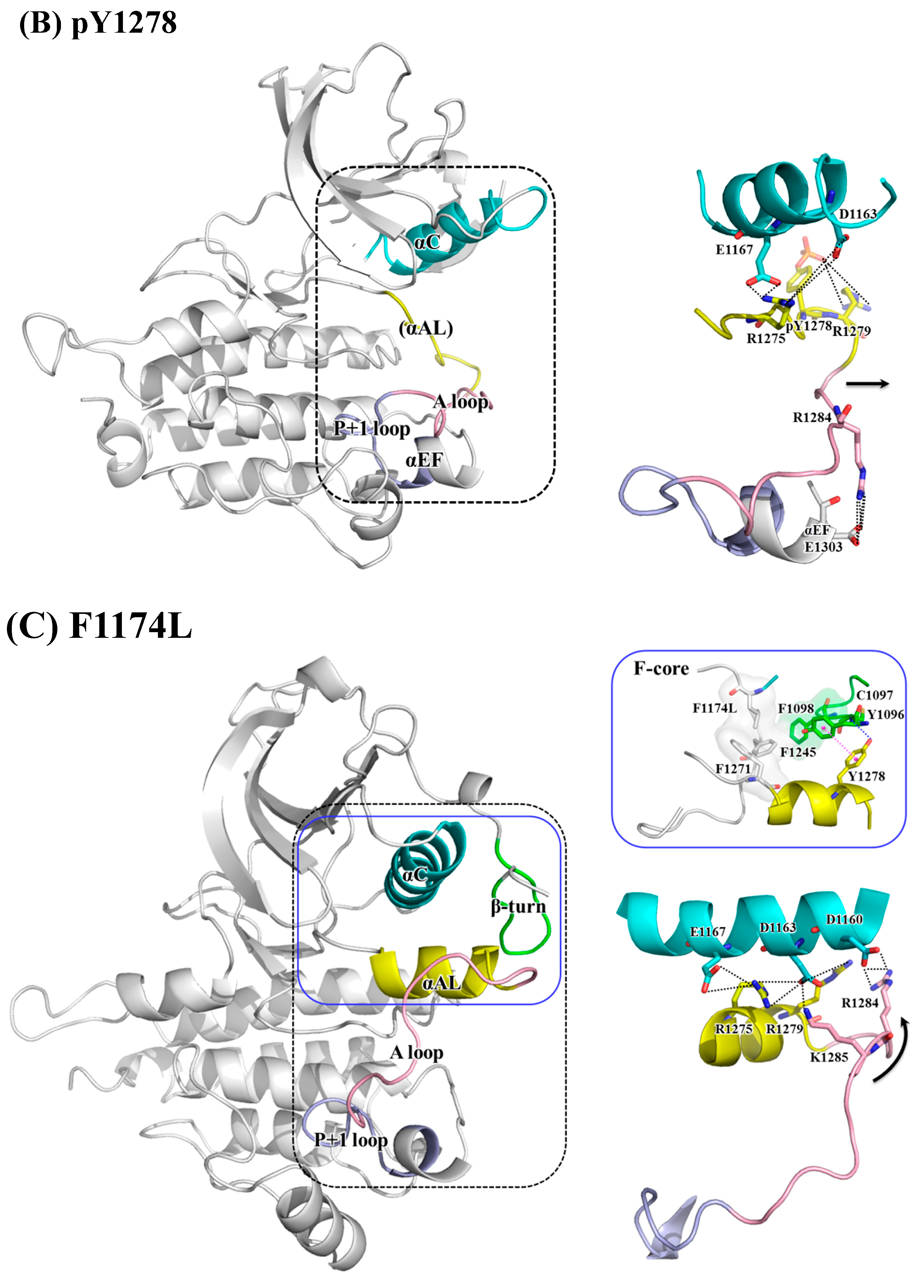
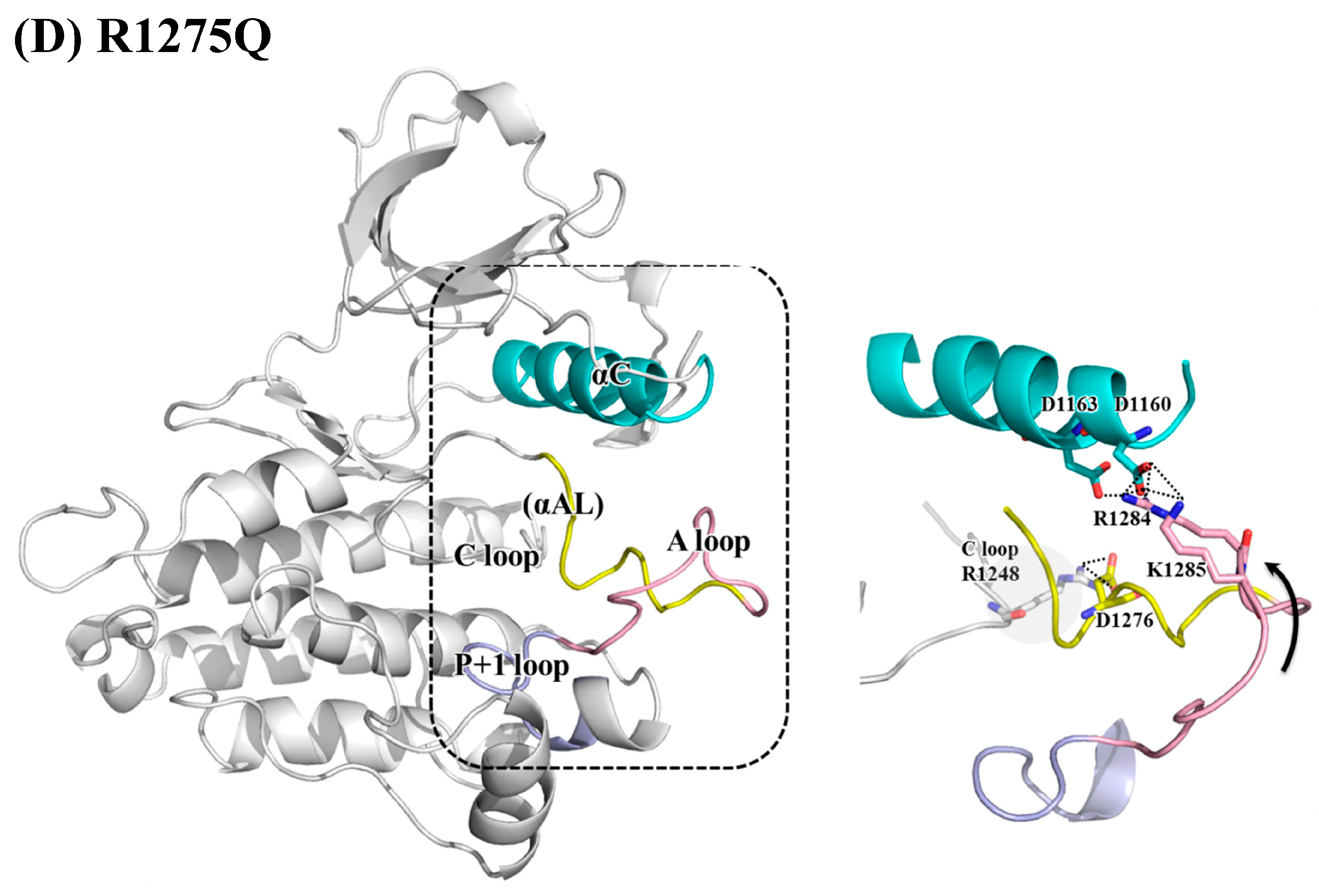
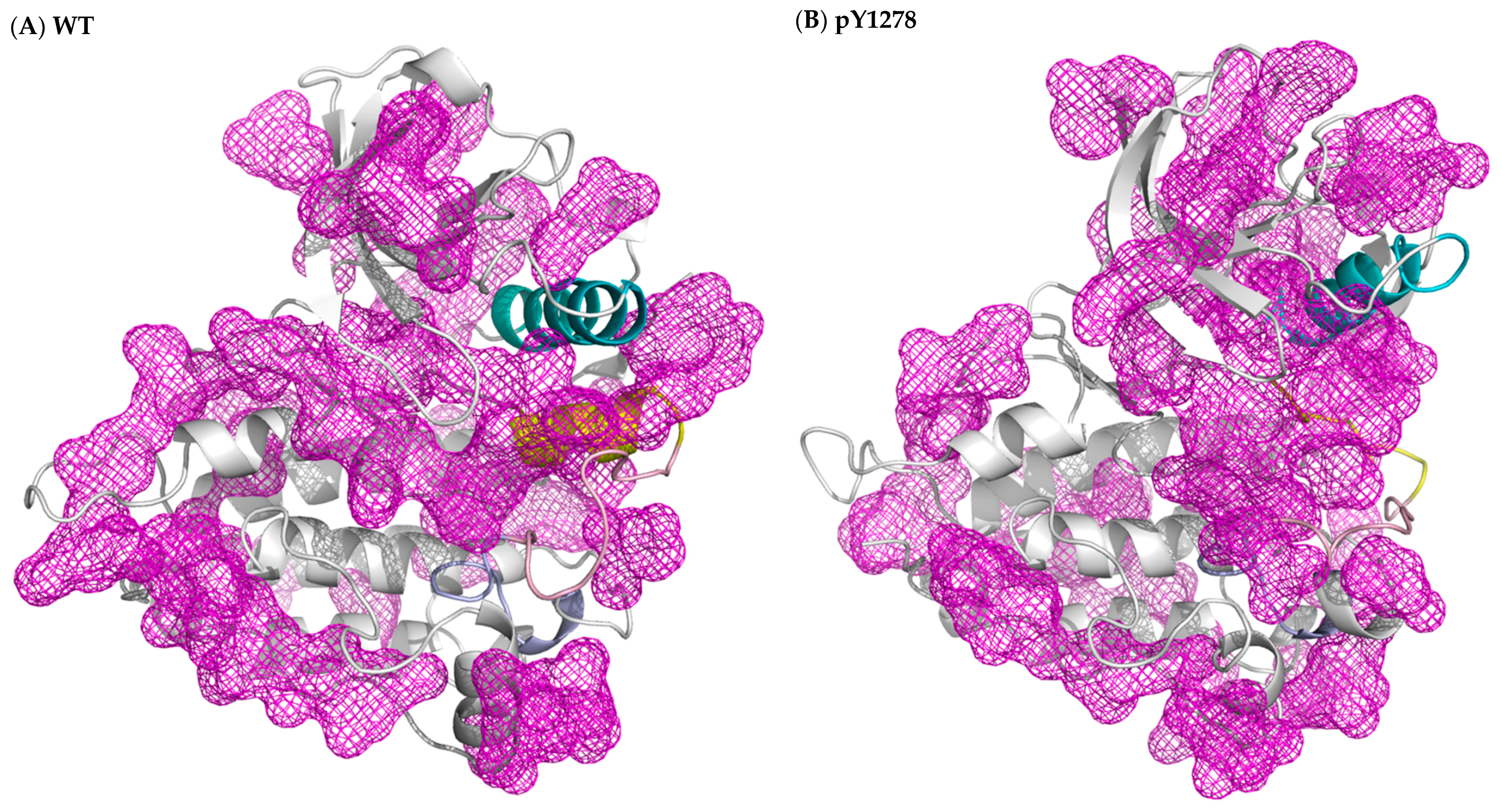
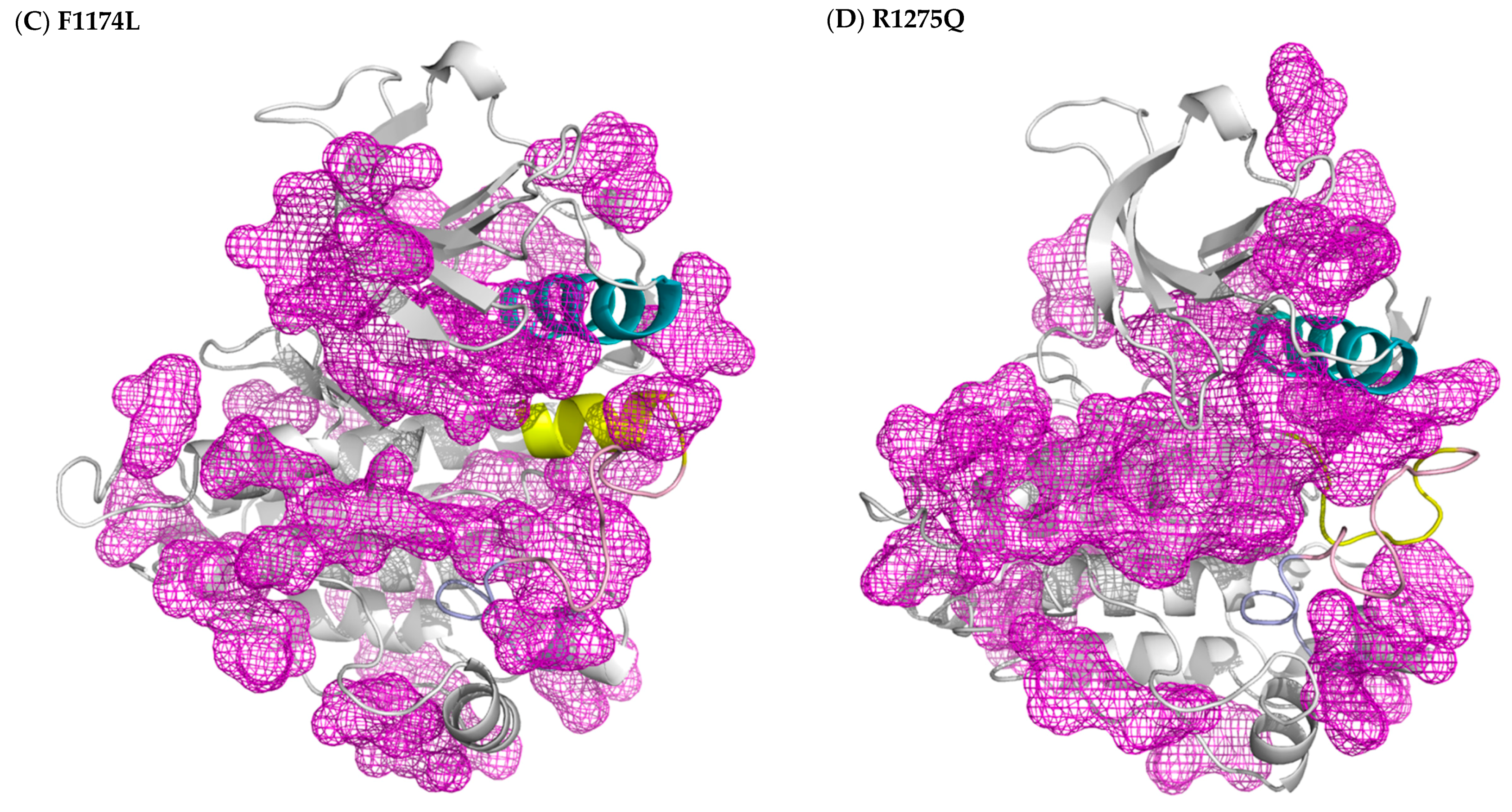
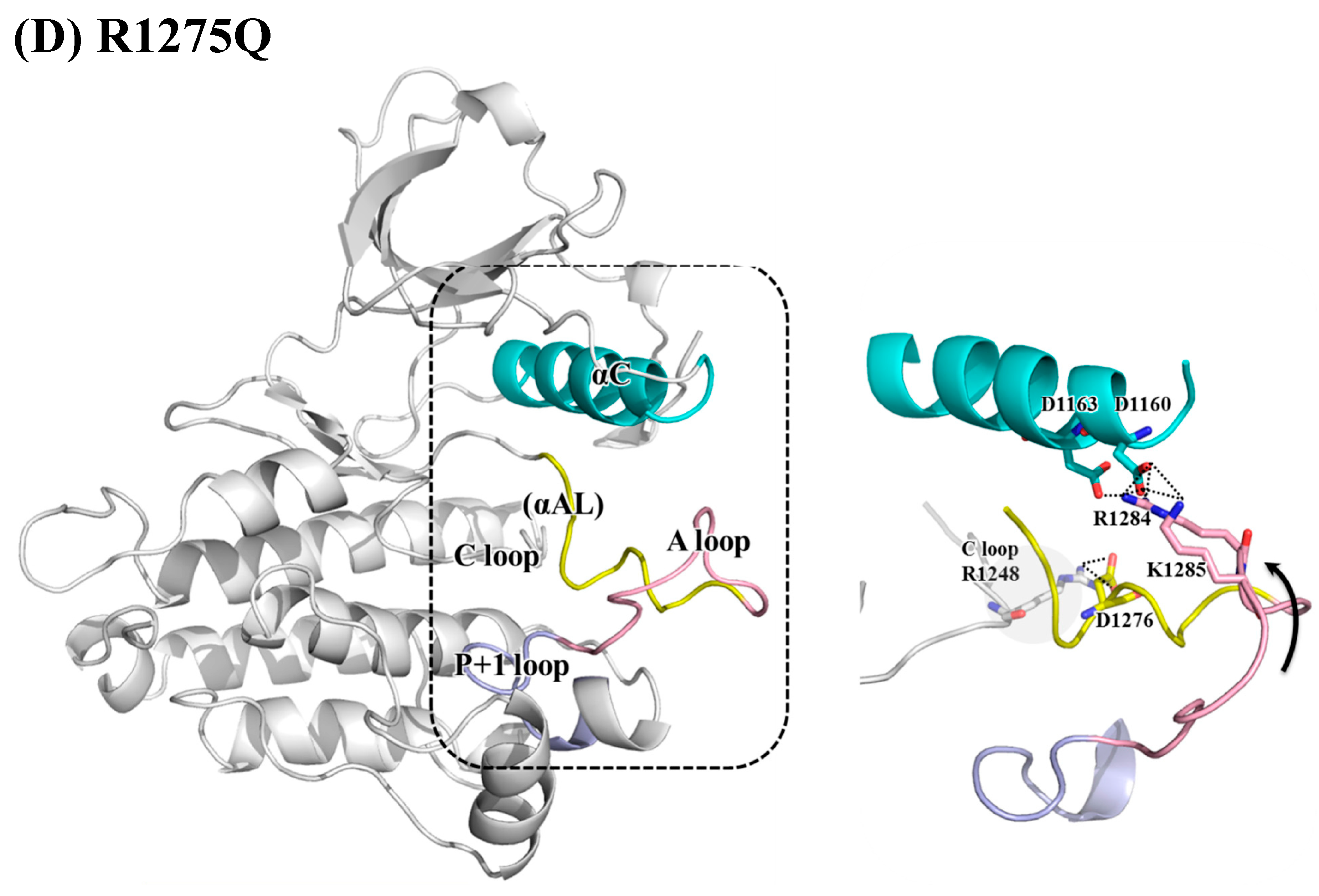
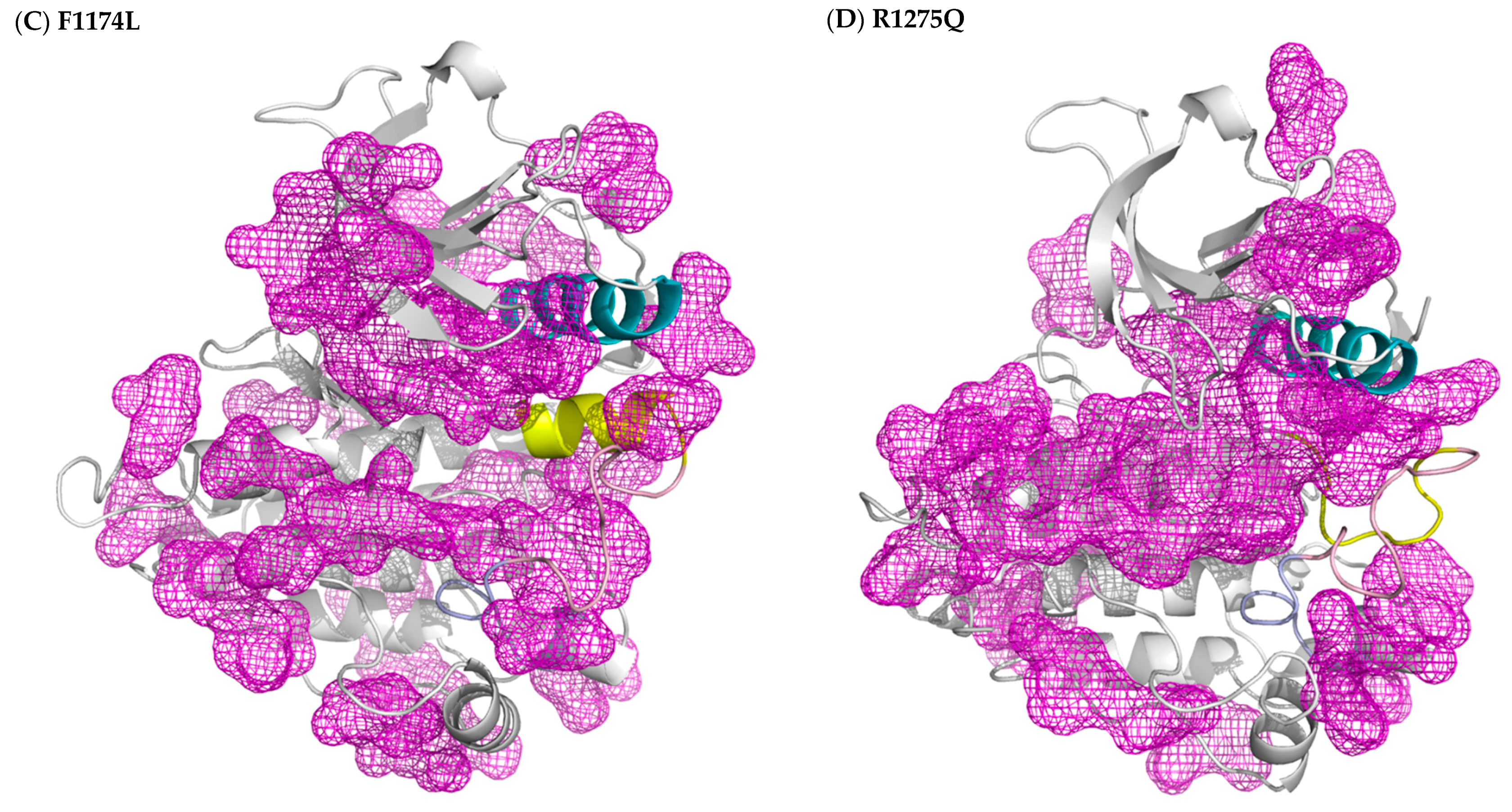
3.2. Structural Variance on the A-loop
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Beà, S.; Lopez-Guillermo, A.; Lespinet, V.; Pinyol, M.; Burkhardt, B.; Lamant, L.; Zettl, A.; Horsman, D.; Gascoyne, R. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br. J. Haematol. 2008, 140, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018, 109, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Powers, C.; Bowden, E.T.; Sale, E.B.; Riegel, A.T.; Wellstein, A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J. Biol. Chem. 2002, 277, 35990–35998. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Aigner, A.; Sunitha, I.; Souttou, B.; Malerczyk, C.; Caughey, D.J.; Wen, D.; Karavanov, A.; Riegel, A.T. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J. Biol. Chem. 2001, 276, 16772–16779. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Biol. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.-Y.; Hua, S.; Chen, C.; Kim, S. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ma, J.; Li, W.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Miller, W.T.; Mohammadi, M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell 2007, 27, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songyang, Z.; Lu, K.P.; Kwon, Y.T.; Tsai, L.-H.; Filhol, O.; Cochet, C.; Brickey, D.A.; Soderling, T.R.; Bartleson, C.; Graves, D.J. A structural basis for substrate specificities of protein Ser/Thr kinases: Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 1996, 16, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Blechner, S.; Hoagland, N.; Hoekstra, M.F.; Piwnica-Worms, H.; Cantley, L.C. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 1994, 4, 973–982. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Chatterjee, S.; Chaudhuri, M.; White, M.F. YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 2027–2031. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, L.; Rastelli, G. αC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Taylor, S.S. Defining the conserved internal architecture of a protein kinase. BBA Proteins Proteom. 2010, 1804, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Till, J.H.; Becerra, M.; Watty, A.; Lu, Y.; Ma, Y.; Neubert, T.A.; Burden, S.J.; Hubbard, S.R. Crystal structure of the MuSK tyrosine kinase: Insights into receptor autoregulation. Structure 2002, 10, 1187–1196. [Google Scholar] [CrossRef]
- Taylor, S.S.; Zhang, P.; Steichen, J.M.; Keshwani, M.M.; Kornev, A.P. PKA: Lessons learned after twenty years. BBA Proteins Proteom. 2013, 1834, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114–108ra114. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.F.; Chen, H.; Emkey, R.; Whittington, D.A. The R1275Q neuroblastoma mutant and certain ATP-competitive inhibitors stabilize alternative activation loop conformations of anaplastic lymphoma kinase. J. Biol. Chem. 2012, 287, 37447–37457. [Google Scholar] [CrossRef] [PubMed]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A. The ALKF1174L mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Neumann, P.; Konevega, A.L.; Bock, L.V.; Ficner, R.; Rodnina, M.V.; Stark, H. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by C s-corrected cryo-EM. Nature 2015, 520, 567. [Google Scholar] [PubMed]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. BBA Proteins Proteom. 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Caliman, A.D.; McCammon, J.A. Allosteric effects of sodium ion binding on activation of the M3 muscarinic G-protein-coupled receptor. Biophys. J. 2015, 108, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Verma, A.; Piana, S.; Lindorff-Larsen, K.; Cerminara, M.; Santiveri, C.M.; Shaw, D.E.; De Alba, E.; Munñoz, V. Interaction networks in protein folding via atomic-resolution experiments and long-time-scale molecular dynamics simulations. J. Am. Chem. Soc. 2015, 137, 6506–6516. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Govender, A.; Permaul, K.; Singh, S.; Bisetty, K. Thermostable chitinase II from Thermomyces lanuginosus SSBP: Cloning, structure prediction and molecular dynamics simulations. JTBio 2015, 374, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Chuang, Y.C.; Liu, Y.L.; Yang, C.N. A molecular dynamics simulation study for variant drug responses due to FMS-like tyrosine kinase 3 G697R mutation. RSC Adv. 2017, 7, 29871–29881. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Walker, R.; Zhang, W.; Merz, K. Assisted Model Building with Energy Refinement (AMBER) 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. JCoCh 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Van Gunsteren, W.; Berendsen, H. Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]
- Munson, M.; Balasubramanian, S.; Fleming, K.G.; Nagi, A.D.; O’Brien, R.; Sturtevant, J.M.; Regan, L. What makes a protein a protein? Hydrophobic core designs that specify stability and structural properties. Protein Sci. 1996, 5, 1584–1593. [Google Scholar] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.-H.; Huang, C.-X.; Chen, Y.-J.; Chuang, Y.-C.; Huang, B.-Y.; Yang, C.-N. Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase. Molecules 2018, 23, 1610. https://doi.org/10.3390/molecules23071610
Jiang C-H, Huang C-X, Chen Y-J, Chuang Y-C, Huang B-Y, Yang C-N. Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase. Molecules. 2018; 23(7):1610. https://doi.org/10.3390/molecules23071610
Chicago/Turabian StyleJiang, Cheng-Han, Chong-Xian Huang, Ya-Jyun Chen, Yu-Chung Chuang, Bo-Yen Huang, and Chia-Ning Yang. 2018. "Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase" Molecules 23, no. 7: 1610. https://doi.org/10.3390/molecules23071610