
Detection of Protein Complexes Based on Penalized Matrix Decomposition in a Sparse Protein–Protein Interaction Network

Abstract
:1. Introduction
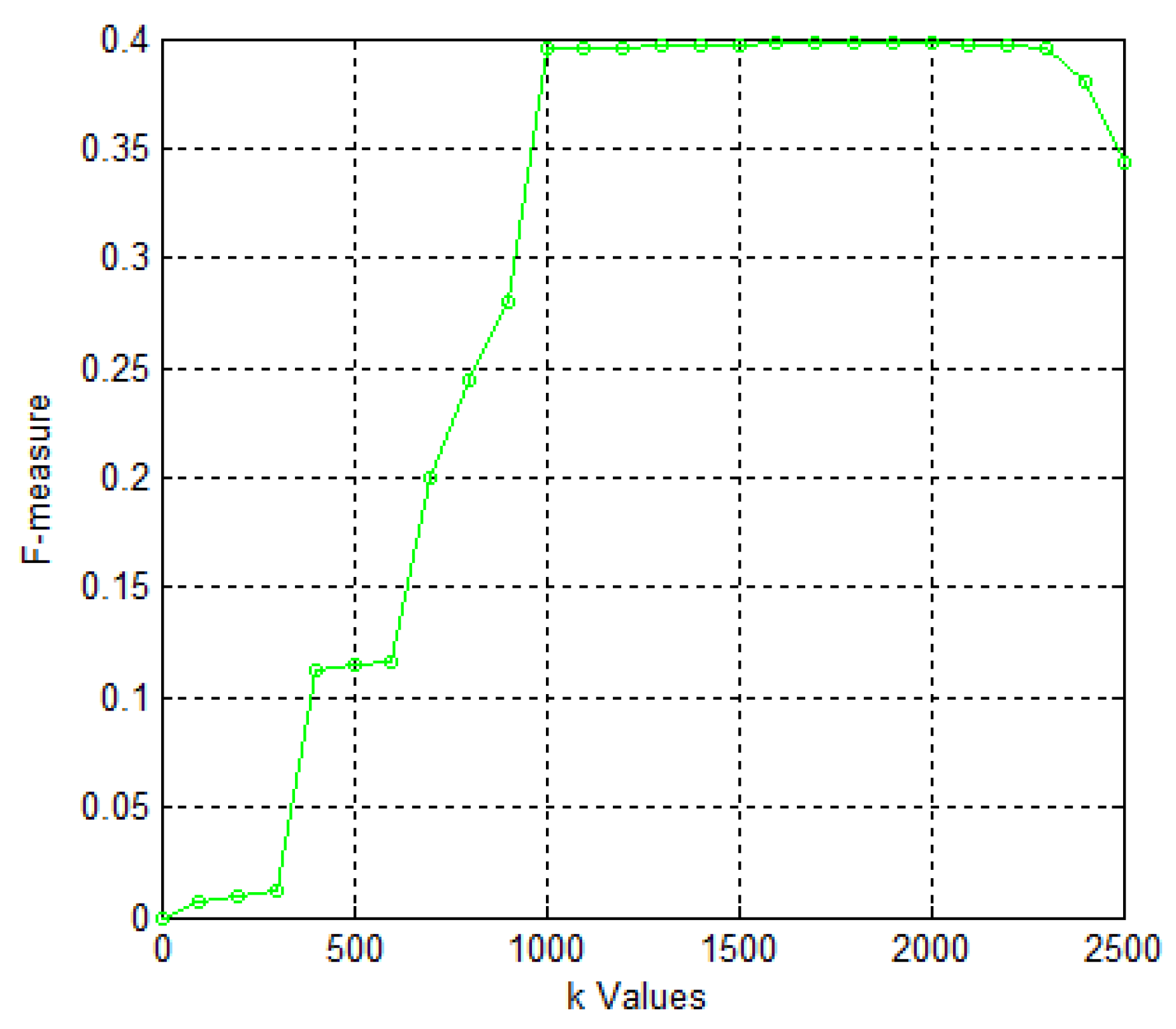
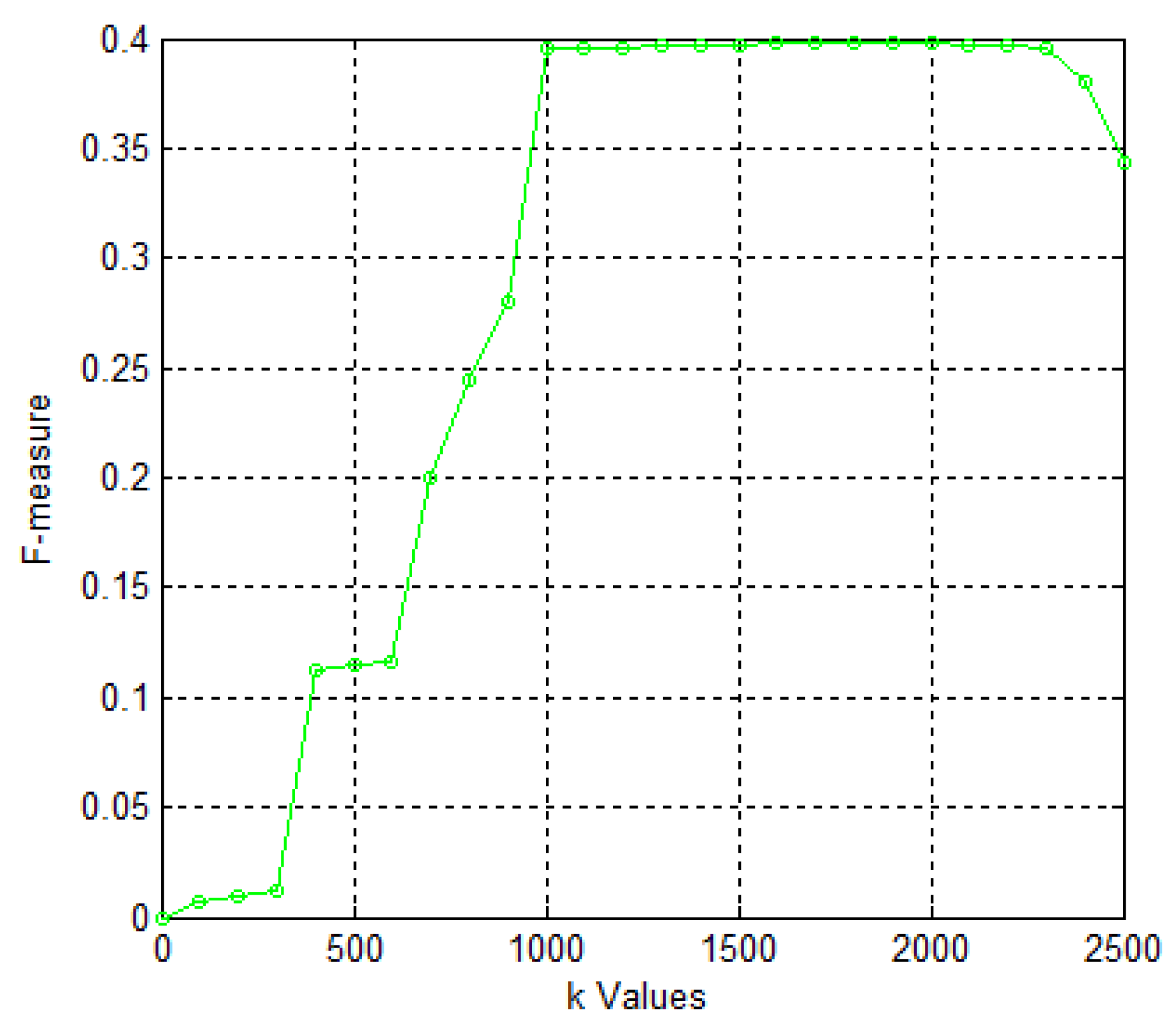
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Datasets
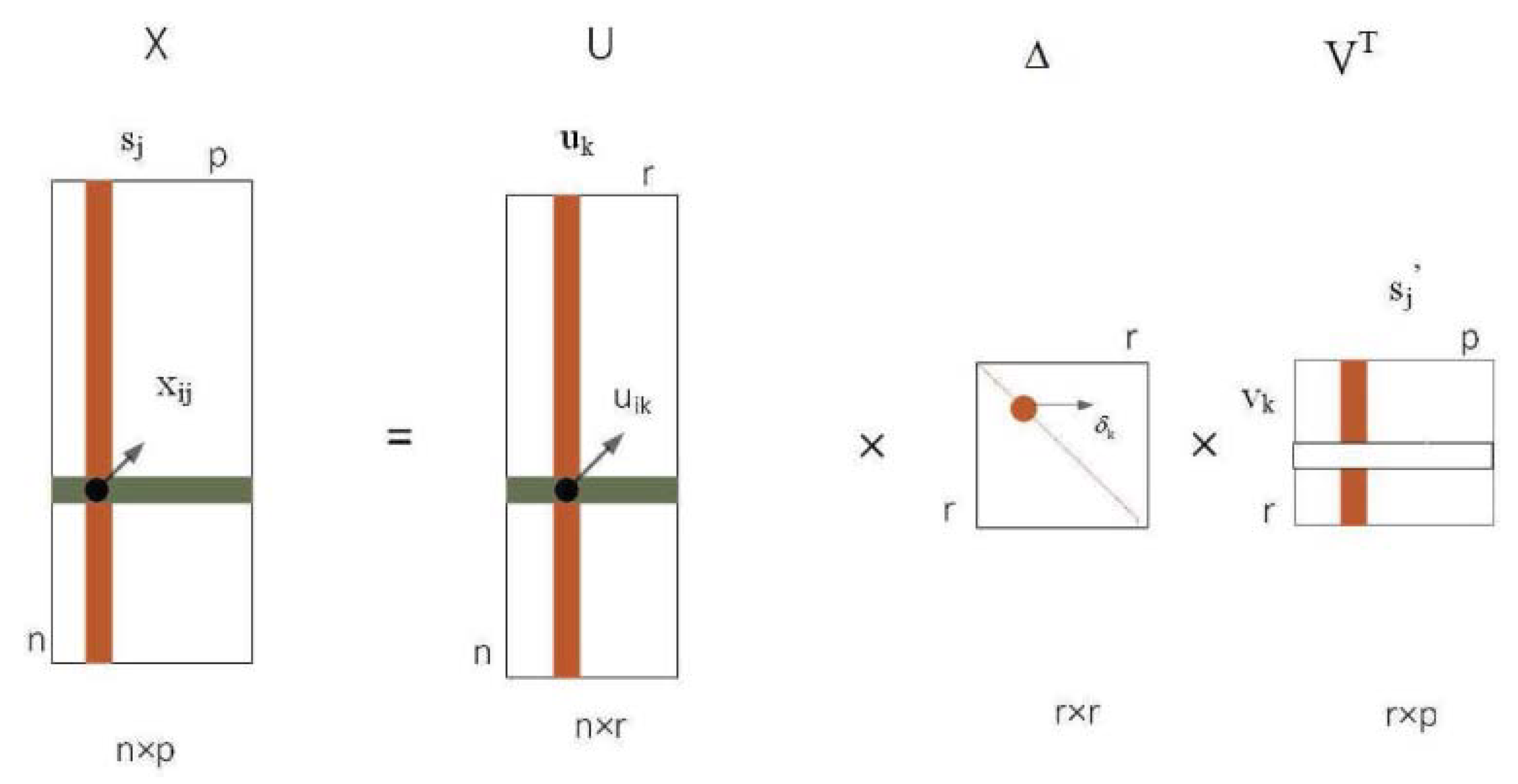
3.2. Methods
| Algorithm 1. Calculating the single factor of PMD. |
| Step1. Initialize and let unit . Step2. Interate until convergence:
Step3. |
| Algorithm 2. Calculating the factor of PMD. |
| Step1.; Step2. For
|
| Algorithm 3. The optimization process of the single factor PMD. |
| Step1. Initialize and let unit . Step2. Iterate until convergence:
Step3. |
3.3. Detection of Protein Complexes Using
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Adamcsek, B.; Palla, G.; Farkas, I.J.; Derenyi, I.; Vicsek, T. Cfinder: Locating cliques and overlapping modules in biological networks. Bioinformatics 2006, 22, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Altaf-Ul-Amin, M.; Shinbo, Y.; Mihara, K.; Kurokawa, K.; Kanaya, S. Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinform. 2006, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Sun, P.G.; Song, J. Clustering algorithm for detecting functional modules in protein interaction networks. J. Bioinform. Comput. Biol. 2011, 7, 217–242. [Google Scholar] [CrossRef]
- Pizzuti, C.; Rombo, S.E. A coclustering approach for mining large protein-protein interaction networks. IEEE ACM Trans. Comput. Biol. 2012, 9, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Li, M.; Chen, J.E.; Pan, Y. A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks. IEEE ACM Trans. Comput. Biol. 2011, 8, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, C.G.; Vakil, R.; Bader, J.S. Nemo: Network module identification in cytoscape. BMC Bioinform. 2010, 11 (Suppl. 1), S61. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.Y.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Gao, L.; Dong, J.H.; Yang, X.F. Detecting overlapping protein complexes by rough-fuzzy clustering in protein-protein interaction networks. PLoS ONE 2014, 9, 1856. [Google Scholar] [CrossRef] [PubMed]
- Rhrissorrakrai, K.; Gunsalus, K.C. Mine: Module identification in networks. BMC Bioinform. 2011, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Voevodski, K.; Teng, S.H.; Xia, Y. Finding local communities in protein networks. BMC Bioinform. 2009, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Singh, M. Spici: A fast clustering algorithm for large biological networks. Bioinformatics 2010, 26, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.W.; Luo, J.W.; Liang, C.; Wang, S.L.; Ding, P.J. Pce-fr: A novel method for identifying overlapping protein complexes in weighted protein-protein interaction networks using pseudo-clique extension based on fuzzy relation. IEEE Trans. Nanobiosci. 2016, 15, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Vella, D.; Marini, S.; Vitali, F.; di Silvestre, D.; Mauri, G.; Bellazzi, R. Mtgo: Ppi network analysis via topological and functional module identification. Sci. Rep. 2018, 8, 5499. [Google Scholar] [CrossRef] [PubMed]
- Kouhsar, M.; Zare-Mirakabad, F.; Jamali, Y. Wcoach: Protein complex prediction in weighted ppi networks. Genes Genet. Syst. 2015, 90, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chan, K.C.C. A density-based clustering approach for identifying overlapping protein complexes with functional preferences. BMC Bioinform. 2015, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Luo, J.; Liang, C.; Wang, S. Identifying protein complexes by combining network topology and biological characteristics. J. Comput. Theor. Nanosci. 2016, 13, 1546–1955. [Google Scholar] [CrossRef]
- King, A.D.; Przulj, N.; Jurisica, I. Protein complex prediction via cost-based clustering. Bioinformatics 2004, 20, 3013–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macropol, K.; Can, T.; Singh, A.K. Rrw: Repeated random walks on genome-scale protein networks for local cluster discovery. BMC Bioinform. 2009, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.M.; Wong, L.; Chua, H.N. Complex discovery from weighted ppi networks. Bioinformatics 2009, 25, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, X.L.; Kwoh, C.K.; Ng, S.K. A core-attachment based method to detect protein complexes in ppi networks. BMC Bioinform. 2009, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Maulik, U.; Mukhopadhyay, A.; Bhattacharyya, M.; Kaderali, L.; Brors, B.; Bandyopadhyay, S.; Eils, R. Mining quasi-bicliques from hiv-1-human protein interaction network: A multiobjective biclustering approach. IEEE ACM Trans. Comput. Biol. 2013, 10, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Luo, J.; Liang, C.; Wang, S.; Song, D. Moepga: A novel method to detect protein complexes in yeast protein-protein interaction networks based on multiobjective evolutionary programming genetic algorithm. Comput. Biol. Chem. 2015, 58, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Deng, S.-P.; You, Z.-H.; Huang, D.-S. Identifying spurious interactions in the protein-protein interaction networks using local similarity preserving embedding. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, N.; Ge, L. A collective nmf method for detecting protein functional module from multiple data sources. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; pp. 655–660. [Google Scholar] [CrossRef]
- Zhang, S.H.; Li, Q.J.; Liu, J.; Zhou, X.J. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microrna-gene regulatory modules. Bioinformatics 2011, 27, I401–I409. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.H.; Zhang, L.; Ng, V.T.Y.; Shiu, S.C.K.; Huang, D.S. Molecular pattern discovery based on penalized matrix decomposition. IEEE ACM Trans. Comput. Biol. 2011, 8, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Witten, D.M.; Tibshirani, R.; Hastie, T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 2009, 10, 515–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-X.; Liu, J.; Gao, Y.-L.; Mi, J.-X.; Ma, C.-X.; Wang, D. A class-information-based penalized matrix decomposition for identifying plants core genes responding to abiotic stresses. PLoS ONE 2014, 9, e106097. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yu, Q.; Li, X.L.; Zheng, J.; Huang, J.F.; Kwoh, C.K. Benchmarking human protein complexes to investigate drug-related systems and evaluate predicted protein complexes. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, X.; Wu, M.; Kwoh, C.K.; Ng, S.K. Inferring gene-phenotype associations via global protein complex network propagation. PLoS ONE 2011, 6, e21502. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Navarro, J.D.; Kristiansen, T.Z.; Amanchy, R.; Surendranath, V.; Muthusamy, B.; Gandhi, T.K.; Chandrika, K.N.; Deshpande, N.; Suresh, S.; et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004, 32, D497. [Google Scholar] [CrossRef] [PubMed]
- Luc, P.V.; Tempst, P. Pindb: A database of nuclear protein complexes from human and yeast. Bioinformatics 2004, 20, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microrna-transcription factor regulatory network. PLoS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
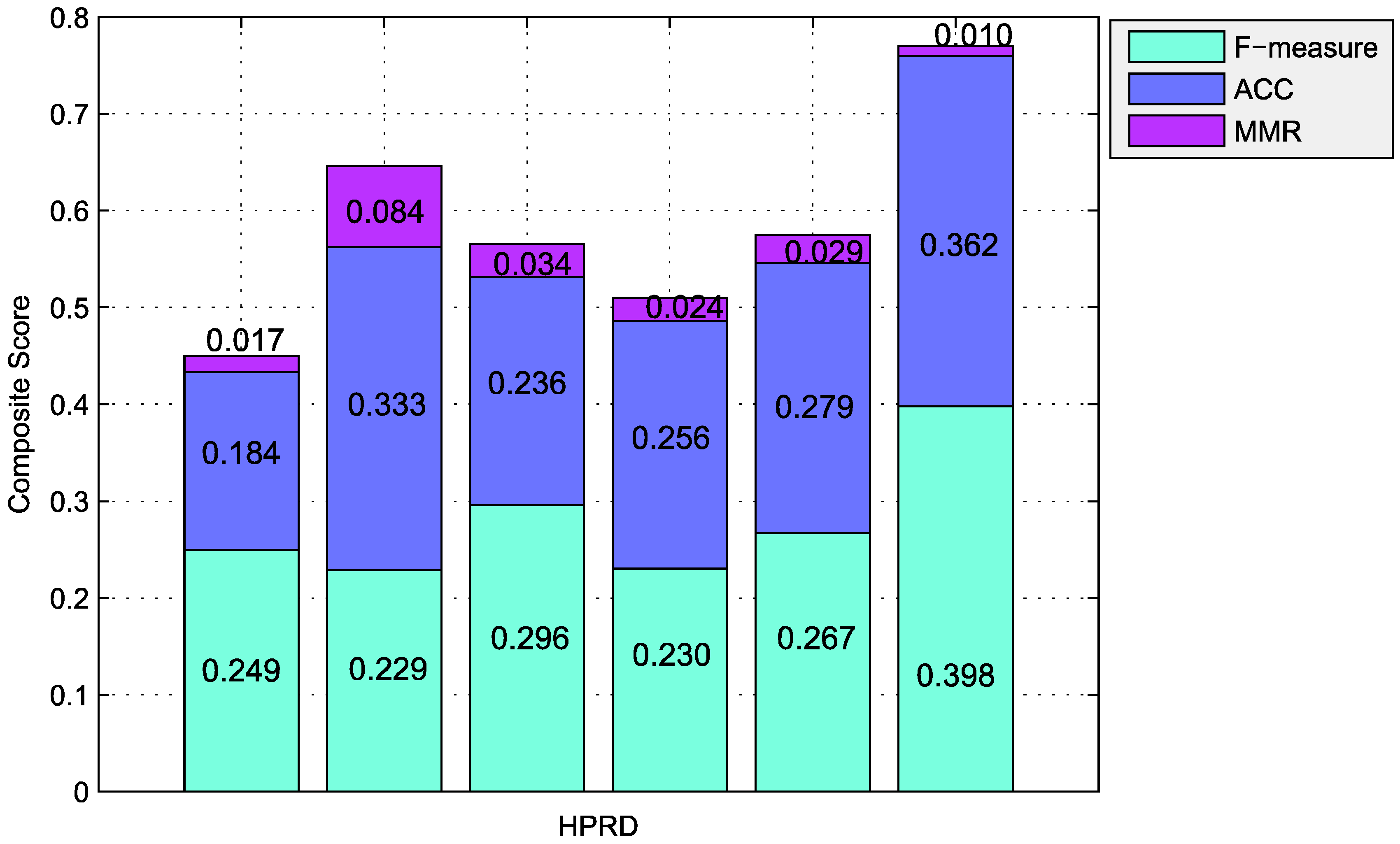
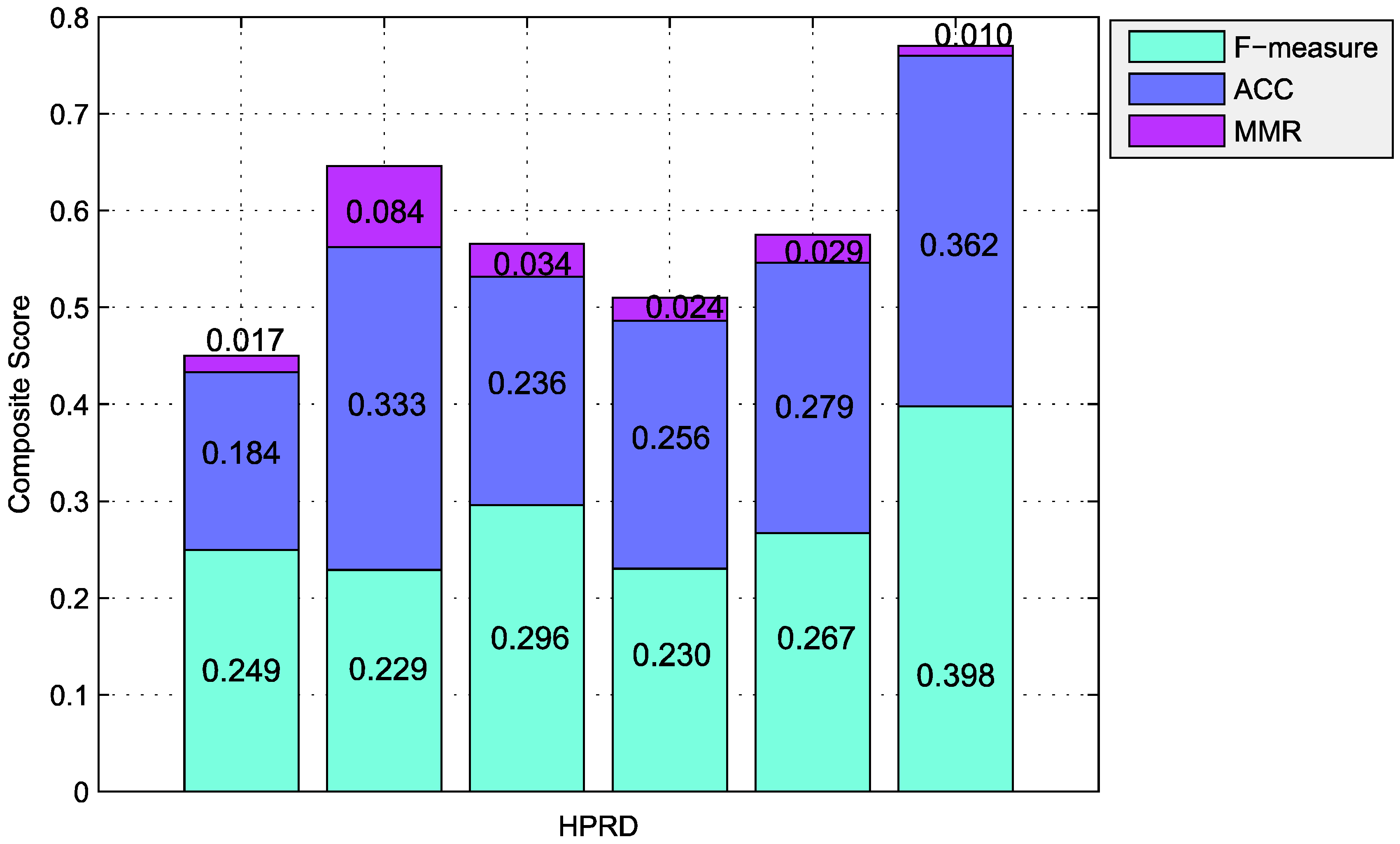
| Algorithms | Number | Precision | Recall | F-Measure | ACC | Sep | MMR | MCC |
|---|---|---|---|---|---|---|---|---|
| CFinder | 49 | 0.959 | 0.143 | 0.249 | 0.184 | 0.165 | 0.017 | 0.327 |
| ClusterONE | 755 | 0.295 | 0.186 | 0.229 | 0.333 | 0.209 | 0.084 | 0.391 |
| RRW | 167 | 0.671 | 0.190 | 0.296 | 0.236 | 0.231 | 0.034 | 0.209 |
| HC-PIN | 99 | 0.646 | 0.140 | 0.230 | 0.256 | 0.233 | 0.024 | 0.196 |
| PCE-FR | 274 | 0.534 | 0.178 | 0.267 | 0.279 | 0.169 | 0.029 | 0.035 |
| 118 | 0.451 | 0.356 | 0.398 | 0.362 | 0.777 | 0.010 | 0.343 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, B.; Deng, S.; Qin, H.; Ding, P.; Chen, S.; Li, G. Detection of Protein Complexes Based on Penalized Matrix Decomposition in a Sparse Protein–Protein Interaction Network. Molecules 2018, 23, 1460. https://doi.org/10.3390/molecules23061460
Cao B, Deng S, Qin H, Ding P, Chen S, Li G. Detection of Protein Complexes Based on Penalized Matrix Decomposition in a Sparse Protein–Protein Interaction Network. Molecules. 2018; 23(6):1460. https://doi.org/10.3390/molecules23061460
Chicago/Turabian StyleCao, Buwen, Shuguang Deng, Hua Qin, Pingjian Ding, Shaopeng Chen, and Guanghui Li. 2018. "Detection of Protein Complexes Based on Penalized Matrix Decomposition in a Sparse Protein–Protein Interaction Network" Molecules 23, no. 6: 1460. https://doi.org/10.3390/molecules23061460