3.2. General Methods
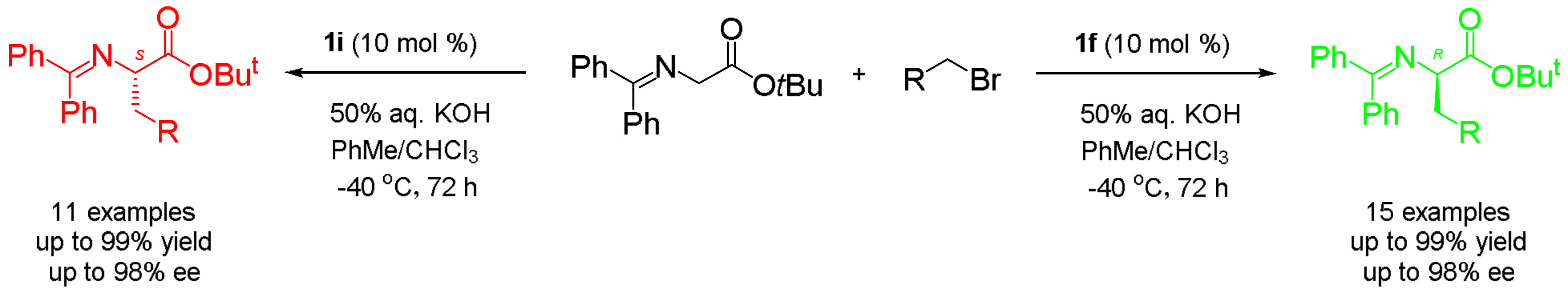
(R)-tert-Butyl N-(diphenylmethylene)-(3,5-dichlorophenyl)alaninate (4a; R=3,5-Cl2-C6H3): A 10 mL reaction tube was charged with 2 (30 mg, 0.1 mmol), 3,5-dichlorobenzyl bromide (119 mg, 0.5 mmol, 5 equivalent), catalyst 1f (6 mg, 0.01 mmol, 0.1 equivalent) and toluene and CHCl3 (1.5 mL, 2:1 v/v), and the mixture was cooled to −40 °C. After the mixture was stirred for 10 min, 50% aq. KOH (28 μL, 0.1 mmol, 5 equivalent) was added, and the whole reaction mixture was stirred at −40 °C for 72 h before being allowed to warm to ambient temperature. The reaction was quenched by adding H2O (2 mL), and the resulting mixture was extracted with EtOAc (3 × 10 mL). The combined extracts were washed with brine (10 mL) and dried (anhydrous Na2SO4), and the crude product was purified by flash column chromatography (eluting with hexane/EtOAc, 50:1) to afford 4a (43 mg, 95% yield) as light yellow liquid. 97% ee; [α 178.8° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.57 (s, 1H), 7.55 (d, J = 1.4 Hz, 1H), 7.40–7.29 (m, 6H), 7.16 (t, J = 1.7 Hz, 1H), 6.95 (d, J = 1.7 Hz, 2H), 6.76 (d, J = 6.1 Hz, 2H), 4.12 (dd, J = 8.9, 4.6 Hz, 1H), 3.19–3.08 (m, 2H), 1.45 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 171.1, 170.2, 141.7, 139.2, 136.1, 134.4, 130.3, 128.7, 128.6, 128.3, 128.3, 128.0, 127.6, 126.4, 81.6, 66.9, 38.9, 28.0; HRMS (ESI, positive): Calcd. for C26H26Cl2NO2 [M + H]+ 454.1335, found: 454.1333. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3,5-dichlorophenyl)alaninate (4a’; R=3,5-Cl2-C6H3): Under the same reaction conditions for 4a except that catalyst 1f was replaced with 1i, enantiomer 4a’ was obtained as light yellow liquid. 98% yield; 97% ee; [α −183.0° (c = 1.0, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3,5-difluorophenyl)alaninate (4b; R=3,5-F2-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3,5-difluorobenzyl bromide, 4b was obtained as white solid. M.p. 35–37 °C; 98% yield; 94% ee; [α 150.2° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.58 (s, 1H), 7.56 (d, J = 1.3 Hz, 1H), 7.39–7.25 (m, 6H), 6.78 (d, J = 6.4 Hz, 2H), 6.63–6.59 (m, 3H), 4.13 (dd, J = 8.9, 4.4 Hz, 1H), 3.23–3.10 (m, 2H), 1.44 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 170.9, 170.2, 163.9, 163.8, 161.5, 161.3, 142.4, 142.3, 142.2, 139.2, 136.1, 130.3, 128.7, 128.5, 128.2, 128.0, 127.6, 112.6, 112.6, 112.4, 112.3, 101.9, 101.6, 101.4, 81.5, 67.1, 39.3, 28.0; HRMS (ESI, positive): calcd. for C26H26F2NO2 [M + H]+ 422.1926, found: 422.1925. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 98:2, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3,5-difluorophenyl)alaninate (4b’; R=3,5-F2-C6H3): Under the same reaction conditions for 4b except that catalyst 1f was replaced with 1i, enantiomer 4b’ was obtained as white solid. 99% yield; 94% ee; [α −154.6° (c = 1.0, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3,5-dibromophenyl)alaninate (4c; R=3,5-Br2-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3,5-dibromobenzyl bromide, 4c was obtained as colorless liquid. 95% yield; 93% ee; [α 174.6° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.54 (d, J = 1.2 Hz, 1H), 7.46 (s, 1H), 7.39–7.25 (m, 6H), 7.15 (d, J = 1.4 Hz, 2H), 6.76 (d, J = 5.3 Hz, 2H), 4.10 (dd, J = 8.8, 4.6 Hz, 1H), 3.18–3.07 (m, 1H), 1.46 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ 171.1, 170.1, 142.3, 139.2, 136.1, 131.8, 131.6, 130.3, 128.7, 128.6, 128.3, 128.0, 127.6, 122.4, 81.6, 66.9, 38.8, 28.0; HRMS (ESI, positive): Calcd. for C26H26Br2NO2 [M + H]+ 542.0325, 544.0304, found: 542.0326, 544.0306. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3,5-dibromophenyl)alaninate (4c’; R=3,5-Br2-C6H3): Under the same reaction conditions for 4c except that catalyst 1f was replaced with 1i, enantiomer 4c’ was obtained as colorless liquid. 83% yield, 95% ee; [α −86° (c = 1.3, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3-chloro-5-fluorophenyl)alaninate (4d; R=3-Cl-5-F-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3-chloro-5-fluorophenyl bromide, 4d was obtained as colorless liquid. 93% yield; 97% ee; [α 169.0° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.57 (d, J = 7.2 Hz, 2H), 7.41–7.28 (m, 6H), 6.89 (d, J = 8.4 Hz, 1H), 6.85 (s, 1H), 6.78 (d, J = 5.6 Hz, 2H), 6.71 (d, J = 9.2 Hz, 1H), 4.13 (q, J = 4.4 Hz, 1H), 3.31–3.10 (m, 2H), 1.45 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 171.1, 170.3, 163.7, 161.2, 142.3, 142.2, 139.3, 136.3, 134.5, 134.4, 130.5, 128.8, 128.7, 128.4, 128.1, 127.7, 125.9, 125.9, 115.4, 115.2, 114.2, 114.0, 81.7, 67.1, 39.2, 28.1; HRMS (ESI, positive): Calcd. for C26H25ClFNaNO2 [M + Na]+ 460.1450, found: 460.1450. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-chloro-5-fluorophenyl)alaninate (4d’; R=3-Cl-5-F-C6H3): Under the same reaction conditions for 4d except that catalyst 1f was replaced with 1i, enantiomer 4d’ was obtained as colorless liquid. 97% yield, 98% ee; [α −168.2° (c = 1.0, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(2-chloro-6-fluorophenyl)alaninate (4e; R=2-Cl-6-F-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 2-chloro-6-fluorophenyl bromide, 4e was obtained as colorless liquid. 68% yield; 98% ee; [α 274.2° (c = 0.9, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.60–7.57 (m, 2H), 7.36–7.26 (m, 6H), 7.11–7.01 (m, 2H), 6.88–6.81 (m, 1H), 6.68 (d, J = 6.9 Hz, 2H), 4.39–4.34 (m, 1H), 3.52–3.45 (m, 1H), 3.36–3.29 (m, 1H), 1.45 (s, 9H); 13C-NMR (75 MHz, CDCl3): δ 170.8, 170.6, 163.6, 160.3, 139.6, 136.2, 136.1, 136.0, 130.2, 129.0, 128.5, 128.2, 128.1, 128.0, 127.8, 125.1, 125.0, 124.7, 124.5, 114.0, 113.7, 81.4, 64.6, 30.1, 28.1; HRMS (ESI, positive): Calcd. for C26H25ClFNO2Na [M + Na]+ 460.1450, found: 460.1447. HPLC analysis: Daicel Chiralcel IC, n-hexane/isopropanol = 98:2, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(2-chloro-6-fluorophenyl)alaninate (4e’; R=2-Cl-6-F-C6H3): Under the same reaction conditions for 4e except that catalyst 1f was replaced with 1i, enantiomer 4e’ was obtained as colorless liquid. 74% yield, 99% ee; [α −231.0° (c = 0.8, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3-chloro-4-fluorophenyl)alaninate (4f; R=3-Cl-4-F-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3-chloro-4-fluorophenyl bromide, 4f was obtained as colorless liquid. 86% yield; 97% ee; [α 178.5° (c = 1.1, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.59–7.55 (m, 2H), 7.41–7.27 (m, 6H), 7.07 (d, J = 7.2 Hz, 1H), 6.97 (s, 1H), 6.94 (d, J = 1.2 Hz, 1H), 6.73 (d, J = 6.0 Hz, 1H), 4.10 (q, J = 4.9 Hz, 1H), 3.20–3.07 (m, 2H), 1.45 (s, 9H); 13C-NMR (75 MHz, CDCl3): δ 170.9, 170.5, 158.5, 155.2, 139.4, 136.3, 135.5, 131.8, 130.5, 129.7, 128.8, 128.6, 128.4, 128.1, 127.7, 120.5, 120.3, 116.2, 116.0, 81.6, 67.5, 38.6, 28.2; HRMS (ESI, positive): Calcd. for C26H25ClFNO2Na [M + Na]+ 460.1450, found: 460.1445. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-chloro-4-fluorophenyl)alaninate (4f’; R=3-Cl-4-F-C6H3): Under the same reaction conditions for 4f except that catalyst 1f was replaced with 1i, enantiomer 4f’ was obtained as colorless liquid. 88% yield, 97% ee; [α −170.0° (c = 1.2, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3-bromo-5-fluorophenyl)alaninate (4g; R=3-Br-5-F-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3-bromo-5-fluorophenyl bromide, 4g was obtained as colorless liquid. 83% yield; 96% ee; [α 151.7° (c = 1.3, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.57 (d, J = 7.6 Hz, 2H), 7.41–7.29 (m, 6H), 7.05 (d, J = 8.0 Hz, 1H), 7.00 (s, 1H), 6.75 (d, J = 8.4 Hz, 3H), 4.12 (q, J = 4.4 Hz, 1H), 3.21–3.09 (m, 2H), 1.45 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 171.1, 170.3, 163.4, 161.2, 142.7, 142.6, 139.3, 136.3, 130.5, 130.2, 128.9, 128.8, 128.8, 128.7, 128.4, 128.1, 127.7, 122.1, 122.0, 117.1, 116.8, 116.0, 115.8, 81.7, 67.1, 39.1, 28.1; HRMS (ESI, positive): Calcd. for C26H26BrFNO2 [M + H]+ 482.1125, found: 482.1119. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-bromo-5-fluorophenyl)alaninate (4g’; R=3-Br-5-F-C6H3): Under the same reaction conditions for 4g except that catalyst 1f was replaced with 1i, enantiomer 4g’ was obtained as colorless liquid. 86% yield, 98% ee; [α −154.4° (c = 1.3, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3,5-dimethoxyphenyl)alaninate (4h; R=3,5-(MeO)2-C6H3): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3,5-dimethoxybenzyl bromide, 4h was obtained as colorless liquid. 85% yield; 96% ee; [α 160.2° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.60 (s, 1H), 7.58 (d, J = 1.4 Hz, 1H), 7.38–7.26 (m, 6H), 6.65 (d, J = 1.7 Hz, 2H), 6.27 (q, J = 2.2 Hz, 1H), 6.21 (d, J = 2.2 Hz, 2H), 4.12 (dd, J = 9.3, 4.2 Hz, 1H), 3.63 (s, 6H), 3.19–3.08 (m, 2H), 1.46 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 170.8, 170.2, 160.4, 140.5, 139.5, 136.3, 130.1, 128.7, 128.2, 127.9, 127.9, 127.7, 107.4, 99.1, 81.3, 67.7, 55.1, 39.8, 28.0; HRMS (ESI, positive): Calcd. for C28H32NO4 [M + H]+ 446.2326, found: 446.2327. HPLC analysis: Daicel Chiralcel IA, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3,5-dimethoxyphenyl)alaninate (4h’; R=3,5-(MeO)2-C6H3): Under the same reaction conditions for 4h except that catalyst 1f was replaced with 1i, enantiomer 4h’ was obtained as colorless liquid. 98% yield, 98% ee; [α −211.4° (c = 1.0, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(3-fluorophenyl)alaninate (
4i; R=3-F-C
6H
4): Under the same reaction conditions for
4a except that 3,5-dichlorophenyl bromide was replaced with 3-fluorobenzyl bromide,
4i was obtained as colorless liquid. 77% yield; 95% ee (Lit 90% ee [
34]); [α
169.2° (
c = 1.1, CH
2Cl
2);
1H-NMR (400 MHz, CDCl
3): δ 7.57 (d,
J = 7.2 Hz, 2H), 7.38–7.24 (m, 6H), 7.14 (dd,
J =14.1, 7.8 Hz, 1H), 6.86–6.83 (m, 2H), 6.75 (d,
J = 9.9 Hz, 1H), 6.69 (d,
J = 6.3 Hz, 2H), 4.12 (dd,
J = 9.0, 4.4 Hz, 1H), 3.25–3.13 (m, 2H), 1.44 (s, 9H);
13C-NMR (75 MHz, CDCl
3): δ 170.6, 170.5, 163.8, 161.4, 140.9, 140.8, 139.3, 136.2, 130.2, 129.4, 129.4, 128.7, 128.3, 128.1, 127.9, 127.6, 125.5, 125.5, 116.6, 116.4, 113.1, 112.9, 81.3, 67.5, 39.2, 28.0. HRMS (ESI, positive): Calcd. for C
26H
26FNNaO
2 [M + Na]
+ 426.1840, found: 426.1846. HPLC analysis: Daicel Chiralcel AD-H,
n-hexane/isopropanol = 97:3, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-fluorophenyl)alaninate (
4i’; R=3-F-C
6H
4): Under the same reaction conditions for
4i except that catalyst
1f was replaced with
1i, enantiomer
4i’ was obtained as colorless liquid. 82% yield; 95% ee (Lit 96% ee [
35]); [α
−156.7° (
c = 0.9, CH
2Cl
2).
(R)-tert-Butyl N-(diphenylmethylene)-(3-chlorophenyl)alaninate (
4j; R=3-Cl-C
6H
4): Under the same reaction conditions for
4a except that 3,5-dichlorophenyl bromide was replaced with 3-chlorobenzyl bromide,
4j was obtained as light yellow liquid. 76% yield; 96% ee (Lit 95% ee [
30]); [α
227.4° (
c = 1.0, CH
2Cl
2);
1H-NMR (400 MHz, CDCl
3): δ 7.56 (d,
J = 7.2 Hz, 2H), 7.38–7.25 (m, 6H), 7.15–7.09 (m, 2H), 7.02 (s, 1H), 6.97 (d,
J = 7.0 Hz, 2H), 6.67 (d,
J = 6.3 Hz, 2H), 4.11 (dd,
J = 9.0, 4.4 Hz, 1H), 3.22–3.11 (m, 2H), 1.45 (s, 9H);
13C-NMR (100 MHz, CDCl
3): δ 170.7, 170.5, 140.4, 139.3, 136.2, 133.8, 130.2, 129.8, 129.3, 128.7, 128.4, 128.1, 128.1, 128.0, 127.6, 126.3, 81.3, 67.4, 39.1, 28.0; HRMS (ESI, positive): Calcd. for C
26H
27ClNO
2 [M + H]
+ 420.1725, found: 420.1724. HPLC analysis: Daicel Chiralcel OD-H,
n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-chlorophenyl)alaninate (
4j’; R=3-Cl-C
6H
4): Under the same reaction conditions for
4j except that catalyst
1f was replaced with
1i, enantiomer
4j’ (1.6 g, 92% yield) was obtained as light yellow oil, and used for synthesis of
13. 97% ee (Lit 91% ee [
36], Lit 92% ee [
37,
38]); [α
−223.4° (
c = 1.0, CH
2Cl
2); [Lit [α
−16.3° (
c = 0.2, CHCl
3) [
30]].
(R)-tert-Butyl N-(diphenylmethylene)-(3-bromophenyl)alaninate (
4k; R=3-Br-C
6H
4): Under the same reaction conditions for
4a except that 3,5-dichlorophenyl bromide was replaced with 3-bromobenzyl bromide,
4k was obtained as colorless liquid. 98% yield, 95% ee (Lit 92% ee [
30]); [α
185.4° (
c = 1.0, CH
2Cl
2);
1H-NMR (400 MHz, CDCl
3): δ 7.56 (d,
J = 7.0 Hz, 2H), 7.36–7.30 (m, 7H), 7.18 (s, 1H), 7.07–7.03 (m, 2H), 6.67 (s, 2H), 4.12–4.09 (m, 1H), 3.21–3.10 (m, 2H), 1.45 (s, 9H);
13C-NMR (400 MHz, CDCl
3): δ 170.7, 170.4, 140.7, 139.3, 136.2, 132.7, 130.2, 129.6, 129.2, 128.7, 128.6, 128.4, 128.2, 128.0, 127.5, 122.1, 81.4, 67.4, 39.1, 18.0; HRMS (ESI, positive): Calcd. for C
26H
26BrNaNO
2 [M + Na]
+ 486.1039, found: 486.1040. HPLC analysis: Daicel Chiralcel OD-H,
n-hexane/isopropanol = 98:2, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-bromophenyl)alaninate (4k’; R=3-Br-C6H4): Under the same reaction conditions for 4k except that catalyst 1f was replaced with 1i, enantiomer 4k’ was obtained as colorless liquid. 93% yield, 93% ee; [α −188.2° (c = 1.0, CH2Cl2).
(R)-tert-Butyl N-(diphenylmethylene)-(4-bromophenyl)alaninate (4l; R=4-Br-C6H4): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 4-bromobenzyl bromide, 4l was obtained as colorless liquid. 95% yield; 95% ee; [α 127.2° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.58 (s, 1H), 7.56 (d, J = 1.5 Hz, 2H), 7.40–7.29 (m, 8H), 6.93 (d, J = 8.3 Hz, 2H), 6.67 (d, J = 6.4 Hz, 2H), 4.09 (dd, J = 9.0, 4.4 Hz, 1H), 3.19–3.08 (m, 2H), 1.44 (s, 9H); 13C-NMR (300 MHz CDCl3): δ 170.6, 170.5, 139.3, 137.4, 136.2, 131.6, 130.2, 128.7, 128.3, 128.1, 128.0, 127.6, 120.0, 81.3, 67.5, 38.9, 28.0; HRMS (ESI, positive): Calcd. for C26H27BrNO2 [M + H]+ 464.1220, found: 464.1220. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 98:2, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(4-bromophenyl)alaninate (
4l’; R=4-Br-C
6H
4): Under the same reaction conditions for
4l except that catalyst
1f was replaced with
1i, enantiomer
4l’ was obtained as colorless liquid. 95% yield; 95% ee (Lit 80% ee [
32]); [α
−134.4° (
c = 1.0, CH
2Cl
2); [Lit [α
−110.1° (
c = 1.09, CHCl
3) [
32]].
(R)-tert-Butyl N-(diphenylmethylene)-(3-iodophenyl)alaninate (4m; R=3-I-C6H4): Under the same reaction conditions for 4a except that 3,5-dichlorophenyl bromide was replaced with 3-iodobenzyl bromide, 4m was obtained as colorless liquid. 82% yield, 96% ee; [α 235.6° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ 7.56 (d, J = 7.2 Hz, 2H), 7.49 (d, J = 7.8 Hz, 1H), 7.38–7.25 (m, 7H), 7.05 (d, J = 7.6 Hz, 1H), 6.92 (t, J = 7.7 Hz, 1H), 6.65 (d, J = 5.3 Hz, 2H), 4.09 (dd, J = 4.8, 4.4 Hz, 1H), 3.18–3.07 (m, 2H), 1.45 (s, 9H); 13C-NMR (100 MHz, CDCl3): δ 170.7, 170.4, 140.8, 139.3, 138.6, 136.2, 135.2, 130.2, 129.8, 129.2, 128.7, 128.3, 128.2, 127.9, 127.6, 94.1, 81.4, 67.4, 39.0, 28.0; HRMS: Calcd. for C26H27INO2+ [M + H]+ 512.1081, found: 512.1083. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 95:5, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(3-iodophenyl)alaninate (
4m’; R=3-I-C
6H
4): Under the same reaction conditions for
4m except that catalyst
1f was replaced with
1i, enantiomer
4m’ was obtained as colorless liquid. 86% yield; 94% ee (Lit 95% ee [
39]); [α
−191.4° (
c = 1.0, CH
2Cl
2).
(R)-tert-Butyl N-(diphenylmethylene)-(4-nitrophenyl)alaninate (
4n; R=4-NO
2-C
6H
4): Under the same reaction conditions for
4a except that 3,5-dichlorophenyl bromide was replaced with 4-nitrobenzyl bromide,
4n was obtained as light yellow solid. M.p. 129–131 °C; 99% yield; 95% ee (Lit 90% ee [
30]); [α
184.8° (
c = 1.0, CH
2Cl
2);
1H-NMR (400 MHz, CDCl
3): δ 8.06 (dd,
J = 7.0, 1.8 Hz, 2H), 7.58 (s, 1H), 7.56 (d,
J = 1.5 Hz, 1H), 7.41–7.36 (m, 2H), 7.34–7.29 (m, 4H), 7.26 (d,
J = 2.5 Hz, 1H), 7.24 (s, 1H), 6.71 (d,
J = 6.8 Hz, 2H), 4.18 (dd,
J = 8.1, 5.2 Hz, 1H), 3.35–3.25 (m, 2H), 1.45 (s, 9H);
13C-NMR (100 MHz, CDCl
3): δ 170.9, 170.0, 146.5, 146.4, 139.0, 135.9, 130.6, 130.4, 128.7, 128.6, 128.3, 128.0, 127.4, 123.2, 81.6, 66.9, 39.3, 28.0; HRMS (ESI, positive): Calcd. for C
26H
27N
2O
4 [M + H]
+ 431.1965, found: 431.1962. HPLC analysis: Daicel Chiralcel IA,
n-hexane/isopropanol = 94:6, flow rate = 0.5 mL/min.
(S)-tert-Butyl N-(diphenylmethylene)-(4-nitrophenyl)alaninate (
4n’; R=4-NO
2-C
6H
4): Under the same reaction conditions for
4n except that catalyst
1f was replaced with
1i, enantiomer
4n’ was obtained as light yellow solid. 96% yield; 94% ee (Lit 99% ee [
39]); [α
−165.4° (
c = 1.0, CH
2Cl
2).
(R)-tert-Butyl 2-((diphenylmethylene)amino)-3-(1-naphthyl)propanoate (
4o; R=α-naphthyl): Under the same reaction conditions for
4a except that 3,5-dichlorophenyl bromide was replaced with 1-(bromomethyl)naphthalene,
4o was obtained as colorless liquid. 81% yield; 96% ee (Lit 96% ee [
40], Lit 99% ee [
33]); [α
331.6° (
c = 1.0, CH
2Cl
2); [Lit [α
343.7° (
c = 1.19, CHCl
3) [
33]];
1H-NMR (400 MHz, CDCl
3): δ 7.78 (d,
J = 8.0 Hz, 1H), 7.72–7.67 (m, 2H), 7.51 (d,
J =7.6 Hz, 2H), 7.38 (t,
J = 7.4 Hz, 1H), 7.33–7.22 (m, 6H), 7.11 (t,
J = 7.5 Hz, 1H), 6.95 (t,
J = 7.5 Hz, 2H), 6.24 (s, 2H), 4.32 (dd,
J = 9.5, 4.0 Hz, 1H), 3.80 (dd,
J = 13.7, 4.0 Hz, 1H), 3.50 (dd,
J = 13.7, 9.6 Hz, 1H), 1.46 (s, 9H);
13C-NMR (75 MHz, CDCl
3): δ 171.0, 170.2, 139.3, 135.8, 134.1, 133.6, 132.2, 130.0, 128.6, 128.4, 128.2, 128.2, 127.8, 127.6, 127.2, 127.0, 125.6, 125.2, 125.2, 123.6, 81.1, 66.5, 36.6, 28.0; HRMS (ESI, positive): Calcd. for C
30H
29NNaO
2+ [M + Na]
+ 458.2091, found: 458.2091. HPLC analysis: Daicel Chiralcel AD-H,
n-hexane/isopropanol = 97:3, flow rate = 0.5 mL/min.
(S)-tert-Butyl 2-((diphenylmethylene)amino)-3-(1-naphthyl)propanoate (
4o’; R=α-naphthyl): Under the same reaction conditions for
4o except that catalyst
1f was replaced with
1i, enantiomer
4o’ was obtained as colorless liquid. 85% yield; 97% ee (Lit 98% ee [
41]); [α
−295.3° (
c = 0.9, CH
2Cl
2).
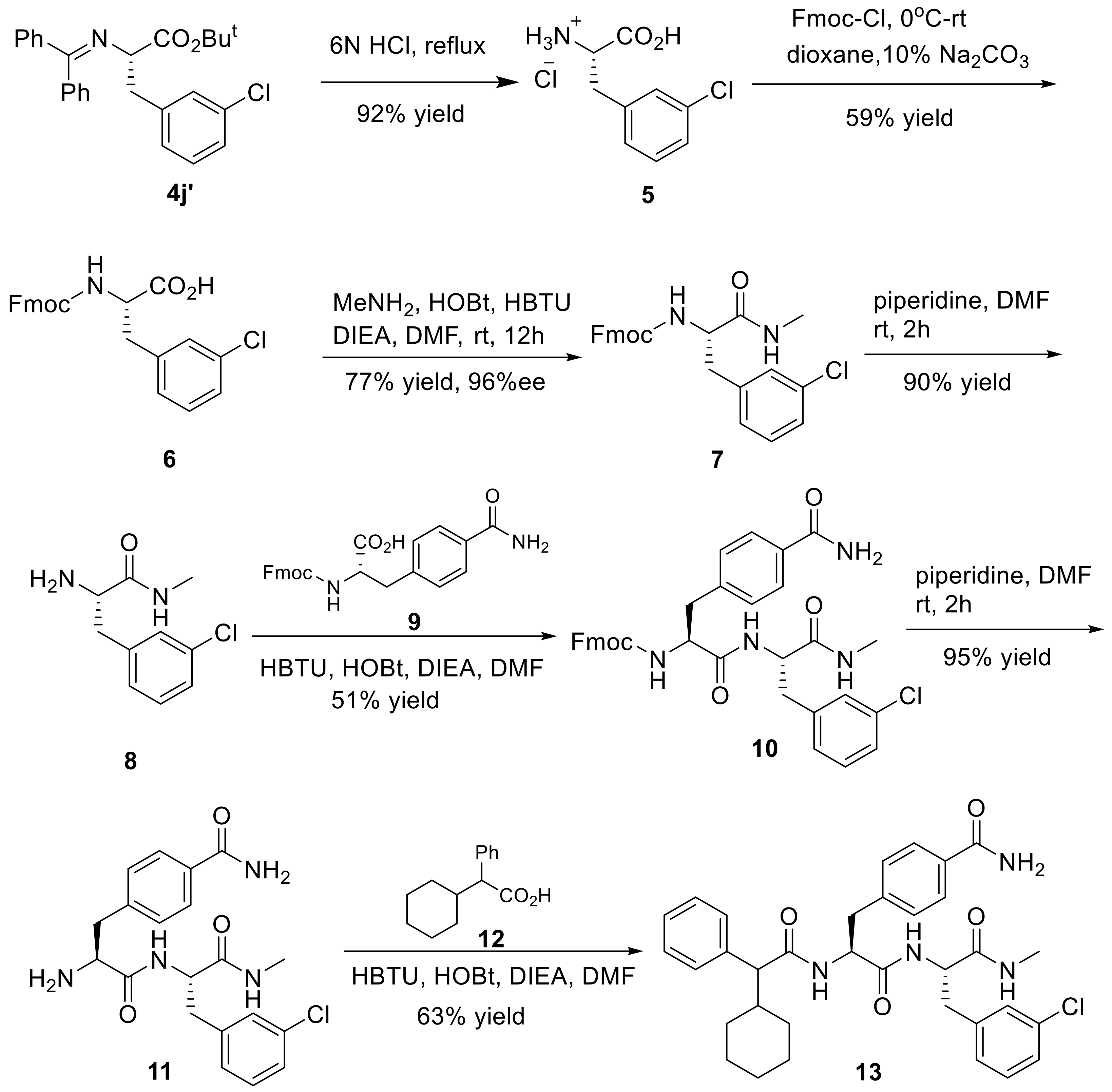
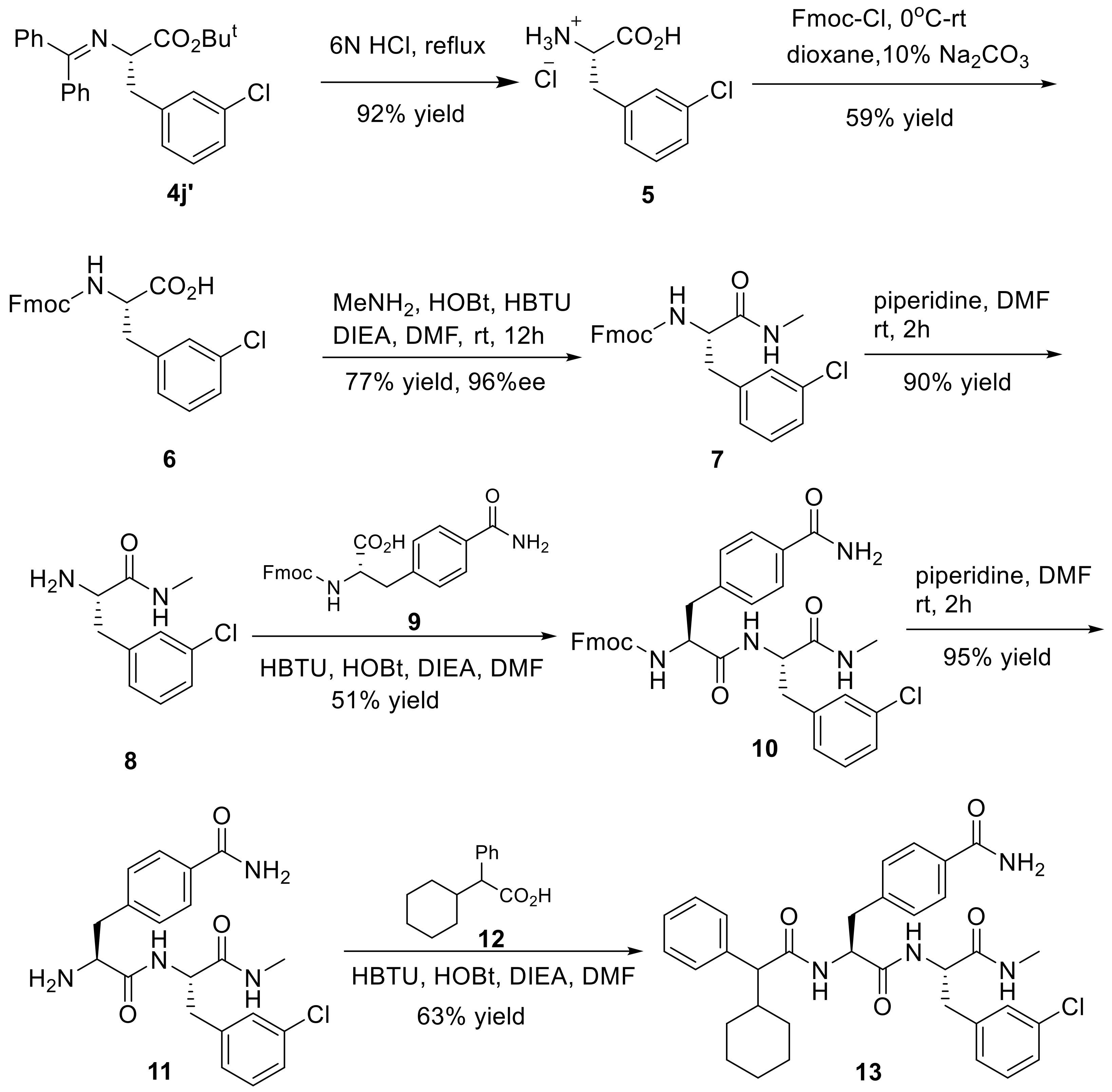
(S)-3-Chloro phenylalanine hydrochloride (5): A mixture of 4j’ (1.2 g, 2.8 mmol) and 6 M HCl (6 mL) was heated at 100 °C for 3 h, and then cooled to ambient temperature, resulting in white precipitates. 5 (525 mg, 92% yield) was obtained by suction filtration as white solid, and was used for the next step without further purification. M.p. 264–266 °C; 1H-NMR (300 MHz, DMSO-d6): δ 3.18 (d, J = 6.2 Hz, 2H), 4.17 (s, 1H), 7.26–7.39 (m, 4H), 8.62 (s, 3H), 13.88 (s, 1H); 13C-NMR (75 MHz, DMSO-d6): δ 35.1, 52.9, 127.2, 128.5, 129.5, 130.4, 133.1, 137.7, 170.2; HRMS (ESI, positive): calcd. for C9H11ClNO2 [M + H]+ 200.0473, found: 200.0474.
(9H-Fluoren-9-yl)methyl (S)-(3-chloro)phenylalanine (6): To an ice-cold solution of 5 (525 mg, 2.6 mmol), dioxane (3 mL) and 10% Na2CO3 aqueous solution (6 mL) was added dropwise a solution of Fmoc-Cl (673 mg, 2.6 mmol) in dioxane (3 mL). The mixture was stirred at 0 °C for 4 h, and then warmed to ambient temperature with the stirring continued for an additional 18 h. The reaction was quenched by adding 2 M HCl (5 mL) and H2O (40 mL). The resulting mixture was extracted with EtOAc (2 × 60 mL), and the combined extracts were washed with brine (2 × 30 mL) and dried. The crude product was purified by flash column chromatography (eluting with hexane/EtOAc, 20:1) to give 6 (644 mg, 59% yield) as white solid. M.p. 123–125 °C; 1H-NMR (300 MHz, DMSO-d6): δ 2.88 (q, J = 7.7 Hz, 1H), 3.17 (s, 1H), 4.09–4.17 (m, 3H), 4.25 (q, J = 4.8 Hz, 1H), 7.20–7.39 (m, 9H), 7.61 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 7.5 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 36.5, 46.6, 55.9, 65.6, 120.2, 125.2, 125.4, 126.3, 127.1, 127.7, 128.0, 129.2, 129.9, 132.7, 140.6, 140.7, 141.2, 143.8, 143.9, 155.9, 173.9; HRMS (ESI, positive): calcd. for C24H20ClNO4Na [M + Na]+ 444.0973, found: 444.0967.
(9H-Fluoren-9-yl)methyl(S)-(3-(3-chlorophenyl)-1-(methylamino)-1-oxopropan-2-yl)carbamate (7): Methyl-amine hydrochloride (202 mg, 3 mmol) and N,N-diisopropylethylamine (DIEA, 388 mg, 3 mmol) was added successively to an ice-cold stirred solution of the substituted 6 (624 mg, 1.5 mmol), HOBt (405 mg, 3 mmol) and HBTU (1.1 g, 3 mmol) in DMF (6 mL) at 0 °C. The reaction mixture was stirred for 30 min at the same temperature, and then allowed to warm to ambient temperature while the stirring continued for an additional 12 h. The solvents and volatiles were removed under reduced pressure, and the residue was dissolved in EtOAc (150 mL) and then washed with saturated NaHCO3 solution (50 mL) and brine (2 × 50 mL) and finally dried (anhydrous Na2SO4). After the solvent was concentrated, the crude product was crystallized from EtOAc to give 7 (504 mg, 77% yield) as white solid. M.p. 179–181 °C; 96% ee; [α −0.6° (c = 0.7, CH2Cl2); 1H-NMR (300 MHz, DMSO-d6): δ 2.57 (d, J = 4.5 Hz, 3H), 2.76–2.98 (m, 4H), 3.16 (s, 1H), 4.46 (d, J = 5.0 Hz, 1H), 7.14–7.29 (m, 5H), 7.38 (d, J = 1.7 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.91 (d, J = 4.3 Hz, 2H), 8.12 (d, J = 7.8 Hz, 1H); 13C-NMR (75 MHz, DMSO-d6): δ 26.0, 37.6, 47.0, 56.6, 66.1, 120.6, 125.7, 125.8, 126.8, 127.5, 128.1, 128.4, 129.6, 130.3, 133.1, 141.0, 141.1, 141.4, 144.2, 144.2, 156.3, 172.1; HRMS (ESI, positive): calcd. for C25H24ClN2O3 [M + H]+ 435.1470, found: 435.1470. HPLC analysis: Daicel Chiralcel OD-H, n-hexane/isopropanol = 85:15, flow rate = 1.0 mL/min.
(S)-2-Amino-3-chloro-N-methylpropanamides (8): To a stirred solution of 7 (480 mg, 1.1 mmol) in DMF (4 mL) was added piperidine (2 mL) at room temperature. The reaction mixture was stirred at ambient temperature and under nitrogen atmosphere for 2 h. The solvent and volatiles were removed under reduced pressure, and the residue was purified by flash column chromatography (eluting with DCM/MeOH, 20:1) to afford 8 (210 mg, 90% yield) as light yellow liquid; 1H-NMR (300 MHz, DMSO-d6): δ 7.91 (d, J = 4.5 Hz, 1H), 7.32–7.23 (m, 3H), 7.16 (m, 1H), 3.42 (dd, J = 8.1, 5.2 Hz, 1H), 2.92 (dd, J = 13.4, 5.1 Hz, 1H), 2.77 (s, 2H), 2.65 (dd, J = 13.4, 5.1 Hz, 1H), 2.58 (d, J = 4.7 Hz, 3H); 13C-NMR (75 MHz, DMSO-d6): δ 174.3, 141.4, 132.9, 130.0, 129.3, 128.2, 126.3, 56.1, 40.5, 25.6; HRMS (ESI, positive): calcd. for C10H14ClN2O [M + H]+ 213.0789, found: 213.0789.
(9H-Fluoren-9-yl)methyl((S)-1-(((S)-3-(3-chlorophenyl)-1-(methylamino)-1-oxopropan-2-yl)amino)-3-(4-carbamoylphenyl)-1-oxopropan-2-yl)carbamate (10): HOBt (126 mg, 0.9 mmol) and HBTU (354 mg, 0.9 mmol) were added to a stirred solution of 9 (0.52 mmol) in DMF (6 mL) at rt. After the mixture was cooled to 0 °C, 8 (165 mg, 0.8 mmol) and DIEA (1 mmol) were introduced. After the whole reaction mixture was stirred at rt for 12 h, the solvents and volatiles were removed under reduced pressure. The solid residue was crystallized from dichloromethane to give 10 (350 mg, 51% yield) as white solid. M.p. 238–240 °C; 1H-NMR (300 MHz, DMSO-d6): δ 2.57 (d, J = 4.3 Hz, 3H), 2.68–2.88 (m, 2H), 2.95–3.01 (m, 2H), 4.00–4.31 (m, 4H), 4.44–4.51 (m, 1H), 7.16–7.42 (m, 12H), 7.60 (t, J = 7.57 Hz, 3H), 7.78–7.93 (m, 6H), 8.24 (d, J = 8.2 Hz, 1H); 13C-NMR (75 MHz, DMSO-d6): δ 25.6, 37.4, 37.7, 46.6, 53.9, 56.0, 65.8, 120.2, 125.3, 125.4, 126.5, 127.2, 127.4, 127.7, 128.1, 129.1, 130.0, 132.4, 132.8, 140.3, 140.8, 141.6, 143.8, 143.9, 155.8, 167.9, 171.0, 171.3; HRMS (ESI, positive): calcd. for C35H33ClN4O3Na [M + Na]+ 647.2032, found: 647.2035.
4-((S)-2-Amino-3-(((S)-3-(3-chlorophenyl)-1-(methylamino)-1-oxopropan-2-yl)amino)-3-oxopropyl)benzamide (11): Piperidine (2 mL) was added to a stirred solution of 10 (330 mg, 0.5 mmol) dissolved in DMF (4 mL), and the mixture was stirred at rt for 2 h. After the completion of the reaction, the solvent and volatiles were removed under reduced pressure, and the solid residue was crystallized from EtOAc to give 11 as light yellow solid (200 mg, 95% yield). M.p. 237–239 °C; 1H-NMR (300 MHz, DMSO-d6): δ 2.54 (t, J = 10.5 Hz, 3H), 2.79–3.39 (m, 3H), 4.46 (s, 1H), 7.12–7.31 (m, 6H), 7.77 (d, J = 7.8 Hz, 2H), 7.99 (t, J = 9.1 Hz, 2H), 8.20 (d, J = 5.8 Hz, 1H); 13C-NMR (75 MHz, DMSO-d6): δ 25.6, 37.6, 53.3, 56.2, 126.4, 127.4, 128.1, 129.1, 129.2, 129.9, 132.2, 132.7, 140.4, 142.2, 167.8, 171.0, 174.0; HRMS (ESI, positive): calcd. for C20H23ClN4O3Na [M + Na]+ 245.1351, found: 245.1350.
(2S)-3-(3-Chlorophenyl)-2-((2S)-2-(2-cyclohexyl-2-phenylacetamido)-3-phenylpropan-amido)-N-methyl-propanamide (13): HOBt (114 mg, 0.8 mmol) and HBTU (320 mg, 0.8 mmol) were added to a stirred solution of 12 (0.8 mmol) in DMF (4 mL) at rt. After the mixture was cooled to 0 °C, 11 (170 mg, 0.4 mmol) was added, followed by addition of DIEA (1.2 mmol). The reaction mixture was stirred at ambient temperature for 12 h, the solvent and volatiles were evaporated under reduced pressure, and then the solid residue was crystallized from EtOAc to generate 13 (160 mg, 63% yield) as white solid. M.p. 243–245 °C; 1H-NMR (300 MHz, DMSO-d6): δ 0.56–0.64 (m, 3H), 0.81–0.93 (m, 6H), 1.06–1.59 (m, 3H), 2.55 (d, J = 4.5 Hz, 3H), 2.68–3.06 (m, 3H), 3.19 (d, J = 10.8 Hz, 1H), 4.39–4.44 (m, 2H), 6.96 (d, J = 8.4 Hz, 1H), 7.18–7.33 (m, 7H), 7.52 (d, J = 8.1 Hz, 1H), 7.77–7.94 (m, 3H), 8.21–8.25 (m, 3H); 13C-NMR (75 MHz, DMSO-d6): δ 24.9, 25.2, 25.9, 26.0, 30.8, 37.7, 39.1, 42.7, 54.1, 57.3, 121.8, 126.8, 127.5, 128.6, 128.7, 129.3, 129.7, 130.6, 132.2, 132.5, 132.6, 140.8, 141.3, 141.7, 168.1, 171.1, 171.2, 173.0; HRMS (ESI, positive): calcd. for C34H40ClN4O4 [M + H]+ 603.2733, found: 603.2730.




{kind=link}
{kind=link}
{kind=link}
{kind=link}


