Synthesis of the Sex Pheromone of the Tea Tussock Moth Based on a Resource Chemistry Strategy
by

Hong-Li Zhang
1,†, 
Zhi-Feng Sun
2,3,†,
Lu-Nan Zhou
3,
Lu Liu
3,
Tao Zhang
3,* and
Zhen-Ting Du
3,4,* 1
State Key Laboratory of Crop Stress Biology in Arid Areas, Northwest A&F University, Yangling 712100, China
2
Shaanxi Key Laboratory for Catalysis, College of Chemical and Environment Science, Shaanxi University of Technology, Hanzhong 723001, China
3
College of Chemistry and Pharmacy, Northwest A&F University, Yangling 712100, China
4
Key Laboratory of Botanical Pesticide R&D in Shaanxi Province, Yangling 712100, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2018, 23(6), 1347; https://doi.org/10.3390/molecules23061347
Submission received: 16 May 2018
/
Revised: 30 May 2018
/
Accepted: 3 June 2018
/
Published: 4 June 2018
(This article belongs to the Special Issue Synthesis, Study and Utilization of Natural Products)
Abstract
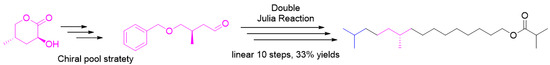
:Synthesis of the sex pheromone of the tea tussock moth in 33% overall yield over 10 steps was achieved. Moreover, the chiral pool concept was applied in the asymmetric synthesis. The synthesis used a chemical available on a large-scale from recycling of wastewater from the steroid industry. The carbon skeleton was constructed using the C4+C5+C8 strategy. Based on this strategy, the original chiral center was totally retained.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Today, safer agricultural produce demands that farming procedures be performed in a greener way. In this context, integrated pest management (IPM) is proposed, which is a broad-based approach that integrates practices for the economic control of pests. Among them, using low-dose pheromones to control pest has been accepted by more and more agricutural providers. In East Asia, a kind of pest called tea tussock moth (Euproctis pseudoconspersa) causes huge destruction in tea orchids. The bit leaves may fall off and decrease the gross product. If this kind of pest was controlled by pheromone, the tea leaves could be provided better than before. The main component of the moth’s sex pheromone was first reported by Wakamura as 10,14-dimethyl-l-pentadecyl isobutyrate [1,2]. Then, a field attraction attempt was tested using crude extract from females and synthetic pheromones [3], and the result revealed that both S and R configurations have similar luring activities. As continuation of our interest in green agrochemicals [4,5,6,7], a synthesis of insect pheromone 1 based on a resource chemistry strategy was envisioned. As a matter of fact, there are several synthetic approaches to this compound in the literature. Ichikawa [8] and Zhao [2] synthesized the tussock moth pheromone (R)-1 from (S)-citronellol or its corresponding bromide. Very recently, a synthesis of tussock pheromone based on a protective-group-free strategy [9] has been reported by our group, in which the Evans’ template was adopted to control the chirality.
Some 4000 tons of sapogenin are produced every year by the Chinese steroid industry. If a new H2O2 oxidation procedure were to be applied to pregna-16,20-diol, a large quantity of (5S)-3-hydroxy-5-methyltetrahydro-2H-pyran-2-one (2) could become available, and 1000 tons of chiral material could be recycled. Tian et al. developed a toolkit of chiral building blocks from this resource chemical [10] and used it to achieve several syntheses of natural compounds [6,11,12,13]. He coined the term “resource chemical”, which refers to large-scale and useful substances. Herein, we discuss the synthesis of the pheromone of the tea tussock moth based on this resource chemical.
2. Results and Discussions
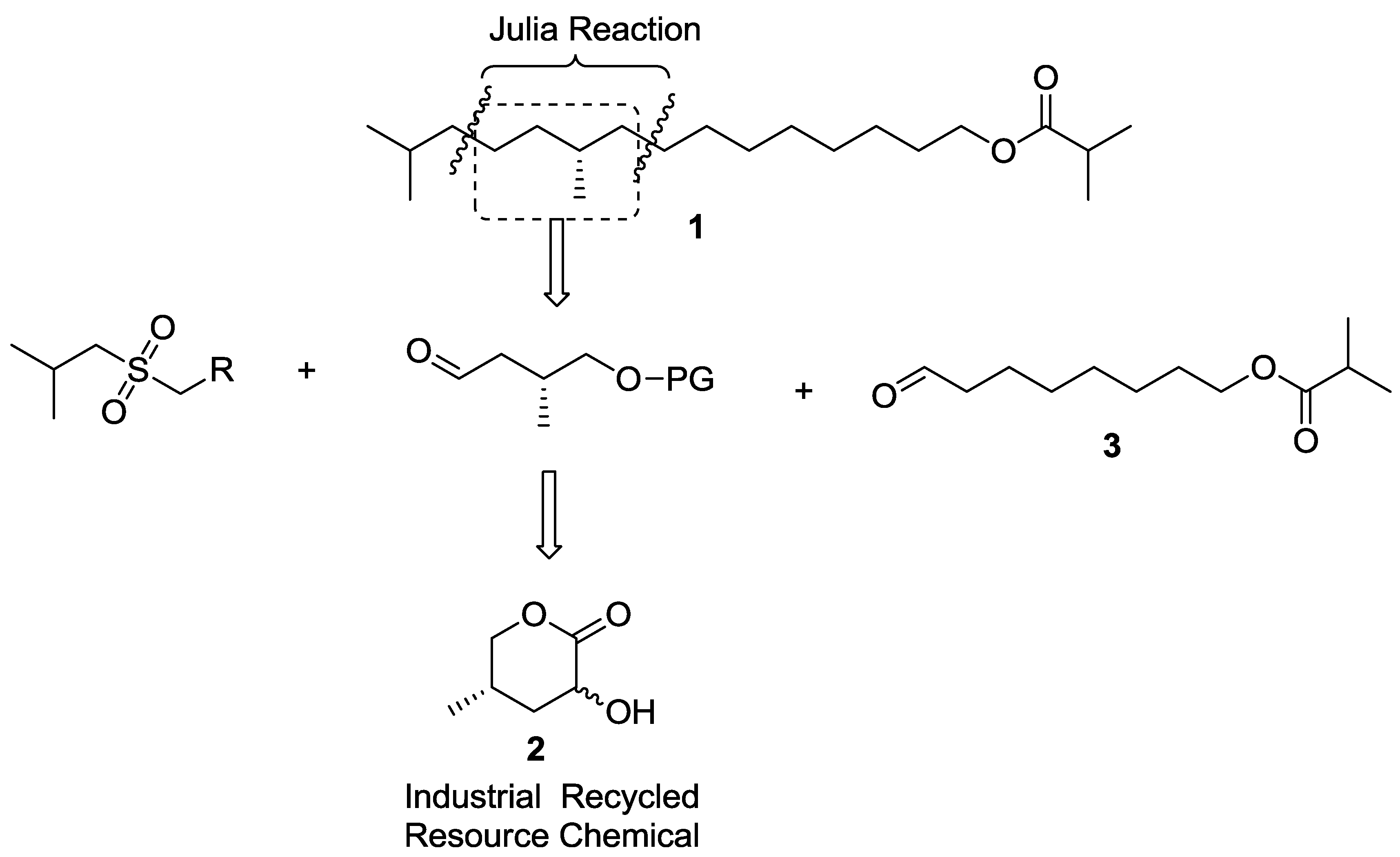
As Scheme 1 shows, the retrosynthesis was based on two Julia coupling reactions, namely a C4+C5+C8 strategy. The left hand C4 synthon was easily obtained, and the right C8 subunit was a protected aldehyde 3. The middle unit can be derived from a chiral methyl aldehyde, which is a large-scale resource chemical derivative.
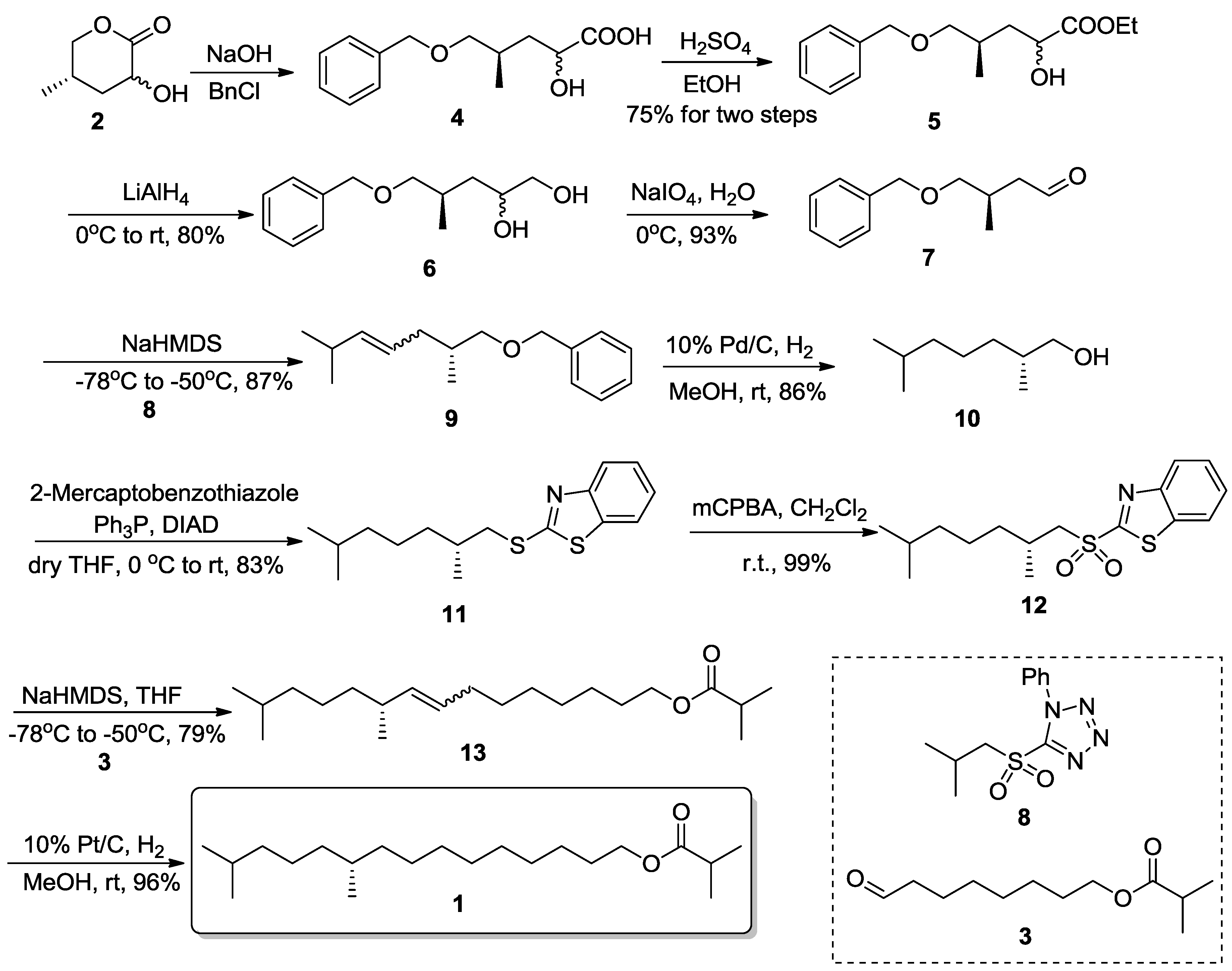
As Scheme 2 shows, the synthesis commenced from chiral lactone 2 which was ring-opened [14,15] under basic conditions and subjected to selective benzylation to give compound 4. In practice it proved laborious to purify the very polar hydroxyacid, so it was used directly without further purification. To reduce the carboxylic acid effectively, the ethyl ester 5 could be obtained in 75% yield for two steps under acid-catalyzed esterification conditions [11], but according to the 1H-NMR and 13C-NMR spectra (supplementary material), it could be observed that the hydroxyl group in the 2-position was partially epimerized. We suspect this resulted from the basic conditions used. We could observe some small peaks and high peaks with a height ratio of c.a. 1:3. Fortunately, the chirality of the desired methyl group didn’t matter. The subsequent reduction by LiAlH4 gave the vicinal diol 6 in 80% yield, which is a good substrate for an aldehyde. Therefore, the chiral aldehyde (R)-4-(benzyloxy)-3-methylbutanal 7 could be produced in 93% yield after a conventional oxidative diol cleavage using NaIO4 in aqueous solvent. Several preparation methods of compound 7 that can be found in the literature. Aside from Wei’s route [11], the rest require more chemical operations [16], an expensive chiral auxiliary such as the Evans template [17,18,19] or poisonous cyanide [20]. The e.e. was determined by comparison of the optical rotation value [α]D25 + 10.6 (c 0.7, CHCl3); lit. [17] [α]D20 + 10.4 (c 0.7, CHCl3). Next, at −78–−50 °C, a Julia-Kocienski reaction was performed between 5-(isobutylsulfonyl)-1-phenyl-1H-tetrazole (8), which was prepared according to the literature [21] and the chiral aldehyde 7, affording the coupling product 9 in 87% yield (E/Z ratio of 1:1). The subsequent hydrogenation of the C=C double bond and removal of the benzyl protecting group was achieved in one-pot in 86% yield. (R)-2,6-dimethylheptan-1-ol (10) was converted into a Julia sulfone reagent 12 through the Mitsunobu protocol [22], followed by an oxidative reaction (83% and 99% yield, respectively). A Julia-Lythgoe reaction between aldehyde 3 and sulfone 12 gave the corresponding alkene product 13 in 79% yield with an E/Z ratio of 2:1. To prevent the isomerization and racemization of the chiral center, alkene 13 was subjected to Pt/C catalytic hydrogenation [23], giving (S)-1 in 99% yield. All the analytic data for synthetic compound 1 are in accordance with that reported in the literature [8,9].
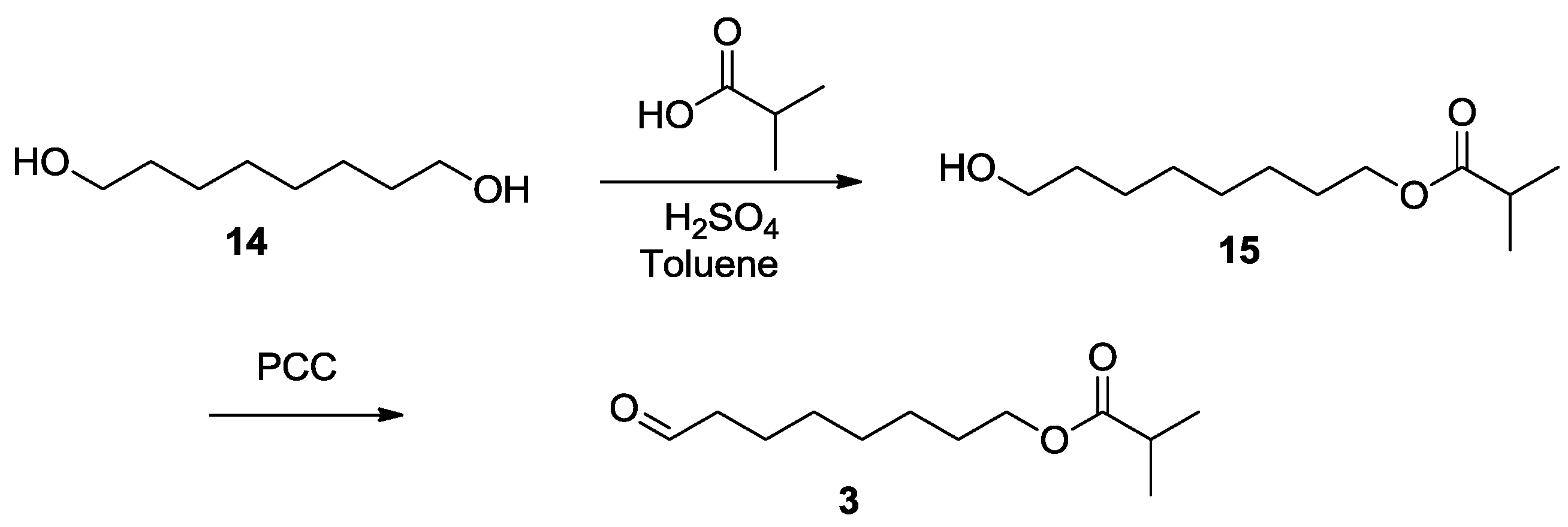
As Scheme 3 shows, similar to the literature [9], octane-1,8-diol was esterified selectively with 70% yield using a stoichiometric amount of isobutyric acid at presence of catalytic sulfuric acid in toluene [24]. Then, a pyridinium chlorochromate (PCC) [25] oxidation was applied, and the aldehyde 3 was produced in 90% yield (Scheme 3).
3. Experimental Section
3.1. General Methods
THF was distilled from sodium/benzophenone and CH2Cl2 was distilled from CaH2 before use. Reactions were monitored by thin-layer chromatography (TLC) on glass plates coated with silica gel with fluorescent indicator. Flash chromatography was performed on silica gel (200–300) with petroleum/EtOAc as the eluent. Optical rotations were measured on a polarimeter with a sodium lamp. HRMS spectra were measured on a LCMS-IT-TOF or LTQ-Orbitrap-XL apparatus. IR spectra were recorded using a Fourier transform infrared spectrometer. NMR spectra were recorded on a AC-500 MHz instrument (Bruker, Madison, WI, USA). Chemical shifts were reported in δ (ppm) and referenced to an internal TMS standard for 1H-NMR and CDCl3 (77.16 ppm) for 13C- NMR (Supplementary material).
3.2. Ethyl (4R)-5-(Benzyloxy)-2-Hydroxy-4-Methylpentanoate (5)
The raw material 2 was obtained from the Pharmaceutical Factory of Shaanxi Academy of Science, Yangling, China as a strong basic, aqueous solution. If the water was removed, a large-scale raw (70–75% purity) brown solid could be obtained. In this paper, this solution was acidified to pH = 1, filtered, and the filtrate was extracted with ethyl acetate to afford crude 2. Compound 2 was purified through column chromatography. Because the straight separation of 2 from proved laborious, the raw lactone 2 was used directly sometimes to save time.
To purified lactone 2 (6.5 g, 50 mmol), toluene (100 mL) and powdered NaOH (8 g, 200 mmol) were added. After the mixture was refluxed for 6 h, at the same time, the produced water was removed through a Dean-Stark trap, and then BnCl (15.8 g, 125 mmol) was added in 3–5 batches, and the reaction mixture was allowed to react for another 12 h. Then, water (100 mL) was added, and the aqueous phase was extracted with Et2O (70 mL × 3). The organic phases were discarded, and the aqueous phase was acidified with concentrated hydrochloric acid to pH = 1. The resulting aqueous phase was extracted with EtOAc (70 mL × 3), and the combined organic layers were dried over MgSO4, filtered, and concentrated to give the crude acid without further purification. The above crude acid was dissolved in EtOH (150 mL), and then H2SO4 (5.00 mL, 98%) was added dropwise. After the mixture was refluxed for 4 h, it was monitored by TLC. Then, the reaction was dispensed into EtOAc (250 mL), and washed with water, saturated NaHCO3, and brine. The solvent was evaporated, and the residue was purified through column chromatography to give the ester 5 with partly epimerized hydroxyl group 8.9 g as a yellowish oil. IR (film) νmax 3450, 2958, 2851, 2359, 1737, 1458, 1211, 1092, 740, 701 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.33–7.37 (m, 5H), 4.22−4.35 (m, 3H), 3.30−3.40 (m, 1H), 3.31−3.38 (m, 2H), 2.04−2.14 (m, 1H), 1.62−1.77 (m, 2H), 1.29 (t, J = 7.1 Hz, 2H), 1.05 (d, J = 6.8 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 175.4, 138.2, 130.2, 128.4, 127.6, 75.8, 73.1, 69.3, 61.5 39.3, 30.6, 16.8, 14.2.
3.3. (R)-4-(Benzyloxy)-3-Methylbutanal (7)
To a suspension of LAH (1.11 g, 30.0 mmol) in THF (45 mL), a solution of ester 5 (5.2 g, 19.5 mmol) in THF (10 mL) was added dropwise in an ice bath. The reaction mixture was warmed to r.t after being stirred for 3 h, followed by another stirring for 10 h. The resulting mixture was carefully quenched with water (1 mL) and aqueous NaOH (5%, 2 mL), and the precipitated salt was filtered. The cake was washed with THF (30 mL), combined with the filtrate, and concentrated to give the crude diol without further purification. From the crude NMR, we could see there were two diastereomers in ca 1:3. At 0 °C, to diol 6 (1.2g 5 mmol) in water (20 mL), sulfuric acid was added to pH = 6. Then NaIO4 (1.6 g, 7.5 mmol) was added portionwise and the resulting mixture was stirred for 1.5 h. After the reaction was complete, the mixture was extracted with Et2O (30 mL × 3), and the combined organic layers were washed with brine. The extract was dried, filtrated, and concentrated to give the aldehyde 7 as a colorless oil with 87% yield in two steps. [α]D25 +10.6 (c 0.7, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 9.80 (t, 1H, J = 3.2Hz), 7.32–7.41 (m, 5H), 4.53 (s, 2H), 3.42 (dd, J = 9.1, 5.2 Hz, 1H), 3.26 (dd, J = 9.1, 7.7 Hz, 1H), 2.56 (ddd, J = 16.2, 6.3, 2.3 Hz, 1H), 2.30–2.44 (m, 1H), 2.28 (ddd, J = 16.2, 7.0, 2.1 Hz, 1H), 0.99 (d, J = 6.8 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 202.4, 138.3, 128.4, 127.6, 127.6, 74.9, 73.1, 48.5, 29.2, 17.1.
3.4. (2R)-(((2,6-Dimethylhept-4-en-1-yl)oxy)Methyl)Benzene (9)
To a stirred solution of compound 8 (2.4 g, 8.9 mmol) in THF (30.0 mL) at –78 °C under argon, NaHMDS (2.0 M in THF, 5.60 mL, 11.3 mmol) was added dropwise. After 30 min, a solution of compound 7 (1.4 g, 7.26mmol) in THF (10.0 mL) was added dropwise. Upon stirring at −78 °C for 2 h, the reaction was warmed to −50 °C overnight. After the reaction was quenched with saturated aqueous NH4Cl at −50 °C, it was extracted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4, concentrated, and purified by flash chromatography on silica gel (hexanes: ethyl acetate = 20:1) to give E/Z ca. 1:1 mixture of compound 9 as a pale-yellow oil (1.9 g, 87%). [α]D25 + 1.32 (c 2.5, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 7.34–7.25 (m, 5H), 5.37–5.22 (m, 2H), 4.5 (d, J = 2.5 Hz, 2H), 3.36–3.23 (m, 2H), 2.6–2.11 (m, 2H), 1.96–1.79 (m, 2H), 0.96–0.9 (m, 9H); 13C-NMR (125 MHz, CDCl3) δ 139.4, 138.7, 128.3, 127.6, 127.6, 127.4, 125.2, 124.8, 75.5, 75.4, 73.0, 73.0, 36.6, 34.0, 33.8, 31.3, 31.1, 26.4, 23.1, 23.1, 22.7, 22.7, 17.0, 16.8.
3.5. (R)-2,6-dimethylheptan-1-ol (10)
To a solution of ether 9 (1.6 g, 6.89 mmol) in methanol (50 mL), Pd/C (10%, 0.3 g) was added and the atmosphere was exchanged with H2. The reaction mixture was stirred for 24 h and monitored by TLC. After completion, the catalyst was filtered through a Celite pad, and the filtrate was evaporated, giving compound 10 after flash column purification as a pale-yellow oil (0.85 g, 86%). [α]D25 + 4.68 (c 2.4, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 3.53-3.49 (m, 1H), 3.44-3.39 (m, 1H), 1.61 (s, 1H), 1.56–1.51 (m, 1H), 1.36–1.08 (m, 7H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ 68.4, 35.8, 33.4, 27.9, 24.7, 22.7, 22.6, 16.6.
3.6. (R)-2-((2,6-Dimethylheptyl)thio)Benzo[d]Thiazole (11)
Under argon, to a flask with (R)-2,6-dimethylheptan-1-ol 10 (0.7 g, 4.85mmol), THF (25 mL), benzo[d]thiazole-2-thiol (1.0 g, 6.0 mmol), and TPP (1.5 g, 6.0 mmol) were added. To this mixture, DIAD (1.1 mL, 6.0mmol) was added dropwise at 0 °C. After the reaction was completed, the volatile was evaporated, and the residue was purified through column chromatography to give compound 11 (1.2 g, 83% yield). [α]D25 –3.63 (c 3.1, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 7.9 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8 Hz, 1H), 7.45 (t, J = 7.3 Hz, 1H), 7.33 (t, J = 7.3 Hz, 1H), 3.48–3.22 (m, 2H), 2.01–1.95 (m, 1H), 1.6–1.3 (m, 5H), 1.24–1.2 (m, 2H), 1.12 (d, J = 6.7Hz, 3H), 0.92 (d, J = 6.6 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ 167.79, 153.38, 135.19, 126.0, 124.09, 121.45, 120.91, 40.78, 39.06, 36.35, 33.3, 27.95, 24.69, 22.71, 22.6, 19.4. HRMS (ESI) m/z calcd. for C16H24NO2S2+ (M + H)+: 326.1248, found 326.1245.
3.7. (R)-2-((2,6-Dimethylheptyl)Sulfonyl)Benzo[d]Thiazole (12)
To a solution of compound 11 (1.0 g, 3.61mmol) in CH2Cl2 (35 mL), mCPBA (4.7 g, 70%, 5.6 eq) was added, and the reaction mixture was stirred for 12 h at r.t. Then, saturated Na2S2O3 was added to quench the reaction, and the reaction was neutralized using saturated Na2CO3 and extracted with CH2Cl2. The combined organic layers were washed with brine, and the extract was dried over MgSO4, filtrated, and concentrated to give compound 12 as a colorless oil with 99% yield. [α]D25 − 2.99 (c 2.6, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 8.21 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.66–7.58 (m, 2H), 3.56 (dd, J = 14.3, 4.7 Hz, 1H), 3.35 (dd, J = 14.3, 8.0 Hz, 1H), 2.31–2.25 (m, 1H), 1.49–1.43 (m, 2H), 1.34–1.25 (m, 3H), 1.14 (d, J = 6.7 Hz, 3H), 1.11–1.06 (m, 2H), 0.82(d, J = 6.5, 3H), 0.81(d, J = 6.5 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 166.83, 152.76, 136.79, 127.98, 127.64, 125.46, 122.37, 60.83, 38.7, 36.89, 28.57, 27.84, 24.08, 22.57, 22.49, 19.91. HRMS (ESI) m/z calcd. for C16H24NO2S2+ (M + H)+: 326.1248, found 326.1245.
3.8. 8-Hydroxyoctyl Isobutyrate 15
To a solution of octane-1,8-diol (7.3 g, 50 mmol) and isobutyric acid (4.2 g, 48 mmol) in toluene (100 mL), four drops of concentrated sulfuric acid were added, and the reaction mixture was warmed to 75 °C for 20 h. The reaction mixture was diluted using ethyl acetate (200 mL) and washed with water, NaHCO3, and brine, then dried over MgSO4. The solvent was evaporated, and the residue was purified through column chromatography to give compound 15 (8.8 g, 85%) as a colorless oil. 1H- NMR (500 MHz, CDCl3) δ 4.04 (t, J = 6.7 Hz, 2H), 3.62 (t, J = 6.6 Hz, 2H), 2.55–2.49 (m, 1H), 2.03 (s, 1H), 1.62–1.52 (m, 4H), 1.32 (s, 8H), 1.15 (d, J = 7.0 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ 177.26, 64.31, 62.93, 34.02, 32.70, 29.24, 29.15, 28.59, 25.79, 25.62, 18.97.
3.9. 8-Oxo-Octyl Isobutyrate (3)
To a solution of 8-hydroxyoctyl isobutyrate 15 (2.8 g, 13 mmol) in CH2Cl2 (30 mL), silica gel (200 mesh, 2 g) and PCC (4.2 g, 19.5 mmol) were added. The above reaction mixture was stirred overnight, followed by the addition of ether (50 mL). Then, the solution of crude aldehyde was decanted and purified through flash column chromatography to give 8-oxooctyl isobutyrate 3 (2.6 g, 94%) as a colorless oil. 1H-NMR (500 MHz, CDCl3) δ 9.76 (t, J = 1.8 Hz, 1H), 4.05 (t, J = 6.7 Hz, 2H), 2.56–2.50 (m, 1H), 2.42 (td, J = 1.7, 7.3 Hz, 2H), 1.64–1.61 (m, 4H), 1.34 (s, 6H), 1.15 (d, J = 7.0 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ 202.69, 177.22, 64.22, 43.83, 34.02, 29.0, 28.95, 28.55, 25.69, 21.94, 18.99.
3.10. (R)-10,14-Dimethylpentadec-8-en-1-yl Isobutyrate (13)
Under argon, NaHMDS (1.6 mL, 2.0 M in THF, 3.2 mmol) was added dropwise to a solution of compound 12 (0.8 g, 2.5 mmol) in anhydrous THF (25 mL) at –78 °C. After half an hour, 8-oxooctyl isobutyrate 3 (0.7 g, 3.3 mmol) in THF (10 mL) was added. The reaction mixture was stirred under the same temperature for 3 h, followed by stirring for another 12 h at –50 °C. After the reaction was quenched with saturated aqueous NH4Cl at –50 °C, it was extracted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4, concentrated, and purified by flash chromatography on silica gel (hexanes: ethyl acetate = 30:1) to give compound 13 as a pale-yellow oil (0.6 g, 79%). [α]D25 + 3.06 (c 2.5, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 7.33–7.25 (m, 5H), 5.36–5.08 (m, 2H), 4.49–4.44 (m, 2H), 4.06–4.03 (m, 2H), 3.49–3.39 (m, 2H), 2.65–2.5 (m, 2H), 2.07–1.93 (m, 2H), 1.7–1.43 (m, 4H), 1.3 (s, 8H), 1.16 (d, J = 7.0 Hz, 6H), 0.98–0.95 (m, 3H); 13C-NMR (125 MHz, CDCl3) δ 177.25, 138.76, 138.73, 135.53, 129.05, 128.34, 127.66, 127.61, 127.47, 72.99, 68.82, 64.4, 37.28, 34.08, 29.85, 29.25, 29.19, 28.68, 28.61, 27.43, 25.93, 25.9, 21.51, 19.05.
3.11. (S)-10,14-Dimethylpentadecyl Isobutyrate (1)
To a solution of compound 13 (0.3 g, 9.3 mmol) in ethanol (15 mL), Pt/C (10%, 6 mg) was added and the atmosphere was exchanged with H2. The reaction mixture was then stirred for 24 h and monitored by TLC. After completion, the catalyst was filtered through a Celite pad, and the filtrate was evaporated. The residue was purified by flash chromatography on silica gel (PE/EA = 30:1) to give compound 1 (0.28 g, 93%) as a colorless oil: [α]D25 − 0.28 (c 2.4, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 4.05 (t, J = 6.8 Hz, 2H), 2.56–2.5 (m, 1H), 1.65–1.59 (m, 2H), 1.55–1.5 (m, 1H), 1.36–1.19 (m, 17H), 1.16 (d, J = 7.1 Hz, 6H), 1.15–1.04 (m, 4H), 0.85 (dd, J = 6.7, 13.2 Hz, 9H); 13C-NMR (500 MHz, CDCl3) δ 177.26, 64.42, 39.4, 37.34, 37.12, 34.08, 32.79, 30.0, 29.62, 29.55, 29.27, 28.68, 28.00, 27.09, 25.93, 24.82, 22.73, 22.64, 19.73, 19.03. HRMS (ESI) m/z calcd. for C21H43O2+ (M + H)+: 327.32576, found 327.32599.
4. Conclusions
Based on a chiral pool strategy, an efficient synthesis of (S)-10,14-dimethylpentadecyl isobutyrate, the sex pheromone of the tea tussock moth, was achieved. The longest linear synthetic step was 10 steps, and the overall yield was 33%. The key step was accomplished by a double Julia coupling. This synthesis will be helpful in research involving the differences between the biological effects of R-1 and S-1. The scale-up and evaluation of the biological activities of the sex pheromone is currently under investigation.
Supplementary Materials
Supplementary Materials are available online.
Author Contributions
T.Z. and Z.-T.D. conceived and designed the experiments; Z.-F.S. and L.-N.Z. performed the experiments; H.-L.Z. and L.L. analyzed the data. Z.-T.D. wrote the paper.
Acknowledgments
Partial financial support from the National Natural Science Foundation of China (31301712, 21502151,) is greatly appreciated. Zhen-Ting Du would like to thank Opening Funds of Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences for the partial financial support for funding this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wakamura, S.; Yasuda, T.; Ichikawa, A.; Fukumoto, T.; Mochizuki, F. Sex attractant pheromone of the tea tussock moth (Euproctis pseudoconspersa): Identification and field attraction. Appl. Entomol. Zool. 1994, 29, 403–411. [Google Scholar] [CrossRef]
- Zhao, C.-H.; Millar, J.G.; Pan, K.-H.; Xu, C.-S. Responses of tea tussock moth, Euproctis pseudoconspersa, to its pheromone, (R)-10,14-dimethylpentadecyl isobutyrate, and to the S-enantiomer of its pheromone. J. Chem. Ecol. 1998, 24, 1347–1353. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, F.; Liu, X.; Feng, F.; Wang, L. Evaluation of mass-trapping for control of tea tussock moth Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae) with synthetic sex pheromone in south China. Int. J. Pest Manag. 2005, 51, 291–298. [Google Scholar]
- Yan, Z.; Guan, C.; Yu, Z.; Tian, W. Fluoroalkanosulfonyl fluorides-mediated cyclodehydration of beta-hydroxy sulfonamides and beta-hydroxy thioamides to the corresponding aziridines and thiazolines. Tetrahedron Lett. 2013, 54, 5788–5790. [Google Scholar] [CrossRef]
- Zhang, T.; Feng, J.; Cai, C.; Zhang, X. Synthesis and Field Test of Three Candidates for Soybean Pod Borer’s Sex Pheromone. Nat. Prod. Commun. 2011, 6, 1323–1326. [Google Scholar] [PubMed]
- Wang, Z.; Xu, Q.; Tian, W.; Pan, X. Stereoselective synthesis of (2S,3S,7S)-3,7-dimethylpentadec-2-yl acetate and propionate, the sex pheromones of pine sawflies. Tetrahedron Lett. 2007, 48, 7549–7551. [Google Scholar] [CrossRef]
- Sun, Z.-F.; Zhou, L.-N.; Zhang, T.; Du, Z.-T. Stereoselective synthesis of the Paulownia bagworm sex pheromone. Chin. Chem. Lett. 2017, 28, 558–562. [Google Scholar] [CrossRef]
- Ichikawa, A.; Yasuda, T.; Wakamura, S. Absolute configuration of sex pheromone for tea tussock moth, Euproctis pseudoconspersa (strand) via synthesis of (R)- and (S)-10,14-dimethyl-1-pentadecyl isobutyrates. J. Chem. Ecol. 1995, 21, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-F.; Zhou, L.-N.; Meng, Y.; Zhang, T.; Du, Z.-T.; Zheng, H. Concise asymmetric synthesis of the sex pheromone of the tea tussock moth. Tetrahedron Asymmetry 2017, 28, 1562–1567. [Google Scholar] [CrossRef]
- Tian, W.S.; Wang, Z.K.; Li, B.; Xu, Q.-H. Synthesis of Thioketal and Application in Synthesis of Optical Pure Diahrotica Undecimpunctata Sex Pheromone. Patent No. CN 101050211, 10 May 2007. [Google Scholar]
- Mao, Z.-Y.; Si, C.-M.; Liu, Y.-W.; Dong, H.-Q.; Wei, B.-G.; Lin, G.-Q. Divergent Synthesis of Revised Apratoxin E, 30-epi-Apratoxin E, and 30S/30R-Oxoapratoxin E. J. Org. Chem. 2017, 82, 10830–10845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-K.; Tian, W.-S.; Pan, X.-F. Practical synthesis of the main sex pheromones of pine sawflies (2S,3S,7S)-3,7-dimethylpentadecan-2-ol esters by utilizing (R)-4-methyl-delta-valerolactone obtained from the industrial waste. Acta Chim. Sin. 2007, 65, 705–710. [Google Scholar]
- Wang, Z.-K.; Tian, W.-S.; Pan, X.-F. A concise synthesis of the sex pheromones of Pine sawflies. Chin. J. Org. Chem. 2007, 27, 866–869. [Google Scholar]
- Chung, J.; Kushner, A.M.; Weisman, A.C.; Guan, Z. Direct correlation of single-molecule properties with bulk mechanical performance for the biomimetic design of polymers. Nat. Mater. 2014, 13, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, L.; Yang, J.; Luo, J.; Hong, R. Stereoselective α-Hydroxylation of Amides Using Oppolzer’s Sultam as Chiral Auxiliary. J. Org. Chem. 2016, 81, 3890–3900. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Hansen, H.J. Synthesis of optically active bifunctional isoprenoid building blocks by rhodium(I)-catalyzed asymmetric allylamine to enamine isomerization. Helv. Chim. Acta 1990, 73, 1258–1275. [Google Scholar] [CrossRef]
- Chorley, D.F.; Chen, J.L.-Y.; Furkert, D.P.; Sperry, J.; Brimble, M.A. Total synthesis of danshenspiroketallactone. Synlett 2012, 23, 128–130. [Google Scholar]
- Hansen, D.B.; Starr, M.-L.; Tolstoy, N.; Joullie, M.M. A stereoselective synthesis of (2S,4R)-δ-hydroxyleucine methyl ester: A component of cyclomarin A. Tetrahedron Asymmetry 2005, 16, 3623–3627. [Google Scholar] [CrossRef]
- Jones, T.K.; Reamer, R.A.; Desmond, R.; Mills, S.G. Chemistry of tricarbonyl hemiketals and application of Evans technology to the total synthesis of the immunosuppressant (−)-FK-506. J. Am. Chem. Soc. 1990, 112, 2998–3017. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Wang, Y.; Kim, J.T. Total Synthesis of Microtubule-Stabilizing Agent (−)-Laulimalide. J. Org. Chem. 2001, 66, 8973–8982. [Google Scholar] [CrossRef] [PubMed]
- DiBlasi, C.M.; Macks, D.E.; Tan, D.S. An Acid-Stable tert-Butyldiarylsilyl (TBDAS) Linker for Solid-Phase Organic Synthesis. Org. Lett. 2005, 7, 1777–1780. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- Li, N.-S.; Scharf, L.; Adams, E.J.; Piccirilli, J.A. Highly Stereocontrolled Total Synthesis of β-d-Mannosyl Phosphomycoketide: A Natural Product from Mycobacterium tuberculosis. J. Org. Chem. 2013, 78, 5970–5986. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Yong, J.-P.; Huang, F.-L.; Bai, S.-Z. A facile synthesis of the sex pheromone of Grapholitha molesta. Chem. Nat. Compd. 2012, 48, 103–105. [Google Scholar] [CrossRef]
- Corey, E.J.; Suggs, J.W. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975, 16, 2647–2650. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds from 1 to 15 are available from the authors. |
Scheme 1.
Retrosynthesis of compound 1 based on Julia olefination and a resource-chemical.

Scheme 2.
Synthesis of (S)-1 through a double Julia approach.

Scheme 3.
Synthesis of compound 3.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, H.-L.; Sun, Z.-F.; Zhou, L.-N.; Liu, L.; Zhang, T.; Du, Z.-T. Synthesis of the Sex Pheromone of the Tea Tussock Moth Based on a Resource Chemistry Strategy. Molecules 2018, 23, 1347. https://doi.org/10.3390/molecules23061347
AMA Style
Zhang H-L, Sun Z-F, Zhou L-N, Liu L, Zhang T, Du Z-T. Synthesis of the Sex Pheromone of the Tea Tussock Moth Based on a Resource Chemistry Strategy. Molecules. 2018; 23(6):1347. https://doi.org/10.3390/molecules23061347
Chicago/Turabian StyleZhang, Hong-Li, Zhi-Feng Sun, Lu-Nan Zhou, Lu Liu, Tao Zhang, and Zhen-Ting Du. 2018. "Synthesis of the Sex Pheromone of the Tea Tussock Moth Based on a Resource Chemistry Strategy" Molecules 23, no. 6: 1347. https://doi.org/10.3390/molecules23061347