
Aromaticity as a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids



Abstract
:1. Introduction
2. Results and Discussion
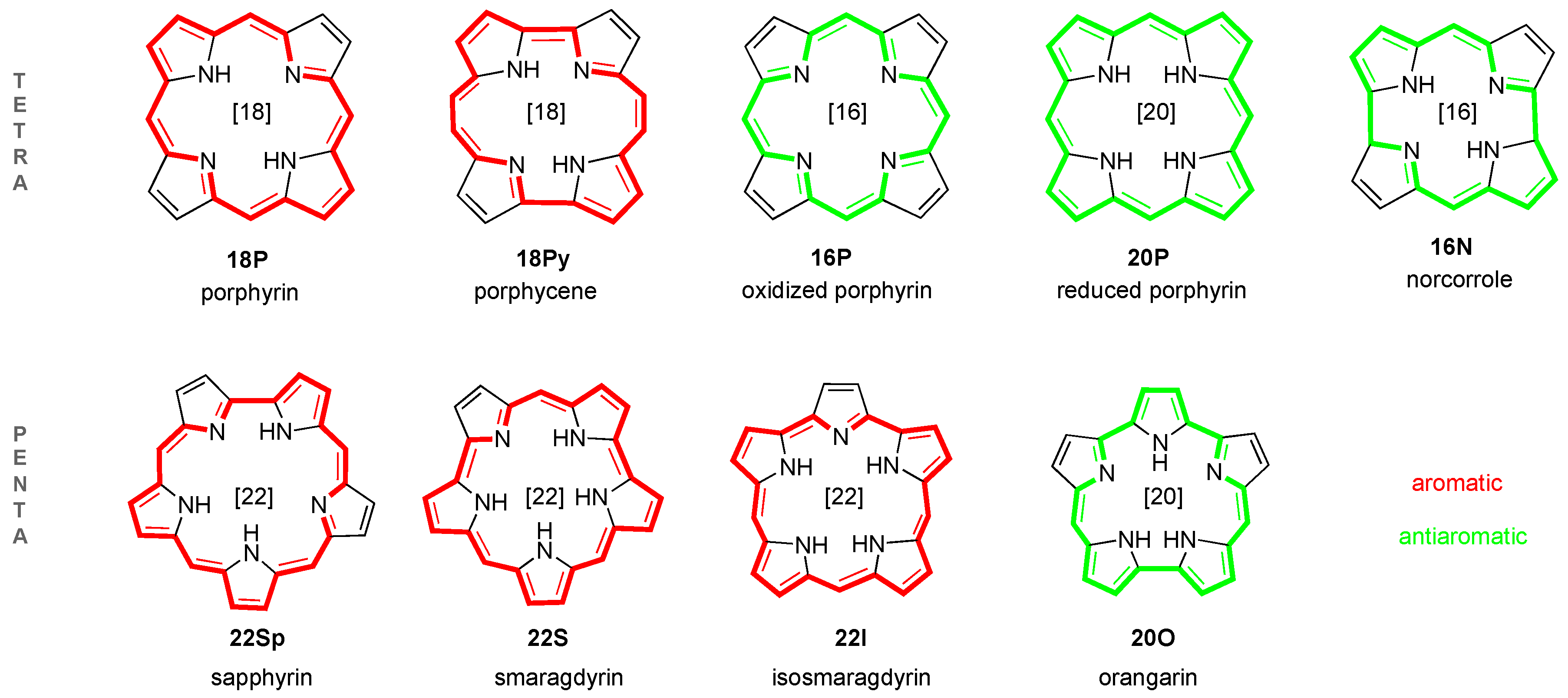
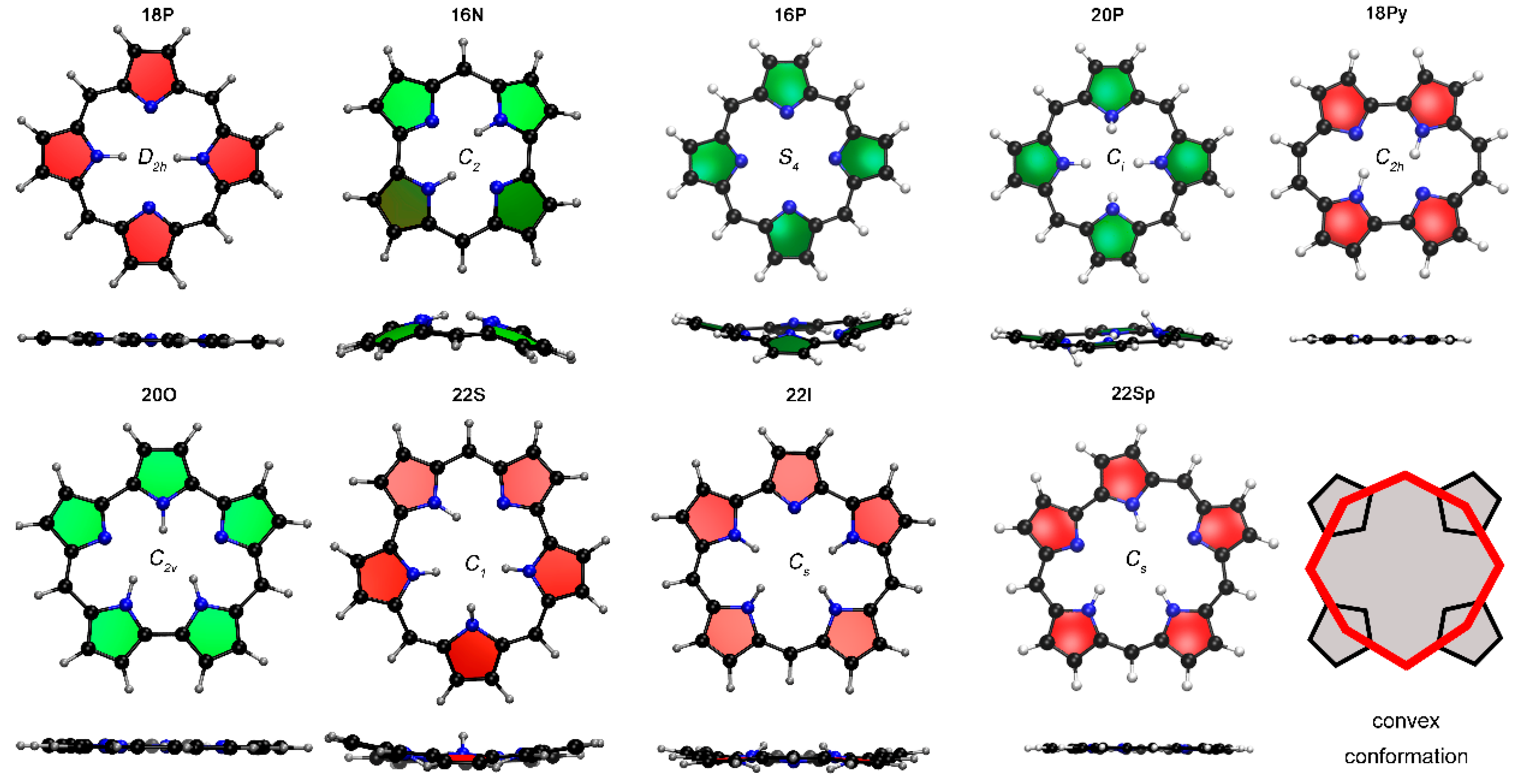
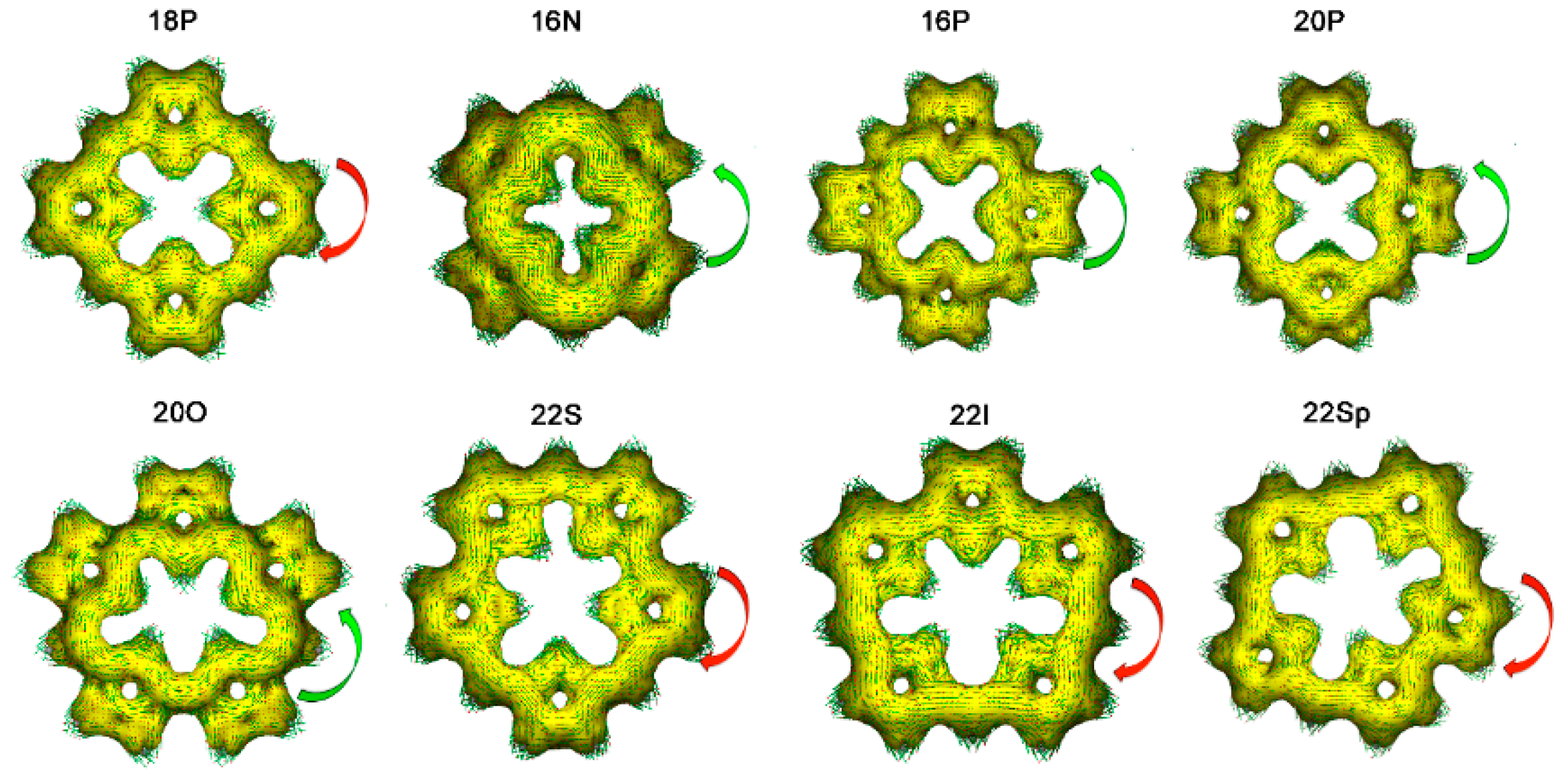
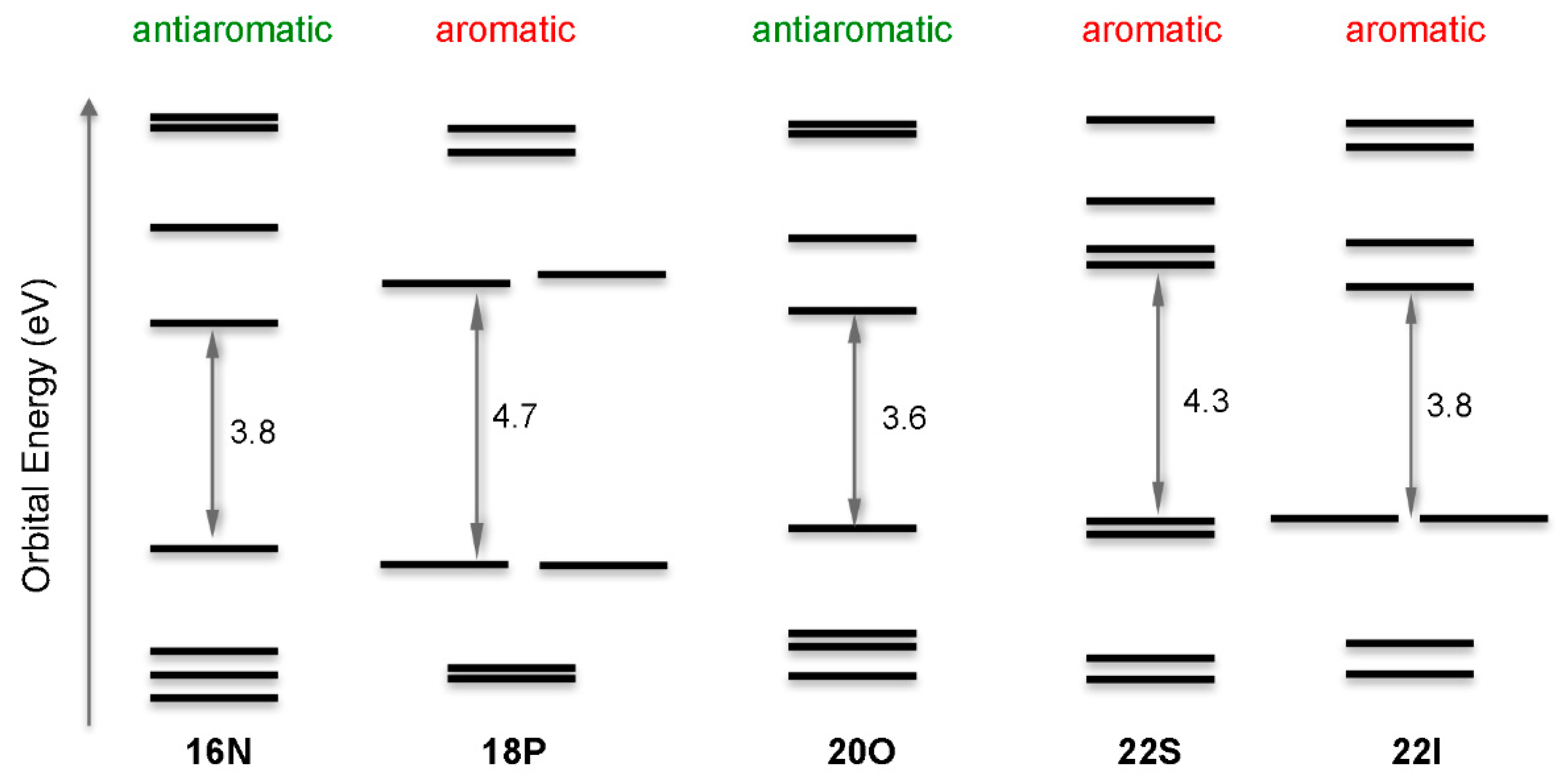
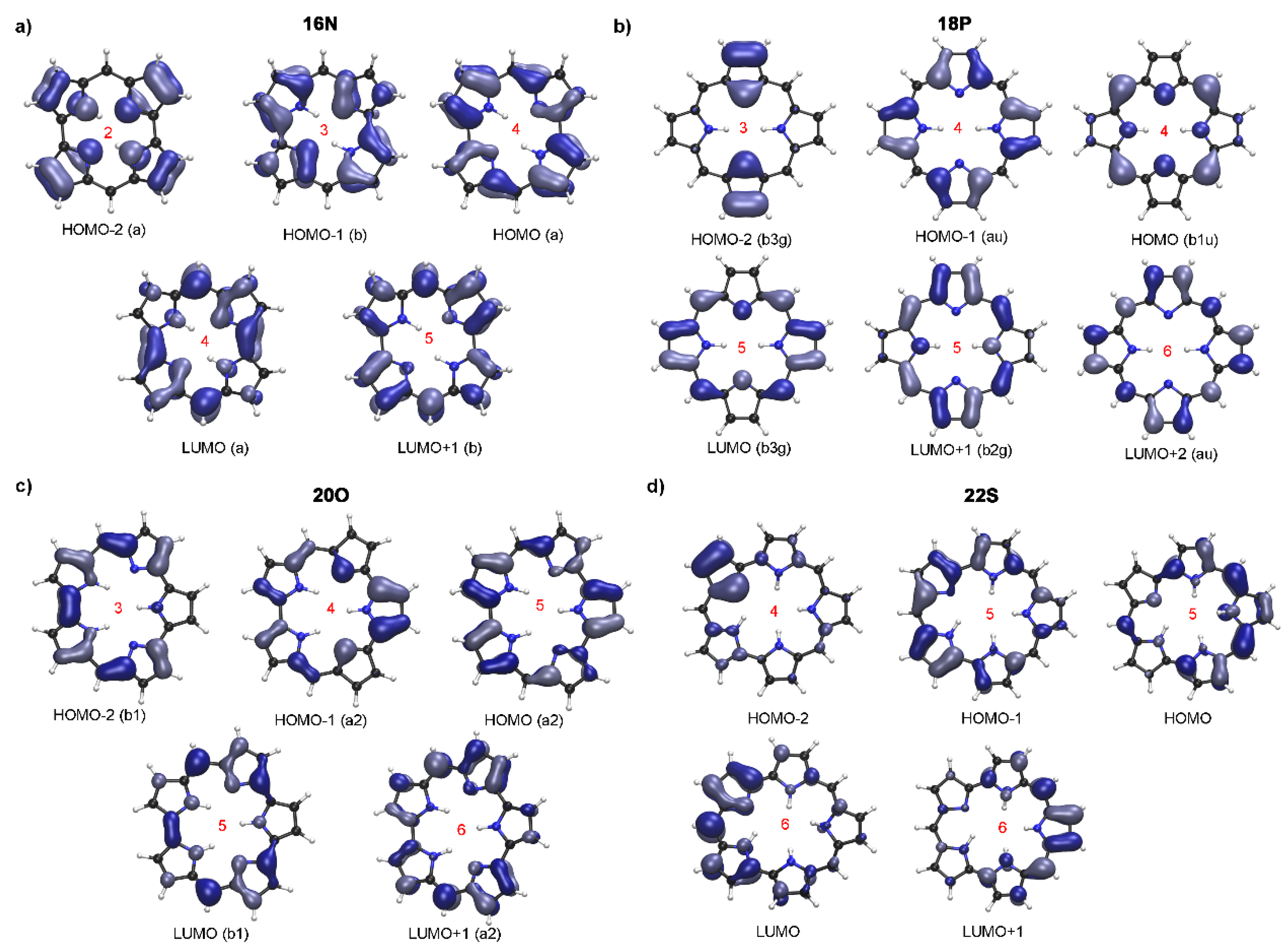
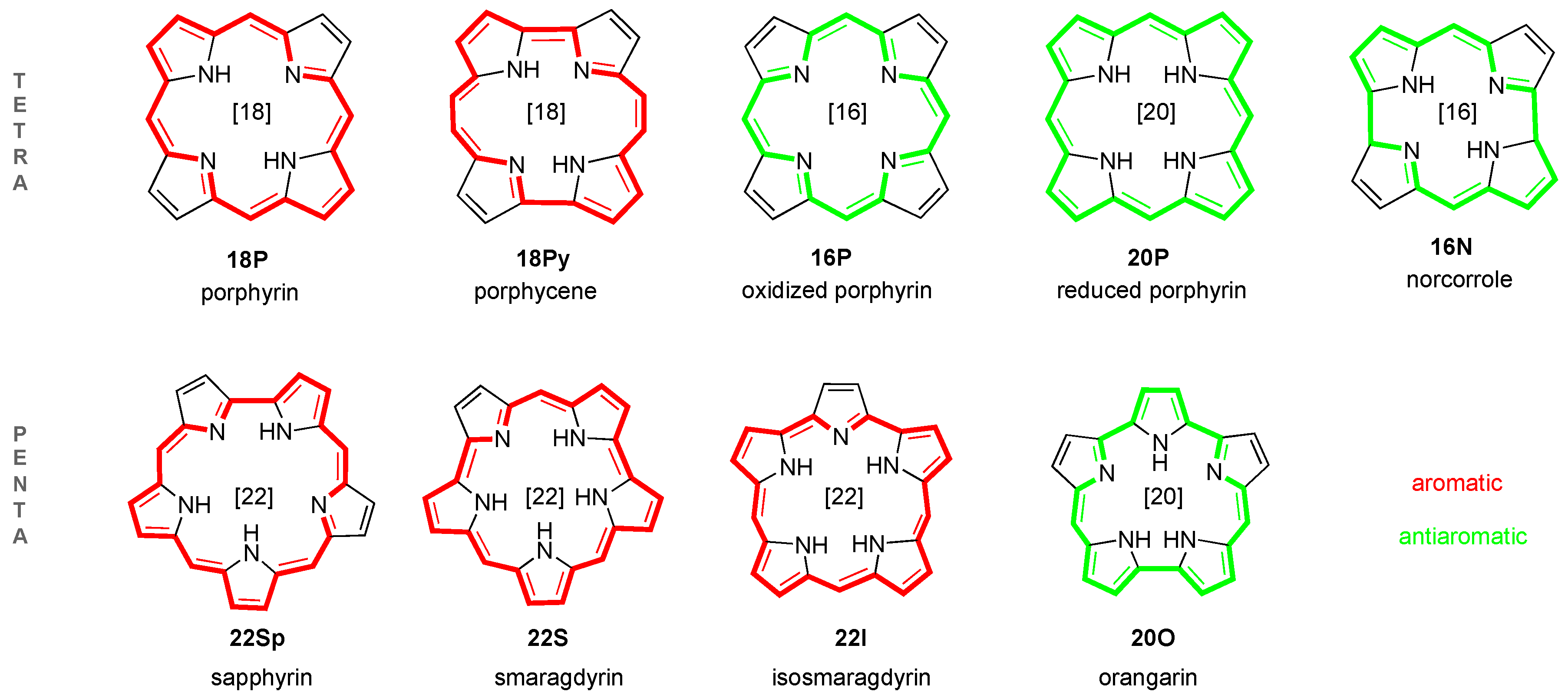
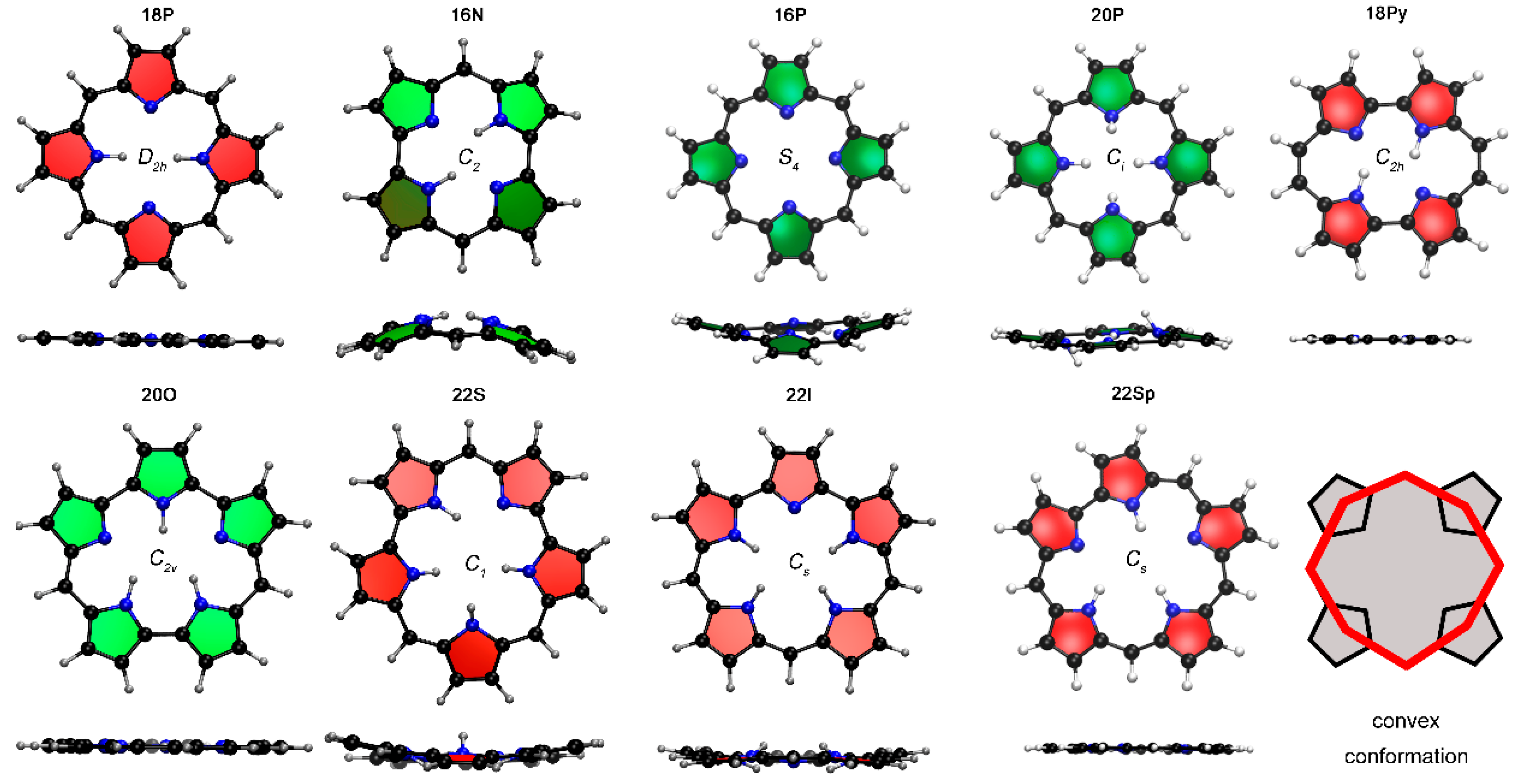
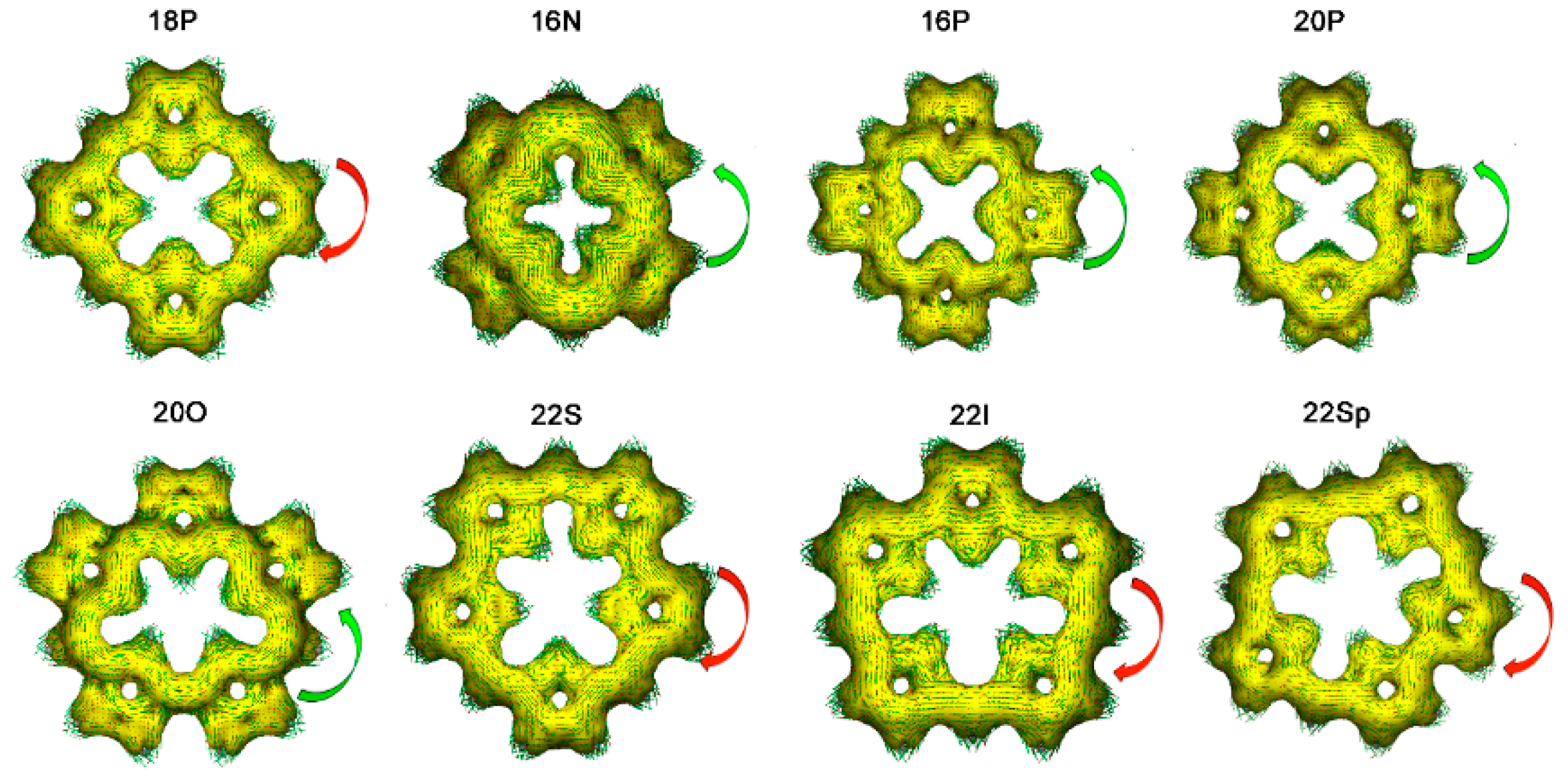
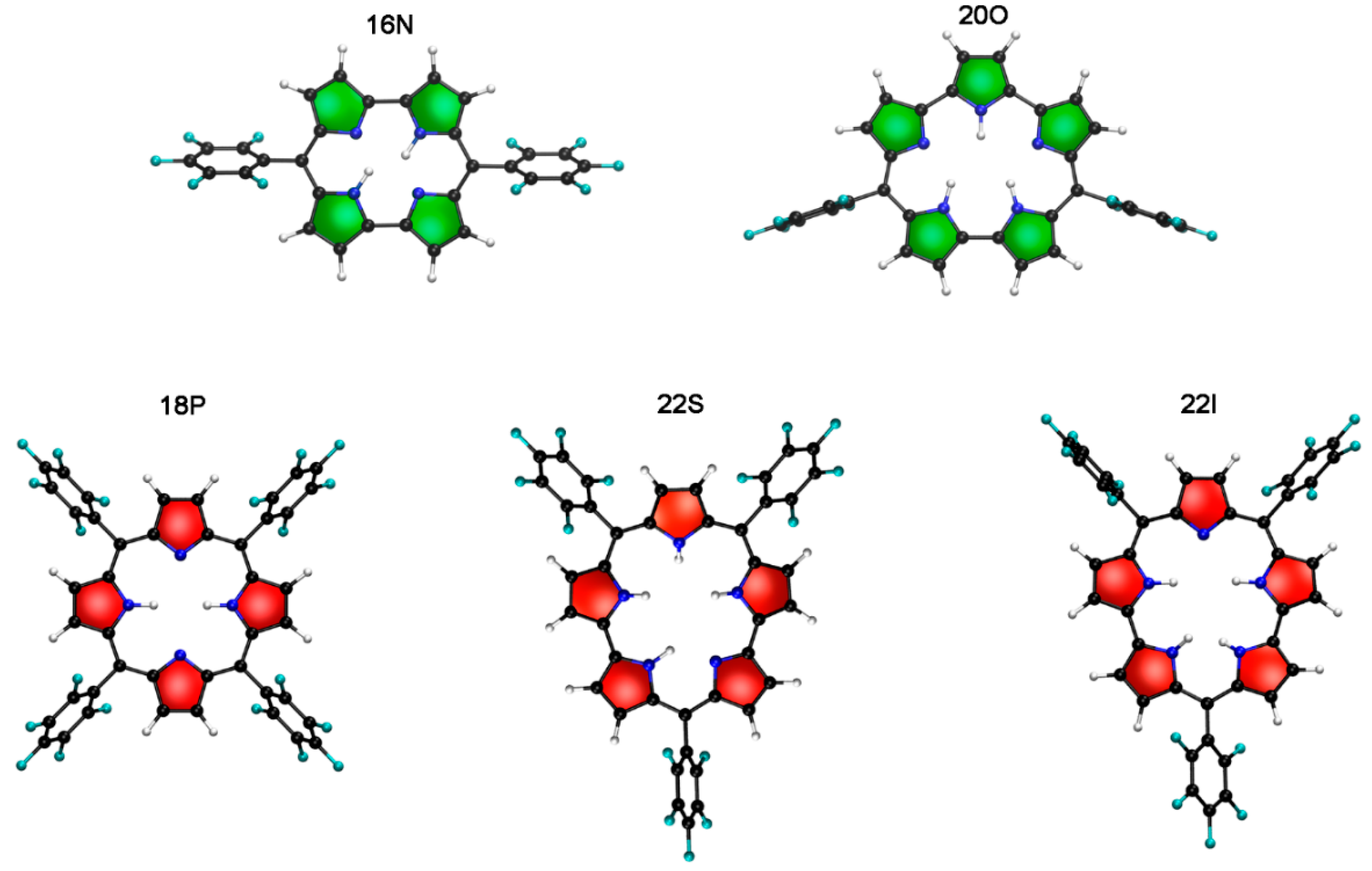
2.1. Structure-Aromaticity Relationship in Hückel Porphyrinoids
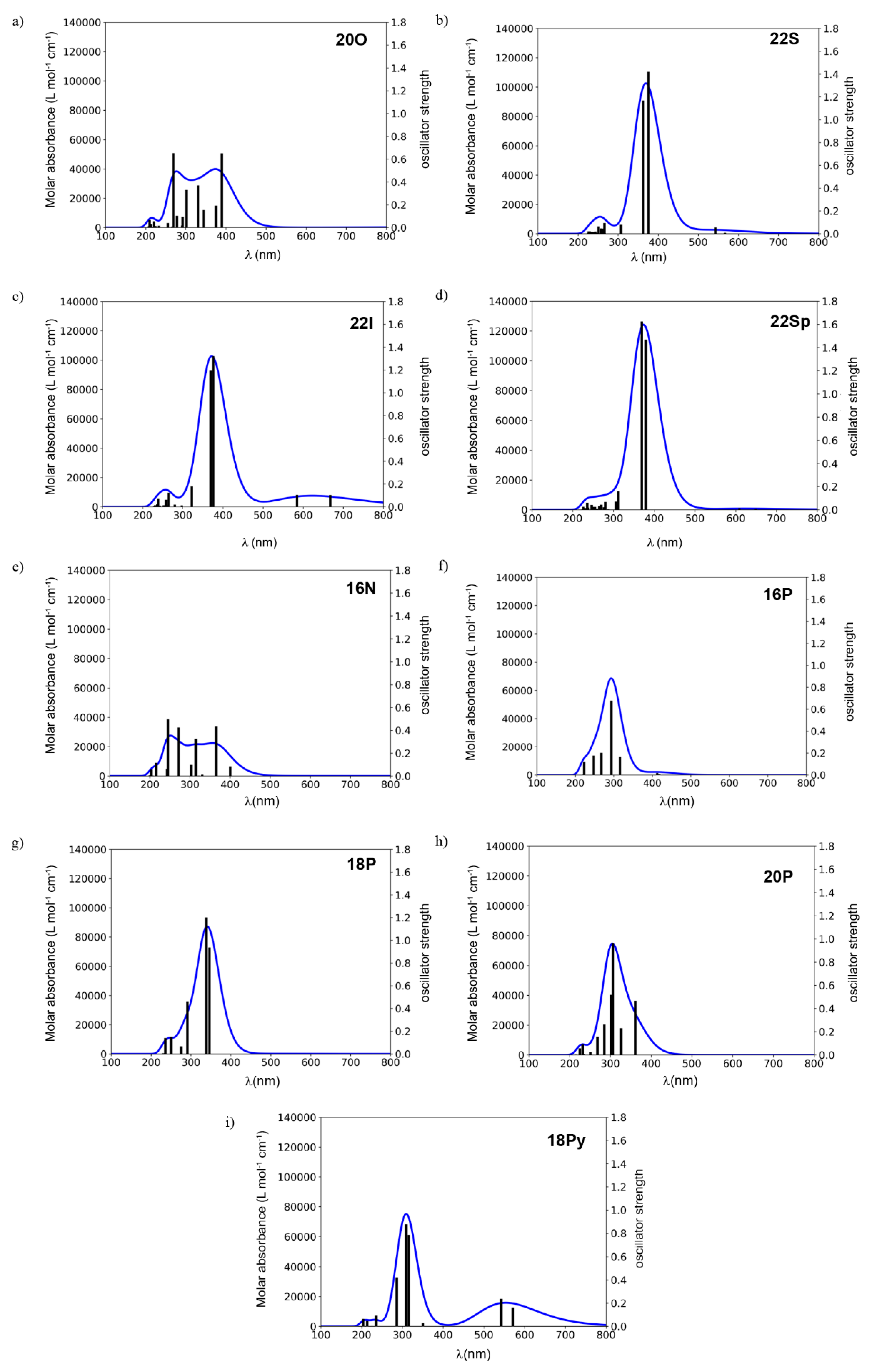
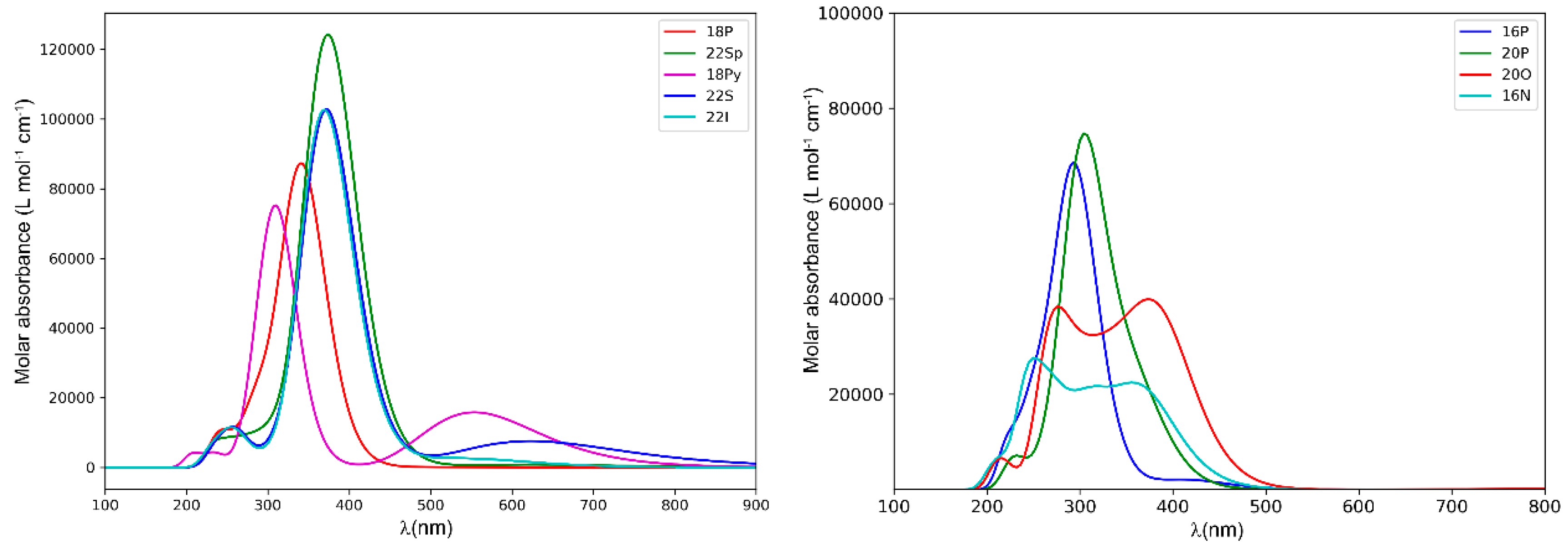
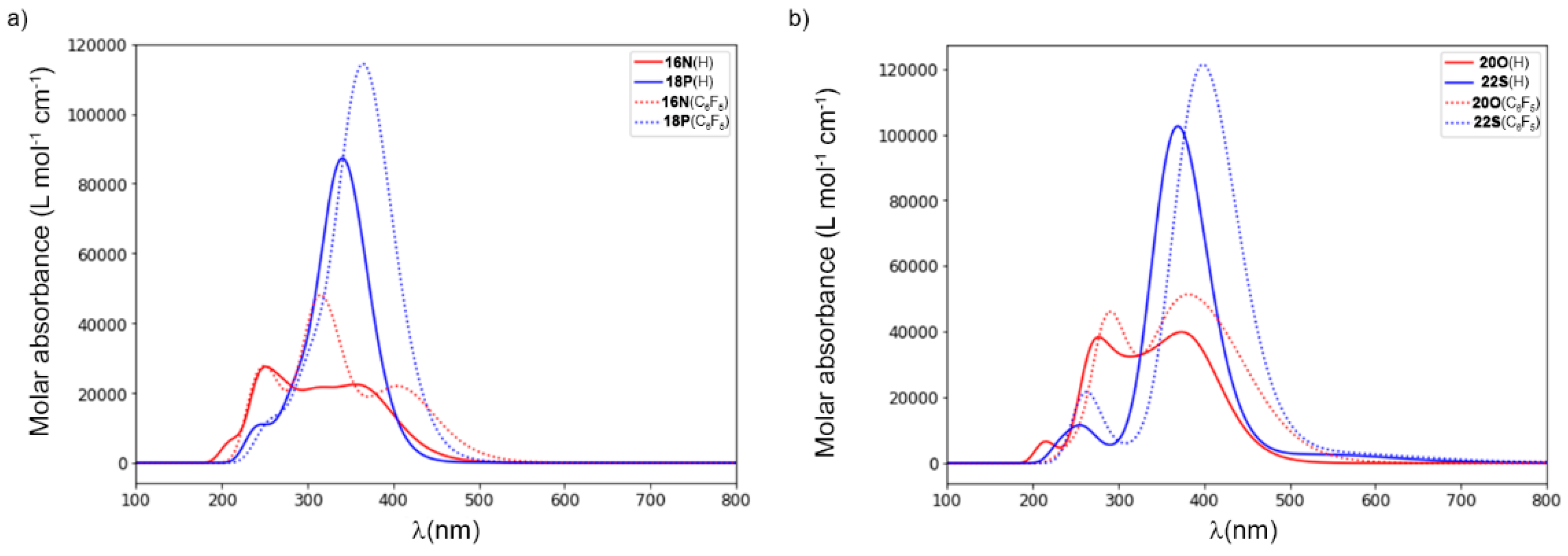
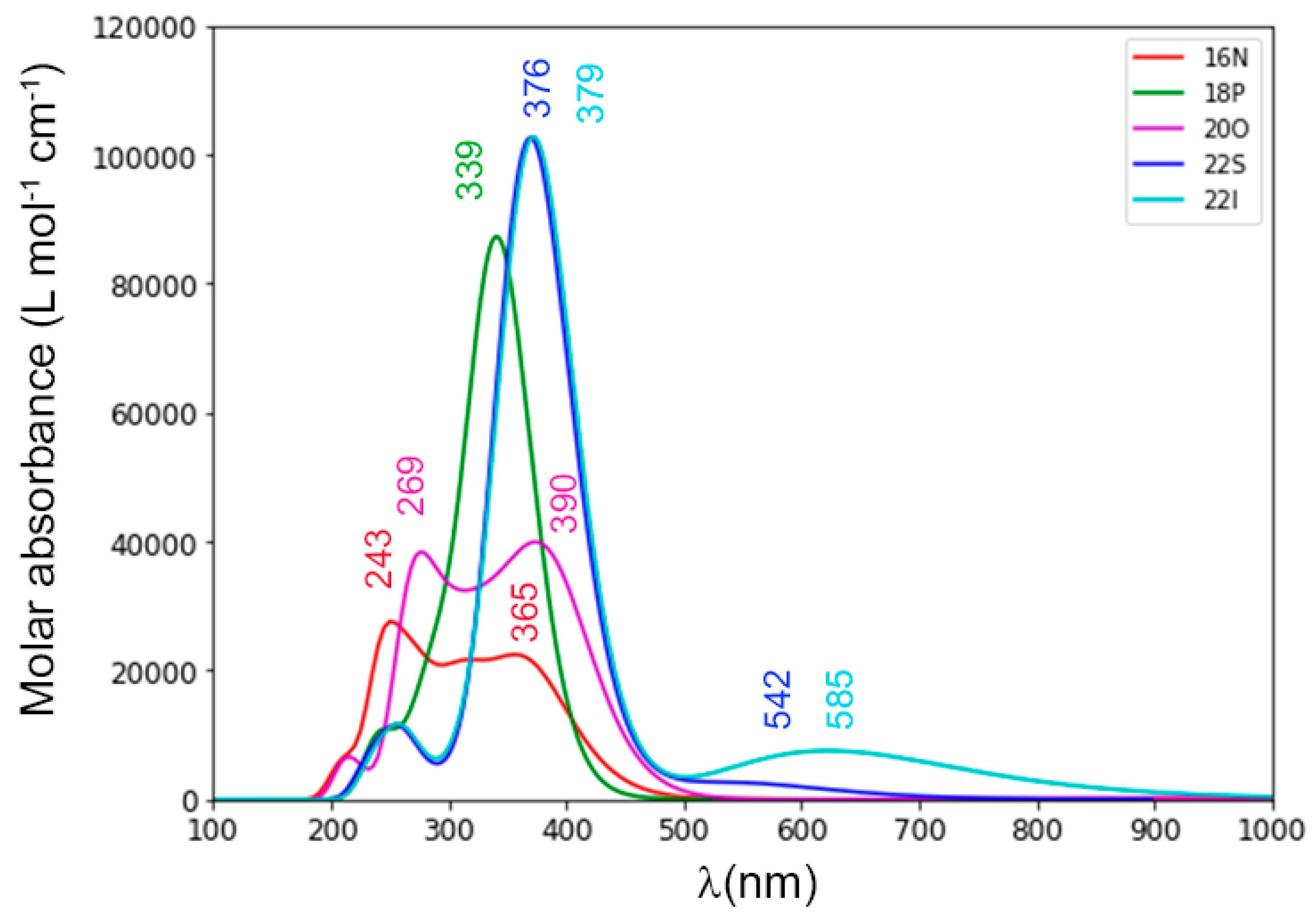
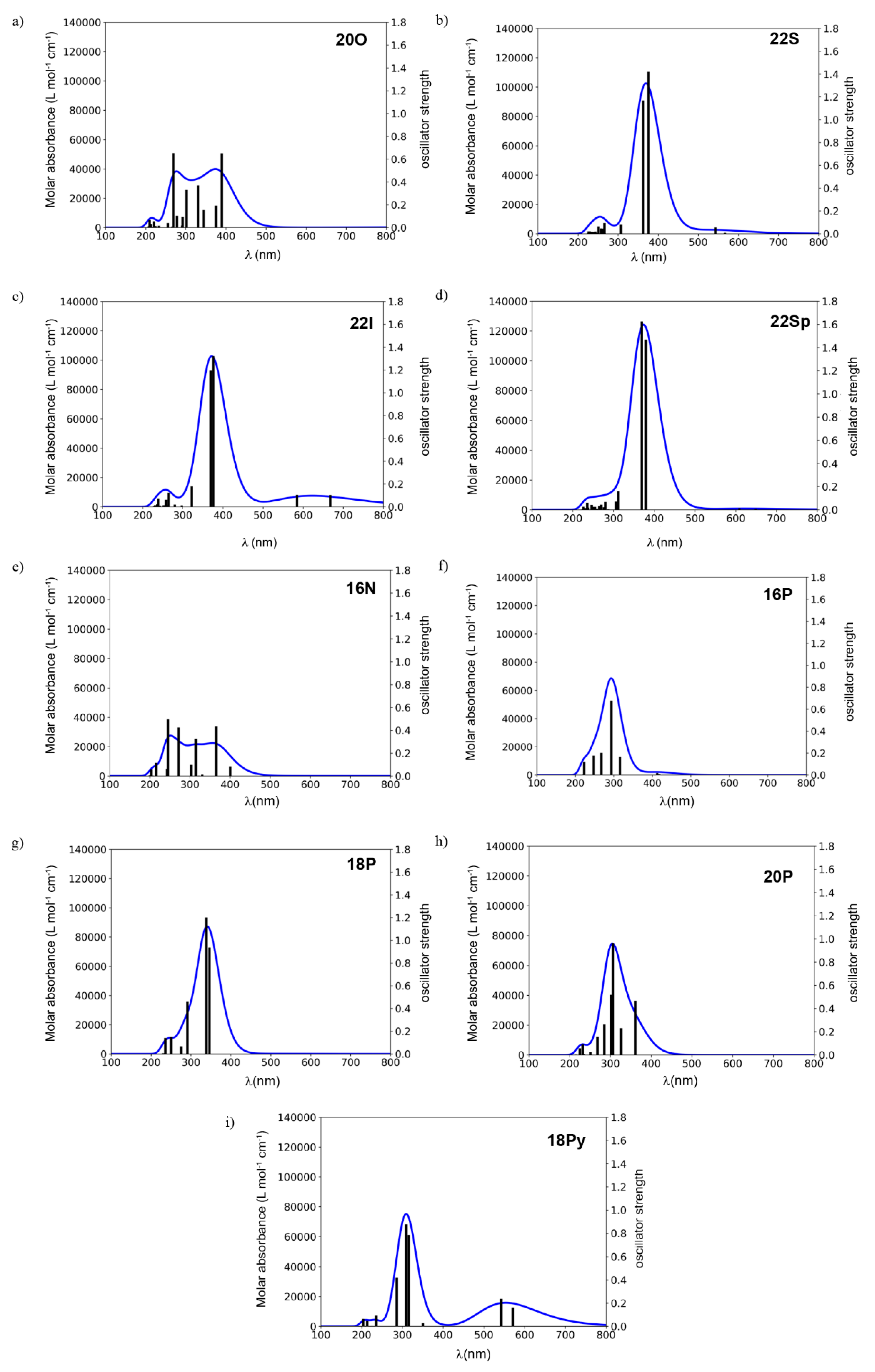
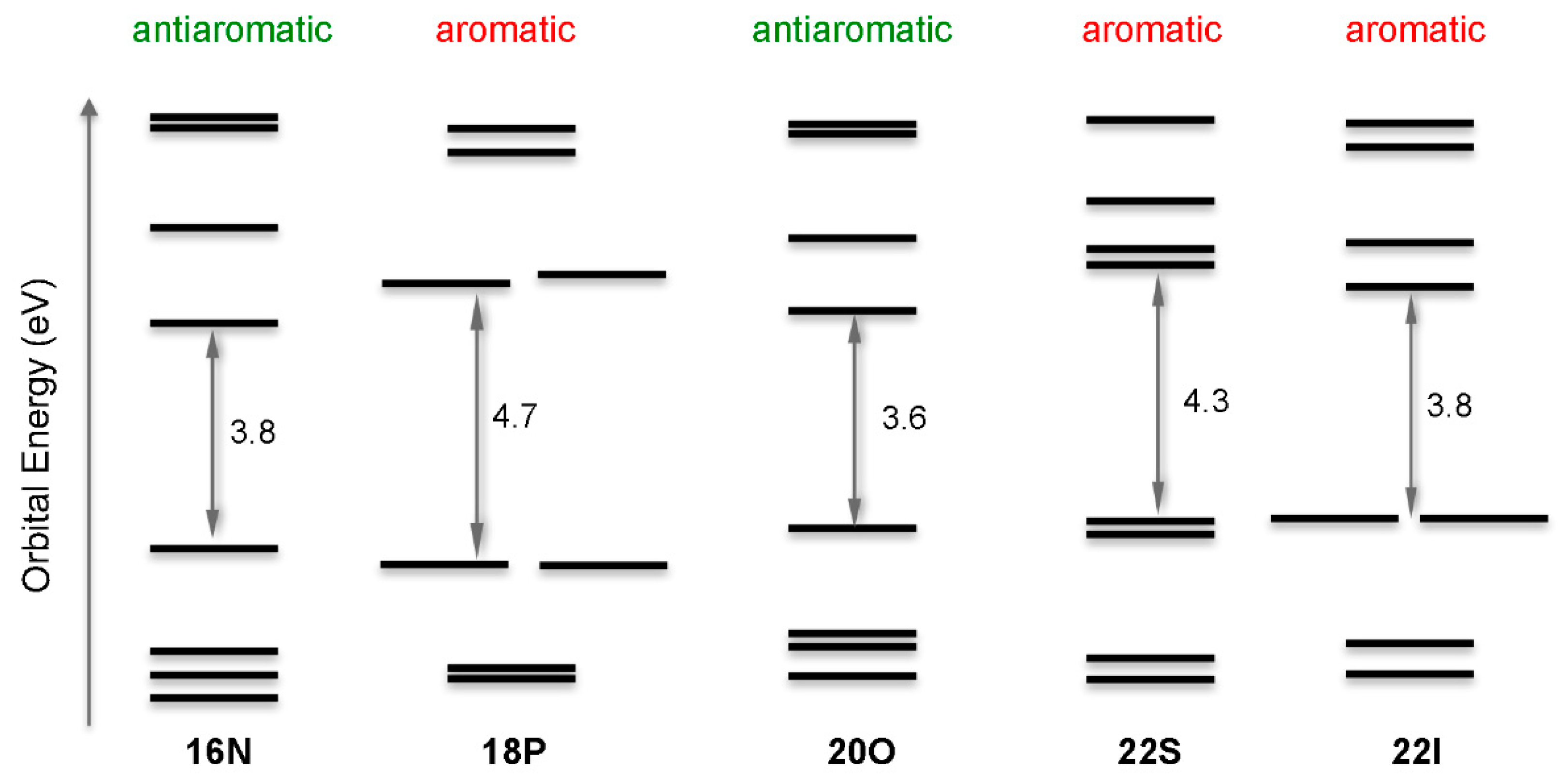
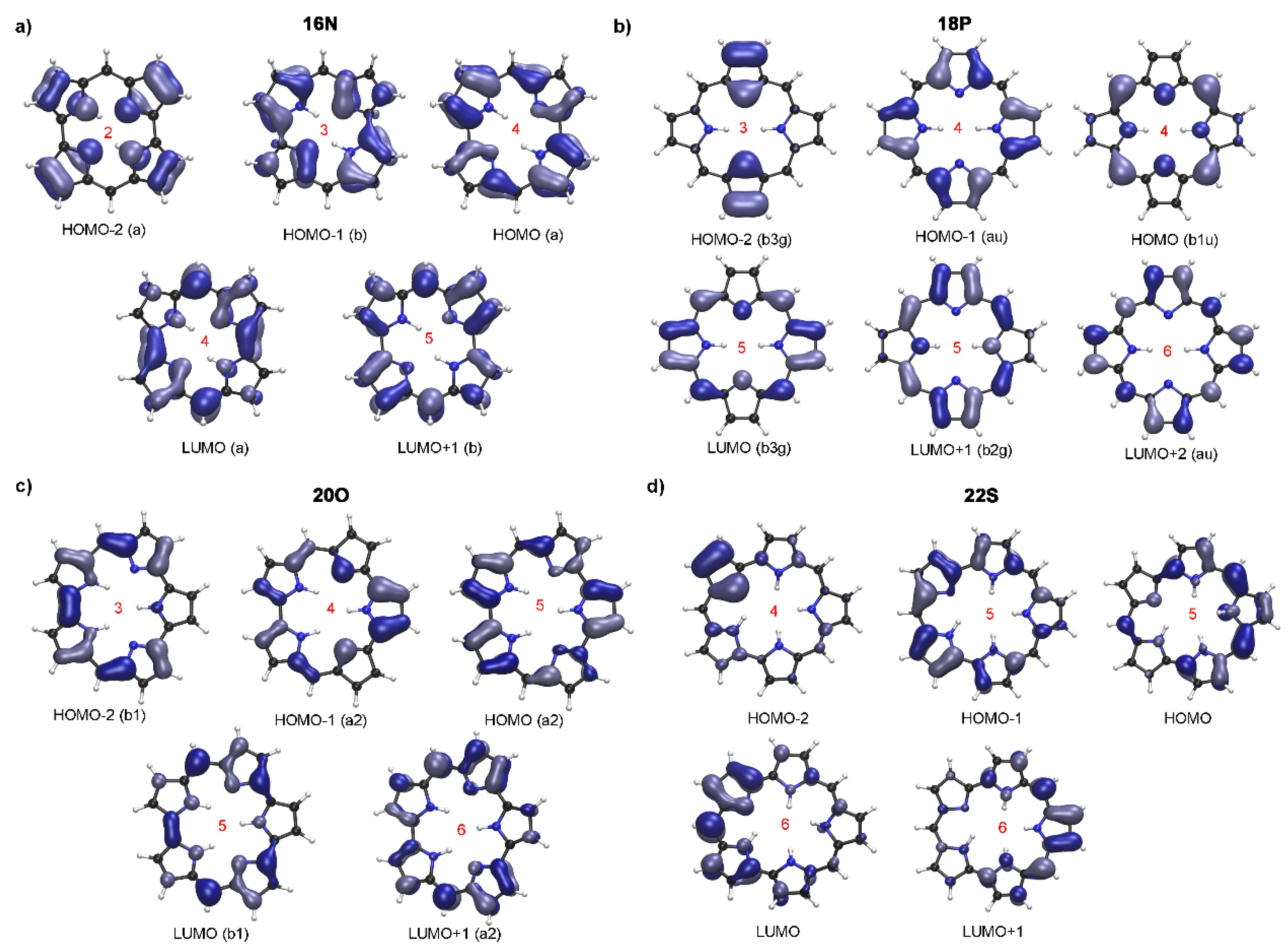
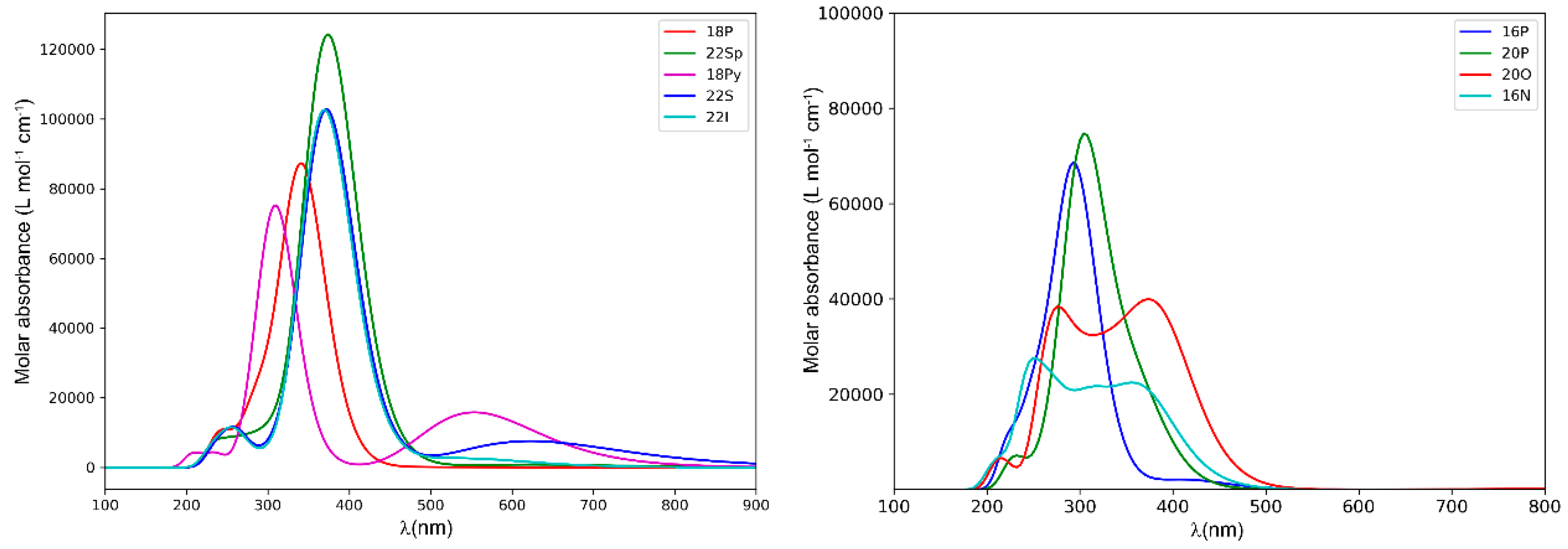
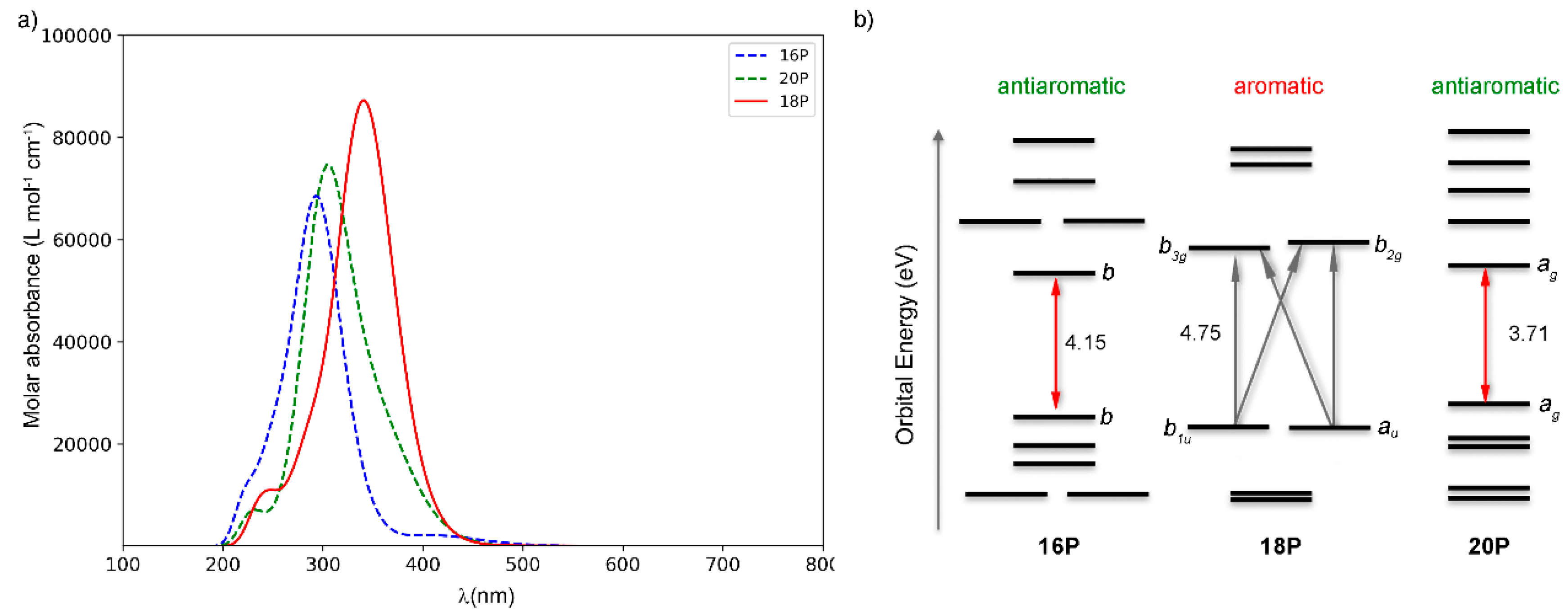
2.2. Photophysical Properties of Porphyrinoids
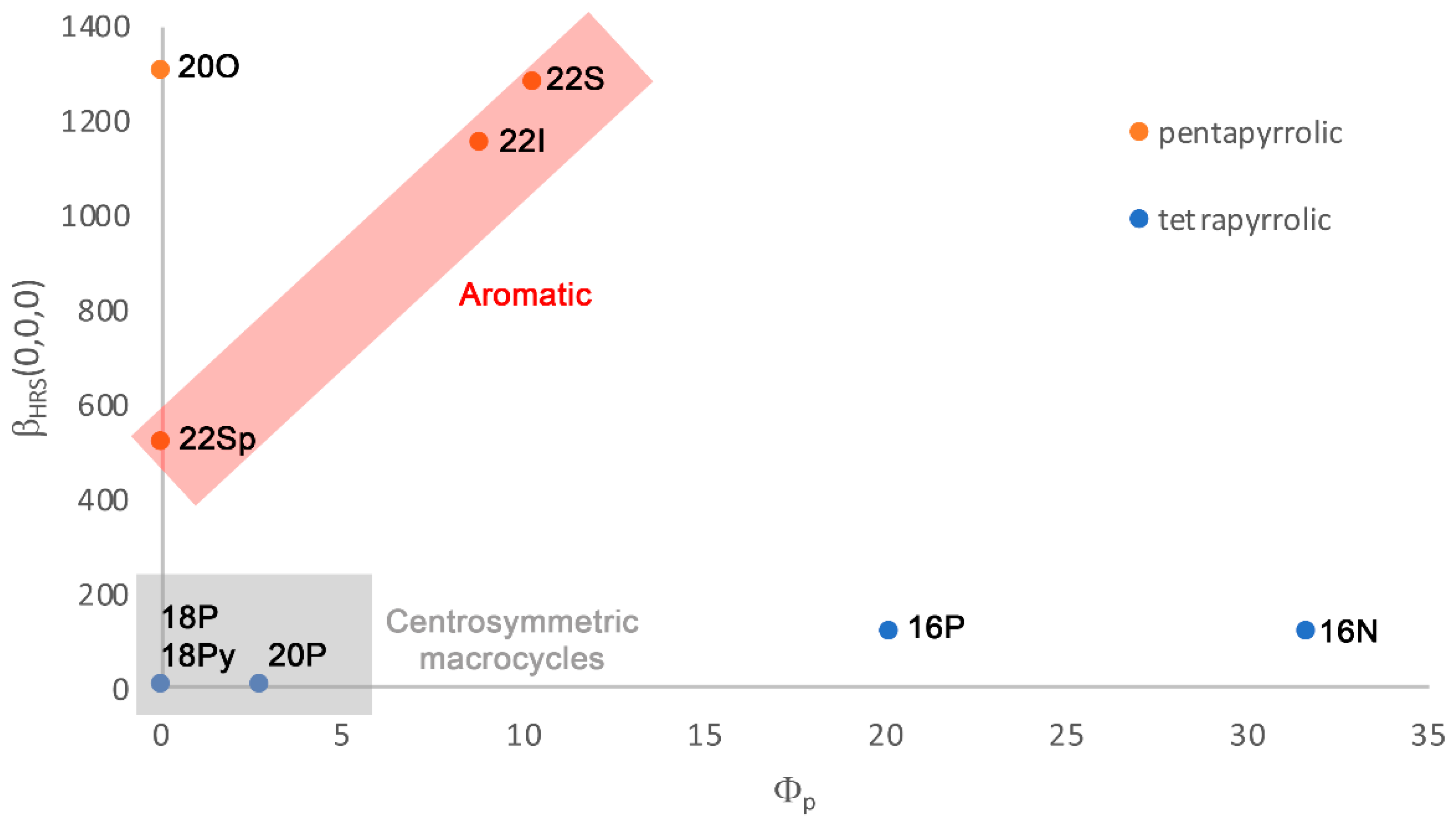
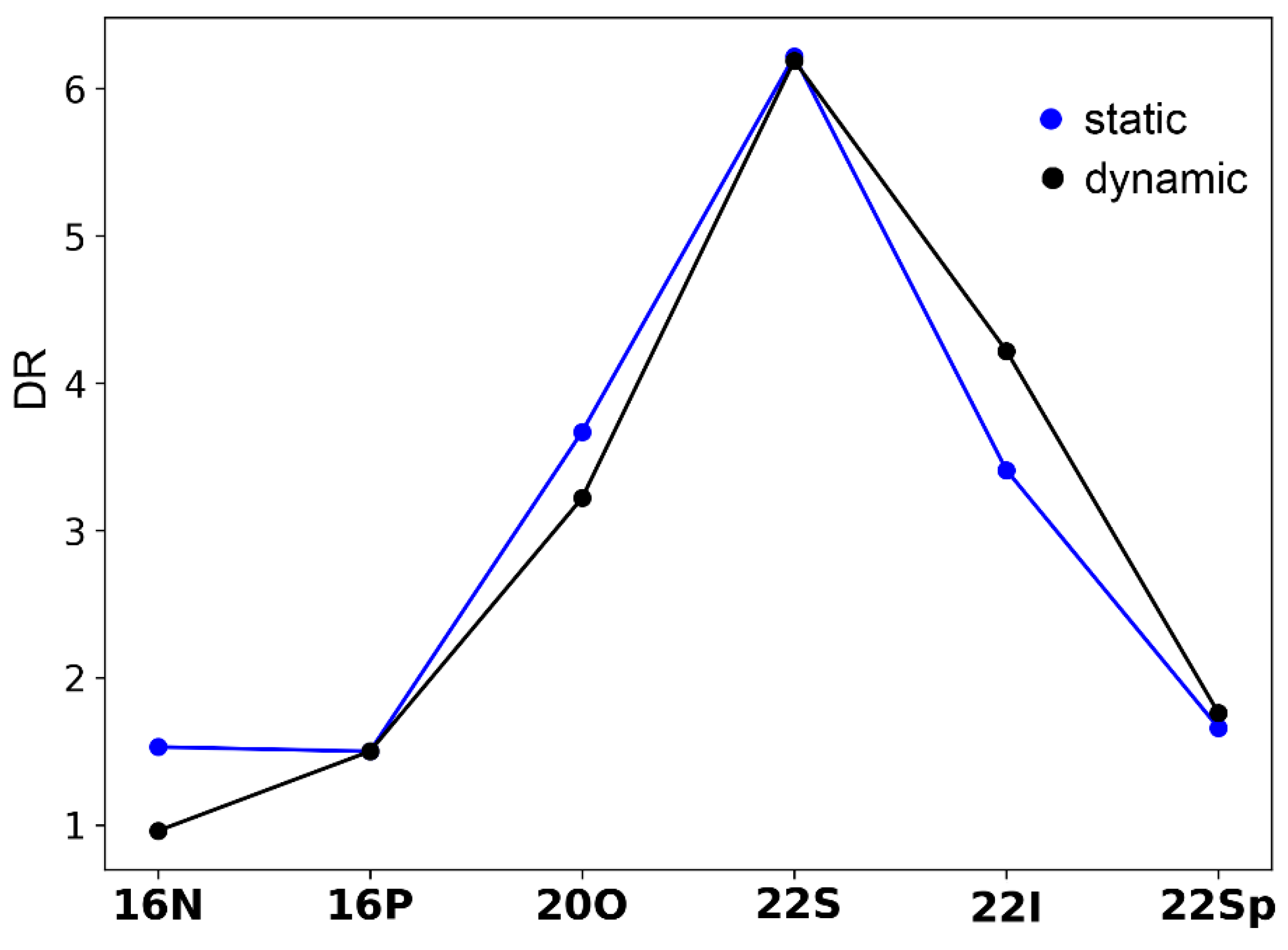
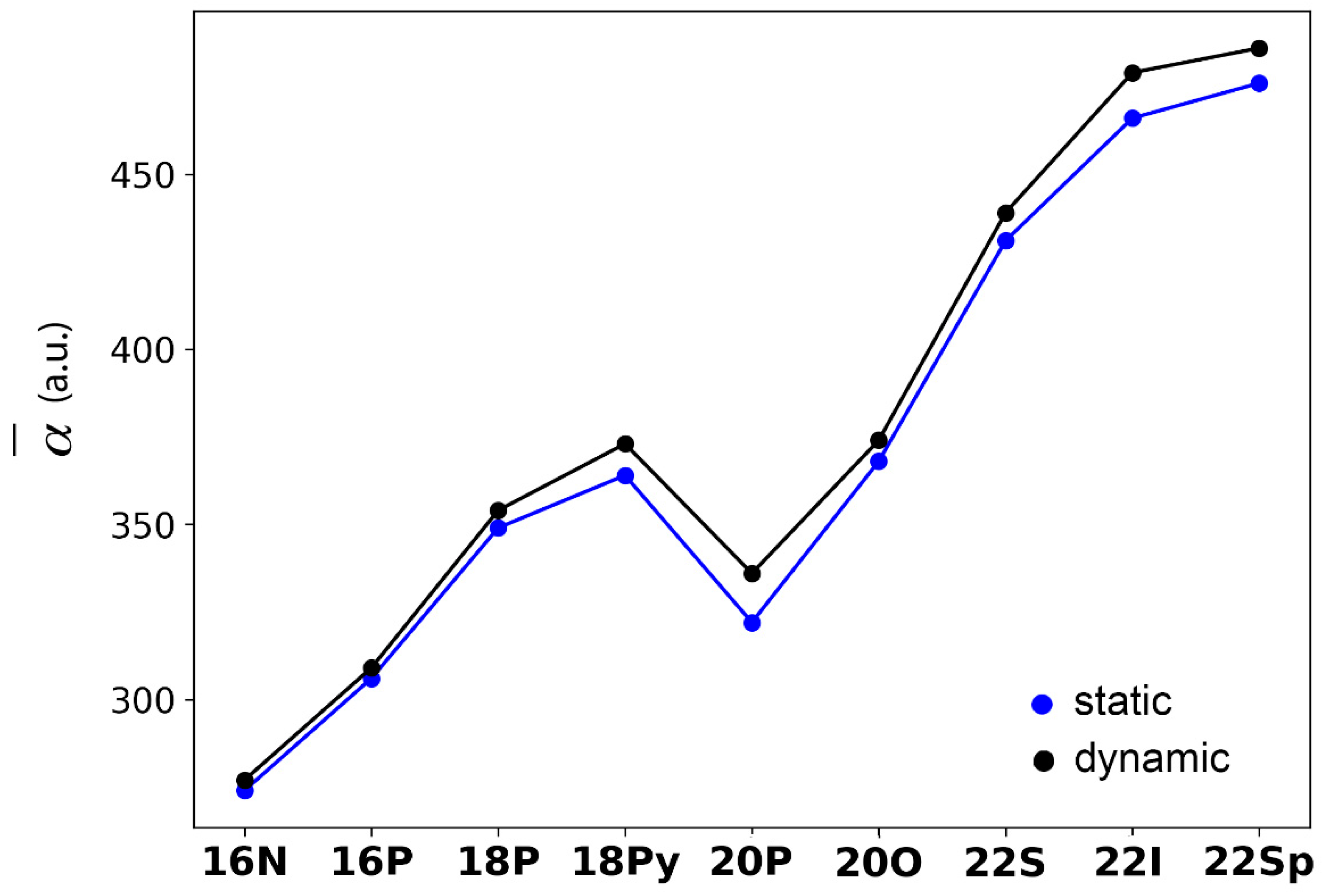
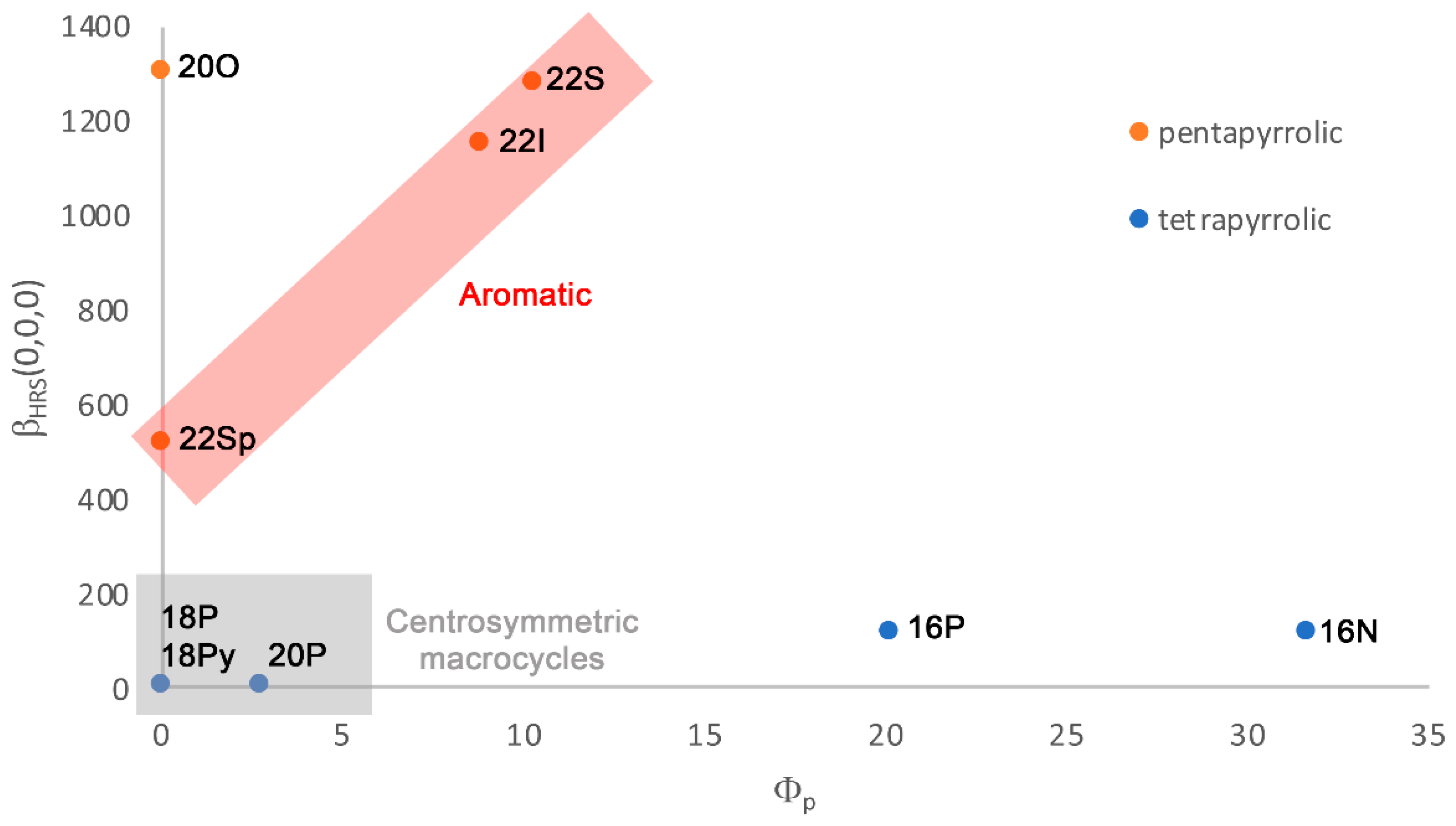
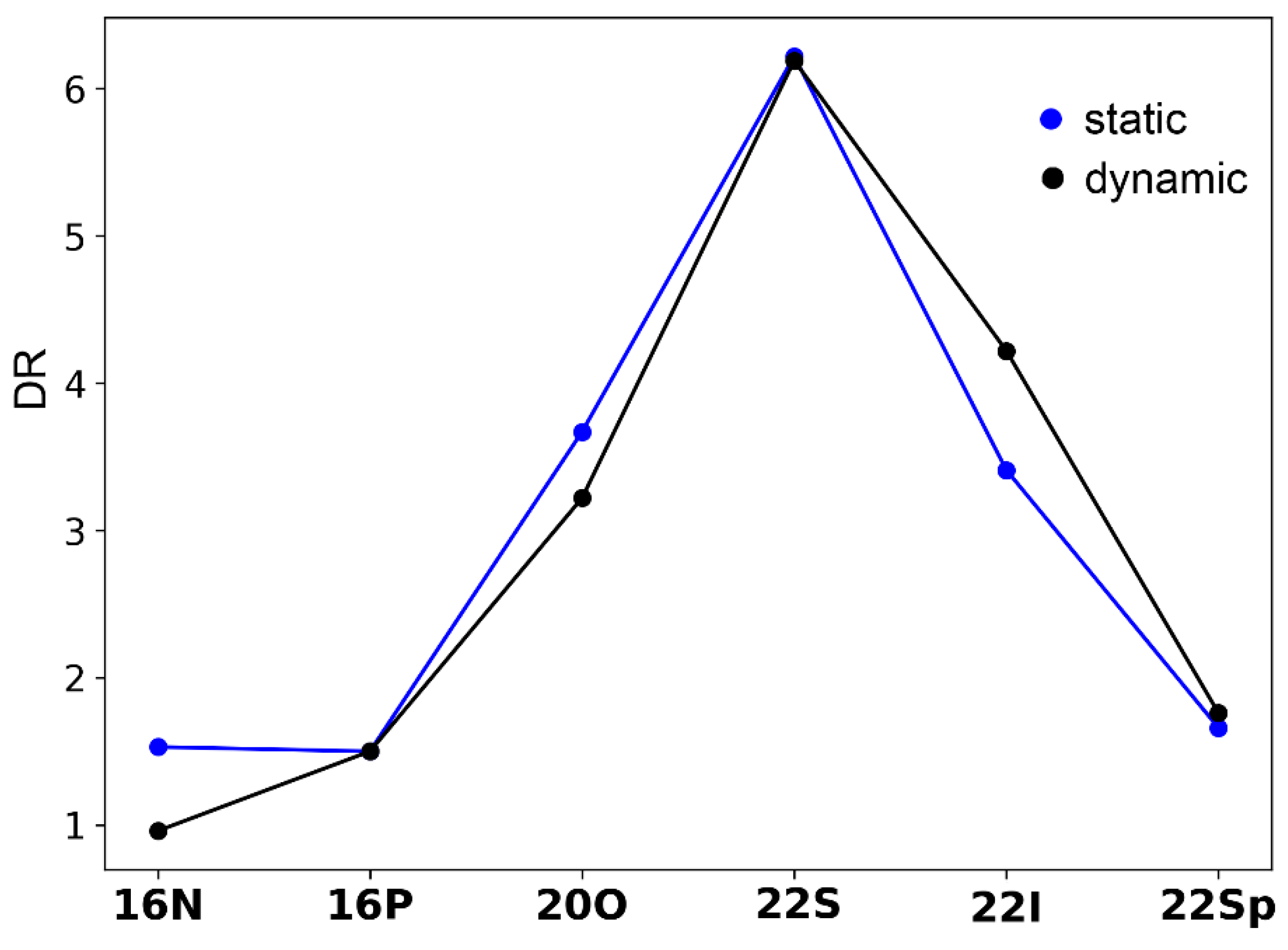
2.3. Polarizability and Nonlinear Optical Properties in Hückel Porphyrinoids
2.4. Substituent Effect on the Structure-Property Relationships
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Stepien, M.; Sprutta, N.; Latos-Grazynski, L. Figure eights, Mobius bands, and more: Conformation and aromaticity of porphyrinoids. Angew. Chem. Int. Ed. 2011, 50, 4288–4340. [Google Scholar] [CrossRef] [PubMed]
- Sessler, J.L.; Seidel, D. Synthetic expanded porphyrin chemistry. Angew. Chem. Int. Ed. 2003, 42, 5134–5175. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Osuka, A. Chemistry of meso-aryl-substituted expanded porphyrins: Aromaticity and Molecular Twist. Chem. Rev. 2017, 117, 2584–2640. [Google Scholar] [CrossRef] [PubMed]
- Szyszko, B.; Białek, M.J.; Pacholska-Dudziak, E.; Latos-Grażyński, L. Flexible porphyrinoids. Chem. Rev. 2017, 117, 2839–2909. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhu, W.-H.; Xie, Y. Development of ion chemosensors based on porphyrin analogues. Chem. Rev. 2017, 117, 2203–2256. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, J.O.; Rusin, O.; Lim, S.; Strongin, R.M. NIR Dyes for bioimaging applications. Curr. Opin. Chem. Biol. 2010, 14, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-photon absorption and the design of two-photon dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.M.; Yoon, Z.S.; Shin, J.-Y.; Kim, K.S.; Yoon, M.-C.; Kim, D. The photophysical properties of expanded porphyrins: Relationships between aromaticity, molecular geometry and non-linear optical properties. Chem. Commun. 2008, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Woller, T.; Ramos-Berdullas, N.; Mandado, M.; Alonso, M.; de Proft, F.; Contreras-Garcia, J. Understanding conductivity in molecular switches: A real space approach in octaphyrins. Phys. Chem. Chem. Phys. 2016, 18, 11829–11838. [Google Scholar] [CrossRef] [PubMed]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular electronic components of functional devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.M.; Oh, J.; Cha, W.-Y.; Kim, W.; Lim, J.M.; Yoon, M.-C.; Kim, D. Control and switching of aromaticity in various all-aza-expanded porphyrins: Spectroscopic and theoretical analyses. Chem. Rev. 2017, 117, 2257–2312. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Kim, K.S.; Yoon, M.C.; Lim, J.M.; Yoon, Z.S.; Osuka, A.; Kim, D. Aromaticity and photophysical properties of various topology-controlled expanded porphyrins. Chem. Soc. Rev. 2010, 39, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yoon, Z.S.; Kim, K.S.; Yoon, M.-C.; Cho, D.-G.; Sessler, J.L.; Kim, D. Defining spectroscopic features of heteroannulenic antiaromatic porphyrinoids. J. Phys. Chem. Lett. 2010, 1, 895–900. [Google Scholar] [CrossRef]
- Yoon, Z.S.; Kwon, J.H.; Yoon, M.-C.; Koh, M.K.; Noh, S.B.; Sessler, J.L.; Lee, J.T.; Seidel, D.; Aguilar, A.; Shimizu, S.; et al. Nonlinear optical properties and excited-state dynamics of highly symmetric expanded porphyrins. J. Am. Chem. Soc. 2006, 128, 14128–14134. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Yoon, M.-C.; Lim, J.M.; Rath, H.; Naoda, K.; Osuka, A.; Kim, D. Reversal of Hückel (anti)aromaticity in the lowest triplet states of hexaphyrins and spectroscopic evidence for Baird’s rule. Nat. Chem. 2015, 7, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Ottosson, H.; Borbas, K.E. Aromaticity: A light-switched yin and yang pair. Nat. Chem. 2015, 7, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Z.S.; Cho, D.-G.; Kim, K.S.; Sessler, J.L.; Kim, D. Nonlinear Optical Properties as a guide to aromaticity in congeneric pentapyrrolic expanded porphyrins: Pentaphyrin, sapphyrin, isosmaragdyrin, and orangarin. J. Am. Chem. Soc. 2008, 130, 6930–6931. [Google Scholar] [CrossRef] [PubMed]
- Naoda, K.; Mori, H.; Aratani, N.; Lee, B.S.; Kim, D.; Osuka, A. Hexaphyrin fused to two anthracenes. Angew. Chem. Int. Ed. 2012, 51, 9856–9859. [Google Scholar] [CrossRef] [PubMed]
- Rath, H.; Prabhuraja, V.; Chandrashekar, T.K.; Nag, A.; Goswami, D.; Joshi, B.S. Aromatic core modified decaphyrins with the largest two-photon absorption cross-sections: Syntheses and characterization. Org. Lett. 2006, 8, 2325–2328. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Nakatsuji, H.; Fukuda, R.; Okamoto, H.; Imai, H.; Matsuda, T.; Tochio, H.; Shirakawa, M.; Tkachenko, N.V.; Hashida, M.; et al. Hexaphyrin as a potential theranostic dye for photothermal therapy and 19F magnetic resonance imaging. ChemBioChem 2017, 18, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Torrent-Sucarrat, M.; Anglada, J.M.; Luis, J.M. Evaluation of the nonlinear optical properties for an expanded porphyrin Hückel-Möbius aromaticity switch. J. Chem. Phys. 2012, 137, 184306. [Google Scholar] [CrossRef] [PubMed]
- Torrent-Sucarrat, M.; Navarro, S.; Marcos, E.; Anglada, J.M.; Luis, J.M. Design of Hückel–Möbius topological switches with high nonlinear optical properties. J. Phys. Chem. C 2017, 121, 19348–19357. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; Anglada, J.M.; Luis, J.M. Evaluation of the nonlinear optical properties for annulenes with Hückel and Möbius topologies. J. Chem. Theory Comput. 2011, 7, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, T.; Perrin, M.; Geerlings, P.; De Proft, F.; Alonso, M. Conductance switching in expanded porphyrins through aromaticity and topology changes. J. Am. Chem. Soc. 2018, 140, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Osuka, A.; Saito, S. Expanded porphyrins and aromaticity. Chem. Commun. 2011, 47, 4330–4339. [Google Scholar] [CrossRef] [PubMed]
- Stanger, A. What is... aromaticity: A critique of the concept of aromaticity-can it really be defined? Chem. Commun. 2009, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R. The many guises of aromaticity. Am. Sci. 2015, 103, 18–22. [Google Scholar] [CrossRef]
- Solà, M. Why Aromaticity Is a Suspicious Concept? Why? Front. Chem. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Cyrański, M.K. Energetic aspects of cyclic pi-electron delocalization: Evaluation of the methods of estimating aromatic stabilization energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef] [PubMed]
- De Proft, F.; Geerlings, P. Conceptual and Computational DFT in the Study of Aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the viewpoint of molecular geometry: Application to planar systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef] [PubMed]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Sola, M. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, M.; Herradón, B. A universal scale of aromaticity for π-organic compounds. J. Comp. Chem. 2010, 31, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. On the performance of some aromaticity indices: A critical assessment using a test set. J. Comp. Chem. 2008, 29, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.I.; Fernández, I.; Schleyer, P.V.R. Description of aromaticity in porphyrinoids. J. Am. Chem. Soc. 2013, 135, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Geerlings, P.; De Proft, F. Viability of Möbius topologies in [26]- and [28]hexaphyrins. Chem. Eur. J. 2012, 18, 10916–10928. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Geerlings, P.; De Proft, F. Topology switching in [32]heptaphyrins controlled by solvent, protonation, and meso-substituents. Chem. Eur. J. 2013, 19, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Geerlings, P.; De Proft, F. Conformational control in [22]- and [24]pentaphyrins(1.1.1.1.1) by meso substituents and their N-fusion reaction. J. Org. Chem. 2013, 78, 4419–4431. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Geerlings, P.; De Proft, F. Exploring the structure-aromaticity relationship in Huckel and Mobius N-fused pentaphyrins using DFT. Phys. Chem. Chem. Phys. 2014, 16, 14396–14407. [Google Scholar] [CrossRef] [PubMed]
- Woller, T.; Contreras-Garcia, J.; Geerlings, P.; De Proft, F.; Alonso, M. Understanding the molecular switching properties of octaphyrins. Phys. Chem. Chem. Phys. 2016, 18, 11885–11900. [Google Scholar] [CrossRef] [PubMed]
- Vogel, E. The porphyrins from the ‘annulene chemist’s’ perspective. Pure Appl. Chem. 1993, 65, 143–152. [Google Scholar] [CrossRef]
- Vogel, E. From small carbocyclic rings to porphyrins. Angew. Chem. Int. Ed. 2011, 50, 4278–4287. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Jones, S.A.; Ferrence, G.M. Synthesis and characterization of tetraphenyl-21,23-dideazaporphyrin: The best evidence yet that porphyrins really are the [18]annulenes of nature. J. Am. Chem. Soc. 2010, 132, 12786–12787. [Google Scholar] [CrossRef] [PubMed]
- Aihara, J.-I.; Nakagami, Y.; Sekine, R.; Makino, M. Validity and limitations of the bridged annulene model for porphyrins. J. Phys. Chem. A 2012, 116, 11718–11730. [Google Scholar] [CrossRef] [PubMed]
- Bröring, M. How should aromaticity be described in porphyrinoids? Angew. Chem. Int. Ed. 2011, 50, 2436–2438. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Maeda, H.; Osuka, A. Confusion, inversion, and creation-a new spring from porphyrin chemistry. Chem. Commun. 2002, 1795–1804. [Google Scholar] [CrossRef]
- Casademont, I.; Woller, T.; Contreras Garcia, J.; Alonso, M.; Torrent-Sucarrat, M.; Matito, E. New Electron Delocalization Tools to Describe the Aromaticity in Porphyrinoids. Phys. Chem. Chem. Phys. 2018, 20, 2787–2796. [Google Scholar] [CrossRef] [PubMed]
- Matito, E. An electronic aromaticity index for large rings. Phys. Chem. Chem. Phys. 2016, 18, 11839–11846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Fernández, C.; Sierda, E.; Abadía, M.; Bugenhagen, B.; Prosenc, M.H.; Wiesendanger, R.; Bazarnik, M.; Ortega, J.E.; Brede, J.; Matito, E.; et al. Exploring the relation between intramolecular conjugation and band dispersion in one-dimensional polymers. J. Phys. Chem. C 2017, 121, 27118–27125. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamamoto, A.; Furuta, S.-Y.; Horie, M.; Kodama, M.; Sato, W.; Akiba, K.-Y.; Tsuzuki, S.; Uchimaru, T.; Hashizume, D.; et al. Synthesis and structure of 16 π octaalkyltetraphenylporphyrins. J. Am. Chem. Soc. 2005, 127, 14540–14541. [Google Scholar] [CrossRef] [PubMed]
- Cissell, J.A.; Vaid, T.P.; Rheingold, A.L. An Antiaromatic Porphyrin Complex: Tetraphenylporphyrinato(silicon)(L)2 (L = THF or Pyridine). J. Am. Chem. Soc. 2005, 127, 12212–12213. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.; Srinivasan, A.; Ravikanth, M.; Chandrasheka, T.K. Smaragdyrins and sapphyrins analogues. Chem. Rev. 2017, 117, 3329–3376. [Google Scholar] [CrossRef] [PubMed]
- Szterenberg, L.; Latos-Grazynski, L. Geometry and tautomerism of sapphyrin—DFT studies. J. Mol. Struct. Theochem 1999, 490, 33–46. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Pühlhofer, F. Recommendations for the evaluation of aromatic stabilization energies. Org. Lett. 2002, 4, 2873–2876. [Google Scholar] [CrossRef] [PubMed]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Herges, R. Topology in chemistry: Designing Möbius molecules. Chem. Rev. 2006, 106, 4820–4842. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, W.-Y.; Qiu, Y.-Q.; Lu, H.-Z. Second-order nonlinear optical response of electron donor–acceptor hybrids formed between corannulene and metallofullerenes. J. Phys. Chem. C 2015, 119, 24965–24975. [Google Scholar] [CrossRef]
- The degeneracy of the orbitals is not perfect. Orbital energy differences lower than 0.1 eV are represented as degenerate orbitals in Figure 7.
- Mack, J. Expanded, contracted, and isomeric porphyrins: Theoretical aspects. Chem. Rev. 2017, 117, 3444–3478. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.R. Classification of spectra of cata-condensed hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Chantrell, S.J.; McAuliffe, C.A.; Munn, R.W.; Pratt, A.C. The status of molecular orbital calculations on porphyrins and their complexes. Coord. Chem. Rev. 1975, 16, 259–284. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hirata, Y.; Kodama, M.; Yamaguchi, T.; Matsukawa, S.; Akiba, K.-Y.; Hashizume, D.; Iwasaki, F.; Muranaka, A.; Uchiyama, M.; et al. Synthesis, reactions, and electronic properties of 16 π-electron octaisobutyltetraphenylporphyrin. J. Am. Chem. Soc. 2010, 132, 12627–12638. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, T.; Clays, K.; Rodriguez, V. Second-Order Nonlinear Optical Characterizations Techniques: An Introduction; CRC Press: New York, NY, USA, 2009. [Google Scholar]
- Castet, F.; Rodriguez, V.; Pozzo, J.-L.; Ducasse, L.; Plaquet, A.; Champagne, B. Design and characterization of molecular nonlinear optical switches. Acc. Chem. Res. 2013, 46, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Plaquet, A.; Guillaume, M.; Champagne, B.; Castet, F.; Ducasse, L.; Pozzo, J.-L.; Rodriguez, V. In silico optimization of merocyanine-spiropyran compounds as second-order nonlinear optical molecular switches. Phys. Chem. Chem. Phys. 2008, 10, 6223–6232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. For a detailed account on these types of basis sets. In Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of pi-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Dauben, H.J.; Wilson, J.D.; Laity, J.L. Diamagnetic susceptibility exaltation as a criterion of aromaticity. J. Am. Chem. Soc. 1968, 90, 811–813. [Google Scholar] [CrossRef]
- De Proft, F.; Geerlings, P. Relative hardness as a measure of aromaticity. Phys. Chem. Chem. Phys. 2004, 6, 242–248. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Which NICS aromaticity index for planar π rings is best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Nenon, S.; Champagne, B.; Spassova, M.I. Assessing long-range corrected functionals with physically-adjusted range-separated parameters for calculating the polarizability and the second hyperpolarizability of polydiacetylene and polybutatriene chains. Phys. Chem. Chem. Phys. 2014, 16, 7083–7088. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






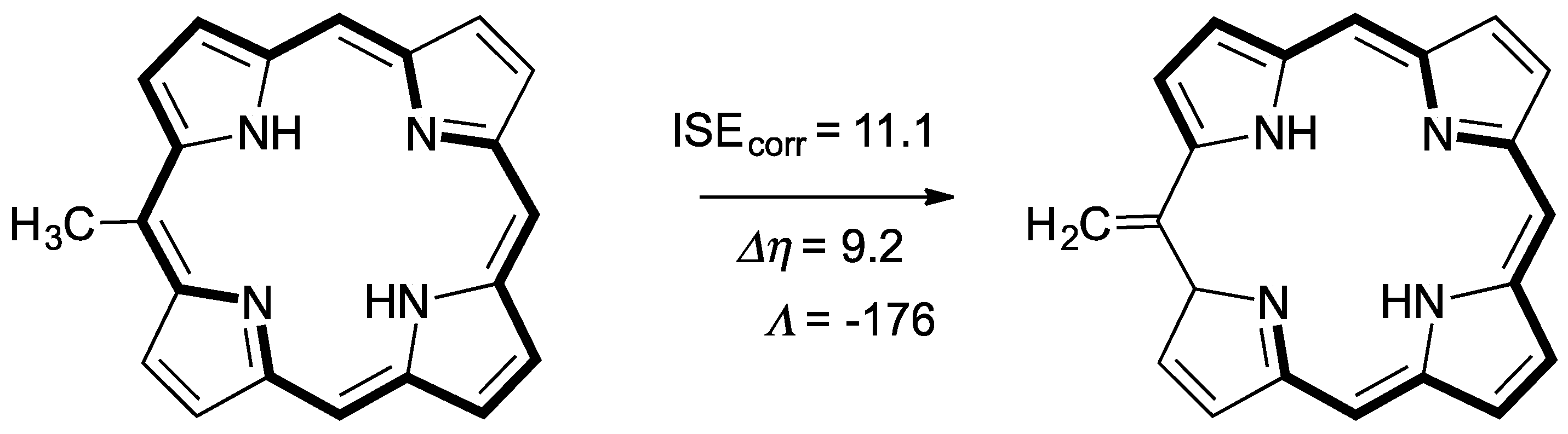

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
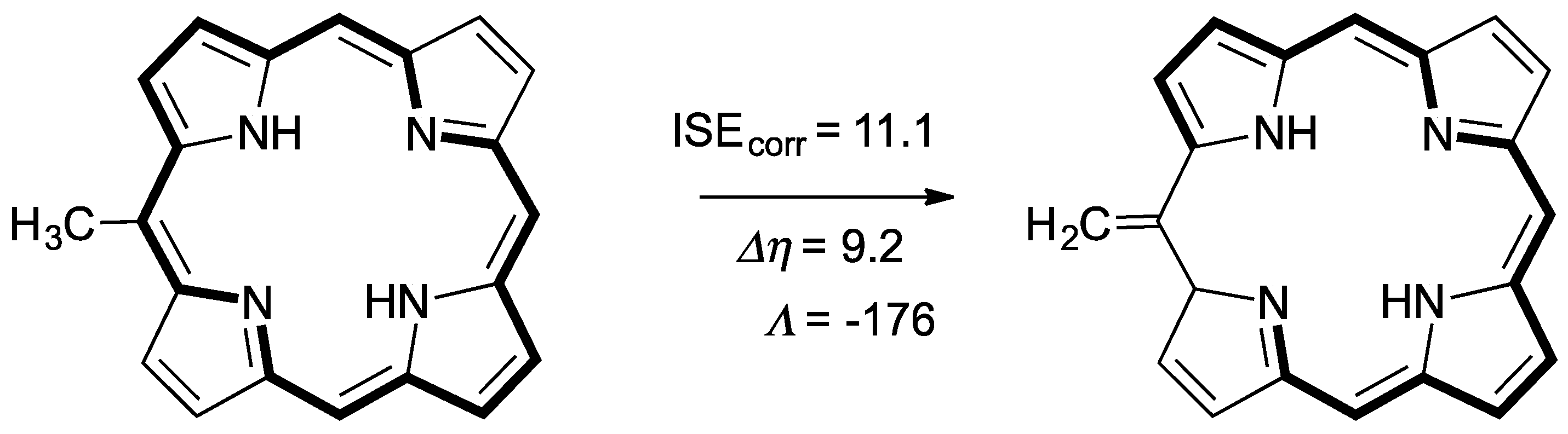{kind=link}
| πe b | Δη | Λ | NICS(0) | NICS(1) | NICSzz(1) | HOMA | Π | Φp | ΔEH-L c | AV1245 d | AV min d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 16N | −3.0 | 78.8 | 23.1 | 18.9 | 62.3 | 0.610 | 0.44 | 31.7 | 3.76 | 1.262 | 0.136 |
| 16P | −11.9 | 27.5 | 9.4 | 7.6 | 29.2 | 0.634 | 0.74 | 20.2 | 4.15 | 0.732 | 0.294 |
| 18P | 9.2 | −175.8 | −14.9 | −13.7 | −38.5 | 0.880 | 1.00 | 0.0 | 4.75 | 2.139 | 1.271 |
| 18Py | 9.8 | −147.0 | −13.3 | −12.5 | −13.9 | 0.878 | 0.99 | 0.0 | 4.07 | 2.114 | 1.399 |
| 20P | −17.8 | 156.3 | 18.7 | 16.7 | 52.4 | 0.641 | 0.93 | 2.8 | 3.71 | 1.226 | 0.856 |
| 20O | −13.9 | 128.6 | 16.3 | 14.0 | 44.6 | 0.780 | 1.00 | 0.0 | 3.27 | 1.352 | 0.267 |
| 22S | 2.2 | −140.6 | −9.6 | −9.2 | −24.6 | 0.892 | 0.88 | 10.3 | 4.25 | 2.066 | 0.794 |
| 22Sp | 13.7 | −249.6 | −14.3 | −13.4 | −37.7 | 0.882 | 1.00 | 0.0 | 4.03 | 2.124 | 1.137 |
| 22I | 1.9 | −249.1 | −13.6 | −12.8 | −35.7 | 0.901 | 0.91 | 8.8 | 3.85 | 2.435 | 0.963 |
| Excitation | Transition | Contribution (%) | λb | foscc | Ed | Polarization | Assignment |
|---|---|---|---|---|---|---|---|
| 1 | H→L | 98.69 | 1052.37 | 0.000 | 1.18 | z | Q |
| 2 | H−4→L−4 | 6.96 | 400.45 | 0.083 | 3.10 | z | |
| H−2→L+1 | 3.33 | x,y | |||||
| H−1→L | 84.53 | x,y | |||||
| 3 | H−7→L | 5.70 | 388.11 | 0.000 | 3.19 | x,y | |
| H−2→L | 85.09 | z | |||||
| H−1→L+1 | 4.69 | z | |||||
| 4 | H−4→L | 2.11 | 365.39 | 0.436 | 3.39 | z | B |
| H→L+2 | 94.22 | z | |||||
| 5 | H−5→L+2 | 2.51 | 330.87 | 0.013 | 3.73 | x,y | |
| H−4→L | 2.36 | z | |||||
| H−3→L | 90.21 | x,y | |||||
| 7 | H→L+4 | 41.38 | 314.52 | 0.327 | 3.38 | z | |
| 10 | H−4→L | 81.08 | 271.66 | 0.426 | 3.81 | z | |
| 13 | H→L+4 | 81.19 | 242.94 | 0.496 | 4.00 | z |
| Excitation | Transition | Contribution (%) | λb | foscc | Ed | Polarization | Assignment |
|---|---|---|---|---|---|---|---|
| 1 | H−1→L | 47.52 | 559.66 | 0.008 | 2.22 | x | Qx |
| H→L+1 | 52.62 | x | |||||
| 2 | H−1→L+1 | 51.15 | 514.30 | 0.002 | 2.41 | y | Qy |
| H→L | 48.60 | y | |||||
| 3 | H−4→L+1 | 12.58 | 346.60 | 0.937 | 3.58 | x | Bx |
| H−1→L | 48.22 | x | |||||
| H→L+1 | 39.86 | x | |||||
| L+1→H | 2.46 | x | |||||
| 4 | H−1→L+1 | 49.12 | 338.53 | 1.202 | 3.66 | y | By |
| H→L | 52.30 | y | |||||
| 5 | H−3→L+2 | 4.98 | 314.18 | 0.000 | 3.95 | / | |
| H−2→L+1 | 91.50 | / | |||||
| H→L+2 | 2.08 | z | |||||
| 6 | H−5→L | 80.03 | 291.26 | 0.461 | 4.26 | y | |
| 16 | H−5→L | 85.33 | 250.14 | 0.149 | 4.96 | y | |
| 18 | H−4→L | 96.34 | 235.80 | 0.140 | 5.26 | x |
| Excitation | Transition | Contribution (%) | λb | foscc | Ed | Polarization | Assignment |
|---|---|---|---|---|---|---|---|
| 1 | H→L | 97.55 | 954.1 | 0.007 | 1.29 | y | Q |
| 2 | H−1→L+1 | 98.45 | 389.6 | 0.652 | 3.18 | z | B |
| 3 | H−1→L | 6.86 | 375.1 | 0.192 | 3.31 | y | |
| H−4→L | 83.88 | y | |||||
| 4 | H−3→L | 42.60 | 344.5 | 0.154 | 3.60 | z | |
| H−2→L | 28.92 | z | |||||
| 5 | H−3→L | 27.26 | 330.3 | 0.369 | 3.75 | z | |
| H−2→L | 66.51 | z | |||||
| 6 | H−4→L | 67.52 | 301.6 | 0.329 | 4.11 | y | |
| 12 | H→L+2 | 93.84 | 269.0 | 0.653 | 4.61 | y |
| Excitation | Transition | Contribution (%) | λb | foscc | Ed | Polarization | Assignment |
|---|---|---|---|---|---|---|---|
| 1 | H−1→L | 52.81 | 566.44 | 0.008 | 2.19 | y | Qy |
| H→L+1 | 42.44 | ||||||
| 2 | H−1→L+1 | 27.12 | 542.5 | 0.056 | 2.29 | x | Qx |
| H→L | 68.23 | ||||||
| 3 | H−1→L | 44.57 | 375.78 | 1.420 | 3.30 | y | By |
| H→L+1 | 55.86 | ||||||
| 4 | H−1→L+1 | 69.40 | 362.30 | 1.168 | 3.42 | x | Bx |
| H→L | 28.69 | ||||||
| 5 | H−3→L | 9.47 | 307.14 | 0.081 | 4.04 | x | |
| H−2→L | 77.50 | ||||||
| 8 | H→L+3 | 61.17 | 265.99 | 0.094 | 4.66 | x | |
| H−1→L+6 | 9.28 | ||||||
| 11 | H−5→L | 24.76 | 257.92 | 0.046 | 4.81 | x | |
| H−2→L+1 | 11.93 | ||||||
| H−9→L | 9.68 | ||||||
| H−1→L+4 | 15.21 |
| Excitation | Transition | Contribution (%) | λb | foscc | Ed | Polarization | Assignment |
|---|---|---|---|---|---|---|---|
| 1 | H−1→L+1 | 28.61 | 667.43 | 0.103 | 1.86 | z | Q |
| H→L | 70.93 | z | |||||
| 2 | H−1→L | 72.54 | 584.78 | 0.105 | 2.12 | x,y | Q |
| H→L+1 | 26.94 | x,y | |||||
| 3 | H−1→L | 28.57 | 375.89 | 1.321 | 3.30 | x,y | B |
| H→L+1 | 73.62 | x,y | |||||
| 4 | H−1→L+1 | 69.35 | 369.59 | 1.195 | 3.35 | z | B |
| H→L | 28.97 | z | |||||
| 5 | H−2→L | 93.02 | 322.38 | 0.180 | 3.85 | z | |
| H−1→L+1 | 2.46 | z | |||||
| 9 | H−4→L | 29.11 | 261.61 | 0.124 | 4.69 | z | |
| H−4→L+4 | 29.54 | x,y | |||||
| H−4→L+5 | 14.13 | x,y | |||||
| 12 | H−8→L | 9.18 | 258.16 | 0.061 | 4.80 | z | |
| H−6→L | 38.48 | z |
| System | (−ω,ω) | Δα (−ω,ω) | ||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.413 | 0.583 | 0.653 | 0 | 0.583 | 0.653 | 0.413 | |
| 16N (C2) | 274 (348) | 275.5 (325) | 277 (327) | 277 (353) | 237 (329) | 239 (301) | 242 (304) | 243 (339) |
| 16P (S4) | 306 (381) | 307 (358) | 308 (385) | 309 (386) | 263 (344) | 264 (322) | 266 (350) | 267 (352) |
| 18P (D2h) | 349 (458) | 333 (424) | 353 (427) | 354 (467) | 333 (465) | 336 (425) | 339 (430) | 341 (479) |
| 18Py (C2h) | 364 (488) | 367 (495) | 371 (502) | 373 (506) | 360 (519) | 365 (529) | 370 (539) | 373 (545) |
| 20P (Ci) | 332 (424) | 333 (427) | 335 (430) | 336 (432) | 310 (428) | 313 (433) | 315 (438) | 317 (440) |
| 20O (C2v) | 368 (472) | 370 (476) | 373 (480) | 374 (483) | 354 (497) | 358 (553) | 361 (504) | 363 (508) |
| 22S (Ci) | 431 (573) | 434 (579) | 438 (585) | 439 (589) | 419 (595) | 424 (604) | 429 (614) | 432 (619) |
| 22I (Cs) | 466 (637) | 471 (647) | 476 (657) | 479 (663) | 475 (693) | 482 (707) | 489 (723) | 493 (732) |
| 22Sp (Cs) | 476 (641) | 480 (649) | 484 (657) | 486 (661) | 487 (698) | 493 (709) | 499 (721) | 502 (728) |
| System | βHRS | DR | β//(−2ω,ω,ω) | γ//(2ω,ω,ω,0) | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 0.653 | 0 | 0.653 | 0 | 0.653 | 0 | 0.653 | |
| 16N (C2) | 115 (211) | 119 (226) | 1.53 (1.54) | 1.15 (1.18) | 25 (−53) | −29 (−59) | 111 (247) | 25 (−1) |
| 16P (S4) | 118 (198) | 134 (229) | 1.50 (1.50) | 1.50 (1.50) | 0 (0) | 0 (0) | 162 (355) | 204 (461) |
| 18P (D2h) | 0 (0) | 0 (0) | -b | -b | 0 (0) | 0 (0) | 89 (20) | 102 (240) |
| 18Py (C2h) | 1 (3) | 2 (6) | -b | -b | 2 (4) | 2 (8) | −34 (−246) | −94 (−528) |
| 20P (Ci) | 0 (0) | 2 (3) | -b | -b | 0 (0) | 0 (1) | 199 (499) | 728 (1722) |
| 20O (C2v) | 1308 (2982) | 23376 c (86032) | 3.67 (3.89) | 1.73 c (1.65) | −1692 (−3956) | −22466 c (−81580) | 254 (649) | 5087 c (37351) |
| 22S (C1) | 1281 (3846) | 1967 (6307) | 6.22 (6.15) | 6.18 (6.29) | −1937 (−5762) | −2947 (−9373) | 322 (976) | 476 (1601) |
| 22I (Cs) | 1150 (3002) | 2255 (6629) | 3.41 (2.77) | 3.83 (2.90) | −1369 (−3201) | −2783 (−1067) | 78 (61) | 65 (−122) |
| 22Sp (Cs) | 516 (1279) | 623 (1652) | 1.66 (1.78) | 1.77 (2.11) | −232 (−686) | 310 (−1061) | 168 (475) | 230 (735) |
| System | π b | NICS(0) | NICS(1) | NICSzz(1) | HOMA | Π | Φp | ΔEH-L |
|---|---|---|---|---|---|---|---|---|
| 16N | 16 | 20.1 | 16.4 | 51.9 | 0.580 | 0.49 | 20.4 | 3.65 |
| 18P | 18 | −13.9 | −12.8 | −35.5 | 0.860 | 1.00 | 1.1 | 4.59 |
| 20O | 20 | 17.2 | 15.1 | 50.0 | 0.780 | 1.00 | 0.0 | 3.52 |
| 22S | 22 | −9.8 | −9.3 | −24.7 | 0.890 | 0.89 | 15.0 | 4.14 |
| 22I | 22 | −17.1 | −15.0 | −40.4 | 0.888 | 0.87 | 12.0 | 3.79 |
| βHRS | DR | β//(−2ω,ω,ω) | γ//(−2ω,ω,ω,0) | |||||
|---|---|---|---|---|---|---|---|---|
| ω | 0 | 0.653 | 0 | 0.653 | 0 | 0.653 | 0 | 0.653 |
| 16N (C2) | 3 (6) | 6 (12) | -b | -b | −1 (−1) | −1 (−1) | 375 (749) | 229 (376) |
| 18P (D2h) | 0 (0) | 0 (1.0) | -b | -b | 0 (0) | 0 (0) | 219 (458) | 268 (580) |
| 20O (C2v) | 1385 (3146) | 3373 (6628) | 3.64 (3.92) | 0.40 (0.27) | 1785 (4185) | −1614 (−2007) | 413 (964) | −363 (−831) |
| 22S (C1) | 2073 (4961) | 3448 (8763) | 3.50 (3.96) | 3.15 (3.69) | −1240 (−5045) | −1810 (−8372) | 825 (2078) | 1324 (3644) |
| 22I (Cs) | 2509 (5771) | 5191 (13727) | 4.14 (3.85) | 4.46 (4.07) | 3314 (7097) | 6725 (16649) | 447 (1058) | 7325 (1946) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woller, T.; Geerlings, P.; De Proft, F.; Champagne, B.; Alonso, M. Aromaticity as a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids. Molecules 2018, 23, 1333. https://doi.org/10.3390/molecules23061333
Woller T, Geerlings P, De Proft F, Champagne B, Alonso M. Aromaticity as a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids. Molecules. 2018; 23(6):1333. https://doi.org/10.3390/molecules23061333
Chicago/Turabian StyleWoller, Tatiana, Paul Geerlings, Frank De Proft, Benoît Champagne, and Mercedes Alonso. 2018. "Aromaticity as a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids" Molecules 23, no. 6: 1333. https://doi.org/10.3390/molecules23061333