
Comparative Targeted Proteomics of the Central Metabolism and Photosystems in SigE Mutant Strains of Synechocystis sp. PCC 6803


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
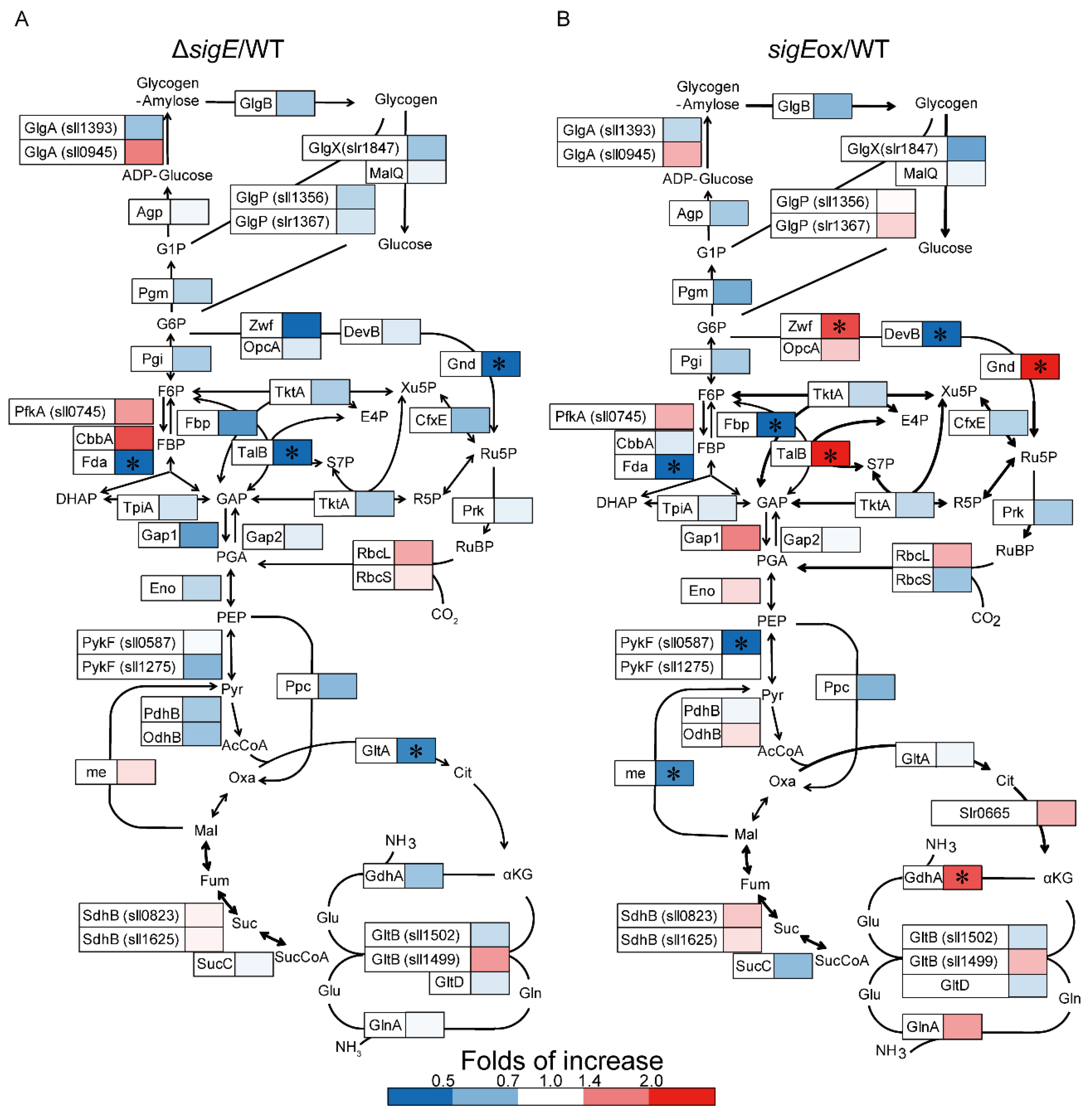
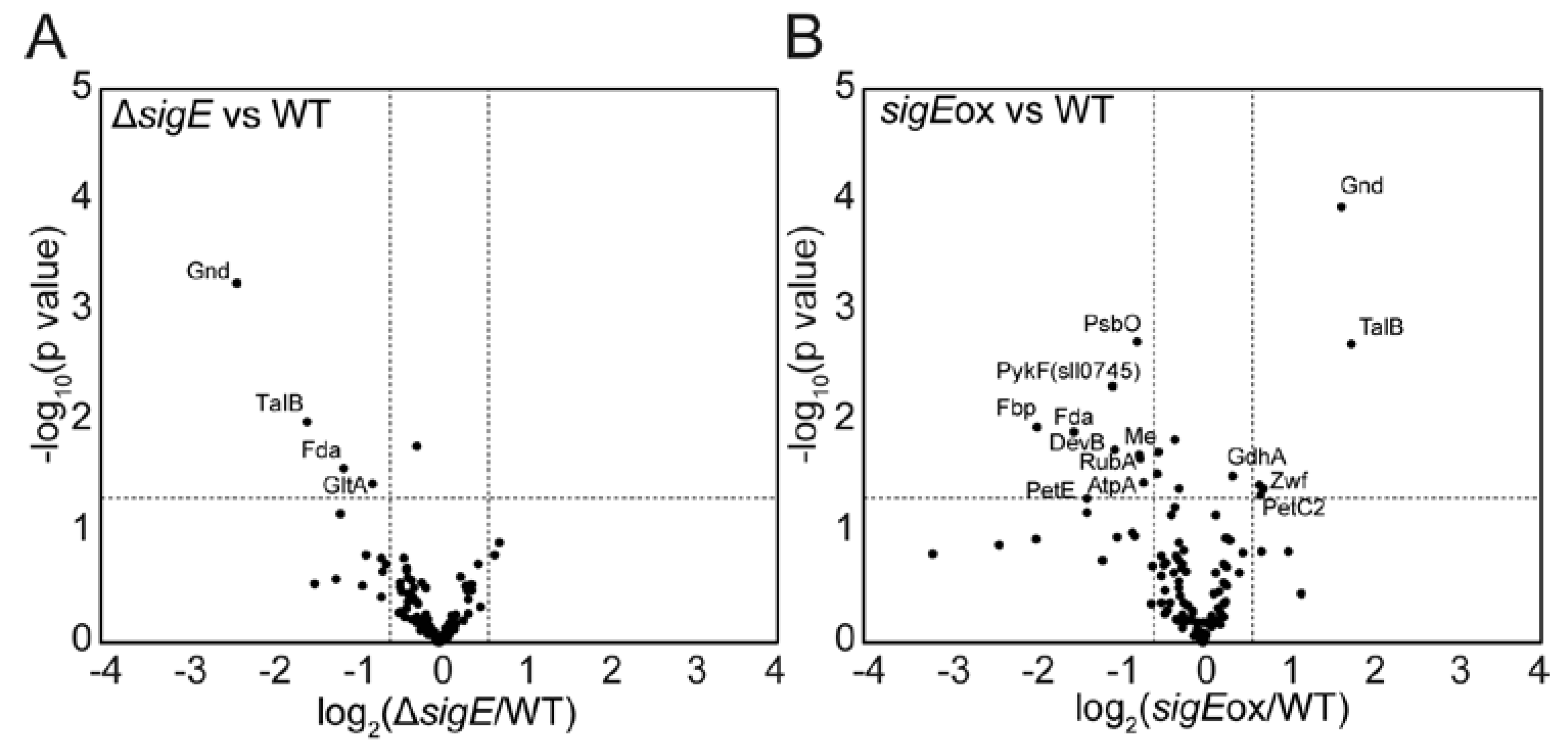
2.1. Targeted Proteome Analysis of Wild-Type, sigE Deleted and Overexpressed Strains of Synechocystis sp. PCC 6803
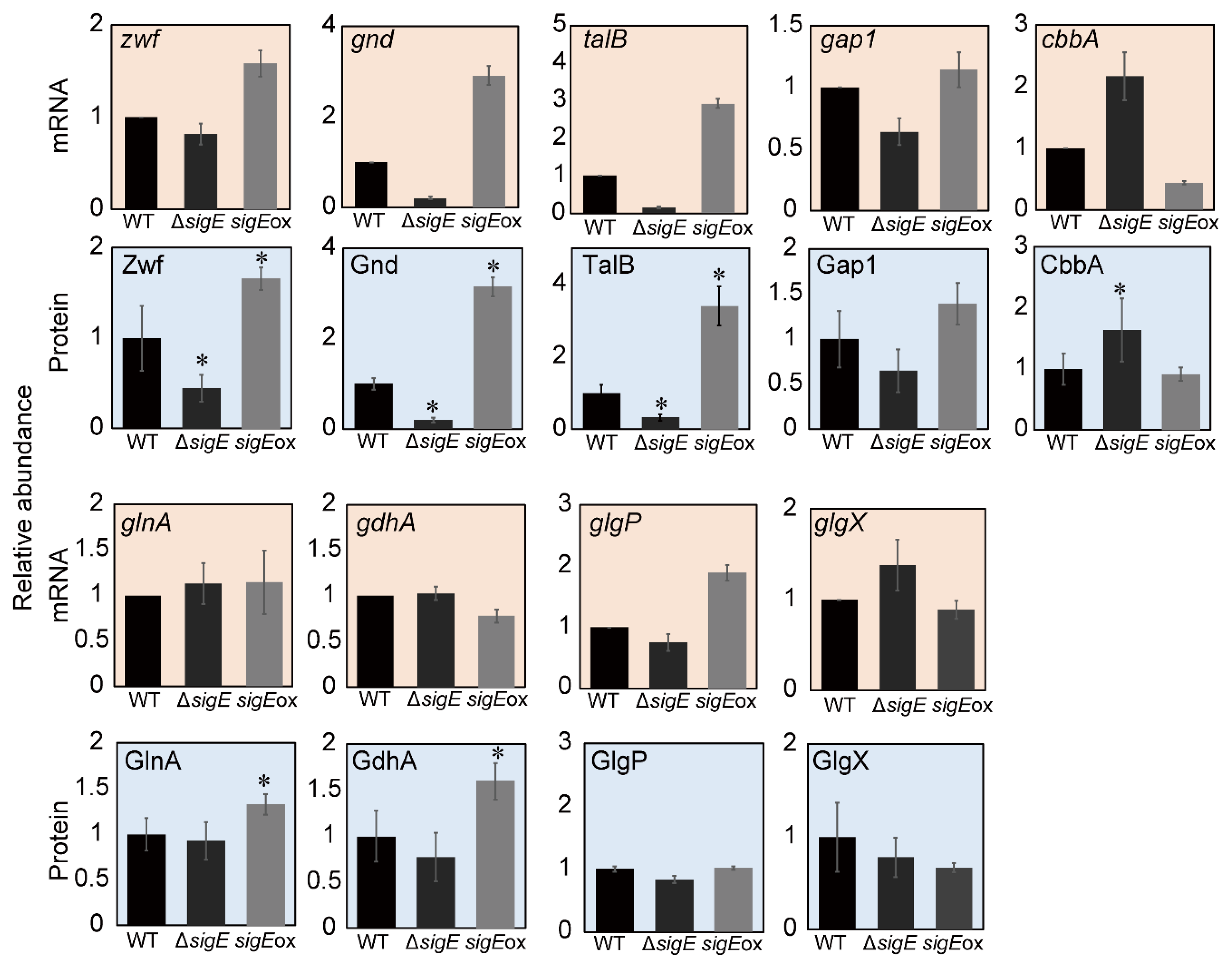
2.2. Comparison with the Gene-Expression Data
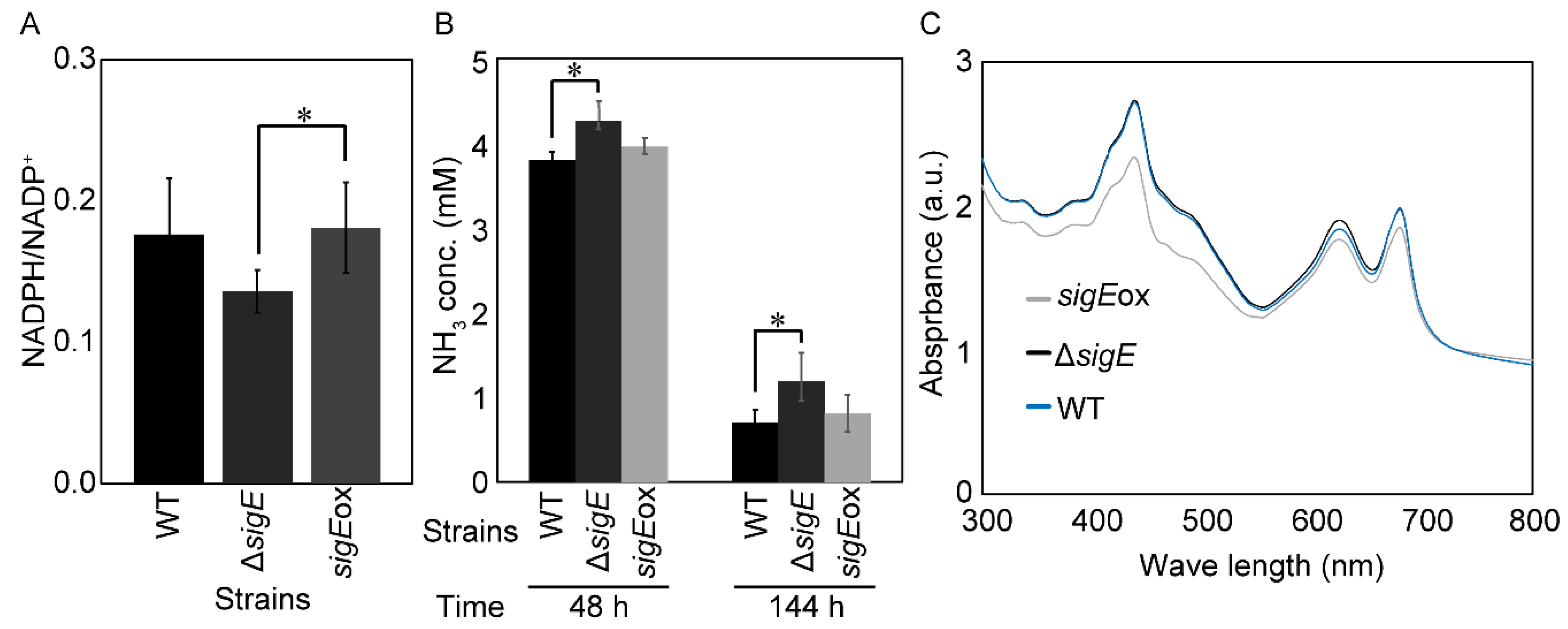
2.3. Effect of sigE Deletion and Overexpression on NADPH/NADP+ Ratio
2.4. SigE-Dependent Regulation of the Nitrogen Assimilation Pathway
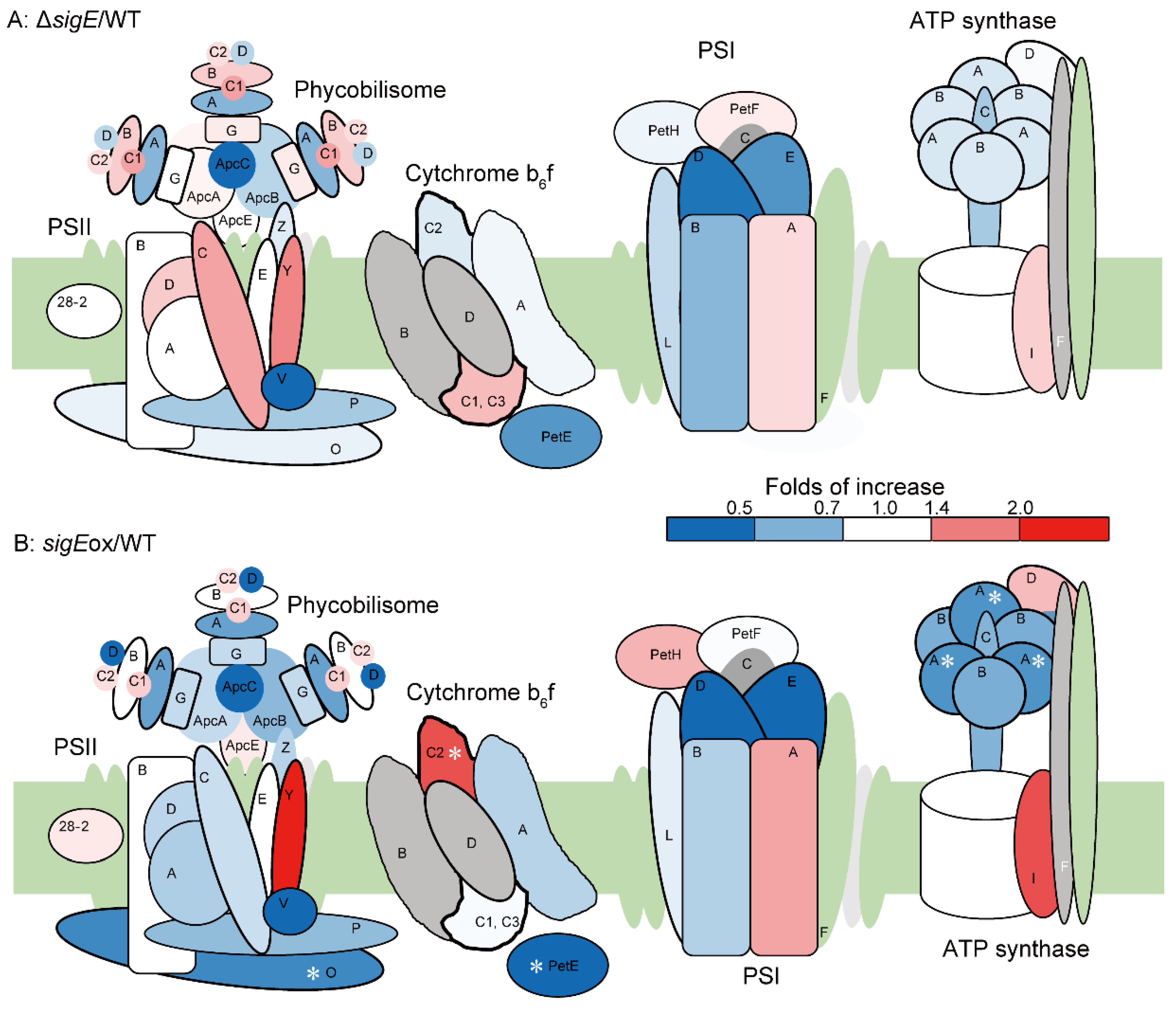
2.5. Protein Abundance Profiles of the Photosynthetic Apparatus
3. Materials and Methods
3.1. Bacterial Strains and Culture Conditions
3.2. Sample Preparation for Proteome Analysis
3.3. Reduction and Alkylation/Methanol Chloroform Precipitation
3.4. Trypsin/LysC Digestion
3.5. Desalting Samples
3.6. Design of Multiple Reaction Monitoring (MRM) Assay
3.7. Nano Liquid-Chromatography–Triple Quadrupole Mass Spectrometry (LC–MS/MS) Analysis by MRM Assay
3.8. Quantification of NH3 in the Medium
3.9. Ultraviolet–Visible (UV–VIS) Spectrum
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dismukes, G.C.; Carrieri, D.; Bennette, N.; Ananyev, G.M.; Posewitz, M.C. Aquatic phototrophs: Efficient alternatives to land-based crops for biofuels. Curr. Opin. Biotechnol. 2008, 19, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Georgianna, D.R.; Mayfield, S.P. Exploiting diversity and synthetic biology for the production of algal biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Leite, G.B.; Abdelaziz, A.E.M.; Hallenbeck, P.C. Algal biofuels: Challenges and opportunities. Bioresour. Technol. 2013, 145, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondo, S.; Takahashi, M.; Osanai, T.; Matsuda, M.; Hasunuma, T.; Tazuke, A.; Nakahira, Y.; Chohnan, S.; Hasegawa, M.; Asayama, M. Genetic engineering and metabolite profiling for overproduction of polyhydroxybutyrate in cyanobacteria. J. Biosci. Bioeng. 2015, 120, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Mikkat, S.; Huang, F.; Huckauf, J.; Marin, K.; Norling, B.; Hagemann, M. Proteome analysis of salt stress response in the cyanobacterium Synechocystis sp. strain PCC 6803. Proteomics 2006, 6, 2733–2745. [Google Scholar] [CrossRef] [PubMed]
- Kurian, D.; Phadwal, K.; Mäenpää, P. Proteomic characterization of acid stress response in Synechocystis sp. PCC 6803. Proteomics 2006, 6, 3614–3624. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Aikawa, S.; Sasaki, K.; Teramura, H.; Hasunuma, T.; Matsuda, F.; Osanai, T.; Hirai, M.Y.; Kondo, A. Rre37 stimulates accumulation of 2-oxoglutarate and glycogen under nitrogen starvation in Synechocystis sp. PCC 6803. FEBS Lett. 2014, 588, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Azuma, M.; Tanaka, K. Sugar catabolism regulated by light- and nitrogen-status in the cyanobacterium Synechocystis sp. PCC 6803. Photochem. Photobiol. Sci. 2007, 6, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Imamura, S.; Asayama, M.; Shirai, M.; Suzuki, I.; Murata, N.; Tanaka, K. Nitrogen induction of sugar catabolic gene expression in Synechocystis sp. PCC 6803. DNA Res. 2006, 13, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Kanesaki, Y.; Nakano, T.; Takahashi, H.; Asayama, M.; Shirai, M.; Kanehisa, M.; Suzuki, I.; Murata, N.; Tanaka, K. Positive regulation of sugar catabolic pathways in the cyanobacterium Synechocystis sp. PCC 6803 by the group 2 σ factor SigE. J. Biol. Chem. 2005, 280, 30653–30659. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Sato, S.; Tabata, S.; Tanaka, K. Identification of PamA as a PII-binding membrane protein important in nitrogen-related and sugar-catabolic gene expression in Synechocystis sp. PCC 6803. J. Biol. Chem. 2005, 280, 34684–34690. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Imamura, S.; Tanaka, K.; Shirai, M.; Asayama, M. Cooperation of group 2 σ factors, SigD and SigE for light-induced transcription in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett. 2007, 581, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Oikawa, A.; Azuma, M.; Tanaka, K.; Saito, K.; Hirai, M.Y.; Ikeuchi, M. Genetic engineering of group 2 σ factor SigE widely activates expressions of sugar catabolic genes in Synechocystis species PCC 6803. J. Biol. Chem. 2011, 286, 30962–30971. [Google Scholar] [CrossRef] [PubMed]
- Hakkila, K.; Antal, T.; Gunnelius, L.; Kurkela, J.; Matthijs, H.C.P.; Tyystjärvi, E.; Tyystjarvi, T. Group 2 sigma factor mutant ΔsigCDE of the cyanobacterium Synechocystis sp. PCC 6803 reveals functionality of both carotenoids and flavodiiron proteins in photoprotection of photosystem II. Plant Cell Physiol. 2013, 54, 1780–1790. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Numata, K.; Oikawa, A.; Kuwahara, A.; Iijima, H.; Doi, Y.; Tanaka, K.; Saito, K.; Hirai, M.Y. Increased bioplastic production with an RNA polymerase sigma factor SigE during nitrogen starvation in Synechocystis sp. PCC 6803. DNA Res. 2013, 20, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Huokko, T.; Muth-Pawlak, D.; Battchikova, N.; Allahverdiyeva, Y.; Aro, E.-M. Role of type 2 NAD(P)H dehydrogenase NdbC in redox regulation of carbon allocation in Synechocystis. Plant Physiol. 2017, 174, 1863–1880. [Google Scholar] [CrossRef] [PubMed]
- Veit, S.; Takeda, K.; Tsunoyama, Y.; Baymann, F.; Nevo, R.; Reich, Z.; Rögner, M.; Miki, K.; Rexroth, S. Structural and functional characterisation of the cyanobacterial PetC3 Rieske protein family. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Pade, N.; Mikkat, S.; Hagemann, M. Ethanol, glycogen and glucosylglycerol represent competing carbon pools in ethanol-producing cells of Synechocystis sp. PCC 6803 under high-salt conditions. Microbiology (UK) 2017, 163, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Borirak, O.; de Koning, L.J.; van der Woude, A.D.; Hoefsloot, H.C.J.; Dekker, H.L.; Roseboom, W.; de Koster, C.G.; Hellingwerf, K.J. Quantitative proteomics analysis of an ethanol- and a lactate-producing mutant strain of Synechocystis sp. PCC6803. Biotechnol. Biofuels 2015, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Ge, H.; Huang, X.; Liu, Y.; Lu, M.; Wang, J.; Chen, W.; Xu, W.; Wang, Y. Trophic mode-dependent proteomic analysis reveals functional significance of light-independent chlorophyll synthesis in Synechocystis sp. PCC 6803. Mol. Plant 2017, 10, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, J.; Ge, H.; Fang, L.; Zhang, Y.; Huang, X.; Wang, Y. Toward the complete proteome of Synechocystis sp. PCC 6803. Photosynth. Res. 2015, 126, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Plohnke, N.; Seidel, T.; Kahmann, U.; Rögner, M.; Schneider, D.; Rexroth, S. The proteome and lipidome of Synechocystis sp. PCC 6803 cells grown under light-activated heterotrophic conditions. Mol. Cell. Proteom. 2015, 14, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Shi, M.; Chen, L.; Wang, J.; Zhang, W. Integrated proteomic and metabolomic characterization of a novel two-component response regulator Slr1909 involved in acid tolerance in Synechocystis sp. PCC 6803. J. Proteom. 2014, 109, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Liebler, D.C.; Zimmerman, L.J. Targeted quantitation of proteins by mass spectrometry. Biochemistry 2013, 52, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Tachikawa, M.; Obuchi, W.; Hoshi, Y.; Tomioka, Y.; Ohtsuki, S.; Terasaki, T. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: Application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood-brain barrier in ddY, FVB, and C57BL/6J mice. Fluids Barriers CNS 2013, 10, 21. [Google Scholar] [PubMed]
- Vuorijoki, L.; Isojärvi, J.; Kallio, P.; Kouvonen, P.; Aro, E.M.; Corthals, G.L.; Jones, P.R.; Muth-Pawlak, D. Development of a quantitative SRM-based proteomics method to study iron metabolism of Synechocystis sp. PCC 6803. J. Proteome Res. 2016, 15, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Vuorijoki, L.; Kallio, P.; Aro, E.M. SRM dataset of the proteome of inactivated iron-sulfur cluster biogenesis regulator SufR in Synechocystis sp. PCC 6803. Data BR 2017, 11, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Vuorijoki, L.; Tiwari, A.; Kallio, P.; Aro, E.M. Inactivation of iron-sulfur cluster biogenesis regulator SufR in Synechocystis sp. PCC 6803 induces unique iron-dependent protein-level responses. Biochim. Biophys. Acta 2017, 1861, 1085–1098. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, K.; Yamamoto, H.; Miyake, C.; Yokota, A. Purification and characterization of class-I and class-II fructose-1,6-bisphosphate aldolases from the cyanobacterium Synechocystis sp. PCC6803. Plant Cell Physiol. 2003, 44, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Kajihata, S.; Yoshikawa, K.; Matsuda, F.; Furusawa, C.; Hirasawa, T.; Shimizu, H. Integrated metabolic flux and omics analysis of Synechocystis sp. PCC 6803 under mixotrophic and photoheterotrophic conditions. Plant Cell Physiol. 2014, 55, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- You, L.; He, L.; Tang, Y.J. Photoheterotrophic fluxome in Synechocystis sp. strain PCC 6803 and its implications for cyanobacterial bioenergetics. J. Bacteriol. 2015, 197, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Muro-Pastor, M.I.; Reyes, J.C.; Florencio, F.J. Ammonium assimilation in cyanobacteria. Photosynth. Res. 2005, 83, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Von Wobeser, E.A.; Ibelings, B.W.; Bok, J.; Krasikov, V.; Huisman, J.; Matthijs, H.C.P. Concerted changes in gene expression and cell physiology of the cyanobacterium Synechocystis sp. strain PCC 6803 during transitions between nitrogen and light-limited growth. Plant Physiol. 2011, 155, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Chávez, S.; Reyes, J.C.; Chauvat, F.; Florencio, F.J.; Candau, P. The NADP-glutamate dehydrogenase of the cyanobacterium Synechocystis 6803: Cloning, transcriptional analysis and disruption of the gdhA gene. Plant Mol. Biol. 1995, 28, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Schriek, S.; Aguirre-Von-Wobeser, E.; Nodop, A.; Becker, A.; Ibelings, B.W.; Bok, J.; Staiger, D.; Matthijs, H.C.P.; Pistorius, E.K.; Michel, K.P. Transcript profiling indicates that the absence of PsbO affects the coordination of C and N metabolism in Synechocystis sp. PCC 6803. Physiol. Plant. 2008, 133, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Zhao, J.; Reimer, S.K.; Antonkine, M.L.; Cai, Q.; Weiland, S.M.; Golbeck, J.H.; Bryant, D.A. Assembly of photosystem I: I. Inactivation of the rubA gene encoding a membrane-associated rubredoxin in the cyanobacterium Synechococcus sp. PCC 7002 causes a loss of photosystem I activity. J. Biol. Chem. 2002, 277, 20343–20354. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Jiang, H.B.; Zhang, R.; Qiu, B.S. Inactivation of the petE gene encoding plastocyanin causes different photosynthetic responses in cyanobacterium Synechocystis PCC 6803 under light-dark photoperiod and continuous light conditions. FEMS Microbiol. Lett. 2013, 341, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Kuwahara, A.; Iijima, H.; Toyooka, K.; Sato, M.; Tanaka, K.; Ikeuchi, M.; Saito, K.; Hirai, M.Y. Pleiotropic effect of sigE over-expression on cell morphology, photosynthesis and hydrogen production in Synechocystis sp. PCC 6803. Plant J. 2013, 76, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Coruzzi, G.M.; Zhou, L. Carbon and nitrogen sensing and signaling in plants: Emerging “matrix effects”. Curr. Opin. Plant Biol. 2001, 4, 247–253. [Google Scholar] [CrossRef]
- Richaud, C.; Zabulon, G.; Joder, A.; Thomas, J.C. Nitrogen or sulfur starvation differentially affects phycobilisome degradation and expression of the nblA gene in Synechocystis strain PCC 6803. J. Bacteriol. 2001, 183, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Krasikov, V.; Aguirre von Wobeser, E.; Dekker, H.L.; Huisman, J.; Matthijs, H.C.P. Time-series resolution of gradual nitrogen starvation and its impact on photosynthesis in the cyanobacterium Synechocystis PCC 6803. Physiol. Plant. 2012, 145, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Elvitigala, T.; Bhattacharyya-Pakrasi, M.; Aurora, R.; Ghosh, B.; Pakrasi, H.B. Integration of carbon and nitrogen metabolism with energy production is crucial to light acclimation in the cyanobacterium Synechocystis. Plant Physiol. 2008, 148, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Antal, T.; Kurkela, J.; Parikainen, M.; Kårlund, A.; Hakkila, K.; Tyystjärvi, E.; Tyystjärvi, T. Roles of group 2 sigma factors in acclimation of the cyanobacterium Synechocystis sp. PCC 6803 to nitrogen deficiency. Plant Cell Physiol. 2016, 57, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Muro-Pastor, A.M.; Herrero, A.; Flores, E. Nitrogen-regulated group 2 sigma factor from Synechocystis sp. strain PCC 6803 involved in survival under nitrogen stress. J. Bacteriol. 2001, 183, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, B.; Hakkila, K.; Georg, J.; Tyystjärvi, T.; Hess, W.R.; Axmann, I.M.; Dienst, D. 6S RNA plays a role in recovery from nitrogen depletion in Synechocystis sp. PCC 6803. BMC Microbiol. 2017, 17, 229. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.K. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. Methods Enzymol. 1988, 167, 766–778. [Google Scholar]
- Schuurmans, R.M.; Matthijs, J.C.P.; Hellingwerf, K.J. Transition from exponential to linear photoautotrophic growth changes the physiology of Synechocystis sp. PCC 6803. Photosynth. Res. 2017, 132, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flügge, U.I. A Method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Rappsilber, J.; Mann, M. Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 2006, 5, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, F.; Tomita, A.; Shimizu, H. Prediction of hopeless peptides unlikely to be selected for targeted proteome analysis. Mass Spectrom. (Tokyo) 2017, 6, A0056. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Mori, N.; Moriyama, T.; Misumi, O.; Sato, N. Analysisi of triacylglycerol accumulation under nitrogen deprivation in the red alga Cyanidioschyzon merolae. Microbiology 2016, 803–812. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors upon request. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokumaru, Y.; Uebayashi, K.; Toyoshima, M.; Osanai, T.; Matsuda, F.; Shimizu, H. Comparative Targeted Proteomics of the Central Metabolism and Photosystems in SigE Mutant Strains of Synechocystis sp. PCC 6803. Molecules 2018, 23, 1051. https://doi.org/10.3390/molecules23051051
Tokumaru Y, Uebayashi K, Toyoshima M, Osanai T, Matsuda F, Shimizu H. Comparative Targeted Proteomics of the Central Metabolism and Photosystems in SigE Mutant Strains of Synechocystis sp. PCC 6803. Molecules. 2018; 23(5):1051. https://doi.org/10.3390/molecules23051051
Chicago/Turabian StyleTokumaru, Yuma, Kiyoka Uebayashi, Masakazu Toyoshima, Takashi Osanai, Fumio Matsuda, and Hiroshi Shimizu. 2018. "Comparative Targeted Proteomics of the Central Metabolism and Photosystems in SigE Mutant Strains of Synechocystis sp. PCC 6803" Molecules 23, no. 5: 1051. https://doi.org/10.3390/molecules23051051