
1-(Acylamino)alkylphosphonic Acids—Alkaline Deacylation
by
 , ,
, ,
Marek Cypryk
1 ,
,
Jozef Drabowicz
1,2,*,
Bartlomiej Gostynski
1,
Marcin H. Kudzin
3,
Zbigniew H. Kudzin
4,* and
Pawel Urbaniak
4 1
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 120a, Lodz 90-363, Poland
2
Department of Chemistry and Environment Protection, Jan Dlugosz University, Czestochowa 42-200, Poland
3
Textile Research Institute, Brzezinska 5/15, Lodz 92-103, Poland
4
Faculty of Chemistry, University of Lodz, Tamka 12, Lodz 91-403, Poland
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(4), 859; https://doi.org/10.3390/molecules23040859
Submission received: 5 March 2018
/
Revised: 26 March 2018
/
Accepted: 28 March 2018
/
Published: 9 April 2018
(This article belongs to the Special Issue Organophosphorus Chemistry 2018)
Abstract
:The alkaline deacylation of a representative series of 1-(acylamino)alkylphosphonic acids [(AC)-AAP: (AC) = Ac, TFA, Bz; AAP = GlyP, AlaP, ValP, PglP and PheP] in an aqueous solution of KOH (2M) was investigated. The results suggested a two-stage reaction mechanism with a quick interaction of the hydroxyl ion on the carbonyl function of the amide R-C(O)-N(H)- group in the first stage, which leads to instant formation of the intermediary acyl-hydroxyl adducts of R-C(O−)2-N(H)-, visible in the 31P NMR spectra. In the second stage, these intermediates decompose slowly by splitting of the RC(O−)2-N(H)- function with the subsequent formation of 1-aminoalkylphosphonate and carboxylate ions.
1. Introduction
Aminoalkylphosphonic acids (AAP) are structural analogues of amino acids (AAC) [1], some of which are of natural origin [2]. Due to the structural analogy, they present similar biological properties to the class and are important inhibitors of enzymes of the amino acids metabolism [1,3] (Table 1).
Several papers reflected the complexing abilities of this class of compounds [16,17,18] and their pharmacological [19,20,21,22], agro-chemical [23] and industrial [11,24,25] applications. Therefore, the studies on the synthesis [26,27,28,29,30] and physico-chemical properties [31,32,33,34,35,36,37,38,39,40] of AAP and their derivatives constitute an important topic in chemistry and biochemistry also in addition to material science (e.g., Self-assembled monolayers agents, SAMs [41]).
The 1-(acylamino)alkylphosphonic acids (AC)-AAP belong to the interesting group of compounds that is of potential pharmacological importance since the corresponding 1-N-aminoacylamino derivatives of AAC-AAP (mixed P-terminal phosphono-dipeptides, e.g., Alaphosphaline) exhibit antibacterial activity [1,23] (Table 1).
1-(Acylamino)alkylphosphonic acids [36,37] and esters [42] are easily formed from starting aminoalkylphosphonic acids, the latter being substrates for selective deacylation to O,O-dialkyl aminoalkylphosphonates [43]. Both of these groups are substrates for stereoselective enzymatic hydrolysis [44,45,46,47,48] (Figure 1). 1-(Acylamino)alkylphosphonic acids are also formed in so-called Engelman-Pikl-Oleksyszyn method synthesis of 1-aminoalkylphosphonic and 1-aminoalkylphosphonic acids, which starts from appropriate amides and phosphorus chlorides [10,27,49,50] ( Figure 2). As some secondary 1-aminoalkanephosphonates are unstable in the process of acidic degradation [40], alkaline hydrolysis can present an alternative method in such procedures.
The susceptibility of (AC)-AAP to hydrolysis/solvolysis constitutes an important factor influencing their biological activity, especially during penetration through cell membranes. Therefore, the detailed studies on deacylation of these compounds should be helpful in obtaining a deeper understanding of this phenomenon. To tackle this problem, we have decided to carry out studies devoted to deacylation of (AC)-AAP under different reaction conditions. Our preliminary results of experiments were carried out in aqueous media in the pH range of 0–6.5, which occurs namely in an aqueous 2M HCl and buffer solutions. These are briefly described in a recently published article [51]. As an obvious and necessary continuation of this topic, this paper presents our results on the deacylation of representative types of 1-(acylamino)alkylphosphonic acids (AC)-AAP in aqueous 2M KOH solution. These include 1-(acetylamino)alkylphosphonic Ac-AAP, 1-(trifluoroacetylamino)alkylphosphonic acids TFA-AAP and 1-(benzoylamino)alkylphosphonic Bz-AAP, derived from representative 1-aminoalkylphosphonic acids AAP (GlyP, AlaP, ValP, PglP and PheP).
2. Results and Discussion
It is generally known that amides can be hydrolyzed with either acidic or basic catalysis, with products being the free acid and the ammonium/substituted ammonium ions or the salts of the acid and ammonia/amine, respectively. Both the acid- and base-catalyzed hydrolyses are essentially irreversible, since salts are formed in both cases [52,53,54,55]. Water alone is not sufficient to hydrolyze most of amides [56]. The very low rate of amide hydrolysis by water has been measured by Kahne & Still [57].
A kinetic study has been conducted on the alkaline hydrolyses of several types of amides, including formanilides [58,59], N-methylformanilides and N-acetanilides [60], 1,8-bis(trifluoroacetylamino)-naphthalene [61] also in addition to N-methylacetamide, N-methylbenzamide and acetanilide [62]. A mild protocol for the alkaline hydrolysis of secondary and tertiary amides in non-aqueous conditions at room temperature or under reflux has been recently described [63].
The structural analogy of 1-(acylamino)alkylphosphonic acids (AC)-AAP and 1-(acylamino)-alkanoic acids (AC)-AAC or amides causes the mechanism of hydrolytic scission of the amide linkage R-C(O)-N in common amides and in 1-(acylamino)alkylphosphonic acids. These should be ruled by similar if not the same mechanisms.
2.1. Investigations on the Deacylation Course of 1-(acylamino)alkylphosphonic Acids
The (AC)-AAP contain amidic functions, which has a hydrolytic sensitivity that should be dependent on the structure of their acyl moieties and on the type of applied hydrolytic medium [49]. For conducting an inquiry into the hydrolytic stability of (AC)-AAP, we undertook deacylation investigations of these compounds in aqueous 2M KOH solutions.
The application of 31P NMR monitoring of the reaction course enabled identification and quantification of phosphoric components of the reaction mixtures formed. Essentially, monitoring of the deacylation course of Ac-GlyP in 2M KOH solution reveals the formation of GlyP (Figure 3). The results of 31P NMR monitoring of the deacylation of the various types of (AC)-AAP (Ac-AAP, Bz-AAP and TFA-AAP) at a temperature of 25 °C and exposition period up to 240 h are presented in Figure 4 (Ac-AAP), Figure 5 (TFA-AAP) and Figure 6 (TFA-AAP), respectively.
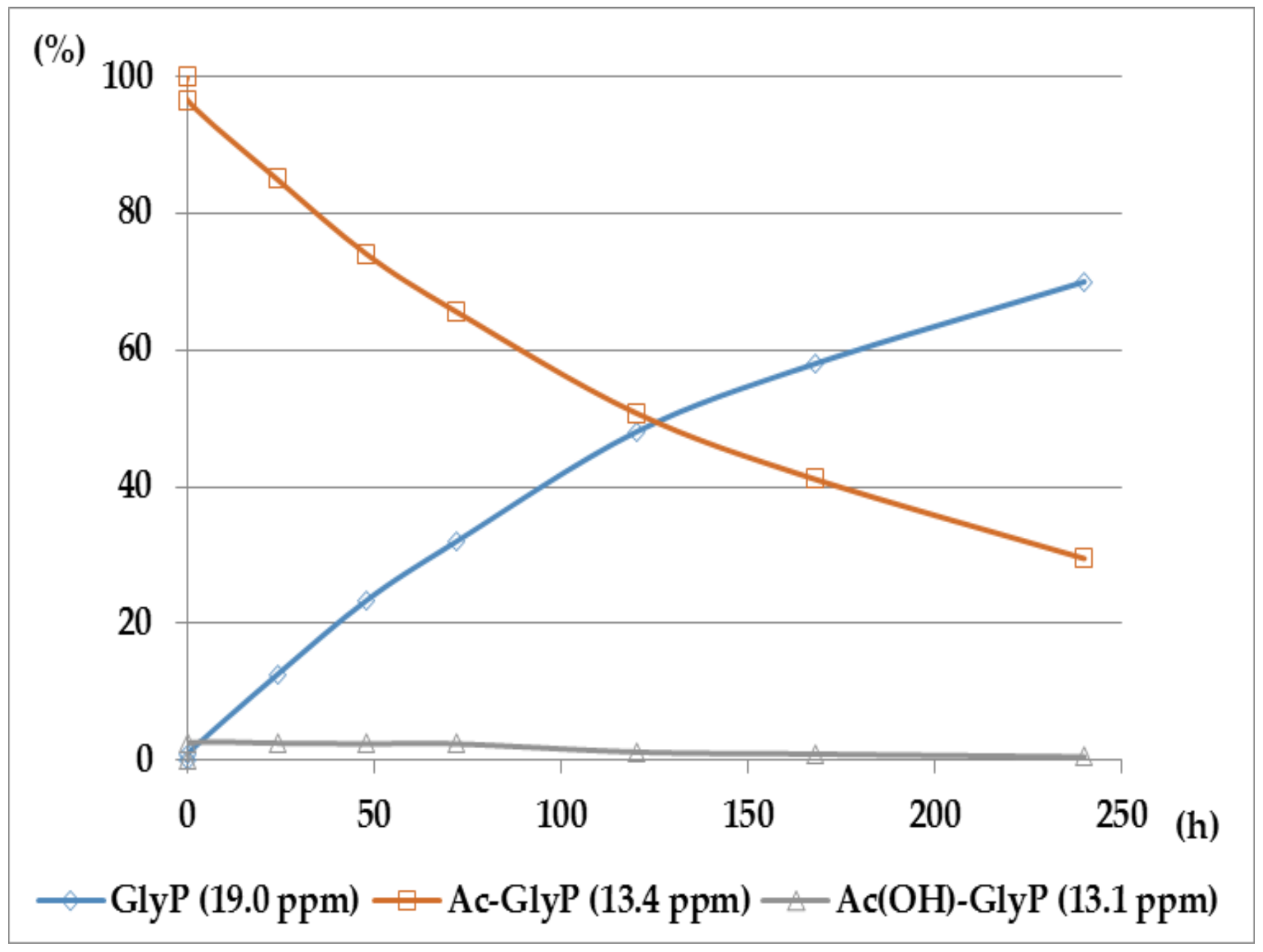
The comparison of hydrolytic stability of TFA-GlyP, Ac-GlyP and Bz-GlyP in 2M KOH is presented in Figure 7.
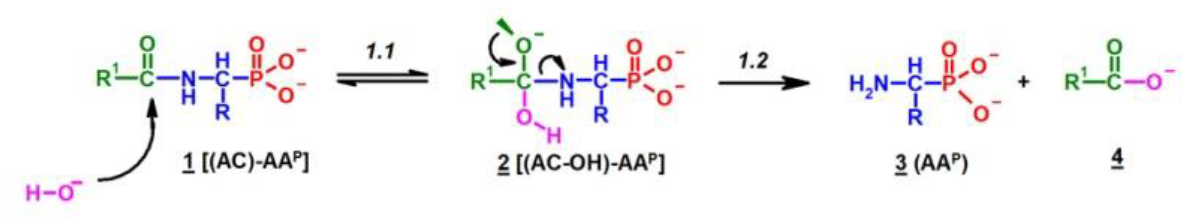
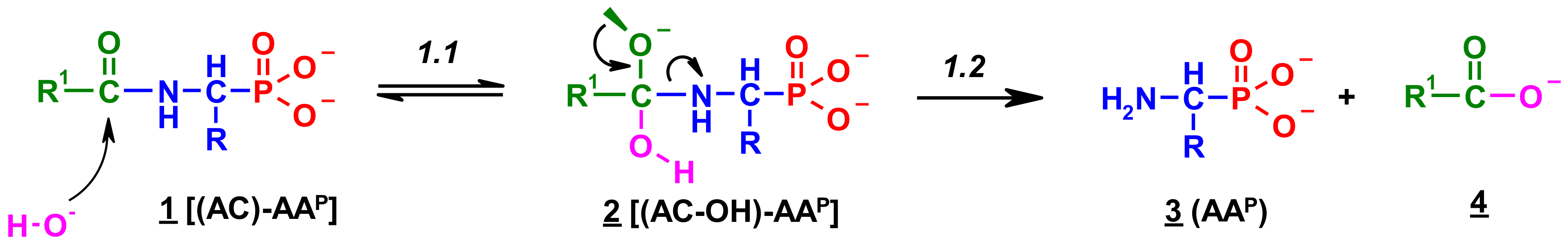
Since no products of the scission of the C-P bond in all investigated cases were observed, the deacylation mechanism of (AC)-AAP in alkaline solutions can be illustrated by an analogy to the corresponding basic deacylation of 1-(acylamino)alkanoic acids (Scheme 1) [54,62,63].
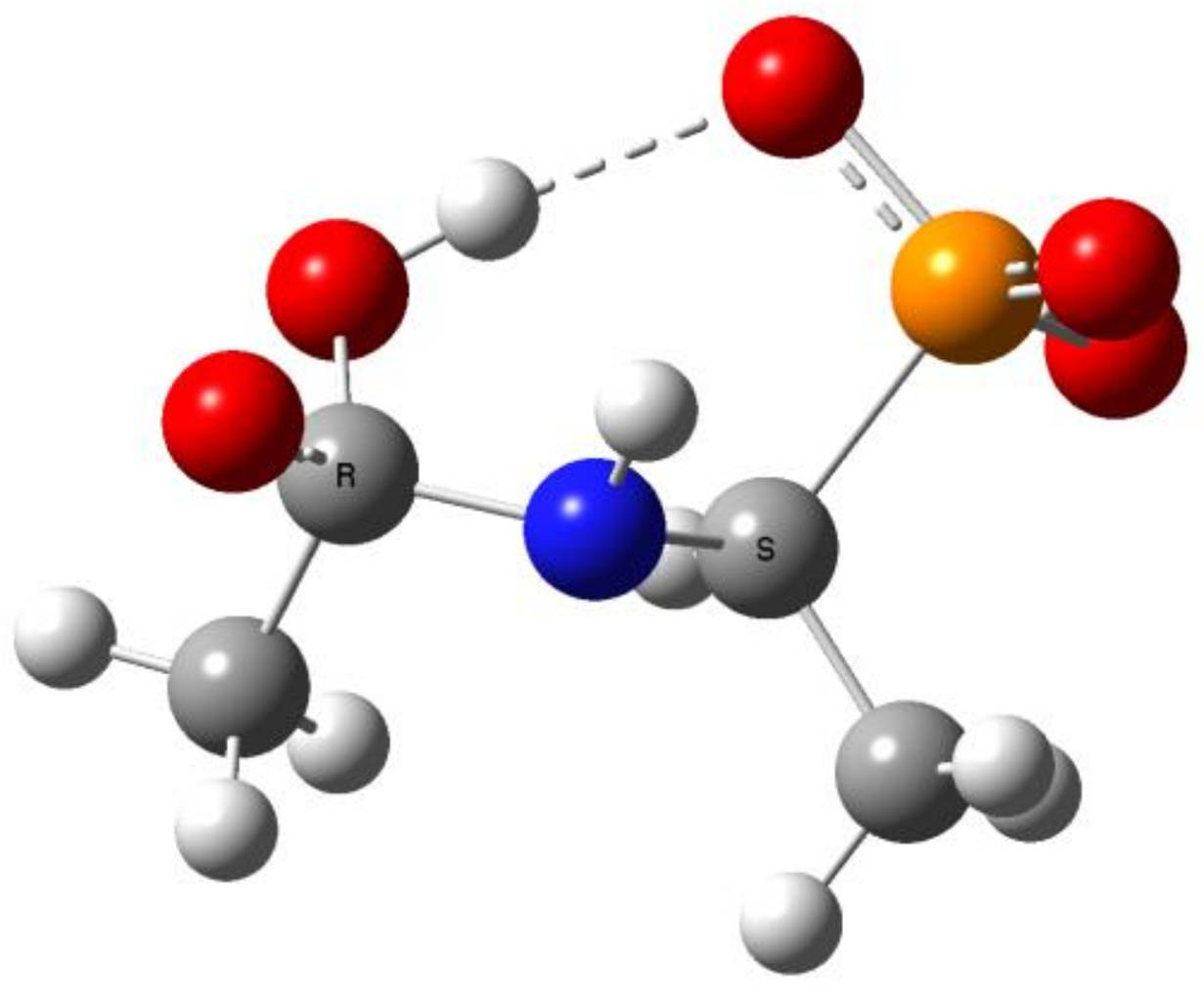
This includes the prior interaction of hydroxyl ion on the carbonyl carbon of 1, which occurs with the formation of the tetrahedral intermediate state 2 (Scheme 1). These intermediates were observed on 31P NMR spectra as signals down-shifted in comparison with the parent signals of AC-AAP (e.g., Figure 2). The 31P NMR chemical shifts of (AC)-AAP (1) and the corresponding adduct compounds AC(OH)-AAP (2) formed are listed in Table 2.
These compounds [AC(OH)]-AAP] slowly disappeared during the deacylation progress (Figure 8).
The rehybridization (sp3→sp2) of the carbonyl carbon in 2 enforces the splitting of the amide bond R-C(O)-N and the subsequent formation of the aminophosphonate 3.
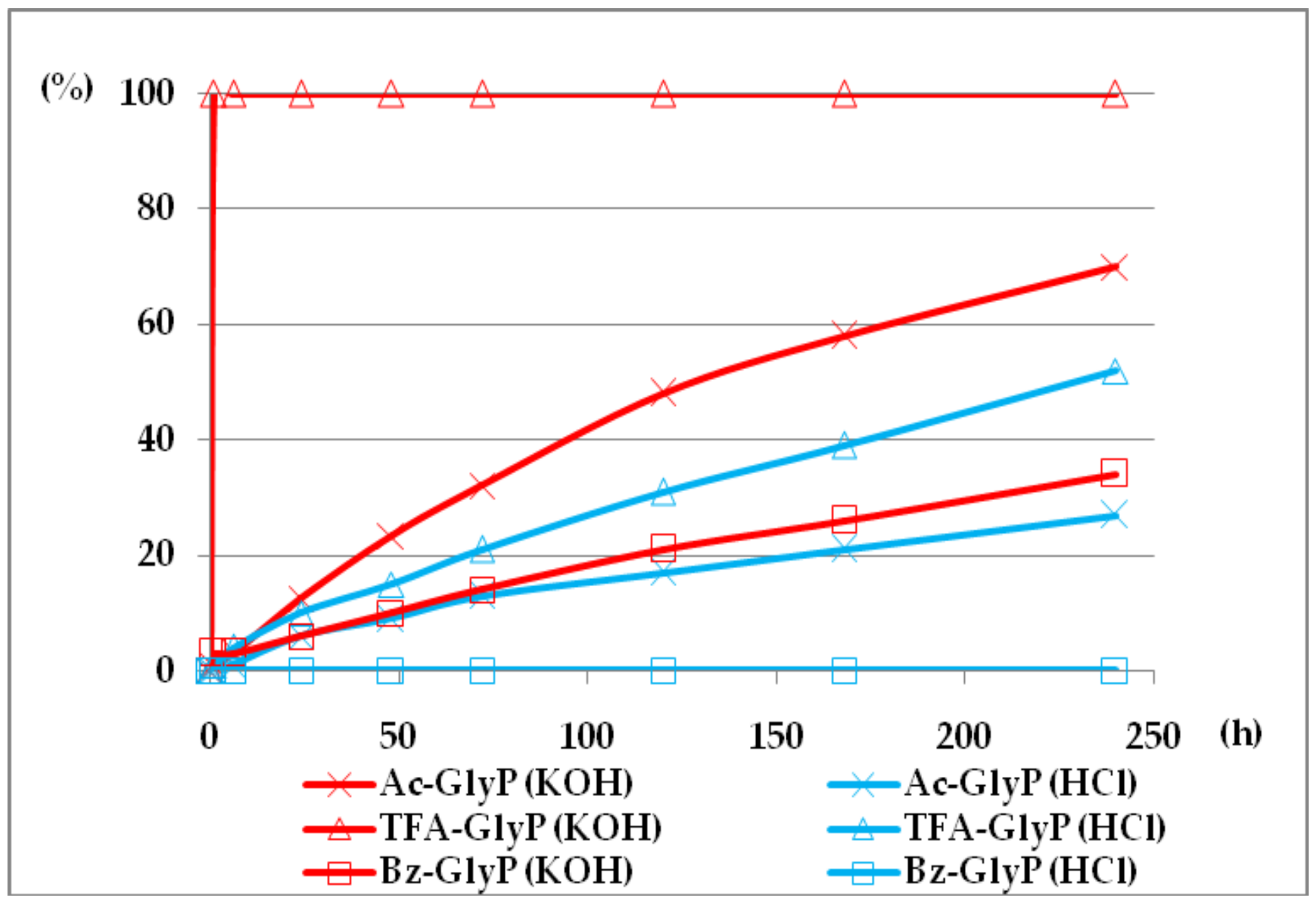
The comparison of (AC)-AAP stability in aqueous 2M KOH and 2M HCl solutions is given in Figure 9.
2.2. Quantum Chemical Calculations for Deacylation of (AC)-AAP in Basic Conditions
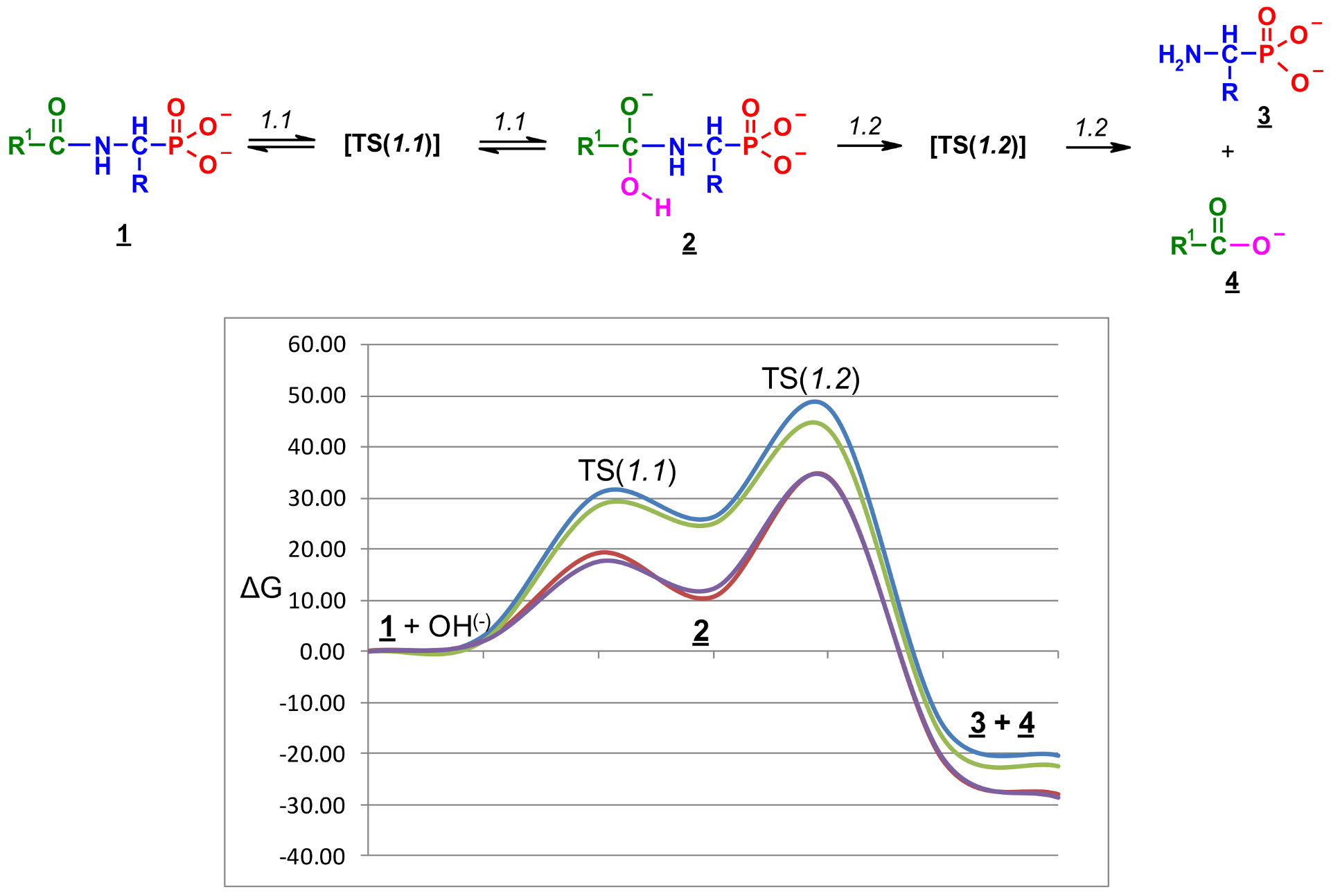
The reaction illustrated in Scheme 1 was divided into two stages (reactions 1.1 and 1.2, respectively), which were studied separately. Stage 1.1 concerns the addition of hydroxide to amidophosphonate anion 1 with the formation of trianion 2 (the example structure of the addition product 2 for Ac-AlaP (R=R1=Me) is given in Figure 10). Stage 1.2 involves dissociation of the trianion 2 into aminophosphonate dianion 3 and carboxylate anion 4. We assumed that the transfer of proton from OH to the amide nitrogen in 2 proceeds simultaneously with dissociation. The thermodynamics of this two-stage process was studied by density functional theory (DFT) methods in the gas phase and in aqueous solution. Gas phase calculations were performed only for comparison with the aqueous system, which was the real reaction system to illustrate the essential role of the solvation effect.
This reaction stage presumably proceeds by the attack of a lone pair of negatively charged oxygen atoms on sp2 carbon in the carbonyl group. This process is highly unfavorable since it involves the reaction of two anions with the formation of the molecule with a total charge of −3. The energy of this reaction in the gas phase is very high, so this process seems to be thermodynamically very unlikely (Table 3).
However, when carried out in aqueous solutions, this reaction is much less unfavorable due to the stabilization of ionic structures by the polar solvent (Table 2). Similarly, this reaction should also be facilitated by counterions, although these were omitted in order to keep the model system simple. The values of the Gibbs free energy ΔG298 (kcal/mol) for reaction 1.1 in gas phase and in aqueous phase are listed in Table 3 and Table 4, respectively.
The free energy barriers, ΔG‡ (298 K), calculated for reaction 1.1 in a water solution are shown in Table 5. The lowest energy barriers for OH− addition (Scheme 1, r. 1.1) occur when R1 = CF3, which results from stabilization of the negative charge by the electron-withdrawing CF3 group.
Stage 2: Dissociation of trianion 2 into aminophosphonate dianion 3 and carboxylate anion 4 (Scheme 1, reaction 1.2).
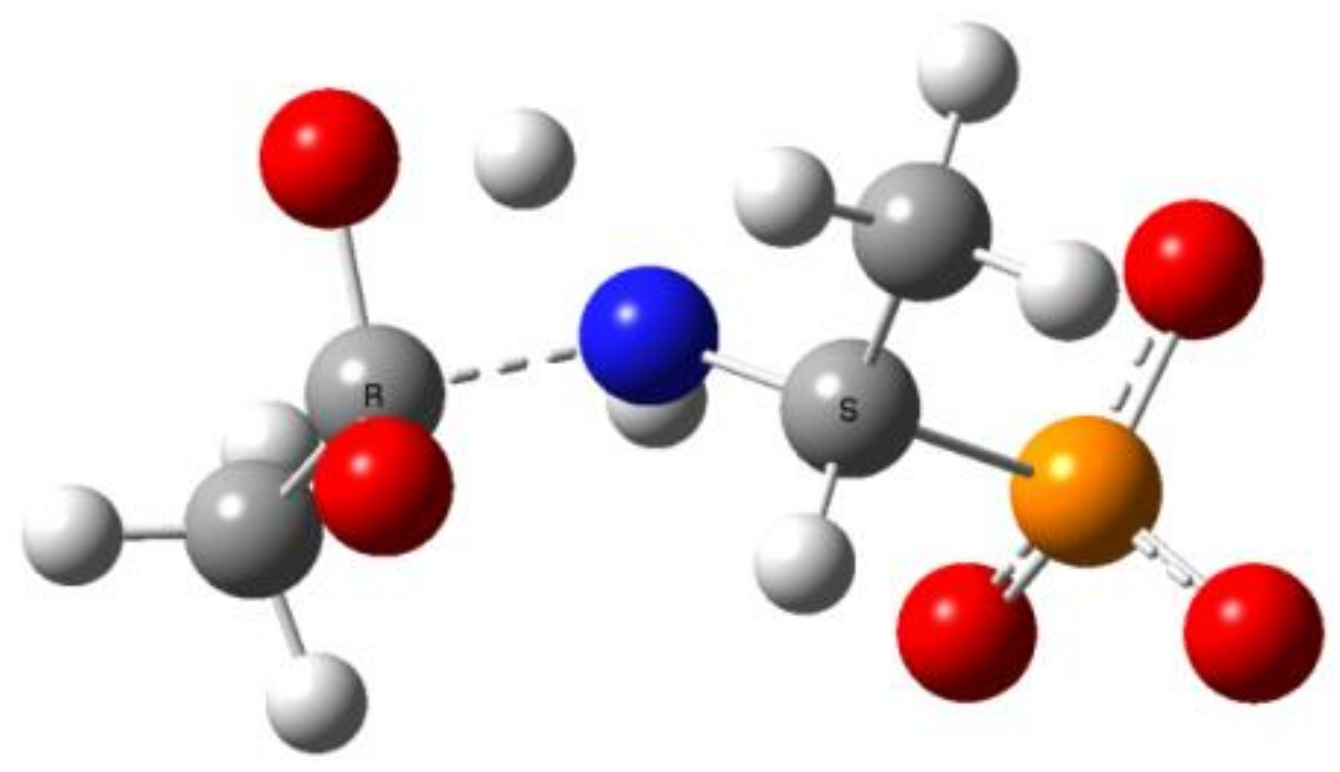
This reaction stage presumably involves the proton transfer from the geminal hydroxyl group to nitrogen with dissociation of the C-N bond. This hypothesis is supported by the transition structures found for this reaction. The example of this structure for the proton transfer/dissociation of the C-N bond in Ac-AlaP(3−) is shown in Figure 11. Vibrational analysis indicates that the imaginary vibration is associated with proton transfer from OH to NH. In the structure of the transition state shown in Figure 11, the geminal C-O bond distance is 1.284 Å, which corresponds well to C=O double bond. Furthermore, the C-N distance is 1.802 Å compared to 1.503 Å in the starting amide, which indicates that the bond breaking in the transition state is significantly advanced.
The Gibbs free energies for the dissociation reaction 1.2 in the gas phase and in water are presented in Table 6 and Table 7, respectively.
Comparing the thermodynamic quantities in Table 3 and Table 6, it is evident that the total free energy of the reaction 1 is negative in all cases by approximately −30 kcal/mol and thus, the entire process 1 is thermodynamically feasible. However, the very high energy barrier for the first step of reaction 1.1 (Table 3) prevents this reaction from proceeding in the gas phase.
Analogously, the reaction 1 in water is equally thermodynamically favorable (ΔG = −20 to −30 kcal/mol, Table 4 and Table 7), but the energy barrier associated with the first step 1.1 is much lower in this case (Table 4). Table 8 presents the results of free energy barrier calculations for reaction 1.2 in water.
The total Gibbs free energy profiles of entire process 1 for two selected pairs of 1-(acylamino)alkylphosphonic acids [Ac-AlaP(2−), TFA-AlaP(2−) , Bz-PglP(2−) and TFA-PglP(2−)] in water solution are shown in Figure 12.
The total Gibbs free energies and reaction barriers for the entire process 1 are shown in Table 9. Moreover, Figure 12 reveals that the energy profiles for both TFA-substituted phosphonic acids are essentially identical and all stationary points are significantly lower in energy than Ac-substituted phosphonic acids.
Therefore, the presence of a TFA group is the only factor determining the free energy barrier of the reaction. From the energy profile, it is obvious that the overall energy barrier in water ΔG‡ is G298[TS (1.2)] − (G298(1) + G298(OH−)), while the net Gibbs energy effect for the reaction in water solution can be calculated as ΔGr298 = G298(3) + G298(4) −[(G298(1) + G298(OH−)] = ΔG298(1.1) + ΔG‡(1.2).
The total energetic effect of the process 1 in water is negative, which means that the overall reaction is thermodynamically feasible (Table 8). The free energy of the process when R1=CF3 is also approximately 8 kcal/mol more favorable than for R1=Me. The comparison of values in Table 9 also reveals that the transformation 1 of substrates with R1=Ph are thermodynamically more favored by approximately 3 kcal/mol than R1=Me. Thus, the thermodynamic effect of reaction 1 depends on the R1 substituents in the order of: CH3 < Ph < CF3. Table 9 also leads to the conclusion that the substrates with a trifluoromethyl group in the R1 position systematically show free energy barriers that are approximately 10–15 kcal/mol lower than those for other substituents R1, due to the additional stabilization of the trianion 2 by the inductive effect of the strongly electron-withdrawing CF3 group. This suggests that trifluoromethyl-substituted anions should be more reactive than the other derivatives.
3. Materials and Methods
3.1. General Information
The 31P NMR spectra were recorded on a Bruker AV 200 spectrometer (Rheinstetten, Germany) operating at 81.01 MHz and on a Bruker Avance III 600 spectrometer (Bruker BioSpin, Rheinstetten, Germany) operating at 242.9 MHz. The1H NMR spectra were recorded on a Bruker Avance III 600 spectrometer operating at 600 MHz. The positive chemical shift values of 31P were reported for compounds that were absorbing at lower fields than H3PO4. The purity of the 1-aminoalkylphosphonic acids and corresponding 1-(acylamino)alkylphosphonic acids were determined by pH-metric titration using a computer aided automatic titrator connected to the EMU-meter (Wroclaw University of Science and Technology, Wrocław, Poland), which was fitted with a combined glass microelectrode Crison 5028.
3.2. Investigated Compounds
Phosphonoglycine (GlyP) was obtained according to a previous study [64]. The 1-Aminoalkylphosphonic acids of phosphonoalanine (AlaP); phosphonovaline (ValP); phosphonophenylglycine (PglP) and phosphonophenylalanine (PheP) have been prepared according to a previous study [65]. 1-(Acetylamino)alkylphosphonic acids (Ac-AAP: Ac-GlyP; Ac-AlaP; Ac-ValP; Ac-PglP and Ac-PheP) and 1-(benzoylamino)alkylphosphonic acids (Bz-AAP: Bz-GlyP; Bz-AlaP; Bz-ValP; Bz-PglP and Bz-PheP) were synthesized according to a previous study [36]. 1-(Trifluoroacetylamino)alkylphosphonic acids (TFA-AAP: TFA-GlyP; TFA-AlaP; TFA-ValP; TFA-PglP and TFA-PheP) were synthesized according to a previous study [37]. (Structures, names and abbreviations of aminophosphonic acids and corresponding 1-(acylamino)phosphonic acids discussed in this work are given in Table 10). Purity of all (AC)-AAP synthesized was determined by 31P NMR and 1H NMR in addition to potentiometric titration. Analytical standards (fixanals) and other reagents were purchased from Aldrich–Sigma (Poznań, Poland).
3.3. Reaction of Deacylation of 1-(acylamino)alkylphosphonic Acids
A sample of (AC)-AAP (0.5 mmol) was placed into 5-mL V-vials, before being dissolved in 5 mL of appropriate solution fortified with D2O (2 M KOH). The vials were maintained at 25 °C ± 0.2 °C. At various time intervals, the vials were removed from the baths and the samples of 0.5 mL were taken for 3lP NMR analysis. The concentration of the substrate and formed products were determined from the integration of its NMR signal.
3.4. Computational Methods
All quantum mechanical calculations were performed using the Gaussian 09 suite of programs [66]. Geometries of model compounds in the gas phase and in water solution were optimized using the B3LYP hybrid functional containing three-parameter Becke (B3) exchange and Lee-Yang-Parr (LYP) correlation functional and the 6–31+G(d) basis set. All stationary points were identified as stable minima by frequency calculations. The vibrational analysis also provided the thermal enthalpy and entropy corrections at 298 K within the rigid rotor/harmonic oscillator/ideal gas approximation. Computed frequencies were scaled by a factor of 0.98. Transition states were verified by the intrinsic reaction path (IRC) procedure to confirm if they did link the substrates and products along the reaction path.
Free energies of the reaction in water were calculated within a continuum (implicit) solvent approximation using the Conductor-like Polarizable Continuum Model (CPCM) with UFF (Universal Force Field) cavities (SCRF = CPCM option as defined in Gaussian 09 program) [67]. This approximation assumes that molecule of the solute is placed in a cavity within the solvent, which is treated as a dielectric continuum (self-consistent reaction field). Thermodynamic functions in solution were calculated based on vibrational analysis, which was described above.
4. Conclusions
The analysis of 31P NMR results presented above leads to the following conclusions:
- The alkaline deacylation of (AC)-AAP occurs through the hydroxyl adduct intermediates, which was observed on 31P NMR spectra of the reaction mixtures;
- The deacylation ability of investigated (AC)-AAP derivatives exhibited strong dependence of electron-acceptor character of the acyl group, with CF3-C(O)- > CH3-C(O)-;
- The 1-(acylamino)alkylphosphonic acids are present in aqueous solutions with substantial stability at ambient temperatures, which decreases with temperature elevation;
- The deacylation of (AC)-AAP increases substantially in basic (2 M KOH) solutions;
- The lowest deacylation ability (highest stability) was found for the 1-(benzoylamino acids Bz-AAP;
- For the same type of 1-(acylamino)-derivatives (AC)-AAP, the highest stability was found in(AC)-ValP and (AC)-PheP; lower stability in (AC)-AlaP; and the lowest in (AC)-GlyP and (AC)-PglP;
- All examined 31P NMR spectra of deacylation mixtures did not reveal any trace of H3PO3 and/orH3PO4, which represent the products of dephosphonylation or oxidative dephosphonylation of (AC)-AAP.
The theoretical calculations lead to the following conclusions:
The substrates with the trifluoromethyl group in R1 position are predicted to be significantly more reactive than the other phosphonic amino acids studied, which is consistent with the experimental results. The intermediates and products when R1=CF3 are also more thermodynamically favored than the other derivatives. This difference in reactivity is due to additional stabilization of the trianion 2 by the inductive effect of the strongly electron-withdrawing CF3 group. The calculations did not provide unequivocal information about relative reactivities of other AA derivatives as the calculated reaction energy barriers are similar (within a computational error), which is shown in Table 9 and Figure 12. The calculation results are consistent with those reported for acidic deacylation of 1-(acylamino)alkylphosphonic acids [51]. As CF3 is strongly electron-withdrawing, this group plays an activating role in basic media, increasing the acidity of carbonyl moiety. In contrast, it has a deactivating effect in acidic media, decreasing the basicity of carbonyl oxygen.
Acknowledgments
The studies at the Textile Research Institute, Łódź, in 2017 were partly financed by the Polish Ministry of Science and Higher Education within statutory research founds (for M.H.K.). DFT calculations were supported by the PL-Grid infrastructure. We would like to thank the National Science Center (Krakow, Poland) for the support in the frame of the grant UMO 2014/15/B/ST5/05329 (for J.D.). Dedicated to the memory of Professor Andrzej Okruszek.
Author Contributions
J.D. and Z.H.K. designed the research study and contributed to the data interpretation and to the manuscript drafting and revisions. M.C. and B.G. carried out the Quantum Chemical Calculations and contributed to writing the manuscript. M.H.K. performed the synthesis and purification of investigated 1-(acylamino)alkylphosphonic acids and was involved in the conception of the research study, analyzed the data, and contributed to writing the manuscript. P.U. recorded NMR spectra and analyzed the experimental data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kukhar, V.P.; Hudson, H.R. (Eds.) Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; J. Wiley & Sons, Ltd.: Chichester, UK; New York, NY, USA; Weinheim, Germany; Brisbane, Australia; Singapore; Toronto, ON, Canada, 2000; ISBN 0-471-89149-5. [Google Scholar]
- Kittredge, J.S.; Roberts, E. A carbon-phosphorus compounds in nature. Science 1969, 164, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P.; Mastalerz, P. Aminophosphonates, Natural Occurrence, Biochemistry and Biological Properties; Beiträge zur Wirkstofforschung; Institut für Wirkstofforschung: Berlin, Germany, 1984; Volume 24, pp. 1–110. [Google Scholar]
- Horiguchi, M.; Kandatsu, M. Isolation of 2-aminoethyl phosphonic amid from rumen protozoa. Nature 1959, 184, 901–902. [Google Scholar] [CrossRef] [PubMed]
- Kittredge, J.S.; Hughes, R.R. Occurence of α-amino-β-phosphono-propionic acid in the zoanthid, zoanthussociatus, and the ciliate, Tetrahymena pyrifornis. Biochemistry 1964, 3, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.D.; Deaborn, D.G.; Falles, H.M.; Sokoloski, E.A. A major polysaccharide constituents of the amoeba plasma membrane contains 2-aminoethylphosphonic acid and 1-hydroxy-2-aminoethyl-phosphonic acid. J. Biol. Chem. 1973, 248, 2257–2259. [Google Scholar] [PubMed]
- Kido, Y.; Hamakado, T.; Anno, M.; Miyagawa, E.; Motoki, Y.; Wakamiya, T.; Shiba, T. Isolation and characterization of 15112, a new phosphorus containing inhibitor of angiotensin I converting enzyme produced by Actinomadura sp. J. Antibiot. 1984, 37, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.; Gugel, K.H.; Haegele, K.; Hagenmaier, H.; Jessipov, S.; Koenig, W.A.; Zaehner, H. Metabolic products of microorganisms. 98. Phosphinothricine and phosphinothricylo-alanylo-alanine. Helv. Chim. Acta 1972, 55, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Franz, J.E. Herbicidal compositions and methods employing esters of N-phosphonoglycine. U.S. Patent US 3997860, 14 December 1976. [Google Scholar]
- Mastalerz, P. Inhibition of glutamine synthetase by phosphonic analogs of glutamic acid. Arch. Immun. Ter. Dośw. 1959, 7, 201–210. [Google Scholar]
- Engelmann, M.; Pikl, J. Phosphonic Acids Derived from Organic Acylamidomethyl Compounds. U.S. Patent 2304156, 8 December 1942. [Google Scholar]
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassal, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.E.; Bartlett, P.A. A phosphonamidate dipeptide analog as an inhibitor of carboxypeptidase A. J. Am. Chem. Soc. 1981, 103, 654–657. [Google Scholar] [CrossRef]
- McLeod, D.A.; Brinkworth, R.; Ashley, J.A.; Janda, K.D.; Wirsching, P. Phosphonamidates and phosphonamidate esters as HIV-1 protease inhibitors. Bioorg. Med. Chem. Lett. 1991, 1, 653–658. [Google Scholar] [CrossRef]
- Drabowicz, J.; Jakubowski, H.; Kudzin, M.H.; Kudzin, Z.H. The nomenclature of 1-aminoalkylphosphonic acids and derivatives: Evolution of the code system. Acta Biochim. Pol. 2015, 62, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Kabachnik, M.I.; Medved, T.Y.; Dyatlova, N.M.; Archipova, O.G.; Rudomino, M.W. Phosphoroorganic complexones. Usp. Khim. 1974, 43, 1554–1574. [Google Scholar] [CrossRef]
- Rizkalla, E.N. Metal chelates of phosphonate-containing ligands. Rev. Inorg. Chem. 1983, 5, 223–304. [Google Scholar]
- Nowack, B. Environmental chemistry of phosphonates. Water Res. 2003, 37, 2533–2546. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B. Aminophosphonic acids of potential medical importance. Curr. Med. Chem. Anti-Cancer Agents 2001, 1, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Lejczak, B.; Kafarski, P. Biological activity of aminophosphonic acids and their short peptides. Top. Heterocycl. Chem. 2009, 20, 31–63. [Google Scholar] [CrossRef]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the α-amino-phosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, J.A.; Logush, E.W. Aliphatic carbon-phosphorus compound as herbicides. In Handbook in Organophosphorus Chemistry; Engel, R., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1988; Volume Chapter 15, pp. 737–806. [Google Scholar]
- Maier, L. Phosphoroorganic detergents. Chimia 1969, 23, 323–330. [Google Scholar]
- Petrov, K.A.; Chauzov, V.A.; Erokhina, T.E. Aminoalkyl organo-phosphorus compounds. Usp. Khim. 1974, 43, 2045–2087. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; de los Santos, J.M. Synthesis of β-amino-phosphonates and -phosphinates. Chem. Rev. 2005, 105, 899–931. [Google Scholar] [CrossRef] [PubMed]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J.; Stevens, C. Aminophosphonic acids—Phosphorus analogues of natural amino acids. P. 1: Syntheses of α-amino-phosphonic acids. Curr. Org. Chem. 2011, 15, 2015–2071. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J. Thioureidoalkylphosphonates in the synthesis of 1-aminoalkylphosphonic acids. The Ptc-aminophosphonate method. Arkivoc 2011, 6, 227–269. [Google Scholar] [CrossRef]
- Ma, J.-N. Catalytic asymmetric synthesis of α- and β-amino phosphonic acid derivatives. Chem. Soc. Rev. 2006, 35, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, M.; Rojas-Cabrera, H.; Cativiela, C. An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef] [PubMed]
- Kukhar, V.P.; Solodenko, V.A. The phosphorus analogs of aminocarboxylic acids. Usp. Khim. 1987, 56, 1504–1532. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Mokrzan, J.; Skowroński, R. Long chain 1-aminothia-alkanephosphonates, their sulphinyl and sulphonyl derivatives. A new class of complexane type surfactants. Phosphorus Sulfur Silicon Relat. Elem. 1989, 42, 41–46. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Skrzypek, S.W.; Skowroński, R.; Ciesielski, W.; Cristau, H.J.; Plenat, F. Polarographic investigations of functionalized alkanephosphonic acids. Part II. Phosphorus Sulfur Silicon Relat. Elem. 1996, 119, 201–207. [Google Scholar] [CrossRef]
- Chęcińska, L.; Kudzin, Z.H.; Małecka, M.; Nazarski, R.B.; Okruszek, A. [(Diphenoxyphosphinyl)methylidene]triphenylphosphorane—The double P+-stabilisedcarboanion: A crystallographic, computational and solution NMR comperative study on the ylidic bonding. Tetrahedron 2003, 59, 7681–7693. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Saganiak, M.; Andrijewski, G.; Drabowicz, J. Oxidation of phosphonocysteine and phosphonohomocysteine. Synthesis of phosphonocysteic and phosphonohomocysteic acids. Pol. J. Chem. 2005, 79, 529–539. [Google Scholar]
- Kudzin, Z.H.; Depczyński, R.; Andrijewski, G.; Drabowicz, J.; Łuczak, J. 1-(N-acylamino)alkanephosphonates. IV. N-acylation of 1-aminoalkanephosphonic acids. Pol. J. Chem. 2005, 79, 499–513. [Google Scholar]
- Kudzin, Z.H.; Depczyński, R.; Kudzin, M.H.; Drabowicz, J.; Łuczak, J. 1-(N-Trifluoroacetylamino)alkylphosphonic acids. Synthesis and properties. Amino Acids 2007, 33, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Kudzin, Z.H.; Depczyński, R.; Kudzin, M.H.; Drabowicz, J. 1-(N-Chloroacetylamino)alkylphosphonic acids—Synthetic precursors of glycylophosphono-peptides and related compounds. Amino Acids 2008, 34, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Drabowicz, J.; Jordan, F.; Kudzin, M.H; Kudzin, Z.H.; Urbaniak, P.; Stevens, C. Reactivity of aminophosphonic acids. oxidative dephosphonylation of 1-aminoalkylphosphonic acids by aqueous halogens. Dalton Trans. 2016, 45, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Gancarz, R.; Kiersnowska, A.D. Associative vs. dissociative mechanism of P–C bond breaking in α-aminophosphonates leading to phosphoric acid [P(V)] derivatives. Arkivoc 2017, 2, 285–302. [Google Scholar] [CrossRef]
- Aldrich-Sigma Catalog 2016. Aminophosphonic Self-Assembled Monolayers Agents (e.g., 795798, SIK 7701-10; SIK 7701-101). Available online: https://www.sigmaaldrich.com/china-mainland.html/ (accessed on 9 April 2018).
- Kudzin, Z.H.; Łuczak, J. A facile conversion of aminoalkanephosphonic acids into O,O-dialkyl 1-(N-acylamino)alkanephosphonate derivatives. Synthesis 1995, 509–511. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Łyżwa, P.; Łuczak, J.; Andrijewski, G. Aminoalkanephosphonates. P.II. Facile conversion of 1-aminoalkanephosphonic acids their O,O-diester derivatives. Synthesis 1997, 44–47. [Google Scholar] [CrossRef]
- Svedas, V.K.; Kozlova, E.V.; Mironenko, D.A.; Kukhar, V.P.; Kasheva, T.N.; Solodenko, V.A.; Belozersky, A.N. Enzymatic hydrolysis of N-acylated 1-aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 1990, 51, 411. [Google Scholar] [CrossRef]
- Solodenko, V.; Kasheva, T.; Kukhar, V.; Kozlova, E.; Mironenko, D.; Svedas, V. Preparation of optically active 1-aminoalkylphosphonic acids by stereoselective enzymatic hydrolysis of racemic N-acylated 1-aminoalkyl-phosphonic acids. Tetrahedron 1991, 47, 3989–3998. [Google Scholar] [CrossRef]
- Solodenko, V.; Belik, M.; Galushko, S.; Kukhar, V.; Kozlova, E.; Mironenko, D.; Svedas, V. Enzymatic preparation of both l- and d-enantiomers of phosphonic and phosphonous analogues of alanine using penicillin acylase. Tetrahedron Asymmetry 1993, 4, 1965–1968. [Google Scholar] [CrossRef]
- Zielinska, K.; Mazurkiewicz, R.; Szymanska, K.; Jarzebski, A.; Magierac, S.; Erfurt, K. Penicillin G acylase-mediated kinetic resolution of racemic 1-(N-acylamino)alkylphosphonic and 1-(N-acylamino)alkylphosphinic acids and their esters. J. Mol. Catal. B Enzym. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Zielinska, K.; Szymanska, K.; Mazurkiewicz, R.; Jarzebski, A. Batch and in-flow kinetic resolution of racemic 1-(N-acylamino)alkylphosphonic and 1-(N-acylamino)alkylphosphinic acids and their esters using immobilized penicillin G acylase. Tetrahedron Asymmetry 2017, 28, 146–152. [Google Scholar] [CrossRef]
- Oleksyszyn, J. An amidoalkylation of trivalent phosphorous compounds with P(O)H functions including acetic acid solutions of PCl3, RPCl2 or R2PCl, diesters of phosphorous acid and phosphorous-III-acids. J. Prakt. Chem. 1987, 329, 19–29. [Google Scholar] [CrossRef]
- Soroka, M. The synthesis of 1-aminoalkylphosphonic acids. A revised mechanism of the reaction of phosphorus trichloride, amides and aldehydes or ketones in acetic acid solution (Oleksyszyn reaction). Liebigs Ann. Chem. 1990, 4, 331–334. [Google Scholar] [CrossRef]
- Cypryk, M.; Drabowicz, J.; Gostynski, B.; Kudzin, M.H.; Kudzin, Z.H.; Urbaniak, P. 1-(N-Acylamino)-alkylphosphonic acids. Deacylation in aqueous solutions. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 651–658. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume Chapters 16–60, pp. 1408–1411. ISBN 13-978-0-471-72091-1. [Google Scholar]
- O’Connor, C. Acidic and basic amide hydrolysis. Q. Rev. Chem. Soc. 1970, 24, 553–564. [Google Scholar] [CrossRef]
- Brown, R.S.; Bennet, A.J.; Slebocka-Tilk, H. Recent perspectives concerning the mechanism of H3O+ and OH− promoted amide hydrolysis. Acc. Chem. Res. 1992, 25, 481–488. [Google Scholar] [CrossRef]
- Bagno, A.; Lovato, G.; Scorrano, G. Thermodynamics of protonation and hydration of aliphatic amides. J. Chem. Soc. Perkin Trans. 1993, 2, 1091–1098. [Google Scholar] [CrossRef]
- Zahn, D. On the role of water in amide hydrolysis. Eur. J. Org. Chem. 2004, 4020–4023. [Google Scholar] [CrossRef]
- Kahne, D.; Still, W.C. Hydrolysis of a peptide bond in neutral water. J. Am. Chem. Soc. 1988, 110, 7529–7534. [Google Scholar] [CrossRef]
- DeWolfe, R.H.; Newcombe, R.C. Hydrolysis of formanilides in alkaline solutions. J. Org. Chem. 1971, 36, 3870–3878. [Google Scholar] [CrossRef]
- Desai, S.D.; Kirsch, L.E. The ortho effect on the acidic and alkaline hydrolysis of substituted formanilides. Int. J. Chem. Kinet. 2015, 47, 471–488. [Google Scholar] [CrossRef]
- Bowden, K.; Bromley, K. Reactions of carbonyl compounds in basic solutions. Part 14. The alkaline hydrolysis of substituted N-methylformanilides, N-methylacetanilides, 1-phenylazetidin-2-ones, 1-phenyl-2-pyrrolidones, and 1-phenyl-2-piperidones. J. Chem. Soc. Perkin Trans. 1990, 2, 2103–2109. [Google Scholar] [CrossRef]
- Hibbert, F.; Malana, M.A. Kinetics of the alkaline hydrolysis of 1,8-bis(trifluoroacetylamino)-naphthalene to 1-amino-8-trifluoroacetylaminonaphthalene in 70%, 80% and 90%(v/v) Me2SO–H2O. J. Chem. Soc. Perkin Trans. 1992, 2, 755–759. [Google Scholar] [CrossRef]
- Cheshmedzhieva, D.; Ilieva, S.; Hadjieva, B.; Galabov, B. The mechanism of alkaline hydrolysis of amides: A comparative computational and experimental study of the hydrolysis of N-methylacetamide, N-methylbenzamide, and acetanilide. J. Phys. Org. Chem. 2009, 22, 619–631. [Google Scholar] [CrossRef]
- Theodorou, V.; Paraskevopoulos, G.; Skobridis, K. A mild alkaline hydrolysis of N- and N,N-substituted amides and nitriles. Arkivoc 2015, 7, 101–112. [Google Scholar] [CrossRef]
- Soroka, M. Comments on the synthesis of aminomethylphosphonic acid. Synthesis 1989, 547–548. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Stec, W.J. Synthesis of 1-aminoalkanephosphonic acids via thioureidoalkanephosphonates. Synthesis 1978, 469–472. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all 1-(acylamino)phosphonic acids discussed in this paper are available from the authors. Calculated geometries of all modeled species are available upon request. |
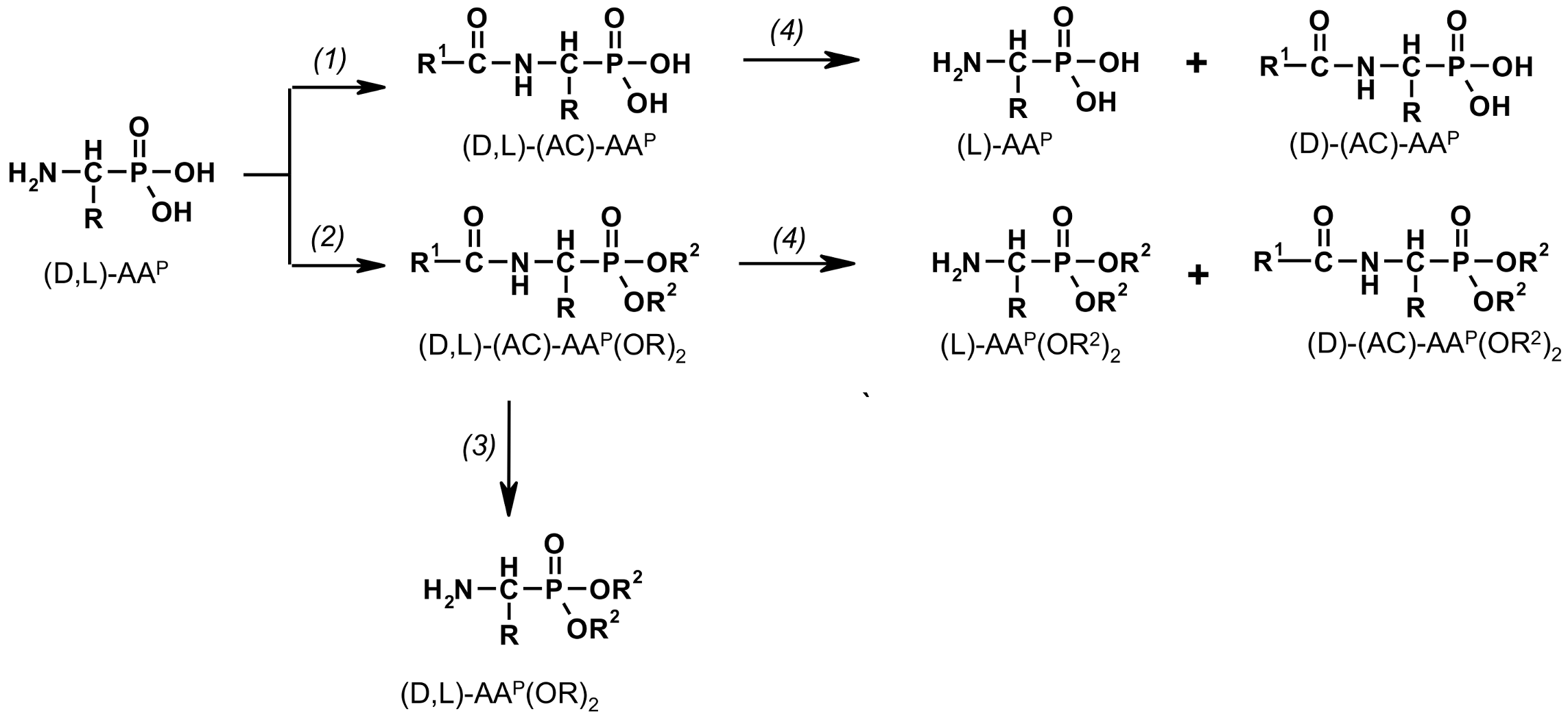
Figure 1.
Transformation of 1-aminoalkylphosphonic acids via N-acyl-derivatives [(1) R1-C(O)OH/[R1-C(O)]2O [36,37,38]; (2) R1-C(O)OH/[R1-C(O)]2O/HC(OR2)3 [42]; (3) NaBH4/MeOH [43]; (4) enzymatic hydrolysis of racemic (D,L)-(AC)-AAP and (D,L)-(AC)-AAP(OR)2 [44,45,46,47,48]].

Figure 2.
Scheme of Engelmann-Pikl-Oleksyszyn methods for synthesis of secondary 1-aminoalkylphosphonic (R2=OH) and 1-aminoalkylphosphinic (R2=H, alkyl) acids (1) aldehyde, R-PCl2/AcOH; and (2) 5M HCl-H2O, Δ8–10 h.
Figure 2.
Scheme of Engelmann-Pikl-Oleksyszyn methods for synthesis of secondary 1-aminoalkylphosphonic (R2=OH) and 1-aminoalkylphosphinic (R2=H, alkyl) acids (1) aldehyde, R-PCl2/AcOH; and (2) 5M HCl-H2O, Δ8–10 h.

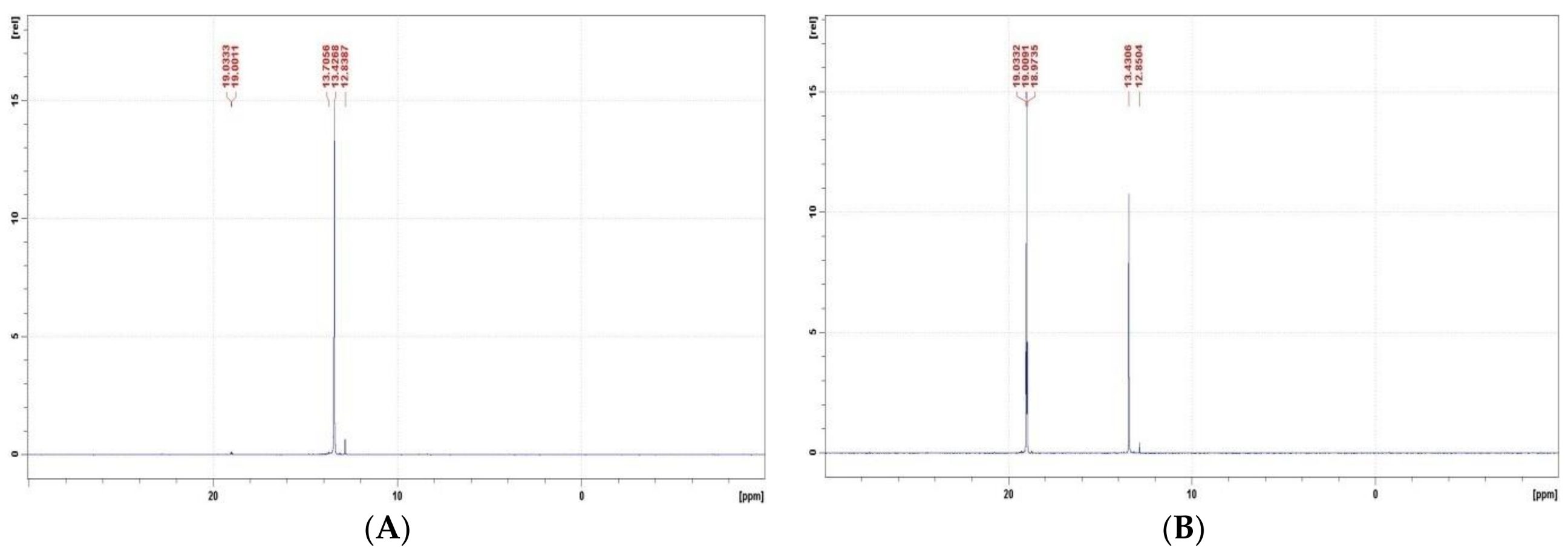
Figure 3.
31P NMR spectra of Ac-GlyP recorded in 2 M KOH. (A) Immediately after mixing the reaction mixture, and (B) After 192 h of exposition of the reaction mixture. [Ac-GlyP: 13.4 ppm; GlyP: 19.0 ppm].
Figure 3.
31P NMR spectra of Ac-GlyP recorded in 2 M KOH. (A) Immediately after mixing the reaction mixture, and (B) After 192 h of exposition of the reaction mixture. [Ac-GlyP: 13.4 ppm; GlyP: 19.0 ppm].

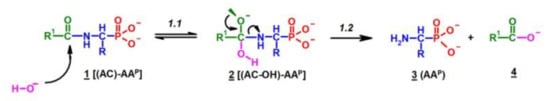
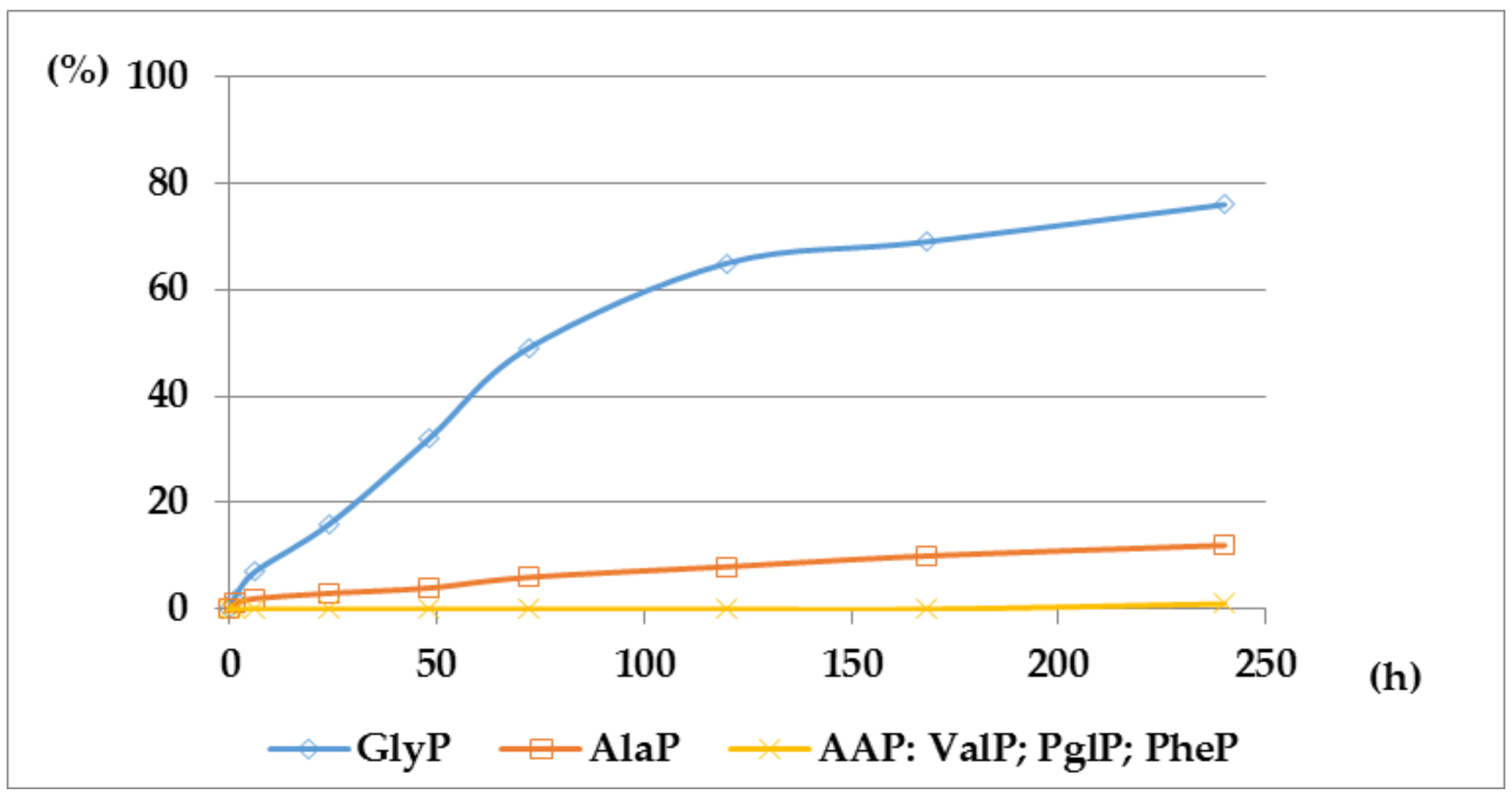
Figure 4.
A solution of 0.1 M 1-(acetylamino)alkylphosphonic acids Ac-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)].
Figure 4.
A solution of 0.1 M 1-(acetylamino)alkylphosphonic acids Ac-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)].
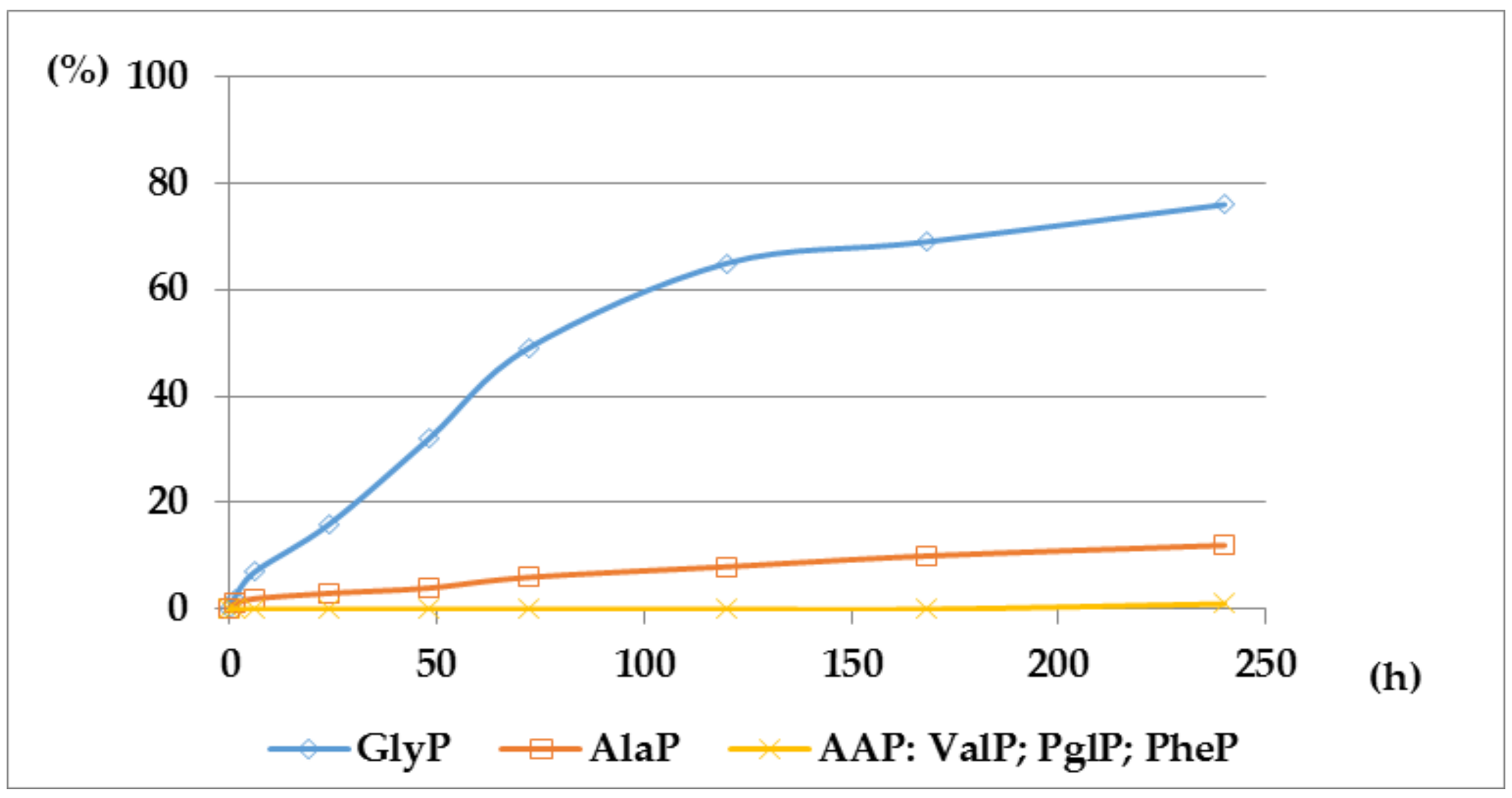
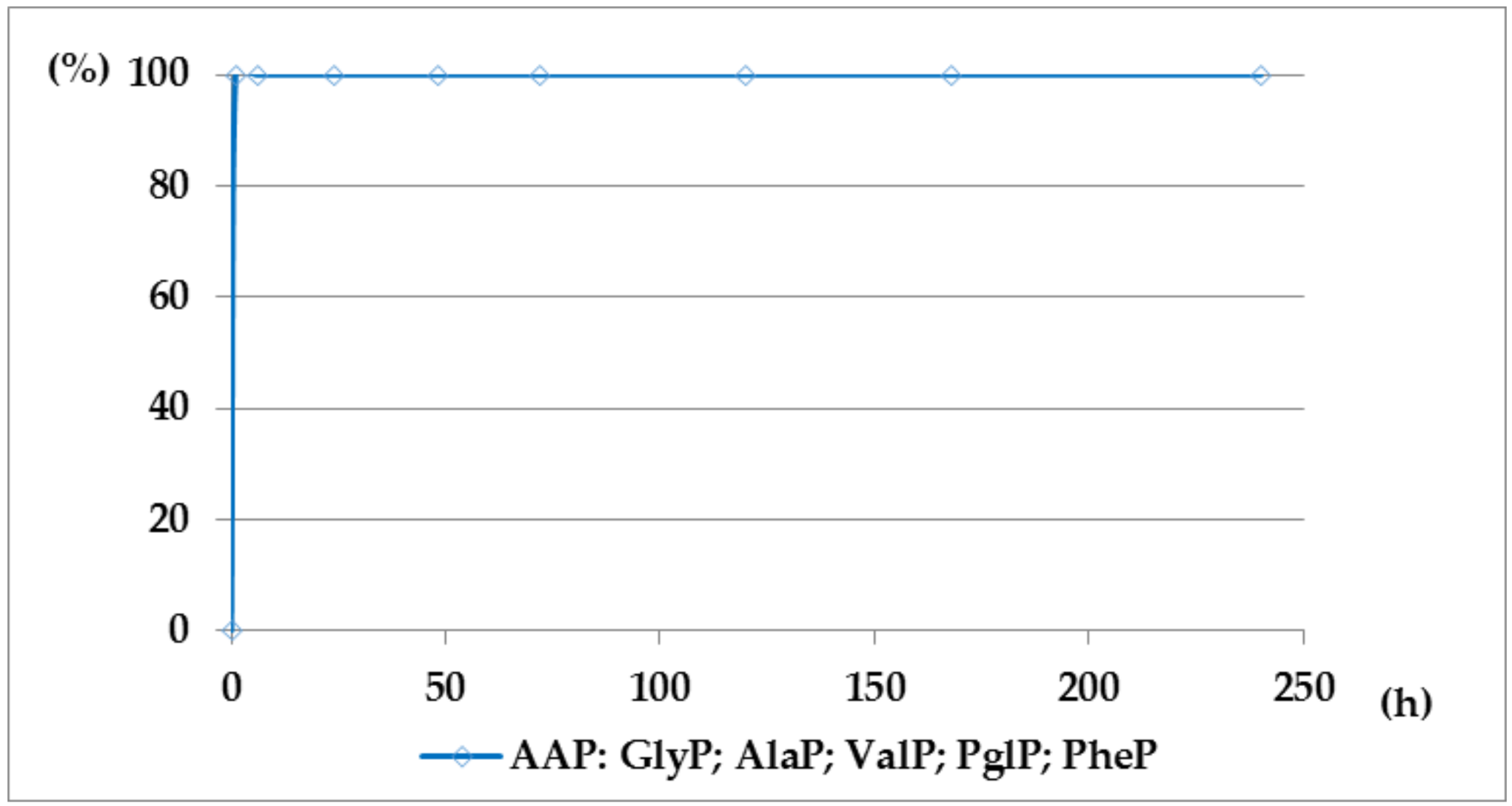
Figure 5.
A solution of 0.1 M 1-(trifluoroacetylamino)alkylphosphonic acids TFA-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)].
Figure 5.
A solution of 0.1 M 1-(trifluoroacetylamino)alkylphosphonic acids TFA-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)].
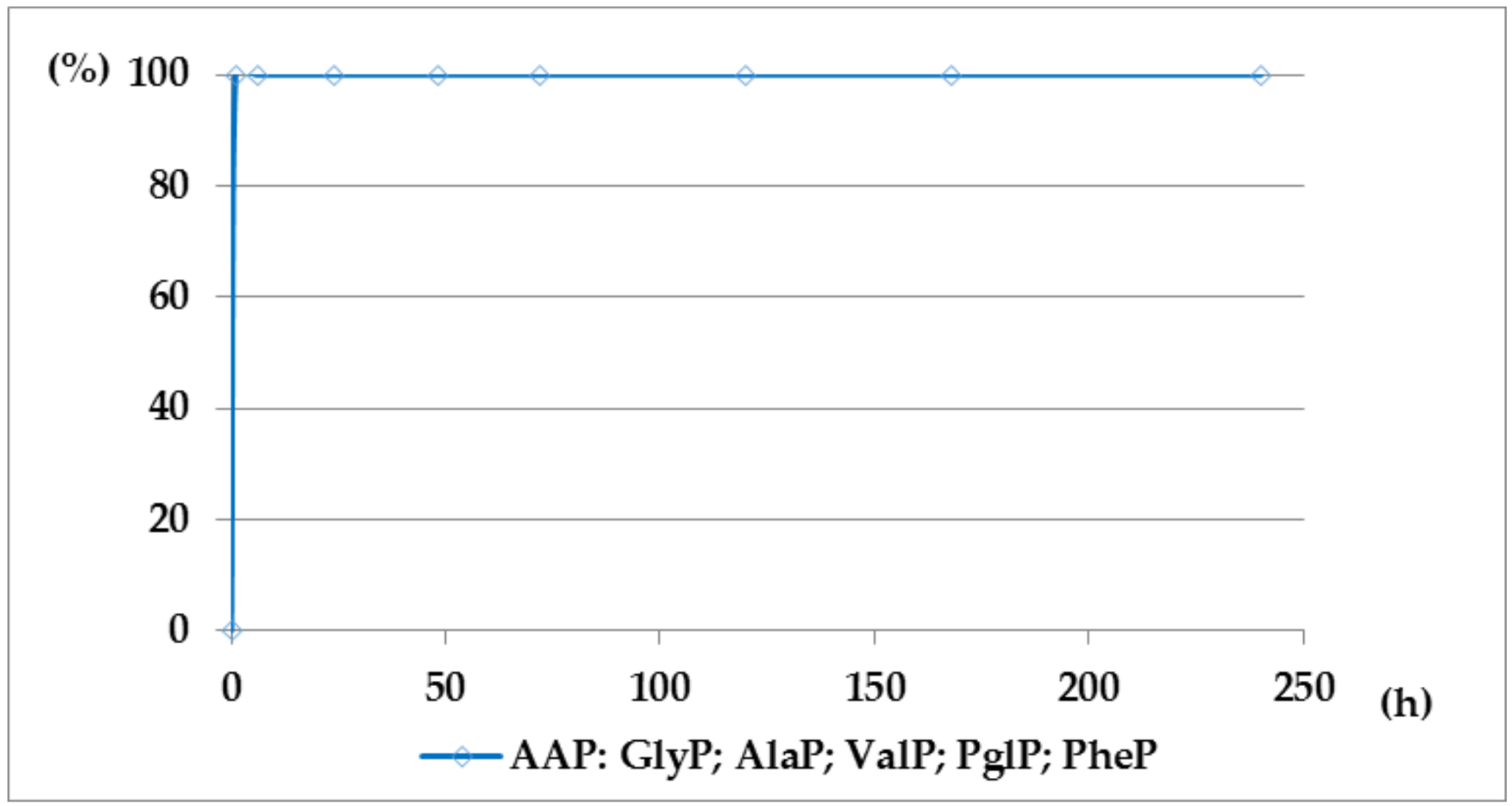
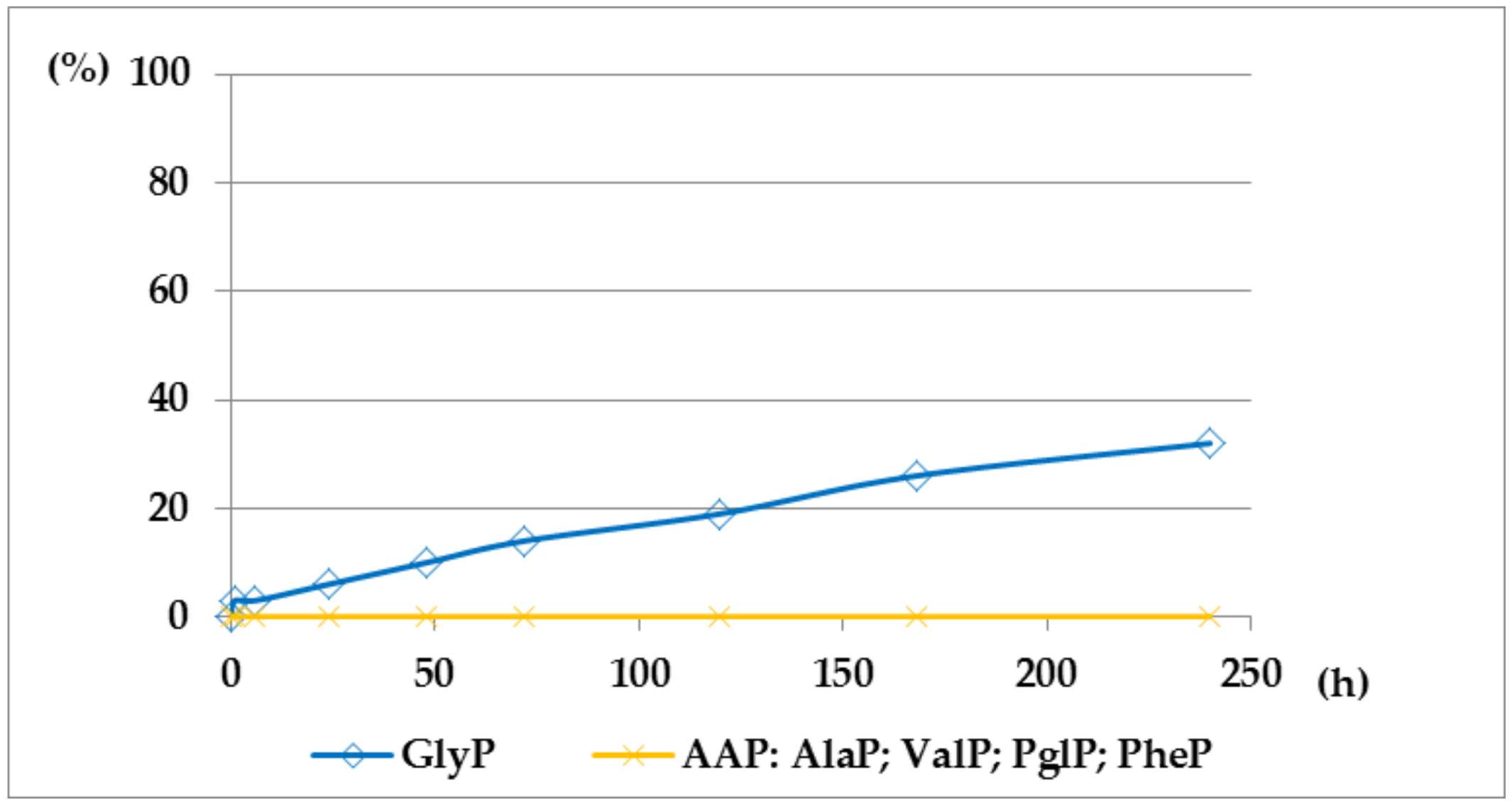
Figure 6.
A solution of 0.1 M 1-(benzoylamino)alkylphosphonic acids Bz-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)]. Thus, the presented graphs illustrate much higher stability of benzoyl-derivatives Bz-AAP (Bz-GlyP: DD200h ~30%; for Bz-AlaP and Bz-PglP: DD200h < 3%) in comparison with acetyl-derivatives Ac-AAP (Ac-GlyP: DD200h ~70%; for Ac-AlaP: DD200h ~20%; and Ac-PglP: DD200h < 7%).
Figure 6.
A solution of 0.1 M 1-(benzoylamino)alkylphosphonic acids Bz-AAP in 2 M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)]. Thus, the presented graphs illustrate much higher stability of benzoyl-derivatives Bz-AAP (Bz-GlyP: DD200h ~30%; for Bz-AlaP and Bz-PglP: DD200h < 3%) in comparison with acetyl-derivatives Ac-AAP (Ac-GlyP: DD200h ~70%; for Ac-AlaP: DD200h ~20%; and Ac-PglP: DD200h < 7%).

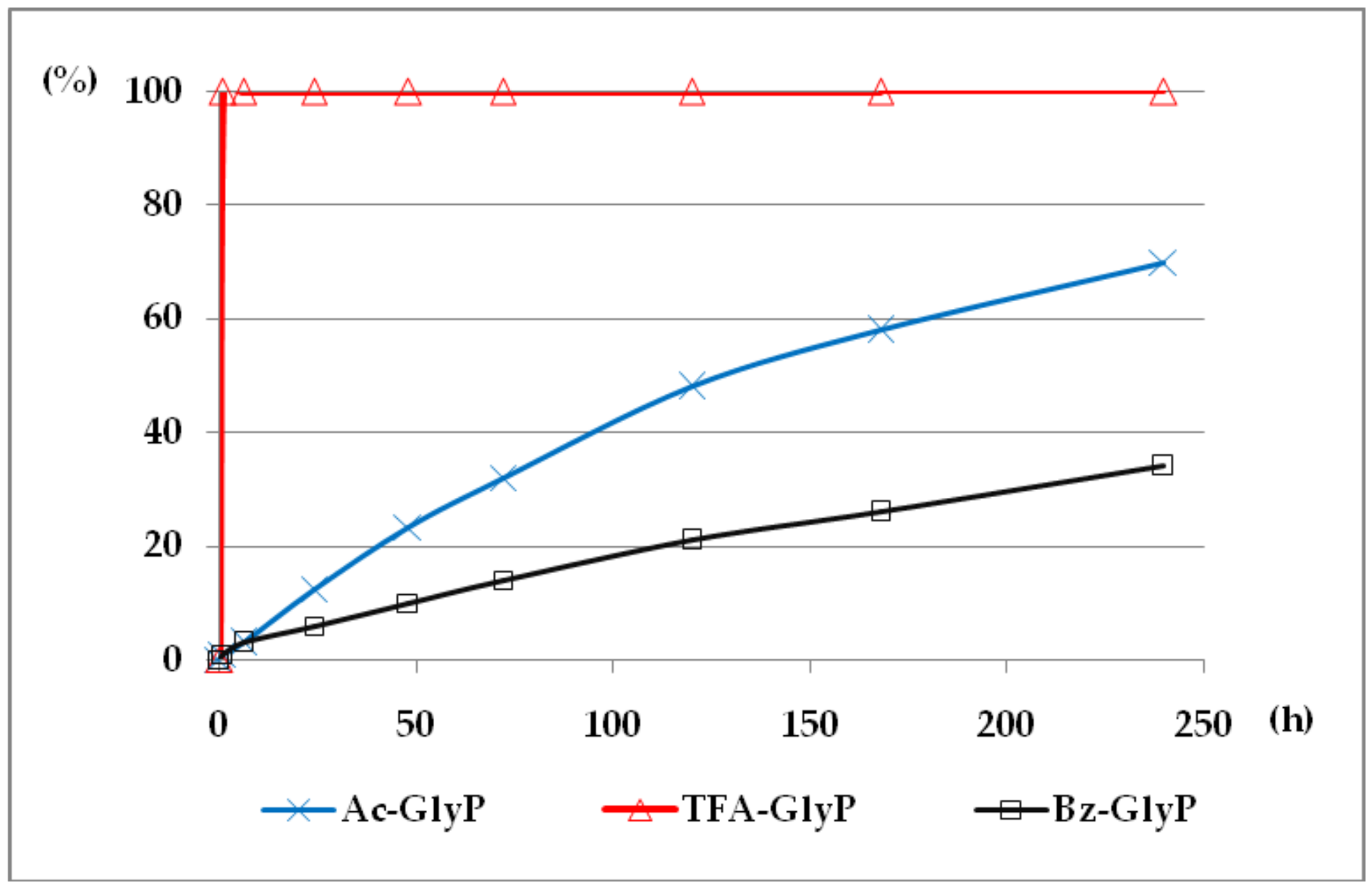
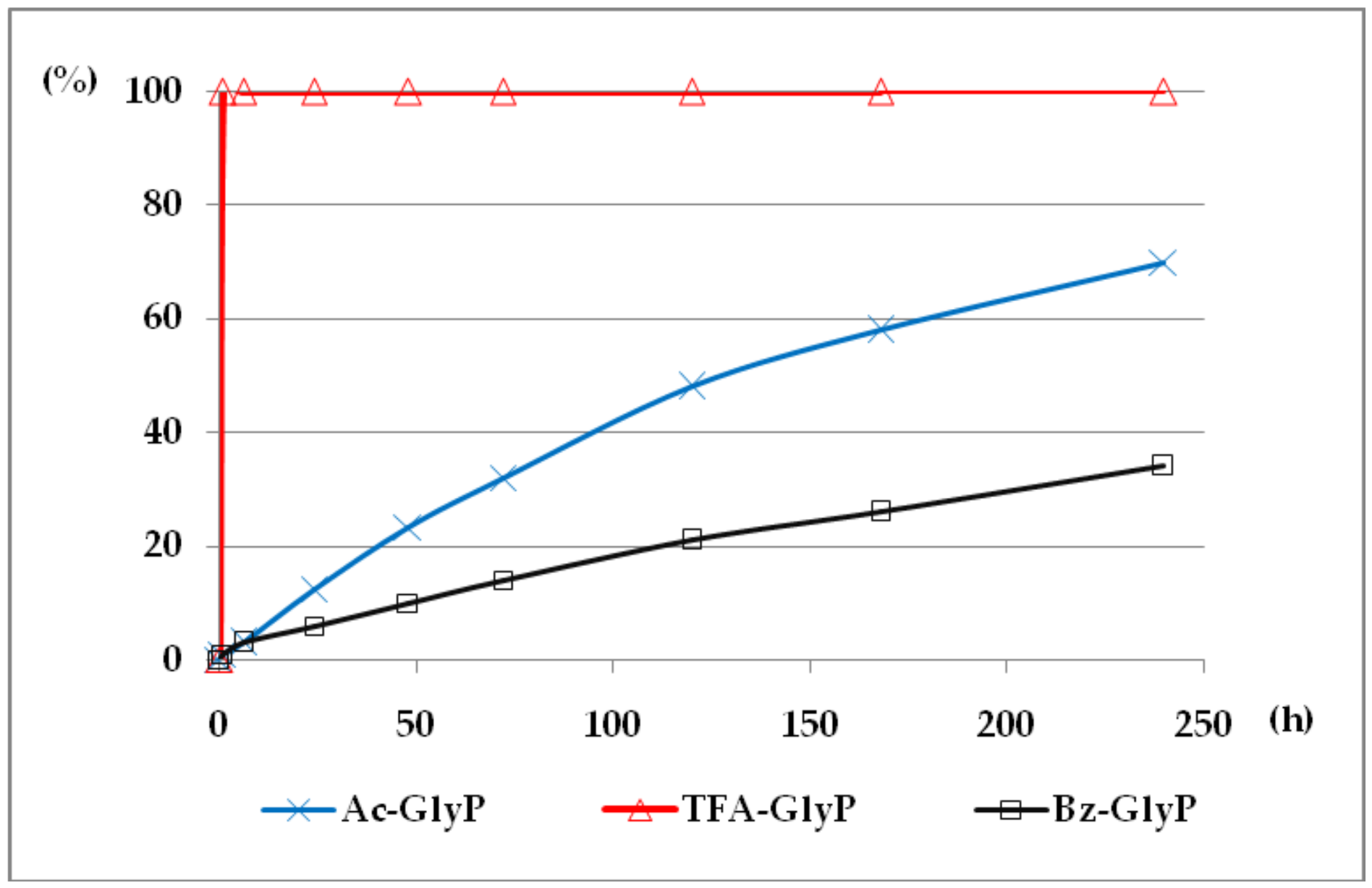
Figure 7.
A solution of 0.1 M 1-(acylamino)methylphosphonic acids (AC)-GlyP (AC: Ac, TFA, Bz) in 2M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)]. Points on the plots corresponds to GlyP released from the parent (AC)-GlyP, which means GlyP from Ac-GlyP, GlyP from Bz-GlyP and GlyP from TFA-GlyP.
Figure 7.
A solution of 0.1 M 1-(acylamino)methylphosphonic acids (AC)-GlyP (AC: Ac, TFA, Bz) in 2M KOH reacted at 25 °C, which was monitored by 31P NMR [DD (%) vs. time (h)]. Points on the plots corresponds to GlyP released from the parent (AC)-GlyP, which means GlyP from Ac-GlyP, GlyP from Bz-GlyP and GlyP from TFA-GlyP.
Scheme 1.
Hypothetical mechanism of deacylation of (AC)-AAP in 2 M KOH solution.
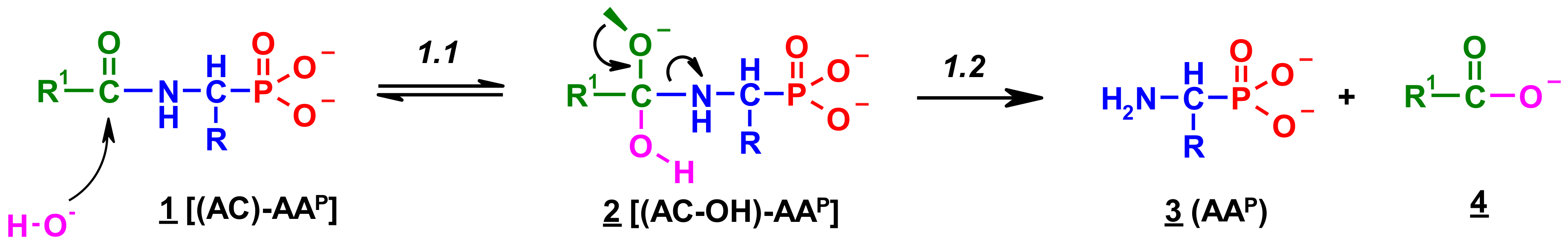
Figure 8.
Ac-GlyP hydrolysis in 2M KOH [DD (%) vs. time (h)].

Figure 9.
(AC)-GlyP(Ac-GlyP, TFA-GlyP, TFA-GlyP) hydrolysis in aqueous solutions of 2M KOH and 2M HCl [DD (%) vs. time (h)].
Figure 9.
(AC)-GlyP(Ac-GlyP, TFA-GlyP, TFA-GlyP) hydrolysis in aqueous solutions of 2M KOH and 2M HCl [DD (%) vs. time (h)].
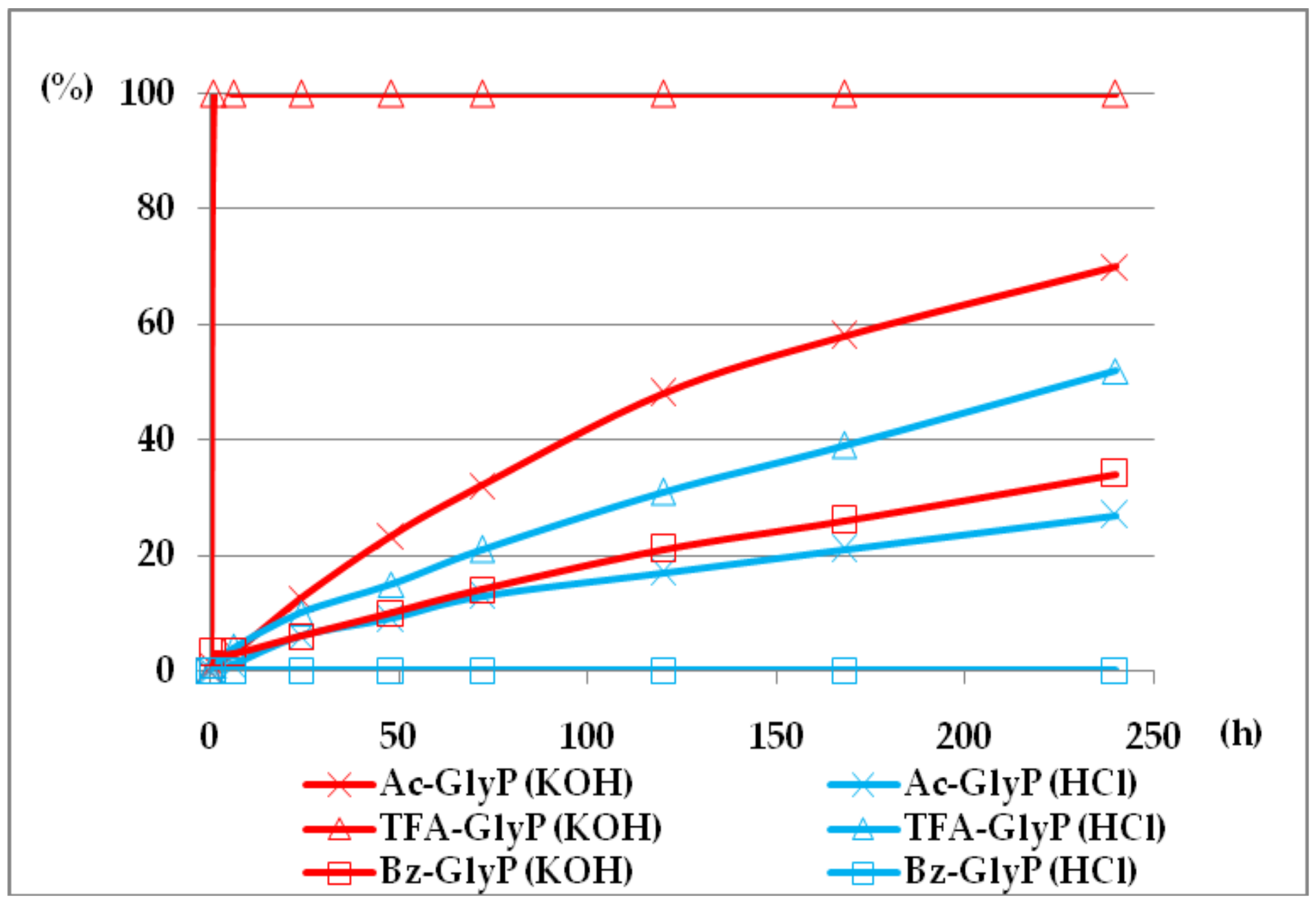
Figure 10.
The structure of the addition product 2 (R=R1=Me, Ac(OH)-AlaP) with intramolecular hydrogen bond between OH and phosphoryl group (orange—phosphorus, blue—nitrogen, red—oxygen).
Figure 10.
The structure of the addition product 2 (R=R1=Me, Ac(OH)-AlaP) with intramolecular hydrogen bond between OH and phosphoryl group (orange—phosphorus, blue—nitrogen, red—oxygen).
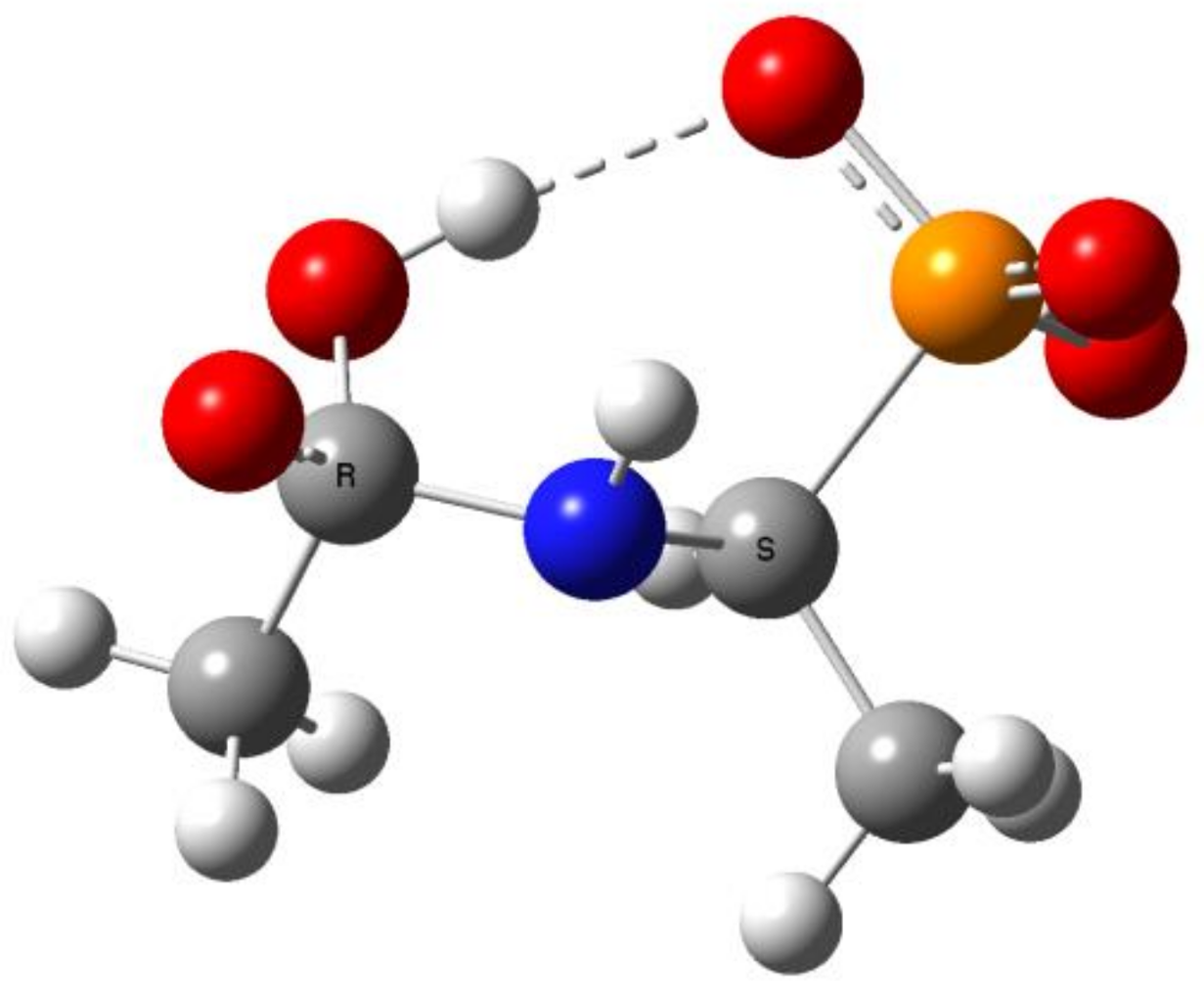
Figure 11.
The transition state for proton transfer/dissociation of the C-N bond in Ac(OH)-AlaP(3−).
Figure 11.
The transition state for proton transfer/dissociation of the C-N bond in Ac(OH)-AlaP(3−).
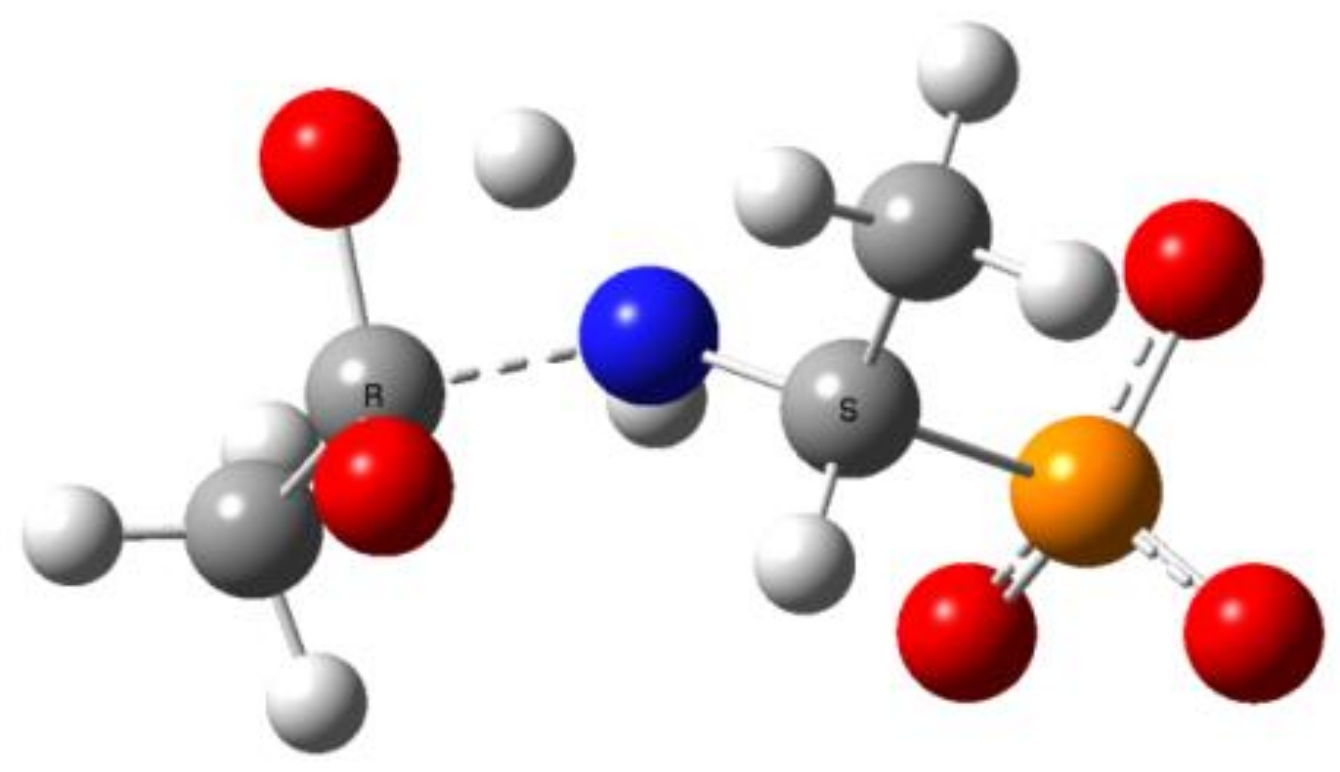
Figure 12.
Free energy profiles ΔG298 (kcal/mol) for the reaction of Ac-AlaP(2−) (blue), TFA-AlaP(2−) (red), Bz-PglP(2−) (green), and TFA-PglP(2−) (purple) with hydroxyl ions in water solution (the general reaction scheme is given above).
Figure 12.
Free energy profiles ΔG298 (kcal/mol) for the reaction of Ac-AlaP(2−) (blue), TFA-AlaP(2−) (red), Bz-PglP(2−) (green), and TFA-PglP(2−) (purple) with hydroxyl ions in water solution (the general reaction scheme is given above).


{kind=link}
{kind=link}
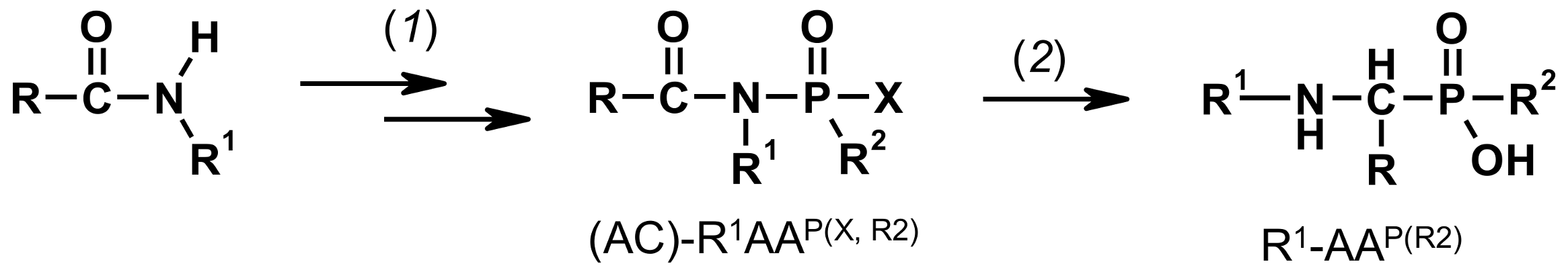{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structures of aminophosphonic acids (AAP)—phosphonic analogs of amino acids (AAC) and 1-(acylamino)alkylphosphonic acids (AC)-AAP and phosphonopeptides AAC-AAP and AAP-AAC [4,5,6,7,8,9,10,11,12,13,14].
 |  |  |  |  |
| AAC | ω-AAC | AAP | ω-AAP | AAP–R |
| Naturally occurring aminophosphonic acids. | ||||
 |  |  |  |  |
| β-AlaP [4] | Aspβ–P [5] | IserP [6] | TyrP [7] | Glu−γ–P(Me) [8] |
| Representative, biologically active aminophosphonates. | ||||
 |  |  |  |  |
| PMG [9] | Gluγ–P [10] | (AC)-AAP[R=H, (AC)-GlyP] [11] | AAC-AAP(R=R1=Me, Ala-AlaP) [12] | AAP-AAC [13,14] |
| Abbreviation of AAP and (AC)-AAP follow the general rules elaborated by Kudzin et al. [15]. | ||||
Table 2.
31P NMR chemical shifts of (AC)-AAP and the corresponding adduct compounds (AC)(OH)-AAP formed immediately after mixing the reaction mixture. (A) 31P NMR chemical shifts of Ac-AAP and the corresponding adduct compounds Ac(OH)-AAP formed. (B) 31P NMR chemical shifts of (AC)-GlyP and the corresponding adduct compounds [AC(OH)]-GlyP formed.
Table 2.
31P NMR chemical shifts of (AC)-AAP and the corresponding adduct compounds (AC)(OH)-AAP formed immediately after mixing the reaction mixture. (A) 31P NMR chemical shifts of Ac-AAP and the corresponding adduct compounds Ac(OH)-AAP formed. (B) 31P NMR chemical shifts of (AC)-GlyP and the corresponding adduct compounds [AC(OH)]-GlyP formed.
| (A) | |||||||||||
| Ac-GlyP | Ac-AlaP | Ac-NvaP | Ac-ValP | Ac-PglP | |||||||
| Ac-GlyP | Ac(OH)-GlyP | Ac-AlaP | Ac(OH)-AlaP | Ac-NvaP | Ac(OH)-NvaP | Ac-ValP | Ac(OH)-ValP | Ac-PglP | Ac(OH)-PglP | ||
| δ | 13.4 | 12.8 | 17.0 | 16.5 | 15.9 | 15.2 | 16.8 | 16.1 | 13.3 | 12.7 | |
| % | 97.1 | 2.9 | 97.1 | 2.9 | 96.3 | 3.7 | 97.0 | 3.0 | 97.0 | 3.0 | |
| (B) | |||||||||||
| Ac-GlyP | Prp-GlyP | Bz-GlyP | |||||||||
| GlyP | Ac-GlyP | Ac(OH)-GlyP | GlyP | Prp-GlyP | Prp(OH)-GlyP | GlyP | Bz-GlyP | Bz(OH)-GlyP | |||
| δ | 19.1 | 13.4 | 12.8 | 19.3 | 13.4 | 12.8 | 19.3 | 13.5 | 13.2 | ||
| % | - | 97.1 | 2.9 | - | 98.3 | 1.7 | - | 99.5 | <0.5 | ||
Table 3.
Gibbs free energies ΔG (298 K) for reaction 1.1 in gas phase (kcal/mol).
 | ||||
|---|---|---|---|---|
| (AC)-AAP (1Xx) | R | R1-C(O)- (1Xx) | ||
| CH3- (1Ax) | CF3- (1Bx) | Ph- (1Cx) | ||
| (AC)-GlyP (1Xa) | H- | 139.43 | 123.16 | 126.87 |
| (AC)-AlaP (1Xb) | CH3- | 135.19 | 119.26 | 124.82 |
| (AC)-ValP (1Xc) | (CH3)2CH- | 133.61 | 117.48 | 132.28 |
| (AC)-PglP (1Xd) | Ph- | 125.63 | 121.43 | 117.08 |
| (AC)-PheP (1Xe) | PhCH2- | 139.31 | 122.06 | 129.57 |
Table 4.
Gibbs free energies ΔG (298 K) for reaction 1.1 in aqueous phase (kcal/mol).
 | ||||
|---|---|---|---|---|
| (AC)-AAP (1Xx) | R | R1-C(O)- (1Xx) | ||
| CH3- (1Ax) | CF3- (1Bx) | Ph- (1Cx) | ||
| (AC)-GlyP (1Xa) | H- | 26.70 | 11.77 | 25.70 |
| (AC)-AlaP (1Xb) | CH3- | 26.26 | 10.54 | 25.45 |
| (AC)-ValP (1Xc) | (CH3)2CH- | 26.15 | 10.90 | 26.57 |
| (AC)-PglP (1Xd) | Ph- | 25.47 | 12.27 | 24.96 |
| (AC)-PheP (1Xe) | PhCH2- | 24.22 | 7.73 | 24.53 |
Table 5.
Free energy barriers (ΔG‡ (298 K), kcal/mol) for OH− addition (reaction 1.1) in water.
 | ||||
|---|---|---|---|---|
| (AC)-AAP (1Xx) | R | R1-C(O)- (1Xx) | ||
| CH3- (1Ax) | CF3- (1Bx) | Ph- (1Cx) | ||
| (AC)-GlyP (1Xa) | H- | 29.99 | 19.51 | 25.70 |
| (AC)-AlaP (1Xb) | CH3- | 30.85 | 19.12 | 26.75 |
| (AC)-ValP (1Xc) | (CH3)2CH- | 31.40 | 18.47 | 29.68 |
| (AC)-PglP (1Xd) | Ph- | 28.57 | 17.60 | 28.44 |
| (AC)-PheP (1Xe) | PhCH2- | 28.85 | 18.66 | 30.23 |
Table 6.
Gibbs free energy ΔG (298 K) for the reaction 1.2 in gas phase (kcal/mol).
 | ||||
|---|---|---|---|---|
| (AC-OH)-AAP (2-Xx) | R | R1-C(O−)(OH)- (2Xx) | ||
| CH3- (2Ax) | CF3- (2Bx) | Ph- (2Cx) | ||
| (AC-OH)-GlyP (2Xa) | H- | −165.22 | −153.75 | −152.91 |
| (AC-OH)-AlaP (2Xb) | CH3- | −159.50 | −149.37 | −149.31 |
| (AC-OH)-ValP (2Xc) | (CH3)2CH- | −162.42 | −152.74 | −161.46 |
| (AC-OH)-PglP (2Xd) | Ph- | −157.24 | −160.78 | −150.14 |
| (AC-OH)-PheP (2Xe) | PhCH2- | −168.00 | −158.21 | −159.56 |
Table 7.
Gibbs free energy ΔG (298 K) for the reaction 1.2 in an aqueous phase (kcal/mol).
 | ||||
|---|---|---|---|---|
| (AC-OH)-AAP (2-Xx) | R | R1-C(O−)(OH)- (2Xx) | ||
| CH3- (2Ax) | CF3- (2Bx) | Ph- (2Cx) | ||
| (AC-OH)-GlyP (2Xa) | H- | −46.41 | −40.32 | −48.51 |
| (AC-OH)-AlaP (2Xb) | CH3- | −46.62 | −38.58 | −48.56 |
| (AC-OH)-ValP (2Xc) | (CH3)2CH- | −44.45 | −37.28 | −47.29 |
| (AC-OH)-PglP (2Xd) | Ph- | −45.60 | −40.90 | −47.44 |
| (AC-OH)-PheP (2Xe) | PhCH2- | −47.74 | −39.80 | −50.89 |
Table 8.
Free energy barriers (ΔG‡ (298 K), kcal/mol) for proton transfer/dissociation of the C-N bond in water (reaction 1.2).
Table 8.
Free energy barriers (ΔG‡ (298 K), kcal/mol) for proton transfer/dissociation of the C-N bond in water (reaction 1.2).
 | ||||
|---|---|---|---|---|
| (AC-OH)-AAP (2-Xx) | R | R1-C(O−)(OH)- (2Xx) | ||
| CH3- (2Ax) | CF3- (2Bx) | Ph- (2Cx) | ||
| (AC-OH)-GlyP (2Xa) | H- | 20.76 | 21.54 | 16.91 |
| (AC-OH)-AlaP (2Xb) | CH3- | 21.47 | 23.43 | 19.00 |
| (AC-OH)-ValP (2Xc) | (CH3)2CH- | 23.73 | 24.91 | 20.56 |
| (AC-OH)-PglP (2Xd) | Ph- | 20.05 | 21.74 | 18.51 |
| (AC-OH)-PheP (2Xe) | PhCH2- | 22.50 | 25.00 | 18.99 |
Table 9.
Total Gibbs free energiesΔG298(1.1) + ΔG298(1.2) for the overall deacylation process in the aqueous phase and total Gibbs free energy barriers (ΔG298(1.1) + ΔG‡(1.2), in parentheses) (kcal/mol).
Table 9.
Total Gibbs free energiesΔG298(1.1) + ΔG298(1.2) for the overall deacylation process in the aqueous phase and total Gibbs free energy barriers (ΔG298(1.1) + ΔG‡(1.2), in parentheses) (kcal/mol).
 | ||||
|---|---|---|---|---|
| (AC)-AAP (1Xx) | R | R1-C(O)- (1Xx) | ||
| CH3- (1Ax) | CF3- (1Bx) | Ph- (1Cx) | ||
| (AC)-GlyP (1Xa) | H- | −19.72 (47.5) | −28.56 (31.3) | −22.82 (42.6) |
| (AC)-AlaP (1Xb) | CH3- | −20.36 (47.7) | −28.04 (34.0) | −23.11 (44.5) |
| (AC)-ValP (1Xc) | (CH3)2CH- | −18.30 (49.9) | −26.38 (35.8) | −20.72 (47.1) |
| (AC)-PglP (1Xd) | Ph- | −20.13 (45.5) | −28.63 (34.0) | −22.48 (43.5) |
| (AC)-PheP (1Xe) | PhCH2- | −23.53 (46.7) | −32.08 (32.7) | −26.36 (43.5) |
Table 10.
Structures, names and abbreviations of aminophosphonic acids and corresponding 1-(acylamino)phosphonic acids discussed in this work a.
Table 10.
Structures, names and abbreviations of aminophosphonic acids and corresponding 1-(acylamino)phosphonic acids discussed in this work a.
| Name | Trivial Name | Abbr./(No) | Ref. | ||
 | Aminoalkyl-phosphonic acid | Phosphono-Amino Acid | AAP (3) | ||
| R | |||||
| H | Aminomethyl-phosphonic acid | Phosphono-glycine | GlyP (3a) | [64] | |
| Me | 1-Aminoethyl-phosphonic acid | Phosphono-alanine | AlaP (3b) | [65] | |
| iPr | 1-Amino-1-methylethyl-phosphonic acid | Phosphono-valine | ValP (3c) | ||
| Ph | 1-Amino-1-phenylmethyl-phosphonic acid | Phosphono-phenyl-glycine | PglP (3d) | ||
| PhCH2 | 1-Amino-1-phenylethyl-phosphonic acid | Phosphono-phenyl-alanine | PheP (3e) | ||
 | 1-(Acylamino)alkyl-phosphonic acid | Acylamino-Phosphono Acid | (AC)-AAP (2) | ||
| R1 | R | ||||
| CH3 | H, Me, iPr, Ph, PhCH2 | 1-(Acetylamino)alkyl-phosphonic acid | Acetylamino-Phosphono Acid | Ac-AAP (2A) | [36] |
| CH3 | H | 1-(Acetylamino)methyl-phosphonic acid | Phosphono-Acetylamino Glycine | Ac-GlyP (2Aa) | |
| CH3 | Ph | 1-(Acetylamino)-1-phenyl-methyl-phosphonic acid | Phosphono-Acetyl-amino-PhenylGlycine | Ac-PglP (2Ad) | |
| CF3 | H, Me, iPr, Ph, PhCH2 | 1-(Trifluoroacetylamino)-alkyl-phosphonic acid | Phosphono-Trifluoroacetyl-amino Acid | TFA-AAP (2B) | [37] |
| CF3 | iPr | 1-(Trifluoroacetylamino)-2-methylethyl-phosphonic acid | Phosphono-Trifluoroacetyl-Valine | TFA-ValP (2Bc) | |
| Ph | H, Me, iPr, Ph, PhCH2 | 1-(Benzoylamino)alkyl-phosphonic acid | Phosphono-Benzoylamino Acid | Bz-AAP (2C) | [36] |
| Ph | PhCH2 | 1-(Benzoylamino)-2-phenyl-ethyl-phosphonic acid | Phosphono-Benzoyl-Phenyl-alanine | Bz-PheP (2Ce) | |
a Applied names were in accordance with the IUPAC rules, and the abbreviations were in agreement with the general rules elaborated by Kudzin et al. [15].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cypryk, M.; Drabowicz, J.; Gostynski, B.; Kudzin, M.H.; Kudzin, Z.H.; Urbaniak, P. 1-(Acylamino)alkylphosphonic Acids—Alkaline Deacylation. Molecules 2018, 23, 859. https://doi.org/10.3390/molecules23040859
AMA Style
Cypryk M, Drabowicz J, Gostynski B, Kudzin MH, Kudzin ZH, Urbaniak P. 1-(Acylamino)alkylphosphonic Acids—Alkaline Deacylation. Molecules. 2018; 23(4):859. https://doi.org/10.3390/molecules23040859
Chicago/Turabian StyleCypryk, Marek, Jozef Drabowicz, Bartlomiej Gostynski, Marcin H. Kudzin, Zbigniew H. Kudzin, and Pawel Urbaniak. 2018. "1-(Acylamino)alkylphosphonic Acids—Alkaline Deacylation" Molecules 23, no. 4: 859. https://doi.org/10.3390/molecules23040859