
Exosome-Mediated Transfer of Cancer Cell Resistance to Antiestrogen Drugs


 , , ,
, , , 
Abstract
:1. Introduction
2. Results
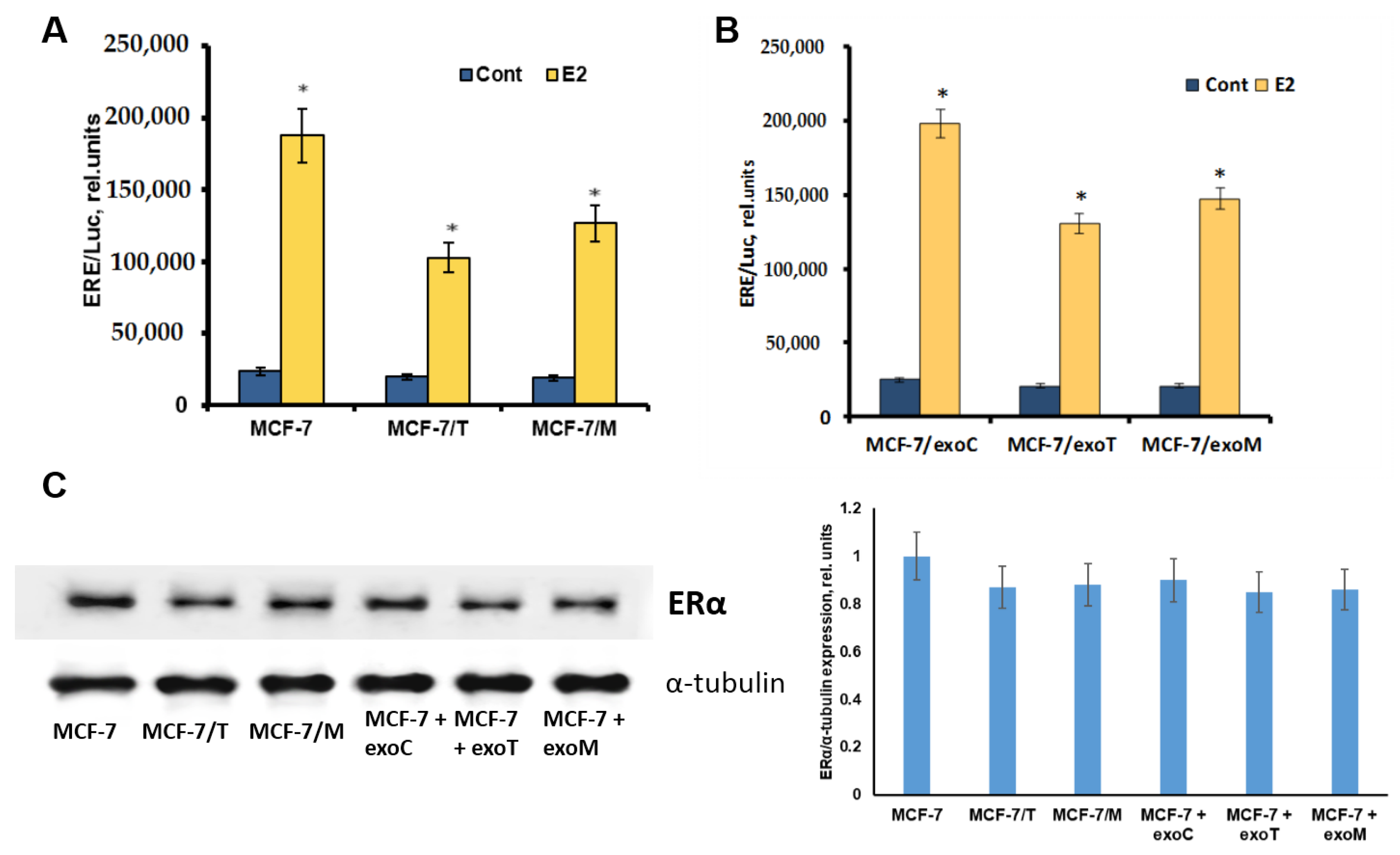
2.1. Development and Characteristics of MCF-7 Resistant Derivatives
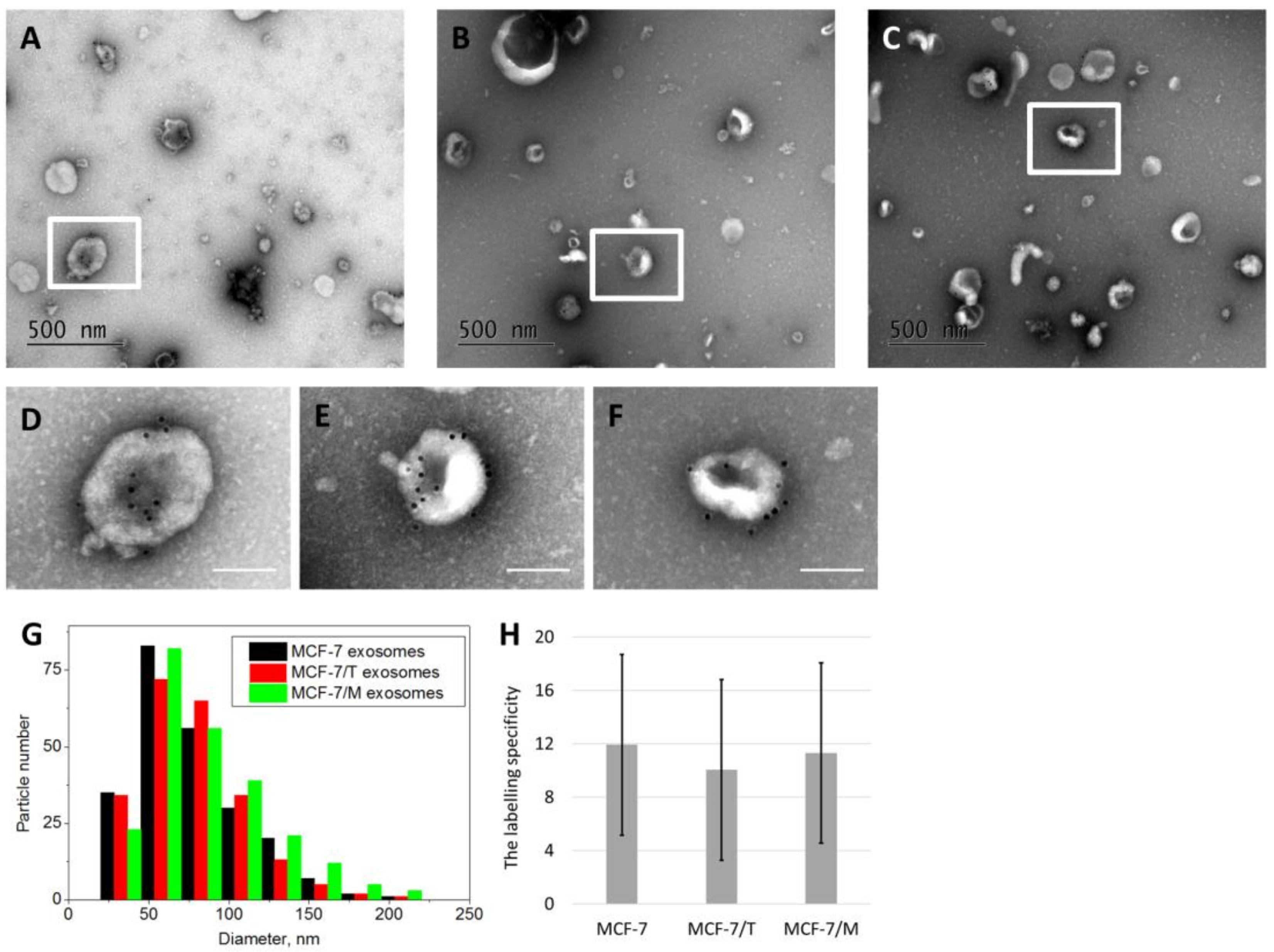
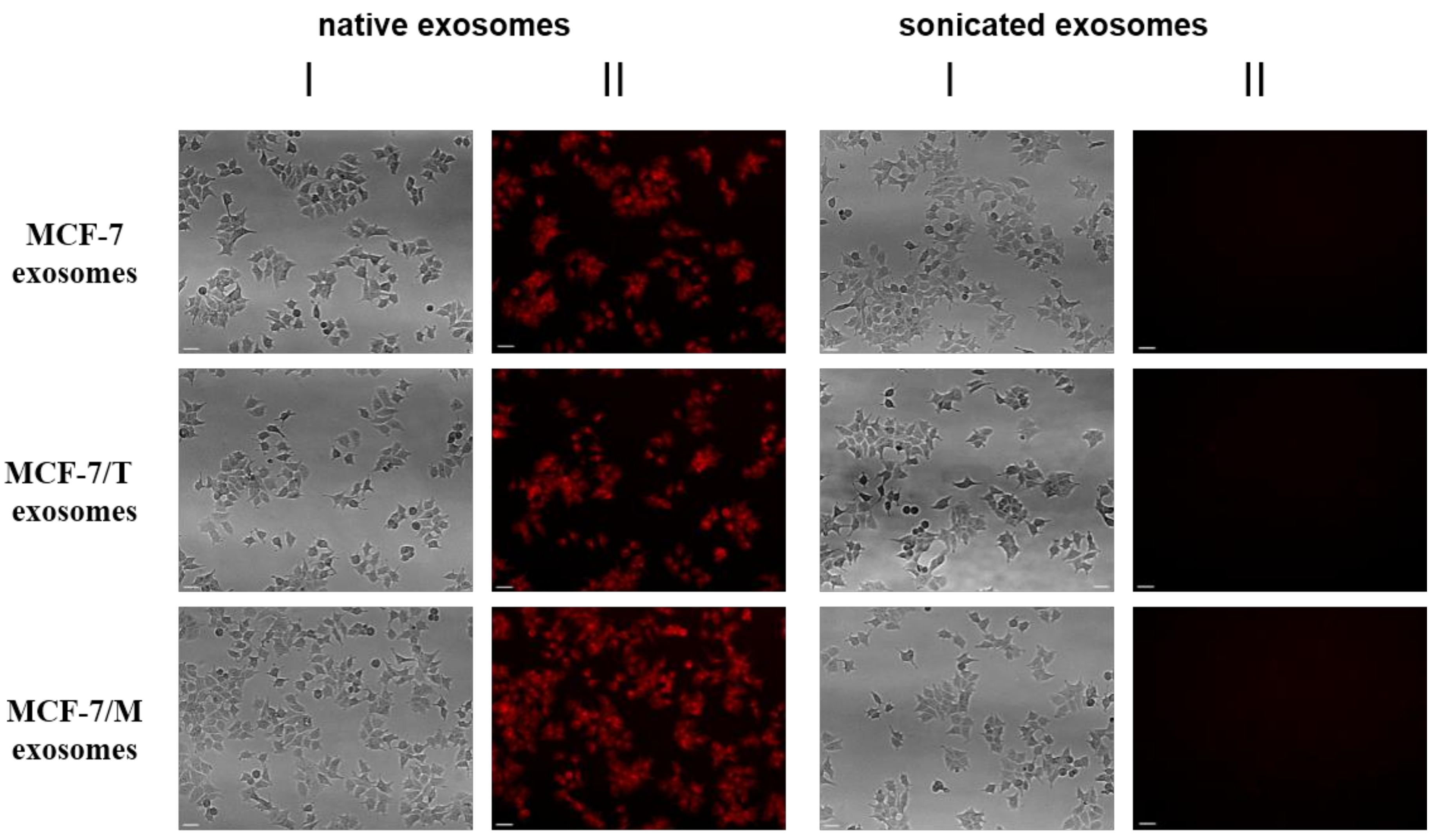
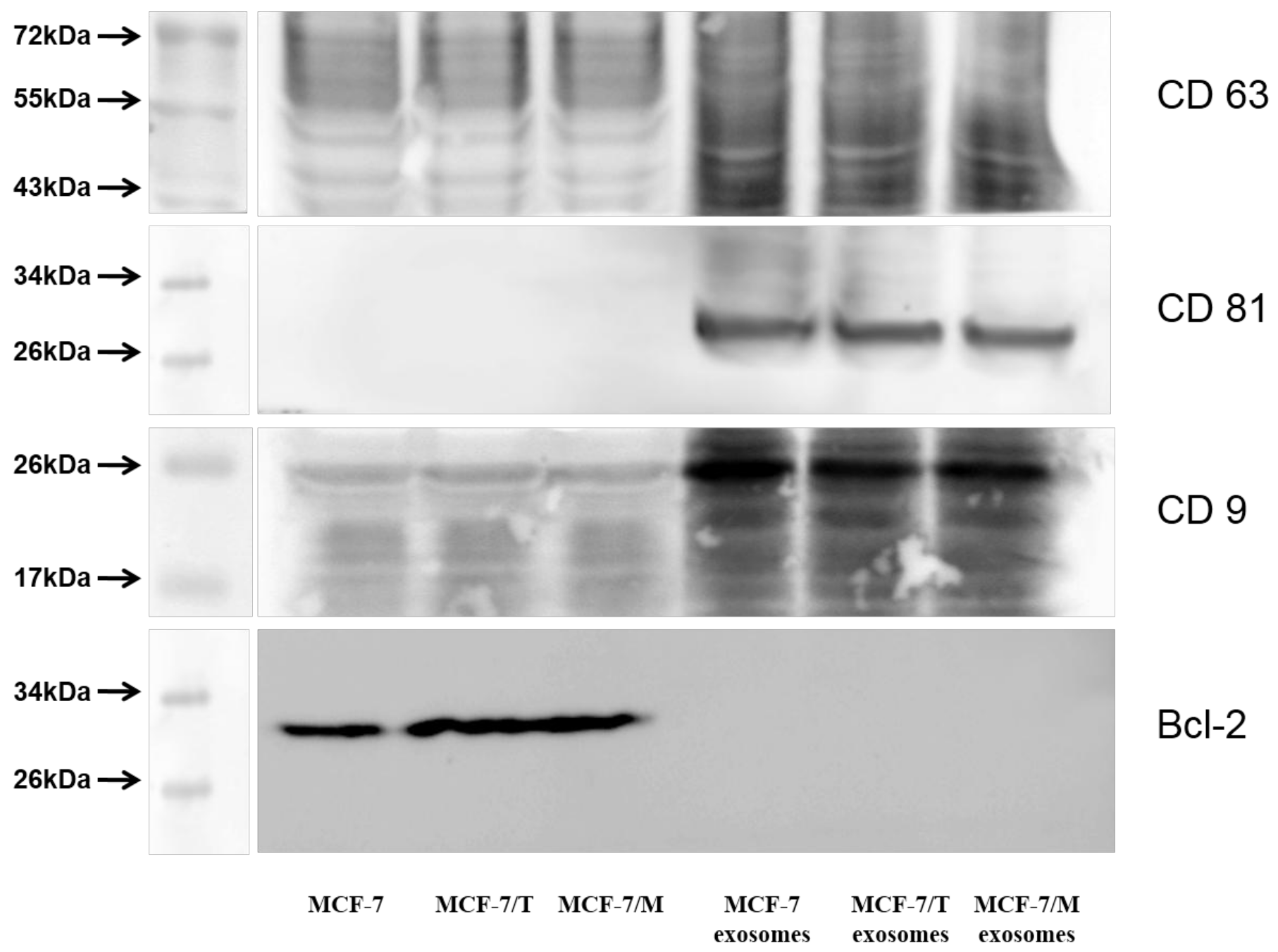
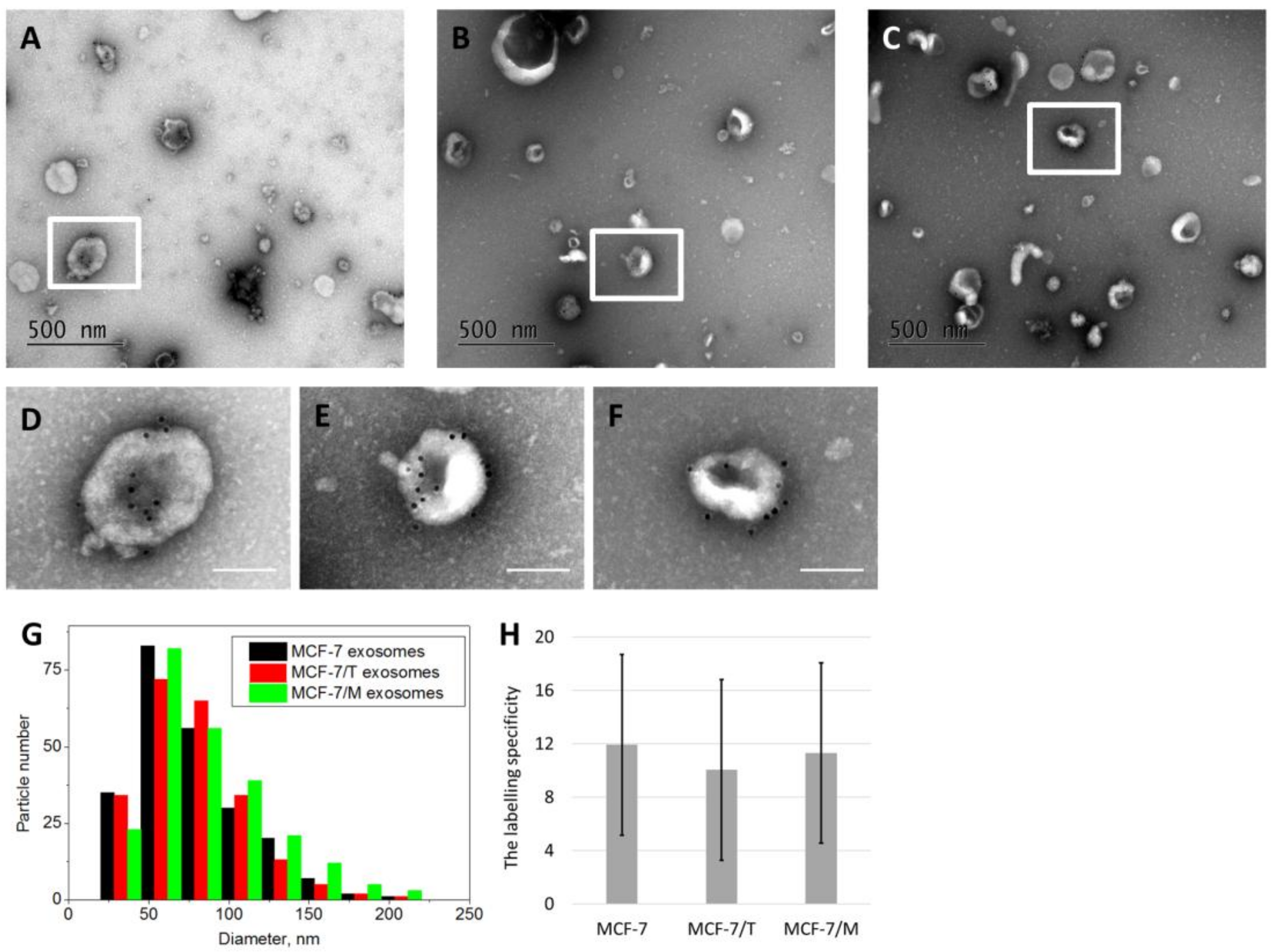
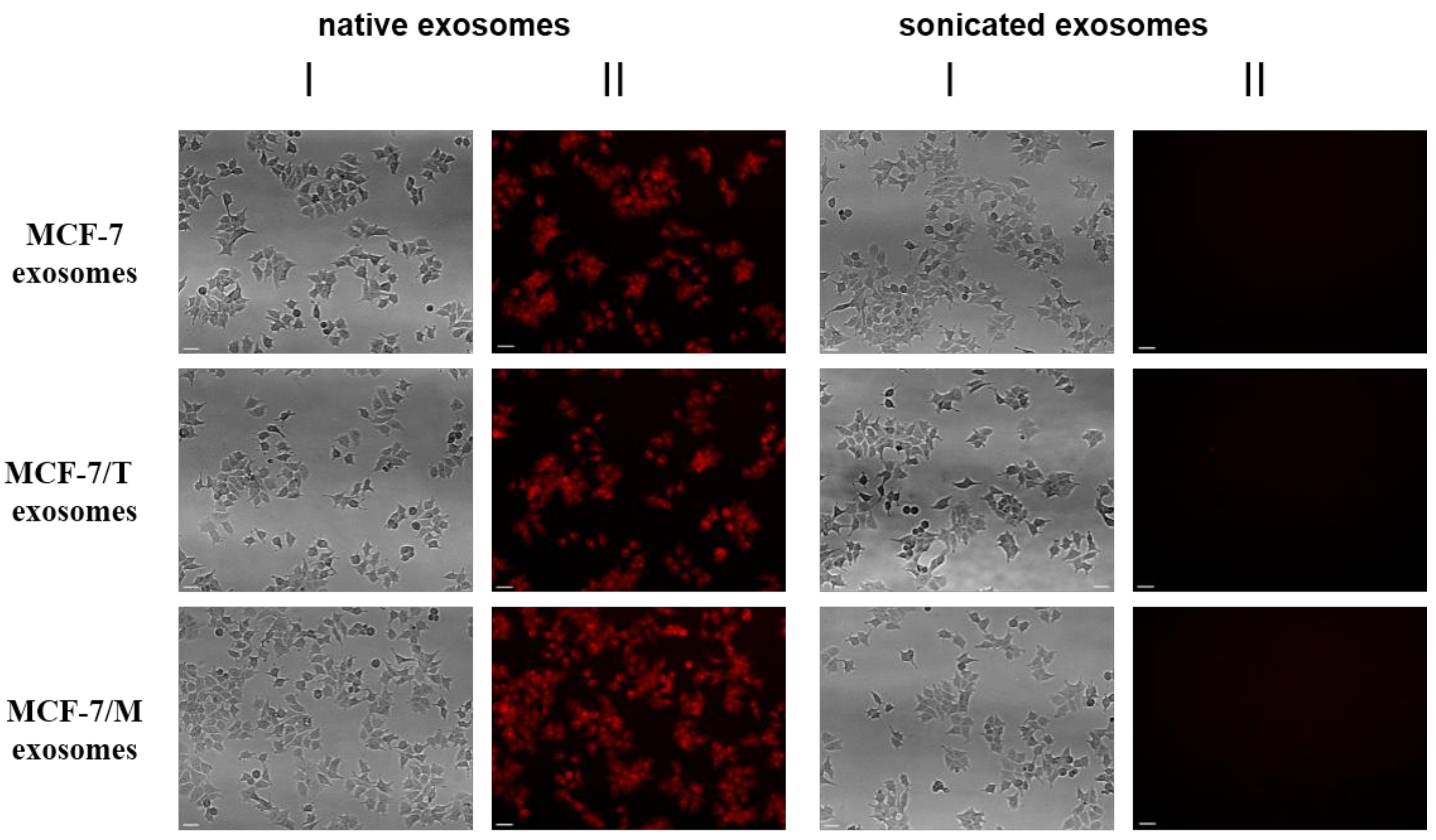
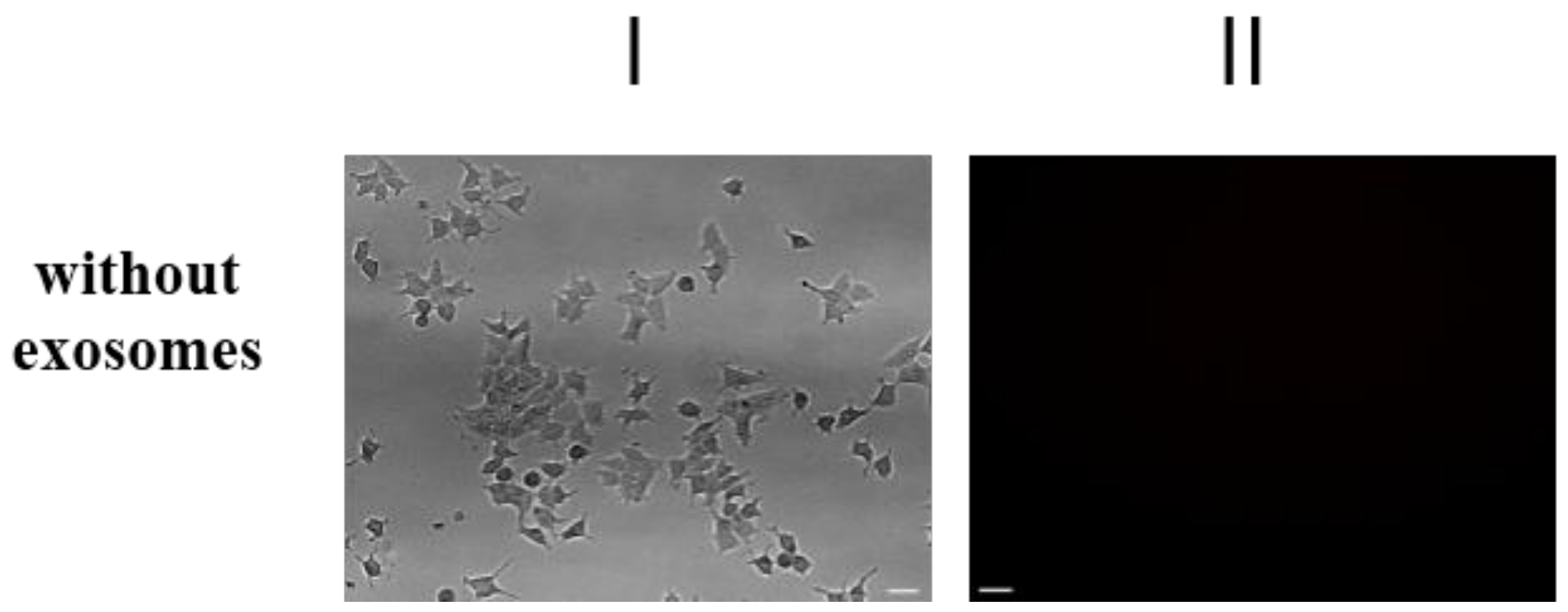
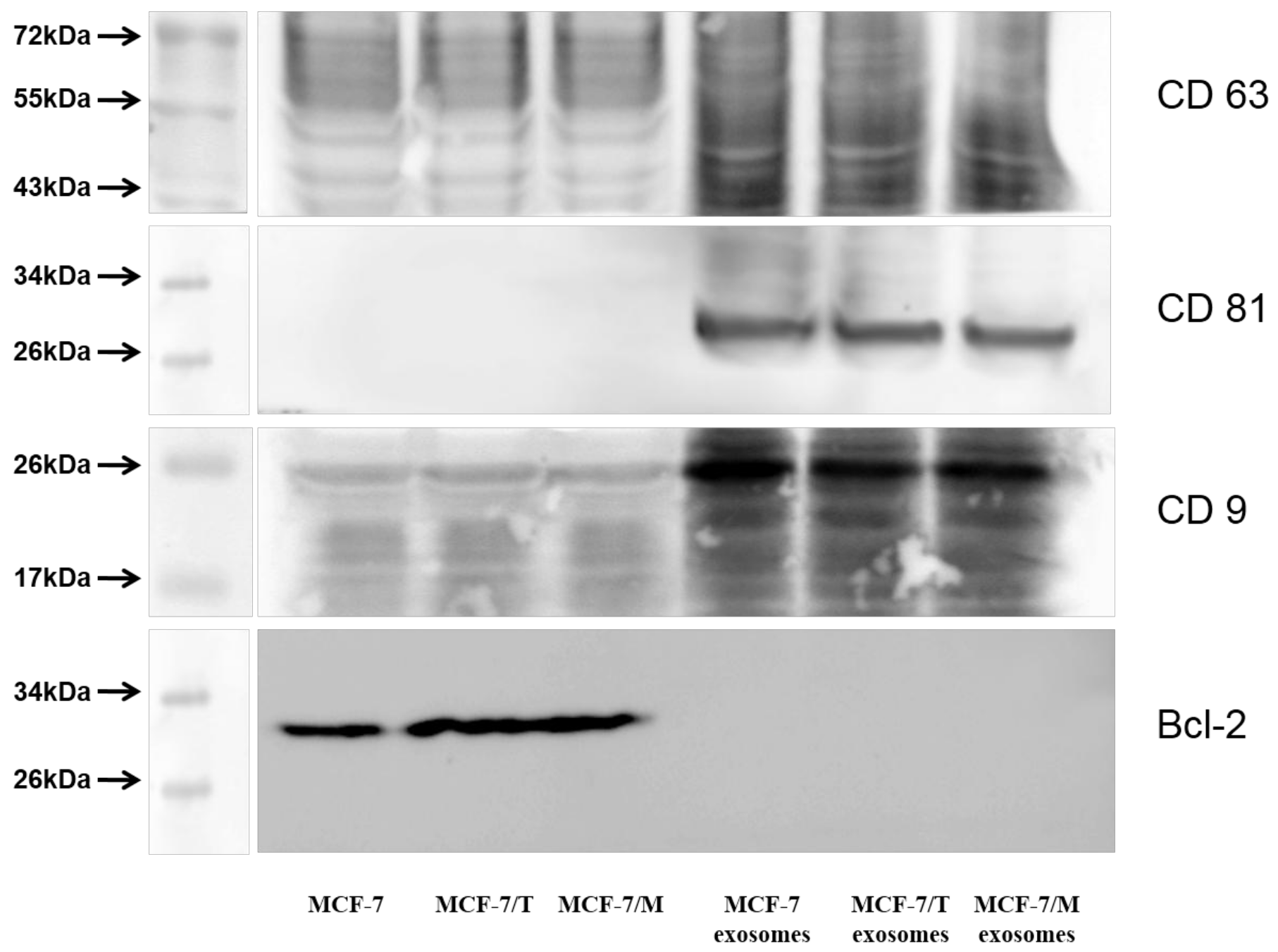
2.2. Purification and Characterization of the Exosomes
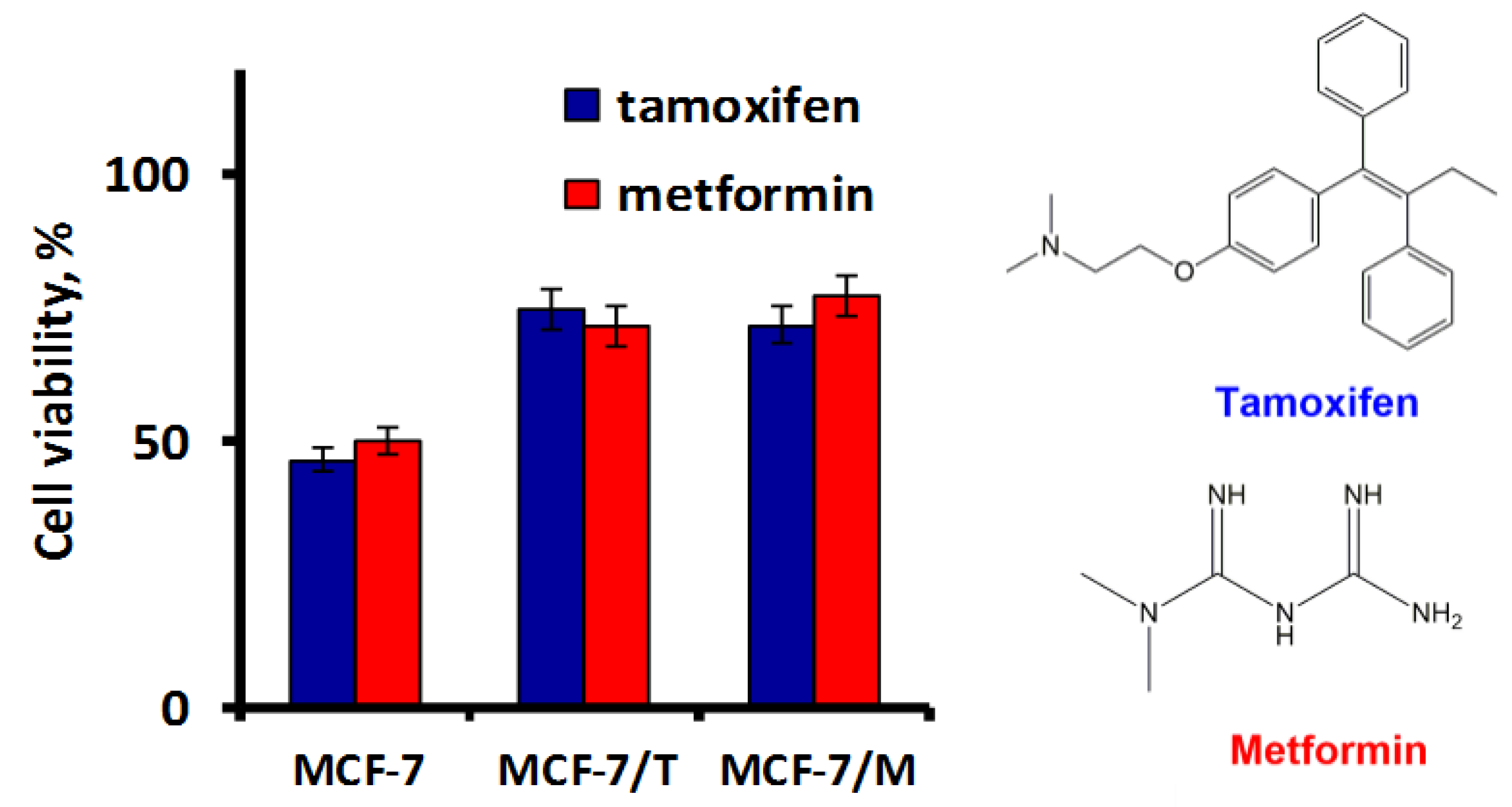
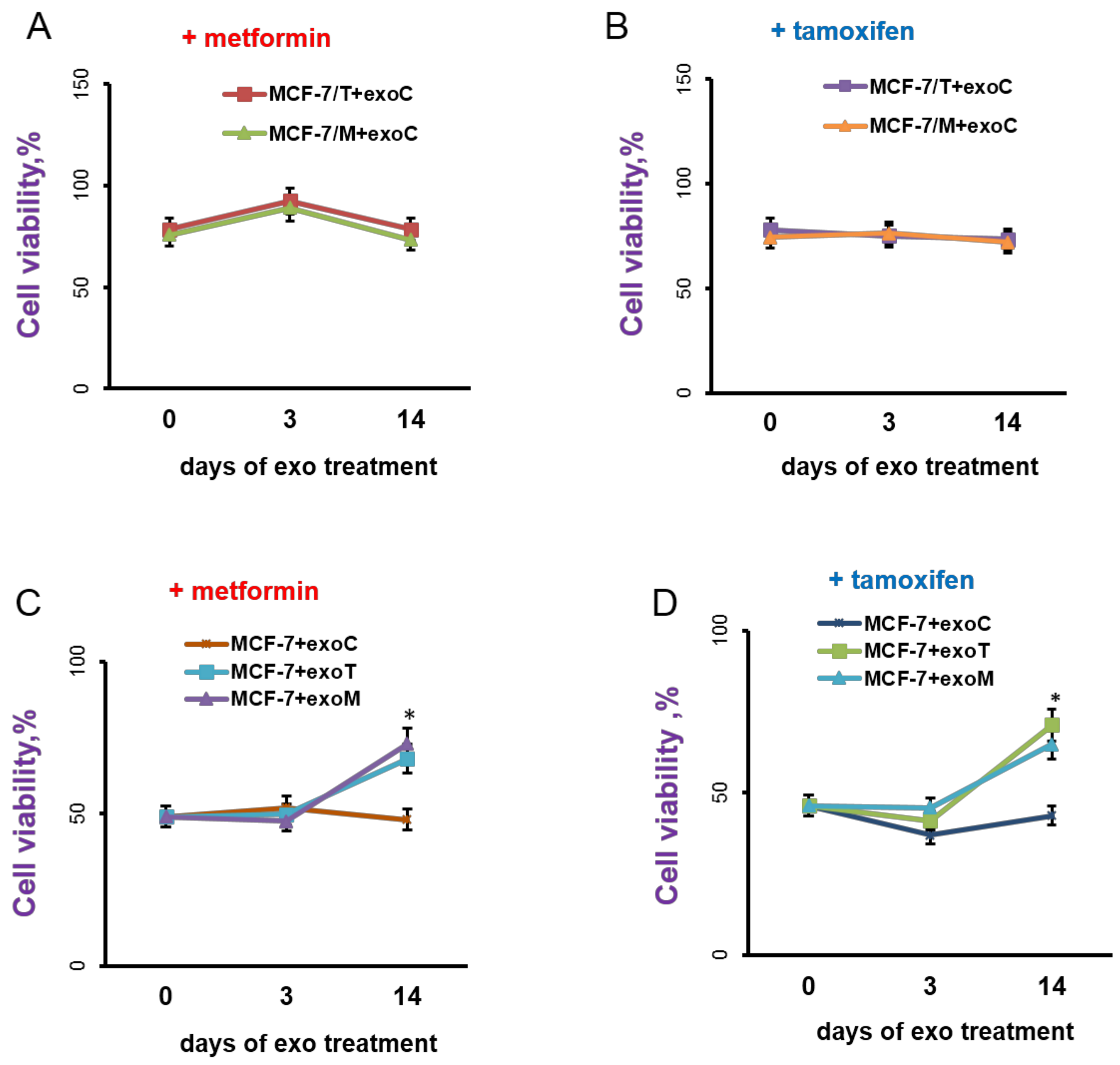
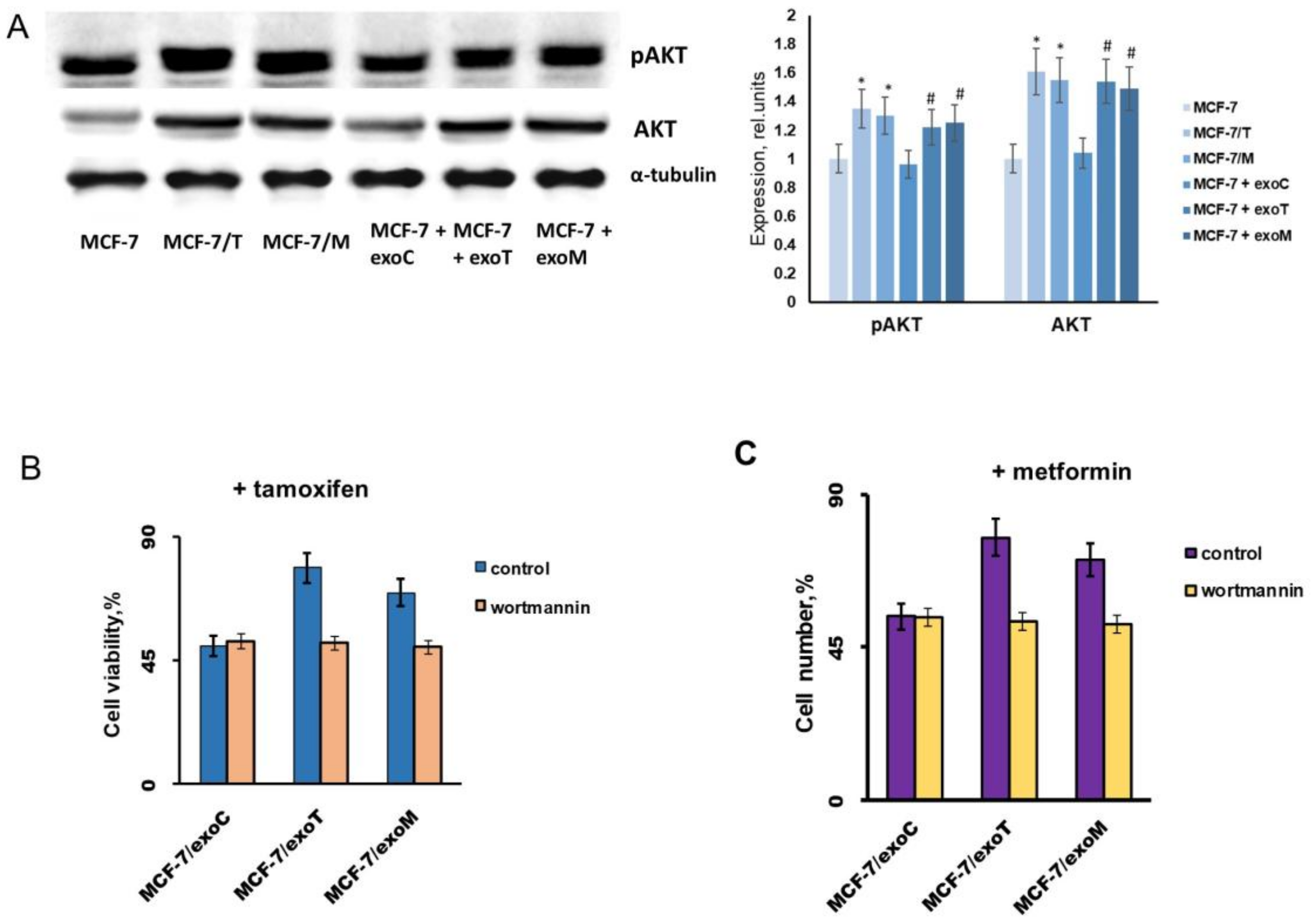
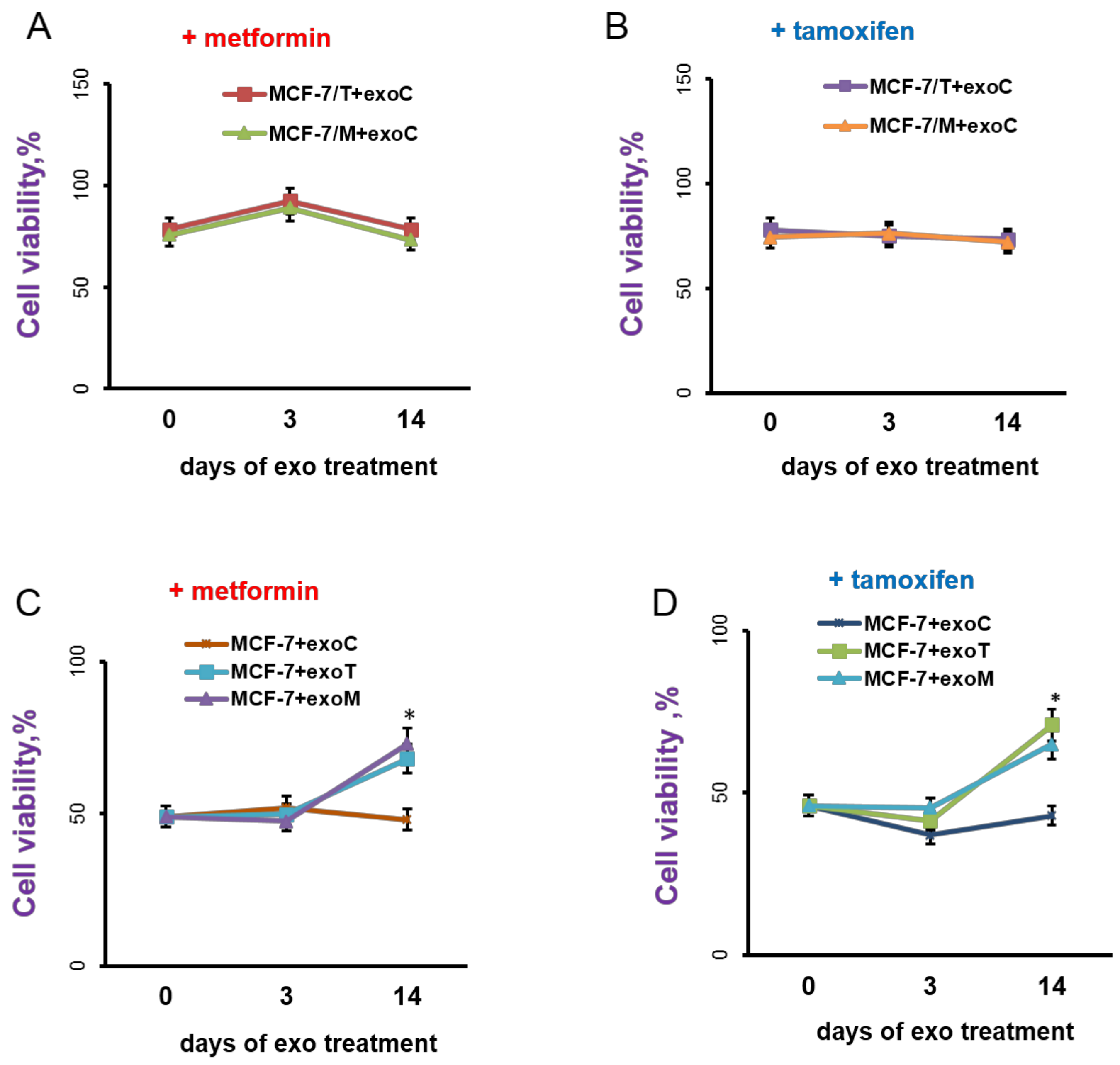
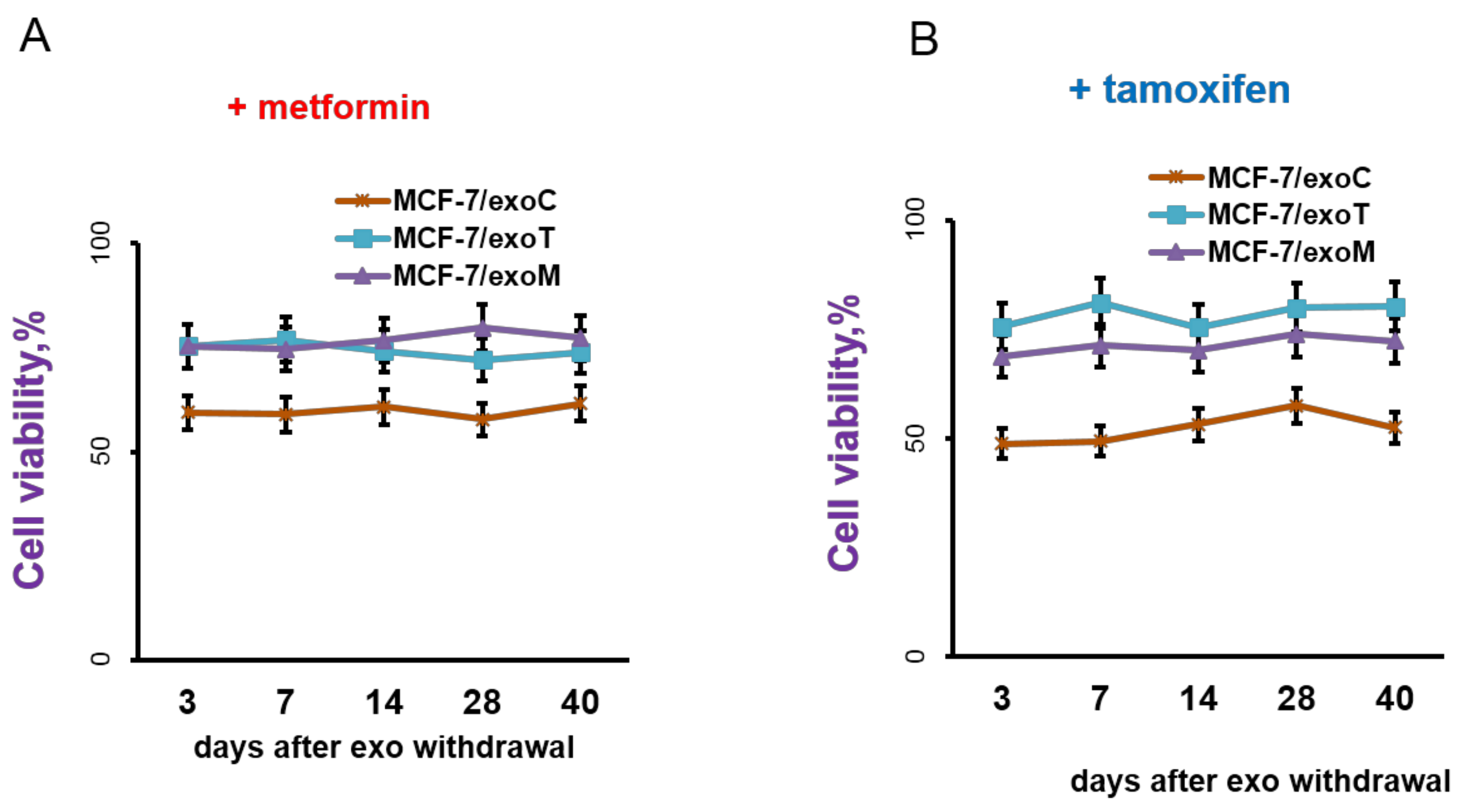
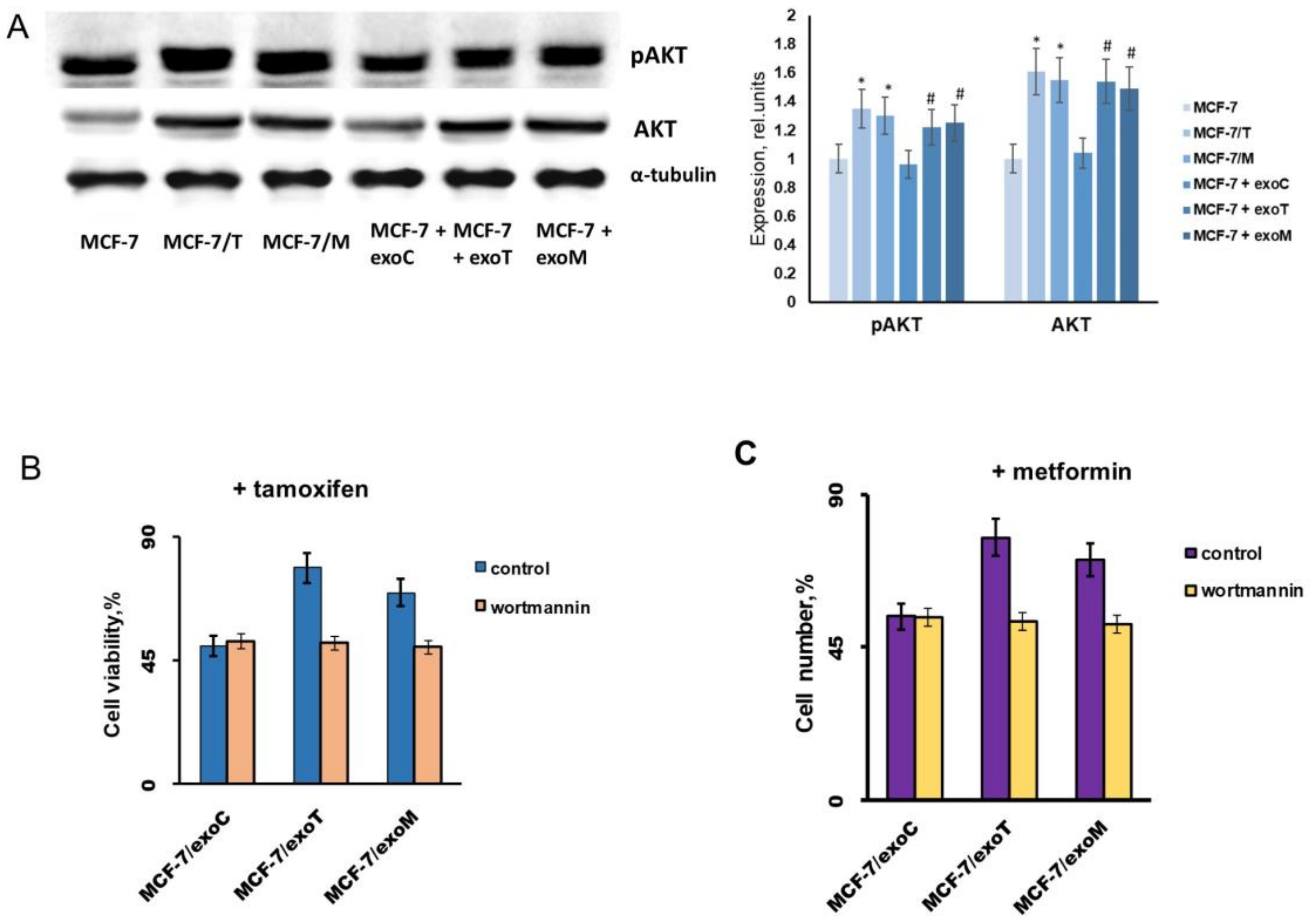
2.3. Exosomes Influence on the Cell Response to Tamoxifen and Metformin
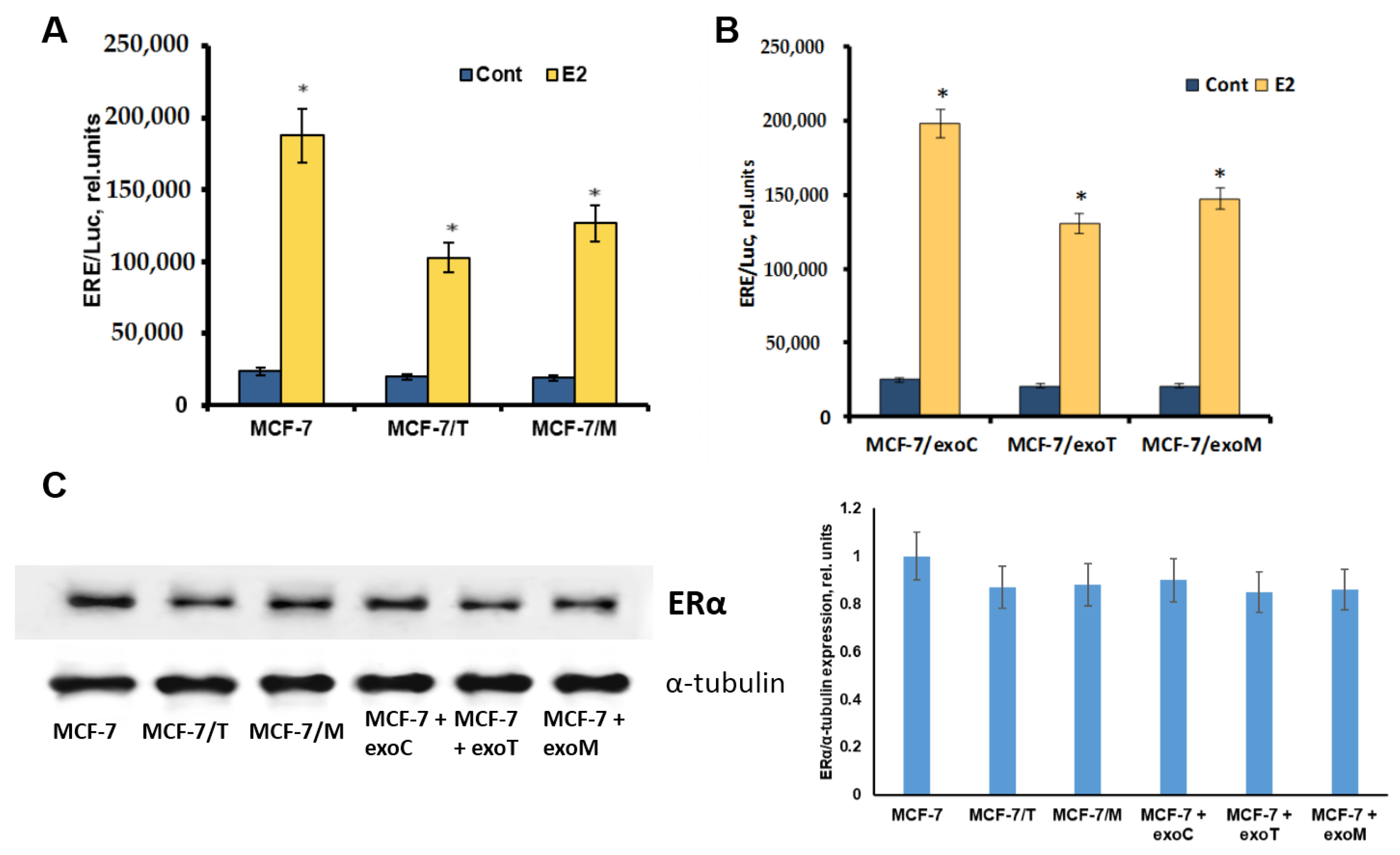
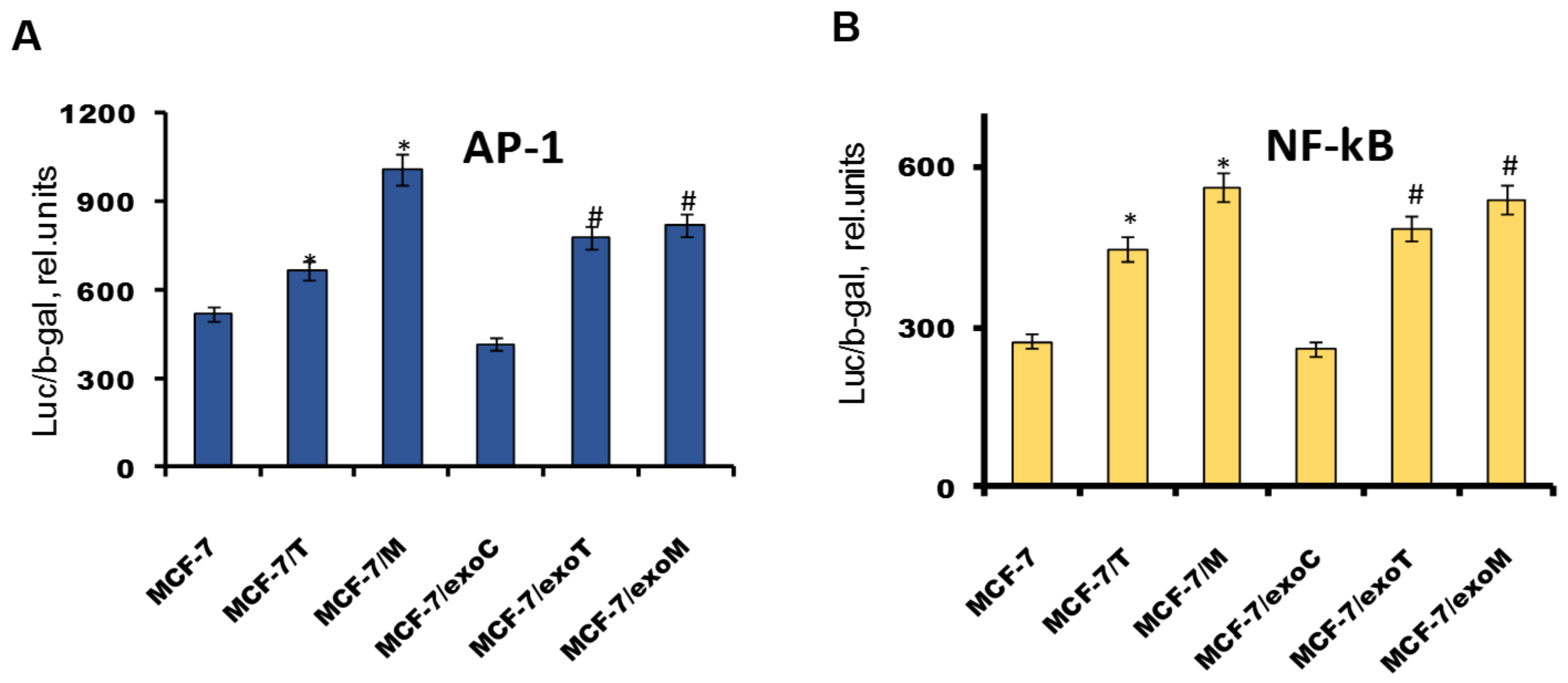
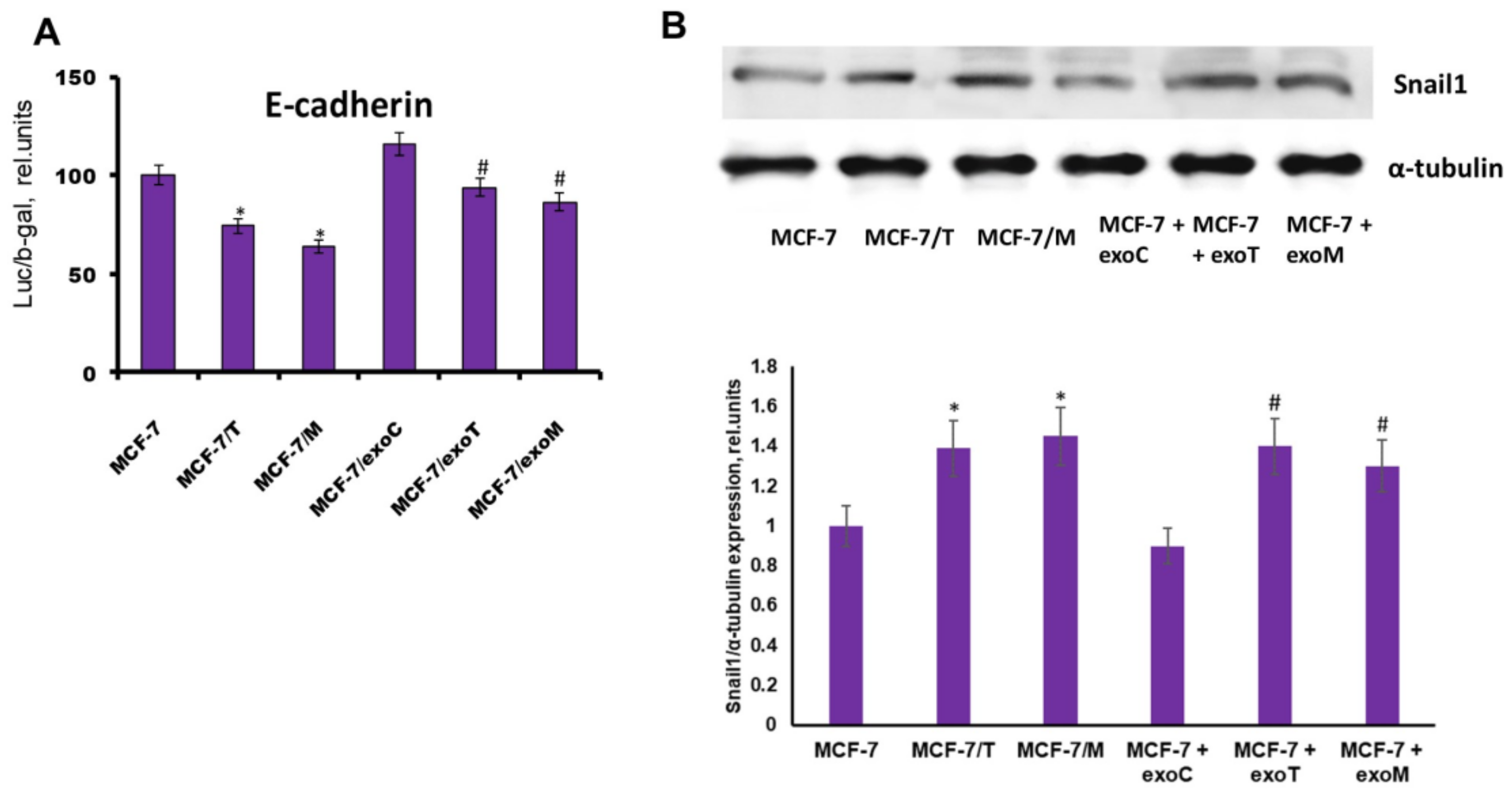
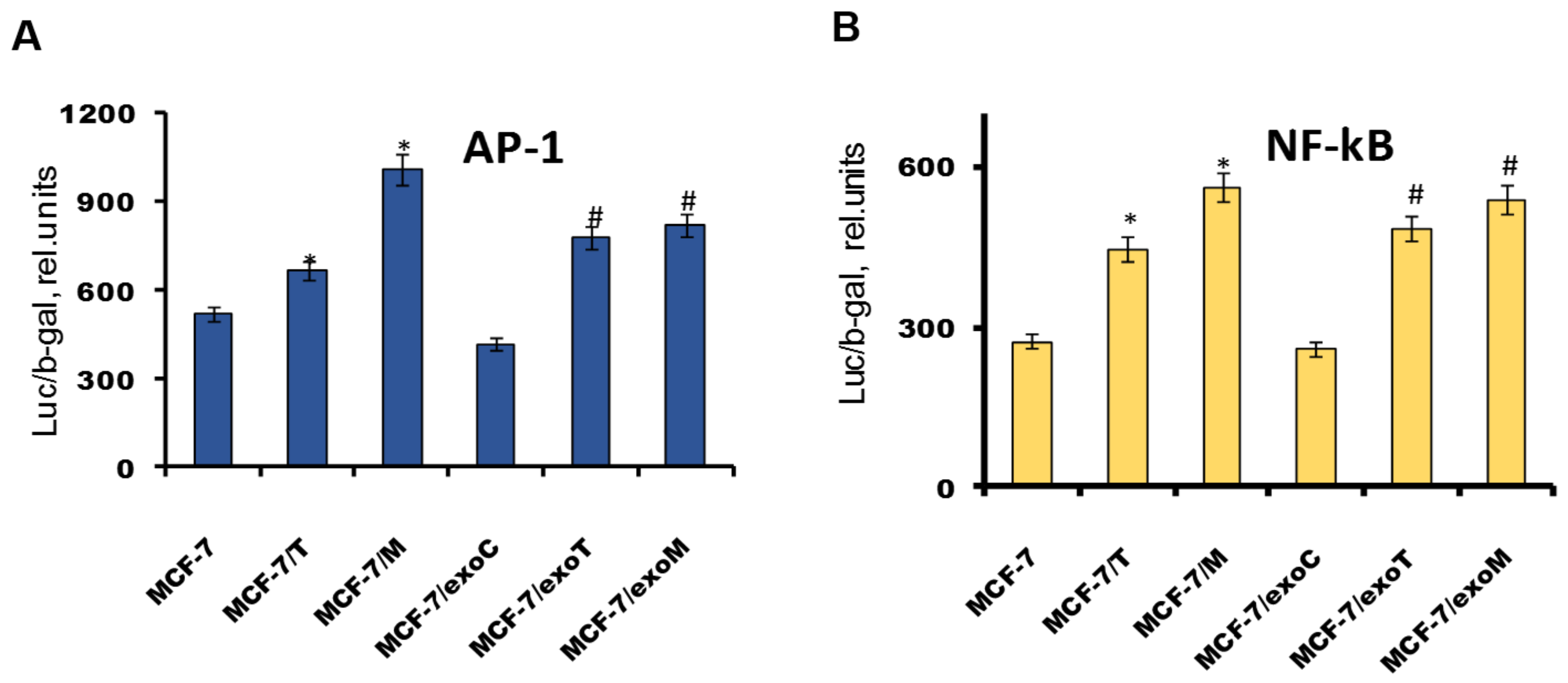
2.4. Protein Signature of the Exosome-Generated Resistant Cells
3. Discussion and Conclusions
4. Materials and Methods
4.1. Cell Cultures and Development of Drug Resistant Derivatives
4.2. Next Generation Sequencing
4.2.1. DNA Preparation
4.2.2. Library Preparation for NGS. Clonal Amplification
4.2.3. Sequencing
4.2.4. NGS Mapping
4.3. Transient Transfection and Measurement of Reporter Gene Activity
4.4. Western Blot Analysis of Cell Lysates
4.5. Exosomes Purification and Analysis
4.5.1. Exosomes Isolation by Ultracentrifugation
Western Blot Analysis of Exosomes
4.5.2. Transportation of Fluorescent-Labeled Compounds and Peptides
4.5.3. Extracellular Vesicles Size and Concentration Measurements
4.5.4. Transmission Electron Microscopy (TEM)
4.5.5. Cells Treatment with Exosome Preparations
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DMSO | Dimethylsulfoxide |
| STR | Short Tandem Repeats |
| DMEM | Dulbecco’s Modified Eagle Medium |
| ERE | Estrogen-responsive elements |
| FBS | Fetal Bovine Serum |
| SDS | Sodium Dodecyl Sulfate |
| TBS | Tris-Buffered Saline |
| E2 | 17β-Estradiol |
References
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.P.; Ma, C.X. Endocrine resistance in breast cancer: Molecular pathways and rational development of targeted therapies. Future Oncol. 2012, 8, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Arteaga, C.L.; Miller, T.W. Abrogating endocrine resistance by targeting ERalpha and PI3K in breast cancer. Front. Oncol. 2012, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Riggins, R.B.; Bouton, A.H.; Liu, M.C.; Clarke, R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam. Horm. 2005, 71, 201–237. [Google Scholar] [PubMed]
- Krasil’nikov, M.A. Modern approaches to estrogen-independent growth of breast tumor. Vopr. Onkol. 2004, 50, 399–405. [Google Scholar] [PubMed]
- Arpino, G.; De Angelis, C.; Giuliano, M.; Giordano, A.; Falato, C.; De Laurentiis, M.; De Placido, S. Molecular mechanism and clinical implications of endocrine therapy resistance in breast cancer. Oncology 2009, 77 (Suppl. 1), 23–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Eppenberger-Castori, S.; Eppenberger, U.; Benz, C.C. The NFkappaB pathway and endocrine-resistant breast cancer. Endocr. Relat. Cancer 2005, 12 (Suppl. 1), S37–S46. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Awasthi, S.; Peterson, J.R.; Hamburger, A.W. Regulation of tamoxifen sensitivity by a PAK1-EBP1 signalling pathway in breast cancer. Br. J. Cancer 2013, 108, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Andreeva, O.E.; Shatskaya, V.A.; Krasil’nikov, M.A. The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J. Cell. Biochem. 2012, 113, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Magal, P.; Boulange-Lecomte, C.; Webb, G.; Le Foll, F. Consequences of cell-to-cell P-glycoprotein transfer on acquired multidrug resistance in breast cancer: A cell population dynamics model. Biol. Direct. 2011, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.X.; Liu, X.M.; Lv, M.M.; Chen, L.; Zhao, J.H.; Zhong, S.L.; Ji, M.H.; Hu, Q.; Luo, Z.; Wu, J.Z.; et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.; Bebawy, M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.G.; Yang, M.F.; Ren, Y.Q.; Wu, C.H.; Wang, L.Q. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4362–4368. [Google Scholar] [PubMed]
- Min, Q.H.; Wang, X.Z.; Zhang, J.; Chen, Q.G.; Li, S.Q.; Liu, X.Q.; Li, J.; Liu, J.; Yang, W.M.; Jiang, Y.H.; et al. Exosomes derived from imatinib-resistant chronic myeloid leukemia cells mediate a horizontal transfer of drug-resistant trait by delivering miR-365. Exp. Cell Res. 2018, 362, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Abd Elmageed, Z.Y.; Hawke, D.H.; Worner, P.M.; Jansen, D.A.; Abdel-Mageed, A.B.; Alt, E.U.; Izadpanah, R. Molecular characterization of exosome-like vesicles from breast cancer cells. BMC Cancer 2014, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Riches, A.; Campbell, E.; Borger, E.; Powis, S. Regulation of exosome release from mammary epithelial and breast cancer cells—A new regulatory pathway. Eur. J. Cancer 2014, 50, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Villagrasa, A.; Alvarez, P.J.; Osuna, A.; Garrido, J.M.; Aranega, A.; Rodriguez-Serrano, F. Exosomes Derived from Breast Cancer Cells, Small Trojan Horses? J. Mammary Gland Biol. Neoplasia 2014, 19, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed]
- Semina, S.E.; Scherbakov, A.M.; Kovalev, S.V.; Shevchenko, V.E.; Krasil’nikov, M.A. Horizontal Transfer of Tamoxifen Resistance in MCF-7 Cell Derivates: Proteome Study. Cancer Investig. 2017, 35, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Sorokin, D.V.; Tatarskiy, V.V., Jr.; Prokhorov, N.S.; Semina, S.E.; Berstein, L.M.; Krasil’nikov, M.A. The phenomenon of acquired resistance to metformin in breast cancer cells: The interaction of growth pathways and estrogen receptor signaling. IUBMB Life 2016, 68, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.G.; Dickler, M.N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr.-Relat. Cancer 2016, 23, R337–R352. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef] [PubMed]
- Takov, K.; Yellon, D.M.; Davidson, S.M. Confounding factors in vesicle uptake studies using fluorescent lipophilic membrane dyes. J. Extracell. Vesicles 2017, 6, 1388731. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A.; Alam-Eldin, N. Overcoming Resistance to Endocrine Therapy in Breast Cancer: New Approaches to a Nagging Problem. Med. Princ. Pract. 2016, 25 (Suppl. 2), 28–40. [Google Scholar] [CrossRef] [PubMed]
- Gionet, N.; Jansson, D.; Mader, S.; Pratt, M.A. NF-kappaB and estrogen receptor alpha interactions: Differential function in estrogen receptor-negative and -positive hormone-independent breast cancer cells. J. Cell. Biochem. 2009, 107, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Dahlman-Wright, K.; Qiao, Y.; Jonsson, P.; Gustafsson, J.A.; Williams, C.; Zhao, C. Interplay between AP-1 and estrogen receptor alpha in regulating gene expression and proliferation networks in breast cancer cells. Carcinogenesis 2012, 33, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogne, J.M. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Muluhngwi, P.; Klinge, C.M. Identification of miRNAs as biomarkers for acquired endocrine resistance in breast cancer. Mol. Cell. Endocrinol. 2017, 456, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Leotta, M.; Bellizzi, D.; Di Martino, M.T.; D’Aquila, P.; Lionetti, M.; Fabiani, F.; Leone, E.; Gulla, A.M.; Passarino, G.; et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012, 3, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Guo, Q.; Fu, F.J.; Wang, Z.; Yin, Z.; Wei, Y.B.; Yang, J.R. The role of miR-29b in cancer: Regulation, function, and signaling. Onco Targets Ther. 2015, 8, 539–548. [Google Scholar] [PubMed]
- Zhang, H.P.; Wang, Y.H.; Cao, C.J.; Yang, X.M.; Ma, S.C.; Han, X.B.; Yang, X.L.; Yang, A.N.; Tian, J.; Xu, H.; et al. A regulatory circuit involving miR-143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol. Med. Rep. 2016, 13, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; You, Y.; Li, Q.; Zeng, C.; Fu, F.; Guo, A.; Zhang, H.; Zou, P.; Zhong, Z.; Wang, H.; et al. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: Implications for donor cell leukemia. Leukemia 2014, 28, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Taylor, J.C.; Black, M.A.; Nicolas, E.; Slifker, M.J.; Azzam, D.J.; Boyd, J. A New Role for ERalpha: Silencing via DNA Methylation of Basal, Stem Cell, and EMT Genes. Mol. Cancer Res. 2017, 15, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, J.; Yin, G.; Zhao, Q.; Elias, D.; Lykkesfeldt, A.E.; Stenvang, J.; Brunner, N.; Wang, J.; Yang, H.; et al. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 2013, 15, R119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, A.; Valdes-Mora, F.; Gee, J.M.; Farrow, L.; McClelland, R.A.; Fiegl, H.; Dutkowski, C.; McCloy, R.A.; Sutherland, R.L.; Musgrove, E.A.; et al. Tamoxifen-induced epigenetic silencing of oestrogen-regulated genes in anti-hormone resistant breast cancer. PLoS ONE 2012, 7, e40466. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Kaneko, Y.; Nagatomo, T.; Fujii, R.; Hanamura, T.; Gohno, T.; Yamaguchi, Y.; Niwa, T.; Hayashi, S.I. Different epigenetic mechanisms of ERalpha implicated in the fate of fulvestrant-resistant breast cancer. J. Steroid Biochem. Mol. Biol. 2017, 167, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, Y.; Wang, Y.; Fan, Y.; Li, C.; Zhang, Y.; Wang, Y.; Dong, Q.; Ma, Y.; Teng, Y.E.; et al. Tamoxifen reverses epithelial-mesenchymal transition by demethylating miR-200c in triple-negative breast cancer cells. BMC Cancer 2017, 17, 492. [Google Scholar] [CrossRef] [PubMed]
- Milani, A.; Geuna, E.; Mittica, G.; Valabrega, G. Overcoming endocrine resistance in metastatic breast cancer: Current evidence and future directions. World J. Clin. Oncol. 2014, 5, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Iselt, M.; Holtei, W.; Hilgard, P. The tetrazolium dye assay for rapid in vitro assessment of cytotoxicity. Arzneimittelforschung 1989, 39, 747–749. [Google Scholar] [PubMed]
- Volkova, Y.A.; Antonov, Y.S.; Komkov, A.V.; Scherbakov, A.M.; Shashkov, A.S.; Menchikov, L.G.; Chernoburova, E.I.; Zavarzin, I.V. Access to steroidal pyridazines via modified thiohydrazides. RSC Adv. 2016, 6, 42863–42868. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Hubner, M.R.; Metivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Yao, Y.J.; Kowalczyk, D.; Lyakh, L.A.; Karseladze, A.; Slaga, T.J.; Budunova, I.V. The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell. Sci. 2002, 115 Pt 1, 141–151. [Google Scholar] [PubMed]
- Scherbakov, A.M.; Komkov, A.V.; Komendantova, A.S.; Yastrebova, M.A.; Andreeva, O.E.; Shirinian, V.Z.; Hajra, A.; Zavarzin, I.V.; Volkova, Y.A. Steroidal Pyrimidines and Dihydrotriazines as Novel Classes of Anticancer Agents against Hormone-Dependent Breast Cancer Cells. Front. Pharmacol. 2018, 8, 979. [Google Scholar] [CrossRef] [PubMed]
- Mruk, D.D.; Cheng, C.Y. Enhanced chemiluminescence (ECL) for routine immunoblotting: An inexpensive alternative to commercially available kits. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3–22. [Google Scholar] [CrossRef]
- ASTM E2834-12. Standard Guide for Measurement of Particle Size Distribution of Nanomaterials in Suspension by Nanoparticle Tracking Analysis (NTA); ASTM International: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | MCF-7 | MCF-7/M | MCF-7/T | |
|---|---|---|---|---|
| Gene | ||||
| PIK3CA | c.1633G > A Allele fraction: 66% (of 17,311 reads) | c.1633G > A Allele fraction: 67% (of 12,976 reads) | c.1633G > A Allele fraction: 65% (of 11,224 reads) | |
| ALK | NO | NO | NO | |
| EGFR | NO | NO | NO | |
| EGFR-AS1 | NO | NO | NO | |
| ESR1 | NO | NO | NO | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semina, S.E.; Scherbakov, A.M.; Vnukova, A.A.; Bagrov, D.V.; Evtushenko, E.G.; Safronova, V.M.; Golovina, D.A.; Lyubchenko, L.N.; Gudkova, M.V.; Krasil’nikov, M.A. Exosome-Mediated Transfer of Cancer Cell Resistance to Antiestrogen Drugs. Molecules 2018, 23, 829. https://doi.org/10.3390/molecules23040829
Semina SE, Scherbakov AM, Vnukova AA, Bagrov DV, Evtushenko EG, Safronova VM, Golovina DA, Lyubchenko LN, Gudkova MV, Krasil’nikov MA. Exosome-Mediated Transfer of Cancer Cell Resistance to Antiestrogen Drugs. Molecules. 2018; 23(4):829. https://doi.org/10.3390/molecules23040829
Chicago/Turabian StyleSemina, Svetlana E., Alexander M. Scherbakov, Anna A. Vnukova, Dmitry V. Bagrov, Evgeniy G. Evtushenko, Vera M. Safronova, Daria A. Golovina, Ludmila N. Lyubchenko, Margarita V. Gudkova, and Mikhail A. Krasil’nikov. 2018. "Exosome-Mediated Transfer of Cancer Cell Resistance to Antiestrogen Drugs" Molecules 23, no. 4: 829. https://doi.org/10.3390/molecules23040829