Synthesis of Novel C-2- or C-15-Labeled BODIPY—Estrone Conjugates
by
 , ,
, ,
Ildikó Bacsa
1 ,
,
Csilla Konc
1,
Anna Boglárka Orosz
1,
Gábor Kecskeméti
2,
Réka Rigó
3,
Csilla Özvegy-Laczka
3 and
Erzsébet Mernyák
1,* 1
Department of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
2
Department of Medicinal Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
3
Membrane protein research group, Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(4), 821; https://doi.org/10.3390/molecules23040821
Submission received: 6 March 2018
/
Revised: 29 March 2018
/
Accepted: 2 April 2018
/
Published: 3 April 2018
(This article belongs to the Section Organic Chemistry)
Abstract
:Novel BODIPY–estrone conjugates were synthesized via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC). Estrone-alkynes or an estrone-azide as starting compounds were synthesized via Michael addition or Sonogashira reaction as key steps. Fluorescent dyes based on BODIPY-core were provided by azide or alkyne functional groups. Fluorescent labeling of estrone was efficiently achieved at the C-2 or C-15 position. The newly-elaborated coupling procedures might have a broad applicability in the synthesis of fluorescent-labeled estrone conjugates suitable for biological assays.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
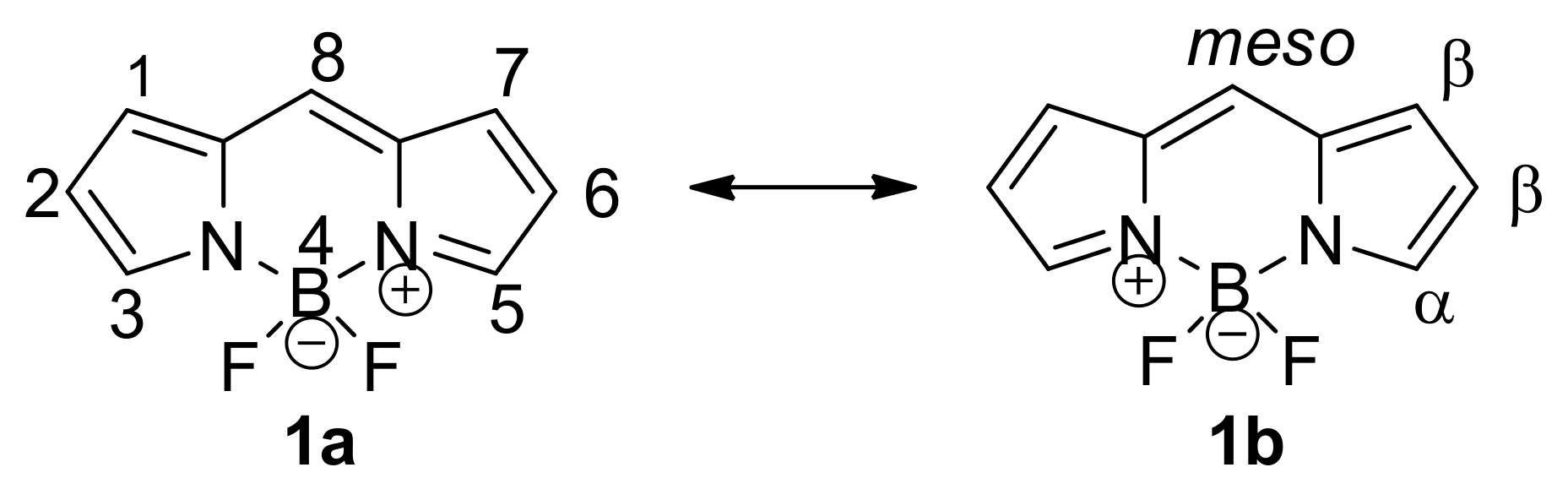
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPY, 1, Figure 1.) are strongly UV-absorbing small dyes with characteristic spectroscopic properties [1,2]. They emit relatively sharp fluorescence peaks with high quantum yields and possess excellent photostability. Additionally, they are stable to physiological conditions owing to their special behavior concerning their insensitivity to pH and the polarity of their environment. Their fluorescence characteristics can be modulated by the directed chemical tuning of the chromophore. An unambiguous trend toward red-shifted absorption and emission maxima with increased substitution at the 1-, 3-, 5- and/or 7-positions is observed. However, alkylation or arylation at the meso position has no substantial effect on the absorption and emission wavelengths. These dyes are extensively used in biomolecule labeling owing to their beneficial properties [3,4,5,6,7,8]. Nevertheless, their biological application is limited because of the limited water solubility and emission wavelength (under 600 nm).
The data from the literature reveals several synthetic strategies for the preparation of BODIPY dyes [1,2]. The dipyrrolylmethene core might be constructed from aromatic aldehydes and pyrroles and the subsequent oxidation of the resulting dipyrrolylmethane [7]. After the preparation of the heterocyclic core, complexation with boron leads to the desired BODIPY derivative. In order to obtain the appropriately substituted compound at the meso position, a reasonable choice of the aromatic aldehyde is needed. With the aim of avoiding the oxidation step, the dipyrrolylmethene core is usually synthesized via the condensation of acyl chlorides with pyrroles [9,10]. In the latter case, the meso substituent is derived from the acyl chloride, thus, the directed selection of the appropriate acyl chloride is crucial. In the case of attaching the BODIPY dye to a biomolecule, the feasibility of the introduction of the desired functional group onto the BODIPY core is of particular interest. The coupling mode of the dye to a biomolecule might be determined with regard to its biological behavior. The development of imaging probes to monitor the mechanism of action of biologically active compounds in living systems is of great interest in medicinal chemistry.
Estrogens belong to a class of natural steroids which possess hormonal activity. However, their chemical modifications may lead to biologically active estrone derivatives lacking hormonal behavior [11,12]. Certain synthetic estrone-derived compounds are described as antitumoral agents, but the mechanism of their action is mostly unknown [11]. In order to investigate and monitor their mechanism, their labeling is essential. Enzymatic or receptorial assays are mainly based on radioisotope labeling. Nowadays, there is a need for the replacement of the harmful radioactive methods for greener and friendlier fluorescent ones. However, to the best of our knowledge, there is no reported general use of BODIPY-labeled estrone derivatives in fluorescent biological assays. BODIPY–estrone conjugates are rarely described in the literature [13]. The compounds known presently are labeled at positions 3-, 7α-, or 17α-, mainly affecting the two oxygen functionalities of the steroid. The coupling procedures involved amide formation, olefin metathesis, the Sonogashira reaction, or the SNAr-type reaction [13]. Certain 7α-conjugates seemed to be able to bind to the estrogen receptor alpha; thus, they might be used in fluorescent receptorial assays. Not only the position of labeling, but the nature of the linking group and the spacing between the two moieties might also have a substantial influence on the biological behavior. There exist BODIPY–estrone conjugates labeled at the 17α-position coupled via Cu(I)-catalyzed azide–alkyne click-reaction (CuAAC) [14]. This is a powerful and convenient route for the conjugation of two molecular entities via metabolically stable triazole linker [15,16]. Click chemistry is emphasized by its high efficiency and tolerance toward several functional groups [15,16].
2. Results and Discussion
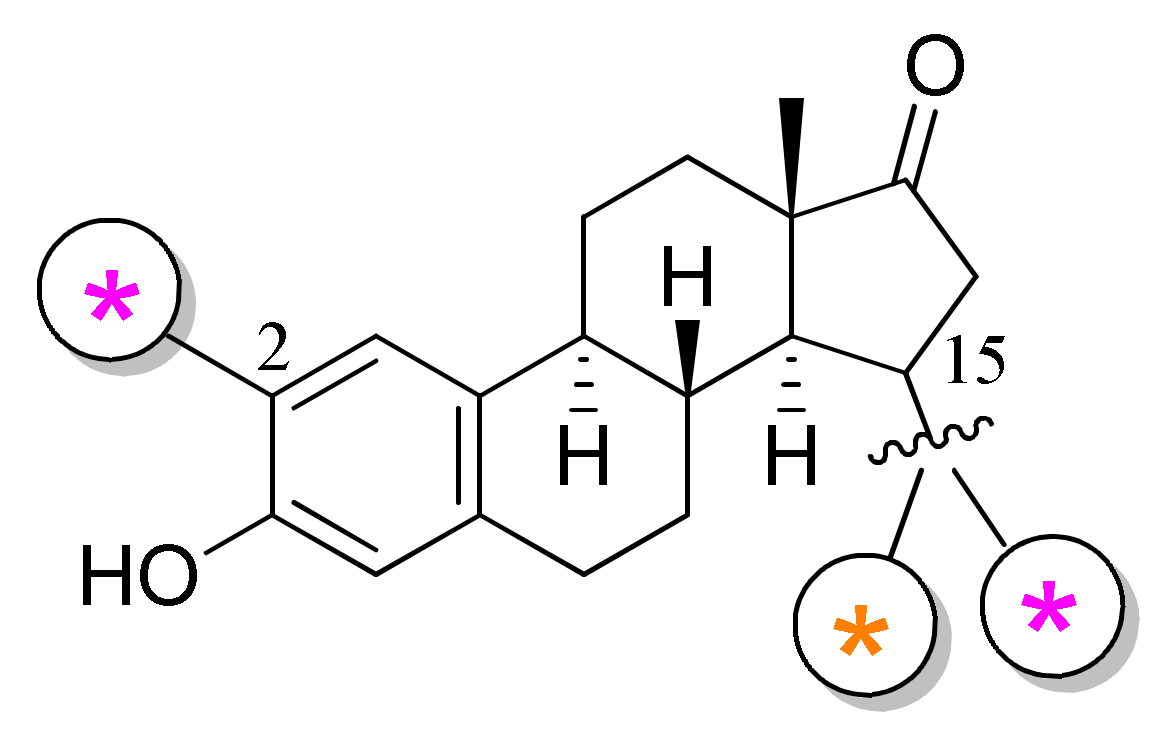
Here we aimed to synthesize BODIPY–estrone conjugates without transforming the two oxygen functionalities of the steroid and by establishing a chemically and metabolically stabile attaching moiety. Therefore, we chose the positions C-2 or C-15 for labeling, thereby avoiding covalent modifications at the C-3 and C-17 positions (Figure 2). Estrone and its 17β-hydroxy counterpart display strong secondary interactions with the enzyme or receptor targets through their keto or hydroxy groups [17,18]. The blocking of these groups may result in biological behavior different than that of estrone or 17β-estradiol. However, all conjugations at sites different than the main functionalities might be of value. According to the literature, there are several 2- or 15-substituted estrone derivatives that are biologically active steroids, displaying important activities such as the antitumoral effect [12,19,20,21,22]. It is known that certain derivatives act through different protein targets, including tubulin or enzymes involved in estrogen biosynthesis [11,12,19,20,21,22]. The fluorescent labeling of compounds potentially interacting with the above-mentioned proteins might have high biological relevance.
Recent advances in chemical synthesis techniques, in particular, in cross coupling and conjugation methods, allow for the accomplishment of reactions which could formerly not be achieved. Concerning the mode of conjugation, clicking via CuAAC reaction is one of the most feasible and effective strategies [15,16,23]. On the basis of our longstanding experience [24,25,26,27,28,29,30,31] in developing steroidal azides and alkynes suitable for the formation of biologically active triazolyl conjugates, here, we chose the CuAAC reaction for attaching the dye to the steroid. An added advantage of the presence of this heterocycle on a steroid core is due to its behavior being similar to that of a peptide bond [32]. Nevertheless, there is a great challenge in the labeling process concerning the establishment of the best reaction sequence after taking into account the reactivity and the sensitivity of the already introduced functional groups.
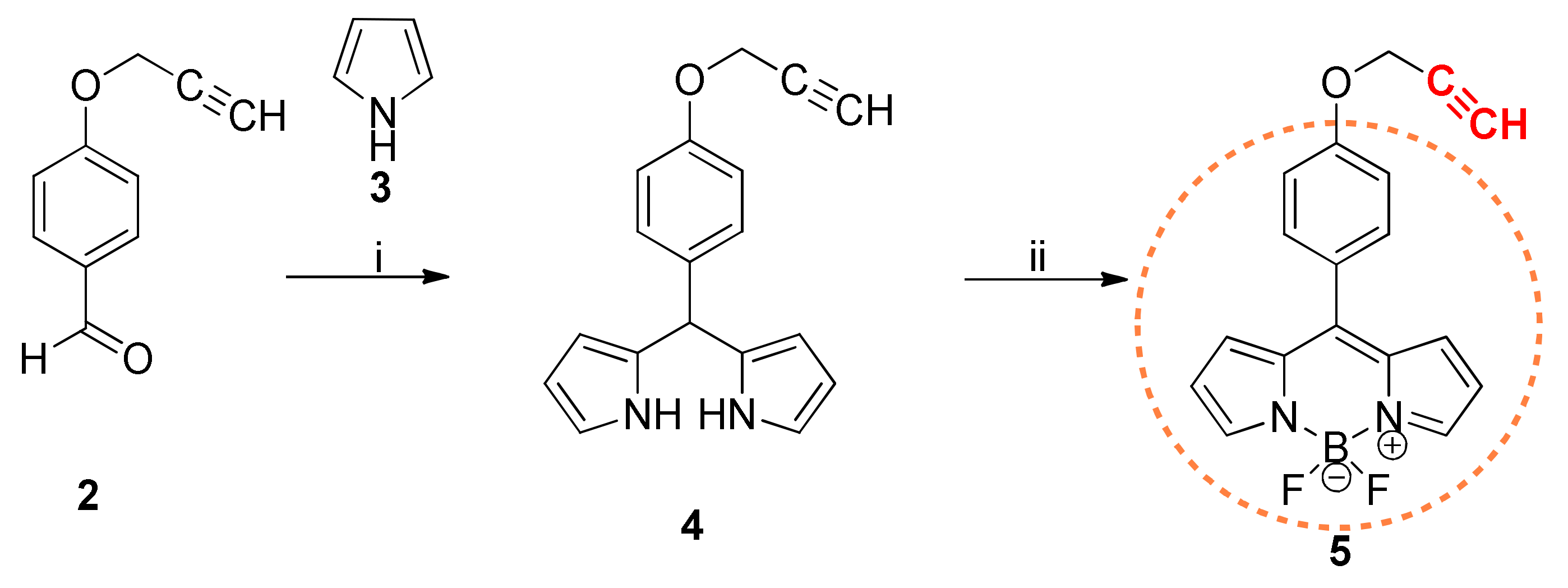
At first, we prepared the building elements for the couplings. That is, the BODIPY derivatives bearing terminal alkyne or azide functions (5, 11) and the estrone derivatives possessing the complementary functions (17, 18, 21) were synthesized. BODIPY-alkyne 5 was synthesized using the aldehyde–pyrrole condensation strategy described above (Scheme 1). The propargylation of p-hydroxybenzaldehyde was efficiently achieved based on our earlier established procedure using propargyl bromide and potassium carbonate [31]. The dipyrrolylmethane core was built via trifluoroacetic acid (TFA) -catalyzed condensation of the aldehyde (2) and the pyrrole (3). The literature procedures were modified and combined in order to avoid the formation of oligomers [33,34]. Accordingly, a large excess of pyrrole was used, which served as both the reactant and the solvent in the condensation, allowing the selective formation of the dipyrrolylmethane. The desired compound 4 could efficiently be purified by flash chromatography by adding triethylamine to the eluent and covering the silica gel column with Al foil. Subsequent oxidation of the dipyrrolylmethane with DDQ (2,3-dichloro-5,6-dicyano-p-benzoquinone) and complexation with boron furnished the desired terminal alkyne (5) in high yield.
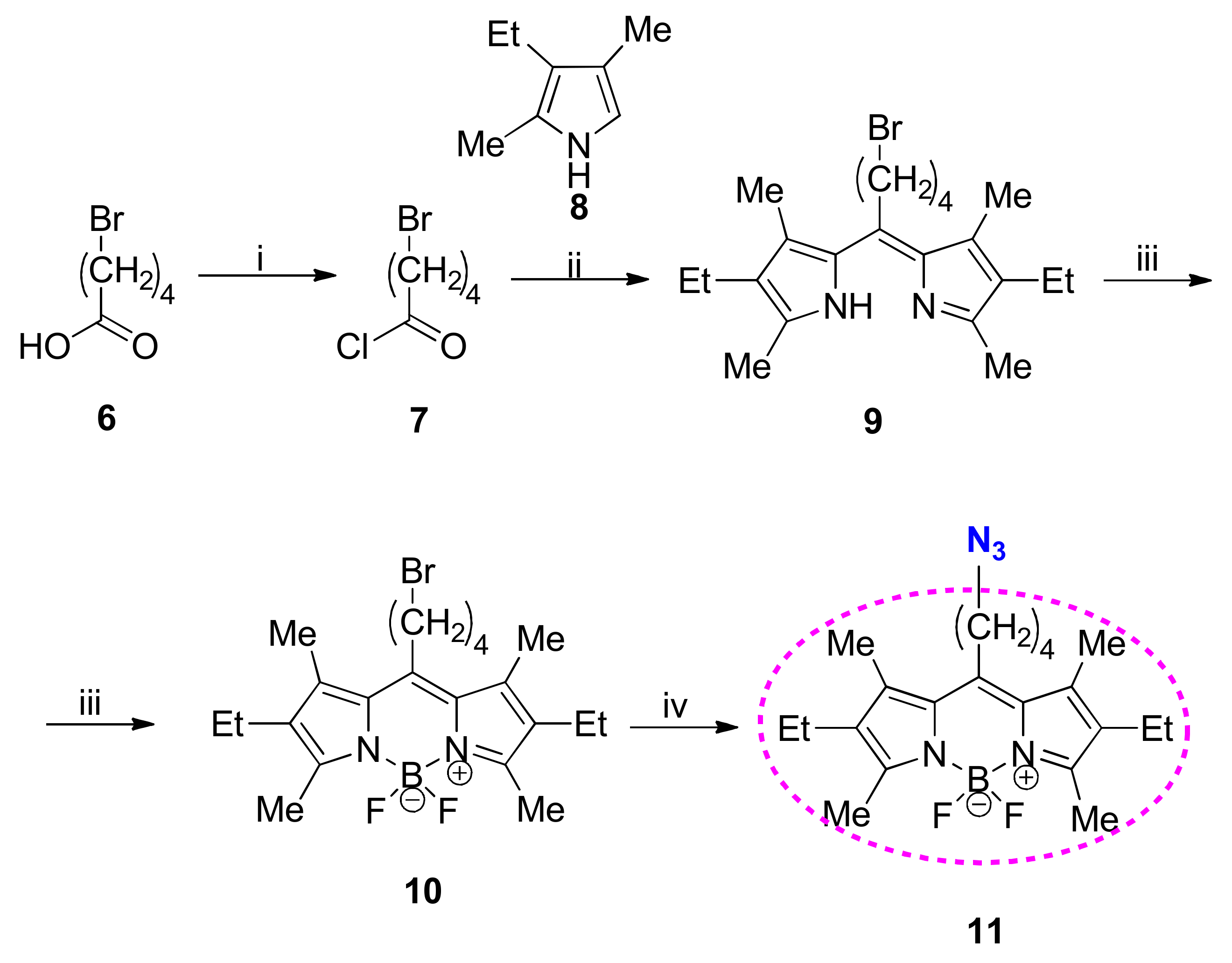
The parent BODIPY derivative bearing azide function (11) was synthesized using the acyl chloride–pyrrole condensation strategy (Scheme 2) [35]. ω-Bromovaleric acid 6 was used as the azide precursor in order to build in a four-carbon-long spacer chain to the conjugate. Acyl chloride 7 was formed in situ with oxalyl chloride and a few drops of DMF. 3-Ethyl-2,4-dimethylpyrrole 8 was used for the condensation as a building block in order to avoid couplings at different positions or the formation of other side-products. Taking into consideration the different methodologies described in the literature [1,2,3,4,5,6,7,8,35], we tried to perform modifications in order to achieve the best yields and the highest chemoselectivity. We found that the order of reagent addition greatly influences the outcome of the condensation. It was found that the optimal conditions of the crucial step involved the use of diluted solutions of acyl chloride (7) and pyrrole (8). Additionally, the dropwise addition of the solution of 7 to that of 8 seemed to be superior to that of the reverse order. Subsequent complexation with BF3·OEt2 led to the bromo-BODIPY product (10) in high yield. The last step in the reaction sequence in the formation of the new azido-BODIPY product (11) was a bromine to azide exchange reaction using NaN3 as an azide source in the dimethyl sulfoxide (DMSO) solvent with a catalytic amount of AcOH.
Next, we shifted our attention to the synthesis of estrone azide 18 and alkyne derivatives 17 and 21 suitable for CuAACs with BODIPY-alkyne 5 or -azide 11. We earlier developed an efficient methodology for the generation of a Δ15-double bond in ring D of 3-O-methyl- and 3-O-benzyl-13α-estrone [36]. The α,β-unsaturated ketone 16 may serve as the key intermediate in the synthesis of 15-substituted estrone derivatives. Herein, we present the synthesis of the α,β-unsaturated ketone 16 in the 13β-estrone series without etherification of its 3-OH function (Scheme 3). We started with the 3-acetylated compound (12) developing a protecting group at C-17 in order to prevent the formation of undesired side-products during the reaction sequence. The next step was the reaction with pyridinium hydrobromide perbromide, which afforded the desired 16α-bromo derivative [37]. The dehydrobromination with potassium tert-butylate was accompanied by the hydrolysis of the acetate ester. The deprotection of the 17-ketal (15) led to the unsaturated ketone (16). The resulting ketone intermediate (16) was transformed into the appropriate alkyne (17) or azide (18) derivative by Michael addition. The nucleophile was stereoselectively introduced to C-15 in both cases, leading to the formation of one stereoisomer. The terminal alkyne function was introduced onto C-15 via O-propargylation with propargyl alcohol in dichloromethane, using a catalytic amount of NaOH as the base. The 15β-azide (18) was synthesized via the addition of HN3 formed in situ from NaN3 and AcOH. The data from the literature indicates 15β-orientation for the nucleophiles introduced via Michael addition onto the 3-protected α,β-unsaturated estrones [38]. We established the β-orientation of the functional group at C-15 in compound 17 through two-dimensional NMR measurements. The α-position of 15-H was deduced from the NOESY spectra since a cross-peak was observed between 15-H and 16α-H.
The terminal alkyne function was introduced not only onto the C-15 but also directly onto the C-2 position via the Sonogashira reaction (Scheme 4). Coupling at C-2 could efficiently be achieved via our microwave-assisted Sonogashira procedure established earlier [39]. The synthesis of the steroidal 2-iodo derivative starting compound 19 has been reported by us recently [40]. It was reacted with trimethylsilylacetylene using 0.1 equiv. of Pd(PPh3)4 and 0.1 equiv. of CuI in tetrahydrofurane (THF) as a solvent in the presence of Et3N as a base at 50 °C for 20 min in a microwave reactor. The resulting 2-trimethylsilylethynyl-estrone (20) was purified by flash chromatography and served as a precursor of 2-ethynyl-estrone (21).
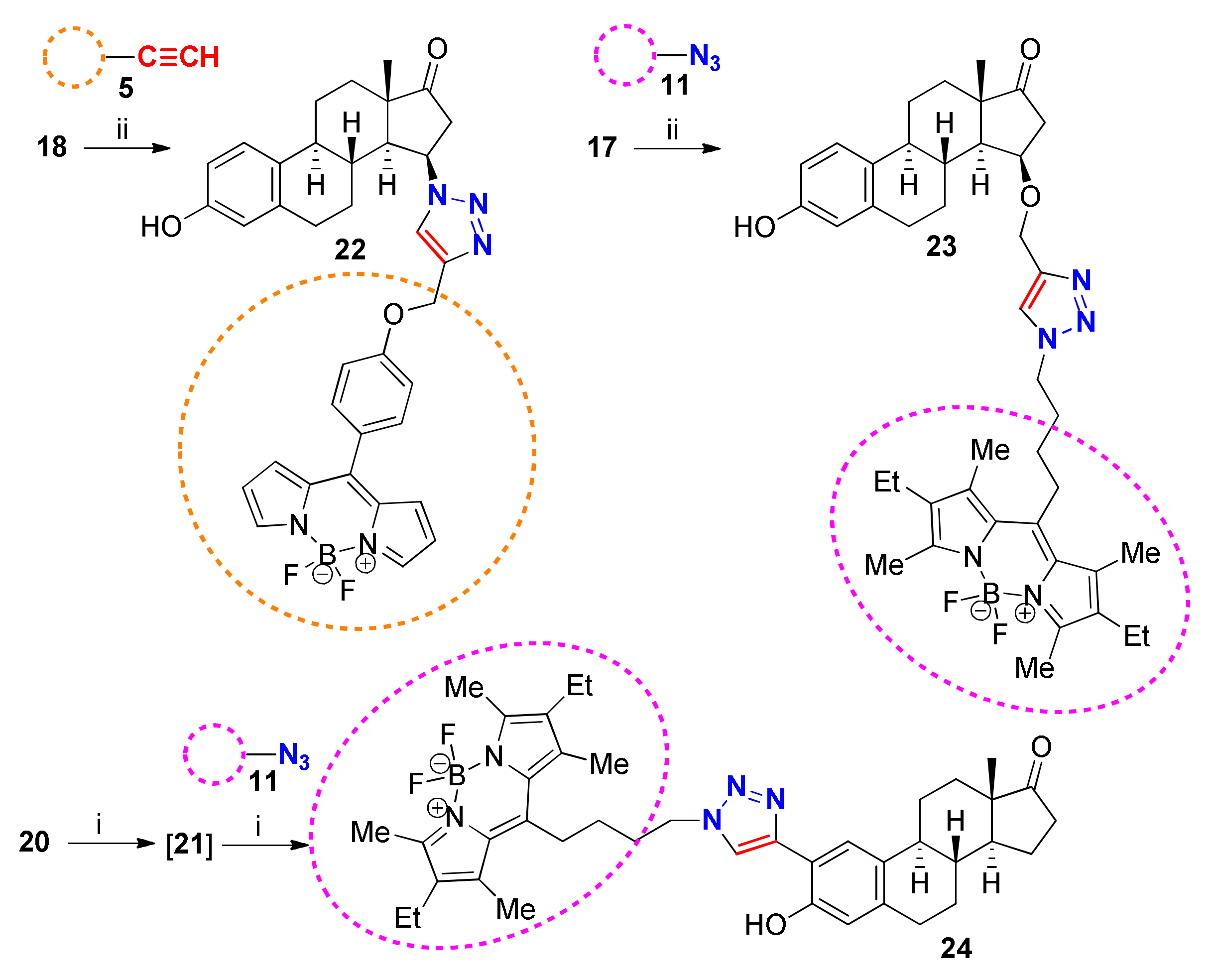
With alkyne (5, 17, 21) and azide (11, 18) complementers in hand, CuAAC reactions were carried out in order to synthesize the desired fluorescent estrone conjugates (Scheme 5). Estrone alkynes (17, 21) or azides (18) were reacted with BODIPY-azide 11 or BODIPY-alkyne 5 under the previously reported CuAAC reaction conditions, using CuI as a catalyst, PPh3 as a ligand, and DIPEA as a base under conventional heating in toluene solvent. This protocol proved to be suitable for the efficient formation of the fluorescent conjugates (22–24). The 2-trimethylsilylethynyl-estrone derivative 20 underwent deprotection with tetrabutylammonium fluoride in situ during the click reaction. The conjugates (22–24) were regioselectively synthesized in high yields. The structures of the newly-synthesized labeled compounds (22–24) and their precursors were confirmed by 1H and 13C-NMR measurements.
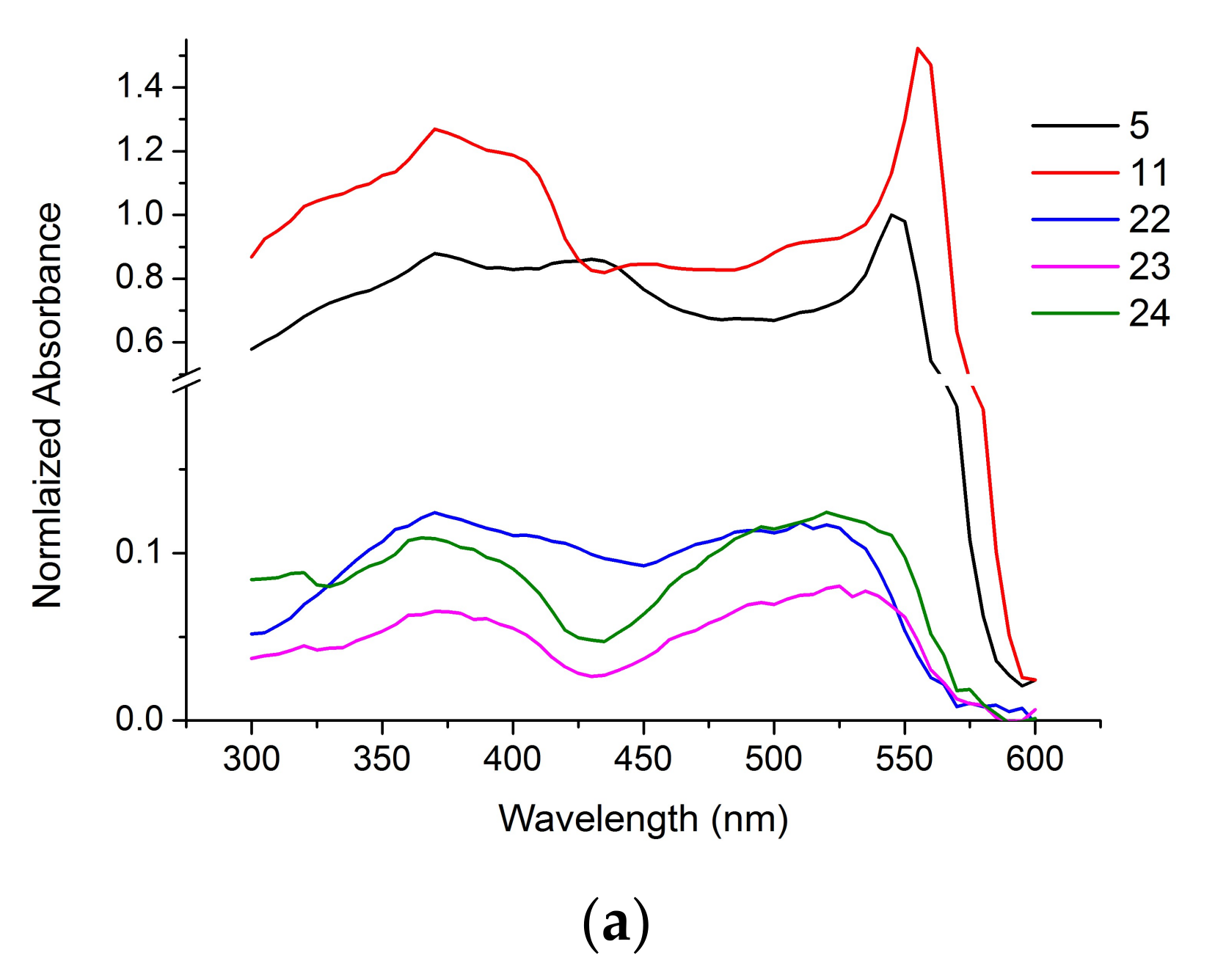
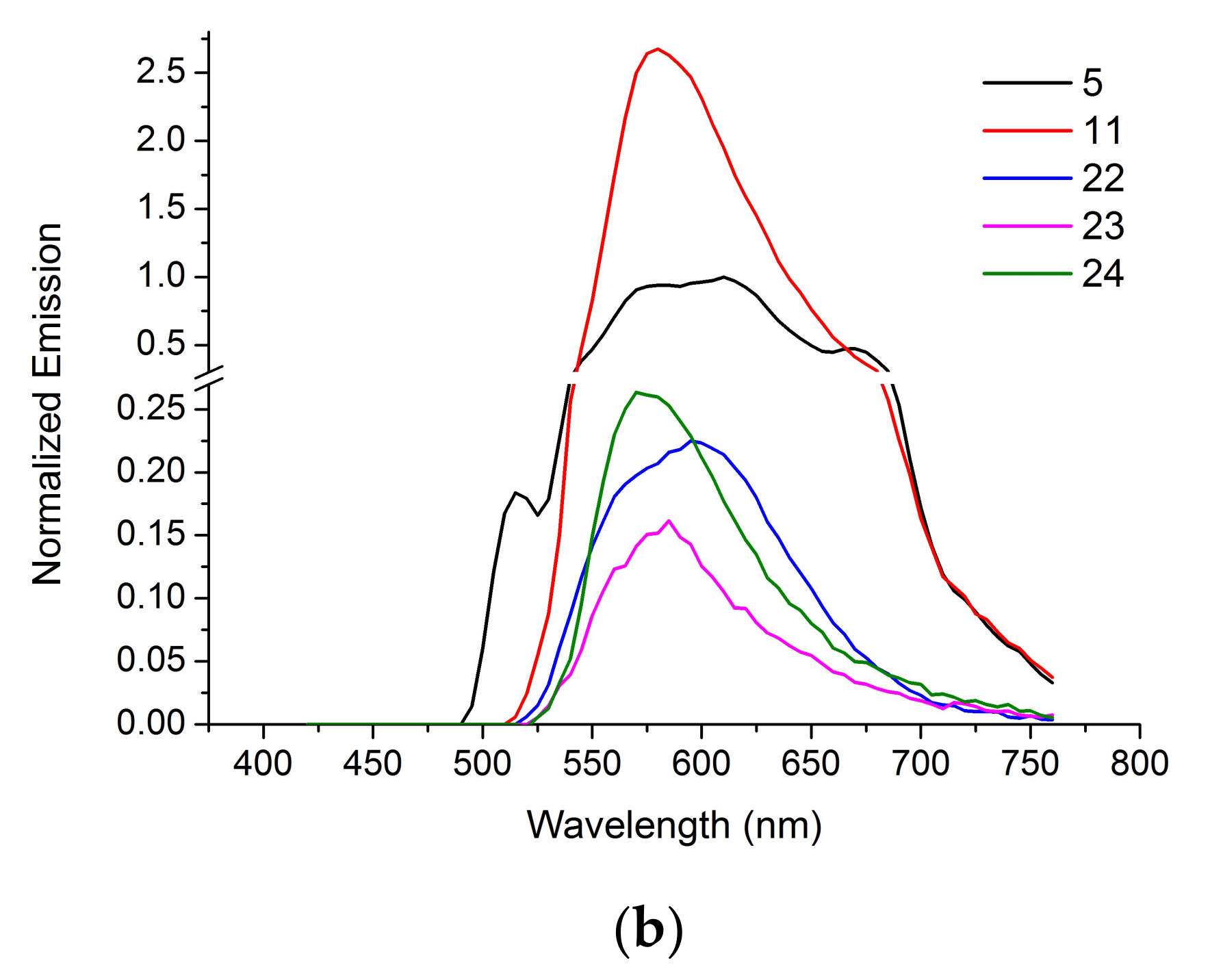
Figure 3 shows the absorption and the fluorescence emission spectra of the non-conjugated BODIPY dyes (5, 11) and the steroid-BODIPY conjugates (22–24). Absorbance and emission spectra were normalized to maximum fluorescence of the unconjugated dye 5 measured at 545 nm emission and 610 nm excitation wavelengths, respectively. The absorption spectra of all the compounds have similar shape with two peaks of maximum absorption at 350–400 and 510–555 nm. The emission spectra of the dyes (5, 11) and the conjugates (22–24) are also similar with a band around 580 nm for compounds 11, 23 and 24; 600 nm for 22 and 610 nm for 5. These data suggest the feasibility of the observation of conjugates 22–24 in living cells and tissues.
3. Materials and Methods
General
The melting points (m.p.) were determined with a Kofler hot-stage apparatus and are uncorrected. Elemental analyses were performed with a Perkin-Elmer CHN analyzer model 2400. Thin-layer chromatography: silica gel 60 F254; layer thickness 0.2 mm (Merck); eluent: a: 10% EtOAc/hexane, b: 2% EtOAc/CH2Cl2, and c: 20% EtOAc/CH2Cl2, if otherwise not stated, detection with I2 or UV (365 nm) after spraying with 5% phosphomolybdic acid in 50% aqueous phosphoric acid and heating at 100–120 °C for 10 min. Flash chromatography: silica gel 60, 40–63 μm (Merck). Reactions under microwave irradiation were carried out with a CEM Corporation focused microwave system, Model Discover SP. The 1H-NMR spectra were recorded in DMSO-d6, a CDCl3 solution with a Bruker DRX-500 instrument at 500 MHz, with Me4Si as an internal standard. The 13C-NMR spectra were recorded with the same instrument at 125 MHz under the same conditions. Mass spectrometry: a full scan mass spectra of the compounds were acquired in the range of 50–1000 m/z with a Finnigan TSQ-7000 triple quadrupole mass spectrometer (Finnigan-MAT, San Jose, CA, USA) equipped with a Finnigan electrospray ionization source. Analyses were performed in the positive ion mode using flow injection mass spectrometry with a mobile phase of 50% aqueous acetonitrile containing 0.1 v/v% formic acid. The flow rate was 0.3 mL/min. A 5-µL aliquot of the samples was loaded into the flow. The ESI capillary was adjusted to 4.5 kV and N2 was used as a nebulizer gas. Compound stocks prepared in DMSO (20 mM) were further diluted in 1× phosphate buffered saline, pH 7.4 to a final concentration of 500 µM. Absorption (at fixed 620 nm emission wavelengths) and emission (at fixed 400 nm excitation wavelength) spectra were measured using an Enspire plate reader (Perkin Elmer, Waltham, MA, USA).
3.1. Synthesis of BODIPY-alkyne 5
3.1.1. Propargylation of 4-Hydroxybenzaldehyde
4-Hydroxybenzaldehyde (12.22 g, 10.0 mmol) was dissolved in acetone (100 mL) and then, propargyl bromide (1.7 mL (80 wt.% in toluene), 15.0 mmol) and K2CO3 (9.68 g, 70 mmol) were added. The reaction mixture was stirred at 70 °C for 24 h, the solvent was then evaporated off, and the residue was purified by flash chromatography with CH2Cl2/hexane = 80/20 as an eluent. Compound 2 was obtained as a white solid (14.9 g, 93%). Compound 2 is identical with that described in the literature [41,42].
3.1.2. Synthesis of Dipyrrolylmethane 4
To a solution of compound 2 (0.48 g, 3 mmol) and pyrrole (9.0 mL, 1.29 mol), trifluoroacetic acid (0.024 mL, 0.3 mmol) was added. The solution was stirred for 10 min at room temperature under a nitrogen atmosphere. The mixture was diluted with CH2Cl2 (200 mL), washed with 0.1 M aq NaOH, water, and dried Na2SO4. The solvent and a great amount of unreacted pyrrole were removed under reduced pressure. The residue was purified by flash chromatography on a silica gel column covered with Al foil with hexane/EtOAc/Et3N = 80/20/1 as an eluent. Compound 4 was obtained as a brown solid (550 mg, 67%). Compound 4 is identical with that described in the literature [41,42].
3.1.3. Synthesis of Boron Dipyrrolylmethene 5
Dipyrrolylmethane (4, 0.6 g, 2.2 mmol) was dissolved in dry CH2Cl2 (30 mL), then DDQ (0.49 g, 2.2 mmol) was added and the reaction mixture was stirred for 1 h at rt (room temperature) under a nitrogen atmosphere. This was followed by the successive addition of Et3N (2 mL, 14.4 mmol) and BF3·Et2O (2.3 mL, 8.6 mmol) and the stirring was continued for 4 h. The reaction was quenched by the addition of a saturated NaHCO3 solution (25 mL). The organic layer was separated and dried over with Na2SO4. The solvents were removed under reduced pressure and the residue was purified by flash chromatography with EtOAc/hexane = 20/80 as an eluent. Compound 5 was obtained as a tawny solid (550 mg, 78%). M.p.: 147–150 °C, Rf = 0.78b. Compound 5 is identical with that described in the literature [34]. 1H-NMR (CDCl3) δ (ppm): 2.60 (s, 1H, C≡CH); 4.80 (s, 2H, OCH2); 6.55 (s, 2H); 6.97 (s, 2H); 7.14 (m, 2H); 7.56 (m, 2H); 7.92 (s, 2H).
3.2. Synthesis of BODIPY-azide 11
3.2.1. Synthesis of BODIPY-bromide 10
DMF (3 drops) and oxalyl chloride (0.5 mL, 5.8 mmol) were added to a solution of 5-bromovaleric acid (6, 0.76 g, 4.2 mmol) in dry CH2Cl2 (20 mL) and the mixture was stirred for 3 h at rt. Then the mixture was evaporated with toluene (3 × 10 mL). The residue was re-dissolved in anhydrous CH2Cl2 (20 mL) and the resulting solution was added dropwise to a stirred solution of 3-ethyl-2,4-dimethylpyrrole (8, 1.23 g, 10 mmol) in dry dichloromethane (20 mL). After heating under reflux for 3 h, the mixture was cooled to rt and triethylamine (11 mL, 80 mmol) was added. After a 30 min reflux, BF3·Et2O (10 mL, 80 mmol) was added and the reaction mixture was stirred for 3 h under reflux. After cooling to rt, the reaction was quenched by the addition of a saturated NaHCO3 solution (30 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried (Na2SO4) and the solvents were removed under a reduced pressure. The residue was purified by flash chromatography with EtOAc/hexane = 10/90 as an eluent. Compound 10 was obtained as a red solid (1.01 g, 55%). M.p.: 160–161 °C, Rf = 0.54a. Compound 10 is identical with that described in the literature without characterization [43]. Anal calcd. for C21H30BBrF2N2: C, 57.43; H, 6.89. Found: C, 57.52; H, 6.73. 1H-NMR (DMSO-d6) δ (ppm): 1.05 (t, 6H, J = 7.5 Hz, 2xCH2CH3); 1.80 (m, 2H), 2.06 (m, 2H); 2.34 and 2.50 (2xs, 2x6H, 4xCH3); 2.40 (q, 4H, J = 7.5 Hz, 2xCH2CH3); 3.02 (m, 2H); 3.45 (m, 2H). 13C-NMR δ (ppm): 12.4 (2C); 13.4 (2C), 14.8 (2C); 17.2 (2C); 27.6; 30.2; 32.7; 33.1; 130.9; 132.7 (2C); 135.5 (2C), 143.5 (2C); 152.4 (2C). MS m/z (%): 419 (100, [M − F]+).
3.2.2. Synthesis of BODIPY-azide 11
A stirred solution of compound 10 (878 mg, 2.0 mmol) in dry DMSO (5 mL) was treated with acetic acid (3 drops) and NaN3 (130 mg, 2.0 mmol). The mixture was stirred for 0.5 h at rt, poured into water (50 mL), and extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried (Na2SO4) and the solvents were removed under a reduced pressure. The residue was purified by flash chromatography with EtOAc/hexane = 10/90 as an eluent. Compound 11 was obtained as a red solid (746 mg, 92%). M.p.: 110–112 °C, Rf = 0.50a. Anal calcd. for C21H30BF2N5: C, 62.85; H, 7.53. Found: C, 62.92; H, 7.47. 1H-NMR (DMSO-d6) δ (ppm): 1.05 (t, 6H, J = 7.5 Hz, 2xCH2CH3); 1.73–1.80 (overlapping multiplets, 4H); 2.33 and 2.50 (2xs, 2x6H, 4xCH3); 2.41 (q, 4H, J = 7.5 Hz, 2xCH2CH3); 3.02 (m, 2H); 3.37 (m, 2H). 13C-NMR δ (ppm): 12.4 (2C); 13.3 (2C), 14.8 (2C); 17.2 (2C); 27.8; 28.8; 29.2; 51.0; 130.9; 132.7 (2C); 135.5 (2C), 143.6 (2C); 152.4 (2C). MS m/z (%): 382 (100, [M − F]+).
3.3. Synthesis of α,β-Unsaturated Ketone Intermediate 16
3.3.1. Synthesis of 16-Bromo-17-ketal 14
To a suspension of 12 (13.75 g, 44 mmol) in triethyl orthoformate (15 mL, 88 mmol) and ethylene glycol (6 mL, 110 mmol), a catalytic amount of p-TsOH was added, and the mixture was gently heated for 0.5 h until the suspension transformed into a clear solution. The warm reaction mixture was then poured into a saturated solution of sodium bicarbonate (100 mL). The white precipitate formed was filtered off, washed with water, and dried in air. To a solution of crude product 13 (10.70 g, 30 mmol) in dry tetrahydrofuran (100 mL), 10 g (30 mmol) of pyridinium hydrobromide perbromide was added. A white precipitate formed immediately from the yellow solution. The reaction mixture was stirred at rt for 2 h, then it was diluted with a saturated aqueous solution of sodium bicarbonate (500 mL). The precipitate formed was filtered off, washed with water, and dried in air. The crude product reacted further without purification. Compound 14 is identical with that described in Reference [44].
3.3.2. Synthesis of α,β-Unsaturated Ketal 15
To a solution of 14 (4.35 g, 10 mmol) in dimethyl sulfoxide (50 mL), potassium tert-butylate (2.24 g, 20 mmol) was added. The suspension was vigorously stirred and heated (80 °C) for 5 h until it became a clear yellow-brown solution. The mixture was poured into ice-water (1 L) and the resulting precipitate was filtered off, washed with water, and dried in air. The crude product was subjected to chromatographic separation with dichloromethane as an eluent. Compound 15 was obtained as a white solid (2.6 g, 83%). Compound 15 is identical with that described in Reference [44].
3.3.3. Synthesis of α,β-Unsaturated Ketone 16
To a solution of 15 (1.25 g, 4 mmol) in acetone (25 mL), formaldehyde (5 mL, 37% in water, 60 mmol formaldehyde) and a catalytic amount of p-TsOH were added. The mixture was stirred for 1.5 h at rt and then poured into water (100 mL). The precipitate formed was filtered off, washed with water, and dried in air. The crude product (16) was subjected to chromatographic separation with EtOAc/CH2Cl2 = 2/98 as an eluent. Compound 16 was obtained as a white solid (1.0 g, 84%). M.p.: 245–246 °C, Rf = 0.16b. Compound 16 is identical with that discussed in the literature without full characterization [44]. Anal calcd. for C18H20O2: C, 80.56; H, 7.51. Found: C, 80.64; H, 7.42. 1H-NMR (DMSO-d6) δ (ppm): 0.99 (s, 3H, 18-H3); 2.79 (m, 2H, 6-H2); 6.05 (d, 1H, J = 2.5 Hz, 16-H); 6.47 (d, 1H, J = 2.2 Hz, 4-H); 6.52 (double doublet 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.04 (d, 1H, J = 8.6 Hz, 1-H); 7.84 (d, 1H, J = 5.6 Hz, 15-H); 9,02 (s, 1H, OH). 13C-NMR δ (ppm): 20.5 (C-18); 25.0; 26.0; 28.6; 28.8; 35.2; 44.3; 50.7; 55.2; 112.7; 114.9; 125.6; 129.8; 130.8; 136.9; 155.0; 159.5; 211.9. MS m/z (%): 310 (100); 269 (66, [M + H]+).
3.4. Synthesis of Steroidal Alkyne 17
To a stirred solution of compound 16 (268.4 mg, 1.0 mmol) in dry CH2Cl2 (15 mL), propargyl alcohol (3.6 mL, 31 mmol) and 5 drops of 5% aq. NaOH were added. The reaction mixture was stirred at rt for 24 h. The reaction was quenched by the addition of water (150 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed under a reduced pressure. The residue was purified by flash chromatography with CH2Cl2 as an eluent. Compound 17 was obtained as a white solid (256 mg, 79%). M.p.: 167–169 °C, Rf = 0.20b. Anal calcd. for C21H24O3: C, 77.75; H, 7.46. Found: C, 77.83; H, 7.38. 1H-NMR (DMSO-d6) δ (ppm): 1.03 (s, 3H, 18-H3); 2.75 (m, 2H, 6-H2); 3.44 (s, 1H, C≡CH); 4.11 and 4.25 (2xm, 2x1H, OCH2); 4.37 (t, 1H, J = 5.1 Hz, 15-H); 6.46 (d, 1H, J = 2.2 Hz, 4-H); 6.51 (d, 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.05 (d, 1H, J = 8.6 Hz, 2-H); 9.03 (s, 1H, OH). 13C-NMR δ (ppm): 17.2 (C-18); 25.3 (2C); 28.9; 32.3; 34.6; 42.3; 43.6; 46.5; 52.9; 55.6 (OCH2); 73.1 (C-15); 76.9 (C≡CH); 80.4 (C≡CH); 112.7 (C-2); 114.9 (C-4); 125.8 (C-1); 129.9 (C-10); 137.0 (C-5); 154.9 (C-3); 218.6 (C-17).
3.5. Synthesis of the Steroidal Azide 18
A stirred solution of compound 16 (268 mg, 1.0 mmol) in dry DMSO (5 mL) was treated with acetic acid (0.24 mL, 4.0 mmol) and NaN3 (65 mg, 1.0 mmol). The mixture was stirred for 0.5 h at rt, poured into water (50 mL), and extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried (Na2SO4) and the solvents were removed under a reduced pressure. The residue was purified by flash chromatography with EtOAc/hexane = 1/99 as an eluent. Compound 18 was obtained as a white solid (252 mg, 81%). M.p.: 138–139 °C, Rf = 0.24b. Anal calcd. for C18H21N3O2: C, 69.43; H, 6.80. Found: C, 69.48; H, 6.72. 1H-NMR (DMSO-d6) δ (ppm): 0.89 (s, 3H, 18-H3); 2.76 (m, 2H, 6-H2); 4.16 (m, 1H, 15-H); 6.44 (d, 1H, J = 2.2 Hz, 4-H); 6.52 (dd, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.05 (d, 1H, J = 8.6 Hz, 1-H); 9.04 (s, 1H, OH). 13C-NMR δ (ppm): 14.7 (C-18); 25.6; 26.3; 29.0; 31.0; 37.7; 42.0; 43.1; 49.7; 53.1; 58.1; 112.8; 114.7; 126.3; 129.3; 136.8; 155.0; 214.4.
3.6. Synthesis of Triazoles 22 and 23
To a stirred solution of 5 (322 mg, 1.0 mmol) or 17 (324 mg, 1.0 mmol) in toluene (5 mL), Ph3P (26 mg, 0.1 mmol), CuI (9.5 mg, 0.05 mmol), DIPEA (0.52 mL, 3.0 mmol), and 18 (311 mg, 1.0 mmol) or 11 (401 mg, 1.0 mmol) were added. The reaction mixture was kept at the reflux temperature for 2 h and then allowed to cool and evaporate in vacuo. The residue was purified by flash chromatography with EtOAc/CH2Cl2 = 10/90 as an eluent.
Compound 22 was obtained as a red solid (557 mg, 88%). M.p.: 273–275 °C, Rf = 0.42c. Anal calcd. for C36H34BF2N5O3: C, 68.55; H, 5.41. Found: C, 68.63; H, 5.33. 1H-NMR (DMSO-d6) δ (ppm): 1.03 (s, 3H, 18-H3); 5.34 (s, 2H, OCH2); 5.42 (m, 1H, 15-H); 6.32 (d, 1H, J = 2.2 Hz, 4-H); 6.50 (dd, 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 6.64 (m, 2H); 7,01 (m, 2H); 7,03 (d, 1H, J = 8.6 Hz, 1-H); 7.27 (m, 2H); 7.64 (m, 2H); 8.09 (s, 2H); 8.59 (s, 1H); 9.00 (s, 1H). 13C-NMR δ (ppm): 14.5 (C-18); 24.4; 28.6; 31.0; 37.7; 43.0; 44.0; 50.0; 53.2; 57.6; 61.4; 112.8; 114.6; 115.1 (2C); 118.9 (2C); 123.4; 125.6; 126.2; 129.1; 131.4 (2C); 132.5 (2C); 133.9; 136.5; 142.8; 143.9 (2C); 146.8; 154.9; 160.5; 213.6. MS m/z (%): 143 (100); 634 (80, [M + H]+).
Compound 23 was obtained as a red solid (588 mg, 81%). M.p.: 177–179 °C, Rf = 0.24c. Anal calcd. for C42H54BF2N5O3: C, 69.51; H, 7.50. Found: C, 69.59; H, 7.40. 1H-NMR (DMSO-d6) δ (ppm): 0.95 (t, 6H, J = 7.5 Hz, 2xCH2CH3); 1.01 (s, 3H, 18-H3); 2.24 (overlapping multiplets, 4H, 2xCH2CH3); 2.33 (overlapping singlets, 12H, 4xCH3); 2.96 (m, 2H, 6-H2); 4.13 (m, 1H, 15-H); 4.41 and 4.61 (2xd, 2x1H, J = 12.5 Hz, OCH2); 4.45 (m, 2H); 6.39 (d, 1H, J = 2.2 Hz, 4-H); 6.50 (dd, 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.01 (d, 1H, J = 8.6 Hz, 1-H); 8.10 (s, 1H, C≡CH); 9.01 (s, 1H, OH). 13C-NMR δ (ppm): 12.7 (2C); 14.0 (2C); 14.6 (2C); 16.4; 17.2 (C-18); 25.3; 25.4; 27.2; 28.0; 28.8; 30.0; 32.3; 34.4; 42.6; 43.3; 46.5; 48.9; 53.0; 54.8; 61.6; 73.0; 112.5; 114.9; 123.9; 125.7; 130.0 (3C); 132.2 (2C); 136.0; 136.1; 137.0; 143.9 (2C); 144.3; 154.9 (2C); 218.7. MS m/z (%): 706 (100, [M − F]+).
3.7. Synthesis of 3-Hydroxy-2-trimethylsilylethynyl-estra-1,3,5(10)-trien-17-one 20
3-Hydroxy-2-iodo-estra-1,3,5(10)-trien-17-one (19, 276 mg, 1.0 mmol), Pd(PPh3)4 (115 mg, 0.01 mmol), CuI (19 mg, 0.01 mmol), and THF (3 mL) were added together under a nitrogen atmosphere. Then, Et3N (0.56 mL, 4.0 mmol) was added and the mixture was stirred at 50 °C for 10 min. Ethynyltrimethylsilane (0.28 mL, 2.0 mmol) was added with a syringe into 10 mL Pyrex pressure vessels (CEM, Part #: 908035) with silicone caps (CEM, Part #: 909210) and the mixture was heated in a CEM microwave reactor at 50 °C for 20 min under stirring. The solvent was evaporated in vacuo and the residue was purified by flash chromatography with diisopropyl ether/hexane = 50/50 as an eluent. Compound 20 was obtained as a white solid (1.0 g, 84%). M.p.: 179–181 °C, Rf = 0.26a. Anal calcd. for C23H30O2Si: C, 75.36; H, 8.25. Found: C, 75.42; H, 8.16 1H-NMR (CDCl3) δ (ppm): 0.26 (s, 9H, 3xSi-CH3); 2.87 (m, 2H, 6-H2); 5.62 (s, 1H, OH); 6.67 (s, 1H, 4-H); 7.25 (s, 1H, 1-H). 13C-NMR δ (ppm): 0.01 (3C, 3xSi-CH3); 13.8 (C-18); 21.6; 25.8; 26.3; 29.5; 31.4; 35.8; 38.1; 43.7; 47.9 (C-13); 50.4; 99.4; 101.3; 106.9, 114.3; 128.5; 131.9; 139.9; 154.8 (C-3); 220.8 (C-17)
3.8. Synthesis of BODIPY–estrone Conjugate 24
To a stirred solution of 20 (367 mg, 1.0 mmol) in toluene (5 mL), Ph3P (26 mg, 0.1 mmol), CuI (9.5 mg, 0.05 mmol), DIPEA (0.52 mL, 3.0 mmol), and 11 (1 equiv.) were added. The reaction mixture was treated at boiling temperature for 0.5 h and TBAF (1 mL, 1 mmol) was added. The heating was continued for 2 h at the reflux temperature. The mixture was allowed to cool and evaporate in vacuo. The residue was purified by flash chromatography with EtOAc/CH2Cl2 = 10/90 as an eluent. Compound 24 was obtained as a red solid (550 mg, 79%). M.p.: 218–220 °C, Rf = 0.26b. Anal calcd. for C41H52BF2N5O2: C, 70.78; H, 7.53. Found: C, 70.85; H, 7.41. 1H-NMR (CDCl3) δ (ppm): 0.95 (s, 3H, 18-H3); 1.01 (t, 6H, J = 7.5 Hz, 2xCH2CH3); 2.26 and 2.47 (2xs, 2x6H, 4xCH3); 2.37 (q, 4H, J = 7.5 Hz, 2xCH2CH3); 2.90 (m, 2H, 6-H2); 3.05 (m, 2H); 4.46(m, 2H, N-CH2); 6.79 (s, 1H, 4-H); 7.29 (s, 1H, 1-H); 7.79 (s, 1H, C≡CH/OH). 13C-NMR δ (ppm): 12.4 (2C); 13.4 (2C); 13.8 (2C); 14.8 (C-18); 17.1; 21.6; 26.0; 26.4; 27.5; 28.6; 29.2; 30.3; 31.5; 35.9; 38.2; 43.8; 48.0; 50.4; 50.5; 111.5; 117.3; 118.6; 122.6; 130.8 (2C); 131.1; 132.9 (2C); 135.3 (2C); 138.7; 142.9 (2C); 147.9; 152.6; 153.7; 220.9(C-17) MS m/z (%): 696 (100, [M + H]+). MS m/z (%): 696 (100, [M + H]+).
4. Conclusions
In conclusion, we described here a facile and efficient synthetic route to C-2- or C-15-labeled BODIPY–estrone conjugates via Sonogashira and/or click chemistry. Our strategy enables variations in the length of linkers, thereby providing a library of fluorescing conjugates. The selection of the site of conjugation as well as the nature of the substituents on the estrone moiety and the use of different linkers allow for the determination of the effect of structural modifications on the biological properties of the labeled compound. The methodologies developed should find extensive applications owing to the great importance of fluorescent labeled biomolecules. The newly synthesized labeled estrone derivatives may serve as good candidates for the development of imaging probes for biological assays.
Supplementary Materials
Supplementary File 1Acknowledgments
The work of Erzsébet Mernyák and Csilla Özvegy-Laczka in this project was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. This work was supported by National Research, Development, and Innovation Office-NKFIH through project OTKA SNN 124329.
Author Contributions
Ildikó Bacsa, Csilla Konc, Anna Boglárka Orosz, Gábor Kecskeméti, and Réka Rigó performed the experiments; Csilla Özvegy-Laczka and Erzsébet Mernyák contributed reagents, materials, and analysis tools; Erzsébet Mernyák and Csilla Özvegy-Laczka conceived and designed the experiments; Erzsébet Mernyák, Csilla Özvegy-Laczka, and Ildikó Bacsa analyzed the data; Erzsébet Mernyák, Ildikó Bacsa, Csilla Özvegy-Laczka, and Réka Rigó wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ulrich, G.; Ziessel, R.; Harriman, A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Karolin, J.; Johansson, L.B.-A.; Strandberg, L.; Ny, T. Fluorescence and Absorption Spectroscopic Properties of Dipyrrometheneboron Difluoride (BODIPY) Derivatives in Liquids, Lipid Membranes, and Proteins. J. Am. Chem. Soc. 1994, 116, 7801–7806. [Google Scholar] [CrossRef]
- Haugland, R.P. Handbook of Fluorescent Probes and Research Chemicals, 6th ed.; Spence, M.T.Z., Johnson, I.D., Eds.; Molecular Probes: Eugene, OR, USA, 1996. [Google Scholar]
- Tan, K.; Jaquinod, L.; Paolesse, R.; Nardis, S.; Di Natale, C.; Di Carlo, A.; Prodi, L.; Montalti, M.; Zaccheroni, N.; Smith, K.M. Synthesis and characterization of β-fused porphyrin-BODIPY® dyads. Tetrahedron 2004, 60, 1099–1106. [Google Scholar] [CrossRef]
- Yee, M.-C.; Fas, S.C.; Stohlmeyer, M.M.; Wandless, T.J.; Cimprich, K.A. A Cell-permeable, Activity-based Probe for Protein and Lipid Kinases. J. Biol. Chem. 2005, 280, 29053–29059. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.W.; Lindsey, J.S. Boron-dipyrromethene dyes for incorporation in synthetic multi-pigment light-harvesting arrays. Pure Appl. Chem. 1996, 68, 1373–1380. [Google Scholar] [CrossRef]
- Metzker, M.L. Substituted 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene Compounds for 8-Color DNA Sequencing. U.S. Patent US20030180769A1, 25 September 2003. [Google Scholar]
- Shah, M.; Thangaraj, K.; Soong, M.L.; Wolford, L.T.; Boyer, J.H.; Politzer, I.R.; Pavlopoulos, T.G. Pyrromethene-BF2 complexes as laser dyes: 1. Heteroat. Chem. 1990, 1, 389–399. [Google Scholar] [CrossRef]
- Boyer, J.H.; Haag, A.M.; Sathyamoorthi, G.; Soong, M.L.; Thangaraj, K.; Pavlopoulos, T.G. Pyrromethene-BF2 complexes as laser dyes: 2. Heteroat. Chem. 1993, 4, 39–49. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, S.B.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Numazawa, M.; Ando, M.; Watari, Y.; Tominaga, T.; Hayata, Y.; Yoshimura, A. Structure–activity relationships of 2-, 4-, or 6-substituted estrogens as aromatase inhibitors. J. Steroid Biochem. Mol. Biol. 2005, 96, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Osati, S.; Ali, H.; van Lier, J.E. BODIPY-steroid conjugates: Syntheses and biological applications. J. Porphyrins Phthalocyanines 2016, 20, 1–15. [Google Scholar] [CrossRef]
- Osati, S.; Ali, H.; Guerin, B.; van Lier, J.E. Synthesis and spectral properties of estrogen- and androgen-BODIPY conjugates. Steroids 2017, 123, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Langhals, H.; Obermeier, A. A Click Reaction for Fluorescent Labelling: Application of the 1,3-Dipolar Cycloaddition Reaction. Eur. J. Org. Chem. 2008, 36, 6144–6151. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Chen, S. Aromatase, estrone sulfatase, and 17β-hydroxysteroid dehydrogenase: structure-function studies and inhibitor development. Mol. Cell. Endocrinol. 2011, 340, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Structure-based design, synthesis and in vitro characterization of potent 17beta-hydroxysteroid dehydrogenase type 1 inhibitors based on 2-substitutions of estrone and d-homo-estrone. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. [Google Scholar] [CrossRef] [PubMed]
- Phan, C.M.; Liu, Y.; Kim, B.M.; Mostafa, Y.; Taylor, S.D. Inhibition of steroid sulfatase with 4-substituted estrone and estradiol derivatives. Bioorg. Med. Chem. 2011, 19, 5999–6005. [Google Scholar] [CrossRef] [PubMed]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Messinger, J.; Johansson, N.; Koskimies, P.; Thole, H.-H.; Husen, B.; Steen, B.J.; Schneider, G.; Hulshof, J.B.E.; Adamski, J. Novel 17β Hydroxysteroid Dehydrogenase Type 1 Inhibitors. WO2005047303 A2, 14 January 2005. [Google Scholar]
- Deobald, A.M.; Camargo, L.R.S.; Alves, D.; Zukerman-Schpector, J.; Corrêa, A.G.; Paixão, M.W. Click Chemistry: An Efficient Synthesis of Heterocycles Substituted with Steroids, Saponins, and Digitalis Analogues. Synthesis 2011, 24, 4003–4010. [Google Scholar] [CrossRef]
- Szabo, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Zupko, I.; Varga, M.; Herman, B.E.; Kalmar, L.; Szecsi, M.; Mernyak, E. Synthesis and in vitro pharmacological evaluation of N-[(1-benzyl-1,2,3-triazol-4-yl)methyl]-carboxamides on d-secoestrone scaffolds. J. Enzym. Inhib. Med. Chem. 2016, 31, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Kadar, Z.; Kovacs, D.; Frank, E.; Schneider, G.; Huber, J.; Zupko, I.; Bartok, T.; Wölfling, J. Synthesis and In Vitro Antiproliferative Activity of Novel Androst-5-ene Triazolyl and Tetrazolyl Derivatives. Molecules 2011, 16, 4786–4806. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Molnar, J.; Zupko, I.; Kadar, Z.; Wölfling, J. Synthesis of novel steroidal 17α-triazolyl derivatives via Cu(I)-catalyzed azide-alkyne cycloaddition, and an evaluation of their cytotoxic activity in vitro. Steroids 2011, 76, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Kadar, Z.; Molnar, J.; Schneider, G.; Zupko, I.; Frank, E. A facile ‘click’ approach to novel 15β-triazolyl-5α-androstane derivatives, and an evaluation of their antiproliferative activities in vitro. Bioorg. Med. Chem. 2012, 20, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Mernyak, E.; Kovacs, I.; Minorics, R.; Sere, P.; Czegany, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Ujfaludi, Z.; Boros, I.; et al. Synthesis of trans-16-triazolyl-13α-methyl-17-estradiol diastereomers and the effects of structural modifications on their in vitro antiproliferative activities. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bodnár, B.; Mernyák, E.; Szabó, J.; Wölfling, J.; Schneider, G.; Zupkó, I.; Kupihár, Z.; Kovács, L. Synthesis and in vitro investigation of potential antiproliferative monosaccharide-d-secoestrone bioconjugates. Bioorg. Med. Chem. Lett. 2017, 27, 1938–1942. [Google Scholar] [CrossRef]
- Bodnár, B.; Mernyák, E.; Wölfling, J.; Schneider, G.; Herman, B.E.; Szécsi, M.; Sinka, I.; Zupkó, I.; Kupihár, Z.; Kovács, L. Synthesis and Biological Evaluation of Triazolyl 13α-Estrone–Nucleoside Bioconjugates. Molecules 2016, 21, 1212. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Pataki, Z.; Wölfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and biological evaluation of 13α-estrone derivatives as potential antiproliferative agents. Steroids 2016, 113, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.S.; Abell, A. 1,2,3-Triazoles in Peptidomimetic Chemistry. Eur. J. Org. Chem. 2011, 13, 2399–2411. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lindsey, J.S. One-flask synthesis of meso-substituted dipyrromethanes and their application in the synthesis of trans-substituted porphyrin building blocks. Tetrahedron 1994, 50, 11427–11440. [Google Scholar] [CrossRef]
- Jurášek, M.; Rimpelová, S.; Pavlíčková, V.; Ruml, T.; Lapčík, O.; Drašar, P.B. Synthesis and biological evaluation of nandrolone–bodipy conjugates. Steroids 2015, 97, 62–66. [Google Scholar] [CrossRef]
- Yalagala, R.S.; Mazinani, S.A.; Maddalena, L.A.; Stuart, J.A.; Yan, F.; Yan, H. Microwave-assisted syntheses of BODIPY-sugar conjugates through click chemistry and conjugate assembly into liposomes. Carbohydr. Res. 2016, 424, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Mernyak, E.; Kozma, E.; Hetenyi, A.; Mark, L.; Schneider, G.; Wolfling, J. Stereoselective synthesis of spiro and condensed pyrazolines of steroidal α,β-unsaturated ketones and nitrilimines by 1,3-dipolar cycloaddition. Steroids 2009, 74, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Teller, D.M. Selective Bromination of a-Ring Aromatic 17-Ketalized Steroids. U.S. Patent US3177206, 6 April 1965. [Google Scholar]
- Černý, I.; Pouzar, V.; Lapcik, O.; Hampl, R. Addition of Azoimide to Unsaturated Ketones in the Steroid Series. Synthesis of N-(17β-Hydroxy-3-oxo-5α-androstan-15β-yl)succinamoic Acid and Its Evaluation as Hapten for Dihydrotestosterone Immunoanalysis. Collect. Czech. Chem. Commun. 1997, 62, 1931–1939. [Google Scholar] [CrossRef]
- Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B.E.; Szécsi, M.; Mernyák, E. Synthesis of novel 13α-estrone derivatives by Sonogashira coupling as potential 17β-HSD1 inhibitors. Beilstein J. Org. Chem. 2017, 13, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Bacsa, I.; Herman, B.E.; Jójárt, R.; Herman, K.S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Rižner, T.L.; Szécsi, M.; et al. Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J. Enzym. Inhib. Med. Chem. submitted.
- Beena, N.; Kumar, R.; Rohilla, K.; Roy, N.; Rawat, D.S. Synthesis and antibacterial activity evaluation of metronidazole-triazole conjugates. Bioorg. Med. Chem. Lett. 2009, 19, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Jurášek, M.; Rimpelová, S.; Kmoníčková, E.; Drašar, P.; Ruml, T. Tailor-made fluorescent trilobolide to study its biological relevance. J. Med. Chem. 2014, 57, 7947–7954. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; Nagai, A.; Chujo, Y. Microwave-assisted preparation of intense luminescent BODIPY-containing hybrids with high photostability and low leachability. J. Mater. Chem. 2010, 20, 2985–2992. [Google Scholar] [CrossRef]
- Nambara, T.; Sudo, K.; Sudo, M. Syntheses of estetrol monoglucuronides. Steroids 1976, 27, 111–122. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
The core structure of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPY) dyes.

Figure 2.
The fluorescent labeling at positions C-2 and C-15 of estrone.

Scheme 1.
Reagents and conditions: (i) 0.1 equiv. TFA, excess of pyrrole, rt (room temperature), 10 min; (ii) 1 equiv. DDQ, CH2Cl2, 45 min, rt; 7 equiv. Et3N, 4 equiv. BF3·OEt2, rt, 3 h.
Scheme 1.
Reagents and conditions: (i) 0.1 equiv. TFA, excess of pyrrole, rt (room temperature), 10 min; (ii) 1 equiv. DDQ, CH2Cl2, 45 min, rt; 7 equiv. Et3N, 4 equiv. BF3·OEt2, rt, 3 h.

Scheme 2.
Reagents and conditions: (i) 1.5 equiv. oxalyl chloride, CH2Cl2, catalytic amount of N,N-dimethylformamide (DMF), rt, 2.5h; (ii) 2.5 equiv. of 8, CH2Cl2, reflux, 4h; (iii) 20 equiv. Et3N, reflux, 30 min, 20 equiv. BF3·OEt2, reflux, 4 h; (iv) 1 equiv. of NaN3, DMSO, catalytic amount of AcOH.
Scheme 2.
Reagents and conditions: (i) 1.5 equiv. oxalyl chloride, CH2Cl2, catalytic amount of N,N-dimethylformamide (DMF), rt, 2.5h; (ii) 2.5 equiv. of 8, CH2Cl2, reflux, 4h; (iii) 20 equiv. Et3N, reflux, 30 min, 20 equiv. BF3·OEt2, reflux, 4 h; (iv) 1 equiv. of NaN3, DMSO, catalytic amount of AcOH.

Scheme 3.
Reagents and conditions: (i) 2.5 equiv. of ethylene glycol; 2 equiv. of triethyl orthoformate; catalytic amount of p-TsOH; rt; 2 h; (ii) 1 equiv. of pyridinium hydrobromide perbromide; tetrahydrofurane (THF); rt; (iii) 2 equiv. of KOtBu; DMSO; 80 °C; 1 h; (iv) 15 equiv. of H2CO; catalytic amount of p-TsOH; acetone; rt; 30 min; (v) propargyl alcohol (31 equiv.), catalytic amount of NaOH (5% aq), CH2Cl2, rt, 24 h; (vi) 1 equiv. of NaN3, DMSO, 4 equiv. of AcOH.
Scheme 3.
Reagents and conditions: (i) 2.5 equiv. of ethylene glycol; 2 equiv. of triethyl orthoformate; catalytic amount of p-TsOH; rt; 2 h; (ii) 1 equiv. of pyridinium hydrobromide perbromide; tetrahydrofurane (THF); rt; (iii) 2 equiv. of KOtBu; DMSO; 80 °C; 1 h; (iv) 15 equiv. of H2CO; catalytic amount of p-TsOH; acetone; rt; 30 min; (v) propargyl alcohol (31 equiv.), catalytic amount of NaOH (5% aq), CH2Cl2, rt, 24 h; (vi) 1 equiv. of NaN3, DMSO, 4 equiv. of AcOH.

Scheme 4.
Reagents and conditions: (i) 0.1 equiv. of Pd(PPh3)4, 0.1 equiv. of CuI, 6 equiv. of Et3N, 2 equiv. of TMS-C≡CH, THF, MW, 50 °C, 20 min; (ii) 1 equiv. tetrabutylammonium fluoride (TBAF), toluene, 5 min.
Scheme 4.
Reagents and conditions: (i) 0.1 equiv. of Pd(PPh3)4, 0.1 equiv. of CuI, 6 equiv. of Et3N, 2 equiv. of TMS-C≡CH, THF, MW, 50 °C, 20 min; (ii) 1 equiv. tetrabutylammonium fluoride (TBAF), toluene, 5 min.

Scheme 5.
Reagents and conditions: (i) CuI (0.05 equiv.), Ph3P (0.1 equiv.), DIPEA (3 equiv.), 1 equiv. TBAF, toluene, reflux, 2 h; (ii) CuI (0.05 equiv.), Ph3P (0.1 equiv.), DIPEA (3 equiv.), toluene, reflux, 2 h.
Scheme 5.
Reagents and conditions: (i) CuI (0.05 equiv.), Ph3P (0.1 equiv.), DIPEA (3 equiv.), 1 equiv. TBAF, toluene, reflux, 2 h; (ii) CuI (0.05 equiv.), Ph3P (0.1 equiv.), DIPEA (3 equiv.), toluene, reflux, 2 h.

Figure 3.
The normalized absorbance (a) and emission (b) spectra of the dyes (5, 11) and the conjugates (22–24).
Figure 3.
The normalized absorbance (a) and emission (b) spectra of the dyes (5, 11) and the conjugates (22–24).


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bacsa, I.; Konc, C.; Orosz, A.B.; Kecskeméti, G.; Rigó, R.; Özvegy-Laczka, C.; Mernyák, E. Synthesis of Novel C-2- or C-15-Labeled BODIPY—Estrone Conjugates. Molecules 2018, 23, 821. https://doi.org/10.3390/molecules23040821
AMA Style
Bacsa I, Konc C, Orosz AB, Kecskeméti G, Rigó R, Özvegy-Laczka C, Mernyák E. Synthesis of Novel C-2- or C-15-Labeled BODIPY—Estrone Conjugates. Molecules. 2018; 23(4):821. https://doi.org/10.3390/molecules23040821
Chicago/Turabian StyleBacsa, Ildikó, Csilla Konc, Anna Boglárka Orosz, Gábor Kecskeméti, Réka Rigó, Csilla Özvegy-Laczka, and Erzsébet Mernyák. 2018. "Synthesis of Novel C-2- or C-15-Labeled BODIPY—Estrone Conjugates" Molecules 23, no. 4: 821. https://doi.org/10.3390/molecules23040821