Synthesis and Physicochemical Evaluation of Entecavir-Fatty Acid Conjugates in Reducing Food Effect on Intestinal Absorption

Abstract
:1. Introduction
2. Results and Discussion
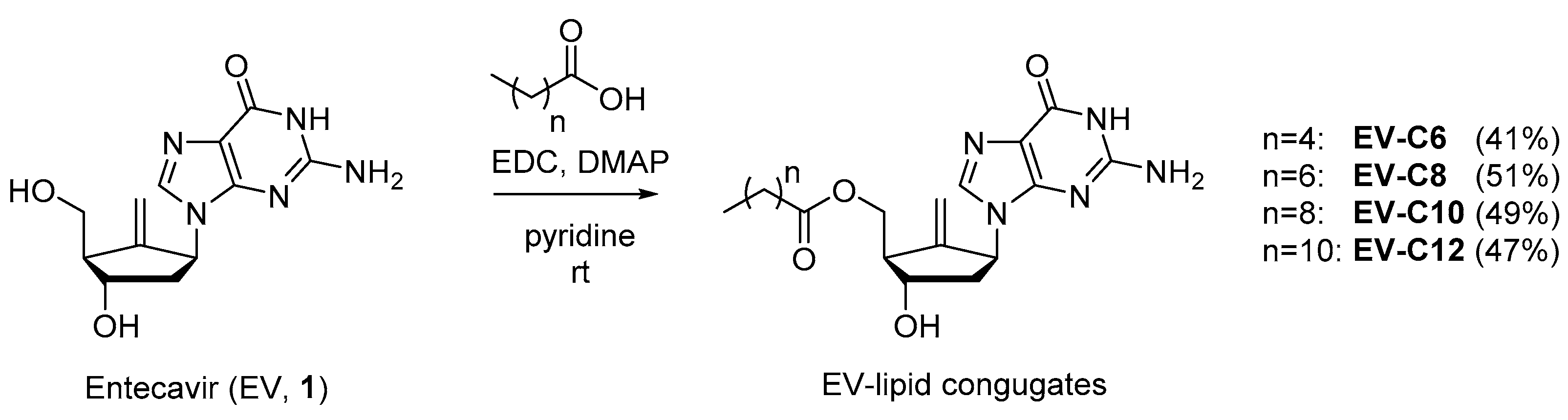
2.1. Chemistry
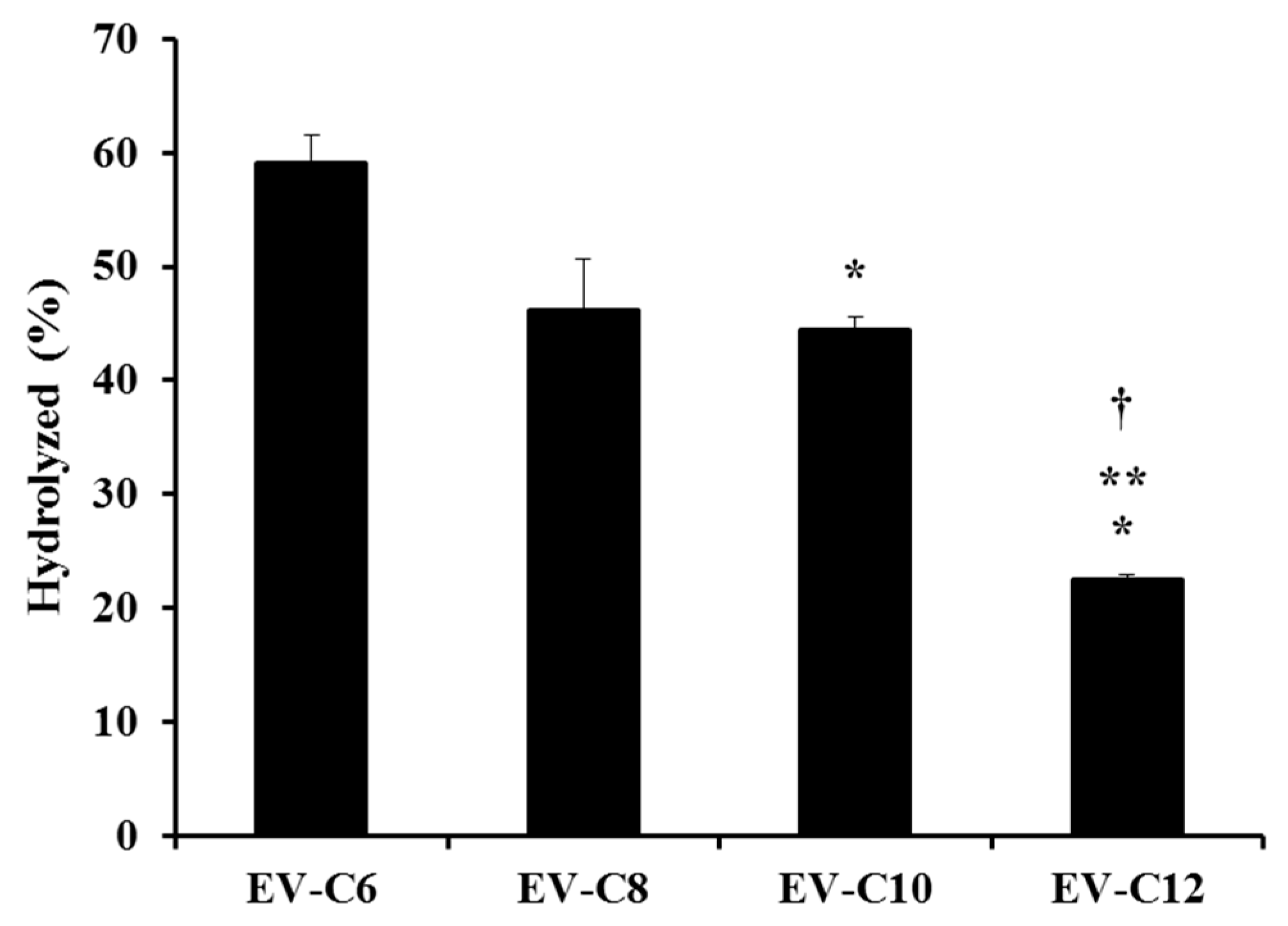
2.2. Determination of Aqueous Solubility, logP, and the Acidic Hydrolysis of EV-FAs
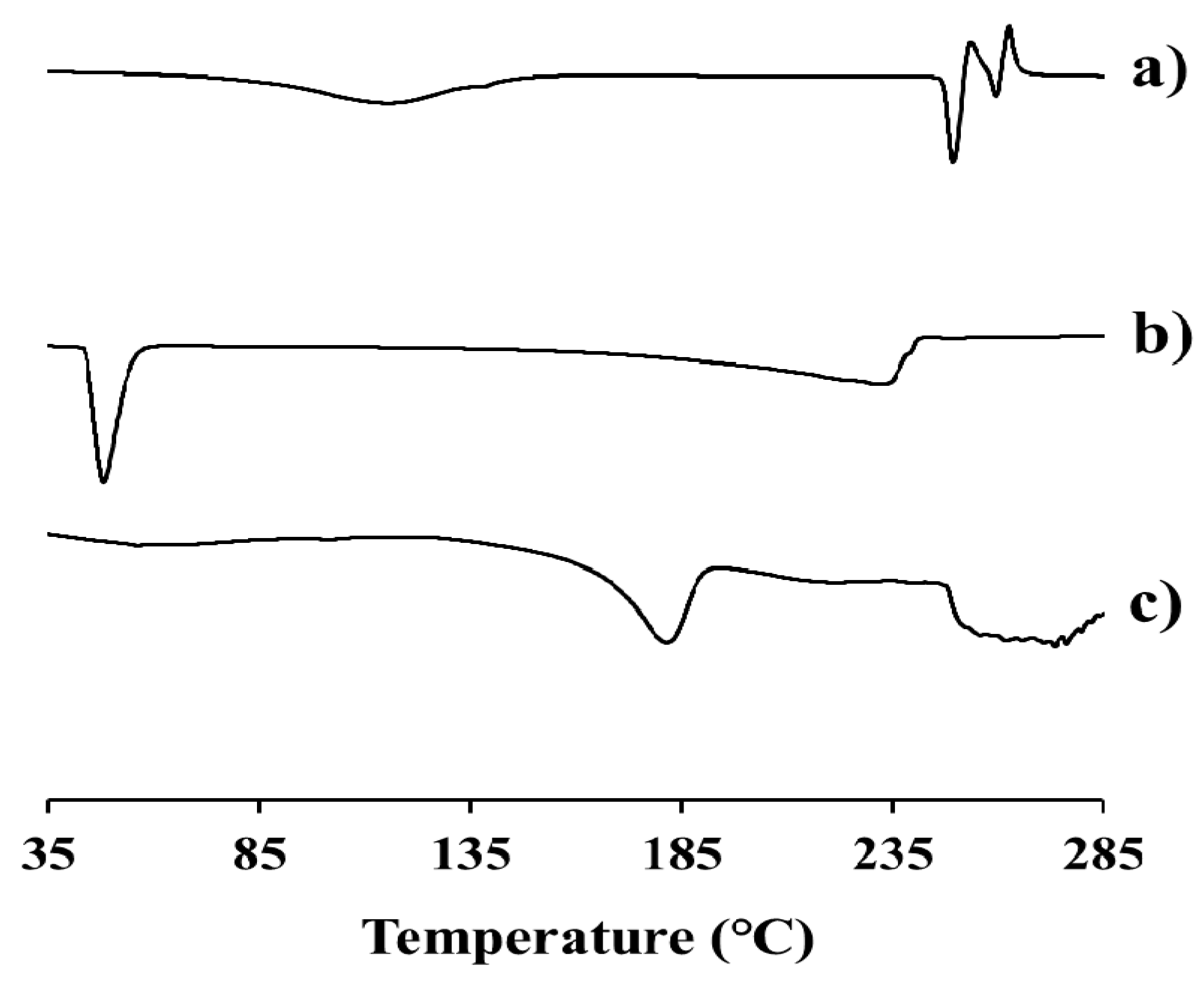
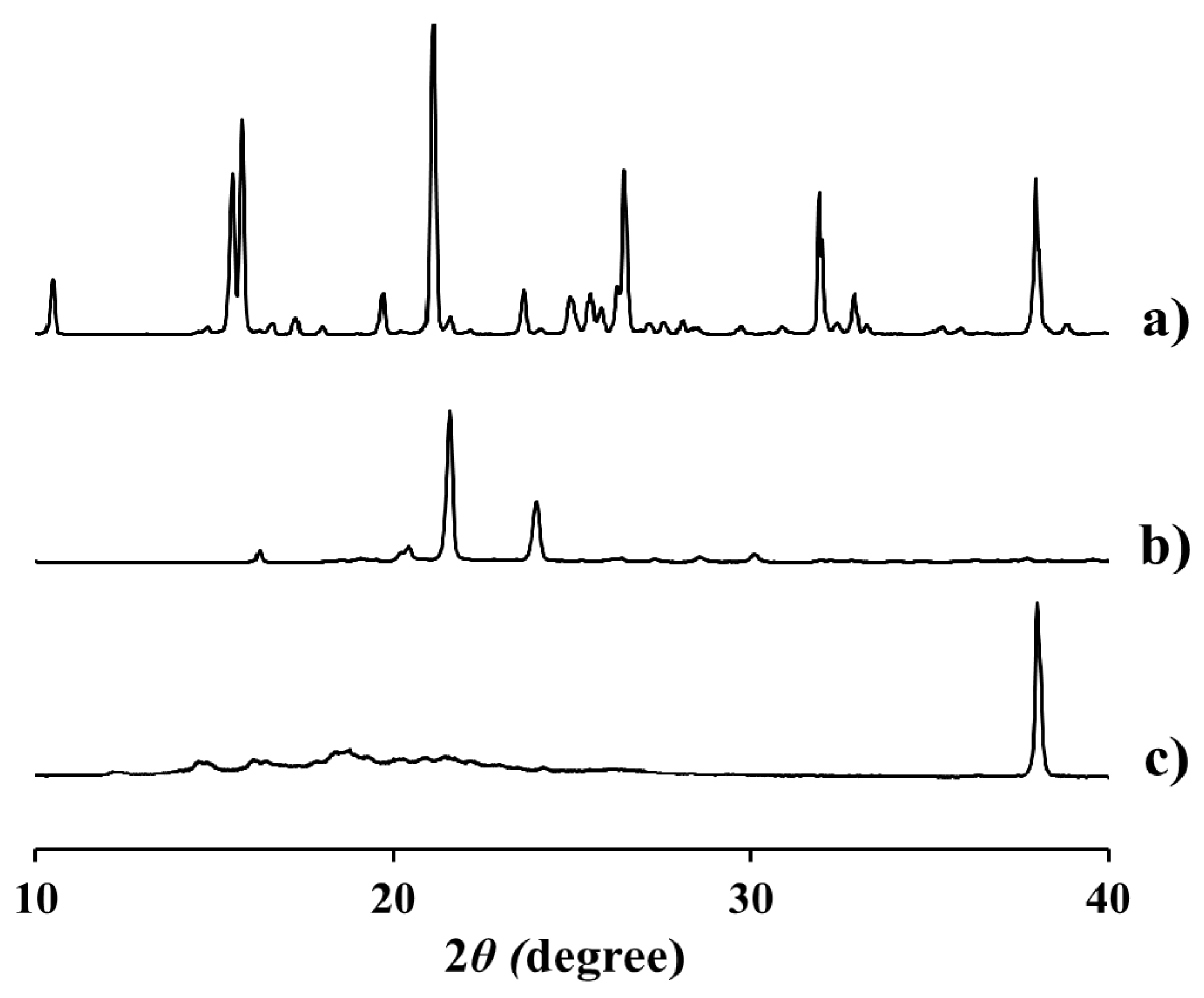
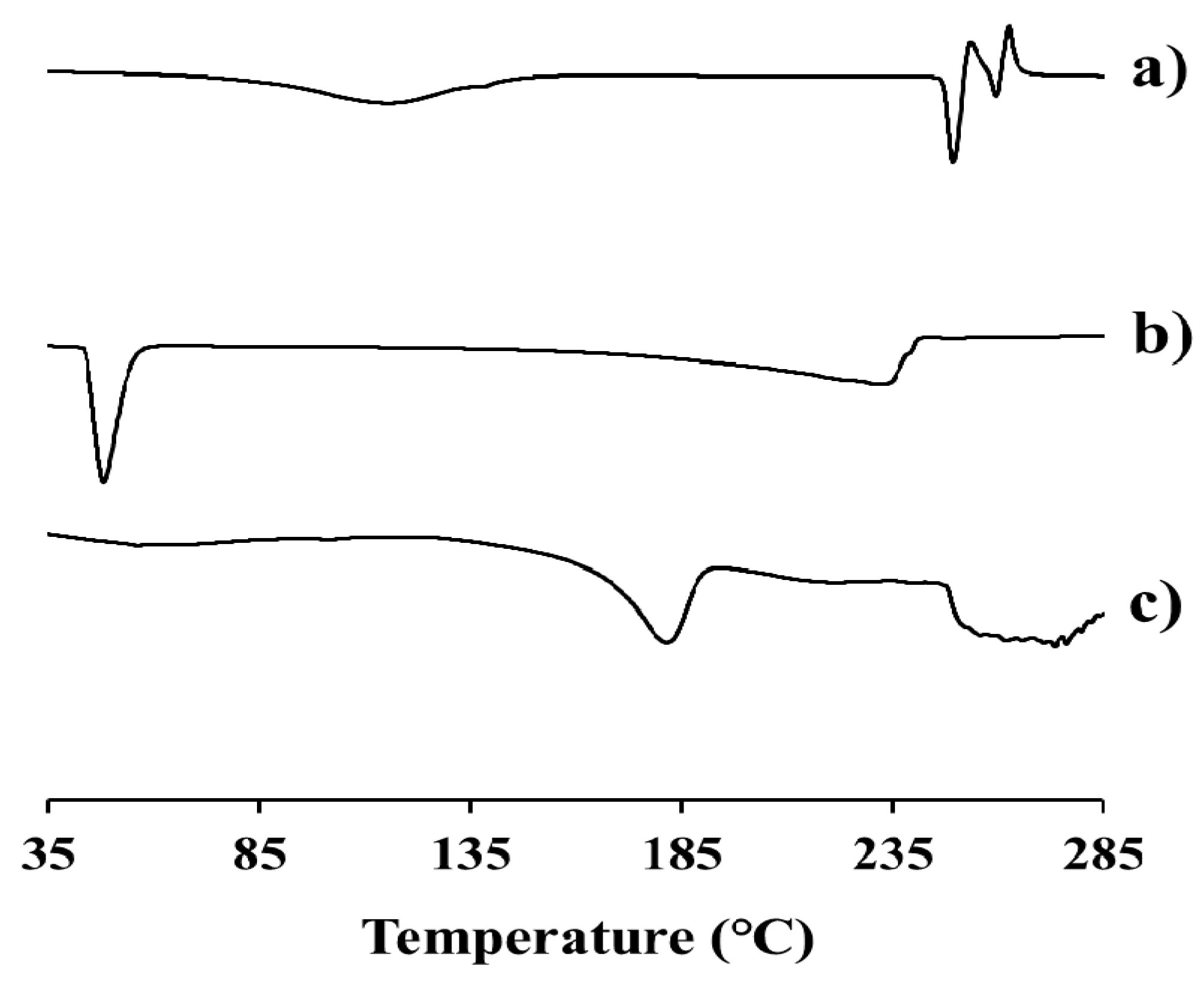
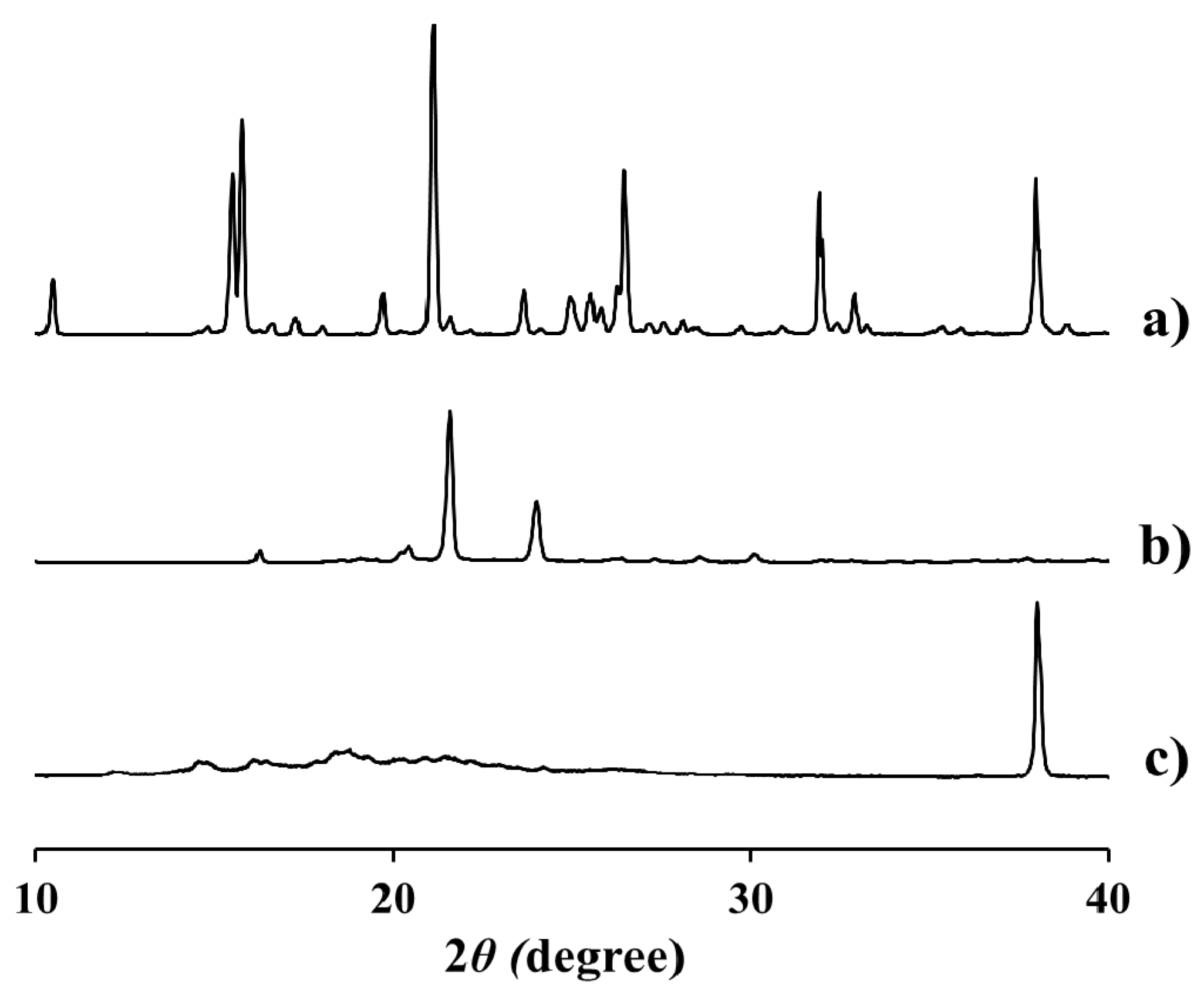
2.3. Physicochemical Characteristics of EV-C12
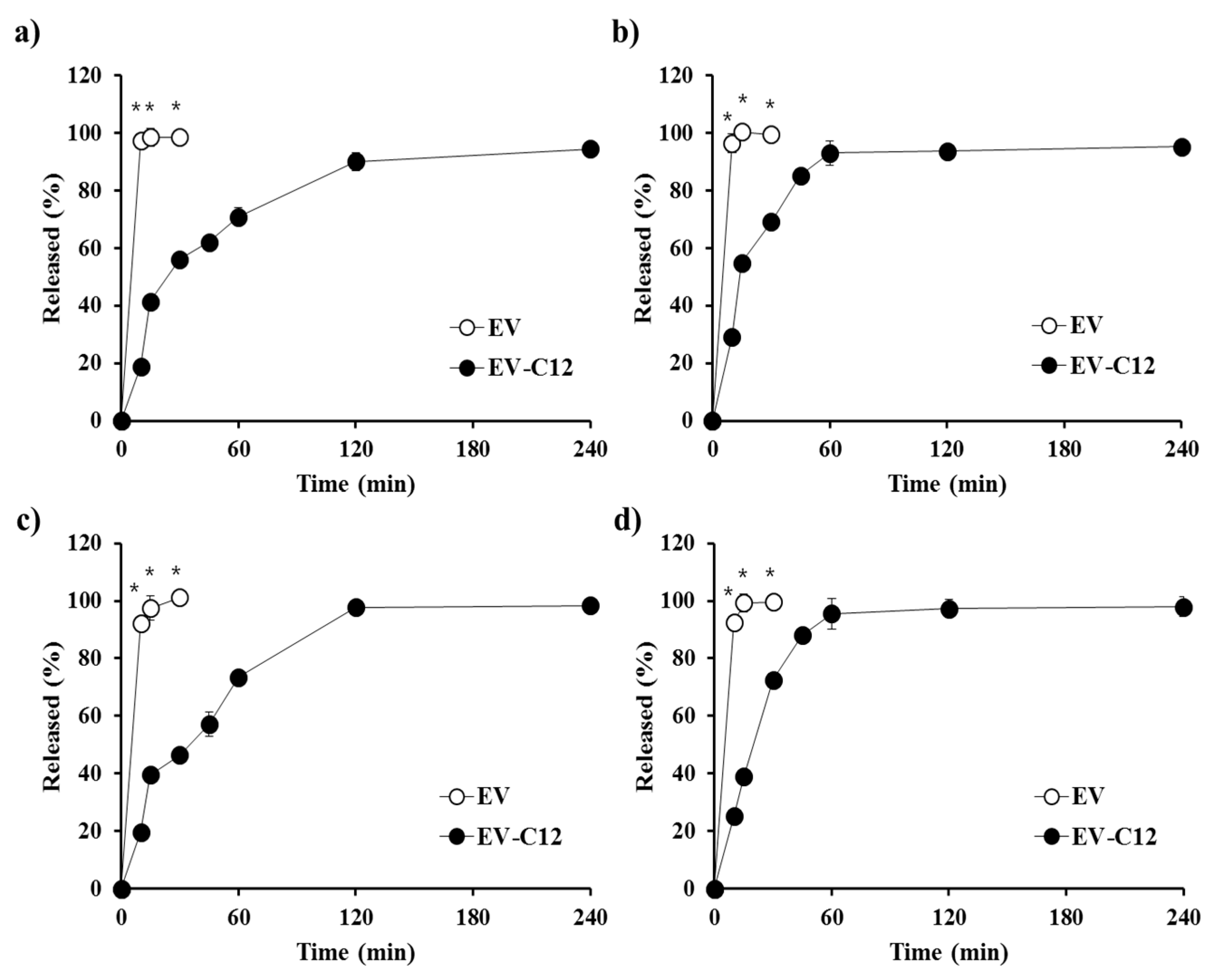
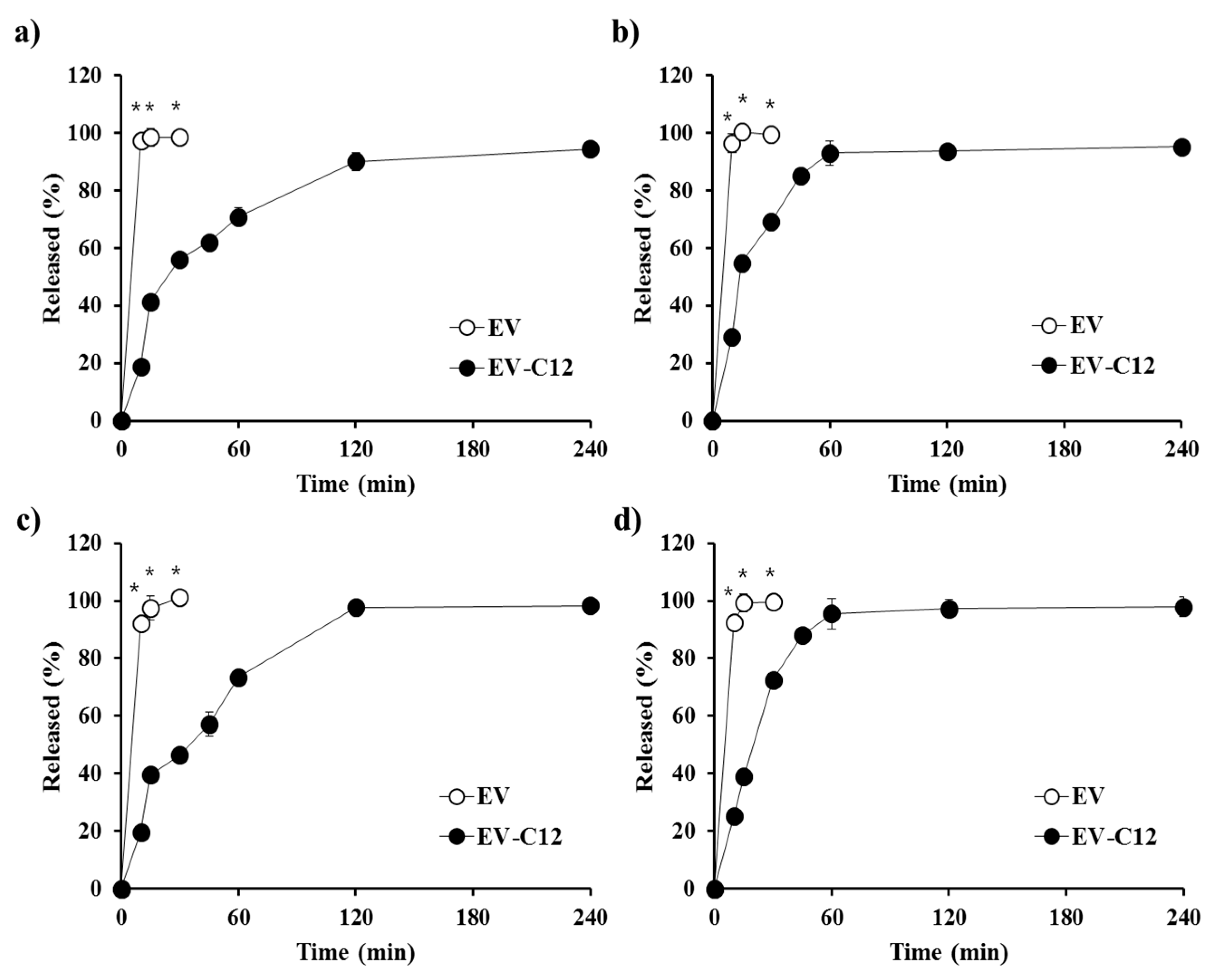
2.4. In Vitro Dissolution Profile in Biorelevant Media
3. Materials and Methods
3.1. Materials
3.2. Synthesis of EV-FA Conjugates
3.2.1. General Synthesis Procedure
3.2.2. (1R,3S,5S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-5-hydroxy-2-methylenecyclopentyl)methyl hexanoate (EV-C6)
3.2.3. ((1R,3S,5S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-5-hydroxy-2-methylenecyclopentyl)methyl octanoate (EV-C8)
3.2.4. ((1R,3S,5S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-5-hydroxy-2-methylenecyclopentyl)methyl decanoate (EV-C10)
3.2.5. ((1R,3S,5S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-5-hydroxy-2-methylenecyclopentyl)methyl dodecanoate (EV-C12)
3.3. HPLC Determination of EV-FA Conjugates
3.4. Determination of Aqueous Solubility, logP, and Acidic Hydrolysis of EV-FAs
3.5. Physical Characterization of EV-C12
3.6. In Vitro Dissolution Test
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lok, A.S.; McMahon, B.J. Chronic hepatitis B: Update 2009. Hepatology 2009, 50, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Liaw, Y.F.; Wu, S.S.; Schiff, E.; Han, K.H.; Lai, C.L.; Safadi, R.; Lee, S.S.; Halota, W.; Goodman, Z.; et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010, 52, 886–893. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Sriplung, H.; Geater, A.; Chongsuvivatwong, V.; Zhuang, L.; Li, Y.L.; Lei, H.; Liu, J.; Chen, H.Y.; Tang, B.Z.; et al. Impact of viral replication inhibition by entecavir on peripheral T lymphocyte subpopulations in chronic hepatitis B patients. BMC Infect. Dis. 2008, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Gish, R.G.; de Man, R.; Gadano, A.; Sollano, J.; Chao, Y.C.; Lok, A.S.; Han, K.H.; Goodman, Z.; Zhu, J.; et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Shouval, D.; Lok, A.S.; Chang, T.T.; Cheinquer, H.; Goodman, Z.; DeHertogh, D.; Wilber, R.; Zink, R.C.; Cross, A.; et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Chen, C.J.; Lu, W.; Ren, H.; Tan, D.; Wang, Y.; Xu, D.; Jiang, M.; Liu, J.; Xu, D.; et al. Entecavir is superior to lamivudine for the treatment of chronic hepatitis B (CHB): Results of a phase 3 Chinese study (ETV-023) in nucleoside-naïve patients. J. Hepatol. 2006, 44, S193. [Google Scholar] [CrossRef]
- Yang, X.; Ma, Z.; Zhou, S.; Weng, Y.; Lei, H.; Zeng, S.; Li, L.; Jiang, H. Multiple drug transporters are involved in renal secretion of entecavir. Antimicrob. Agents Chemother. 2016, 60, 6260–6270. [Google Scholar] [CrossRef] [PubMed]
- Clinical Pharmacology and Biopharmaceutics Review(s) of Entecavir. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/21797_BARACLUDE_biopharmr.PDF (accessed on 8 September 2017).
- Abrahamsson, B.; Albery, T.; Eriksson, A.; Gustafsson, I.; Sjöberg, M. Food effects on tablet disintegration. Eur. J. Pharm. Sci. 2004, 22, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Anneveld, B.; Goudappel, G.J.; Duchateau, G.; Annaert, P.; Augustijns, P.; Zeijdner, E. Food-dependent disintegration of immediate release fosamprenavir tablets: In vitro evaluation using magnetic resonance imaging and a dynamic gastrointestinal system. Eur. J. Pharm. Biopharm. 2011, 77, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cvijic, S.; Parojčić, J.; Langguth, P. Viscosity-mediated negative food effect on oral absorption of poorly permeable drugs with an absorption window in the proximal intestine: In vitro experimental simulation and computational verification. Eur. J. Pharm. Sci. 2014, 61, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Package Inserts of Baraclude® (Entecavir) Tablets. Available online: https://packageinserts.bms.com/pi/pi_baraclude.pdf (accessed on 25 October 2017).
- Stella, V.J. Prodrug Approaches to Enhancing the Oral Delivery of Poorly Permeable Drugs; Prodrugs: Challenges and Rewards; Valentino, S., Ronald, B., Michael, H., Reza, O., Hans, M., Jefferson, T., Eds.; Springer: New York, NY, USA, 2007; pp. 37–82. [Google Scholar]
- Aungst, B.J. Absorption enhancers: Applications and advances. AAPS J. 2012, 14, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.S.; Gupta, D.; Yu, M.; Nguyen, P.; Varghese Gupta, S. Current and evolving approaches for improving the oral permeability of BCS Class III or analogous molecules. Drug Dev. Ind. Pharm. 2017, 43, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control. Release 2008, 126, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sueda, K.; Sadgrove, M.P.; Fitzsimmons, J.M.; Jay, M. Physicochemical characterization of a prodrug of a radionuclide decorporation agent for oral delivery. J. Pharm. Sci. 2012, 101, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Li, P.; Cheng, Y.; Zhang, X.; Sheng, J.; Wang, D.; Li, J.; Zhang, Q.; Zhong, C.; Cao, R.; et al. Enhancing the intestinal absorption of low molecular weight chondroitin sulfate by conjugation with α-linolenic acid and the transport mechanism of the conjugates. Int. J. Pharm. 2014, 465, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Dando, T.; Plosker, G. Adefovir dipivoxil: A review of its use in chronic hepatitis B. Drugs 2003, 63, 2215–2234. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Hansch, C.; Elkins, D. Partition Coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Domini, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Machatha, S.G.; Yalkowsky, S.H. Comparison of the octanol/water partition coefficients calculated by ClogP®, ACDlogP and KowWin® to experimentally determined values. Int. J. Pharm. 2005, 294, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Graffner-Nordberg, M.; Sjödin, K.; Tunek, A.; Hallberg, A. Synthesis and enzymatic hydrolysis of esters, constituting simple models of soft drugs. Chem. Pharm. Bull. 1998, 46, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci. 2000, 11, S15–S27. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Gürakar, A. The effect of fatty acid chain length on the rate of arylester hydrolysis by various albumins. Clin. Chim. Acta 1987, 164, 329–337. [Google Scholar] [CrossRef]
- Hu, X.; Lin, X.; Gu, Y.; Liu, Z.; Tang, Y.; Zhang, Y.; Chen, X.; Wang, Y.; Tang, X. Biocompatible riboflavin laurate long-acting injectable nanosuspensions allowing sterile filtration. Drug Deliv. 2014, 21, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Tian, Z.; Ye, W. Crystal Entecavir, Crystal Entecavir Formulation and Methods for the Preparation Thereof. U.S. Patent US20140220120, 9 August 2016. [Google Scholar]
- Santa Cruz Biotechnology, Inc. Safety Data Sheet of Paliperidone Palmitate. Available online: http://datasheets.scbt.com/sds/eghs/en/sc-478257.pdf (accessed on 25 October 2017).
- Bunaciu, A.A.; Udriştioiu, E.G.; Aboul-Enein, H.Y. X-ray diffraction: Instrumentation and applications. Crit. Rev. Anal. Chem. 2015, 45, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lentz, K.A. Current methods for predicting human food effect. AAPS J. 2008, 10, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Han, S.D.; Jung, S.W.; Jang, S.W.; Son, M.; Kim, B.M.; Kang, M.J. Reduced food-effect on intestinal absorption of dronedarone by self-microemulsifying drug delivery system (SMEDDS). Biol. Pharm. Bull. 2015, 38, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Vig, B.S.; Lorenzi, P.L.; Drach, J.C.; Townsend, L.B.; Amidon, G.L. Amino acid ester prodrugs of the antiviral agent 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole as potential substrates of hPEPT1 transporter. J. Med. Chem. 2005, 48, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FaSSGF (μg/mL) | FaSSIF (μg/mL) | FeSSGF (μg/mL) | FeSSIF (μg/mL) | ClogP 2 | |
|---|---|---|---|---|---|
| EV | 8940 ± 92 | 1624 ± 23 | 2152 ± 13 | 2241 ± 52 | −2.03 |
| EV-C6 | 4.84 ± 0.5 | 5.54 ± 0.6 | 24.9 ± 1.4 | 25.2 ± 2.3 | 0.45 |
| EV-C8 | 4.74 ± 0.3 | 5.74 ± 0.3 | 38.2 ± 1.5 | 36.7 ± 1.2 | 1.51 |
| EV-C10 | 4.41 ± 0.1 | 6.47 ± 0.6 | 61.9 ±3.2 | 66.6 ± 4.4 | 2.56 |
| EV-C12 | 4.82 ± 0.2 | 6.50 ± 0.2 | 76.6 ± 4.0 | 78.8 ± 7.7 | 3.62 |
| Mobile Phase | Calibration Curve | Linearity (r2 Value) | |
|---|---|---|---|
| EV | ACN:DW = 4:96 (pH 2.8) 1 | 1.0000 | |
| EV-C6 | IPA:DW = 30:70 | 0.9995 | |
| EV-C8 | IPA:DW = 40:60 | 0.9996 | |
| EV-C10 | IPA:DW = 40:60 | 0.9998 | |
| EV-C12 | IPA:DW = 50:50 | 0.9998 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.J.; Ho, M.J.; Ahn, S.; Han, Y.T.; Kang, M.J. Synthesis and Physicochemical Evaluation of Entecavir-Fatty Acid Conjugates in Reducing Food Effect on Intestinal Absorption. Molecules 2018, 23, 731. https://doi.org/10.3390/molecules23040731
Jung HJ, Ho MJ, Ahn S, Han YT, Kang MJ. Synthesis and Physicochemical Evaluation of Entecavir-Fatty Acid Conjugates in Reducing Food Effect on Intestinal Absorption. Molecules. 2018; 23(4):731. https://doi.org/10.3390/molecules23040731
Chicago/Turabian StyleJung, Hyuck Jun, Myoung Jin Ho, Sungwan Ahn, Young Taek Han, and Myung Joo Kang. 2018. "Synthesis and Physicochemical Evaluation of Entecavir-Fatty Acid Conjugates in Reducing Food Effect on Intestinal Absorption" Molecules 23, no. 4: 731. https://doi.org/10.3390/molecules23040731