Synthesis of Benzofuran-2-One Derivatives and Evaluation of Their Antioxidant Capacity by Comparing DPPH Assay and Cyclic Voltammetry

 , , ,
, , ,  ,
,  and
and
Abstract
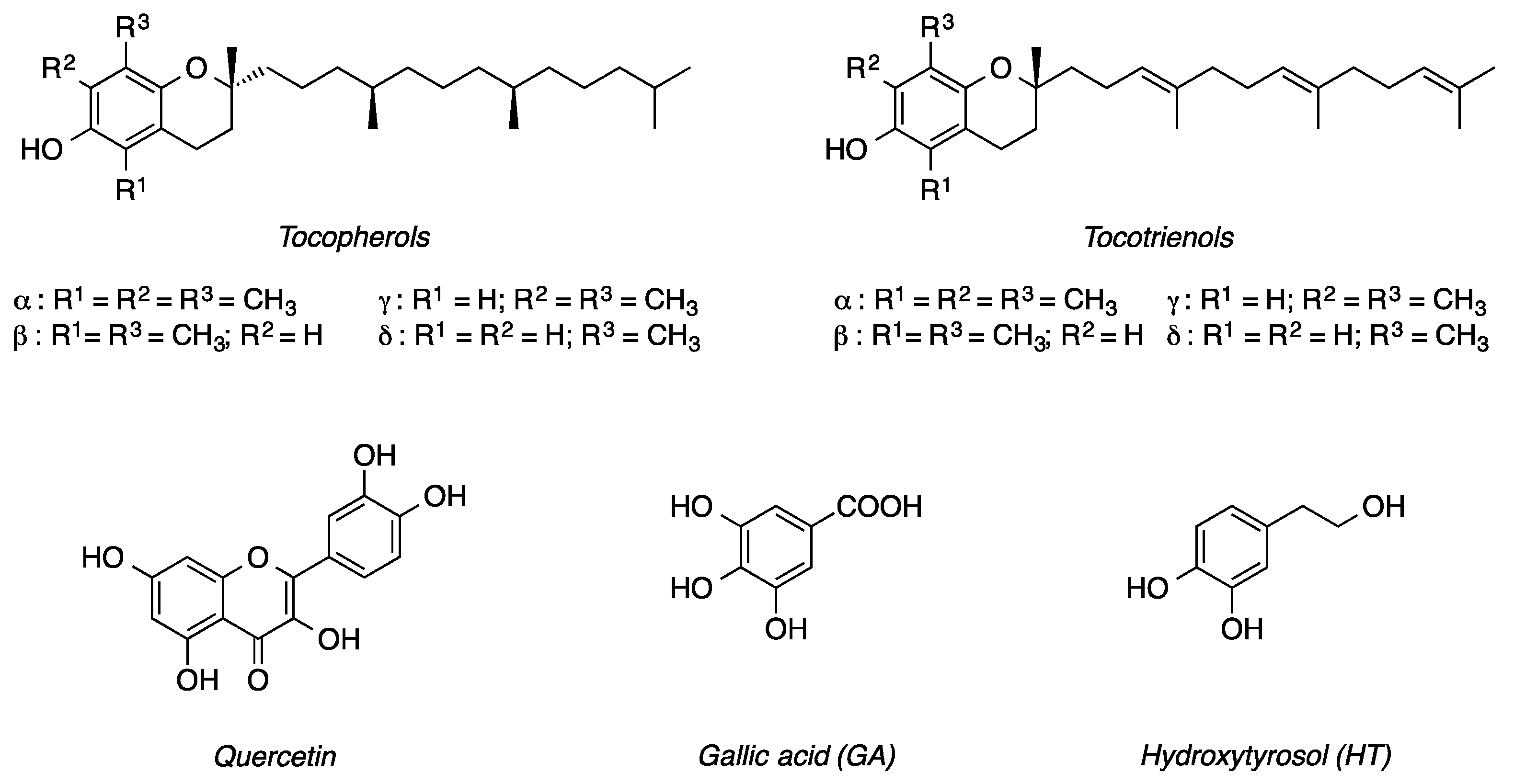
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Antioxidant Capacity Evaluation
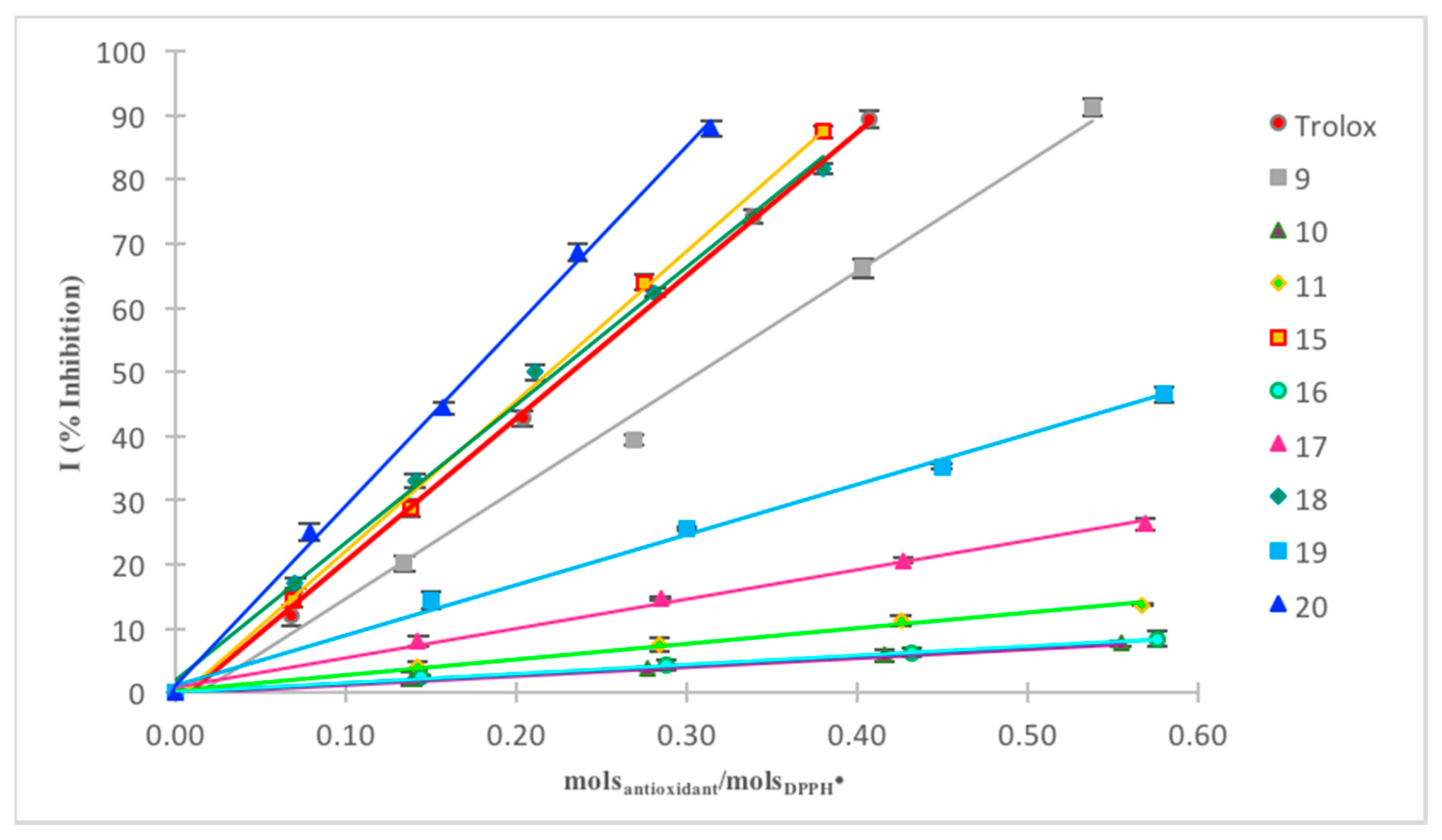
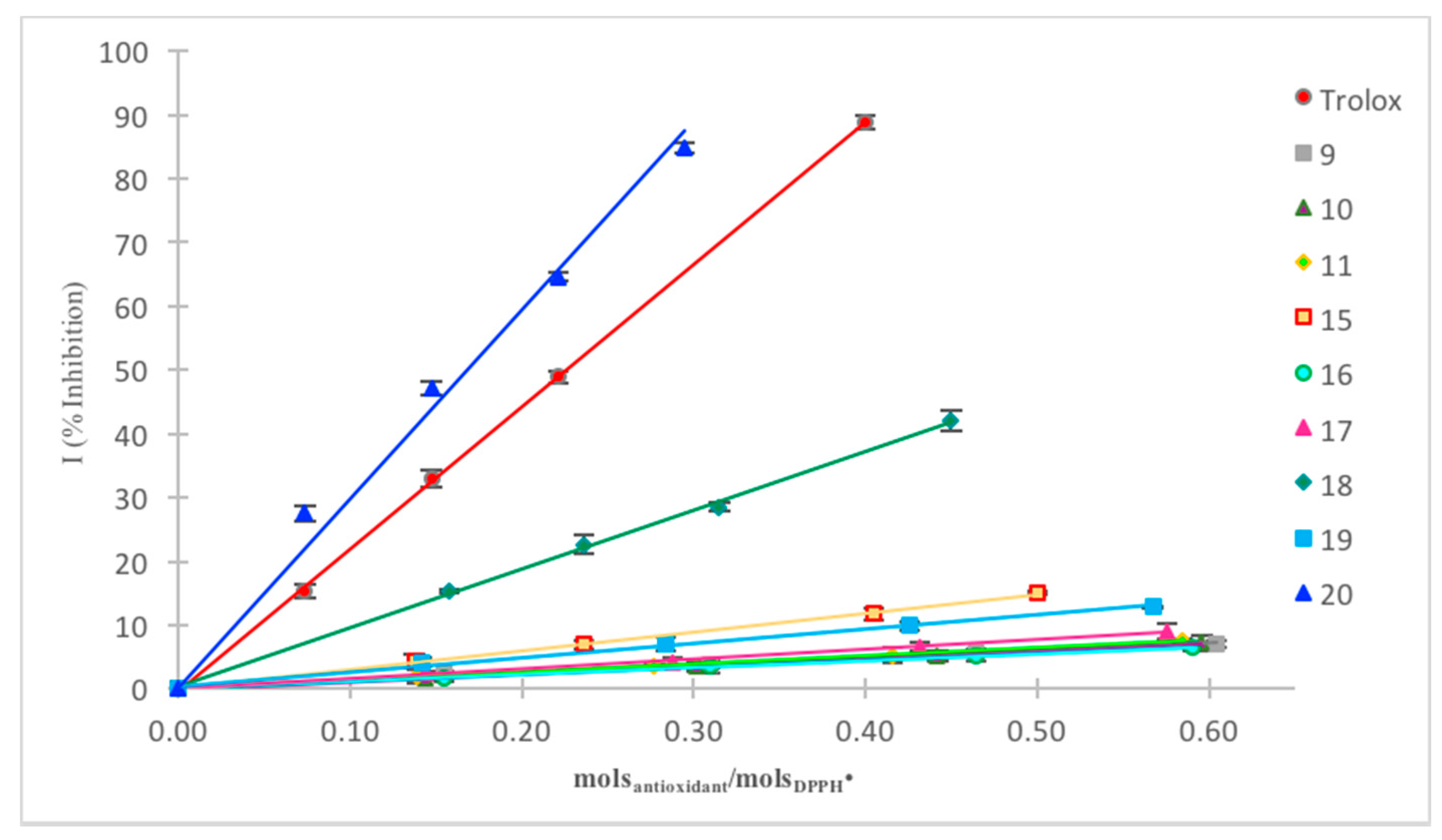
2.2.1. DPPH Assay
DPPH Assay in Methanol
DPPH Assay in Acetonitrile
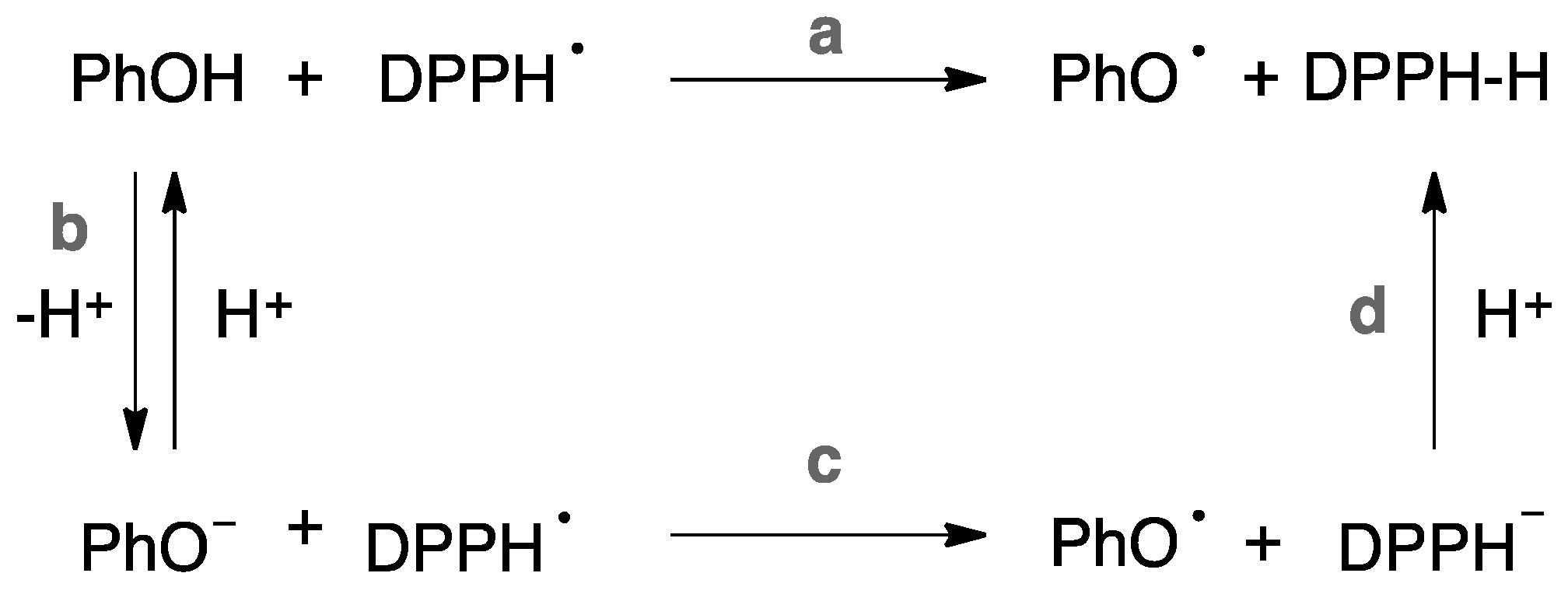
2.2.2. Measurement of Rate Constants for the Reaction of Compounds 9, 15, 18, and 20 with DPPH•
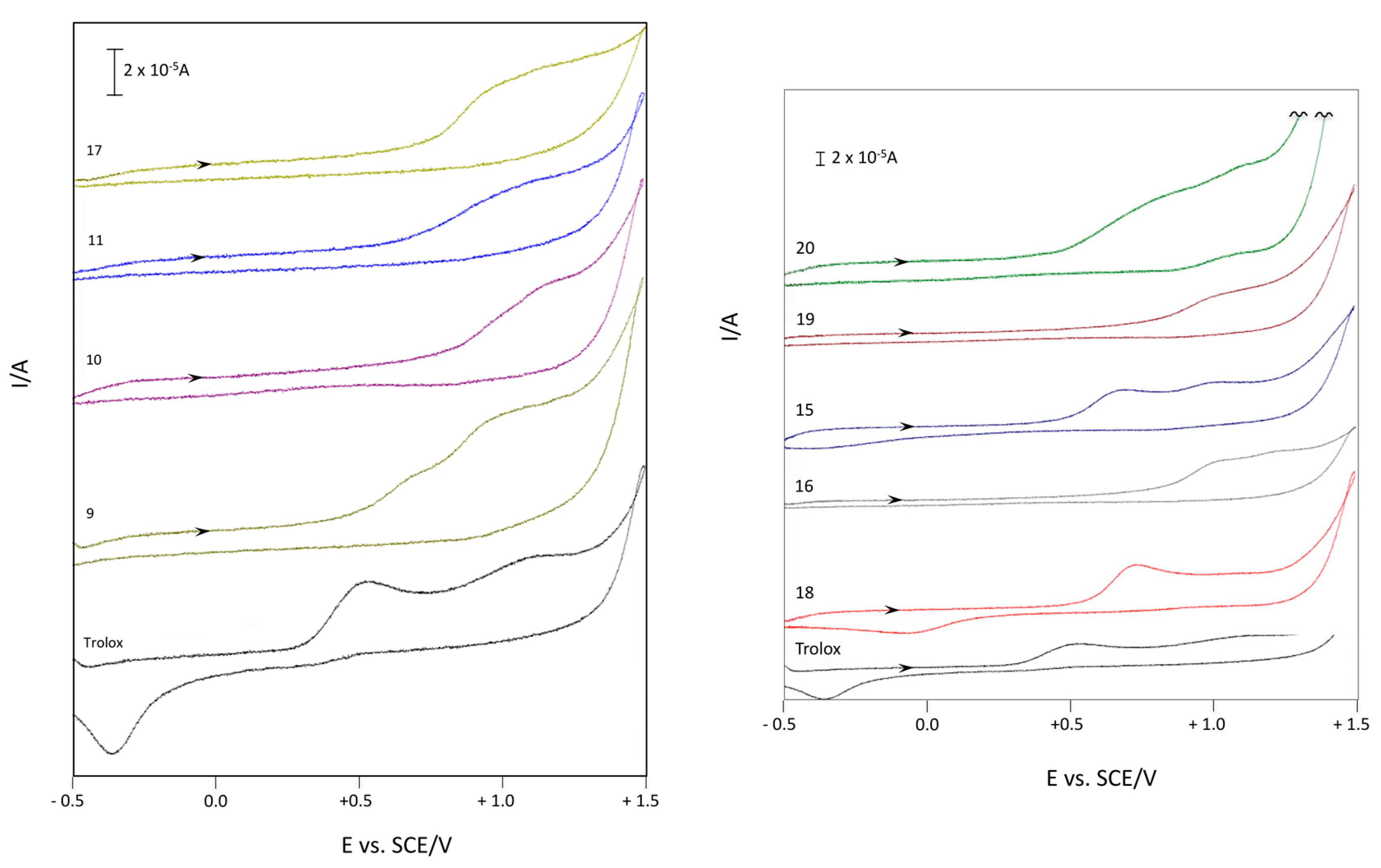
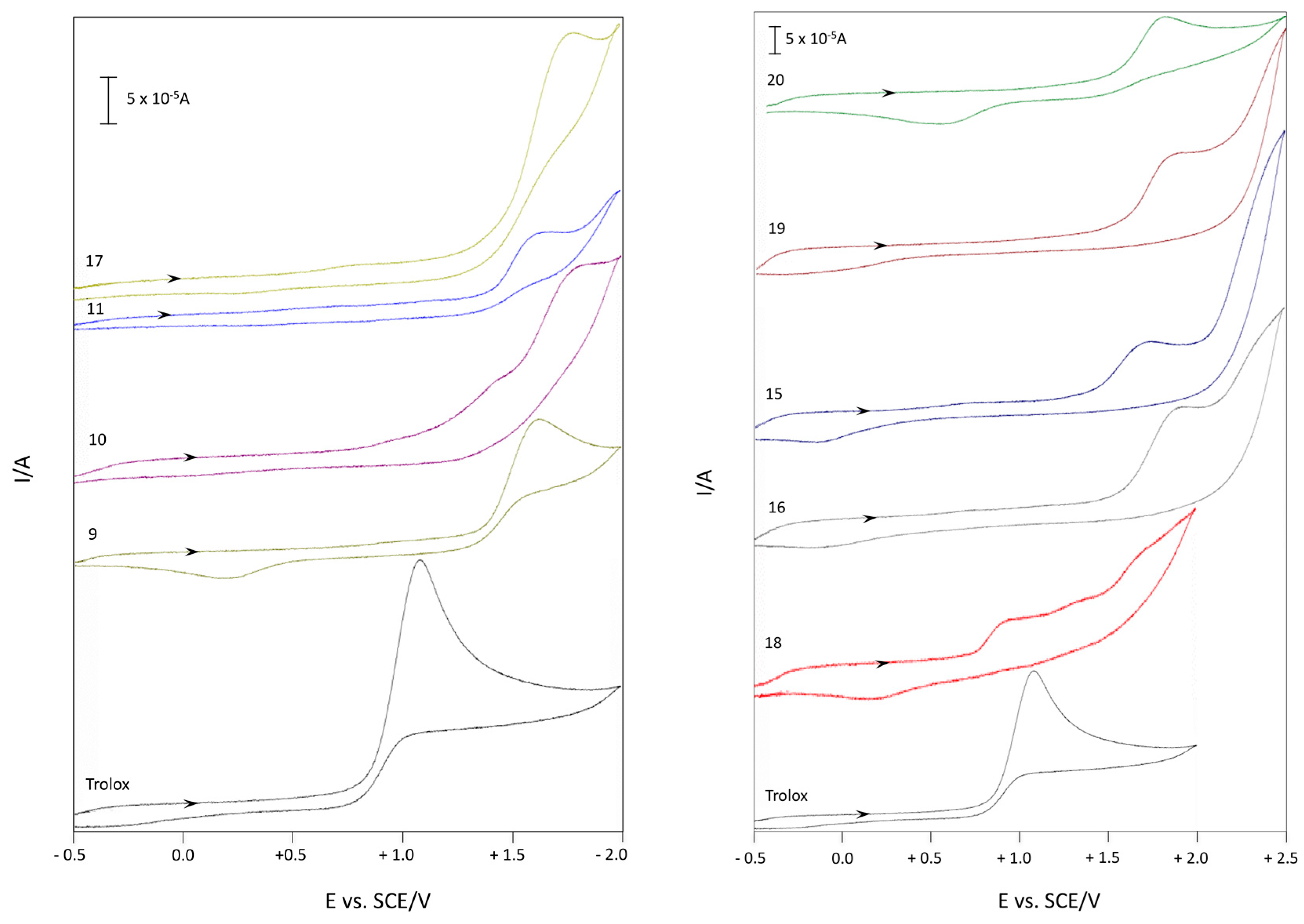
2.2.3. Cyclic Voltammetry
Cyclic Voltammetry in Aqueous Medium of Compounds 9–11 and 15–20
Cyclic Voltammetry in Acetonitrile of Compounds 9–11 and 15–20
3. Materials and Methods
3.1. General Procedure for the Lewis-Acid-Catalysed Friedel–Crafts/Lactonization Reaction
3.2. Characterization Data for Benzofuran 9–11
3.3. General Procedure for the Domino Friedel-Crafts/Lactonization Reaction Performed in AcOH
3.4. Characterization Data for Benzofuran-2(3H)-One 15–20
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halliwell, B. Antioxidants in human health and disease. Ann. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Guerra-Araiza, C.; Alvarez-Mejia, A.L.; Sanchez-Torres, S.; Farfan-Garcia, E.; Mondragon-Lozano, R.; Pinto-Almazan, R.; Salgado-Ceballos, H. Effect of natural exogenous antioxidants on aging and on neurodegenerative diseases. Free Radic. Res. 2013, 47, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 2nd ed.; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Patel, A.K.; Shah, N.; Choudhary, A.K.; Jha, U.K.; Yadav, U.C.; Gupta, P.K.; Pakuwal, U. Oxidative stress and antioxidants in disease and cancer: A review. Asian Pac. J. Cancer Prev. 2014, 15, 4405–4409. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.P.; Li, Y.; Meng, X.; Zhou, T.; Zhou, Y.; Zheng, J.; Zhang, J.J.; Li, H.B. Natural antioxidants in foods and medicinal plants: Extraction, assessment and resources. Int. J. Mol. Sci. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Roy, S. Tocotrienols: Vitamin e beyond tocopherols. Life Sci. 2006, 78, 2088–2098. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.; Nesaretnam, K. Vitamin E tocotrienols: Life beyond tocopherols. Genes Nutr. 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Mazumdar, K.; Mishra, B.K.; Dastidar, S.G.; Park, J.-H. Isolation and identification of a flavone (quercetin) from Butea frondosa bark. Pharm. Chem. J. 2007, 41, 37–39. [Google Scholar] [CrossRef]
- Valentova, K.; Kanova, K.; Di Meo, F.; Pelantova, H.; Chambers, C.S.; Rydlova, L.; Petraskova, L.; Krenkova, A.; Cvacka, J.; Trouillas, P.; et al. Chemoenzymatic preparation and biophysical properties of sulfated quercetin metabolites. Int. J. Mol. Sci. 2017, 18, 2231. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Mateos, R.; Collantes de Teran, L.; Espartero, J.L.; Cert, R.; Jover, M.; Alcudia, F.; Bautista, J.; Cert, A.; Parrado, J. Lipophilic hydroxytyrosyl esters. Antioxidant activity in lipid matrices and biological systems. J. Agric. Food Chem. 2006, 54, 3779–3785. [Google Scholar] [CrossRef] [PubMed]
- Kubo, I.; Xiao, P.; Fujita, K. Antifungal activity of octyl gallate: Structural criteria and mode of action. Bioorg. Med. Chem. Lett. 2001, 11, 347–350. [Google Scholar] [CrossRef]
- Kubo, I.; Masuoka, N.; Xiao, P.; Haraguchi, H. Antioxidant activity of dodecyl gallate. J. Agric. Food Chem. 2002, 50, 3533–3539. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.R.; Albrecht, C.; Cortez, M.V.; Soria, E.A. Pharmacology and toxicology of polyphenols with potential as neurotropic agents in non-communicable diseases. Curr. Drug Targets 2018, 19, 97–110. [Google Scholar] [PubMed]
- Justino, G.C.; Correia, C.F.; Mira, L.; Dos Santos, R.M.B.; Simoes, J.A.M.; Silva, A.M.; Santos, C.; Gigante, B. Antioxidant activity of a catechol derived from abietic acid. J. Agric. Food Chem. 2006, 54, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Li, W.C.; Hu, Y.; Wang, B.; Ren, G.; Feng, L.H. Synthesis of the intermediate for fumimycin: A natural peptide deformylase inhibitor. Res. Chem. Int. 2013, 39, 3049–3054. [Google Scholar] [CrossRef]
- Gross, P.J.; Furche, F.; Nieger, M.; Braese, S. Asymmetric total synthesis of (+)-fumimycin via 1,2-addition to ketimines. Chem. Commun. 2010, 46, 9215–9217. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Snyder, S.A.; Huang, X.H.; Simonsen, K.B.; Koumbis, A.E.; Bigot, A. Studies toward diazonamide A: Initial synthetic forays directed toward the originally proposed structure. J. Am. Chem. Soc. 2004, 126, 10162–10173. [Google Scholar] [CrossRef] [PubMed]
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Kemertelidze, E.; Pizza, C.; Piacente, S. Gloriosaols A and B, two novel phenolics from Yucca gloriosa: Structural characterization and configurational assignment by a combined NMR-quantum mechanical strategy. Tetrahedron 2007, 63, 148–154. [Google Scholar] [CrossRef]
- Fedorova, T.E.; Ivanova, S.Z.; Fedorov, S.V.; Babkin, V.A. Larisinol, a new spirobiflavonoid from Larix gmelinii bark. Chem. Nat. Compd. 2007, 43, 208–209. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Tomic, G.; Nikolic, I.; Nerantzaki, A.A.; Sayyad, N.; Stosic-Grujicic, S.; Stojanovic, I.; Gerothanassis, I.P.; Tzakos, A.G. Phytochemical profile of Rosmarinus officinalis and Salvia officinalis extracts and correlation to their antioxidant and anti-proliferative activity. Food Chem. 2013, 136, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, A.; Cimmino, A.; Vurro, M.; Berestetskiy, A.; Troise, C.; Zonno, M.C.; Motta, A.; Evidente, A. Agropyrenol and agropyrenal, phytotoxins from Ascochyta agropyrina var. nana, a fungal pathogen of Elitrigia repens. Phytochemistry 2012, 79, 102–108. [Google Scholar] [PubMed]
- Chien, Y.-C.; Lin, C.-H.; Chiang, M.Y.; Chang, H.-S.; Liao, C.-H.; Chen, I.-S.; Peng, C.-F.; Tsai, I.-L. Secondary metabolites from the root of Ehretia longiflora and their biological activities. Phytochemistry 2012, 80, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, T.; Punzi, P.; Migliorini, A.; Tofani, D. An organocatalytic approach to the synthesis of six-membered heterocycles. Curr. Org. Chem. 2011, 15, 2098–2146. [Google Scholar] [CrossRef]
- Piacente, S.; Montoro, P.; Oleszek, W.; Pizza, C. Yucca schidigera bark: Phenolic constituents and antioxidant activity. J. Nat. Prod. 2004, 67, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Piacente, S.; Pizza, C.; Oleszek, W.; Stochmal, A.; Pinto, A.; Sorrentino, R.; Autore, G. Inhibition of inducible nitric oxide synthase expression by yuccaol C from Yucca schidigera roezl. Life Sci. 2004, 75, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Olas, B.; Wachowicz, B.; Stochmal, A.; Oleszek, W. Inhibition of blood platelet adhesion and secretion by different phenolics from Yucca schidigera roezl. Bark. Nutrition 2005, 21, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, C.; Felice, F.; Piacente, S.; Pizza, C.; Montoro, P.; Oleszek, W.; Visciano, V.; Balestrieri, M.L. Relative effects of phenolic constituents from Yucca schidigera roezl. Bark on kaposi’s sarcoma cell proliferation, migration, and PAF synthesis. Biochem. Pharmacol. 2006, 71, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Wenzig, E.M.; Oleszek, W.; Stochmal, A.; Kunert, O.; Bauer, R. Influence of phenolic constituents from Yucca schidigera bark on arachidonate metabolism in vitro. J. Agric. Food Chem. 2008, 56, 8885–8890. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, R.M.; Mazziotta, A.; de Sant’Ana, D.P.; Palumbo, C.; Gasperi, T. Active methylene compounds in asymmetric organocatalytic synthesis of natural products and pharmaceutical scaffolds. Curr. Org. Chem. 2012, 16, 2231–2289. [Google Scholar] [CrossRef]
- Gross, P.J.; Braese, S. The total synthesis of (±)-fumimycin. Chem. Eur. J. 2010, 16, 12660–12667. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-J.; Sohn, M.-J.; Zheng, C.-J.; Kim, W.-G. Fumimycin: A peptide deformylase inhibitor with an unusual skeleton produced by Aspergillus fumisynnematus. Org. Lett. 2007, 9, 2449–2451. [Google Scholar] [CrossRef] [PubMed]
- Vetica, F.; Pelosi, A.; Gambacorta, A.; Loreto, M.A.; Miceli, M.; Gasperi, T. Catalytic Friedel-Crafts/lactonization domino reaction: Facile access to 3-hydroxybenzofuran-2-one scaffold. Eur. J. Org. Chem. 2014, 2014, 1899–1906. [Google Scholar] [CrossRef]
- Vetica, F.; de Figueiredo, R.M.; Cupioli, E.; Gambacorta, A.; Loreto, M.A.; Miceli, M.; Gasperi, T. First asymmetric organocatalyzed domino Friedel-Crafts/lactonization reaction in the enantioselective synthesis of the GABAB receptor modulator (S)-BHFF. Tetrahedron Lett. 2016, 57, 750–753. [Google Scholar] [CrossRef]
- Dyachenko, V.I.; Kolomiets, A.F.; Fokin, A.V. Preparation of substituted 3-hydroxy-3-trifluoromethyl-2(3H)benzofuranones. Bull. Acad. Sci. USSR Div. Chem. Sci. 1987, 36, 2332–2337. [Google Scholar] [CrossRef]
- Dyachenko, V.I.; Galakhov, M.V.; Kolomiets, A.F.; Fokin, A.V. 2-hydroxy-2-trifluoromethyl-2,3H-1,4-benzoxazinone-3. Bull. Acad. Sci. USSR Div. Chem. Sci. 1988, 37, 1056. [Google Scholar] [CrossRef]
- Prior, R.L.; Cao, G.H. In vivo total antioxidant capacity: Comparison of different analytical methods. Free Radic. Biol. Med. 1999, 27, 1173–1181. [Google Scholar] [CrossRef]
- Sanchez-Moreno, C. Review: Methods used to evaluate the free radical scavenging activity in foods and biological systems. Food Sci. Technol. Int. 2002, 8, 121–137. [Google Scholar] [CrossRef]
- Aruoma, O.I. Methodological considerations for characterizing potential antioxidant actions of bioactive components in plant foods. Mutation Res.-Fundam. Mol. Mech. Mutagen. 2003, 523, 9–20. [Google Scholar] [CrossRef]
- Huang, D.J.; Ou, B.X.; Prior, R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free-radical method to evaluate antioxidant activity. Food Sci. Technol.-Lebensm.-Wiss. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Foti, M.C. Antioxidant properties of phenols. J. Pharm. Pharmacol. 2007, 59, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.C.; Daquino, C.; Geraci, C. Electron-transfer reaction of cinnamic acids and their methyl esters with the DPPH center dot radical in alcoholic solutions. J. Org. Chem. 2004, 69, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.C.; Daquino, C.; Mackie, I.D.; DiLabio, G.A.; Ingold, K.U. Reaction of phenols with the 2,2-diphenyl-1-picrylhydrazyl radical. Kinetics and DFT calculations applied to determine Aro-H bond dissociation enthalpies and reaction mechanism. J. Org. Chem. 2008, 73, 9270–9282. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L.; Banks, J.T.; Ingold, K.U.; Lusztyk, J. Kinetic solvent effects on hydroxylic hydrogen-atom abstractions are independent of the nature of the abstracting radical—2 extreme tests using vitamin-E and phenol. J. Am. Chem. Soc. 1995, 117, 9966–9971. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Abnormal solvent effects on hydrogen atom abstractions. 1. The reactions of phenols with 2,2-diphenyl-1-picrylhydrazyl (DPPH•) in alcohols. J. Org. Chem. 2003, 68, 3433–3438. [Google Scholar] [CrossRef] [PubMed]
- Litwinienko, G.; Ingold, K.U. Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc. Chem. Res. 2007, 40, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Chevion, S.; Roberts, M.A.; Chevion, M. The use of cyclic voltammetry for the evaluation of antioxidant capacity. Free Radic. Biol. Med. 2000, 28, 860–870. [Google Scholar] [CrossRef]
- Chevion, S.; Chevion, M. Antioxidant status and human health—Use of cyclic voltammetry for the evaluation of the antioxidant capacity of plasma and of edible plants. In Reactive Oxygen Species: From Radiation to Molecular Biology: A Festschrift in Honor of Daniel L. Gilbert; Chiueh, C.C., Ed.; New York Academy of Sciences: New York, NY, USA, 2000; Volume 899, pp. 308–325. [Google Scholar]
- Arteaga, J.F.; Ruiz-Montoya, M.; Palma, A.; Alonso-Garrido, G.; Pintado, S.; Rodriguez-Mellado, J.M. Comparison of the simple cyclic voltammetry (CV) and DDPH assays for the determination of antioxidant capacity of active principles. Molecules 2012, 17, 5126–5138. [Google Scholar] [CrossRef] [PubMed]
- Papanikos, A.; Eklund, J.; Jackson, W.R.; Kenche, V.B.; Campi, E.M.; Robertson, A.D.; Jarrott, B.; Beart, P.M.; Munro, F.E.; Callaway, J.K. Cyclic voltammetry as an indicator of antioxidant activity. Aust. J. Chem. 2002, 55, 205–212. [Google Scholar] [CrossRef]
- Chevion, S.; Berry, E.M.; Kitrossky, N.; Kohen, R. Evaluation of plasma low molecular weight antioxidant capacity by cyclic voltammetry. Free Radic. Biol. Med. 1997, 22, 411–421. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 9–11 and 15–20 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R2 | Product | Yield (%) b | ||
| 1 | 1 |  | CO2Et | 9 |  | 70 |
| 2 | 2 |  | CO2Et | 10 |  | 62 |
| 3 | 3 |  | CO2Et | 11 |  | 81 |
| 4 | 4 |  | CO2Et | 12 |  | 4 (22 c) |
| 5 | 5 |  | CO2Et | 13 |  | 7 (18 c) |
| 6 | 6 |  | CO2Et | 14 |  | 3 (19 c) |
| 7 | 1 |  | CF3 | 15 |  | 35 |
| 8 | 2 |  | CF3 | 16 |  | 28 |
| 9 | 3 |  | CF3 | 17 |  | 61 |
| 10 | 4 |  | CF3 | 18 |  | 5 |
| 11 | 5 |  | CF3 | 19 |  | 11 |
| 12 | 6 |  | CF3 | 20 |  | 7 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Sub | Catalyst | mol% | Solvent | Temp (°C) | Time (h) | Product | Yield (%) b |
| 1 | 1 | BF3·Et2O | 30 | CH2Cl2 | r.t | 48 | 15 | 5 |
| 2 | 1 | BF3·Et2O | 30 | THF | 60 | 22 | 15 | n.d. |
| 3 | 1 | AlCl3 | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 4 | 1 | PhCO2H | 30 | CHCl3 | 60 | 72 | 15 | n.d. |
| 5 | 1 | PhCO2H | 100 | CHCl3 | 60 | 24 | 15 | n.d. |
| 6 | 1 | CSA | 30 | CHCl3 | 60 | 24 | 15 | n.d. |
| 7 | 1 | CSA | 100 | Toluene | 100 | 10 | 15 | n.d. |
| 8 | 1 | DBU | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 9 | 1 | AcOH | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 10 | 1 | - | - | AcOH | 120 | 12 | 15 | 45 |
| 11 c | 1 | - | - | AcOH | 120 | 10 | 15 | 55 |
| 12 d | 1 | - | - | AcOH | 120 | 4 | 15 | 80 |
| 13 d | 2 | - | - | AcOH | 120 | 4 | 16 | 72 |
| 14 d | 3 | - | - | AcOH | 120 | 6 | 17 | 86 |
| 15 d | 4 | - | - | AcOH | 120 | 4 | 18 | 78 |
| 16 d | 5 | - | - | AcOH | 120 | 5 | 19 | 66 |
| 17 d | 6 | - | - | AcOH | 120 | 4 | 20 | 76 |
| Entry | Antioxidant | rIC50 (molsantiox.t/molsDPPH∙) | N° DPPH• Reduced b |
|---|---|---|---|
| 1 | Trolox | 0.23 | 2.16 |
| 2 | 9 | 0.31 | 1.63 |
| 3 | 10 | 3.62 | 0.14 |
| 4 | 11 | 2.03 | 0.25 |
| 5 | 15 | 0.22 | 2.28 |
| 6 | 16 | 3.52 | 0.14 |
| 7 | 17 | 1.07 | 0.47 |
| 8 | 18 | 0.24 | 2.07 |
| 9 | 19 | 0.62 | 0.80 |
| 10 | 20 | 0.18 | 2.72 |
| Entry | Antiox. | rIC50 (molsantiox./molsDPPH∙) | N° DPPH• Reduced b |
|---|---|---|---|
| 1 | Trolox | 0.22 | 2.24 |
| 2 | 9 | 4.26 | 0.12 |
| 3 | 10 | 4.12 | 0.12 |
| 4 | 11 | 3.92 | 0.13 |
| 5 | 15 | 1.69 | 0.30 |
| 6 | 16 | 4.47 | 0.11 |
| 7 | 17 | 3.25 | 0.15 |
| 8 | 18 | 0.54 | 0.93 |
| 9 | 19 | 2.23 | 0.22 |
| 10 | 20 | 0.17 | 3.02 |
| Entry | Antiox. | ks (M−1·s−1) b | rIC50 (molsantioxidant/molsDPPH•) | ks (M−1·s−1) b | rIC50 (molsantioxidant/molsDPPH•) |
|---|---|---|---|---|---|
| MeOH | MeOH | ACN | ACN | ||
| 1 | 9 | 3.26 | 0.31 | 8.54 | 4.26 |
| 2 | 15 | 3.77 | 0.22 | 1.26 | 1.69 |
| 3 | 18 | 1.40 | 0.24 | 0.23 | 0.54 |
| 4 | 20 | 0.77 | 0.18 | 2.22 | 0.17 |
| Entry | Compounds | Epox (V) 1 (H2O) | Epox (V) 1 (ACN) |
|---|---|---|---|
| 1 | Trolox | 0.52 | 1.08 |
| 2 | 9 | 0.72 | 1.62 |
| 3 | 10 | 1.13 | 1.44 |
| 4 | 11 | 1.11 | 1.65 |
| 5 | 15 | 0.62 | 1.72 |
| 6 | 16 | 1.01 | 1.92 |
| 7 | 17 | 1.05 | 1.77 |
| 8 | 18 | 0.73 | 0.92 |
| 9 | 19 | 1.03 | 1.88 |
| 10 | 20 | 0.85 | 1.81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, M.; Roma, E.; Rosa, P.; Feroci, M.; Loreto, M.A.; Tofani, D.; Gasperi, T. Synthesis of Benzofuran-2-One Derivatives and Evaluation of Their Antioxidant Capacity by Comparing DPPH Assay and Cyclic Voltammetry. Molecules 2018, 23, 710. https://doi.org/10.3390/molecules23040710
Miceli M, Roma E, Rosa P, Feroci M, Loreto MA, Tofani D, Gasperi T. Synthesis of Benzofuran-2-One Derivatives and Evaluation of Their Antioxidant Capacity by Comparing DPPH Assay and Cyclic Voltammetry. Molecules. 2018; 23(4):710. https://doi.org/10.3390/molecules23040710
Chicago/Turabian StyleMiceli, Martina, Elia Roma, Paolo Rosa, Marta Feroci, M. Antonietta Loreto, Daniela Tofani, and Tecla Gasperi. 2018. "Synthesis of Benzofuran-2-One Derivatives and Evaluation of Their Antioxidant Capacity by Comparing DPPH Assay and Cyclic Voltammetry" Molecules 23, no. 4: 710. https://doi.org/10.3390/molecules23040710