Silyl Ketene Acetals/B(C6F5)3 Lewis Pair-Catalyzed Living Group Transfer Polymerization of Renewable Cyclic Acrylic Monomers


State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(3), 665; https://doi.org/10.3390/molecules23030665
Submission received: 30 January 2018
/
Revised: 11 March 2018
/
Accepted: 14 March 2018
/
Published: 15 March 2018
(This article belongs to the Special Issue Lewis Pair Polymerization for New Reactivity and Structure in Polymer Synthesis)
Abstract
:This work reveals the silyl ketene acetal (SKA)/B(C6F5)3 Lewis pair-catalyzed room-temperature group transfer polymerization (GTP) of polar acrylic monomers, including methyl linear methacrylate (MMA), and the biorenewable cyclic monomers γ-methyl-α-methylene-γ-butyrolactone (MMBL) and α-methylene-γ-butyrolactone (MBL) as well. The in situ NMR monitored reaction of SKA with B(C6F5)3 indicated the formation of Frustrated Lewis Pairs (FLPs), although it is sluggish for MMA polymerization, such a FLP system exhibits highly activity and living GTP of MMBL and MBL. Detailed investigations, including the characterization of key reaction intermediates, polymerization kinetics and polymer structures have led to a polymerization mechanism, in which the polymerization is initiated with an intermolecular Michael addition of the ester enolate group of SKA to the vinyl group of B(C6F5)3-activated monomer, while the silyl group is transferred to the carbonyl group of the B(C6F5)3-activated monomer to generate the single-monomer-addition species or the active propagating species; the coordinated B(C6F5)3 is released to the incoming monomer, followed by repeated intermolecular Michael additions in the subsequent propagation cycle. Such neutral SKA analogues are the real active species for the polymerization and are retained in the whole process as confirmed by experimental data and the chain-end analysis by matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF MS). Moreover, using this method, we have successfully synthesized well-defined PMMBL-b-PMBL, PMMBL-b-PMBL-b-PMMBL and random copolymers with the predicated molecular weights (Mn) and narrow molecular weight distribution (MWD).
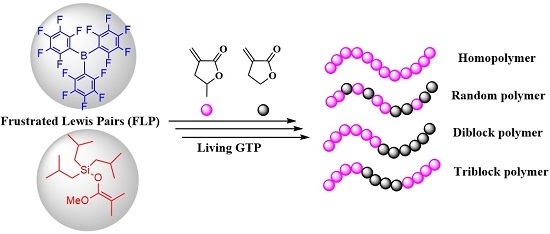
1. Introduction
Lewis pair (LP) polymerization has emerged and attracted intense investigations of the cooperative (or synergistic) catalytic effects of Lewis acid (LA) and Lewis base (LB) pairs on the polymerization of conjugated polar alkenes [1,2,3,4,5,6,7,8], since the seminal works on Frustrated Lewis Pairs (FLPs) by Stephan and Erker in small molecule chemistry [9,10,11,12]. For instance, LPs based on strongly acidic, bulky E(C6F5)3 (E = B, Al) LAs and bulky LBs including phosphines and N-heterocyclic carbenes (NHCs) have been employed to initiate rapid addition polymerization of conjugated polar vinyl monomers such as methyl methacrylate (MMA), cyclic and the naturally renewable monomers α-methylene-γ-butyrolactone (MBL), and γ-methyl-α-methylene-γ-butyrolactone (MMBL) [13,14,15] and monomers bearing the C=C–C=N functionality such as 2-vinylpyridine and 2-isopropenyl-2-oxazoline as well [16,17]. In such polymerizations, the cooperativity of the LA and LB sites of Lewis pairs is essential to achieve an effective polymerization system, which was demonstrated by the borane/phosphine LPs that showed that interacting LPs, and even classical Lewis adducts (CLAs), can be highly active for the polymerization [18]. This was further nicely demonstrated by Rieger and co-workers, showing the high activity and high degree of control over the polymerization of Michael-type and extended Michael monomer systems by the highly interacting organoaluminum and phosphine LPs [19]. Extending beyond the commonly employed NHC and phosphine LBs, in 2014, Lu and co-workers first reported the N-heterocyclic olefin-based LPs for the polymerization of acrylamides, (meth)acrylates and disymmetric divinyl polar monomers as well [20,21]. Although FLPs or CLAs exhibited high activity for polymerization of conjugated polar alkenes, the application of such polymerization is hampered by both low initiation efficiencies and chain-termination side reactions [18,21], evidenced by the much higher observed Mn than the calculated Mn and broad MWD of the resulting polymers (high Ð values), thus giving rise to low initiation efficiencies (I*) and rendering the inability to produce well-defined block copolymers. Based on the above facts, it is really difficult to achieve LP-catalyzed living/controlled polymerization of polar acrylic monomers. More recently, Taton and co-workers reported the direct employment of organic LPs based on phosphine and Me3SiNTf2 for living polymerization of MMA, which proceeds through a similar mechanism compared to the group transfer polymerization (GTP) of MMA [22].
GTP is a successful, commercialized strategy for living polymerization of polar vinyl monomers proceeding through Mukaiyama-Michael reactions [23,24,25]. Such GTP process could be catalyzed by nucleophilic anionic bases, such as SiMe3F2− [23,26,27], HF2− [23,26,27,28], F− [27,28,29], CN− [23,27,28,29], N3− [23,28], oxyanions[30,31], hydrogen bioxyanions [30,31,32]. Recently, neutral Lewis bases such as NHC [33,34,35,36,37,38], phosphorus-based neculeophiles [39,40] and phosphazene superbases [39,41,42,43,44] have been demonstrated be efficient catalysts for living GTP of acrylic monomers. On the other hand, Brønsted acids have been employed as activators for GTP as well [45], while GTP of acrylates catalyzed by Lewis acidic catalysts such as zinc halide [46], mercury iodide [47,48], or organoaluminun [46] requires a much higher catalyst loading (typically 10 mol % based on monomer) to achieve a reasonable degree of polymerization control. Through the oxidative activation of SKAs with a catalytic amount of [Ph3C][B(C6F5)4] (TTPB) as low as 0.025 mol % (based on monomer), we have successfully achieved high-speed living GTP of polar vinyl monomers catalyzed by the silylium ion R3Si+, including (meth)acrylates [49,50] and renewable MBL and MMBL [51]. Covalently linked, unimolecular silyl enolate/silylium nucleophile/electrophile bifunctional active species rendered a rate enhancement by a factor of >40 and high stereoselectivity (at low temperature), as compared to the mononuclear SKA system [52]. More recently, a tandem (FLP and LA) activation method was developed for GTP involving the in-situ generation of SKA initiators by 1,4-hydrosilylation of a methacrylate monomer. In such a process, the catalysts, such as highly electron deficient silylium cations “R3Si+” [53] or strong LAs E(C6F5)3 (E = B, Al) [5,6,54,55], play dual role in both hydrosilylation (via frustrated Lewis Pair (FLP)-type activation) and activation of monomer (classical LA activation). However, such system was shown to be much less effective for E(C6F5)3 (E = B, Al)-catalyzed GTP at high monomer to initiator ratios [6,53] and ill-controlled for Al(C6F5)3-catalyzed MMA polymerization. In this context, herein we report the SKA/B(C6F5)3 FLP-catalyzed GTP of polar vinyl monomers at room temperature, including MMA, renewable MMBL and MBL. More specifically, a highly interacting iBuSKA/B(C6F5)3 LP system promotes the living GTP of MMBL and copolymerization of MMBL and MBL to produce well-defined (co)polymers with predicted molecular weights, and narrow molecular weight distributions. More importantly, this SKA/B(C6F5)3 LP system enabled us to isolate key reaction intermediates and perform kinetic and mechanistic studies, thereby providing the much-needed insights into the polymerization mechanism.
2. Results and Discussion
2.1. Slow MMA Polymerization by R3SiH/B(C6F5)3 and SKA/B(C6F5)3
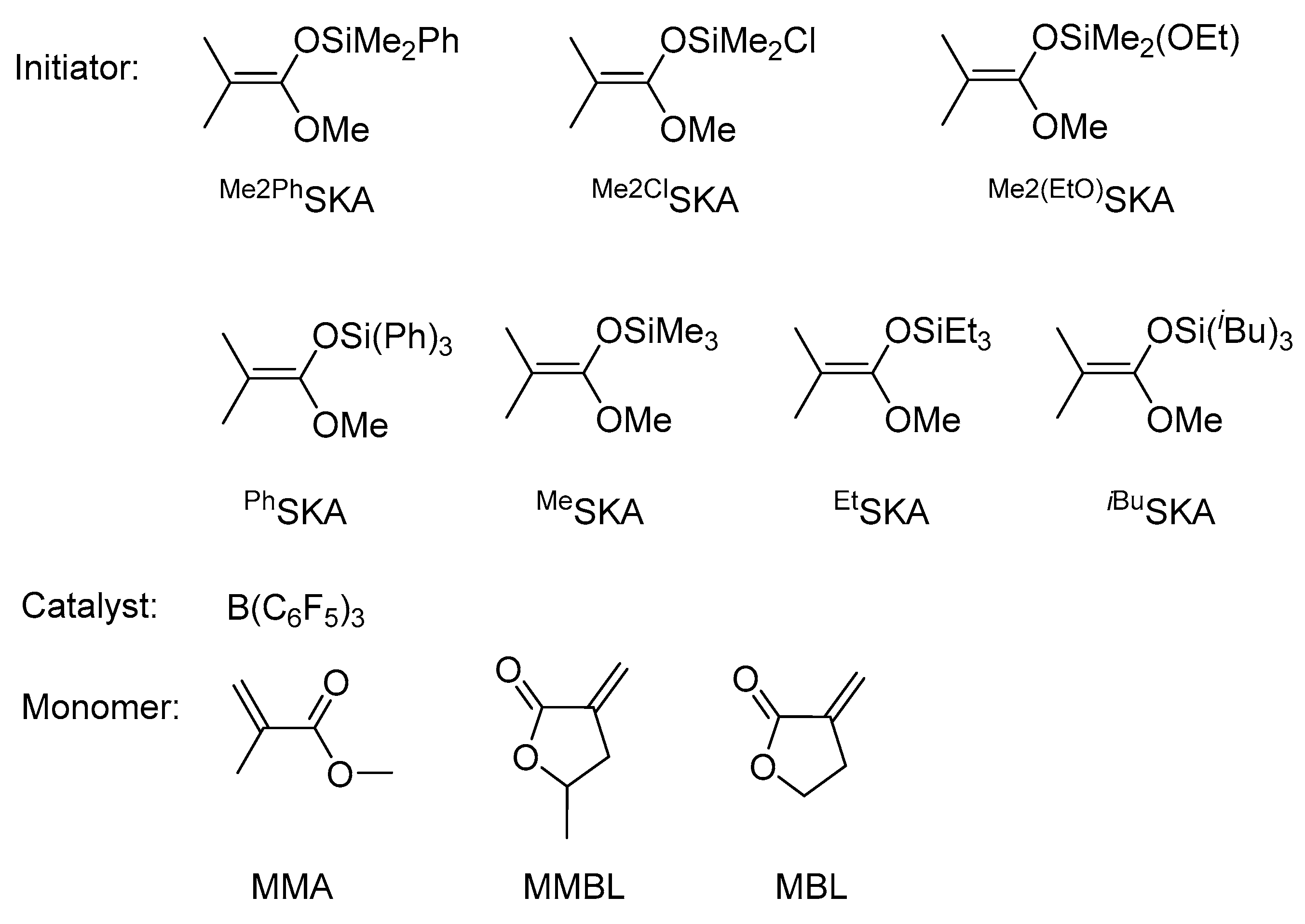
Without B(C6F5)3, all six hydrosilanes, including Et3SiH, Ph3SiH, iBu3SiH, Me2ClSiH, Me2EtSiH, and Me2PhSiH, showed negligible activity for the polymerization of MMA in CH2Cl2 at room temperature. In the presence of B(C6F5)3, the in-situ generation of SKA initiators from the B(C6F5)3-catalyzed hydrosilylation of the monomer MMA with R3SiH exhibited very low polymerization activity and incomplete monomer conversions for various ratios of [MMA]0/[R3SiH]0/[B(C6F5)3]0 for up to 24 h (runs 1–18, Table S1). On the other hand, the direct use of SKA (Scheme 1) as initiator and B(C6F5)3 as catalyst did not lead to obvious enhancement in MMA polymerization activity compared to R3SiH/B(C6F5)3 systems (runs 19–39, Table S1). The corresponding PMMA polymers all possessed syndio-biased tacticities with a methyl triad distribution of around 70% rr, 28% mr and 2% mm, due to the chain-end control nature of this polymerization system.
2.2. Controlled MMBL Polymerization by R3SiH/B(C6F5)3 and SKA/B(C6F5)3
Next, we investigated the effectiveness of the R3SiH/B(C6F5)3 and SKA/B(C6F5)3 systems for renewable monomer MMBL, which is a cyclic analogue of MMA and is readily prepared in a two-step process from the cellulosic biomass-derived levulinic acid. Compared to the MMA polymerization, all R3SiH/B(C6F5)3 and SKA/B(C6F5)3 systems are more effective and controlled for polymerization of MMBL, achieving noticeably better initiation efficiency and narrower molecular weight distribution (Table 1 vs. Table S1). Among the silanes screened, Me2EtSiH and Me2PhSiH achieved quantitative monomer conversion within 30 min (Mn = 48.1 kg·mol−1, Ð = 1.26, I* = 47% for run 1, Table 1; Mn = 57.4 kg·mol-1, Ð = 1.39, I* = 39% for run 2, Table 1). Replacing the Ph group in the R3SiHstructure with the electron-withdrawing Cl (i.e., Me2ClSiH) rendered the polymerization reaching completion in 2 h and producing PMMBL with a Mn of 78.6 kg·mol−1 and a higher Ð value of 1.44 (run 3, Table 1). Interestingly, R3SiH with three Et substituents, Et3SiH, when combined with B(C6F5)3, exhibited comparable polymerization activity with that for Me2EtSiH, producing PMMBL with a Mn of 33 kg·mol−1 and a Ð value of 1.27, thus yielding an enhanced initiation efficiency of 68% (run 4, Table 2). With the increase of steric hindrance of substituents, the corresponding R3SiH exhibited drastically decreased polymerization activity (6 h for Ph3SiH and 24 h for iBu3SiH to reach full monomer conversion) and initiation efficiency (21% for Ph3SiH and 7% for iBu3SiH).
When switching to the SKA/B(C6F5)3 system, both Me2PhSKA/B(C6F5)3 and Me2(EtO)SKA/B(C6F5)3 system achieved quantitative monomer conversion in 30 min and similar initiation efficiency (50% vs. 54%) under our current standard polymerization conditions {[MMBL]0:[SKA]0:[B(C6F5)3]0 = 200:1:1, 0.25 mL MMBL, 2.25 mL CH2Cl2, RT}. Replacing the phenyl group with a chlorine atom, the rate of MMBL polymerization was drastically decreased and quantitative monomer conversion was obtained in 6 h for Me2ClSKA/B(C6F5)3 system, producing PMMBL with a Mn of 85.1 kg·mol−1, a broader Ð value of 1.82 (Table 1, run 9), and thus giving a lower I*% of 27. The combination of PhSKA with B(C6F5)3 produced a polymerization system that is comparable with that of Me2ClSKA/B(C6F5)3 (run 10 vs. 9, Table 1). Interestingly, by replacing the three Ph groups with three electron-donating alkyl groups in SKA, we observed a significantly enhanced initiation efficiency I* (%) (71 for MeSKA, run 11; 61 for EtSKA, run 12; 70 for iBuSKA, run 13).
Since both MeSKA/B(C6F5)3 and iBuSKA/B(C6F5)3 system exhibited the highest initiation efficiency for the polymerization of MMBL (around 70%) under our current condition, therefore, we employed both systems to examine their efficacies for polymerization of the homologues of MMBL, MBL or tulipalin A, which is a natural substance found in tulips and the MBL ring is an integral building block of many natural products. For polymerization with a 200:1:1 [MBL]:[MeSKA]:[B(C6F5)3] ratio, it took 24 h for both MeSKA/B(C6F5)3 and iBuSKA/B(C6F5)3 system to reach complete monomer consumption. However, it should be noted that PMBL produced by MeSKA/B(C6F5)3 system exhibited a bimodal MWD. For polymerization with 100:1:1 [MBL]:[SKA]:[B(C6F5)3] ratio, only 30 min is needed to achieve quantitative monomer conversion for both systems, affording PMBL with predicted Mn and small Ð values (MeSKA: Mn = 12.7 kg·mol−1, Ð = 1.12, I* = 78%, Run 16; iBuSKA: Mn = 13.4 kg·mol−1, Ð = 1.05, I* = 74%, Run 18, Table 1). These results indicated iBuSKA/B(C6F5)3 system exhibited better control on the polymerization than that for MeSKA/B(C6F5)3 system.
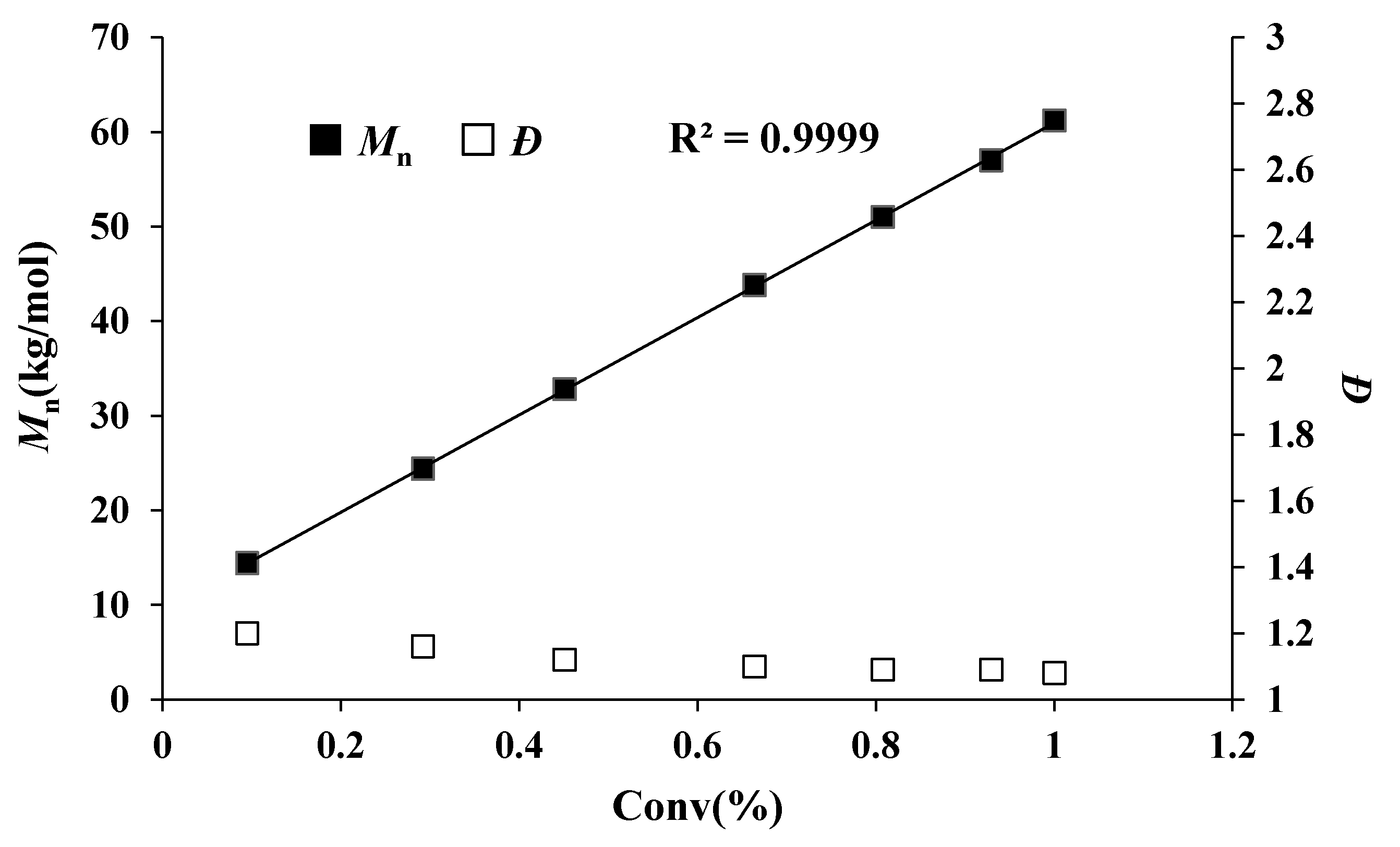
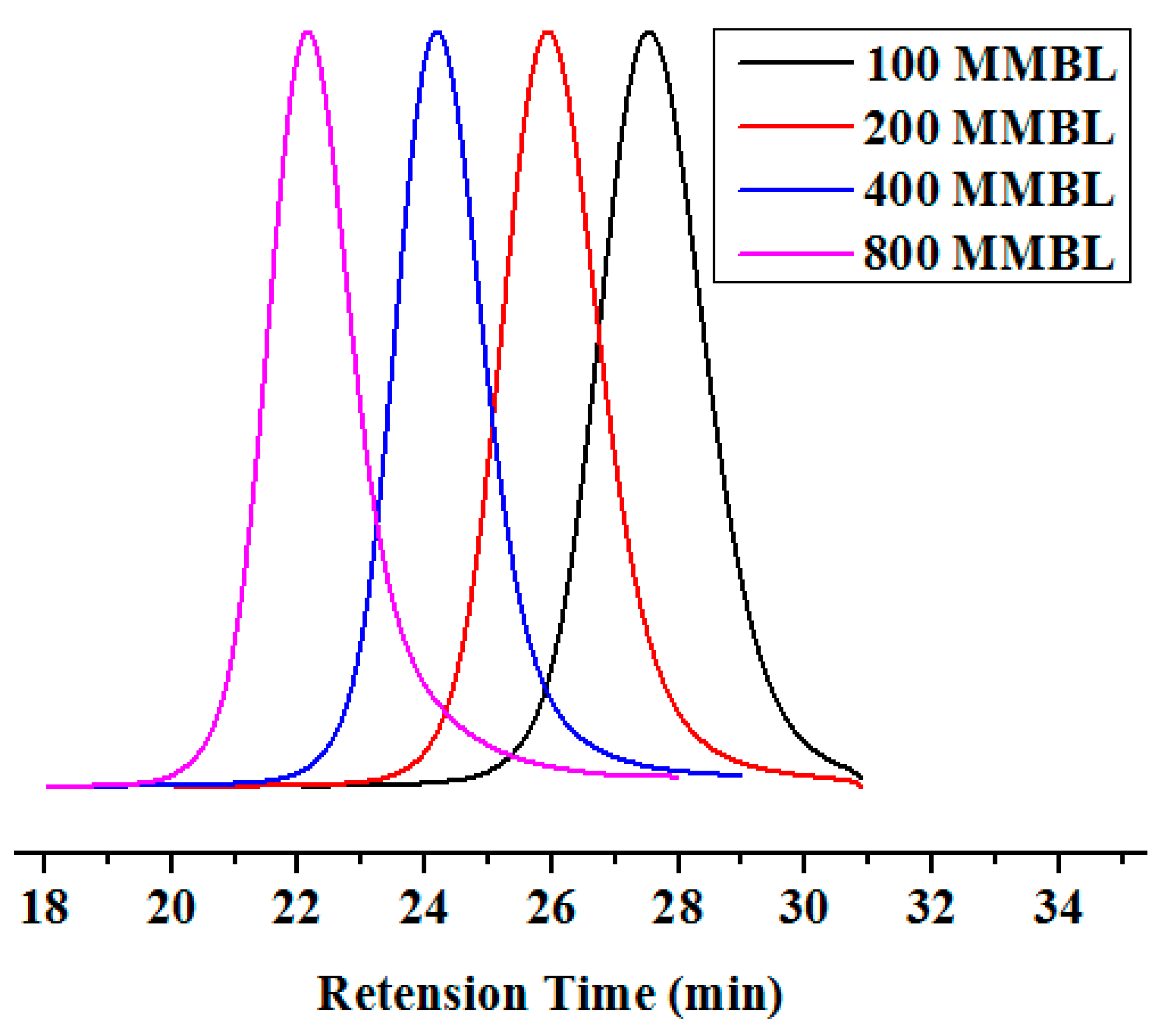
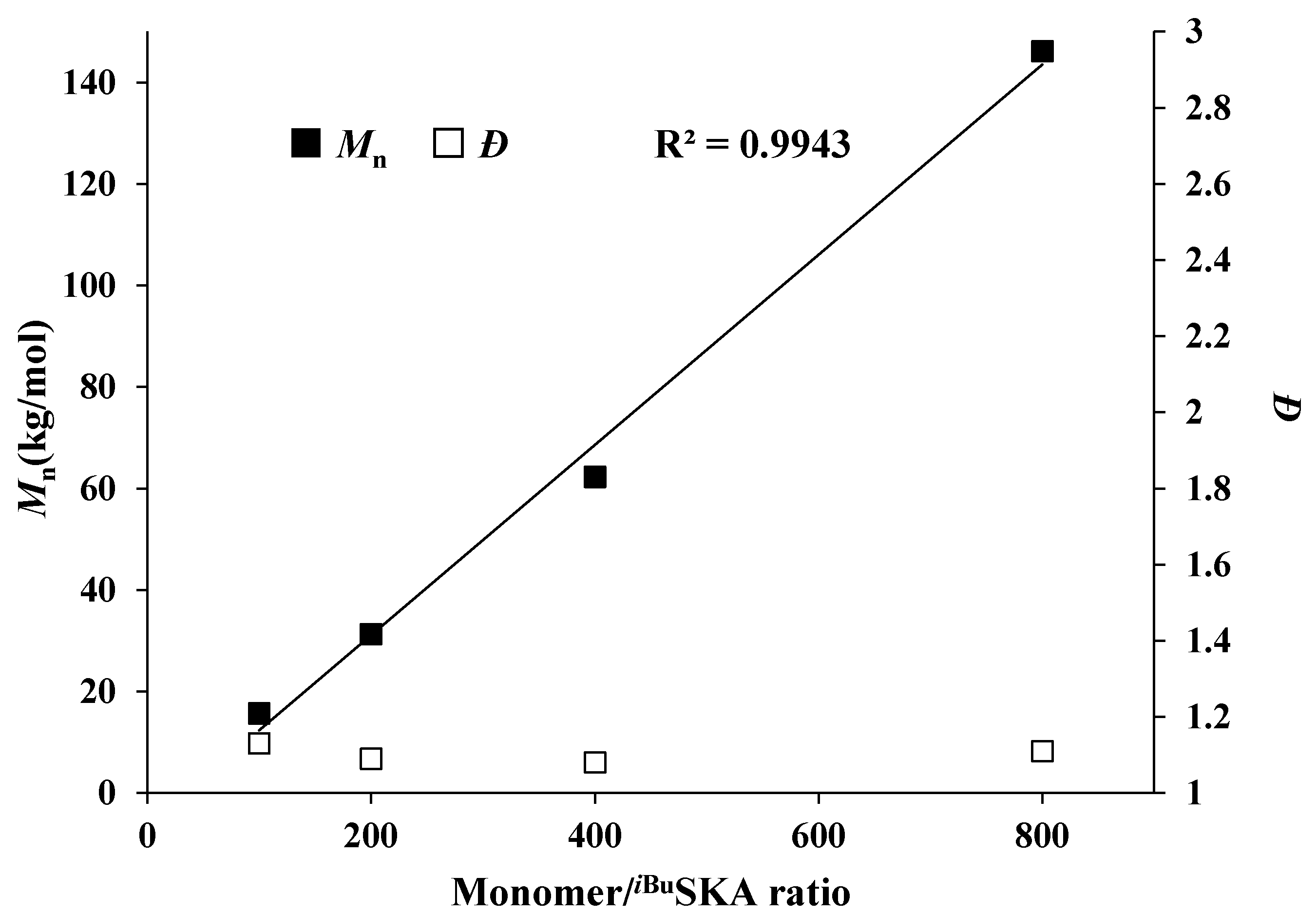
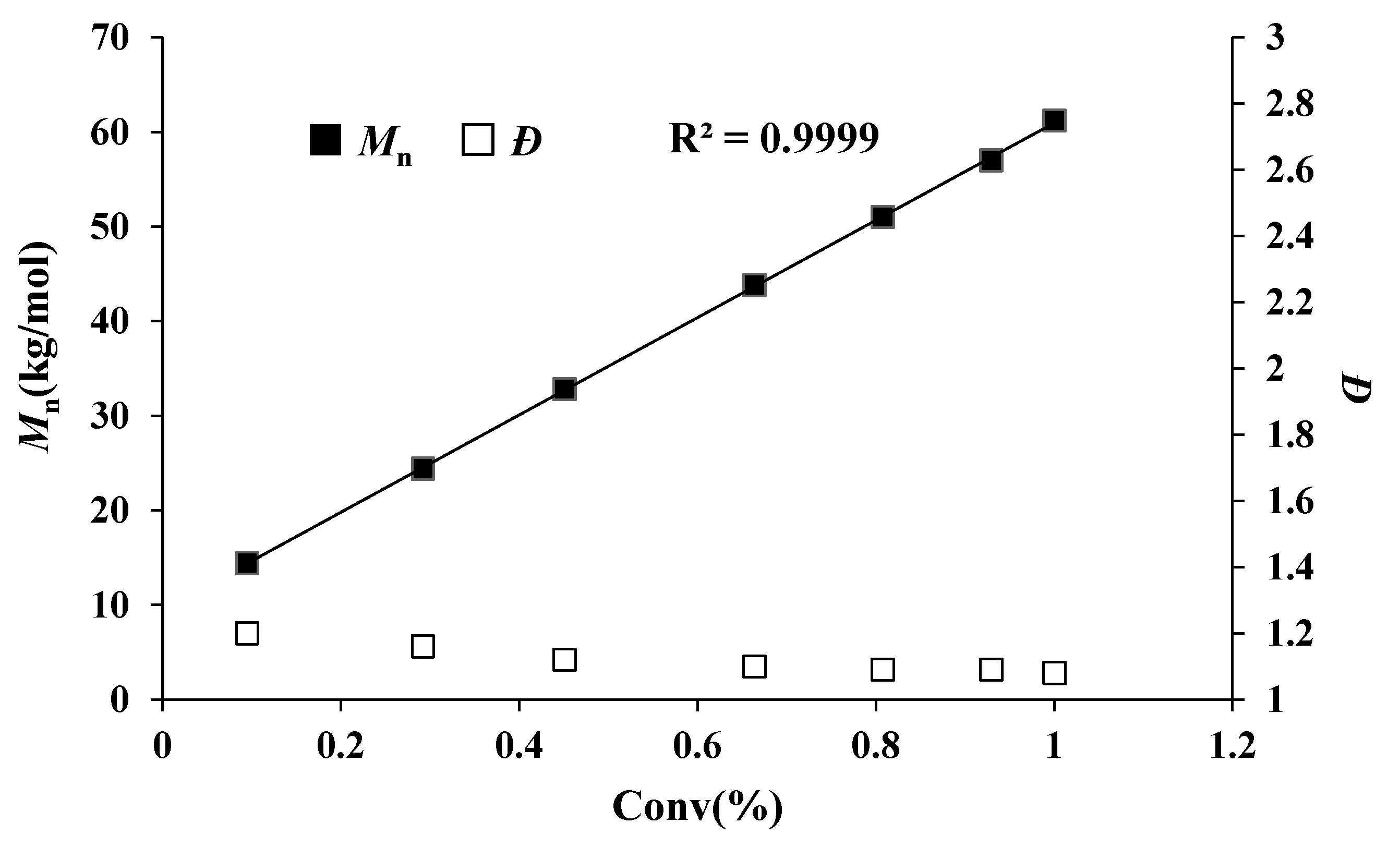
In fact, the MMBL polymerization by iBuSKA/B(C6F5)3 system is living and controlled, and near quantitative monomer conversion was achieved for polymerization with varied [MMBL]/[iBuSKA] ratio from 100 to 800. GPC traces of PMMBL produced by iBuSKA/B(C6F5)3 system also exhibited the gradual shift to the high-molar-mass region with an increase in the [M]/[I] ratio from 100 to 800 and maintained a narrow and unimodal MWD (Figure 1). Although increasing the catalyst loading of B(C6F5)3 would enhance the polymerization rate, the polymer MW is dependent on the concentration of initiator [iBuSKA] and independent on the concentration of catalyst B(C6F5)3 (vide infra), which is evidenced by the linear increase of the Mn values of PMMBL produced by iBuSKA/B(C6F5)3 system with an increase in the [MMBL]/[iBuSKA] ratio from 100 to 800 (Figure 2, R2 = 0.994), while Ð values remained in the very narrow range of 1.08 to 1.13 (Figure 2). It is noted that Mn value of PMMBL obtained for polymerization with different [MMBL]/[B(C6F5)3] ratio maintained around 60 kg·mol−1 (Figure S21), which revealed that the concentration of [B(C6F5)3] has an effect the polymerization rate, but not on both polymer MW and MWD. Therefore, B(C6F5)3 is not the initiator but the catalyst for the activation of monomers. In addition to the aforementioned linearly increased polymer MW with the increase of varied monomer to initiator ratio, the living characteristics of MMBL polymerization by iBuSKA/B(C6F5)3 was also confirmed by a plot of the PMMBL Mn vs monomer conversion at a fixed [MMBL]/[iBuSKA]/[B(C6F5)3] ratio of 400:1:1, which clearly show a straight line (R2 = 0.999) with very narrow Ð value in the range of 1.08–1.2 (Figure 3).
2.3. Characterization of Key Intermediates
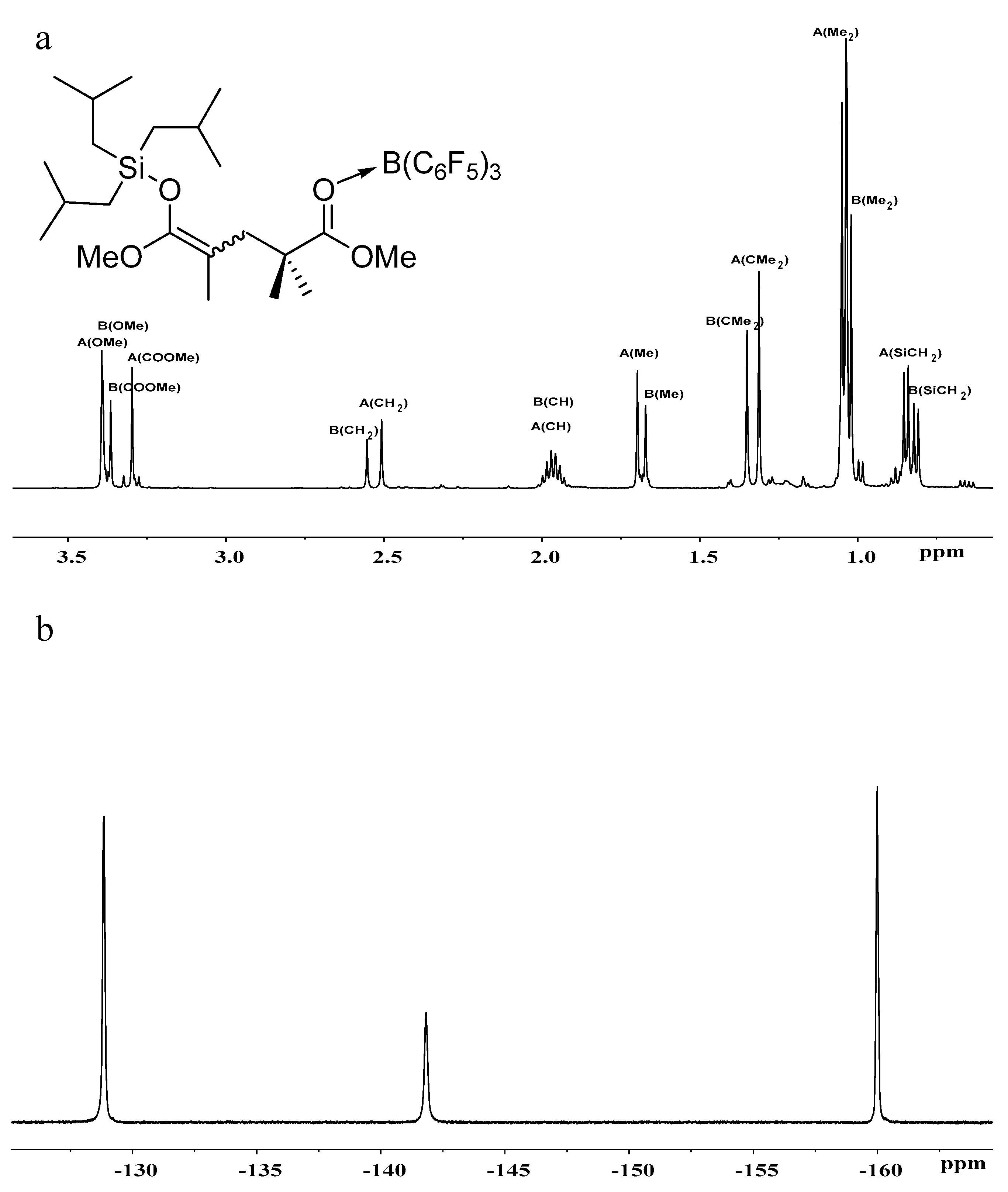
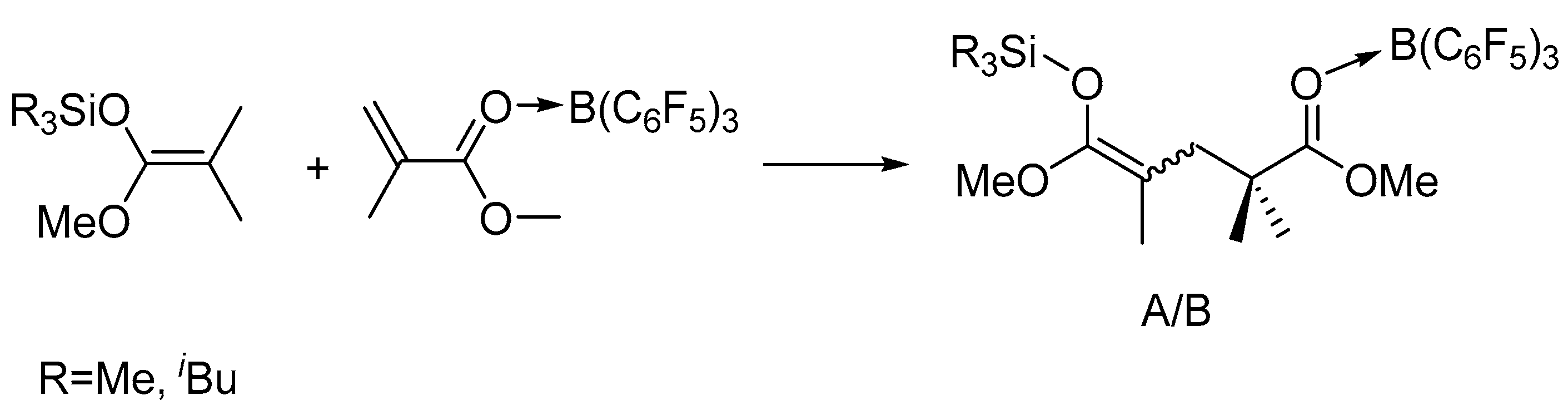
To gain more insights into the polymerization, we studied the in situ NMR reactions of MeSKA with B(C6F5)3 in 1:1 ratio and observed the formation of FLP, which is evidenced by the fact that there is no obvious interaction observed in the reaction of MeSKA with B(C6F5)3 (Figures S11 and S12). Similar FLP was generated in the reaction of iBuSKA with B(C6F5)3 in 1:1 ratio (Figures S13 and S14), too. We also prepared B(C6F5)3·MMA (Figures S7 and S8) and B(C6F5)3·MMBL (Figures S9 and S10) adduct for the study of their reaction with SKA, respectively. The reaction of MeSKA with B(C6F5)3·MMA at room temperature in C6D6 generated major products of dimeric SKA analogue Me3SiO(OMe)C=C(Me)CH2CMe2C(OMe)=O· · ·B(C6F5)3 as two isomers (Z/E) in a 3:2 ratio due to the cis-trans isomerism of MMA (Scheme 2), which could readily derived from the analysis of 1H- and 13C-NMR spectra (Figures S15 and S17).The characteristic proton signals at 0.18 ppm for major isomer and 0.16 ppm for minor isomer attributed to the trimethyl substituents of silyl group in the dimeric SKA analogue is the result of the high field shift from the original 0.18 ppm for neutral MeSKA, which is different from that for Me3Si+ generated by vinylogous hydride abstraction of MeSKA with Ph3C+, exhibiting signal at 0.65 ppm [49]. This result suggests that the silyl group in the dimeric SKA analogue was neutral rather than Me3Si+. In combination with the fact that 19F NMR spectrum exhibiting similar signals [δ −129.03 (br, 6F, o-F), −142.24 (br, 3F, p-F), −160.09 (s, 6F, m-F))] (Figure S16) with that for B(C6F5)3·MMA adduct (Figure S8), we proposed the structure for corresponding dimeric SKA analogues as shown in Scheme 2. Similarly, the reaction of iBuSKA with B(C6F5)3·MMA cleanly afforded the single-monomer-addition product (Scheme 2), as revealed by the 1H-, 19F- and 13C-NMR spectra (Figure 4a,b, and Figure S20). However, we did not observe the formation of such dimeric SKA analogue for reaction of SKA with B(C6F5)3·MMBL probably due to high reactivity of MMBL.
2.4. Kinetics and Mechanism of Polymerization
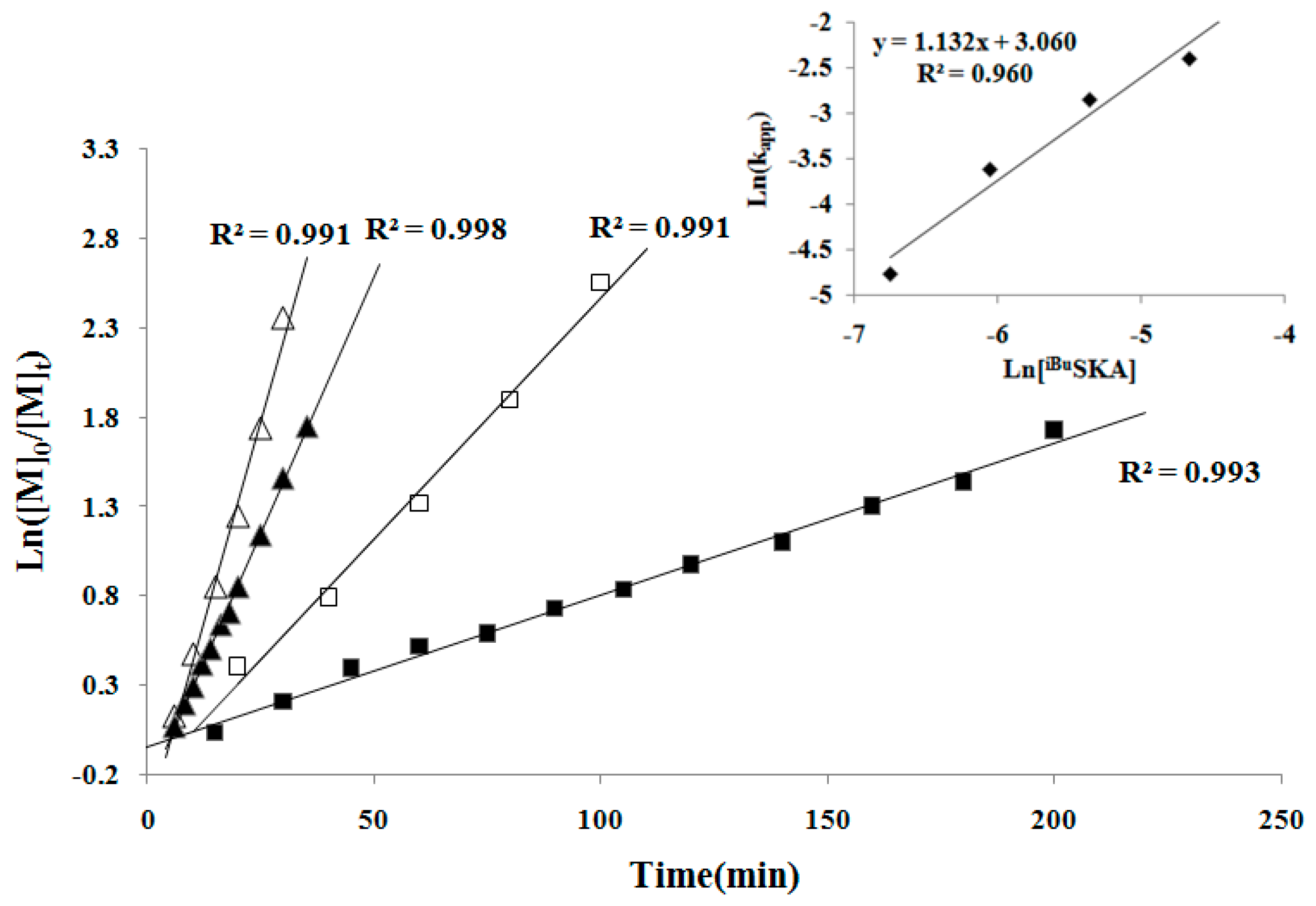
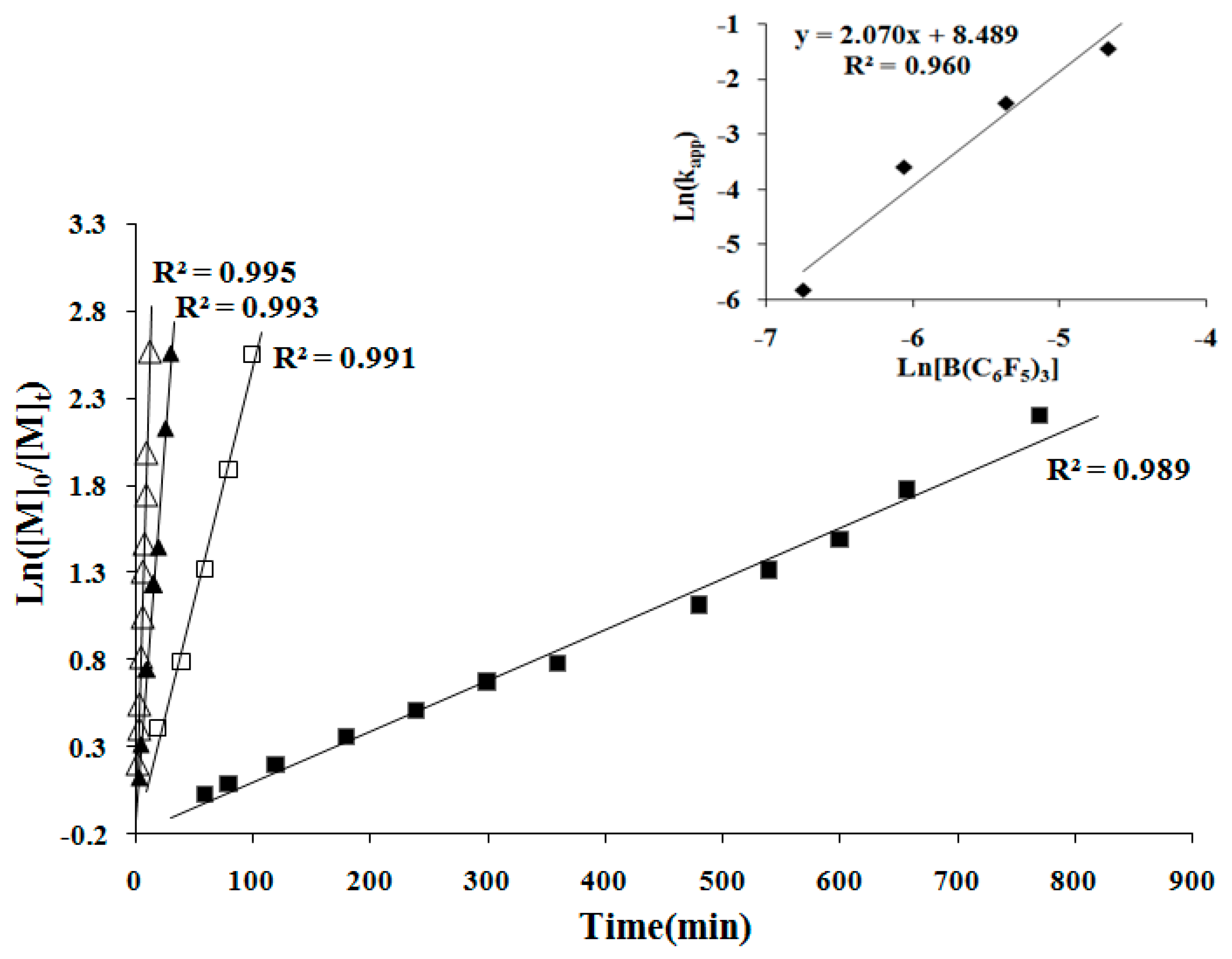
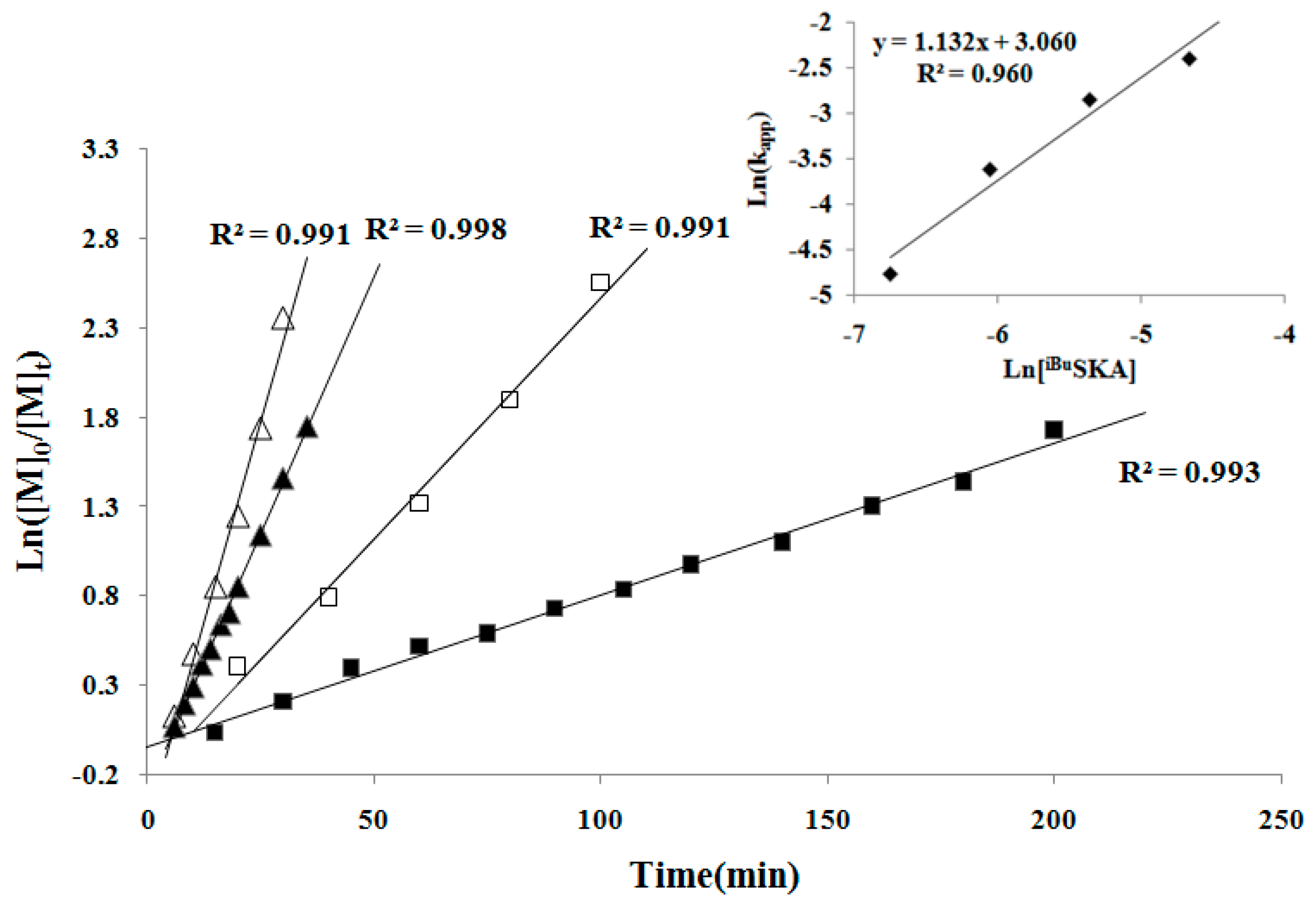
To continue the investigation of the mechanistic aspects of the polymerization, we next examined the kinetics of the MMBL polymerization by the [iBuSKA]/[B(C6F5)3]. The kinetic experiments employed [MMBL]0/[iBuSKA]0/[B(C6F5)3]0 with varied ratio of 400:1:0.5, 400:1:1, 400:1:2, 400:1:4 and a fixed [MMBL]0/[iBuSKA]0 ratio of 400. As can be seen from the representative kinetic plots of Ln([MMBL]0/[MMBL]t) vs. time, the polymerization clearly showed a first-order dependence on [MMBL] for all ratios investigated (Figure 5). Furthermore, a double logarithm plot (insert) of the apparent rate constants (Kapp) obtained from the slopes of the best-fit lines to the plots of Ln([MMBL]0/[MMBL]t) vs. time as a function of ln[B(C6F5)3] was fit to a straight line (R2 = 0.960) with a slop of 2.07. Therefore, the kinetic order with respect to the [B(C6F5)3], given by the slopes of ~2, reveals that the polymerization is second-order in B(C6F5)3 catalyst concentration; these kinetics results suggest a bimolecular propagation.
In the second set of kinetic experiments, with a fixed [MMBL]0/[B(C6F5)3]0 ratio of 400, the kinetic experiments was carried out with varied [MMBL]0/[B(C6F5)3]0/[iBuSKA]0 ratio of 400:1:0.5, 400:1:1, 400:1:2, 400:1:4. The same first-order dependence was observed for all ratios investigated in this study (Figure 6). A double logarithm plot (insert) of the apparent rate constants (kapp), obtained from the slopes of the best-fit lines to the plots of Ln([MMBL]0/[MMBL]t) vs. time, as function of ln[iBuSKA] was fit to a straight line (R2 = 0.960) with a slop of 1.132, revealing that the propagation is first-order in the concentration of [iBuSKA]. Overall, the polymerization by the [iBuSKA]/[B(C6F5)3] systems follows a bimolecular, activated monomer propagation mechanism.
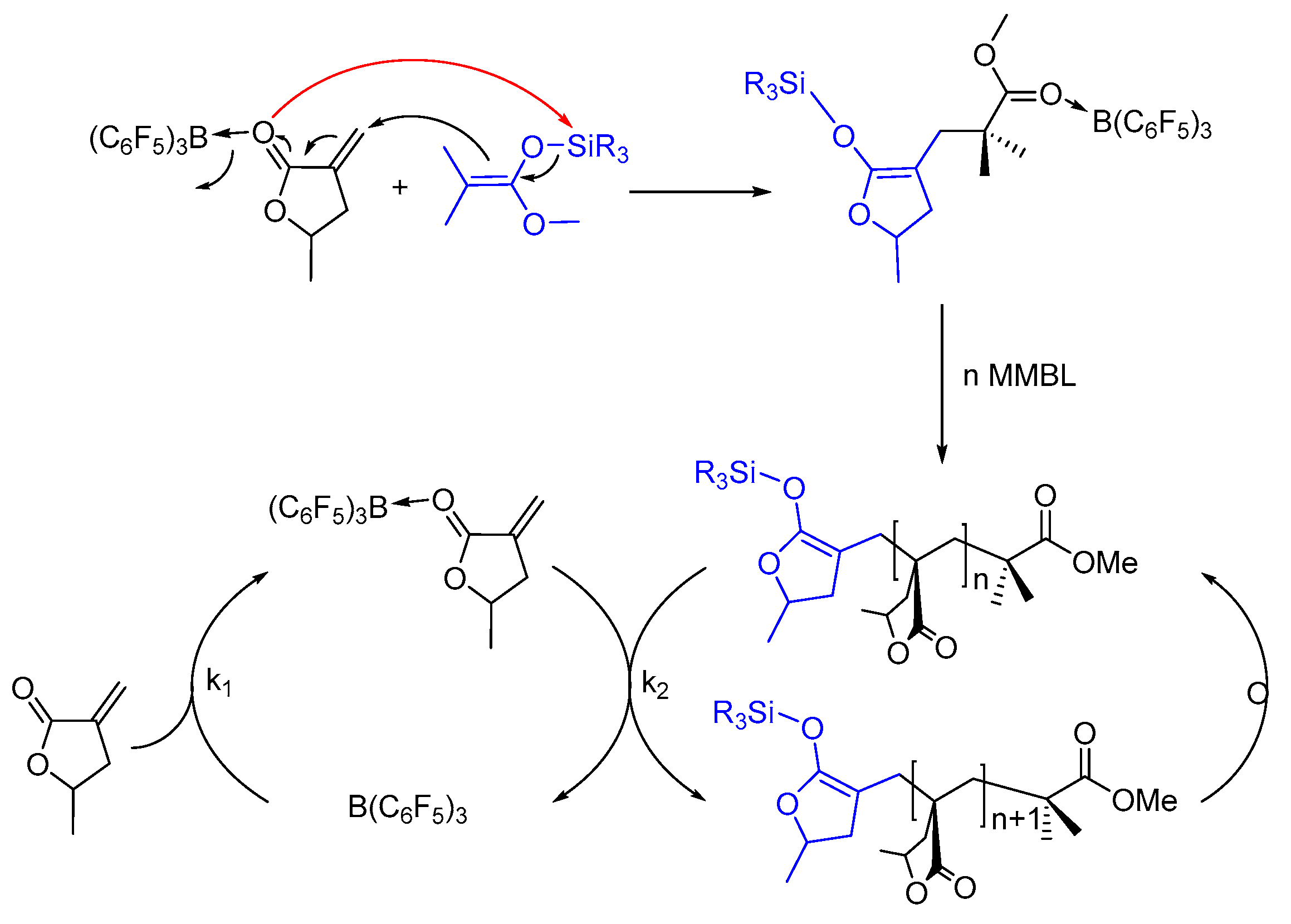
On the basis of the above kinetic results, coupled with mechanistic insights obtained through monitoring the polymerization and characterization of the reaction intermediates (vide supra), we propose the following initiation and propagation mechanism for polymerization of polar vinyl monomers by the SKA/B(C6F5)3 LP system taking MMBL as example (Scheme 3). In this mechanism, the reaction was initiated with the intermolecular Michael addition of the ester enolate groups of the SKA to the vinyl group of B(C6F5)3-activated monomer, meanwhile the Si-O bond of SKA was cleaved and the silyl group was transferred to the carbonyl group of the B(C6F5)3-activated monomer to generate the single-monomer-addition species or the active propagating species. In the propagation cycle, the B(C6F5)3 catalyst is released from the propagating chain to the incoming monomer, followed by the subsequent intermolecular Michael addition to generate the polymeric SKA intermediate (Scheme 3). Our kinetic studies indicated that the polymerization is first-order dependent on both monomer and iBuSKA initiator concentration, but second-order dependent on B(C6F5)3 catalyst concentrations, which revealed that the rate (k2) of the intermolecular Michael addition of the SKA or its homologues to the B(C6F5)3-activated monomer is comparable with that (k1) for the release of the catalyst B(C6F5)3 from the ester group of the growing polymer chain to the incoming monomer for monomer activation (k1 ≈ k2).
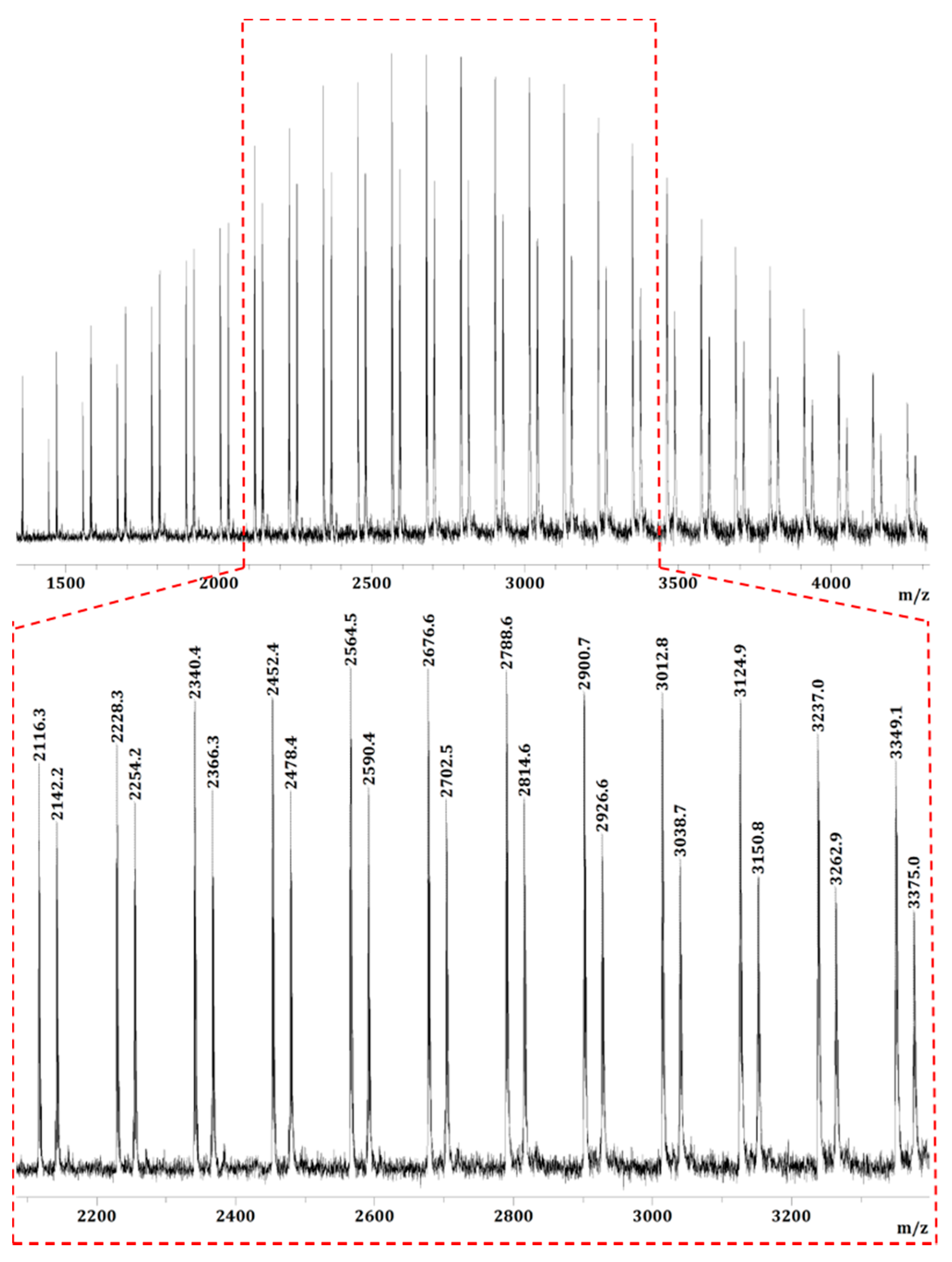
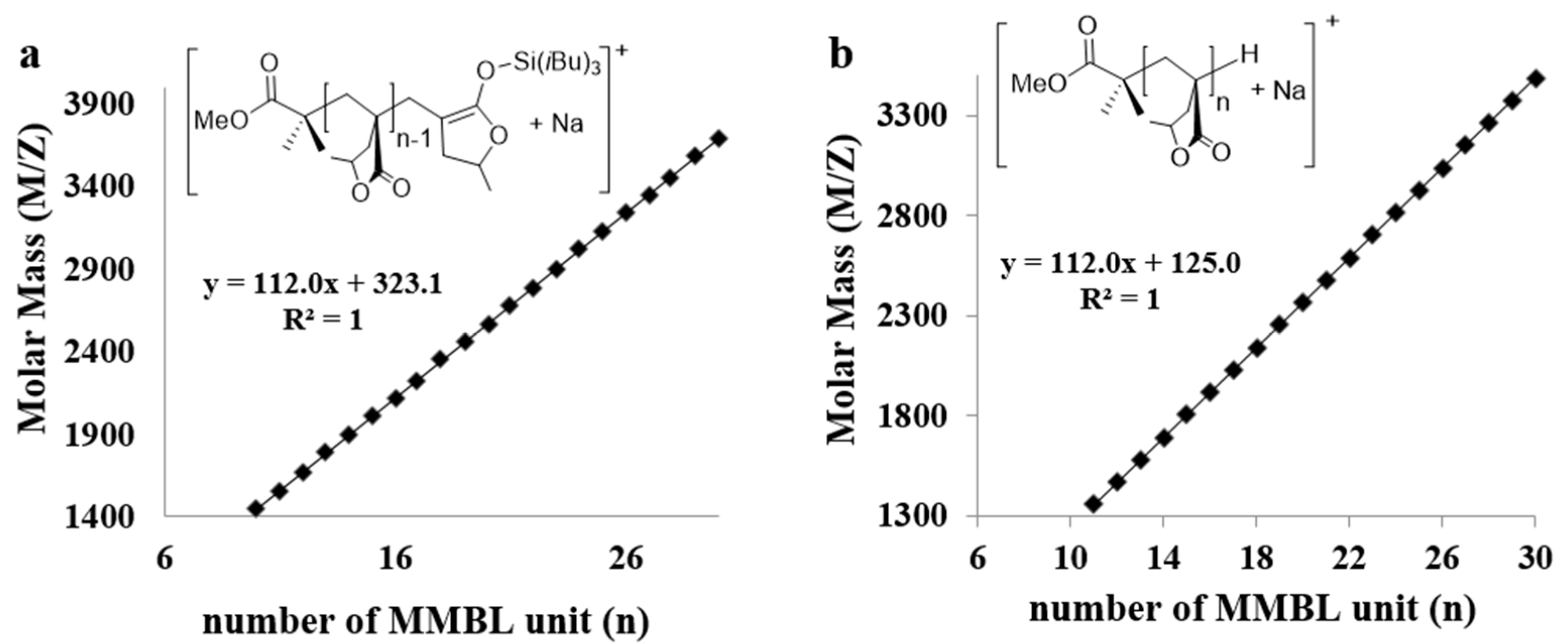
The low MW PMMBL produced by the iBuSKA/B(C6F5)3 system was analyzed by matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF MS). As can be seen from Figure 7, The MS spectrum consisted of two series of molecular mass ions. A plot (Figure 8a) of m/z values for the major series vs the number of MMBL repeat units (n) yielded a straight line with a slope of 112 (mass of MMBL) and an intercept of 323 corresponding to the sum of end groups: MeOC(=O)C(Me)2/Si(iBu)3 + Na+. This analysis yielded a polymer chain structure of MeOC(=O)C(Me)2-(MMBL)n-Si(iBu)3, with both the initiating (the ester enolate group) and termination (the silyl group) chain ends being derived from iBuSKA. Furthermore, a linear plot of m/z values for the minor series in the MS spectrum vs the MMBL repeat units (n) gave the same slope but a different intercept of 125 (Figure 8b), which corresponds to the sum of end groups: MeOC(=O)C(Me)2-(MMBL)n-H + Na+. Formation of such enolate/H chain end is presumably a result of desilylation during the preparation of the MALDI-TOF sample.
2.5. Random and Block Copolymerizations of MMBL with MBL
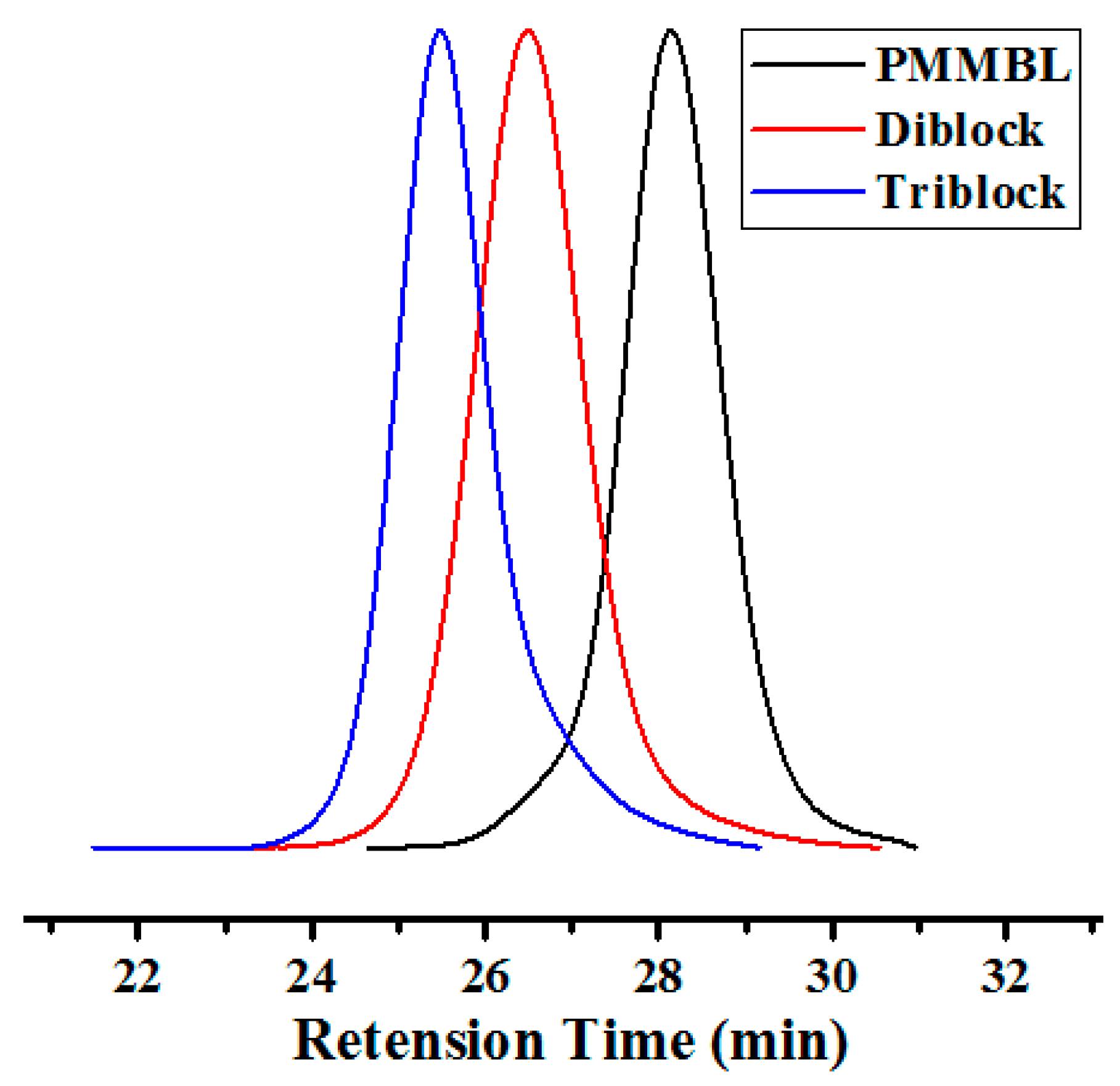
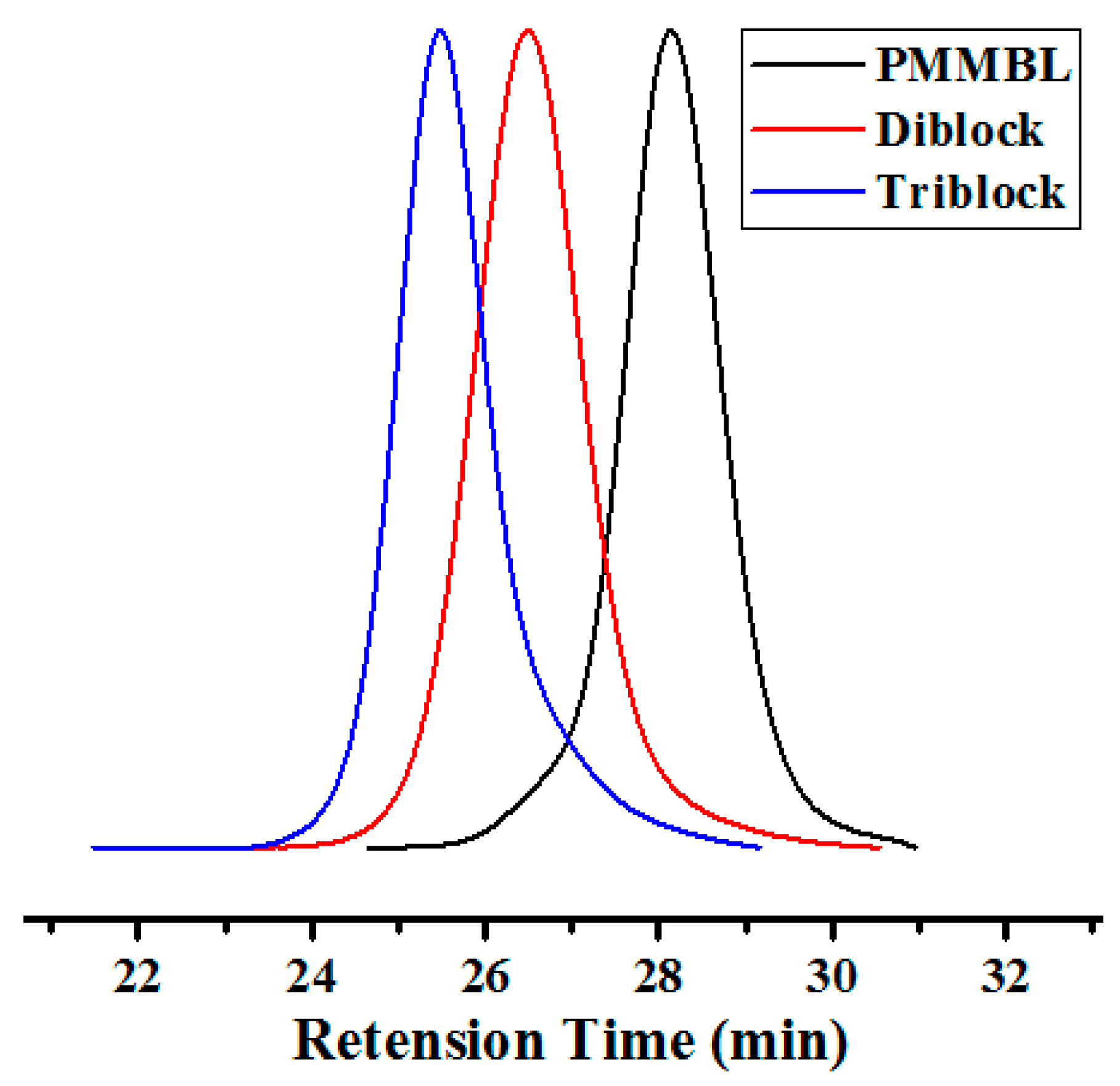
The living features of polymerization of MMBL and MBL by iBuSKA/B(C6F5)3 system, as demonstrated by the above described experiments, also enabled the synthesis of well-defined copolymers. As shown in Table 2, when both monomers were added simultaneously, the copolymerization of MBL and MMBL produced a randomly sequenced copolymer with Mn = 28.3 kg·mol−1 and Ð = 1.03 (run 1, Table 2, Figure S23). On the other hand, sequential block copolymerization by polymerizing MMBL first with [MMBL]/[iBuSKA]/[B(C6F5)3]=100:1:1 without quenching, followed by addition of another 100 equiv. of MBL, successfully afforded linear diblock copolymer PMMBL-b-PMBL. As can be seen from Figure 9, the GPC traces for the PMMBL produced during the initial MMBL polymerization shifted to a higher molecular weight region with low MWD value of 1.07, while the Mn increase from 13.2 kg·mol−1 (black trace) for the homopolymer PMMBL to 25.7 kg·mol−1 (red trace) for the diblock copolymer PMMBL-b-PMBL (run 2, Table 2), which provided further evidence for the formation of well-defined block copolymer by iBuSKA/B(C6F5)3 system. Through the same sequential monomer addition method, well-defined triblock copolymer PMMBL-b-PMBL-b-PMMBL was also successfully prepared with a Mn value of 37.1 kg/mol and a Đ value of 1.06 (Figure 9, blue trace, run 3, Table 2). We also characterized both block copolymers and random copolymers by 13C-NMR analysis (Figure S21). In contrast to diblock copolymer PMMBL-b-PMBL showing two signals corresponding to the PMBL and PMMBL, the yielded random copolymer PMMBL-r-PMBL only exhibited one broad peak in the carbonyl signals, which is ranging between the signals for homopolymers PMBL and PMMBL. The combination of such conclusive 13C NMR results and GPC analyses should provide sufficient evidence for the formation of random and block copolymer from statistical and sequential block polymerization, respectively.
3. Metarials and Methods
3.1. Materials, Reagents and Methods
All syntheses and manipulations of air- and moisture-sensitive materials were carried out in flamed Schlenk-type glassware on a dual-manifold Schlenk line, or an inert gas-filled glovebox. Toluene, benzene, THF and hexane were refluxed over sodium/potassium alloy distilled under nitrogen atmosphere, then stored over molecular sieves 4 Å. CH2Cl2 was refluxed over CaH2 distilled under nitrogen atmosphere, then stored over molecular sieves 4 Å. Benzene-d6 was dried over molecular sieves 4 Å. NMR spectra were recorded using an Avance II 500 (500 MHz, 1H; 126 MHz, 13C; 471 MHz, 19F) instrument (Bruker, Karlsruhe, Baden-Württemberg, Germany) in appropriate deuterated solvents at room temperature. Chemical shifts for 1H and 13C spectra were referenced to internal solvent resonances and are reported as parts per million relative to SiMe4, whereas 19F-NMR spectra were referenced to external CFCl3. Air sensitive NMR samples were conducted in Teflon-valve sealed J. Young-type NMR tubes.
Methyl methacrylate (MMA) was purchased from J&K (Beijing, China), while α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (MMBL) were purchased from TCI (Tokyo, Japan). These monomers were first degassed by three freeze-pump-thaw cycles and dried over CaH2 for 12 h, followed by vacuum distillation. Further purification of MMA involved titration with tri(n-octyl)aluminum (Strem Chemicals, Newburyport, MA, USA) to a yellow end point [56], followed by distillation under reduced pressure. All purified monomers were stored in brown bottles inside a glovebox freezer at −30 °C. Dimethylphenylsilane (Me2PhSiH), and triphenylsilane (Ph3SiH) were purchased from TCI. Triisobutylsilane (iBu3SiH), dimethylethylsilane (Me2EtSiH), dimethylketene methyl trimethylsilyl acetal (MeSKA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Butylated hydroxytoluene (BHT-H, 2,6-di-tert-butyl-4-methylphenol), triethylsilane (Et3SiH), chlorodimethyl-silane (Me2ClSiH), bromopentafluorobenzene were purchased from J&K. Boron trichloride (1.0 M solution in hexanes) and n-BuLi (2.5 M solution in hexanes) were purchased from Energy Chemical (Shanghai, China). Tris(pentafluorophenyl)borane, B(C6F5)3, was prepared according to literature procedures.[57,58] Literature procedures were also employed for the preparation of the following compounds: dimethylketene methyl triethylsilyl acetal (EtSKA) [49], dimethylketene methyl triphenylsilyl acetal Me2C=C(OMe)OSiPh3 (PhSKA) [50], dimethylketene nethyl triisobutylsilyl acetal Me2C=C(OMe)OSi(iBu)3 (iBuSKA) [50],dimethylketene methyl dimethyl-phenylsilyl acetal Me2C=C(OMe)OSiMe3 (Me2PhSKA) [54].
3.2. Synthesis of Dimethylketene Methyl Chlorodimethylsilyl Acetal Me2C=C(OMe)OSiMe2Cl (Me2ClSKA)
In an inert gas-filled glovebox, solution containing B(C6F5)3 (128 mg, 0.250 mmol) and MMA (2.65 mL, 25.0 mmol) in dry CH2Cl2 (50 mL) was charged in a 100 mL Schlenk flask equipped with a stir bar. This flask was sealed with a rubber septum, removed from the glovebox, interfaced to a Schlenk line, and brought to −78 °C for at least 15 min. Chlorodimethylsilane (2.78 mL, 25.0 mmol) was added dropwise to the above flask under nitrogen atmosphere. The resulting reaction mixture was allowed to warm up to room temperature slowly and then subjected to vacuum. The residue was purified by vacuum distillation to afford the final product as colorless oil. Yield: 3.84 g (79%).1H-NMR (500 MHz, benzene-d6) δ 3.35 (s, 3H, OMe), 1.64 (s, 3H, =CMe), 1.61 (s, 3H, =CMe), 0.36 (s, 6H, SiMe2Cl). 13C-NMR (126 MHz, benzene-d6) δ 149.2, 92.8, 57.2, 17.1, 16.4, 2.3.
3.3. Synthesis of Chloro(ethoxy)dimethylsilane Me2(EtO)SiCl
Literature procedures [59] were modified for the preparation of Me2(EtO)SiCl. In an inert gas-filled glovebox, solution of B(C6F5)3 (461 mg, 0.90 mmol) and EtOH (5.08 mL, 90.0 mmol) in dry CH2Cl2 (100 mL) was charged in a 200 mL Schlenk flask equipped with a stir bar. This flask was sealed with a rubber septum, removed from the glovebox, interfaced to a Schlenk line, and brought to −78 °C for at least 20 min. Chlorodimethylsilane(10.0 mL, 90.0 mmol) was added dropwise to the flask under nitrogen atmosphere. The resulting mixture was allowed to warm up to room temperature slowly and then subjected to vacuum. The residue was purified by distillation to afford the product as colorless oil. Yield: 12.48 g (91%).1H-NMR (500 MHz, benzene-d6) δ 3.61 (q, J = 7.0 Hz, 2H, OCH2), 1.06 (t, J = 7.0 Hz, 3H, OCH2CH3), 0.26 (s, 6H, SiMe2). 13C-NMR (126 MHz, benzene-d6) δ 59.1, 18.1, 2.0.
3.4. Synthesis of Dimethylketene Methyl Ethoxydimethylsilyl Acetal Me2C=C-(OMe)OSiMe2(EtO) (Me2(EtO)SKA)
Literature procedures for the general synthesis of ketene trialkylsilyl acetals [60,61] were modified for the preparation of Me2(EtO)SKA. In an inert gas-filled glovebox, a solution of diisopropylamine (7.05 mL, 5.06 g, 50.0 mmol) in dry THF (100 mL) was charged in a 200 mL Schlenk flask equipped with a stir bar. This flask was sealed with a rubber septum, removed from the glovebox, interfaced to a Schlenk line, and placed in a 0 °C ice-water bath for at least 15 min. n-Butyllithium (32.0 mL, 1.6 M in hexane, 51.2 mmol) was added dropwise to the above flask under nitrogen atmosphere. After stirring at 0 °C for 30 min, methyl isobutyrate (5.74 mL, 5.11 g, 50.0 mmol) was added slowly. The reaction mixture was stirred at this temperature for 30 min, after which Chloro(ethoxy)dimethylsilane (7.61 g, 50.0 mmol) was added. The mixture was allowed to warm slowly to room temperature and stirred overnight, after which all volatiles were removed in vacuum and hexanes (50 mL) was added. The resulting precipitates were filtered off under nitrogen atmosphere, the solvent of the filtrate was removed under reduced pressure. The residue was purified by distillation under vacuum to afford the product as colorless oil. Yield: 8.79 g (86%). 1H-NMR (500 MHz, benzene-d6) δ 3.72 (q, J = 7.0 Hz, 2H, OCH2CH3), 3.39 (s, 3H, OMe), 1.72 (s, 3H, =CMe), 1.68 (s, 3H, =CMe), 1.13 (t, J = 7.0 Hz, 3H, OCH2CH3), 0.21 (s, 6H, SiMe2). 13C-NMR (126 MHz, benzene-d6) δ 149.8, 90.7, 58.6, 56.6, 18.5, 17.1, 16.5, −2.5.
3.5. Isolation of Adduct B(C6F5)3·MMA
To a solution of B(C6F5)3 (512 mg, 1.0 mmol) in hexane (15 mL) was added MMA (110 mg, 1.1 mmol) at room temperature with reacting for 30 min. The solution was brought to −30 °C in glovebox freezer for 1 h. After filtration and removal of organic solvents in vacuo, the B(C6F5)3·MMA was obtained as a white powder (546 mg, 89%).1H-NMR (benzene-d6) δ 5.91 (s, 1H, =CH), 5.05–5.03 (s, 1H, =CH), 3.29 (s, 3H, OMe), 1.63 (s, 3H, Me).19F-NMR (benzene-d6) δ −130.18 (d, J = 20.2 Hz, 6F, o-F), −144.90 (t, J = 20.8 Hz, 3F, p-F), −160.70 (m, 6F, m-F).
3.6. Isolation of Adduct B(C6F5)3·MMBL
The B(C6F5)3·MMBL adduct was isolated as a white powder in 91% yield using the same procedure as described for the isolate of the adduct B(C6F5)3·MMA. 1H-NMR (500 MHz, benzene-d6) δ 6.06 (t, J = 3.0 Hz, 1H, =CH), 4.86 (t, J = 2.7 Hz, 1H, =CH), 3.67 (ddq, J = 8.0, 6.3, 5.7 Hz, 1H, OCH), 1.56 (ddt, J = 17.3, 8.0, 2.7 Hz, 1H, CH2), 1.10 (ddt, J = 17.3, 5.7, 2.9 Hz, 1H, CH2), 0.26 (d, J = 6.3 Hz, 3H, Me). 19F- NMR (471 MHz, benzene-d6) δ −134.81 (dd, J = 23.2, 7.3 Hz, 6F, o-F), −156.72 (t, J = 20.7 Hz, 3F, p-F), −163.93 (m, 6F, m-F).
3.7. NMR Reaction of Me2C=C(OMe)OSiMe3 (MeSKA) with B(C6F5)3
In an inert gas-filled glovebox, solution of MeSKA (1.74 mg, 0.01 mmol) in 0.3 mL of C6D6 was charged in a Teflon-valve-sealed J. Young-type NMR tube. A 0.3 mL C6D6 solution of B(C6F5)3 (5.12 mg, 0.01 mmol) was added to this tube via pipet at room temperature and allowed to react for ~15 min before the NMR spectra were recorded, which showed there were no reaction between MeSKA and B(C6F5)3. 1H-NMR (500 MHz, benzene-d6) δ 3.33 (s, 3H, OMe), 1.73 (s, 3H, =CMe), 0.65 (s, 3H, =CMe), 0.18 (s, 9H, SiMe3). 19F-NMR (471 MHz, benzene-d6) δ −128.82 (d, 21.7 Hz, 6F, o-F), −141.81 (t, J = 21.3 Hz, 3F, p-F), −160.06 (m, 6F, m-F).
3.8. NMR Reaction of MeSKA with B(C6F5)3·MMA
In an inert gas-filled glovebox, solution of MeSKA (1.74 mg, 0.01 mmol) in 0.3 mL of C6D6 was charged in a Teflon-valve-sealed J. Young-type NMR tube. A 0.3 mL C6D6 solution of B(C6F5)3·MMA (6.12 mg, 0.01 mmol) was slowly added to this tube via pipet at room temperature and allowed to react for ~15 min before the NMR spectra were recorded. The resulting mixture showed the clean formation of the species Me3SiO(OMe)C=C(Me)CH2CMe2C(OMe)=O···B(C6F5)3 (1) as two isomers (Z/E) in 3:2 ratio: major isomer 1A and minor isomer 1B, plus a small amount of unreacted starting materials. 1A: 1H-NMR (500 MHz, benzene-d6) δ 3.395 (s, 3H, OMe), 3.25 (s, 3H, COOMe), 2.53(s, 2H, CH2), 1.65 (s, 3H, Me), 1.31 (s, 6H, Me2), 0.18 (s, 9H, SiMe3); 1B: δ 3.392 (s, 3H, OMe), 3.30 (s, 3H, COOMe), 2.46 (s, 2H, CH2), 1.72 (s, 3H, Me), 1.32 (s, 6H, Me2), 0.16 (s, 9H, SiMe3); 19F-NMR (471 MHz, benzene-d6) δ −129.03 (br, 6F, o-F), −142.24 (br, 3F, p-F), −160.09 (s, 6F, m-F). 13C-NMR (126 MHz, benzene-d6) δ 178.30, 178.2, 153.3, 152.2, 149.4, 147.4, 138.7, 136.7, 92.1, 91.6, 57.2, 55.6, 51.4, 51.3, 43.14, 43.05, 42.5, 42.4, 25.8, 16.5, 15.4, 0.3, 0.1.
3.9. NMR Reaction of Me2C=C(OMe)OSi(iBu)3 (iBuSKA) with B(C6F5)3
This reaction was carried out in the same manner as the reaction of MeSKA with B(C6F5)3, which shown there were no reaction between iBuSKA and B(C6F5)3. 1H-NMR (500 MHz, benzene-d6) δ 3.38 (s, 3H, OMe), 1.98 (sept, J = 6.6 Hz, 3H, CHMe2), 1.70 (s, 3H, =CMe), 1.69 (s, 3H, =CMe), 1.05 (d, J = 6.6 Hz, 18H, CHMe2), 0.84 (d, J = 6.8 Hz, 6H, SiCH2). 19F-NMR (471 MHz, benzene-d6) δ −128.82 (d, J = 21.7, 6F, o-F), −141.83 (t, J = 20.9 Hz, 3F, p-F), −160.07 (m, 6F, m-F).
3.10. NMR Reaction of iBuSKA with B(C6F5)3·MMA
This reaction was carried out in the same manner as the reaction of MeSKA with B(C6F5)3·MMA, forming cleanly iBu3SiO(OMe)C=C(Me)CH2CMe2C(OMe)=O···B(C6F5)3 (3) as two isomers (Z/E) in 4:3 ratio: major isomer 2A and minor isomer 2B. 2A: 1H-NMR (500 MHz, benzene-d6) δ 3.401 (s, 3H, OMe), 3.30 (s, 3H, COOMe), 2.52 (s, 2H, CH2), 1.98 (m, 3H, CHMe2), 1.71 (s, 3H, Me), 1.32 (s, 6H, CMe2), 1.05 (d, J = 6.5 Hz, 18H, CHMe2), 0.85 (d, J = 6.9 Hz, 6H, SiCH2); 2B: δ 3.397 (s, 3H, OMe), 3.37 (s, 3H, COOMe), 2.56 (s, 2H, CH2), 1.98 (m, 3H, CHMe2), 1.68 (s, 3H, Me), 1.36 (s, 6H, CMe2), 1.04 (d, J = 6.8 Hz, 18H, CHMe2), 0.82 (d, J = 6.9 Hz, 6H, SiCH2); 19F-NMR (471 MHz, benzene-d6) δ −128.84 (br, 6F, o-F), −141.82 (br,3F,p-F), −159.99 (m, 6F, m-F). 13C-NMR (126 MHz, benzene-d6) δ 149.4, 147.4, 138.7, 136.7, 92.5, 58.6, 56.9, 51.3, 51.3, 43.0, 42.4, 42.3, 26.8, 26.7, 26.5, 26.4, 25.9, 25.7, 24.5, 16.6, 15.1.
3.11. General Polymerization Procedures
Polymerizations were performed in 20 mL glass reactors inside the inert gas-filled glovebox for ambient temperature (ca. 25 °C) runs. In a typical procedure, a predetermined amount of B(C6F5)3 and monomer (1 mL for MMA, 250 μL for MMBL or 205 μL for MBL, 200 equiv. relative to the SKA) were dissolved in dry CH2Cl2. A solution of a SKA (1 equiv. of a LA) in 1.0 mL of solvent were added rapidly via a gastight syringe to the solution of B(C6F5)3-monomer. The amount of the monomer was fixed for all polymerizations. After stirring for the measured time, a 0.1 mL aliquot was withdrawn from the reaction mixture and quickly quenched into 0.6 mL of undried “wet” CDCl3 stabilized by 250 ppm of BHT-H; the quenched aliquots were later analyzed by 1H-NMR to obtain the monomer conversion. After the polymerization was stirred for the stated reaction time then the reactor was taken out of the glovebox, and quenched by addition of 10 mL of 5% HCl-acidified methanol. The quenched mixture was isolated by filtration and dried in vacuo overnight at room temperature to a constant weight.
3.12. Polymerization Kinetics
Kinetic experiments were carried out in a stirred glass reactor at ambient temperature (ca. 25 °C) inside an inert gas-filled glovebox using the polymerization procedure already described above, with the [B(C6F5)3]/[iBuSKA] ratio was 0.5/1, 1/1, 2/1, 4/1, [MMBL]0 was fixed at 0.936 M and [iBuSKA]0 was fixed at 2.34 mM, where [B(C6F5)3] = 1.17, 2.34, 4.68, 9.36 mM in 2.5 mL mixture solutions. At appropriate time intervals, 0.1 mL aliquots were taken from the reaction mixture and quickly quenched into 0.6 mL of undried “wet” CDCl3 mixed with 250 ppm BHT-H. The quenched aliquots were analyzed by 1H-NMR for determining the [MMBL]t at a given time t and Ln([MMBL]0: [MMBL]t). Apparent rate constants (kapp) were extracted from the slopes of the best fit lines to the plots of Ln([MMBL]0: [MMBL]t) vs. time. Another set of kinetic experiments were carried out to determine the kinetic order with respect to [iBuSKA]. In these experiments, with the [B(C6F5)3]/[iBuSKA] ratio was 1/0.5, 1/1, 1/2, 1/4, [MMBL]0 was fixed at 0.936 M and [B(C6F5)3]0 was fixed at 2.34 mM, where [iBuSKA] = 1.17, 2.34, 4.68, 9.36 mM in 2.5 mL mixture solutions. The rest of the procedure was same as the described above.
3.13. Polymer Characterizations
Number-average molecular weight (Mn) and molecular weight distributions (Ð = Mw/Mn) of polymers were measured by gel permeation chromatography (GPC) at 35 °C and a flow rate of 1 mL/min, with DMF (HPLC grade, containing 50 mmol/L LiBr) as an eluent on a Waters 1515 instrument (Milford, MA, USA) equipped with a Waters 4.6 × 30 mm guard column and three Waters WAT054466, WAT044226, WAT044223 columns (Polymer Laboratories, Milford, MA, USA; linear range of molecular weight = 500 to 4 × 106). The instrument was calibrated with 10 PMMA standards, and chromatograms were processed with Waters Breeze 2 software (version: 6.01.2154.026; Milford, MA, USA). Tacticities of PMMA was measured by 1H-NMR in CDCl3, while 13C-NMR of P(M)MBL (co)polymers were recorded in DMSO-d6.
The isolated low-MW polymer samples were analyzed by matrix-assissted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS); the experiment was performed on a BrukerAutoflex speed TOF/TOF mass spectrometer (Karlsruhe, Baden-Württemberg, Germany) in linear, positive ion, reflector mode using a 25 KV accelerating voltage. A thin layer of 1% CF3COONa solution was first deposited on the target plate, followed by 0.6 μL of both sample and matrix (trans-2-[3-(4-tertbutylphenyl)-2-methyl-2-propylidene]malonitrile (DCTB), 20 mg/mL in THF). The spectra of the sample was externally calibrated using a peptide calibration mixture (4–6 peptides). The raw data were processed in the Bruker Daltonics FlexAnalysis 3.3.80.0 software (Karlsruhe, Baden-Württemberg, Germany).
4. Conclusions
In summary, FLPs based on bulky organo-LA, B(C6F5)3, and SKAs either preformed or in-situ generated from the hydrosilylation with R3SiH, were employed for GTP of polar vinyl monomers, including linear MMA as well as the cyclic renewable monomers MMBL and MBL. Although the polymerizations of MMA by both R3SiH/B(C6F5)3 and SKA/B(C6F5)3 systems are sluggish, SKA/B(C6F5)3 system promotes the living/controlled GTP of MMBL and MBL, achieving polymers with predicted Mn values and narrow Đ values.
The formation of FLP was confirmed by in situ NMR reactions of SKA with B(C6F5)3. Detailed investigations into the characterization of key reaction intermediates, polymerization kinetics and polymer structures have led to a GTP polymerization mechanism, in which the reaction was initiated with the intermolecular Michael addition of the SKA enolate group to vinyl group of B(C6F5)3-activated monomer, while the silyl group is transferred to the carbonyl group of the monomer to generate the SKA analogues and release B(C6F5)3 which reenters the next catalytic cycle for the activation of incoming monomer. The living features of such iBuSKA/B(C6F5)3FLP system enabled us to successfully synthesized well-defined diblock, triblock copolymer PMMBL-b-PMBL and PMMBL-b-PMBL-b-PMMBL with predicted Mn by using such iBuSKA/B(C6F5)3 FLP system.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 21422401, 21374040, 21774042), 1000 Young Talent Plan of China funds, Talents Fund of Jilin Province, and the startup funds from Jilin University.
Author Contributions
Lu Hu carried out the synthesis of all silyl ketene acetals (SKAs), and SKA/B(C6F5)3 Lewis Pair-catalyzed (co)polymerization of all monomers, investigation of polymerization kinetics, characterization through in situ NMR reaction and MALDI-TOF MS analyses and draft the manuscript. Wuchao Zhao measured both the number average molecular weight (Mn) and molecular weight distributions (Ð = Mw/Mn) of yielded polymers by GPC. Jianghua He analyzed and summarized all data. Yuetao Zhang supervised the whole research work, and finalized the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xu, P.F.; Xu, X. Homoleptic Rare-Earth Aryloxide Based Lewis Pairs for Polymerization of Conjugated Polar Alkenes. ACS Catal. 2018, 8, 198–202. [Google Scholar] [CrossRef]
- Xu, P.F.; Yao, Y.M.; Xu, X. Frustrated Lewis Pair-Like Reactivity of Rare-Earth Metal Complexes: 1,4-Addition Reactions and Polymerizations of Conjugated Polar Alkenes. Chem. Eur. J. 2017, 23, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, Y.; Takasu, A.; Matsuoka, S.; Hayashi, M. N-Heterocyclic Carbene Initiated Anionic Polymerization of (E,E)-Methyl Sorbate and Subsequent Ring-Closing to Cyclic Poly(alkyl sorbate). J. Am. Chem. Soc. 2017, 139, 15005–15012. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.R.; Chen, E.Y.-X. Chemoselective Lewis Pair Polymerization of Renewable Multivinyl-Functionalized γ-Butyrolactones. Phil. Trans. R. Soc. A 2017, 375, 20170003. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Chen, Y.; Kitano, K.; Sato, S.; Satoh, T.; Kakuchi, T. B(C6F5)3-Catalyzed Group Transfer Polymerization of N,N-Disubstituted Acrylamide Using Hydrosilane: Effect of Hydrosilane and Monomer Structures, Polymerization Mechanism, and Synthesis of α-End-Functionalized Polyacrylamides. Macromolecules 2016, 49, 3049–3060. [Google Scholar] [CrossRef]
- Chen, J.W.; Chen, E.Y.-X. Elusive Silane-Alane Complex [Si-H···Al]: Isolation, Characterization, and Multifaceted Frustrated Lewis Pair Type Catalysis. Angew. Chem. Int. Ed. 2015, 54, 6842–6846. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Chen, E.X.-Y. Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules 2015, 20, 9575–9590. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Chen, E.X.-Y. Lewis Pair Polymerization of Acrylic Monomers by N-Heterocyclic Carbenes and B(C6F5)3. Israel J. Chem. 2015, 55, 216–225. [Google Scholar] [CrossRef]
- Stephan, D.W. The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, 1248. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis Pairs: From Concept to Catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pairs I & II. In Topics in Current Chemistry; Springer: New York, NY, USA, 2013; Volume 332, 334. [Google Scholar]
- Chen, E.Y.-X. Polymerization by Classical and Frustrated Lewis Pairs. In Frustrated Lewis Pairs II; Erker, G., Stephan, D.W., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 334, pp. 239–260. [Google Scholar]
- Zhang, Y.T.; Miyake, G.M.; John, M.G.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Lewis pair polymerization by classical and frustrated Lewis pairs: Acid, base and monomer scope and polymerization mechanism. Dalton Trans. 2012, 41, 9119–9134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Miyake, G.M.; Chen, E.Y.X. Alane-Based Classical and Frustrated Lewis Pairs in Polymer Synthesis: Rapid Polymerization of MMA and Naturally Renewable Methylene Butyrolactones into High-Molecular-Weight Polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef] [PubMed]
- He, J.H.; Zhang, Y.T.; Chen, E.Y.-X. Synthesis of Pyridineand 2-Oxazoline- Functionalized Vinyl Polymers by Alane-Based Frustrated Lewis Pairs cluster. Synlett 2014, 25, 1534–1538. [Google Scholar] [CrossRef]
- He, J.H.; Zhang, Y.T.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Chain Propagation and Termination Mechanisms for Polymerization of Conjugated Polar Alkenes by [Al]-Based Frustrated Lewis Pairs. Macromolecules 2014, 47, 7765–7774. [Google Scholar] [CrossRef]
- Xu, T.; Chen, E.Y.-X. Probing Site Cooperativity of Frustrated Phosphine/Borane Lewis Pairs by a Polymerization Study. J. Am. Chem. Soc. 2014, 136, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Knaus, M.G.M.; Giuman, M.M.; Pöthig, A.; Rieger, B. End of Frustration: Catalytic Precision Polymerization with Highly Interacting Lewis Pairs. J. Am. Chem. Soc. 2016, 138, 7776–7781. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.B.; Wang, Y.B.; Ren, W.M.; Xu, T.; Wang, J.; Lu, X.B. Mechanistic Aspects of Initiation and Deactivation in N-Heterocyclic Olefin Mediated Polymerization of Acrylates with Alane as Activator. Macromolecules 2014, 47, 1966–1972. [Google Scholar] [CrossRef]
- Jia, Y.B.; Ren, W.M.; Liu, S.J.; Xu, T.; Wang, Y.B.; Lu, X.B. Controlled Divinyl Monomer Polymerization Mediated by Lewis Pairs: A Powerful Synthetic Strategy for Functional Polymers. ACS Macro Lett. 2014, 3, 896–899. [Google Scholar] [CrossRef]
- Ottou, W.N.; Conde-Mendizabal, E.; Pascual, A.; Wirotius, A.-L.; Bourichon, D.; Vignolle, J.; Robert, F.; Landais, Y.; Sotiropoulos, J.-M.; Miqueu, K.; et al. Organic Lewis Pairs Based on Phosphine and Electrophilic Silane for the Direct and Controlled Polymerization of Methyl Methacrylate: Experimental and Theoretical Investigations. Macromolecules 2017, 50, 762–774. [Google Scholar] [CrossRef]
- Webster, O.W.; Hertler, W.R.; Sogah, D.Y.; Farnham, W.B.; Rajanbabu, T.V. Group-Transfer Polymerization. 1. A New Concept for Addition Polymerization with Organo-Silicon Initiators. J. Am. Chem. Soc. 1983, 105, 5706–5708. [Google Scholar] [CrossRef]
- Webster, O.W. The Discovery and Commercialization of Group Transfer Polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2855–2860. [Google Scholar] [CrossRef]
- Webster, O.W. Group Transfer Polymerization: A Critical Review of Its Mechanism and Comparison with Other Methods for Controlled Polymerization of Acrylic Monomers. Adv. Polym. Sci. 2003, 167, 1–34. [Google Scholar]
- Schubert, W.; Sitz, H.D.; Bandermann, F. Group Transfer Polymerization of Methyl-Methacrylate and Methyl Acrylate in Tetrahydrofuran with Tris(piperidino)sulfonium Bifluoride as Catalyst. Makromol. Chem. 1989, 190, 2193–2201. [Google Scholar] [CrossRef]
- Schubert, W.; Bandermann, F. Group Transfer Polymerization of Methyl Acrylate in Acetonitrile. Makromol. Chem. 1989, 190, 2161–2171. [Google Scholar] [CrossRef]
- Sogah, D.Y.; Hertler, W.R.; Webster, O.W.; Cohen, G.M. Group Transfer Polymerization—Polymerization of Acrylic-Monomers. Macromolecules 1987, 20, 1473–1488. [Google Scholar] [CrossRef]
- Schubert, W.; Bandermann, F. Group Transfer Polymerization of Methyl Acrylate in Tetrahydrofuran with Tetrabutylammonium Fluoride Trihydrate and Tetrabutylammonium Cyanide as Catalysts. Makromol. Chem. 1989, 190, 2721–2726. [Google Scholar] [CrossRef]
- Schmalbrock, U.; Bandermann, F. Group-Transfer Polymerization of Methyl- Methacrylate and Butyl Acrylate in Tetrahydrofuran with Oxy Anions as Catalysts. Makromol. Chem. 1993, 194, 2543–2551. [Google Scholar] [CrossRef]
- Dicker, I.B.; Cohen, G.M.; Farnham, W.B.; Hertler, W.R.; Laganis, E.D.; Sogah, D.Y. Oxyanions Catalyze Group-Transfer Polymerization to Give Living Polymers. Macromolecules 1990, 23, 4034–4041. [Google Scholar] [CrossRef]
- Patrickios, C.S.; Hertler, W.R.; Abbott, N.L.; Hatton, T.A. Diblock, ABC Triblock, and Random Methacrylic Polyampholytes -Synthesis by Group-Transfer Polymerization and Solution Behavior. Macromolecules 1994, 27, 930–937. [Google Scholar] [CrossRef]
- Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Gnanou, Y.; Taton, D. Harnessing the Potential of N-Heterocyclic Carbenes for the Rejuvenation of Group-Transfer Polymerization of (Meth)acrylics. Angew. Chem. Int. Ed. 2008, 47, 5390–5393. [Google Scholar] [CrossRef] [PubMed]
- Scholten, M.D.; Hedrick, J.L.; Waymouth, R.M. Group Transfer Polymerization of Acrylates Catalyzed by N-Heterocyclic Carbenes. Macromolecules 2008, 41, 7399–7404. [Google Scholar] [CrossRef]
- Raynaud, J.; Gnanou, Y.; Taton, D. Group Transfer Polymerization of (Meth)acrylic Monomers Catalyzed by N-Heterocyclic Carbenes and Synthesis of All Acrylic Block Copolymers: Evidence for an Associative Mechanism. Macromolecules 2009, 42, 5996–6005. [Google Scholar] [CrossRef]
- Raynaud, J.; Liu, N.; Gnanou, Y.; Taton, D. Expanding the Scope of Group Transfer Polymerization Using N-Heterocyclic Carbenes as Catalysts: Application to Miscellaneous (Meth)acrylic Monomers and Kinetic Investigations. Macromolecules 2010, 43, 8853–8861. [Google Scholar] [CrossRef]
- Raynaud, J.; Liu, N.; Fevre, M.; Gnanou, Y.; Taton, D. No Matter the Order of Monomer Addition for the Synthesis of Well-Defined Block Copolymers by Sequential Group Transfer Polymerization using N-Heterocyclic Carbenes as Catalysts. Polym. Chem. 2011, 2, 1706–1712. [Google Scholar] [CrossRef]
- Fevre, M.; Pinaud, J.; Gnanou, Y.; Vignolle, J.; Taton, D. N-Heterocyclic Carbenes (NHCs) as Organocatalysts and Structural Components in Metal-Free Polymer Synthesis. Chem. Soc. Rev. 2013, 42, 2142–2172. [Google Scholar] [CrossRef] [PubMed]
- Kakuchi, T.; Chen, Y.G.; Kitakado, J.; Mori, K.; Fuchise, K.; Satoh, T. Organic Superbase as an Efficient Catalyst for Group Transfer Polymerization of Methyl Methacrylate. Macromolecules 2011, 44, 4641–4647. [Google Scholar] [CrossRef]
- Fevre, M.; Vignolle, J.; Heroguez, V.; Taton, D. Tris(2,4,6-trimethoxyphenyl) phosphine (TTMPP) as Potent Organocatalyst for Group Transfer Polymerization of Alkyl (Meth)acrylates. Macromolecules 2012, 45, 7711–7718. [Google Scholar] [CrossRef]
- Chen, Y.G.; Fuchise, K.; Narumi, A.; Kawaguchi, S.; Satoh, T.; Kakuchi, T. Core-First Synthesis of Three-, Four-, and Six-Armed Star-Shaped Poly(methyl methacrylate)s by Group Transfer Polymerization Using Phosphazene Base. Macromolecules 2011, 44, 9091–9098. [Google Scholar] [CrossRef]
- Hsu, J.C.; Chen, Y.G.; Kakuchi, T.; Chen, W.C. Synthesis of Linear and Star-Shaped Poly[4-(diphenylamino)benzyl methacrylate]s by Group Transfer Polymerization and Their Electrical Memory Device Applications. Macromolecules 2011, 44, 5168–5177. [Google Scholar] [CrossRef]
- Kikuchi, S.; Chen, Y.; Fuchise, K.; Takada, K.; Kitakado, J.; Sato, S.; Satoh, T.; Kakuchi, T. Thermoresponsive Properties of 3-, 4-, 6-, and 12-Armed Star-Shaped Poly[2-(dimethylamino)ethyl methacrylate]s Prepared by Core-First Group Transfer Polymerization. Polym. Chem. 2014, 5, 4701–4709. [Google Scholar] [CrossRef]
- Chen, Y.; Takada, K.; Kubota, N.; Eric, O.T.; Ito, T.; Isono, T.; Satoh, T.; Kakuchi, T. Synthesis of End-Functionalized Poly-(methyl methacrylate) by Organocatalyzed Group Transfer Polymerization Using Functional Silyl Ketene Acetals and Alpha-Phenylacrylates. Polym. Chem. 2015, 6, 1830–1837. [Google Scholar] [CrossRef]
- Fuchise, K.; Chen, Y.G.; Satoh, T.; Kakuchi, T. Recent Progressin Organocatalytic Group Transfer Polymerization. Polym. Chem. 2013, 4, 4278–4291. [Google Scholar] [CrossRef]
- Hertler, W.R.; Sogah, D.Y.; Webster, O.W.; Trost, B.M. Group Transfer Polymerization. 3. Lewis Acid Catalysis. Macromolecules 1984, 17, 1415–1419. [Google Scholar] [CrossRef]
- Zhuang, R.; Müller, A.H.E. Group Transfer Polymerization of n-Butyl Acrylate with Lewis Acid Catalysts. 1. Kinetic Investigation Using HgI2 as a Catalyst in Toluene. Macromolecules 1995, 28, 8035–8042. [Google Scholar] [CrossRef]
- Zhuang, R.; Müller, A.H.E. Group Transfer Polymerization of n-Butyl Acrylate with Lewis Acid Catalysts. 2. Kinetic Investigation Using the HgI2/Me3SiI Catalyst System in Toluene and Methylene Chloride. Macromolecules 1995, 28, 8043–8050. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, E.Y.-X. Controlled Polymerization of Methacrylates to High Molecular Weight Polymer Using Oxidatively Activated Group Transfer Polymerization Initiators. Macromolecules 2008, 41, 36–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, E.Y.-X. Structure-Reactivity Relationships in Bimolecular-Ativated Monomer Polymerization of (Meth)acrylates Using Oxidatively Activated Group 14 Ketene Acetals. Macromolecules 2008, 41, 6353–6360. [Google Scholar] [CrossRef]
- Miyake, G.M.; Zhang, Y.T.; Chen, E.Y.-X. Living Polymerization of Naturally Renewable Butyrolactone-Based Vinylidene Monomers by Ambiphilic Silicon Propagators. Macromolecules 2010, 43, 4902–4908. [Google Scholar] [CrossRef]
- Zhang, Y.; Gustafson, L.O.; Chen, E.Y.-X. Dinuclear Silylium-Enolate Bifunctional Active Species: Remarkable Activity and Stereoselectivity toward Polymerization of Methacrylate and Renewable Methylene Butyrolactone Monomers. J. Am. Chem. Soc. 2011, 133, 13674–13684. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Chen, E.Y.-X. Silylium Dual Catalysis in Living Polymerization of Methacrylates via In Situ Hydrosilylation of Monomer. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 1895–1903. [Google Scholar] [CrossRef]
- Chen, Y.G.; Kitano, K.; Tsuchida, S.; Kikuchi, S.; Takada, K.; Satoh, T.; Kakuchi, T. B(C6F5)3-Catalyzed Group Transfer Polymerization of Alkyl Methacrylates with Dimethylphenylsilane through In Situ Formation of Silyl Ketene Acetal by B(C6F5)3-Catalyzed 1,4-Hydrosilylation of Methacrylate Monomer. Polym. Chem. 2015, 6, 3502–3511. [Google Scholar] [CrossRef]
- Fuchise, K.; Tsuchida, S.; Takada, K.; Chen, Y.G.; Satoh, T.; Kakuchi, T. B(C6F5)3-Catalyzed Group Transfer Polymerization of n-Butyl Acrylate with Hydrosilane through In Situ Formation of Initiator by 1,4-Hydrosilylation of n-Butyl Acrylate. ACS Macro Lett. 2014, 3, 1015–1019. [Google Scholar] [CrossRef]
- Allen, R.D.; Long, T.E.; McGrath, J.E. Preparation of high purity, anionic polymerization grade alkyl methacrylate monomers. Polym. Bull. 1986, 15, 127–134. [Google Scholar] [CrossRef]
- Lehmann, M.; Schulz, A.; Villinger, A. Bissilylated Halonium Ions: [Me3Si−X−SiMe3][B(C6F5)4] (X = F, Cl, Br, I). Angew. Chem. Int. Ed. 2009, 48, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Karsch, M.; Lund, H.; Schulz, A.; Villinger, A.; Voss, K. Molecular Networks Based on CN Coordination Bonds. Eur. J. Inorg. Chem. 2012, 2012, 5542–5553. [Google Scholar] [CrossRef]
- Chen, J.; Gowda, R.R.; He, J.; Zhang, Y.; Chen, E.Y.-X. Controlled or High-Speed Group Transfer Polymerization by Silyl Ketene Acetals without Catalyst. Macromolecules 2016, 49, 8075–8087. [Google Scholar] [CrossRef]
- Oisaki, K.; Suto, Y.; Kanai, M.; Shibasaki, M. A New Method for the Catalytic Aldol Reaction to Ketones. J. Am. Chem. Soc. 2003, 125, 5644–5645. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, C.; Chen, F.; Kuo, Y.-N. Ketene alkyltrialkylsilyl acetals: Synthesis, pyrolysis and NMR studies. J. Organomet. Chem. 1972, 46, 59–71. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Scheme 1.
Chemical structures of initiators, catalyst and monomers employed in this study.

Figure 1.
GPC traces of PMMBL samples produced by iBuSKA/B(C6F5)3 with various [MMBL]0/[iBuSKA]0 ratios at RT. Conditions: [MMBL]0/[iBuSKA]0/[B(C6F5)3]0 = 400:4:1, 400:2:1, 400:1:1, 400:0.5:1, [MMBL]0 = 0.936 M.
Figure 1.
GPC traces of PMMBL samples produced by iBuSKA/B(C6F5)3 with various [MMBL]0/[iBuSKA]0 ratios at RT. Conditions: [MMBL]0/[iBuSKA]0/[B(C6F5)3]0 = 400:4:1, 400:2:1, 400:1:1, 400:0.5:1, [MMBL]0 = 0.936 M.
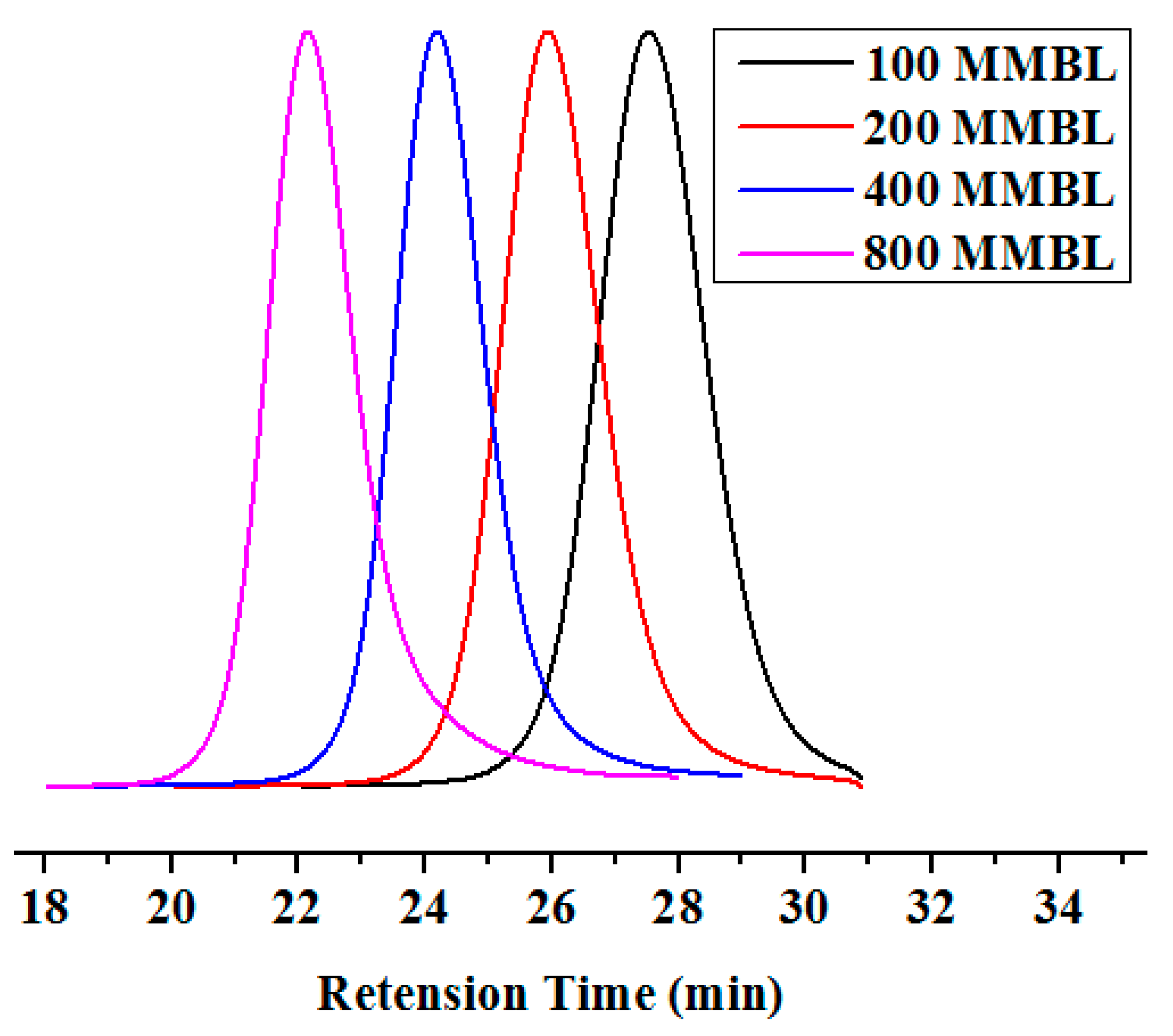
Figure 2.
Plots of Mn and Ð values of PMMBL samples vs [MMBL]0/[iBuSKA]0 ratio at RT. Conditions: [MMBL]0/[iBuSKA]0/[B(C6F5)3]0 = 400:4:1, 400:2:1, 400:1:1, 400:0.5:1, [MMBL]0 = 0.936 M.
Figure 2.
Plots of Mn and Ð values of PMMBL samples vs [MMBL]0/[iBuSKA]0 ratio at RT. Conditions: [MMBL]0/[iBuSKA]0/[B(C6F5)3]0 = 400:4:1, 400:2:1, 400:1:1, 400:0.5:1, [MMBL]0 = 0.936 M.

Figure 3.
Plots of Mn and Ð values of PMMBL samples vs MMBL conversion by iBuSKA/B(C6F5)3 system with MMBL/iBuSKA/B(C6F5)3 = 400/1/1 ratio at RT.
Figure 3.
Plots of Mn and Ð values of PMMBL samples vs MMBL conversion by iBuSKA/B(C6F5)3 system with MMBL/iBuSKA/B(C6F5)3 = 400/1/1 ratio at RT.
Scheme 2.
Generation of first-monomer-addition intermediate or active propagating species (A or B denotes Z/E isomers).
Scheme 2.
Generation of first-monomer-addition intermediate or active propagating species (A or B denotes Z/E isomers).
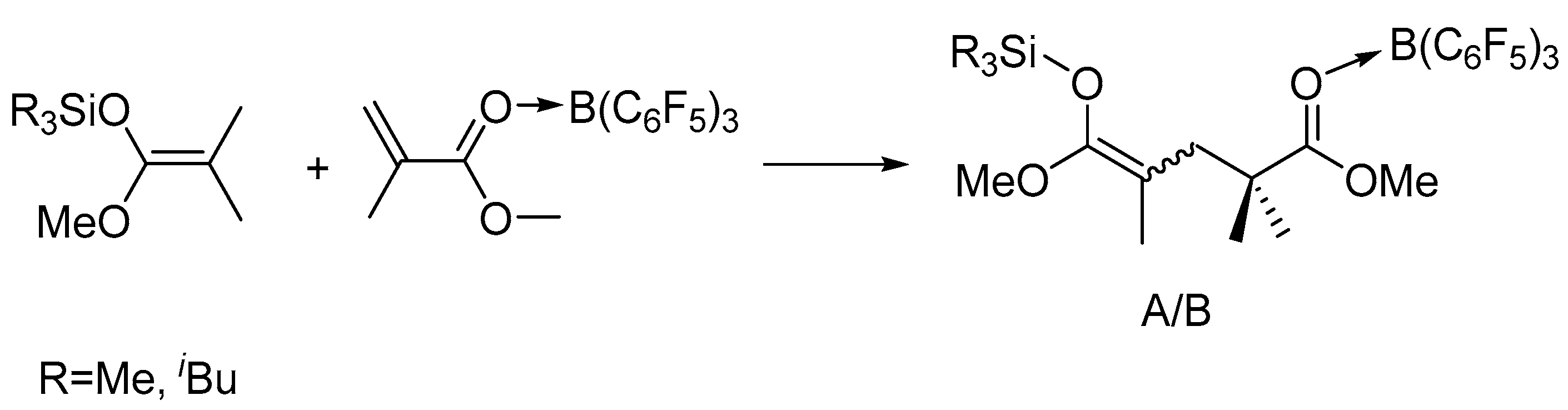
Figure 4.
(a) 1H- and (b) 19F-NMR spectra of iBu3SiO(OMe)C=C(Me)CH2CMe2C(OMe)=O···B(C6F5)3.
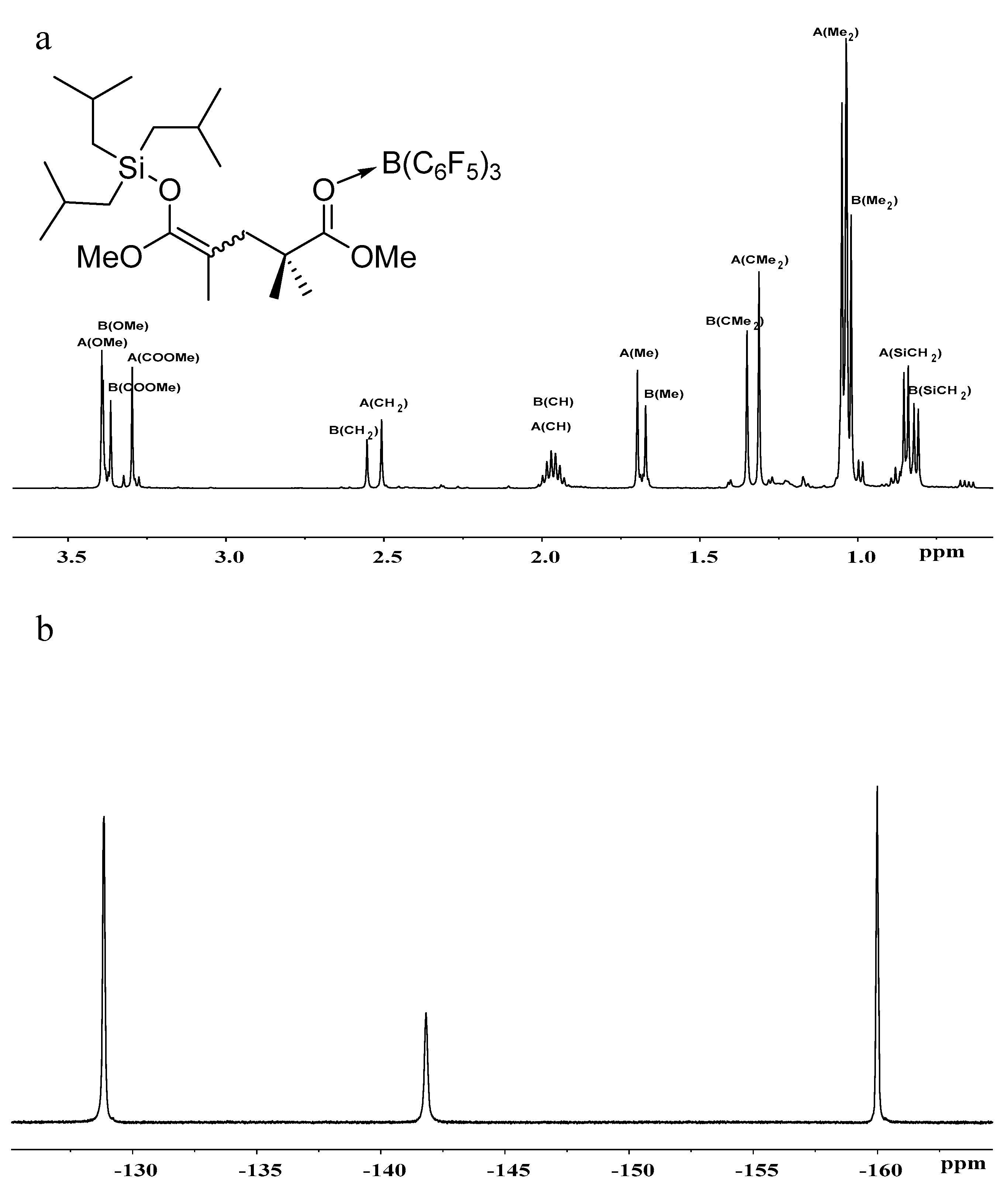
Figure 5.
First-order kinetic plots for MMBL polymerization by iBuSKA/B(C6F5)3 in CH2Cl2 at RT: [MMBL]0 = 0.936 M; [SKA]0 = 2.34 mM; [B(C6F5)3]0 = 9.36 mM (∆), 4.68 mM (▲), 2.34 mM (□), 1.17 mM (■). Inset: plot of ln(kapp) vs. ln[B(C6F5)3].
Figure 5.
First-order kinetic plots for MMBL polymerization by iBuSKA/B(C6F5)3 in CH2Cl2 at RT: [MMBL]0 = 0.936 M; [SKA]0 = 2.34 mM; [B(C6F5)3]0 = 9.36 mM (∆), 4.68 mM (▲), 2.34 mM (□), 1.17 mM (■). Inset: plot of ln(kapp) vs. ln[B(C6F5)3].

Figure 6.
First-order kinetic plots for MMBL polymerization by iBuSKA/B(C6F5)3 in CH2Cl2 at RT: [MMBL]0 = 0.936 M; [B(C6F5)3]0 = 2.34 mM; [SKA]0 = 9.36 mM (∆), 4.68 mM (▲), 2.34 mM (□), 1.17 mM (■). Inset: plot of ln(kapp) vs. ln[iBuSKA].
Figure 6.
First-order kinetic plots for MMBL polymerization by iBuSKA/B(C6F5)3 in CH2Cl2 at RT: [MMBL]0 = 0.936 M; [B(C6F5)3]0 = 2.34 mM; [SKA]0 = 9.36 mM (∆), 4.68 mM (▲), 2.34 mM (□), 1.17 mM (■). Inset: plot of ln(kapp) vs. ln[iBuSKA].
Scheme 3.
Proposed mechanism for MMBL polymerization by SKA/B(C6F5)3.
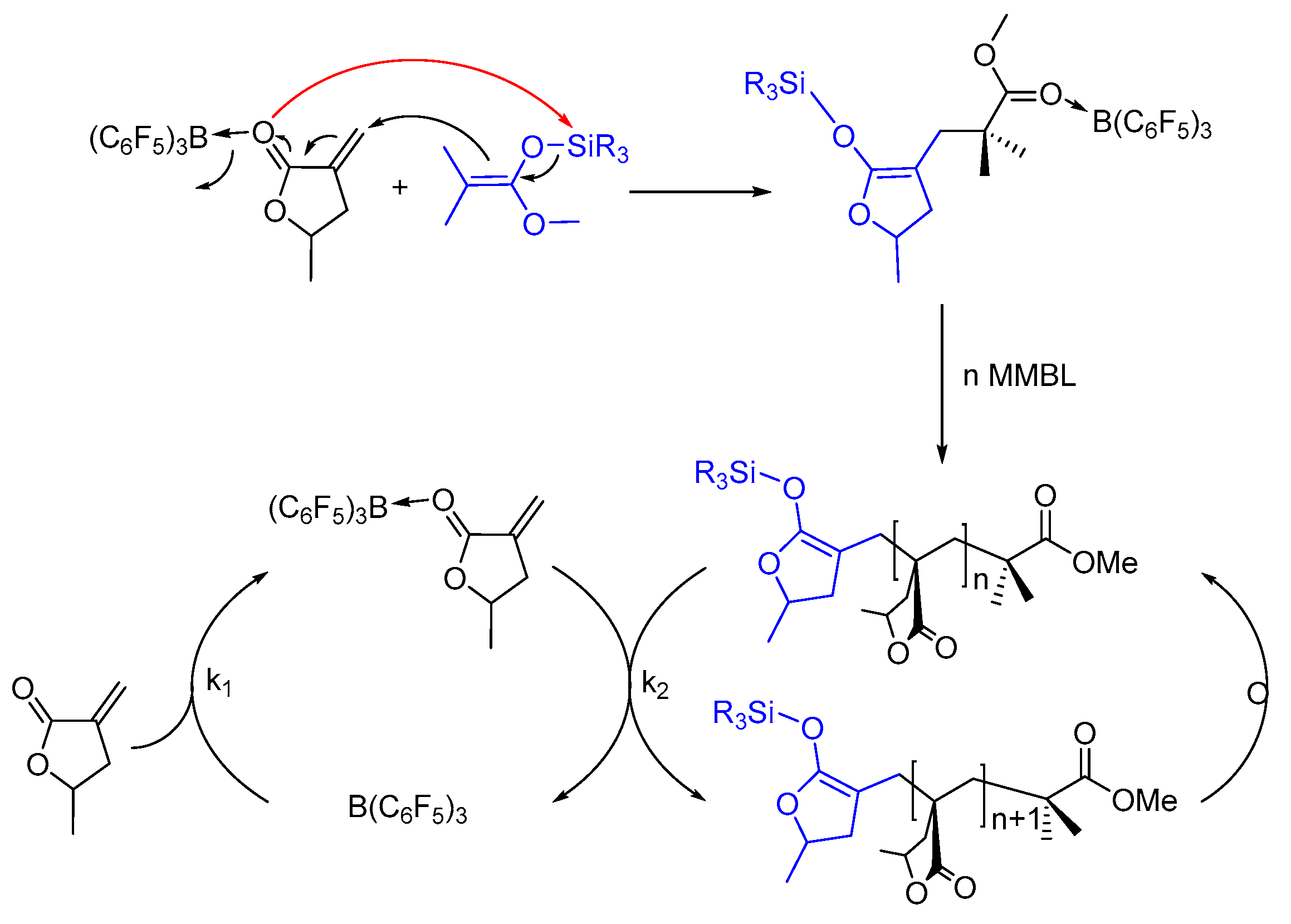
Figure 7.
MALDI-TOF mass spectrum of the low-MW PMMBL sample produced by iBuSKA/B(C6F5)3 in CH2Cl2 at RT.
Figure 7.
MALDI-TOF mass spectrum of the low-MW PMMBL sample produced by iBuSKA/B(C6F5)3 in CH2Cl2 at RT.
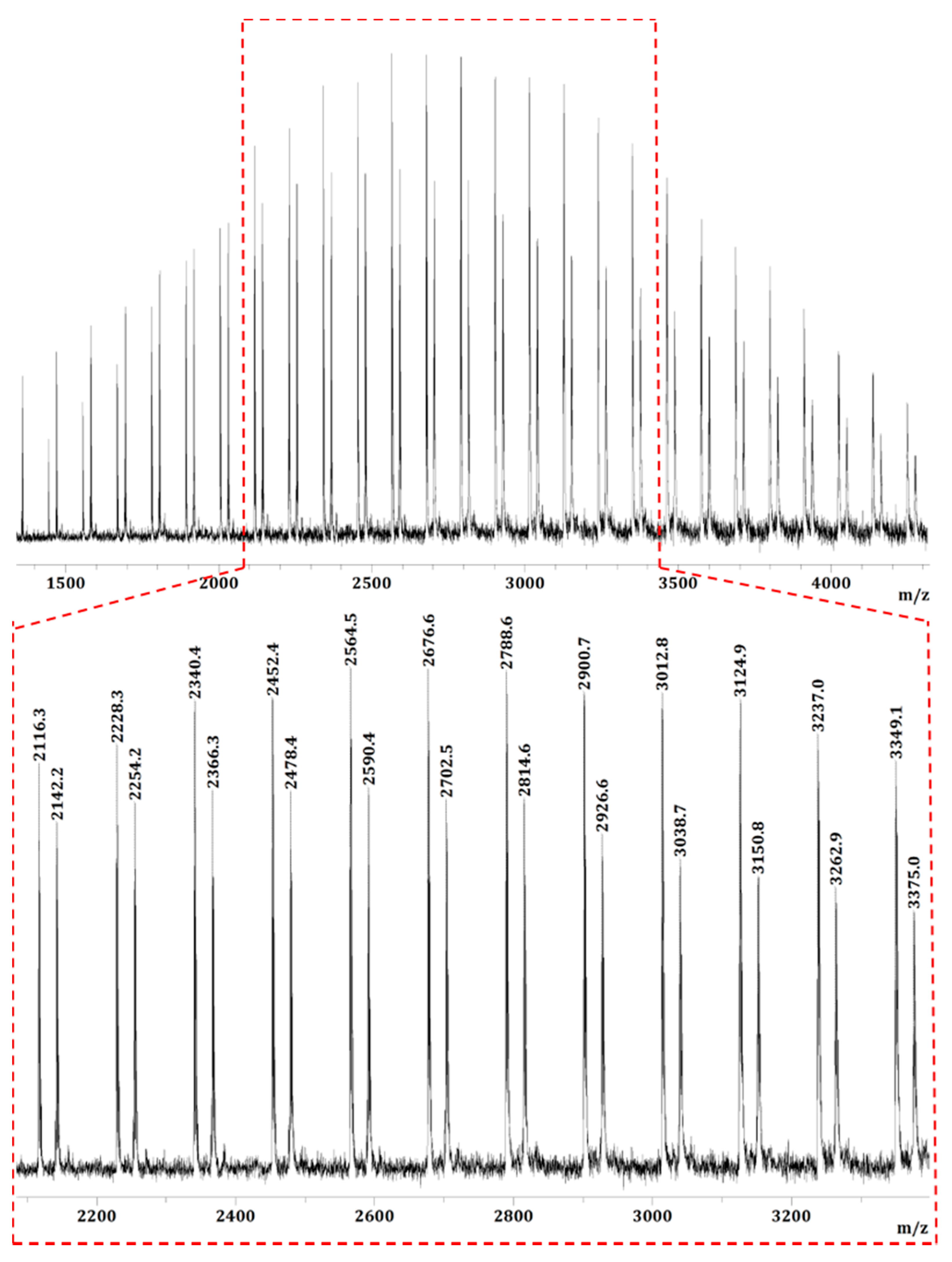
Figure 8.
Plot of m/z values from Figure 7 vs. the number of MMBL repeat units (n) for major series (a) and minor series (b).
Figure 8.
Plot of m/z values from Figure 7 vs. the number of MMBL repeat units (n) for major series (a) and minor series (b).
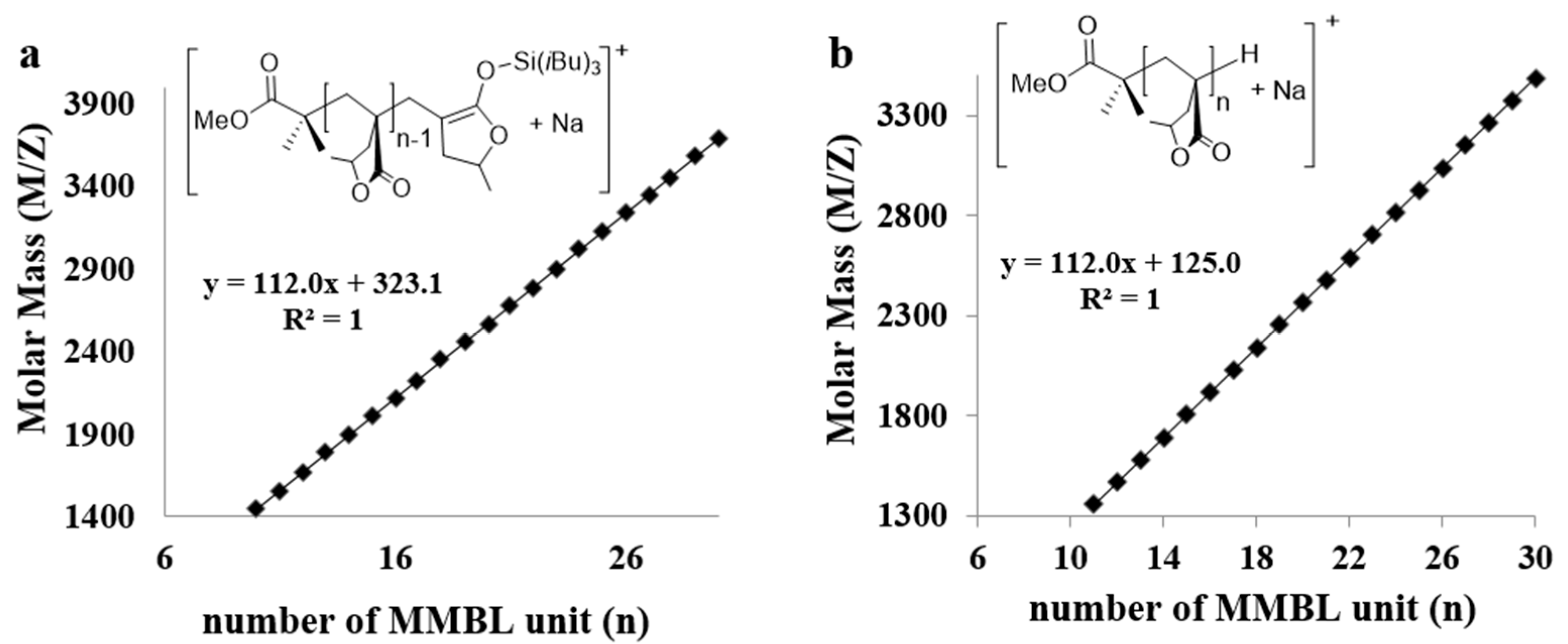
Figure 9.
GPC traces of homopolymer PMMBL (black), diblock copolymer PMMBL-b-PMBL (red), and triblock copolymer PMMBL-b-PMBL-b-PMMBL (blue) produced by iBuSKA/B(C6F5)3 in CH2Cl2 at RT.
Figure 9.
GPC traces of homopolymer PMMBL (black), diblock copolymer PMMBL-b-PMBL (red), and triblock copolymer PMMBL-b-PMBL-b-PMMBL (blue) produced by iBuSKA/B(C6F5)3 in CH2Cl2 at RT.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
B(C6F5)3-catalyzed polymerization of (M)MBL a.
| Run No. | Initiator (I) | Monomer (M) | [M]:[I]:[B] b | Time (h) | Conv. c (%) | Mn. d (kg/mol) | Ð | I* e (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Me2EtSiH | MMBL | 200:1:1 | 0.5 | 100 | 48.1 | 1.26 | 47 |
| 2 | Me2PhSiH | MMBL | 200:1:1 | 0.5 | 100 | 57.4 | 1.39 | 39 |
| 3 | Me2ClSiH | MMBL | 200:1:1 | 2 | 100 | 78.6 | 1.44 | 29 |
| 4 | Et3SiH | MMBL | 200:1:1 | 0.5 | 100 | 33 | 1.27 | 68 |
| 5 | Ph3SiH | MMBL | 200:1:1 | 6 | 100 | 105 | 1.61 | 21 |
| 6 | iBu3SiH | MMBL | 200:1:1 | 24 | 100 | 345 | 1.51 | 7 |
| 7 | Me2PhSKA | MMBL | 200:1:1 | 0.5 | 100 | 45.5 | 1.33 | 50 |
| 8 | Me2(EtO)SKA | MMBL | 200:1:1 | 0.5 | 100 | 41.5 | 1.37 | 54 |
| 9 | Me2ClSKA | MMBL | 200:1:1 | 6 | 100 | 85.1 | 1.82 | 27 |
| 10 | PhSKA | MMBL | 200:1:1 | 2 | 100 | 68.8 | 1.37 | 33 |
| 11 | MeSKA | MMBL | 200:1:1 | 1 | 100 | 31.9 | 1.08 | 71 |
| 12 | EtSKA | MMBL | 200:1:1 | 0.5 | 100 | 37 | 1.21 | 61 |
| 13 | iBuSKA | MMBL | 200:1:1 | 1 | 100 | 32.2 | 1.09 | 70 |
| 14 | iBuSKA | MMBL | 100:1:1 | 0.17 | 100 | 13.2 | 1.08 | 86 |
| 15 | MeSKA | MBL | 200:1:1 | 24 | 100 | 14.6 (58%) 98.0 (42%) | 1.20, 1.80 | - |
| 16 | MeSKA | MBL | 100:1:1 | 0.5 | 100 | 12.7 | 1.12 | 78 |
| 17 | iBuSKA | MBL | 200:1:1 | 24 | 100 | 23.4 | 1.95 | 84 |
| 18 | iBuSKA | MBL | 100:1:1 | 0.5 | 100 | 13.4 | 1.05 | 74 |
a Carried out in 2.25 mL CH2Cl2 at room temperature, where [MMBL]0 or [MBL]0 = 0.936M. b [M] = [Monomer], [I] = [Initiator], and [B] = [B(C6F5)3]. c Monomer conversions measured by 1H NMR. d Mn and Ð determined by GPC relative to PMMA standards in DMF. e Initiator efficiency I* (%) = Mn(calcd)/Mn(exptl) × 100, where Mn(calcd) = [MW(MMBL)] × ([MMBL]0/[I]0) (conversion) + MW of chain-end groups.
Table 2.
B(C6F5)3-catalyzed copolymerization of MMBL and MBL by iBuSKA/B(C6F5)3 system a.
| Run No. | M1/M2 | Conv. b(%) | Mw c (kg/mol) | Đ |
|---|---|---|---|---|
| 1 d | 100 MMBL + 100 MBL | MBL:100 MMBL:100 | 28.3 | 1.03 |
| 2 | 100 MMBL/100 MBL | MBL:100 MMBL:100 | 25.7 | 1.07 |
| 3 | 100 MMBL/100 MBL/100 MMBL | MBL:100 MMBL:100 | 37.1 | 1.06 |
a Carried out in 2.25 mL CH2Cl2 at room temperature, where [MMBL]0 = [MBL]0 = 0.936 M and addition method: catalyst and monomer were premixed, followed by adding initiator to start the polymerization. b Monomer conversions measured by 1H-NMR. c Mn and MWD determined by GPC relative to PMMA standards in DMF. d MBL and MMBL was added at the same time.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hu, L.; Zhao, W.; He, J.; Zhang, Y. Silyl Ketene Acetals/B(C6F5)3 Lewis Pair-Catalyzed Living Group Transfer Polymerization of Renewable Cyclic Acrylic Monomers. Molecules 2018, 23, 665. https://doi.org/10.3390/molecules23030665
AMA Style
Hu L, Zhao W, He J, Zhang Y. Silyl Ketene Acetals/B(C6F5)3 Lewis Pair-Catalyzed Living Group Transfer Polymerization of Renewable Cyclic Acrylic Monomers. Molecules. 2018; 23(3):665. https://doi.org/10.3390/molecules23030665
Chicago/Turabian StyleHu, Lu, Wuchao Zhao, Jianghua He, and Yuetao Zhang. 2018. "Silyl Ketene Acetals/B(C6F5)3 Lewis Pair-Catalyzed Living Group Transfer Polymerization of Renewable Cyclic Acrylic Monomers" Molecules 23, no. 3: 665. https://doi.org/10.3390/molecules23030665