In Vitro ADME Properties of Two Novel Antimicrobial Peptoid-Based Compounds as Potential Agents against Canine Pyoderma

 , and
, and
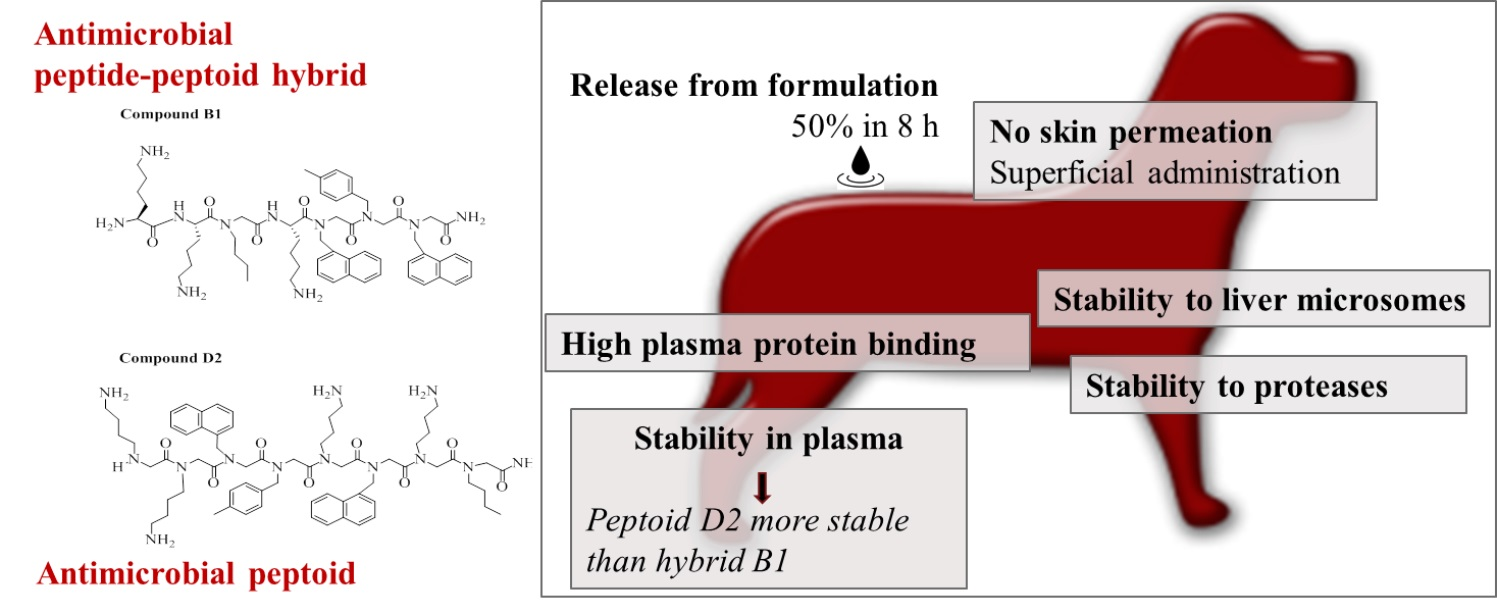
Abstract
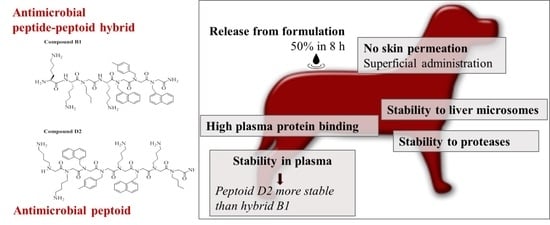
:
1. Introduction
2. Results
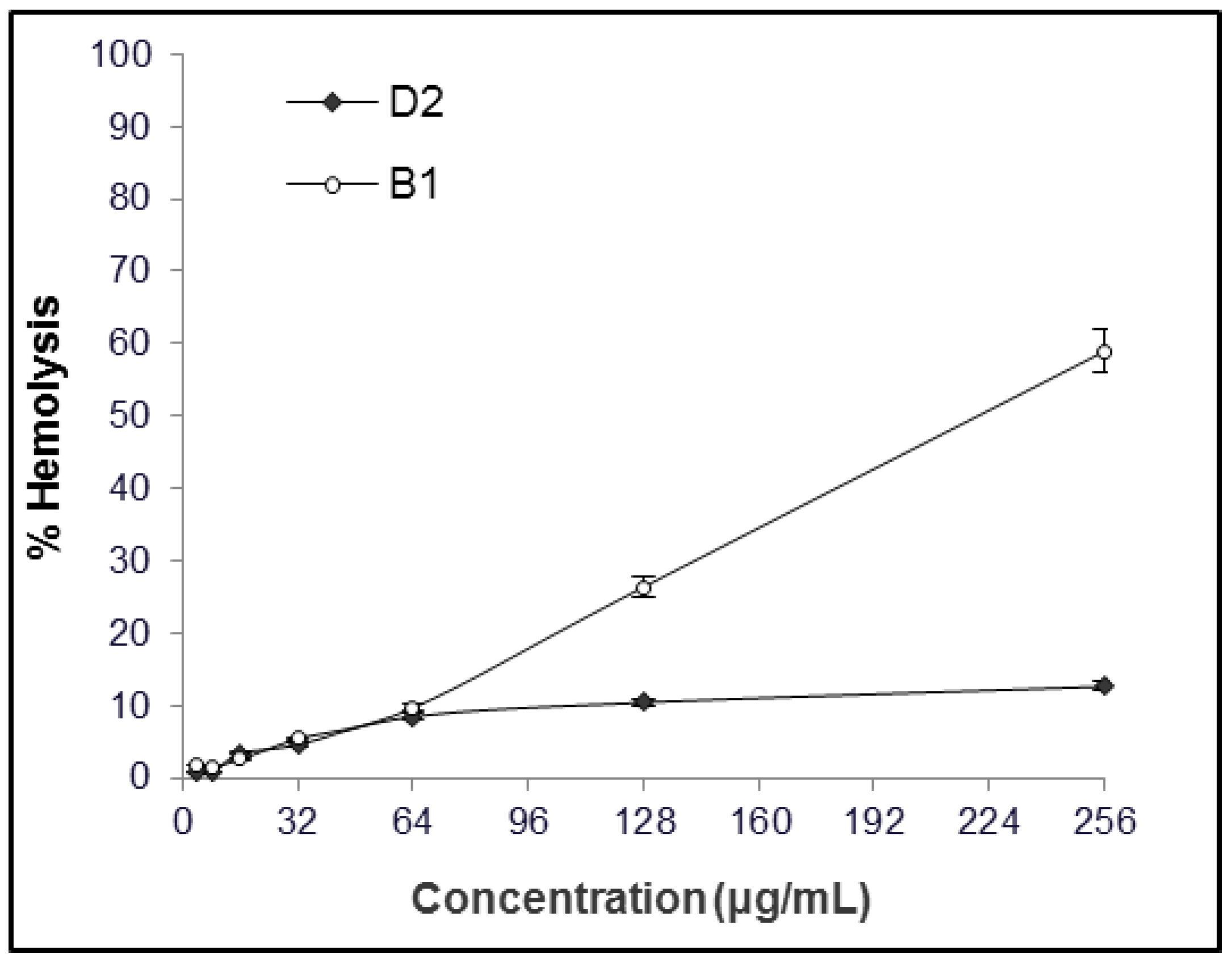
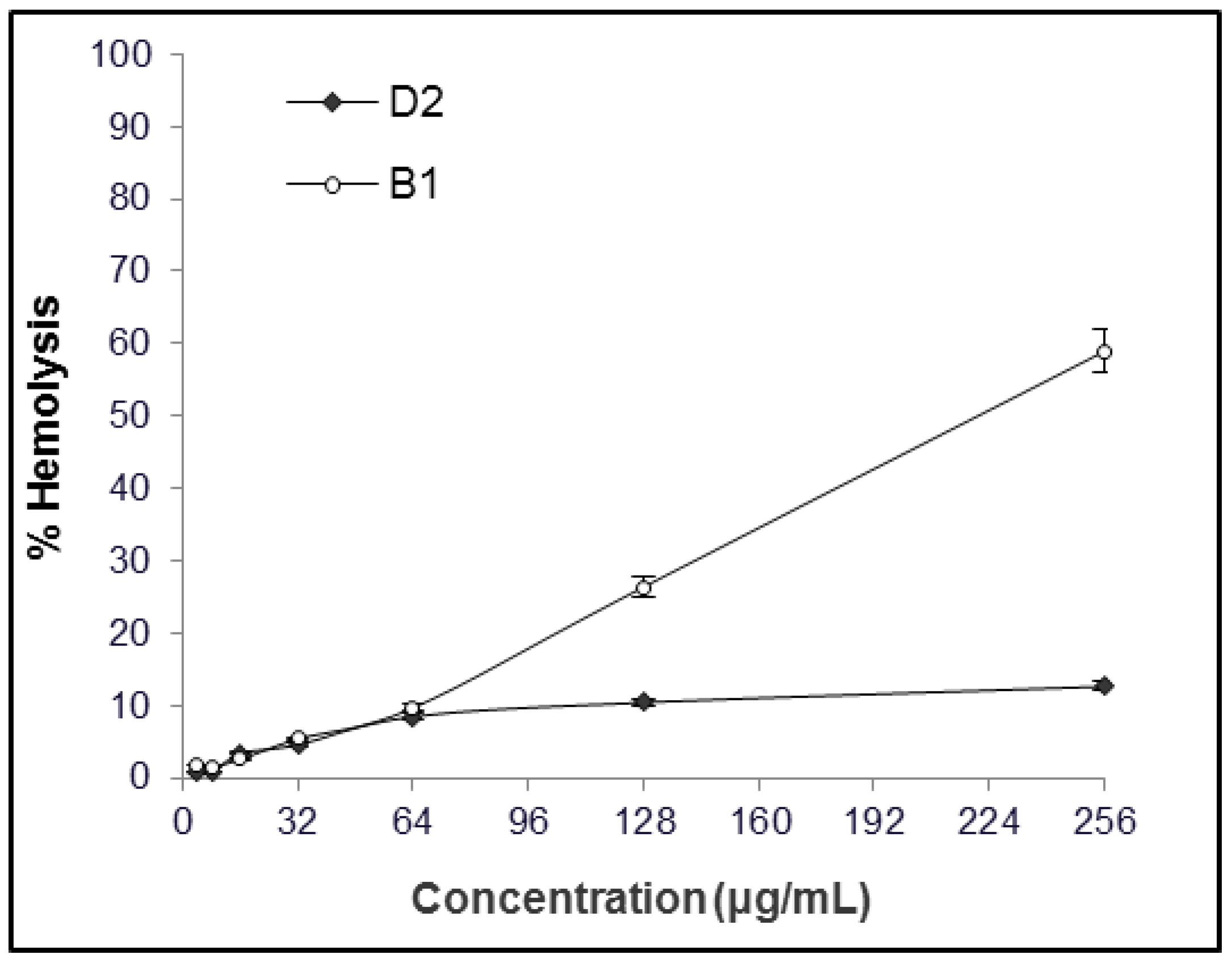
2.1. MIC and Hemolytic Activity In Vitro
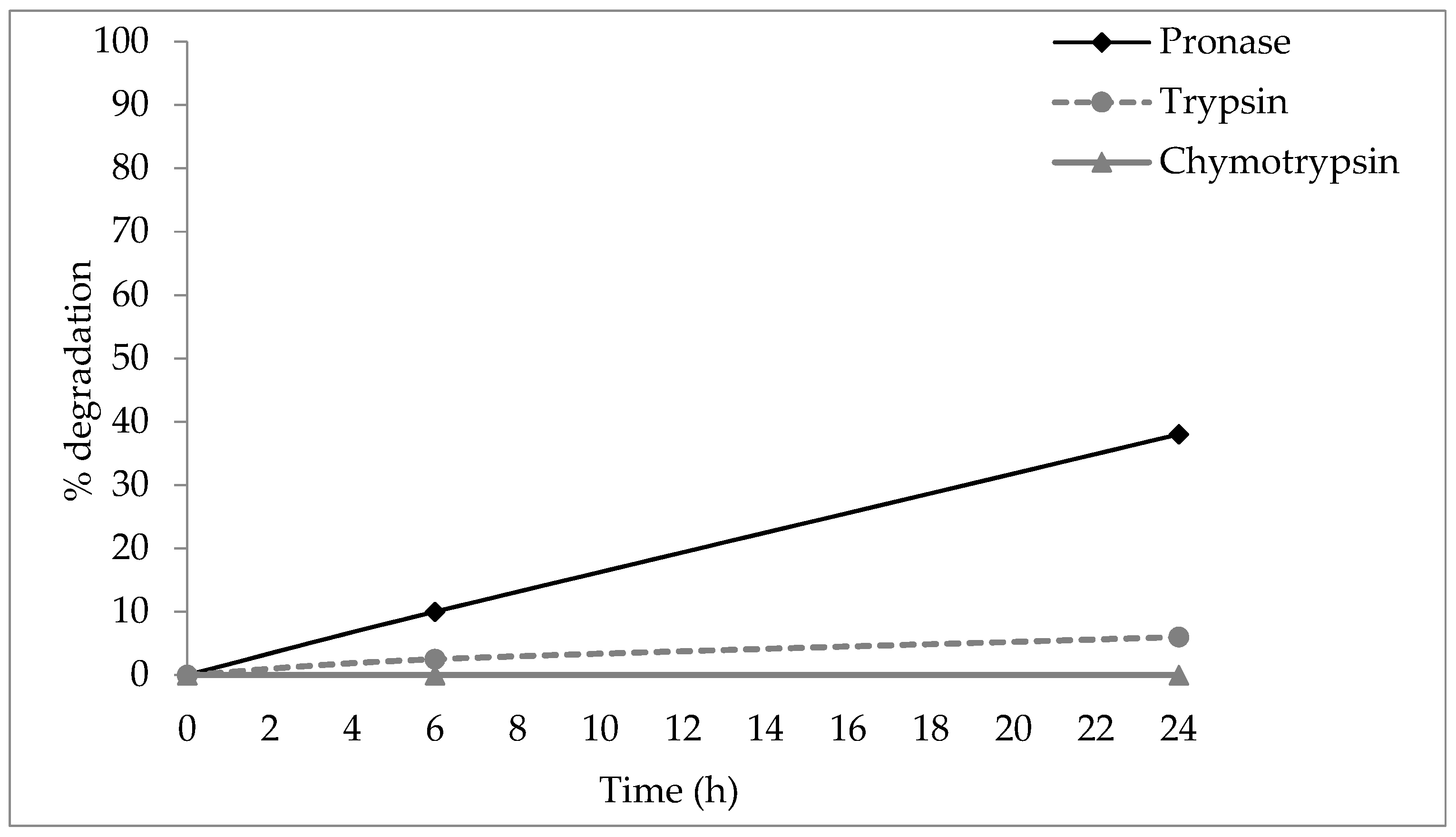
2.2. Stability to Proteases
2.3. Stability in Plasma
2.4. Stabilty in Liver Microsomes
2.5. Plasma Protein Binding
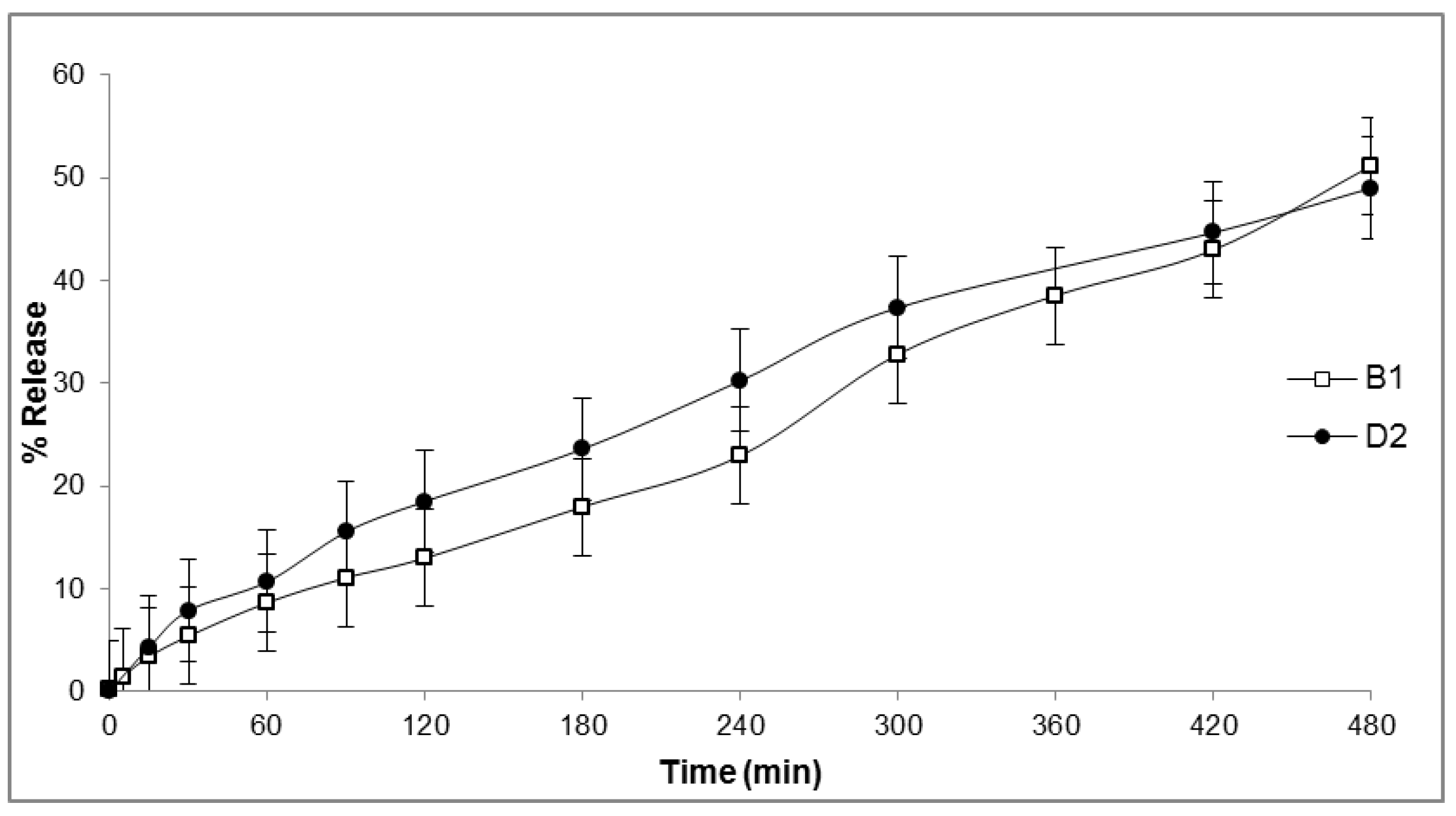
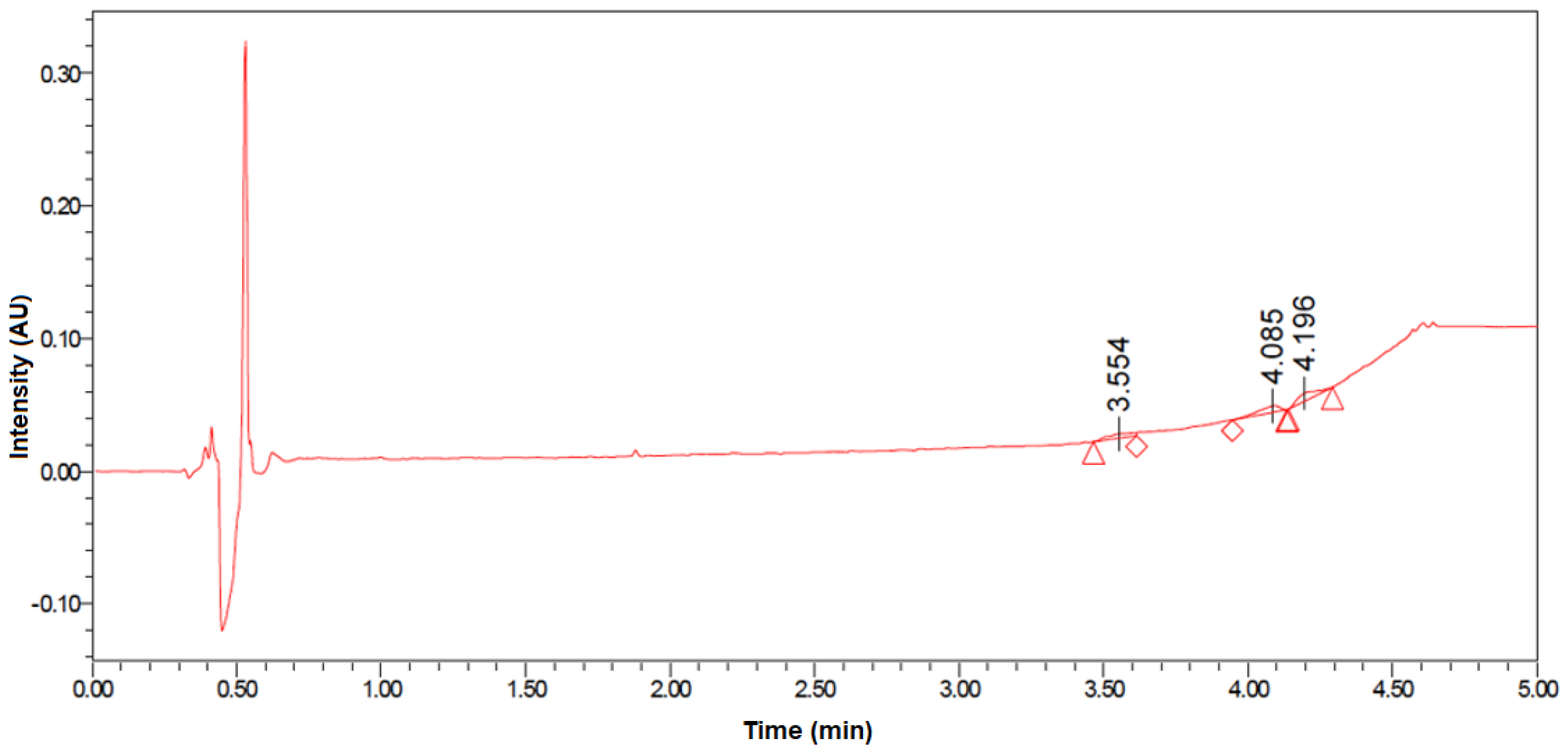
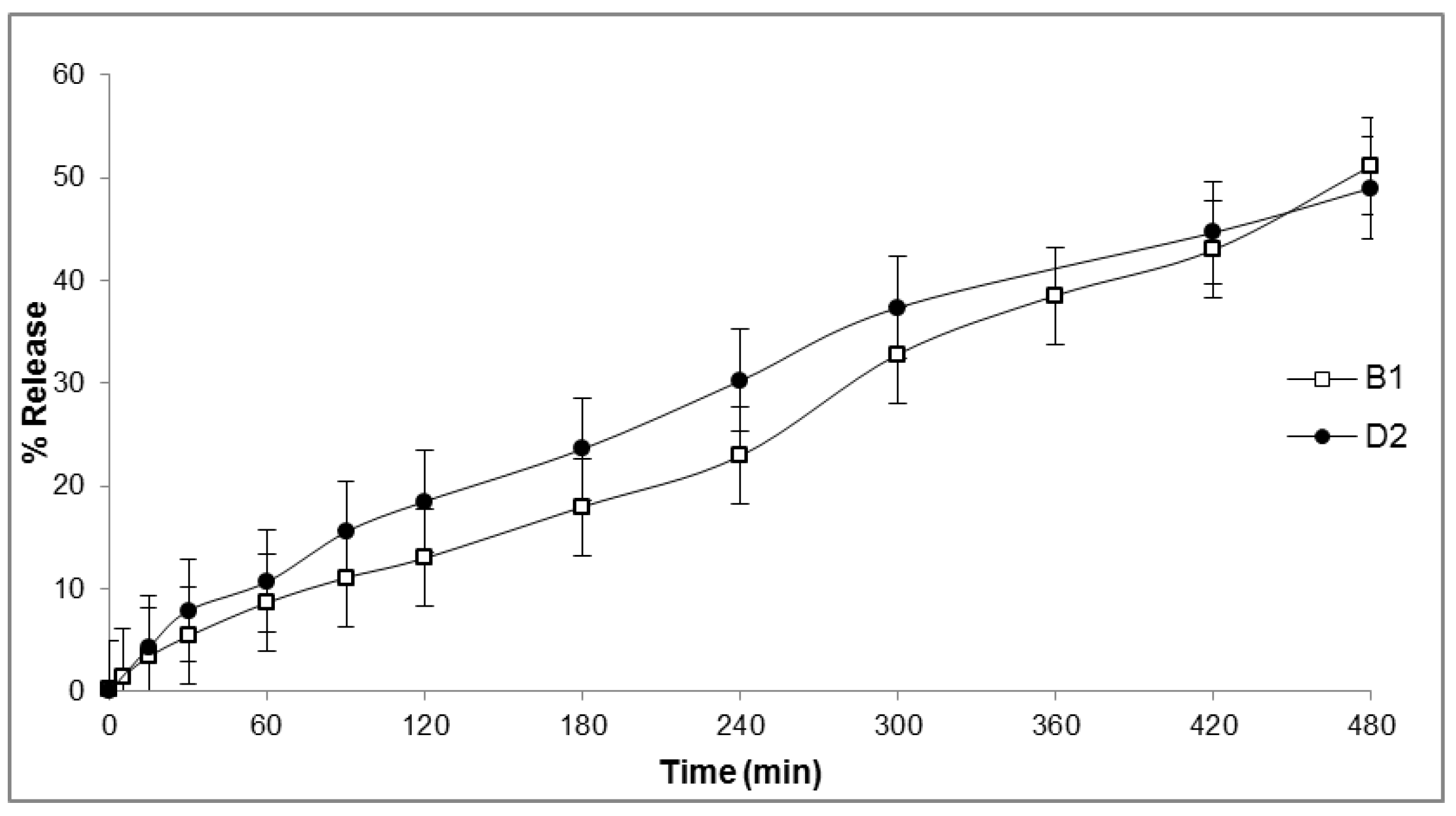
2.6. In Vitro Release
2.7. Skin Permeability
3. Discussion
4. Materials and Methods
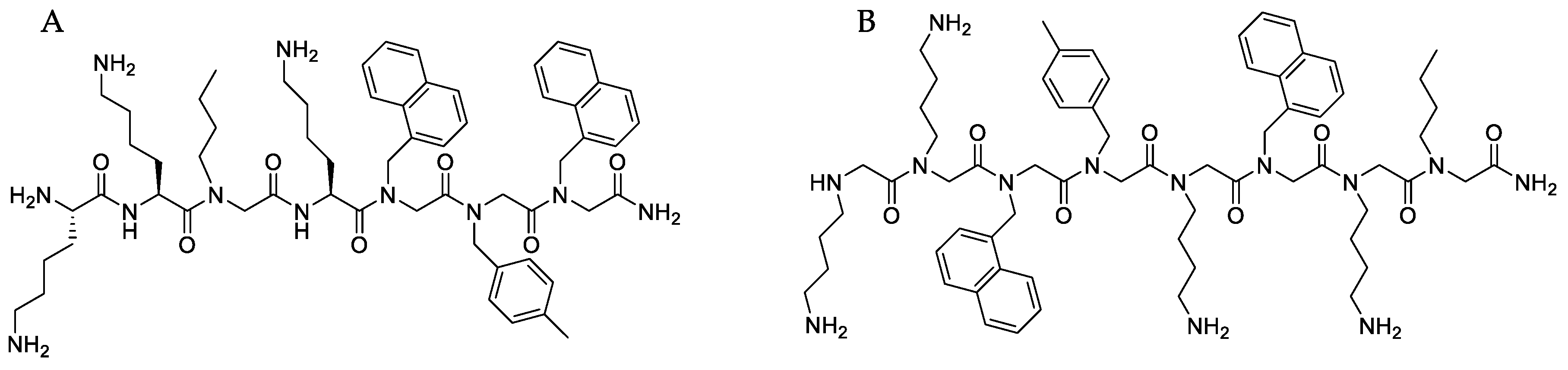
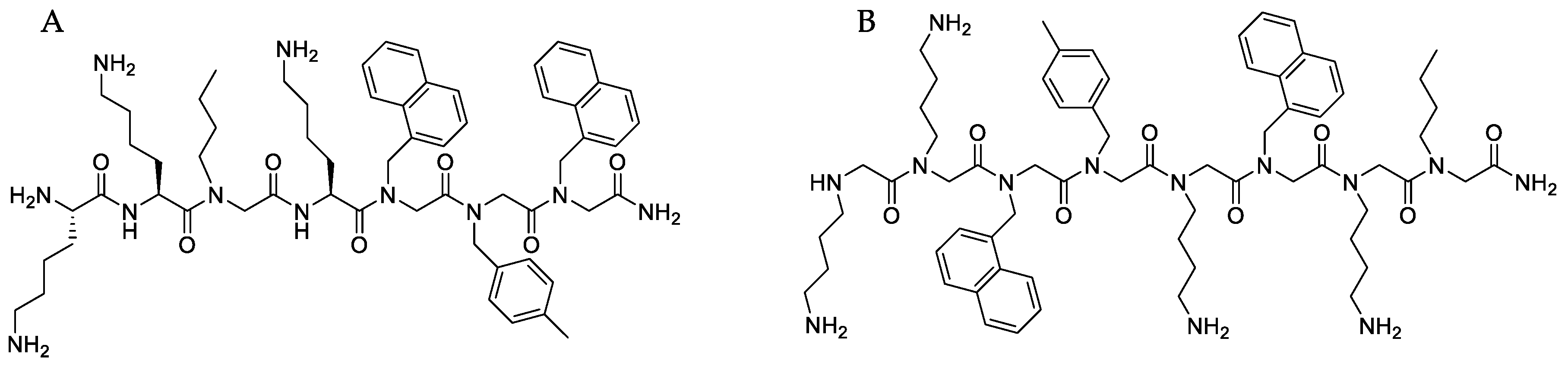
4.1. Synthesis and Characterization of Peptide-Peptoid Hybrid (B1) and Peptoid (D2)
4.2. Minimum Inhibitory Concentration
4.3. Hemolytic Activity
4.4. Stability of Peptide-Peptoid Hybrid (B1) and Peptoid (D2)
4.4.1. Stability to Proteases
4.4.2. Stability in Plasma
4.4.3. Stability to Liver Microsomes
4.5. Plasma Protein Binding
4.6. Formulation Preparation
4.7. In Vitro Drug Release
4.8. Skin Permeation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Antimicrobial Resistance; Global Report on Surveillance; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- European Centre for Disease, Prevention and Control. Antimicrobial Resistance in Europe in 2014; Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net); ECDC: Stockholm, Sweden, 2015. [Google Scholar]
- Rantala, M.; Holso, K.; Lillas, A.; Huovinen, P.; Kaartinen, L. Survey of condition-based prescribing of antimicrobial drugs for dogs at a veterinary teaching hospital. Vet. Rec. 2004, 155, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.D.; Walker, R.D.; Bowman, M.M., II; Schott, H.C.; Rosser, E.J., Jr. Frequency of Isolation and Antimicrobial Susceptibility Patterns of Staphylococcus intermedius and Pseudomonas aeruginosa Isolates From Canine Skin and Ear Samples Over a 6-Year Period (1992–1997). J. Am. Anim. Hosp. Assoc. 2002, 38, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Perreten, V.; Kadlec, K.; Schwarz, S.; Grönlund Andersson, U.; Finn, M.; Greko, C.; Moodley, A.; Kania, S.A.; Frank, L.A.; Bemis, D.A.; et al. Clonal spread of methicillin-resistant Staphylococcus pseudintermedius in Europe and North America: An international multicentre study. J. Antimicrob. Chemother. 2010, 65, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Hillier, A.; Lloyd, D.H.; Weese, J.S.; Blondeau, J.M.; Boothe, D.; Breitschwerdt, E.; Guardabassi, L.; Papich, M.G.; Rankin, S.; Turnidge, J.D.; et al. Guidelines for the diagnosis and antimicrobial therapy of canine superficial bacterial folliculitis (Antimicrobial Guidelines Working Group of the International Society for Companion Animal Infectious Diseases). Vet. Dermatol. 2014, 25, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.K.; O’Neil, D.A. Peptides as the next generation of anti-infectives. Futur. Med. Chem. 2013, 5, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial Peptides: An Introduction. In Antimicrobial Peptides: Methods and Protocols; Hansen, P.R., Ed.; Springer: New York, NY, USA, 2017; pp. 3–22. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Lau, K.; Lushnikova, T.; Golla, R.; Wang, X. Antimicrobial Peptides in 2014. Pharmaceuticals 2015, 8, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Wetzler, M.; Barron, A.E. Functional Synergy between Antimicrobial Peptoids and Peptides against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2011, 55, 5399–5402. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kodadek, T. Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–307. [Google Scholar] [PubMed]
- Fowler, S.A.; Blackwell, H.E. Structure–function relationships in peptoids: Recent advances toward deciphering the structural requirements for biological function. Org. Biomol. Chem. 2009, 7, 1508–1524. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.; Greko, C. Antibiotic resistance-consequences for animal health, welfare, and food production. Upsala J. Med. Sci. 2014, 119, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Guardabassi, L.; Prescott, J.F. Antimicrobial Stewardship in Small Animal Veterinary Practice: From Theory to Practice. Vet. Clin. N. Am. Small Anim. Pract. 2015, 45, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Kahn, L.H.; Kaplan, B. One Health Perspective. ILAR J. 2010, 51, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Pirri, G.; Bozzi, A.; Di Giulio, A.; Aschi, M.; Rinaldi, A.C. Antimicrobial peptides: Natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 2008, 65, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides with Enhanced Activities and Specificity/Therapeutic Index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.; Qi, F.X.; Yarbrough, D.K.; He, J.; Anderson, M.H.; Shi, W.Y. Adding selectivity to antimicrobial peptides: Rational design of a multidomain peptide against Pseudomonas spp. Antimicrob. Agents Chemother. 2006, 50, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Kessler, H. Peptoids—A new approach to the development of pharmaceuticals. Angew. Chem. Int. Ed. 1993, 32, 543–544. [Google Scholar] [CrossRef]
- Dohm, M.T.; Kapoor, R.; Barron, A.E. Peptoids: Bio-Inspired Polymers as Potential Pharmaceuticals. Curr. Pharm. Des. 2011, 17, 2732–2747. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.; Monsignori, A.; Orena, M.; Rinaldi, S.; Castellucci, N.; Tomasini, C. beta-Peptoids: Synthesis of a novel dimer having a fully extended conformation. Amino Acids 2012, 43, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-Activity Relationship Study of Novel Peptoids That Mimic the Structure of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Frimodt-Moller, N.; Hansen, P.R. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy 2008, 54, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Hansen, P.R. Potent antibacterial lysine-peptoid hybrids identified from a positional scanning combinatorial library. Bioorg. Med. Chem. 2006, 14, 4444–4451. [Google Scholar] [CrossRef] [PubMed]
- Godballe, T.; Nilsson, L.L.; Petersen, P.D.; Jenssen, H. Antimicrobial beta-Peptides and alpha-Peptoids. Chem. Biol. Drug Des. 2011, 77, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Nielsen, H.M.; Franzyk, H. Lysine-Based α-peptide/β-peptoid peptidomimetics: Influence of hydrophobicity, fluorination and distribution of cationic charge on antimicrobial activity and cytotoxicity. ChemMedChem 2017, 20, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Knapp, K.M.; Yang, L.; Molin, S.; Franzyk, H.; Folkesson, A. High in vitro antimicrobial activity of beta-peptoid-peptide hybrid oligomers against planktonic and biofilm cultures of Staphylococcus epidermidis. Int. J. Antimicrob. Agents 2013, 41, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Eggimann, G.A.; Jahoda, C.A.B.; Zuckermann, R.N.; Sharples, G.J.; Cobb, S.L. Exploring the links between peptoid antibacterial activity and toxicity. MedChemComm 2017, 8, 886–896. [Google Scholar] [CrossRef]
- Seo, J.; Ren, G.; Liu, H.G.; Miao, Z.; Park, M.; Wang, Y.H.; Miller, T.M.; Barron, A.E.; Cheng, Z. In vivo Biodistribution and Small Animal PET of Cu-64-Labeled Antimicrobial Peptoids. Bioconjugate Chem. 2012, 23, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Zuckermann, R.N.; Reinhard, C.; Frey, W.H. Intranasal administration delivers peptoids to the rat central nervous system. Neurosci. Lett. 2008, 439, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Cai, D.; Archer, C.T.; Kodadek, T. Periodate-triggered cross-linking reveals Sug2/Rpt4 as the molecular target of a peptoid inhibitor of the 19S proteasome regulatory particle. J. Am. Chem. Soc. 2007, 129, 12936–12937. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Hajibeigi, A.; León-Rodríguez, L.M.D.; Öz, O.K.; Sun, X. Peptoid-based PET imaging of vascular endothelial growth factor receptor (VEGFR) expression. Am. J. Nucl. Med. Mol. Imaging 2011, 1, 65–75. [Google Scholar] [PubMed]
- Wang, Y.F.; Lin, H.; Tullman, R.; Jewell, C.F.; Weetall, M.L.; Tse, F.L.S. Absorption and disposition of a tripeptoid and a tetrapeptide in the rat. Biopharm. Drug Dispos. 1999, 20, 69–75. [Google Scholar] [CrossRef]
- Werner, A.H.; Russell, A.D. Mupirocin, fusidic acid and bacitracin: Activity, action and clinical uses of three topical antibiotics. Vet. Dermatol. 1999, 10, 225–240. [Google Scholar] [CrossRef]
- Bonomo, R.A.; Van Zile, P.S.; Li, Q.; Shermock, K.M.; McCormick, W.G.; Kohut, B. Topical triple-anti biotic ointment as a novel therapeutic choice in wound management and infection prevention: A practical perspective. Expert Rev. Anti-Infect. Ther. 2007, 5, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, B.A.; Holroyd, K.J.; Zasloff, M. Topical versus Systemic Antimicrobial Therapy for Treating Mildly Infected Diabetic Foot Ulcers: A Randomized, Controlled, Double-Blinded, Multicenter Trial of Pexiganan Cream. Clin. Infect. Dis. 2008, 47, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Veyries, M.L.; Couarraze, G.; Geiger, S.; Agnely, F.; Massias, L.; Kunzli, B.; Faurisson, F.; Rouveix, B. Controlled release of vancomycin from Poloxamer 407 gels. Int. J. Pharm. 1999, 192, 183–193. [Google Scholar] [CrossRef]
- Czyzewski, A.M.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Chongsiriwatana, N.P.; Yuen, E.; Hancock, R.E.W.; Barron, A.E. In vivo, In Vitro, and In Silico Characterization of Peptoids as Antimicrobial Agents. PLoS ONE 2016, 11, e0135961. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.D.; Meinardi, M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Wise, D. Handbook of Pharmaceutical Controlled Release Technology; CRC Press: Boca Raton, FL, USA, 2000; ISBN 9780824703691. [Google Scholar]
- Mohammed, D.; Hirata, K.; Hadgraft, J.; Lane, M.E. Influence of skin penetration enhancers on skin barrier function and skin protease activity. Eur. J. Pharm. Sci. 2014, 51, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.E. Skin penetration enhancers. Int. J. Pharm. 2013, 447, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.D.; Fahr, A. Synergistic penetration enhancement effect of ethanol and phospholipids on the topical delivery of cyclosporin A. J. Controll. Release 2004, 97, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Fini, A.; Bergamante, V.; Ceschel, G.C.; Ronchi, C.; De Moraes, C.A.F. Control of Transdermal Permeation of Hydrocortisone Acetate from Hydrophilic and Lipophilic Formulations. AAPS PharmSciTech 2008, 9, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.; de Melo Carrasco, L. Novel Formulations for Antimicrobial Peptides. Int. J. Mol. Sci. 2014, 15, 18040–18083. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Proteolytic studies of homologous peptide and N-substituted glycine. Bioorg. Med. Chem. Lett. 1994, 4, 2657–2662. [Google Scholar] [CrossRef]
- Riley, R.J.; McGinnity, D.F.; Austin, R.P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005, 33, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Oesch, F.; Fabian, E.; Oesch-Bartlomowicz, B.; Werner, C.; Landsiedel, R. Drug-metabolizing enzymes in the skin of man, rat, and pig. Drug Metab. Rev. 2007, 39, 659–698. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Barbour, N.P.; Lipper, R.A. Introduction to Biopharmaceutics and its Role in Drug Development. In Biopharmaceutics Applications in Drug Development; Krishna, R., Yu, L., Eds.; Springer: Boston, MA, USA, 2008; pp. 1–25. [Google Scholar] [CrossRef]
- Beer, J.; Wagner, C.C.; Zeitlinger, M. Protein Binding of Antimicrobials: Methods for Quantification and for Investigation of its Impact on Bacterial Killing. AAPS J. 2009, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Burian, A.; Wagner, C.; Stanek, J.; Manafi, M.; Bohmdorfer, M.; Jager, W.; Zeitlinger, M. Plasma protein binding may reduce antimicrobial activity by preventing intra-bacterial uptake of antibiotics, for example clindamycin. J. Antimicrob. Chemother. 2011, 66, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Agerberth, B.; Lothgren, A.; Almstedt, A.; Johansson, J. Apolipoprotein A-I binds and inhibits the human antibacterial/cytotolic peptide LL-37. J. Biol. Chem. 1998, 273, 33115–33118. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals; Approved Standard—Fourth Edition; VET01-A4; CLSI: Wayne, PA, USA, 2013. [Google Scholar]
- Oddo, A.; Nyberg, N.T.; Frimodt-Moller, N.; Thulstrup, P.W.; Hansen, P.R. The effect of glycine replacement with flexible omega-amino acids on the antimicrobial and haemolytic activity of an amphipathic cyclic heptapeptide. Eur. J. Med. Chem. 2015, 102, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Danish Health and Medicines Authority. 2, Appendix 3, Quality of Medicinal Products, Addition to drug form standards. In Danish Medicinal Standards; Danish Health and Medicines Authority: Copenhagen, Denmark, 2013. [Google Scholar]
- Dumortier, G.; Grossiord, J.L.; Agnely, F.; Chaumeil, J.C. A Review of Poloxamer 407 Pharmaceutical and Pharmacological Characteristics. Pharm. Res. 2006, 23, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Plasma (Dog) | Liver Microsomes (Dog) | Liver Microsomes (Human) |
|---|---|---|---|
| B1 | 28.9 | >120 | >120 |
| D2 | >120 | >120 | >120 |
| Peptide-1 | >120 | - | - |
| Peptide-2 | <3 | - | - |
| Peptide-3 | 81.2 | - | - |
| Entry | % Average Recovery | SD 1 | % Unbound | Bound | SD 1 |
|---|---|---|---|---|---|
| B1 | 75.6 | 20.7 | 0.16 | 99.8 | 0.000 |
| D2 | 70.8 | 6.5 | 1.43 | 98.6 | 0.005 |
| Control | 67.1 | 12.6 | 0.27 | 99.7 | 0.001 |
| Formulation | Ingredients | % (w/w) |
|---|---|---|
| Oil-in-water cream | Oil Phase | |
| Polysorbate 80 | 0.5 | |
| Cetostearyl alcohol | 5 | |
| Paraffin oil | 5 | |
| Glycerol monostearate | 6 | |
| Water Phase | ||
| Methyl parahydroxy-benzoate | 0.1 | |
| 85% glycerol | 4 | |
| Sorbitol | 7 | |
| Purified water | 72.4 | |
| Oinment | ||
| Paraffin oil | 20 | |
| Vaselin | 80 | |
| Hydrogel | ||
| Poloxamer 407 | 20 | |
| Purified water | 80 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, I.; Hummel, B.D.; Vasir, J.; Watts, J.L.; Koch, J.; Hansen, J.E.; Nielsen, H.M.; Damborg, P.; Hansen, P.R. In Vitro ADME Properties of Two Novel Antimicrobial Peptoid-Based Compounds as Potential Agents against Canine Pyoderma. Molecules 2018, 23, 630. https://doi.org/10.3390/molecules23030630
Greco I, Hummel BD, Vasir J, Watts JL, Koch J, Hansen JE, Nielsen HM, Damborg P, Hansen PR. In Vitro ADME Properties of Two Novel Antimicrobial Peptoid-Based Compounds as Potential Agents against Canine Pyoderma. Molecules. 2018; 23(3):630. https://doi.org/10.3390/molecules23030630
Chicago/Turabian StyleGreco, Ines, Bernard D. Hummel, Jaspreet Vasir, Jeffrey L. Watts, Jason Koch, Johannes E. Hansen, Hanne Mørck Nielsen, Peter Damborg, and Paul R. Hansen. 2018. "In Vitro ADME Properties of Two Novel Antimicrobial Peptoid-Based Compounds as Potential Agents against Canine Pyoderma" Molecules 23, no. 3: 630. https://doi.org/10.3390/molecules23030630