The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
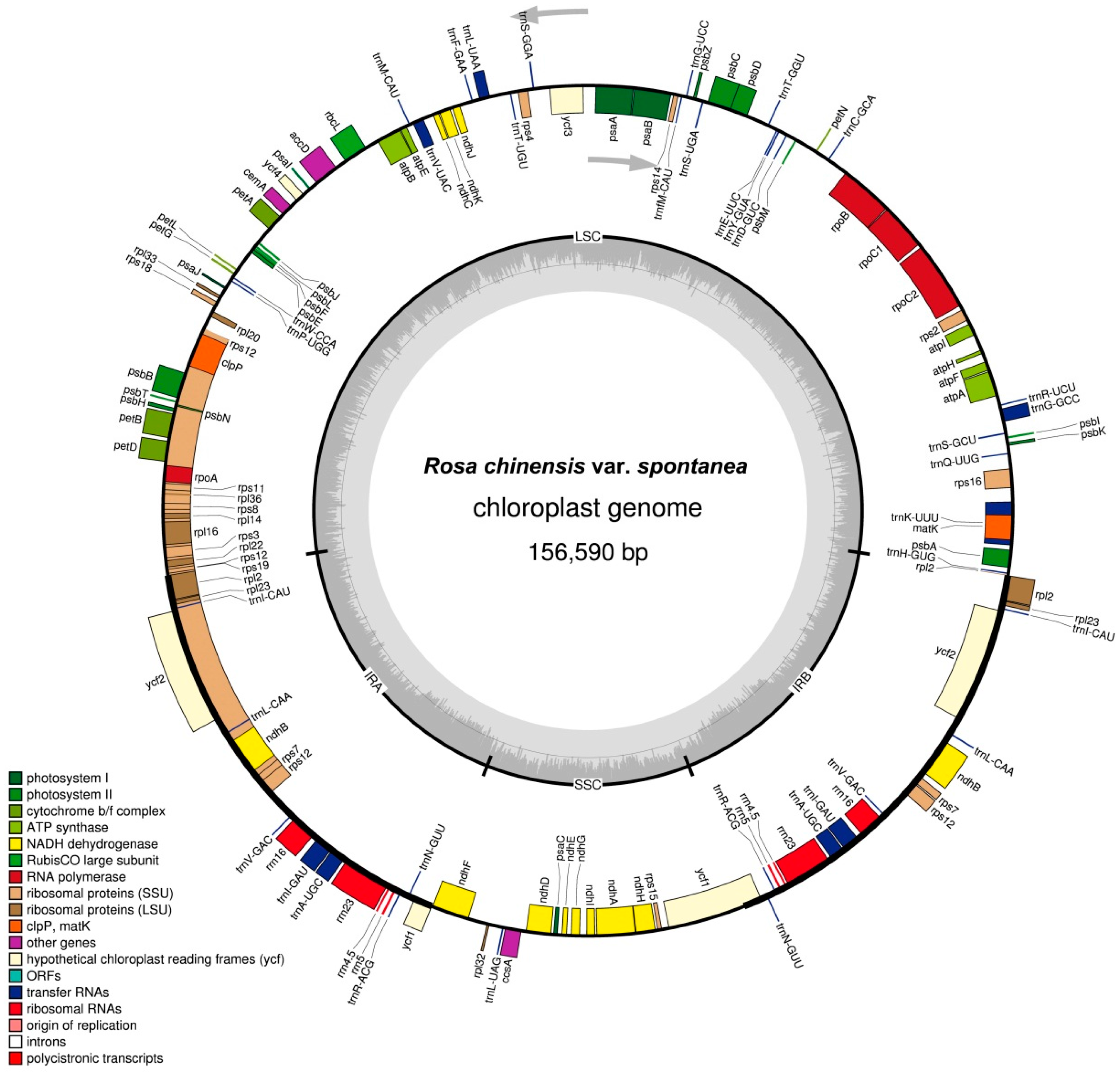
2.1. Characteristics of Chloroplast Genome of R. chinensis var. spontanea
2.2. Repeat and SSR Analysis
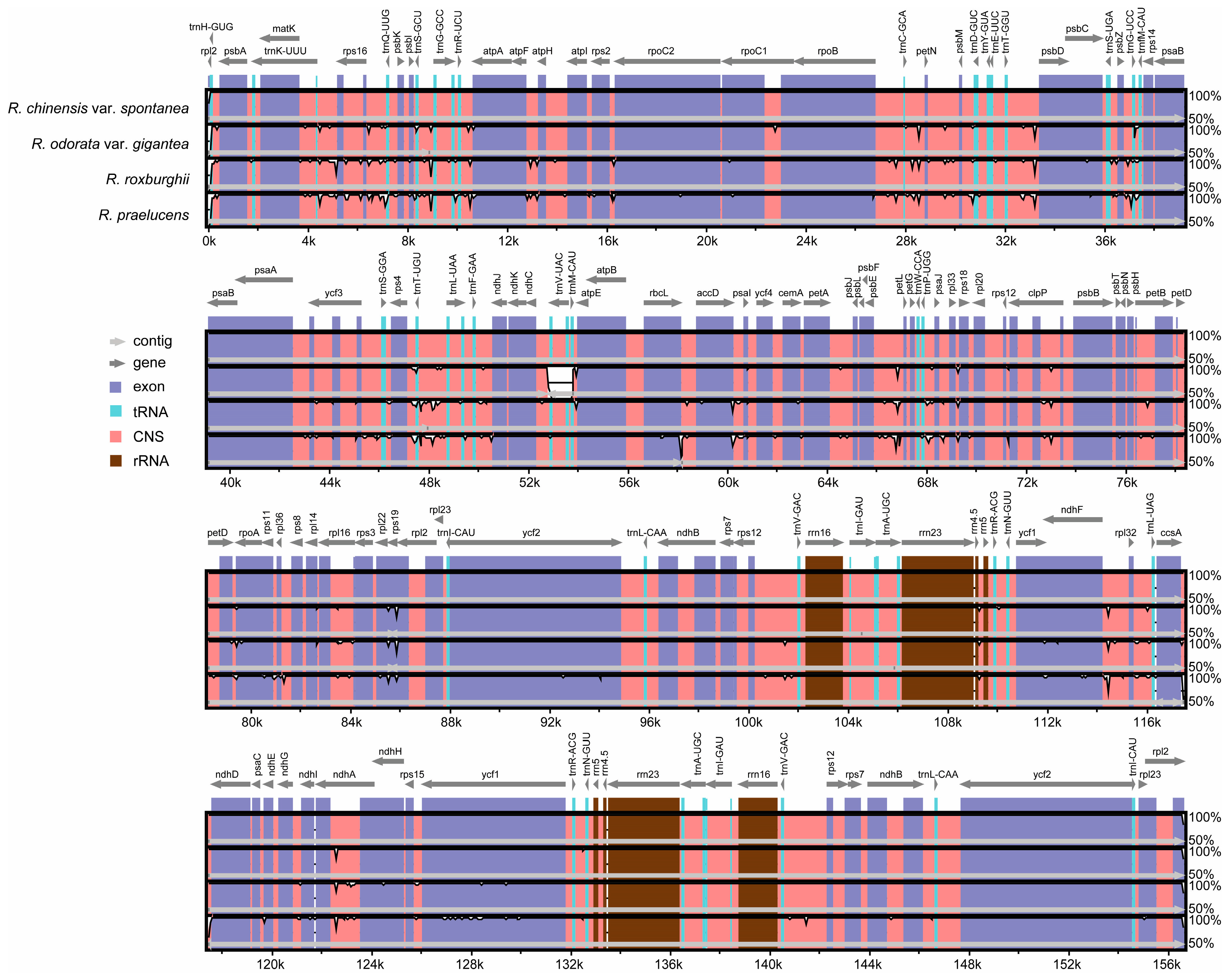
2.3. Comparative Analysis of the Chloroplast Genomes of the Genus Rosa
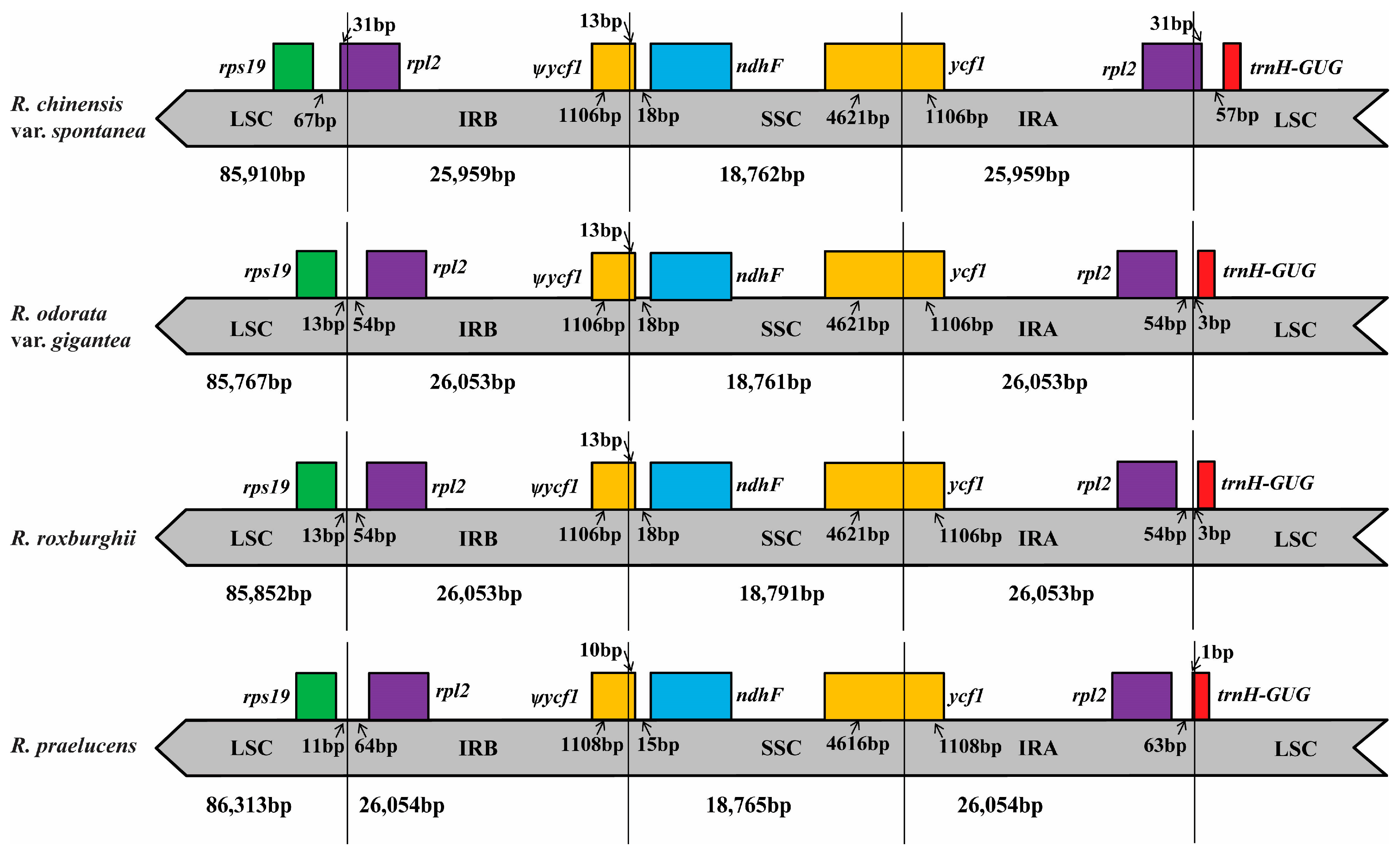
2.4. IR Contraction in the Chloroplast Genome of R. chinensis var. spontanea
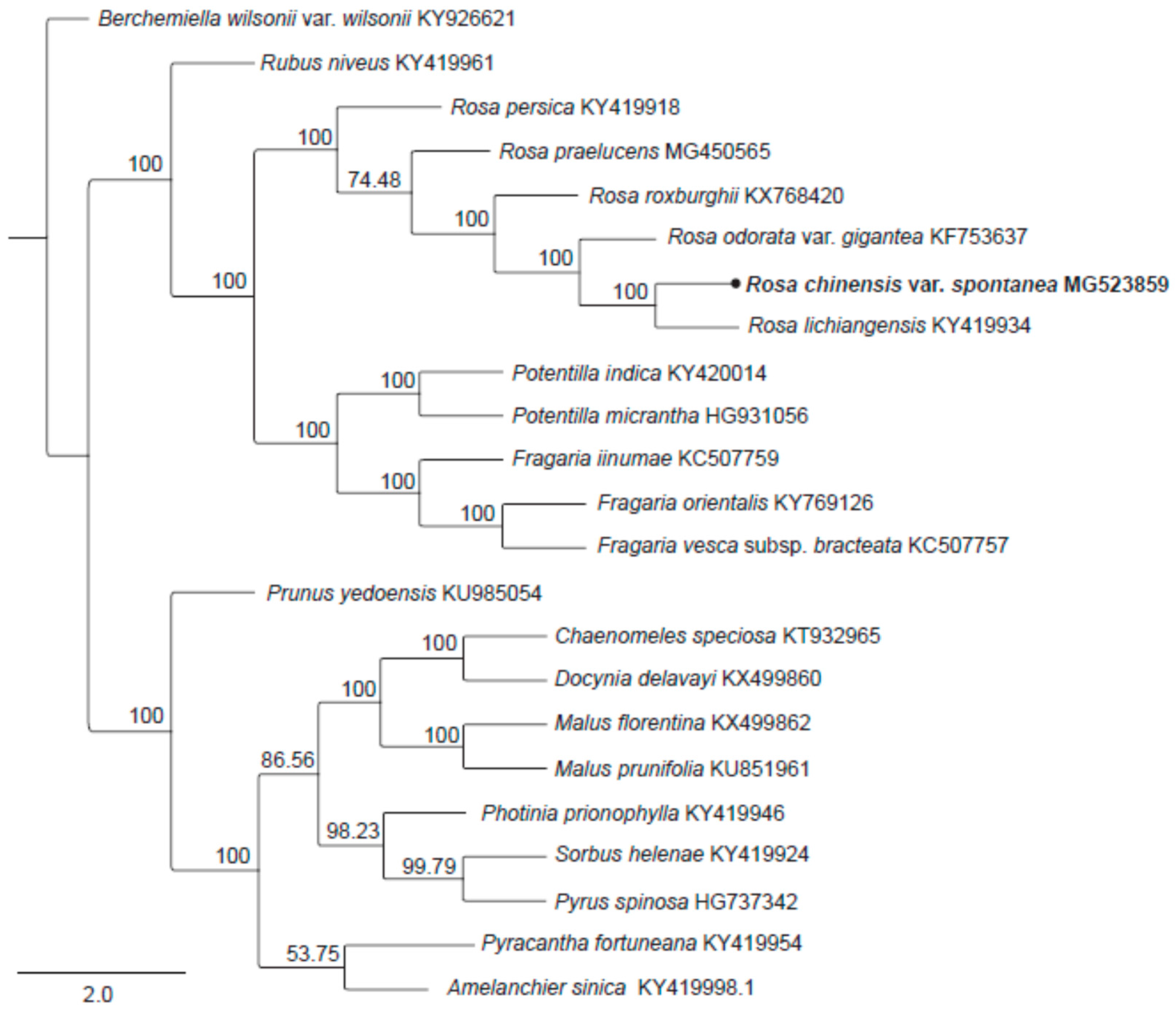
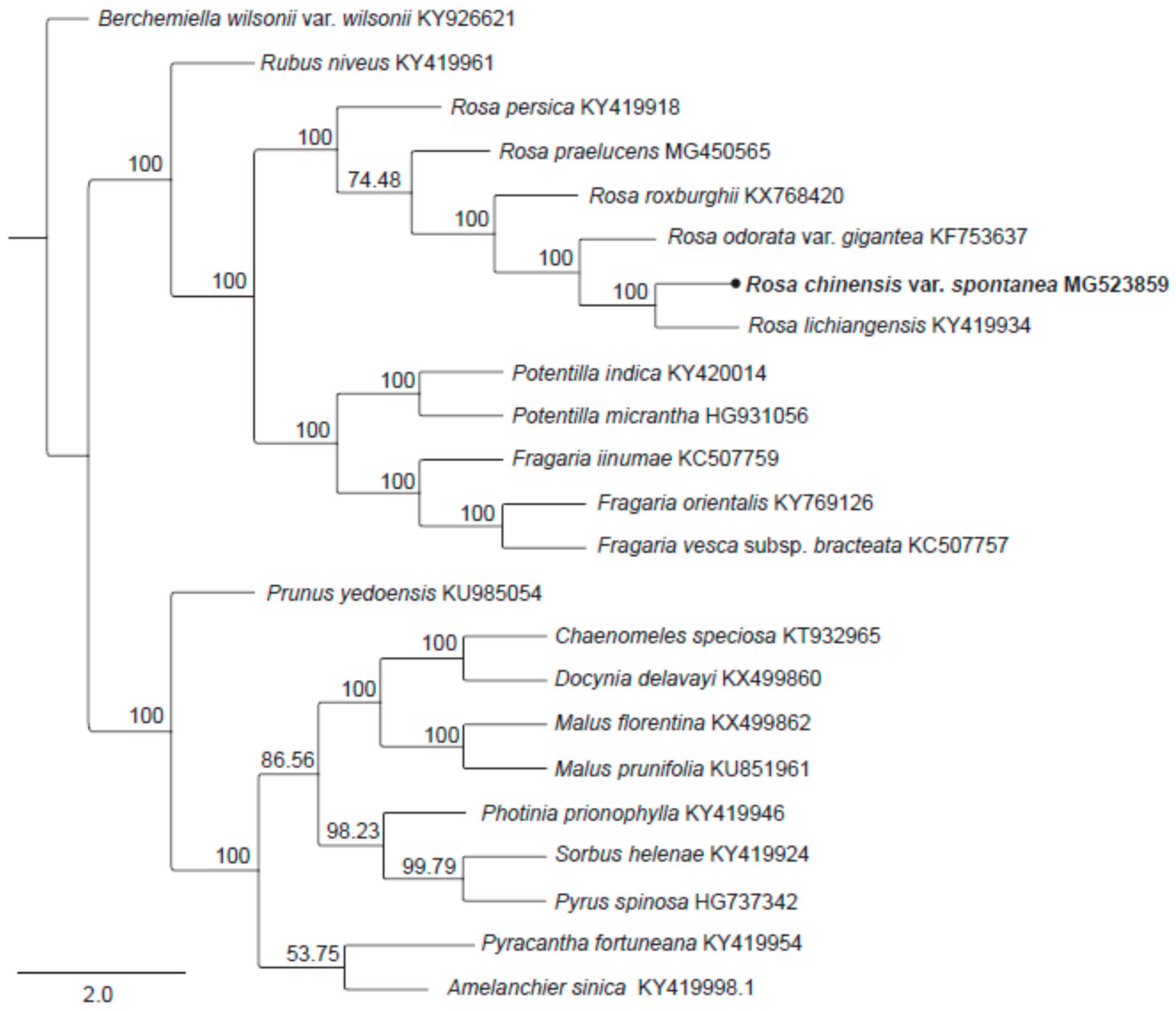
2.5. Phylogenetic Analysis
3. Materials and Methods
3.1. DNA Sequencing and Chloroplast Genome Assembly
3.2. Gene Annotation and Sequence Analysis
3.3. Genome Comparison
3.4. Repeats and Simple Sequence Repeats (SSRs)
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meng, J.; Fougère-Danezan, M.; Zhang, L.B.; Li, D.Z.; Yi, T.S. Untangling the hybrid origin of the Chinese tea roses: Evidence from DNA sequences of single-copy nuclear and chloroplast genes. Plant Syst. Evol. 2011, 297, 157–170. [Google Scholar] [CrossRef]
- Wylie, A. The history of garden roses. J. R. Hortic. Soc. 1954, 79, 555–571. [Google Scholar]
- Rix, M. Rosa chinensis f. spontanea. Curtis’s Bot. Mag. 2005, 22, 214–219. [Google Scholar] [CrossRef]
- Ye, J.Q. Modern Practical Herb; China Press of Traditional Chinese Medicine: Beijing, China, 2015; pp. 129–130. ISBN 9787513223744. [Google Scholar]
- Ku, T.C. Rosa. In Flora Reipublicae Popularis Sinicae; Editorial Board of the Flora Republicae Popularis Sinicae, Ed.; Science Press: Beijing, China, 1985; Volume 37, pp. 360–455. ISBN 7030040171. [Google Scholar]
- Ku, T.C.; Robertson, K.R. Rosa (Rosaceae). In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2003; Volume 9, pp. 339–381. ISBN 9787030130440. [Google Scholar]
- Qin, H.N.; Yang, Y.; Dong, S.Y.; He, Q.; Jia, Y.; Zhao, L.N.; Yu, S.X.; Liu, H.Y.; Liu, B.; Yan, Y.H.; et al. Threatened species list of China’s higher plants. Biodivers. Sci. 2017, 25, 696–744. [Google Scholar] [CrossRef]
- Akasaka, M.; Ueda, Y.; Koba, T. Karyotype analyses of five wild rose species belonging to septet A by fluorescence in situ hybridization. Chromosome Sci. 2002, 6, 17–26. [Google Scholar]
- Yomogida, K. Scent of modern roses. Kouryo 1992, 175, 65–89. [Google Scholar]
- Wu, S.; Watanabe, N.; Mita, S.; Ueda, Y.; Shibuya, M.; Ebizuka, Y. Two O-methytransferases isolated from flower petals of Rosa chinensis var. spontanea involved in scent biosynthesis. J. Biosci. Bioeng. 2003, 96, 119–128. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.Y.; Zhang, S.D.; Zhang, T.; Qiu, X.Q.; Yan, H.J.; Wang, Q.G.; Tang, K.X. Characterization of the complete chloroplast genome of a critically endangered decaploid rose species, Rosa praelucens (Rosaceae). Conserv. Genet. Resour. 2017. [Google Scholar] [CrossRef]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.F.; Wu, M.L.; Liao, B.S.; Liu, Z.X.; Bai, R.; Xiao, S.M.; Li, X.W.; Zhang, B.L.; Xu, J.; Chen, S.L. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Li, X.; Qian, J.; Wang, L.; Ma, L.; Tian, X.; Wang, Y. The complete chloroplast genome sequence of the medicinal plant Swertia mussotii. Using the PacBio RS II platform. Molecules 2016, 21, 1029. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qian, J.; Sun, Z.Y.; Xu, X.L.; Chen, S.L. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Wurdack, K.J.; Kanagaraj, A.; Lee, S.B.; Saski, C.; Jansen, R.K. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II Intron. Theor. Appl. Genet. 2008, 116, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Zhu, H.T.; Xing, Y.; Tan, J.J.; Chen, X.H.; Zhang, J.J.; Peng, H.F.; Xie, Q.J.; Zhang, Z.M. Albino leaf 2 is involved in the splicing of chloroplast group I and II Introns in rice. J. Exp. Bot. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Hübschmann, T.; Börner, T.; Hess, W.R. Splicing and intron-internal RNA editing of trnK-matK transcript in barley plastids: Support for MatK as an essential splice factor 1. J. Mol. Biol. 1997, 270, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 2000, 9, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Flannery, M.L.; Mitchell, F.J.; Coyne, S.; Kavanagh, T.A.; Burke, J.I.; Salamin, N.; Dowding, P.; Hodkinson, T.R. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 2006, 113, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, S.; Zhou, S.L. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 2012, 99, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Guan, Q.; Amin, A.; Wei, Z.; Li, M.; Li, X.; Lin, Z.; Tian, J. Complete plastid genome of Eriobotrya japonica (Thunb.) Lindl and comparative analysis in Rosaceae. SpringerPlus 2016, 5, 2036. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, J.F.; Zhang, H.; Cai, B.H.; Gao, Z.H.; Qiao, Y.S.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3919. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boorem, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174–201. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, A.; Starr, J.R.; Joly, S. Phylogenetic relationships in the genus Rosa: New evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst. Bot. 2007, 32, 366–378. [Google Scholar] [CrossRef]
- Fougère-Danezan, M.; Joly, S.; Bruneau, A.; Gao, X.F.; Zhang, L.B. Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann. Bot. 2015, 115, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.M.; Gao, X.F.; Fougère-Danezan, M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol. Phylogenet. Evol. 2016, 87, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Sotis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Rehder, A. Manual of Cultivated Trees and Shrubs Hardy in North America Exclusive of the Subtropical and Warmed Temperate Regions; Macmillan: New York, NY, USA, 1940. [Google Scholar]
- Wissemann, V. Conventional taxonomy (wild roses). In Encyclopedia of Rose Science; Roberts, A.V., Debener, T., Gudin, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 1, pp. 111–117. ISBN 0-12-227620-5. [Google Scholar]
- Patel, R.K.; Jain, M. NGS QC toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2017, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar Genome-DRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mudunuri, S.B.; Nagarajaram, H.A. IMEx: Imperfect Microsatellite Extractor. Bioinformatics 2007, 23, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sequence data of Rosa chinensis var. spontanea has been deposited into GenBank and are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | A | T (U) | G | C | Length | |
|---|---|---|---|---|---|---|
| LSC | 31.7 | 33.1 | 17.2 | 18.0 | 85,910 | |
| SSC | 34.4 | 34.3 | 15.1 | 16.3 | 18,762 | |
| IRB | 28.7 | 28.5 | 22.2 | 20.6 | 25,959 | |
| IRA | 28.7 | 28.5 | 22.2 | 20.6 | 25,959 | |
| Total | 31.0 | 31.8 | 18.6 | 18.7 | 156,590 | |
| PCGs | 30.6 | 31.4 | 20.3 | 17.7 | 79,773 | |
| 1st position | 30.7 | 24 | 26.9 | 18.7 | 26,591 | |
| 2nd position | 29.5 | 33 | 17.7 | 20.2 | 26,591 | |
| 3rd position | 31.7 | 38 | 16.4 | 14.1 | 26,591 |
| Amino Acid | Codon | Count | RSCU | tRNA | Amino Acid | Codon | Count | RSCU | tRNA |
|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 1015 | 1.3 | Ser | UCU | 580 | 1.62 | ||
| Phe | UUC | 545 | 0.7 | trnF-GAA | Ser | UCC | 370 | 1.03 | trnS-GGA |
| Leu | UUA | 897 | 1.87 | Ser | UCA | 406 | 1.13 | trnS-UGA | |
| Leu | UUG | 580 | 1.21 | trnL-CAA | Ser | UCG | 222 | 0.62 | |
| Leu | CUU | 595 | 1.24 | Pro | CCU | 424 | 1.45 | ||
| Leu | CUC | 217 | 0.45 | Pro | CCC | 241 | 0.82 | ||
| Leu | CUA | 380 | 0.79 | Pro | CCA | 320 | 1.09 | trnP-UGG | |
| Leu | CUG | 202 | 0.42 | Pro | CCG | 187 | 0.64 | ||
| Ile | AUU | 1136 | 1.48 | Thr | ACU | 542 | 1.55 | ||
| Ile | AUC | 451 | 0.59 | trnI-CAU | Thr | ACC | 269 | 0.77 | trnT-GGU |
| Ile | AUA | 716 | 0.93 | Thr | ACA | 418 | 1.19 | trnT-UGU | |
| Met | AUG | 635 | 1 | trnM-CAU | Thr | ACG | 171 | 0.49 | |
| Val | GUU | 550 | 1.44 | Ala | GCU | 645 | 1.76 | ||
| Val | GUC | 193 | 0.5 | trnV-GAC | Ala | GCC | 244 | 0.67 | |
| Val | GUA | 567 | 1.48 | Ala | GCA | 391 | 1.07 | ||
| Val | GUG | 223 | 0.58 | Ala | GCG | 183 | 0.5 | ||
| Tyr | UAU | 798 | 1.6 | Cys | UGU | 237 | 1.48 | ||
| Tyr | UAC | 198 | 0.4 | trnY-GUA | Cys | UGC | 83 | 0.52 | trnC-GCA |
| stop | UAA | 59 | 1.38 | stop | UGA | 21 | 0.49 | ||
| stop | UAG | 48 | 1.13 | Trp | UGG | 484 | 1 | trnW-CCA | |
| His | CAU | 476 | 1.49 | Arg | CGU | 362 | 1.28 | trnR-ACG | |
| His | CAC | 161 | 0.51 | trnH-GUG | Arg | CGC | 120 | 0.43 | |
| Gln | CAA | 734 | 1.51 | trnQ-UUG | Arg | CGA | 385 | 1.36 | |
| Gln | CAG | 236 | 0.49 | Arg | CGG | 144 | 0.51 | ||
| Asn | AAU | 1003 | 1.52 | Ser | AGU | 420 | 1.17 | ||
| Asn | AAC | 317 | 0.48 | Ser | AGC | 156 | 0.43 | trnS-GCU | |
| Lys | AAA | 1082 | 1.48 | Arg | AGA | 488 | 1.73 | trnR-UCU | |
| Lys | AAG | 385 | 0.52 | Arg | AGG | 194 | 0.69 | ||
| Asp | GAU | 890 | 1.62 | Gly | GGU | 612 | 1.3 | ||
| Asp | GAC | 207 | 0.38 | trnD-GUC | Gly | GGC | 209 | 0.44 | trnG-GCC |
| Glu | GAA | 1052 | 1.46 | trnE-UUC | Gly | GGA | 694 | 1.48 | |
| Glu | GAG | 390 | 0.54 | Gly | GGG | 365 | 0.78 |
| ID | Repeat Start 1 | Type | Size (bp) | Repeat Start 2 | Mismatch (bp) | E-Value | Gene | Region |
|---|---|---|---|---|---|---|---|---|
| 1 | 4426 | F | 29 | 45,071 | −2 | 8.74 × 10−5 | IGS; ycf3(intron) | LSC |
| 2 | 4427 | F | 30 | 4428 | −3 | 6.56 × 10−4 | IGS | LSC |
| 3 | 4428 | F | 28 | 45,072 | −3 | 8.47 × 10−3 | IGS | LSC |
| 4 | 4432 | F | 26 | 45,072 | −2 | 4.48 × 10−3 | IGS | LSC |
| 5 | 8329 | F | 29 | 36,077 | −2 | 8.74 × 10−5 | trnS-GCU; trnS-UGA | LSC |
| 6 | 8873 | F | 20 | 8895 | 0 | 6.27 × 10−3 | IGS | LSC |
| 7 | 9804 | F | 27 | 37,135 | −1 | 3.10 × 10−5 | trnG-GCU; trnG-UCC | LSC |
| 8 | 13,510 | F | 20 | 89,606 | 0 | 6.27 × 10−3 | IGS; ycf2 | LSC; IRa |
| 9 | 14,236 | F | 20 | 29,560 | 0 | 6.27 × 10−3 | IGS | LSC |
| 10 | 27,619 | F | 24 | 27,643 | 0 | 2.45 × 10−5 | IGS | LSC |
| 11 | 29,555 | F | 24 | 29,556 | −1 | 1.76 × 10−3 | IGS | LSC |
| 12 | 33,157 | F | 20 | 33,177 | 0 | 6.27 × 10−3 | IGS | LSC |
| 13 | 39,390 | F | 30 | 41,614 | −3 | 6.56 × 10−4 | psaB; psaA | LSC |
| 14 | 42,625 | F | 25 | 147,248 | −1 | 4.59 × 10−4 | IGS | LSC; IRb |
| 15 | 44,406 | F | 39 | 100,262 | 0 | 2.28 × 10−14 | ycf3(intron); IGS | LSC; IRa |
| 16 | 44,406 | F | 38 | 122,332 | 0 | 9.13 × 10−14 | ycf3(intron); ndhA(intron) | LSC; SSC |
| 17 | 45,075 | F | 24 | 142,008 | −1 | 1.76 × 10−3 | ycf3(intron); IGS | LSC; IRb |
| 18 | 47,622 | F | 25 | 47,645 | 0 | 6.13 × 10−6 | IGS | LSC |
| 19 | 58,656 | F | 34 | 58,687 | 0 | 2.34 × 10−11 | IGS | LSC |
| 20 | 66,712 | F | 41 | 66,752 | 0 | 1.43 × 10−15 | IGS | LSC |
| 21 | 66,939 | F | 20 | 66,958 | 0 | 6.27 × 10−3 | IGS | LSC |
| 22 | 68,033 | F | 21 | 68,052 | 0 | 1.57 × 10−3 | IGS | LSC |
| 23 | 71,232 | F | 20 | 84,928 | 0 | 6.27 × 10−3 | IGS | LSC |
| 24 | 80,953 | F | 27 | 80,966 | −2 | 1.21 × 10−3 | IGS | LSC |
| 25 | 83,166 | F | 29 | 122,320 | −3 | 2.36 × 10−3 | rpl16(intron); ndhA(intron) | LSC;SSC |
| 26 | 83,172 | F | 28 | 122,326 | −3 | 8.47 × 10−3 | rpl16(intron); ndhA(intron) | LSC;SSC |
| 27 | 90,610 | F | 29 | 90,631 | −2 | 8.74 × 10−5 | ycf2 | IRa |
| 28 | 97,630 | F | 31 | 144,839 | −3 | 1.81 × 10−4 | ndhB(intron) | IRa; IRb |
| 29 | 100,260 | F | 40 | 122,330 | 0 | 5.70 × 10−15 | IGS; ndhA(intron) | IRa; SSC |
| 30 | 101,012 | F | 23 | 101,033 | 0 | 9.80 × 10−5 | IGS | IRa |
| 31 | 141,437 | F | 30 | 141,458 | −2 | 2.34 × 10−5 | IGS | IRb |
| 32 | 141,444 | F | 23 | 141,465 | 0 | 9.80 × 10−5 | IGS | IRb |
| 33 | 151,840 | F | 29 | 151,861 | −2 | 8.74 × 10−5 | ycf2 | IRb |
| 34 | 6406 | I | 20 | 71,231 | 0 | 6.27 × 10−3 | IGS | LSC |
| 35 | 6408 | I | 24 | 71,228 | −1 | 1.76 × 10−3 | IGS | LSC |
| 36 | 8622 | I | 26 | 45,073 | −2 | 4.48 × 10−3 | IGS | LSC |
| 37 | 8625 | I | 23 | 45,077 | −1 | 6.76 × 10−3 | IGS | LSC |
| 38 | 71,232 | I | 20 | 84,930 | 0 | 6.27 × 10−3 | IGS | LSC |
| ID | Repeat Motif | Length (bp) | Start | End | Region | Gene | ID | Repeat Motif | Length (bp) | Start | End | Region | Gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (A)11 | 11 | 279 | 289 | LSC | 44 | (TTTA)4 | 12 | 50,468 | 50,479 | LSC | ||
| 2 | (T)11 | 11 | 4108 | 4118 | LSC | 45 | (TA)5 | 10 | 52,742 | 52,751 | LSC | ||
| 3 | (A)19 | 19 | 4428 | 4446 | LSC | 46 | (T)10 | 10 | 55,811 | 55,820 | LSC | atpB | |
| 4 | (A)10 | 10 | 4449 | 4458 | LSC | 47 | (AAAT)3 | 12 | 55,911 | 55,922 | LSC | ||
| 5 | (A)10 | 10 | 4887 | 4896 | LSC | 48 | (TAAT)3 | 12 | 58,366 | 58,377 | LSC | ||
| 6 | (T)10 | 10 | 5023 | 5032 | LSC | 49 | (T)14 | 14 | 60,810 | 60,823 | LSC | ||
| 7 | (TATAT)3 | 15 | 6102 | 6116 | LSC | rps16 | 50 | (TC)5 | 10 | 62,280 | 62,289 | LSC | cemA |
| 8 | (T)17 | 17 | 6407 | 6423 | LSC | 51 | (T)11 | 11 | 64,513 | 64,523 | LSC | ||
| 9 | (AATA)3 | 12 | 6525 | 6536 | LSC | 52 | (T)10 | 10 | 69,689 | 69,698 | LSC | ||
| 10 | (AG)5 | 10 | 6755 | 6764 | LSC | 53 | (A)16 | 16 | 69,739 | 69,754 | LSC | ||
| 11 | (A)11 | 11 | 6945 | 6955 | LSC | 54 | (T)18 | 18 | 71,235 | 71,252 | LSC | ||
| 12 | (TAA)4 | 12 | 8257 | 8268 | LSC | 55 | (T)15 | 15 | 71,933 | 71,947 | LSC | clpP | |
| 13 | (A)10 | 10 | 8639 | 8648 | LSC | 56 | (A)10 | 10 | 72,733 | 72,742 | LSC | clpP | |
| 14 | (AT)6 | 12 | 10,093 | 10,104 | LSC | 57 | (AT)6 | 12 | 73,632 | 73,643 | LSC | ||
| 15 | (TAT)4 | 12 | 10,343 | 10,354 | LSC | 58 | (A)12 | 12 | 79,231 | 79,242 | LSC | ||
| 16 | (T)11 | 11 | 12,157 | 12,167 | LSC | 59 | (A)14 | 14 | 79,393 | 79,406 | LSC | ||
| 17 | (T)10 | 10 | 12,915 | 12,924 | LSC | 60 | (T)10 | 10 | 79,429 | 79,438 | LSC | rpoA | |
| 18 | (A)10 | 10 | 13,184 | 13,193 | LSC | 61 | (ATGT)3 | 12 | 79,529 | 79,540 | LSC | rpoA | |
| 19 | (C)10 | 10 | 14,237 | 14,246 | LSC | 62 | (T)11 | 11 | 81,586 | 81,596 | LSC | ||
| 20 | (T)10 | 10 | 14,247 | 14,256 | LSC | 63 | (A)10 | 10 | 82,641 | 82,650 | LSC | ||
| 21 | (T)11 | 11 | 18,361 | 18,371 | LSC | rpoC2 | 64 | (A)12 | 12 | 83,422 | 83,433 | LSC | rpl16 |
| 22 | (TA)5 | 10 | 19,730 | 19,739 | LSC | rpoC2 | 65 | (A)11 | 11 | 83,498 | 83,508 | LSC | rpl16 |
| 23 | (T)10 | 10 | 26,080 | 26,089 | LSC | rpoB | 66 | (T)18 | 18 | 84,931 | 84,948 | LSC | |
| 24 | (T)12 | 12 | 28,925 | 28,936 | LSC | 67 | (TAT)4 | 12 | 86,619 | 86,630 | IRa | rpl2 | |
| 25 | (C)15 | 15 | 29,556 | 29,570 | LSC | 68 | (TAGAAG)3 | 18 | 93,987 | 94,004 | IRa | ycf2 | |
| 26 | (T)10 | 10 | 29,571 | 29,580 | LSC | 69 | (T)11 | 11 | 101,618 | 101,628 | IRa | ||
| 27 | (AAT)4 | 12 | 30,504 | 30,515 | LSC | 70 | (AGGT)3 | 12 | 107,843 | 107,854 | IRa | rrn23 | |
| 28 | (T)14 | 14 | 30,519 | 30,532 | LSC | 71 | (TATT)3 | 12 | 110,028 | 110,039 | IRa | ||
| 29 | (A)10 | 10 | 30,666 | 30,675 | LSC | 72 | (TGT)4 | 12 | 111,869 | 111,880 | SSC | ||
| 30 | (TA)5 | 10 | 36,313 | 36,322 | LSC | 73 | (T)10 | 10 | 115,507 | 115,516 | SSC | ||
| 31 | (T)11 | 11 | 36,473 | 36,483 | LSC | 74 | (TAA)4 | 12 | 115,558 | 115,569 | SSC | ||
| 32 | (AT)5 | 12 | 37,070 | 37,079 | LSC | 75 | (A)13 | 13 | 115,612 | 115,624 | SSC | ||
| 33 | (C)13 | 13 | 37,303 | 37,315 | LSC | 76 | (T)10 | 10 | 120,845 | 120,854 | SSC | ||
| 34 | (A)11 | 11 | 37,316 | 37,326 | LSC | 77 | (AT)7 | 14 | 121,678 | 121,691 | SSC | ||
| 35 | (AT)5 | 10 | 43,682 | 43,691 | LSC | ycf3 | 78 | (A)16 | 16 | 122,551 | 122,566 | SSC | ndhA |
| 36 | (A)15 | 15 | 45,073 | 45,087 | LSC | ycf3 | 79 | (T)15 | 15 | 122,804 | 122,818 | SSC | ndhA |
| 37 | (A)10 | 10 | 45,392 | 45,401 | LSC | 80 | (T)10 | 10 | 129,830 | 129,839 | SSC | ycf1 | |
| 38 | (T)10 | 10 | 45,931 | 45,940 | LSC | 81 | (ATAA)3 | 12 | 132,463 | 132,474 | IRb | ||
| 39 | (A)11 | 11 | 47,296 | 47,306 | LSC | 82 | (CTAC)3 | 12 | 134,645 | 134,656 | IRb | rrn23 | |
| 40 | (TAAT)3 | 12 | 48,112 | 48,123 | LSC | 83 | (A)11 | 11 | 140,873 | 140,883 | IRb | ||
| 41 | (T)14 | 14 | 48,306 | 48,319 | LSC | 84 | (CTTCTA)3 | 18 | 148,497 | 148,514 | IRb | ycf2 | |
| 42 | (A)12 | 12 | 48,420 | 48,431 | LSC | 85 | (ATA)4 | 12 | 155,871 | 155,882 | IRb | ||
| 43 | (TA)5 | 10 | 48,500 | 48,509 | LSC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, H.-Y.; Zhang, Y.-H.; Yan, H.-J.; Qiu, X.-Q.; Wang, Q.-G.; Li, S.-B.; Zhang, S.-D. The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species. Molecules 2018, 23, 389. https://doi.org/10.3390/molecules23020389
Jian H-Y, Zhang Y-H, Yan H-J, Qiu X-Q, Wang Q-G, Li S-B, Zhang S-D. The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species. Molecules. 2018; 23(2):389. https://doi.org/10.3390/molecules23020389
Chicago/Turabian StyleJian, Hong-Ying, Yong-Hong Zhang, Hui-Jun Yan, Xian-Qin Qiu, Qi-Gang Wang, Shu-Bin Li, and Shu-Dong Zhang. 2018. "The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species" Molecules 23, no. 2: 389. https://doi.org/10.3390/molecules23020389