3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Substituted Methylamines 2d, 2k and 2m
To a stirred mixture of 1-adamantanecarboxylic acid (
11, 36.05 g, 200 mmol) and DMF (5 drops) in CH
2Cl
2 (360 mL) cooled in an ice-water bath was added thionyl chloride (26.17 g, 220 mmol) in one portion. The resulting mixture was refluxed until the evolution of HCl gas ceased (typically within 2 h). The solvent and excess thionyl chloride were then removed on a rotary evaporator, and the crude acid chloride
12 was dissolved in THF (100 mL). The resulting solution was then slowly added dropwise to stirred concentrated aqueous ammonia (400 mL) cooled in an ice-water bath. After addition, the resulting white slurry was stirred at room temperature for another 2 h, and the white precipitates were collected via vacuum filtration, washed with cold
n-hexane and dried in vacuo to afford 1-adamantanecarboxamide (
13). White solid; 34.06 g (95%); m.p. 177–179 °C (literature value, 185.5–191.7 °C [
48]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 6.89 (brs, 1H), 6.62 (brs, 1H), 1.93 (s, 3H), 1.73–1.74 (m, 6H), 1.60–1.68 (m, 6H).
A mixture of 1-methylnaphthalene (14, 28.44 g, 200 mmol) and NBS (39.16 g, 220 mmol) in MeCN (300 mL) was stirred at 30-40°C until completion of the reaction as indicated by TLC analysis (typically within 12 h). On cooling to room temperature, the reaction mixture was poured into ice-water (600 mL), and the resulting mixture was extracted with CH2Cl2 (500 mL × 3). The combined extracts were washed successively with 5% aqueous Na2S2O3 (200 mL), saturated aqueous Na2CO3 (100 mL × 3) and 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford 1-bromo-4-methylnaphthalene (15). Colorless oil, 42.01 g (98%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.14–8.16 (m, 1H), 8.07–8.09 (m, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.66–7.70 (m, 2H), 7.30 (d, J = 7.6 Hz, 1H), 2.63 (s, 3H).
A mixture of 15, 9k or 9m (190 mmol) and CuCN (49.00 g, 570 mmol) in DMF (500 mL) were stirred at 130 °C (for 15 and 9m) or reflux (for 9k) in N2 atmosphere for 12 h, when TLC analysis indicated completion of reaction. On cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (1000 mL), and the resulting mixture was further stirred for 1 h and filtered off. The filtrate was washed with 5% brine (500 mL × 5), dried (Na2SO4) and evaporated on a rotary evaporator, which was purified by column chromatography to afford 16, 10k or 10m.
4-Methyl-1-naphthonitrile (
16): White solid; 24.78 g (78%); m.p. 54–55 °C (literature value, 53–54 °C [
49]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.20 (d,
J = 8.0 Hz, 1H), 8.11 (d,
J = 8.0 Hz, 1H), 8.04 (d,
J = 7.2 Hz, 1H), 7.72–7.82 (m, 2H), 7.53 (d,
J = 7.2 Hz, 1H), 2.74 (s, 3H).
Benzo[b]furan-5-carbonitrile (
10k): White solid; 23.12 g (85%); m.p. 81–83 °C (literature value, 81–82 °C [
50]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.22 (d,
J = 1.2 Hz, 1H), 8.19 (d,
J = 2.0 Hz, 1H), 7.81 (d,
J = 8.4 Hz, 1H), 7.73–7.75 (m, 1H), 7.08 (d,
J = 1.6 Hz, 1H).
2-Naphthonitrile (
10m): White solid; 20.96 g (72%); m.p. 63–65 °C (literature value, 65–66 °C [
51]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.57 (s, 1H), 8.03–8.11 (m, 3H), 7.65–7.78 (m, 3H).
To a stirred solution of 13, 10k or 10m (130 mmol) in dried THF (150 mL) cooled in an ice-water bath was added portionwise LiAlH4 (6.41 g, 169 mmol). Thereafter the reaction mixture was stirred at room temperature for 1 h and then at reflux for another 5 h, when the reaction completed as indicated by TLC analysis. On cooling to room temperature, the reaction mixture was carefully poured into ice-water (500 mL) while stirring, and the resulting mixture was diluted with CH2Cl2 (300 mL), stirred for 0.5 h and filtered off through Celite. The organic phase was separated from the filtrate, and the aqueous phase was back-extracted with CH2Cl2 (200 mL × 2). The combined organic phases were washed with saturated brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography through a short silica gel column to afford 2d, 2k or 2m. These amines were used directly in the next step without further purification and characterization.
3.2.2. General Procedure for the Synthesis of Substituted Methylamines 2ha–2hi
To a stirred solution of 5-bromothiophene-2-carbaldehyde (
18, 38.22 g, 200 mmol) and phenylboronic acid (26.82 g, 220 mmol) in dioxane/H
2O (9/1 by
v/
v, 600 mL in total), K
2CO
3 (110.56 g, 800 mmol) and Pd(PPh
3)
4 (11.56 g, 10 mmol) were added. The reaction mixture was heated at reflux for 10 h in N
2 atmosphere. On cooling to room temperature, the reaction mixture was filtered off. The organic phase was evaporated on a rotary evaporator, and the residue thus obtained was extracted with CH
2Cl
2 (200 mL × 3). The combined extracts were washed with 5% brine (200 mL), dried (Na
2SO
4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford 5-phenylthiophene-2-carbaldehyde (
10hg). White solid; 36.90 g (98%); m.p. 89–91 °C (literature value, 92–93 °C [
52]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 9.91 (s, 1H), 8.04 (d,
J = 4.0 Hz, 1H), 7.79–7.81 (m, 2H), 7.74 (d,
J = 4.0 Hz, 1H), 7.41–7.50 (m, 3H).
To a stirred solution of 9ha–9hd (200 mmol) in dried DMF (43.86 g, 600 mmol) cooled in an ice-water bath was added dropwise POCl3 (46.00 g, 300 mmol). The resulting mixture was stirred at this temperature for 30 min and then at 100 °C for another 5 h. After cooling to room temperature, the reaction mixture was poured carefully into ice-water (300 mL). The mixture thus obtained was extracted with CH2Cl2 (300 mL × 3), and the combined extracts were washed successively with 5% brine (200 mL), 10% aqueous K2CO3 (200 mL) and 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to yield 10ha–10hd.
5-Chlorothiophene-2-carbaldehyde (
10ha): Colorless oil; 22.00 g (75%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 9.82 (s, 1H), 7.95 (d,
J = 4.0 Hz, 1H), 7.39 (d,
J = 4.0 Hz, 1H). The
1H-NMR data were in good agreement with those reported [
53].
5-Methylthiophene-2-carbaldehyde (
10hb): Colorless oil; 21.20 g (84%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 9.80 (s, 1H), 7.84 (d,
J = 4.0 Hz, 1H), 7.04–7.05 (m, 1H), 2.54 (s, 3H). The
1H-NMR data were in good agreement with those reported [
53].
5-Ethylthiophene-2-carbaldehyde (10hc): Colorless oil; 22.72 g (81%). 1H-NMR (DMSO-d6, 400 MHz), δ: 9.81 (s, 1H), 7.85 (d, J = 3.6 Hz, 1H), 7.06–7.07 (m, 1H), 2.88 (q, J = 7.5 Hz, 2H), 1.25 (t, J = 7.4 Hz, 3H).
3-Methylthiophene-2-carbaldehyde (
10hd): Colorless oil; 22.46 g (89%). An inseparable mixture of regioisomers as indicated by
1H-NMR. The
1H-NMR data were in good agreement with those reported [
38].
To a stirred solution of 9he, 9hh or 9hi (200 mmol) in dried THF (200 mL) held at −78 °C in N2 atmosphere was added dropwise 1.6 M n-BuLi in n-hexane (125 mL, 200 mmol) via syringe, and the resulting solution was stirred at −78 °C for 1 h ( for 9he and 9hh) or returned to room temperature for 5 h (for 9hi) before dropwise addition of dried DMF (21.93 g, 300 mmol) via syringe. Thereafter the reaction mixture was stirred at −78 °C for 0.5 h and at room temperature for another 1 h. The reaction mixture was poured into ice-water (600 mL) while stirring and the resulting mixture was extracted with CH2Cl2 (300 mL × 3). The combined extracts were washed with 5% brine (500 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford the pure product 10he, 10hh or 10hi.
4-Methylthiophene-2-carbaldehyde (
10he): Colorless oil; 23.97 g (95%). An inseparable mixture of regioisomers as indicated by
1H-NMR. The
1H-NMR data were in good agreement with those reported [
38].
Benzo[b]thiophene-2-carbaldehyde (
10hh): Colorless oil; 29.80 g (92%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 10.14 (s, 1H), 8.43 (s, 1H), 8.09–8.12 (m, 2H), 7.56–7.60 (m, 1H), 7.48–7.52 (m, 1H). The
1H-NMR data were in good agreement with those reported [
54].
Bibenzo[b,d]thiophene-4-carbaldehyde (
10hi): White solid; 36.08 g (85%); m.p. 117–119 °C.
1H-NMR (CDCl
3, 400 MHz) δ: 10.27 (s, 1H), 8.40 (d,
J = 7.6 Hz, 1H), 8.17–8.22 (m, 1H), 7.93–7.97 (m, 2H), 7.64 (t,
J = 7.6 Hz, 1H), 7.47–7.54 (m, 2H). The
1H-NMR data were in good agreement with those reported [
55].
To a stirred solution of 10ha–10hi (145 mmol) and NH2OH·HCl (20.15 g, 290 mmol) in 90% EtOH (300 mL) was added dropwise an aqueous solution prepared by dissolving K2CO3 (24.05 g, 174 mmol) in a minimal amount of water. After addition, the mixture was stirred at room temperature until the reaction completed as indicated by TLC analysis (typically within 5 h). The reaction mixture was concentrated to about one-third of its original volume on a rotary evaporator and the residue was poured into ice-water (400 mL). The resulting mixture was adjusted to pH 6–7 with diluted hydrochloric acid and extracted with CH2Cl2 (200 mL × 3). The combined extracts were washed with 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by recrystallization from n-hexane/EtOAc (5/1 by v/v) to give 11ha–11hi. Oximes 11ha–11hi were used directly in the next step without further characterization.
The synthesis of 2ha–2hi from 11ha–11hi by the reduction with LiAlH4 was carried out using the identical operation to that for the synthesis of 2d, 2k and 2m from 13, 10k and 10m, respectively.
3.2.3. General Procedure for the Synthesis of Isothiocyanates 3c–3n and 3ha–3hi
A suspension of 16 (23.40 g, 140 mmol), BPO (5.09 g, 21 mmol) and NBS (29.90 g, 168 mmol) in CCl4 (400 mL) was stirred at reflux in N2 atmosphere until the completion of reaction as indicated by TLC analysis (typically within 12 h; once the reaction commenced, 2.55 g of BPO was added every 5 h until the reaction completed). The reaction mixture was cooled to room temperature and filtered off, and the filtrate was successively washed with saturated aqueous NaHCO3 (200 mL × 5) and 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to yield (4-cyano-1-naphthyl)methyl bromide (17). White solid; 31.35 g (91%); m.p. 113–115 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.34–8.37 (m, 1H), 8.13–8.21 (m, 2H), 7.82–7.89 (m, 3H), 5.27 (s, 2H).
To a stirred solution of 17 (30.76 g, 125 mmol) in DMF (300 mL) was added KSCN (24.30 g, 250 mmol), and the mixture thus obtained was stirred for 4 h at 140 °C in N2 atmosphere until the completion of reaction as indicated by TLC analysis. On cooling to room temperature, the reaction mixture was poured into ice-water (600 mL) while stirring. The resulting mixture was exacted with CH2Cl2 (200 mL × 3). The combined extracts were washed with 5% brine (300 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford (4-cyano-1-naphthyl)methyl isothiocyanate (3l). White solid; 20.47 g (73%); m.p. 113–115 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.18–8.22 (m, 3H), 7.81–7.89 (m, 2H), 7.75 (d, J = 7.6 Hz, 1H), 5.56 (s, 2H).
To a stirred solution of thiophosgene (15.18 g, 132 mmol) in dried CH2Cl2 (150 mL) cooled in an ice-water bath was added dropwise a solution prepared by dissolving 2c–2k, 2m–2n or 2ha–2hi (120 mmol) and DIPEA (46.53 g, 360 mmol) in dried CH2Cl2 (150 mL). The resulting mixture was stirred for 1 h in an ice-water bath and for another 1 h at room temperature. The reaction mixture was then poured into ice-water (300 mL) while stirring. The organic phase was separated, and the aqueous phase was back-extracted with CH2Cl2 (200 mL × 2). The combined organic phases were washed successively with 5% hydrochloric acid (100 mL × 2; for 3f and 3n, the washing with 5% hydrochloric acid is omitted) and saturated brine (300 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford 3c–3k, 3m–3n or 3ha–3hi.
Cyclohexylmethyl isothiocyanate (
3c): Colorless oil; 18.26 g (98%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 3.51 (d,
J = 6.4 Hz, 2H), 1.60–1.70 (m, 6H), 1.17–1.27 (m, 2H), 1.08–1.14 (m, 1H), 0.93–1.03 (m, 2H). The
1H-NMR data were in good agreement with those reported [
56].
1-Adamantylmethyl isothiocyanate (3d): White solid; 22.89 g (92%); m.p. 76–77 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 3.33 (s, 2H), 1.98 (s, 3H), 1.67–1.70 (m, 3H), 1.57–1.60 (m, 3H), 1.50–1.51 (m, 6H).
Benzyl isothiocyanate (
3e): Colorless oil; 15.94 g (89%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 7.34–7.44 (m, 5H), 4.93 (s, 2H). The
1H-NMR data were in good agreement with those reported [
57].
3-Pyridinylmethyl isothiocyanate (
3f): Colorless oil; 10.81 g (ca 61%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.59 (d,
J = 1.6 Hz, 1H), 8.56 (d,
J = 1.2 Hz and 4.8 Hz, 1H), 7.82 (d,
J = 7.6 Hz, 1H), 7.45 (dd,
J = 4.8 Hz and 8.0 Hz, 1H), 4.99 (s, 2H). The
1H-NMR data were in good agreement with those reported [
56].
2-Furanylmethyl isothiocyanate (
3g): Colorless oil; 15.87g (95%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 7.71 (s, 1H), 6.46–6.47 (m, 2H), 4.95 (s, 2H). The
1H-NMR data were in good agreement with those reported [
56].
2-Thiophenylmethyl isothiocyanate (3h): Colorless oil; 16.39 g (88%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.56 (dd, J = 1.2 Hz and 5.2 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 7.03 (dd, J = 3.6 Hz and 5.2 Hz, 1H), 5.12 (s, 2H).
Biphenyl-4-methyl isothiocyanate (
3i): White solid; 23.25 g (86%); m.p. 66–67 °C.
1H-NMR (DMSO-
d6, 400 MHz) δ: 7.66–7.72 (m, 4H), 7.45–7.48 (m, 4H), 7.35–7.39 (m, 1H), 4.97 (s, 2H). The
1H-NMR data were in good agreement with those reported [
56].
Diphenylmethyl isothiocyanate (
3j): Colorless oil; 22.17 g (82%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 7.31–7.43 (m, 10H), 6.50 (s, 1H). The
1H-NMR data were in good agreement with those reported [
58].
Benzo[b]furan-5-methyl isothiocyanate (3k): Colorless oil; 20.21 g (89%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.03 (d, J = 2.4 Hz, 1H), 7.68 (d, J = 1.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.33 (dd, J = 2.0 Hz and 8.4 Hz, 1H), 7.00 (d, J = 0.8 Hz and 2.4 Hz, 1H), 5.00 (s, 2H).
2-Naphthylmethyl isothiocyanate (3m): White solid; 20.33 g (85%); m.p. 62–63 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.90–7.98 (m, 4H), 7.49–7.57 (m, 3H), 5.11 (s, 2H).
Quinoline-6-methyl isothiocyanate (3n): White solid; 18.74 g (78%); m.p. 186–188 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.22 (dd, J = 1.2 Hz and 8.8 Hz, 1H), 9.03 (d, J = 8.4 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.27 (s, 1H), 8.03 (dd, J = 1.6 Hz and 8.8 Hz, 1H), 7.98 (dd, J = 5.2 Hz and 8.4 Hz, 1H), 5.28 (s, 2H).
(5-Chloro-2-thiophenyl)methyl isothiocyanate (3ha): Colorless oil; 19.80 g (87%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.03–7.05 (m, 2H), 5.06 (s, 2H).
(5-Methyl-2-thiophenyl)methyl isothiocyanate (3hb): Colorless oil; 16.66 g (82%). 1H-NMR (DMSO-d6, 400 MHz) δ: 6.92 (d, J = 3.6 Hz, 1H), 6.69–6.70 (m, 1H), 5.01 (s, 2H), 2.43 (s, 3H).
(5-Ethyl-2-thiophenyl)methyl isothiocyanate (3hc): Colorless oil; 18.69 g (92%). 1H-NMR (DMSO-d6, 400 MHz) δ: 6.93 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 3.6 Hz, 1H), 5.02 (s, 2H), 2.78 (q, J = 7.5 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H).
(3-Methyl-2-thiophenyl)methyl isothiocyanate (3hd): Colorless oil; 11.98 g (59%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.43 (d, J = 5.2 Hz, 1H), 6.91 (d, J = 4.8 Hz, 1H), 5.03 (s, 2H), 2.20 (s, 3H).
(4-Methyl-2-thiophenyl)methyl isothiocyanate (3he): Colorless oil; 10.56 g (52%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.12 (s, 1H), 6.95 (s, 1H), 5.05 (s, 2H), 2.18 (s, 3H).
(3-Thiophenyl)methyl isothiocyanate (3hf) : Colorless oil; 15.46 g (83%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.58–7.60 (m, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.13 (dd, J = 0.8 Hz and 5.2 Hz, 1H), 4.90 (s, 2H). (5-Phenyl-2-thiophenyl)methyl isothiocyanate (3hg): White solid; 26.37 g (95%); m.p. 63–65 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.63–7.65 (m, 2H), 7.40–7.44 (m, 3H), 7.30–7.34 (m, 1H), 7.15 (d, J = 4.0 Hz, 1H), 5.14 (s, 2H).
(Benzo[b]thiophen-2-yl)methyl isothiocyanate (3hh): White solid; 23.65 g (96%); m.p. 62–64 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.96–7.98 (m, 1H), 7.84–7.86 (m, 1H), 7.46 (s, 1H), 7.35–7.41 (m, 2H), 5.25 (s, 2H).
(Dibenzo[b,d]thiophen-4-yl)methyl isothiocyanate (3hi): White solid; 28.19 g (92%); m.p. 81–83 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.39–8.42 (m, 2H), 8.09–8.11 (m, 1H), 7.53–7.60 (m, 4H), 5.24 (s, 2H).
3.2.4. General Procedure for the Synthesis of Thiones 5a–5n and 5ha–5hi
A mixture of isothiocyanate 3a–3n or 3ha–3hi (90 mmol) and formic hydrazide (6.49 g, 108 mmol) in THF (150 mL) was stirred at room temperature until the reaction completed as indicated by TLC analysis, when a white slurry formed. The crystals were collected by vacuum filtration and dried in vacuo at room temperature to give the intermediate 4a–4n or 4ha–4hi, which was used directly in the next step without further purification and characterization. Thus, 4a–4n or 4ha–4hi was dissolved in THF (150 mL; for 4a–4b) or DMF (150 mL; for 4c–4n and 4ha–4hi) followed by addition of aqueous K2CO3 prepared by dissolving K2CO3 (12.44 g, 90 mmol) in a minimal amount of water, and the solution thus obtained was stirred at 50 °C until completion of the reaction as indicated by TLC analysis (typically within 5 h). On cooling to room temperature, for 4a–4b: the reaction mixture was directly concentrated on a rotary evaporator to remove THF, and the aqueous residue was adjusted to pH 5–6 and further evaporated to dryness to give a residue, which was purified by column chromatography to afford 5a–5b; for 4c–4n or 4ha–4hi: the reaction mixture was poured into ice-water (300 mL), and the resulting mixture was adjusted to pH 5–6 with diluted hydrochloric acid to give a white slurry. The white crystals were collected by vacuum filtration, washed with water and dried in vacuo to give the crude product 5c–5n or 5ha–5hi as a white solid, which was triturated with EtOAc/n-hexane to yield the pure product 5c–5n or 5ha–5hi.
4-Ethyl-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5a): White solid; 8.08 g (78%); m.p. 169–171 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.61 (brs, 1H), 8.38 (s, 1H), 3.42 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.36, 142.60, 31.27.
4-n-Propyl-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5b): White solid; 8.49 g (73%); m.p. 90–91 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.63 (brs, 1H), 8.45 (s, 1H), 3.91 (q, J = 7.2 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.57, 141.53, 39.32, 13.92.
4-(Cyclohexylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5c): White solid; 11.90 g (67%); m.p. 138–140 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.64 (brs, 1H), 8.42 (d, J = 1.2 Hz, 1H), 3.75 (d, J = 7.2 Hz, 1H), 1.79–1.88 (m, 1H), 1.59–1.68 (m, 3H), 1.50–1.53 (m, 2H), 1.10–1.20 (m, 3H), 0.90–0.99 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.15, 142.36, 49.58, 36.14, 29.69, 25.75, 24.97.
4-(1-Adamantylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5d): White solid; 18.40 g (82%); m.p. 223–225 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.65 (brs, 1H), 8.34 (s, 1H), 3.68 (s, 2H), 1.93 (s, 3H), 1.62–1.65 (m, 3H), 1.47–1.56 (m, 9H). 13C-NMR (DMSO-d6, 100 MHz) δ: 167.29, 142.86, 54.42, 39.85, 36.14, 34.59, 27.48.
4-Benzyl-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5e): White solid; 13.08 g (76%); m.p. 121–123 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.76 (brs, 1H), 8.54 (s, 1H), 7.29–7.38 (m, 5H), 5.16 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.28, 142.04, 135.99, 128.60, 127.85, 127.75, 46.88.
4-(3-Pyridinylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5f): White solid; 12.46 g (72%); m.p. 211–213 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.80 (brs, 1H), 8.62 (d, J = 2.0 H, 1Hz), 8.60 (s, 1H), 8.51–8.52 (m, 1H), 7.75–7.78 (m, 1H), 7.37–7.40 (m, 1H), 5.20 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.22, 149.19, 149.10, 142.02, 135.66, 131.61, 123.62, 44.70.
4-(2-Furanylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5g): White solid; 12.72 g (78%); m.p. 133–135 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.76 (brs, 1H), 8.45 (s, 1H), 7.64 (d, J = 1.6 Hz, 1H), 6.43–6.46 (m, 2H), 5.17 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.04, 148.30, 143.35, 141.74, 110.75, 109.46, 40.41.
4-(2-Thiophenylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5h): White solid; 13.50 g (76%); m.p. 108–110 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.74 (brs, 1H), 8.54 (s, 1H), 7.47 (dd, J = 1.2 Hz and 4.8 Hz, 1H), 7.20 (d, J = 3.2 Hz, 1H), 6.99–7.01 (m, 1H), 5.33 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.84, 141.62, 137.60, 127.85, 126.91, 126.81, 41.73.
4-(Biphenyl-4-methyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5i): White solid; 18.53 g (77%); m.p. 178–180 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.79 (brs, 1H), 8.59 (d, J = 1.2 Hz, 1H), 7.63–7.66 (m, 4H), 7.43–7.47 (m, 4H), 7.36 (t, J = 7.2 Hz, 1H), 5.21 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.29, 142.09, 139.77, 139.59, 135.16, 128.89, 128.40, 127.51, 126.91, 126.63, 46.61.
4-(Diphenylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5j): White solid; 18.04 g (75%); m.p. 166–168 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.94 (brs, 1H), 8.50 (s, 1H), 7.35–7.43 (m, 6H), 7.18–7.19 (m, 4H), 7.08 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.38, 140.77, 138.00, 128.84, 128.15, 127.99, 61.08.
4-(Benzo[b]furan-5-methyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5k): White solid; 14.78 g (71%); m.p. 151–153 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.75 (brs, 1H), 8.54 (s, 1H), 7.99 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 1.2 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.35–7.38 (m, 1H), 6.96 (d, J = 2.8 Hz, 1H), 5.24 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.17, 153.86, 146.64, 142.00, 130.77, 127.39, 124.46, 120.93, 111.41, 106.71, 47.00.
4-((4-Cyano-1-naphthyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5l): White solid; 15.64 g (72%); m.p. 218–220 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.94 (brs, 1H), 8.46 (s, 1H), 8.36 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 7.2 Hz, 1H), 7.79–7.88 (m, 2H), 7.25 (d, J = 7.6 Hz, 1H), 5.74 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.49, 142.16, 137.78, 132.88, 131.56, 129.83, 129.12, 128.35, 125.09, 124.64, 124.30, 117.36, 109.18, 45.06.
4-(2-Naphthylmethyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5m): White solid; 14.77 g (68%); m.p. 165–167 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.80 (brs, 1H), 8.59 (d, J = 1.2 Hz, 1H), 7.88–7.92 (m, 3H), 7.81 (s, 1H), 7.49–7.54 (m, 3H), 5.33 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.35, 142.16, 133.51, 132.67, 132.39, 128.36, 127.72, 127.55, 126.53, 126.45, 126.25, 125.63, 47.12.
4-(Quinoline-6-methyl)-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5n): White solid; 16.36 g (75%); m.p. 272–275 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.82 (brs, 1H), 8.89 (dd, J = 1.6 Hz and 4.4 Hz, 1H), 8.62 (s, 1H), 8.35 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.85 (s, 1H), 7.75 (dd, J = 2.0 Hz and 8.8 Hz, 1H), 7.51–7.55 (m, 1H), 5.37 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.36, 150.76, 147.16, 142.19, 135.97, 134.17, 129.41, 129.15, 127.55, 126.69, 121.81, 46.85.
4-((5-Chloro-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5ha): White solid; 15.02 g (72%); m.p. 176–178 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.79 (brs, 1H), 8.55 (s, 1H), 7.08 (d, J = 3.6 Hz, 1H), 7.01 (d, J = 3.6 Hz, 1H), 5.25 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.77, 141.54, 136.87, 128.71, 128.02, 126.34, 42.01.
4-((5-Methyl-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hb): White solid; 13.31 g (70%); m.p. 131–133 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.72 (brs, 1H), 8.50 (s, 1H), 6.98 (d, J = 3.2 Hz, 1H), 6.66–6.67 (m, 1H), 5.23 (s, 2H), 2.37 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.81, 141.56, 140.31, 135.16, 127.87, 125.00, 42.01, 14.87.
4-((5-Ethyl-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hc): White solid; 15.21 g (75%); m.p. 64–66 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.73 (brs, 1H), 8.51 (s, 1H), 7.00 (d, J = 3.6 Hz, 1H), 6.70 (d, J = 3.2 Hz, 1H), 5.25 (s, 2H), 2.73 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.6 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.84, 147.80, 141.56, 134.82, 127.65, 123.22, 42.06, 22.75, 15.72.
4-((3-Methyl-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hd): White solid; 14.83 g (78%); m.p. 113–115 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.74 (brs, 1H), 8.41 (s, 1H), 7.38 (d, J = 5.2 Hz, 1H), 6.86 (d, J = 5.2 Hz, 1H), 5.26 (s, 2H), 2.28 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.87, 141.59, 136.18, 130.91, 129.96, 125.12, 40.26, 13.64.
4-((4-Methyl-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5he): White solid; 14.45 g (76%); m.p. 98–100 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.73 (brs, 1H), 8.52 (s, 1H), 7.03 (s, 1H), 6.99 (s, 1H), 5.26 (s, 2H), 2.15 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.86, 141.61, 137.39, 136.83, 129.84, 121.71, 41.90, 15.26.
4-((3-Thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hf): White solid; 12.55 g (70%); m.p. 109–110 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.73 (brs, 1H), 8.51 (s, 1H), 7.53 (dd, J = 3.2 Hz and 4.8 Hz, 1H), 7.47 (d, J = 1.6 Hz, 1H), 7.16 (dd, J = 0.8 Hz and 4.8 Hz, 1H), 5.13 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.01, 141.86, 136.42, 127.40, 127.06, 124.18, 42.41.
4-((5-Phenyl-2-thiophenyl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hg): White solid; 16.73 g (68%); m.p. 160–162 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.78 (brs, 1H), 8.58 (s, 1H), 7.58–7.60 (m, 2H), 7.37–7.41 (m, 3H), 7.30 (t, J = 7.4 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H), 5.34 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 165.88, 143.95, 141.64, 137.14, 133.38, 129.18, 129.09, 127.78, 125.27, 123.27, 42.01.
4-((Benzo[b]thiophen-2-yl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hh): White solid; 15.80 g (71%); m.p. 185–188 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.82 (brs, 1H), 8.60 (s, 1H), 7.90–7.92 (m, 1H), 7.81–7.83 (m, 1H), 7.45 (s, 1H), 7.31–7.38 (m, 2H), 5.45 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.19, 141.85, 139.50, 138.90, 138.87, 124.79, 124.66, 124.18, 123.80, 122.56, 42.84.
4-((Dibenzo[b,d]thiophen-4-yl)methyl]-2,3-dihydro-4H-1,2,4-triazoline-3-thione (5hi): White solid; 18.20 g (68%); m.p. 240–242 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.87 (brs, 1H), 8.46 (s, 1H), 8.35–8.40 (m, 2H), 8.05–8.08 (m, 1H), 7.51–7.56 (m, 3H), 7.29 (d, J = 7.2 Hz, 1H), 5.42 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 166.71, 142.14, 138.15, 136.79, 135.78, 134.94, 129.79, 127.32, 126.14, 125.13, 124.95, 123.00, 122.20, 121.67, 46.32.
3.2.5. General Procedure for the Synthesis of Thiones 5o and 5q
A solution of isothiocyanate 3l or 3h (90 mmol) and NH2CH2CH(OMe)2 (11.36 g, 108 mmol) in THF (150 mL) was stirred at room temperature until the reaction completed as indicated by TLC analysis. The reaction mixture was evaporated on a rotary evaporator to give the crude intermediate 4o or 4p, which was used directly in the next step without further purification. Thus, the crude 4o or 4p was suspended in 25% aqueous H2SO4 containing 20% acetic acid (by v/v) and stirred at reflux for 3 h when TLC analysis indicated the reaction was completed. On cooling to room temperature, the reaction mixture was poured into ice-water (450 mL) and the white crystals were collected by vacuum filtration, washed with water and dried in vacuo to give the crude product 5o or 5q, which was triturated with EtOAc/n-hexane to yield the pure product 5o or 5q.
1-((4-Cyano-1-naphthyl)methyl)-2,3-dihydro-1H-imidazoline-2-thione (5o): White solid; 17.43 g (73%); m.p. 287–289 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.33 (brs, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.14–8.18 (m, 2H), 7.85 (t, J = 7.4 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 7.18 (d, J = 7.2 Hz, 1H), 6.97–7.01 (m, 2H), 5.73 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 161.66, 139.23, 132.89, 131.55, 130.03, 129.04, 128.17, 124.97, 124.62, 124.49, 118.38, 117.47, 115.13, 108.77, 46.95.
1-((2-Thiophenyl)methyl)-2,3-dihydro-1H-imidazoline-2-thione (5q): White solid; 12.37 g (70%); m.p. 124–126 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.12 (brs, 1H), 7.43 (d, J = 5.2 Hz, 1H), 7.16 (d, J = 3.2 Hz, 1H), 7.08–7.09 (m, 1H), 6.98 (dd, J = 3.6 Hz and 5.2 Hz, 1H), 6.86–6.87 (m, 1H), 5.31 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 160.94, 138.85, 127.27, 126.68, 126.32, 117.81, 114.64, 43.57.
3.2.6. Procedure for the Synthesis of (4-Cyano-1-naphthyl)(4-mercapto-3-pyridinyl)methane (5p)
A solution of
17 (49.22 g, 200 mmol) and hexamethylenetetramine (56.07 g, 400 mmol) in 50% acetic acid was stirred at reflux for 6 h followed by addition of concentrated hydrochloric acid (10 mL). The reflux was continued for another 0.5 h, when the reaction completed as indicated by TLC analysis. On cooling to room temperature, the reaction mixture was concentrated to half its original volume and poured into ice-water (300 mL) while stirring. The resulting mixture was extracted with CH
2Cl
2 (200 mL × 3), and the combined extracts were washed successively with saturated aqueous NaHCO
3 (200 mL × 3) and 5% brine (300 mL), dried (Na
2SO
4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford 4-cyano-1-naphthaldehyde (
19). Yellow solid; 30.45 g (78%); m.p. 138–141 °C (literature value, 143–144 °C [
59]).
1H-NMR (DMSO-
d6, 400 MHz) 10.54 (s, 1H), 9.17–9.21 (m, 1H), 8.41 (d,
J = 7.2 Hz, 1H), 8.29 (d,
J = 7.2 Hz, 1H), 8.23–8.27 (m, 1H), 7.89–7.93 (m, 2H).
To a stirred solution of 3-bromo-4-chloropyridine (20, 28.86 g, 150 mmol ) in dried THF (350 mL) held at –78 °C in N2 atmosphere was added dropwise 1.6M n-BuLi in n-hexane (93.75 mL, 150 mmol) via syringe. After addition, the resulting mixture was stirred at this temperature for another 0.5 h, followed by addition of a solution of 19 (29.28 g, 150 mmol) in dried THF (150 mL) in a dropwise manner via syringe. The reaction mixture was slowly warmed to room temperature and stirred at room temperature for another 1 h. The reaction mixture was slowly poured into ice-water (600 mL). The mixture thus obtained was extracted with CH2Cl2 (200 mL × 3), and the combined extracts were washed with 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford (4-chloro-3-pyridinyl)(4-cyano-1-naphthyl)methanol (22). White solid; 29.62 g (67%); m.p. 178–180 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.55 (s, 1H), 8.49 (d, J = 5.6 Hz, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.79–7.83 (m, 1H), 7.72–7.76 (m, 1H), 7.58 (d, J = 5.2 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 6.78 (d, J = 5.6 Hz, 1H), 6.62 (d, J = 6.0 Hz, 1H).
To a stirred solution of 22 (26.53 g, 90 mmol) in dried CH2Cl2 (300 mL) cooled in an ice-water bath was added thionyl chloride (12.85 g, 108 mmol). After addition, the resulting mixture was stirred at room temperature until the reaction completed as indicated by TLC analysis (typically within 4 h) and poured into ice-water (600 mL) while stirring. The mixture thus obtained was adjusted to pH 7–8 with saturated aqueous NaHCO3. The organic phase was separated, and the aqueous phase was back-extracted with CH2Cl2 (200 mL × 2). The combined organic phases were washed with 5% brine (300 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give crude (4-chloro-3-pyridinyl)(4-cyano-1-naphthyl)methyl chloride (23) as a residue, which was used directly in the next step without further purification.
To a stirred solution of crude 23 prepared above (deemed to be 90 mmol) in acetic acid (300 mL) was added Zn powder (13.08 g, 200 mmol) in a portionwise manner at room temperature, and after addition, the reaction mixture was stirred at room temperature until the reaction completed as indicated by TLC analysis (typically within 5 h). The reaction mixture was diluted with CH2Cl2 (300 mL) and the resulting mixture was further stirred for 10 min and filtered off through Celite. The filtrate was evaporated on a rotary evaporator to almost dryness, and the residue was dissolved in CH2Cl2 (200 mL). The solution thus obtained was washed successively saturated aqueous NaHCO3 (200 mL) and 5% brine (200 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford (4-chloro-3-pyridinyl)(4-cyano-1-naphthyl)methane (24). White solid; 18.48 g (78%); m.p. 155–157 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.48 (d, J = 5.6 Hz, 1H), 8.39 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.4 Hz, 1H), 7.76–7.80 (m, 1H), 7.63 (d, J = 5.2 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 4.68 (s, 2H).
A solution of 24 (15.33 g, 55 mmol) and Na2S·9H2O (39.63 g, 165 mmol) in DMF (200 mL) was stirred at 110 °C in N2 atmosphere until the reaction completed as indicated by TLC analysis (typically within 12 h). On cooling to room temperature, the reaction mixture was concentrated on a rotary evaporator to about 50 mL and poured into ice-water (600 mL) while stirring, and the mixture thus obtained was adjusted to pH 5–6 and extracted with CH2Cl2 (200 mL × 3). The combined organic phases were washed with 5% brine (100 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford the pure compound 5p. White solid; 10.94 g (72%); m.p. 208–211 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.38 (brs, 1H), 8.08–8.15 (m, 3H), 7.79 (t, J = 7.8 Hz, 1H), 7.69 (t, J = 7.4 Hz, 1H), 7.56 (d, J = 6.4 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 6.8 Hz, 1H), 7.29 (s, 1H), 4.60 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 188.80, 143.10, 137.95, 132.97, 132.87, 131.75, 131.28, 131.19, 130.04, 128.70, 127.81, 126.05, 125.16, 124.92, 117.80, 107.49, 34.86.
3.2.7. General Procedure for the Synthesis of Esters 6a–6x and 6ha–6hi
A mixture of thiones or thiol
5a–
5q or
5ha–
5hi (60 mmol), bromoacetates BrCH
2CO
2Me, (CH
3)
2CBrCO
2Et or ethyl 1-bromocyclobutanecarboxylate (80 mmol) and K
2CO
3 (16.59 g, 120 mmol) in a suitable solvent (200 mL; acetone for
5a–
5b or DMF for the others) was stirred at a temperature specified in
Scheme 1,
Scheme 2,
Scheme 3,
Scheme 4 and
Scheme 5 until the completion of reaction as indicated by TLC analysis (typically within 12 h). On cooling to room temperature (if necessary), for
5a–
5b: the reaction mixture was filtered off and the filtrate was evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford
6a–
6b; for the others: the reaction mixture was poured into ice-water (300 mL), and the resulting mixture was extracted with CH
2Cl
2 (100 mL × 3). The combined extracts were washed with 5% brine (100 mL × 5), dried (Na
2SO
4) and evaporated on a rotary evaporator to afford the crude product as a residue, which was purified by column chromatography to yield the pure product
6c–
6x or
6ha–
6hi.
Methyl 2-((4-ethyl-4H-1,2,4-triazol-3-yl)thio)acetate (6a): White solid; 9.32 g (83%); m.p. 67–69 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.53 (s, 1H), 4.03 (s, 2H), 3.63 (s, 3H), 3.58 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.85, 148.31, 146.26, 52.42, 34.56, 30.73.
Methyl 2-((4-n-propyl-4H-1,2,4-triazol-3-yl)thio)acetate (6b): Colorless oil; 9.42 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.60 (s, 1H), 4.06 (s, 2H), 3.96 (q, J = 7.3 Hz, 2H), 3.63 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.78, 147.56, 145.03, 52.41, 39.37, 34.50, 15.19.
Methyl 2-((4-(cyclohexylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6c): White solid; 13.25 g (82%); m.p. 56–58 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.55 (s, 1H), 4.07 (s, 2H), 3.79 (d, J = 7.2 Hz, 2H), 3.63 (s, 3H), 1.59–1.72 (m, 4H), 1.47–1.50 (m, 2H), 1.09–1.21 (m, 3H), 0.87–0.96 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.67, 148.12, 145.87, 52.35, 49.80, 37.67, 34.50, 29.63, 25.64, 24.94.
Methyl 2-((4-(1-adamantylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6d): White solid; 15.62 g (81%); m.p. 214–216 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.48 (s, 1H), 4.07 (s, 2H), 3.65 (s, 2H), 3.62 (s, 3H), 1.94 (s, 3H), 1.63–1.66 (m, 3H), 1.52–1.55 (m, 3H), 1.42 (s, 6H). 13C-NMR (CDCl3, 100 MHz) δ: 168.96, 150.42, 145.75, 56.82, 52.99, 40.22, 36.49, 35.79, 34.72, 28.06.
Methyl 2-((4-benzyl-4H-1,2,4-triazol-3-yl)thio)acetate (6e): White solid; 12.01 g (76%); m.p. 82–84 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.71 (s, 1H), 7.32–7.40 (m, 3H), 7.22–7.23 (m, 2H), 5.21 (s, 2H), 4.03 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.63, 148.22, 145.77, 135.59, 128.79, 128.06, 127.41, 52.39, 47.34, 34.39.
Methyl 2-((4-(3-pyridinylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6f): Colorless oil; 10.78 g (68%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.74 (s, 1H), 8.53–8.54 (m, 2H), 7.63–7.65 (m, 1H), 7.39–7.42 (m, 1H), 5.27 (s, 2H), 4.05 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.60, 149.36, 148.89, 148.17, 145.75, 135.39, 131.28, 123.80, 52.41, 45.01, 34.40.
Methyl 2-((4-(2-furanylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6g): White solid; 11.40 g (75%); m.p. 67–69 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.63 (s, 1H), 7.65 (d, J = 1.6 Hz, 1H), 6.48 (d, J = 3.2 Hz, 1H), 6.44–6.46 (m, 1H), 5.25 (s, 2H), 4.04 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.71, 148.16, 148.14, 145.54, 143.72, 110.78, 109.57, 52.41, 40.65, 34.56.
Methyl 2-((4-(2-thiophenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6h): Colorless oil; 16.30 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 7.52–7.53 (m, 1H), 7.12–7.13 (m, 1H), 7.01–7.03 (m, 1H), 5.42 (s, 2H), 4.05 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.66, 147.95, 145.35, 137.68, 127.64, 127.21, 127.02, 52.40, 42.25, 34.61.
Methyl 2-((4-(biphenyl-4-methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6i): White solid; 15.48 g (76%); m.p. 152–154 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.76 (s, 1H), 7.64–7.68 (m, 4H), 7.45 (t, J = 7.6 Hz, 2H), 7.37 (d, J = 7.2 Hz, 1H), 7.31–7.34 (m, 2H), 5.26 (s, 2H), 4.05 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.65, 148.24, 145.81, 139.92, 139.43, 134.74, 128.91, 128.05, 127.59, 127.07, 126.64, 52.40, 47.04, 34.40.
Methyl 2-((4-(diphenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6j): Colorless oil; 16.90 g (83%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.30 (s, 1H), 7.36–7.44 (m, 6H), 7.18–7.19 (m, 4H), 6.79 (s, 1H), 4.03 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.64, 148.70, 144.48, 137.65, 128.92, 128.47, 127.99, 62.12, 52.41, 34.58.
Methyl 2-((4-(benzo[b]furan-5-methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6k): White solid; 14.38 g (79%); m.p. 82–85 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.74 (s, 1H), 8.01 (d, J = 2.4 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.54 (s, 1H), 7.22–7.24 (m, 1H), 6.96 (d, J = 1.2 Hz, 1H), 5.29 (s, 2H), 4.04 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.66, 153.88, 148.17, 146.83, 145.73, 130.27, 127.53, 124.05, 120.55, 111.59, 106.71, 52.38, 47.46, 34.37.
Methyl 2-((4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6l): White solid; 16.65 g (82%); m.p. 135–137 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.72 (s, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.81–7.90 (m, 2H), 7.00 (d, J = 7.2 Hz, 1H), 5.87 (s, 2H), 4.07 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.59, 148.78, 146.17, 137.70, 132.92, 131.49, 129.58, 129.27, 128.47, 125.08, 124.17, 123.82, 117.29, 109.30, 52.45, 45.32, 34.53.
Methyl 2-((4-(2-naphthylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6m): White solid; 15.04 g (80%); m.p. 101–103 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.78 (s, 1H), 7.88–7.94 (m, 3H), 7.73 (s, 1H), 7.51–7.55 (m, 2H), 7.37–7.39 (m, 1H), 5.39 (s, 2H), 4.03 (s, 2H), 3.60 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.63, 148.34, 145.90, 133.06, 132.67, 132.42, 128.58, 127.74, 127.58, 126.57, 126.40, 126.27, 125.16, 52.36, 47.55, 34.38.
Methyl 2-((4-(quinoline-6-methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6n): White solid; 14.33 g (76%); m.p. 141–143 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.90 (dd, J = 1.6 Hz and 4.4 Hz, 1H), 8.80 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 0.8 Hz, 1H), 7.62–7.65 (m, 1H), 7.54 (dd, J = 4.4 Hz and 8.4 Hz, 1H), 5.44 (s, 2H), 4.04 (s, 2H), 3.59 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.63, 150.93, 148.38, 147.20, 145.95, 136.00, 133.77, 129.65, 128.72, 127.61, 126.46, 121.94, 52.38, 47.26, 34.43.
Ethyl 2-((4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)-2-methylpropionate (6o): White solid; 14.38 g (63%); m.p. 110–112 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.81 (s, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.82–7.91 (m, 2H), 6.85 (d, J = 7.6 Hz, 1H), 5.86 (s, 2H), 4.06 (q, J = 7.1 Hz, 2H), 1.52 (s, 6H), 1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.09, 146.45, 138.21, 132.96, 131.44, 129.39, 129.28, 129.24, 128.46, 125.11, 123.97, 123.36, 117.22, 109.28, 61.36, 53.28, 45.41, 25.84, 13.67.
Ethyl 1-((4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)cyclobutanecarboxylate (6p): White solid; 15.01 g (58%); m.p. 141–143 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.79 (s, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 7.2 Hz, 1H), 7.82–7.91 (m, 2H), 6.89 (d, J = 7.6 Hz, 1H), 5.85 (s, 2H), 4.05 (q, J = 7.1 Hz, 2H), 2.57–2.65 (m, 2H), 2.24–2.31 (m, 2H), 2.03–2.12 (m, 1H), 1.83–1.91 (m, 1H), 1.09 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.64, 146.97, 146.25, 138.08, 132.98, 131.47, 129.44, 129.33, 128.51, 125.15, 123.99, 123.47, 117.26, 109.30, 61.28, 54.07, 45.37, 31.76, 15.55, 13.72.
Methyl 2-((1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)acetate (6q): White solid; 14.58 g (72%); m.p. 116–118 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.32 (d, J = 8.0 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.81–7.89 (m, 2H), 7.34 (s, 1H), 7.07 (d, J = 1.2 Hz, 1H), 6.81 (d, J = 7.6 Hz, 1H), 5.83 (s, 2H), 3.91 (s, 2H), 3.58 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.09, 139.81, 139.44, 133.02, 131.45, 129.58, 129.51, 129.18, 128.32, 125.04, 124.17, 123.08, 123.06, 117.40, 108.86, 52.26, 47.09, 35.26.
Ethyl 2-((1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (6r): White solid; 17.76 g (78%); m.p. 108–110 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.28 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.10 (d, J = 7.2 Hz, 1H), 7.81–7.89 (m, 2H), 7.46 (d, J = 1.2 Hz, 1H), 7.19 (d, J = 0.4 Hz, 1H), 6.67 (d, J = 7.6 Hz, 1H), 5.86 (s, 2H), 4.04 (q, J = 7.2 Hz, 2H), 1.46 (s, 6H), 1.11 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.56, 139.92, 137.14, 133.05, 131.38, 130.41, 129.38, 129.21, 128.35, 125.06, 124.11, 123.97, 122.69, 117.36, 108.80, 61.08, 52.84, 47.33, 25.62, 13.72.
Ethyl 1-((1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (6s): White solid; 17.85 g (76%); m.p. 135–137 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.28 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.10–8.12 (d, J = 7.6 Hz, 1H), 7.81–7.90 (m, 2H), 7.45 (s, 1H), 7.16 (s, 1H), 6.71 (d, J = 7.6 Hz, 1H), 5.84 (s, 2H), 4.03 (q, J = 7.1 Hz, 2H), 2.53–2.56 (m, 2H), 2.23–2.29 (m, 2H), 1.98–2.07 (m, 1H), 1.76–1.84 (m, 1H), 1.09 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.24, 139.80, 137.62, 133.05, 131.40, 130.32, 129.41, 129.22, 128.37, 125.07, 123.95, 123.88, 122.79, 117.37, 108.83, 61.02, 54.17, 47.28, 31.33, 15.32, 13.75.
Methyl 2-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)acetate (6t): Colorless oil; 15.05 g (72%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.37 (d, J = 5.2 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.12 (s, 1H), 8.07 (d, J = 7.2 Hz, 1H), 7.84 (t, J = 7.4 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 5.2 H, 1Hz), 7.15 (d, J = 7.6 Hz, 1H), 4.56 (s, 2H), 4.12 (s, 2H), 3.65 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.93, 149.63, 147.96, 146.71, 141.46, 132.96, 131.69, 131.07, 130.96, 128.92, 128.07, 125.40, 125.05, 124.84, 119.59, 117.64, 107.90, 52.50, 33.09, 32.16.
Ethyl 2-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)-2-methylpropionate (6u): Colorless oil; 15.93 g (68%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.45 (d, J = 5.2 Hz, 1H), 8.30 (s, 1H), 8.27 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.4 Hz, 1H), 7.78 (t, J = 7.2 Hz, 1H), 7.35 (d, J = 5.2 Hz, 1H), 7.05 (d, J = 3.6 Hz, 1H), 4.64 (s, 2H), 4.12 (q, J = 7.1 Hz, 2H), 1.51 (s, 6H), 1.13 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.82, 150.91, 147.99, 143.04, 142.39, 135.60, 132.96, 131.66, 130.93, 128.94, 128.06, 126.75, 125.55, 125.05, 124.81, 117.61, 107.83, 61.39, 50.98, 33.44, 25.84, 13.75.
Ethyl 1-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)cyclobutanecarboxylate (6v): White solid; 17.87 g (74%); m.p. 123–125 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.36 (d, J = 5.2 Hz, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.15 (s, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.6 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 7.06 (d, J = 5.6 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.79–2.86 (m, 2H), 2.20–2.27 (m, 2H), 2.06–2.17 (m, 1H), 1.90–2.00 (m, 1H), 1.09 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.31, 150.24, 147.90, 145.83, 141.56, 132.94, 131.68, 131.52, 131.04, 128.93, 128.04, 125.33, 125.06, 124.78, 120.76, 117.62, 107.94, 61.47, 50.89, 33.10, 31.74, 16.20, 13.77.
Ethyl 2-((1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (6w): Oil; 16.78 g (89%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.44–7.46 (m, 2H), 7.05–7.06 (m, 2H), 6.96–6.98 (m, 1H), 5.40 (s, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.46 (s, 6H), 1.12 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.56, 139.55, 136.03, 130.07, 126.97, 126.73, 126.27, 123.13, 61.00, 52.80, 44.26, 25.56, 13.70.
Ethyl 1-((1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (6x): Oil; 15.86 g (82%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.46 (dd, J = 1.2 Hz and 5.2 Hz, 1H), 7.06 (d, J = 3.6 Hz, 1H), 7.03 (d, J = 0.8 Hz, 1H), 6.96–6.99 (m, 1H), 5.38 (s, 2H), 4.05 (q, J = 7.1 Hz, 2H), 2.51–2.56 (m, 2H), 2.24–2.31 (m, 2H), 2.04–2.11 (m, 1H), 1.78–1.85 (m, 1H), 1.11 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.31, 139.47, 136.49, 129.99, 126.98, 126.75, 126.30, 122.89, 60.94, 54.26, 44.21, 31.21, 15.27, 13.75.
Methyl 2-((4-((5-chloro-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6ha): Oil; 14.22 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.69 (s, 1H), 7.04 (d, J = 4.0 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 5.36 (s, 2H), 4.07 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.62, 147.94, 145.31, 136.85, 128.88, 127.79, 126.87, 52.41, 42.32, 34.57.
Methyl 2-((4-((5-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hb): Oil; 12.75 g (75%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.66 (s, 1H), 6.91 (d, J = 3.2 Hz, 1H), 6.68–6.69 (m, 1H), 5.31 (s, 2H), 4.05 (s, 2H), 3.63 (s, 3H), 2.38 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.67, 147.88, 145.27, 140.56, 135.17, 127.67, 125.31, 52.40, 42.45, 34.58, 14.87.
Methyl 2-((4-((5-ethyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hc): Colorless oil; 12.85 g (72%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.69 (s, 1H), 6.94 (d, J = 3.2 Hz, 1H), 6.71 (d, J = 3.2 Hz, 1H), 5.34 (s, 2H), 4.06 (s, 2H), 3.63 (s, 3H), 2.74 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.6 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.67, 148.06, 147.92, 145.31, 134.86, 127.47, 123.52, 52.39, 42.53, 34.62, 22.74, 15.64.
Methyl 2-((4-((3-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hd): Colorless oil; 13.26 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.62 (s, 1H), 7.42 (d, J = 5.2 Hz, 1H), 6.88 (d, J = 5.2 Hz, 1H), 5.33 (s, 2H), 4.03 (s, 2H), 3.62 (s, 3H), 2.25 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.67, 147.90, 145.33, 136.17, 130.70, 130.28, 125.29, 52.39, 40.79, 34.65, 13.45.
Methyl 2-((4-((4-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6he): Colorless oil; 11.39 g (67%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.68 (s, 1H), 7.08 (s, 1H), 6.92 (s, 1H), 5.34 (s, 2H), 4.05 (s, 2H), 3.62 (s, 3H), 2.15 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.66, 147.93, 145.35, 137.45, 137.14, 129.60, 121.85, 52.40, 42.40, 34.62, 15.24.
Methyl 2-((4-((3-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hf): Oil; 10.99 g (68%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.68 (s, 1H), 7.55–7.57 (m, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.04–7.05 (m, 1H), 5.19 (s, 2H), 4.04 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.69, 148.02, 145.56, 136.25, 127.50, 126.96, 124.08, 52.40, 42.83, 34.41.
Methyl 2-((4-((5-phenyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hg): White solid; 17.00 g (82%); m.p. 104–106 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.74 (s, 1H), 7.59–7.61 (m, 2H), 7.38–7.41 (m, 3H), 7.28–7.32 (m, 1H), 7.13 (d, J = 4.0 Hz, 1H), 5.43 (s, 2H), 4.07 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.66, 148.01, 145.39, 144.18, 137.10, 133.20, 129.11, 128.95, 127.89, 125.26, 123.54, 52.41, 42.50, 34.61.
Methyl 2-((4-((benzo[b]thiophen-2-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hh): White solid; 12.46 g (65%); m.p. 84–86 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.77 (s, 1H), 7.92–7.94 (m, 1H), 7.82–7.84 (m, 1H), 7.34–7.38 (m, 3H), 5.55 (s, 2H), 4.06 (s, 2H), 3.61 (s, 3H) . 13C-NMR (DMSO-d6, 100 MHz) δ: 168.64, 148.27, 145.56, 139.25, 138.90, 138.74, 124.81, 124.68, 123.87, 123.84, 122.56, 52.41, 43.19, 34.63.
Methyl 2-((4-((dibenzo[b,d]thiophen-4-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (6hi): White solid; 15.30 g (69%); m.p. 144–145 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.76 (s, 1H), 8.37–8.41 (m, 2H), 8.04–8.07 (m, 1H), 7.53–7.57 (m, 3H), 7.26 (d, J = 7.2 Hz, 1H), 5.52 (s, 2H), 4.01 (s, 2H), 3.60 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.60, 148.76, 146.23, 138.04, 136.78, 135.90, 134.81, 129.46, 127.39, 126.17, 125.22, 124.98, 122.96, 122.23, 121.98, 52.37, 46.76, 34.52.
3.2.8. General Procedure for the Synthesis of Brominated Esters 7a–7s, 7w–7x, 7ha–7hd, 7′he, 7hf–7hg, 7′hh and 7hi
A mixture of a substrate to be brominated
6a–
6x or
6ha–
6hi (40 mmol) and a brominating reagent NBS or DBDMH (48 mmol) in a suitable solvent MeCN or CH
2Cl
2 (100 mL) was stirred at a suitable temperature until the completion of reaction as indicated by TLC analysis (typically within 12 h). The brominating agent, solvent and temperature are specified in
Scheme 1,
Scheme 2,
Scheme 3,
Scheme 4 and
Scheme 5.
The reaction mixture was poured into ice-water (200 mL), and the resulting mixture was extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed successively with 5% aqueous Na2S2O3 (100 mL), saturated aqueous Na2CO3 (100 mL × 5) and 5% brine (100 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford the pure product 7a–7s, 7w–7x, 7ha–7hd, 7′he, 7hf–7hg, 7′hh or 7hi.
Methyl 2-((5-bromo-4-ethyl-4H-1,2,4-triazol-3-yl)thio)acetate (7a): White solid; 5.54 g (52%); m.p. 86–88 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 4.04 (s, 2H), 3.64 (s, 3H), 3.54 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.62, 150.98, 130.88, 52.46, 34.54, 31.90.
Methyl 2-((5-bromo-4-n-propyl-4H-1,2,4-triazol-3-yl)thio)acetate (7b): White solid; 9.19 g (82%); m.p. 56–58 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 4.10 (s, 2H), 3.98 (q, J = 7.2 Hz, 2H), 3.64 (s, 3H), 1.25 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.53, 150.45, 129.71, 52.47, 40.57, 34.41, 14.38.
Methyl 2-((5-bromo-4-(cyclohexylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7c): Colorless oil; 10.03 g (72%); 1H-NMR (DMSO-d6, 400 MHz) δ: 4.11 (s, 2H), 3.77 (d, J = 7.2 Hz, 2H), 3.64 (s, 3H), 1.61–1.78 (m, 4H), 1.49–1.52 (m, 2H), 1.11–1.18 (m, 3H), 0.95–1.04 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.46, 151.27, 130.47, 52.46, 50.82, 37.40, 34.33, 29.78, 25.53, 24.98.
Methyl 2-((-4-(1-adamantylmethyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (7d): White solid; 10.05 g (69%); m.p. 120–122 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 4.08 (s, 2H), 3.67 (s, 2H), 3.63 (s, 3H), 1.95 (s, 3H), 1.63–1.66 (m, 3H), 1.55–1.58 (m, 3H), 1.52–1.53 (m, 6H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.53, 152.52, 131.40, 56.34, 52.45, 40.20, 35.86, 35.43, 35.20, 27.50.
Methyl 2-((4-benzyl-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (7e): White solid; 8.90 g (65%); m.p. 87–89 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.31–7.40 (m, 3H), 7.15–7.17 (m, 2H), 5.21 (s, 2H), 4.08 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.41, 151.44, 134.43, 130.64, 128.87, 128.13, 126.79, 52.48, 48.11, 34.25.
Methyl 2-((5-bromo-4-(3-pyridinylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7f): White solid; 7.55 g (55%); m.p. 68–70 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.54–8.56 (m, 1H), 8.51 (d, J = 1.6 Hz, 1H), 7.57–7.59 (m, 1H), 7.41–7.44 (m, 1H), 5.28 (s, 2H), 4.11 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.41, 151.45, 149.16, 148.17, 135.12, 130.56, 130.44, 123.98, 52.51, 45.89, 34.32.
Methyl 2-((5-bromo-4-(2-furanylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7g): White solid; 8.24 g (62%); m.p. 78–80 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.66 (d, J = 1.2 Hz, 1H), 6.50 (d, J = 3.2 Hz, 1H), 6.45–6.47 (m, 1H), 5.22 (s, 2H), 4.08 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.48, 151.22, 147.06, 143.89, 130.33, 110.79, 109.97, 52.48, 41.79, 34.44.
Methyl 2-((5-bromo-4-(2-thiophenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7h): White solid; 10.86 g (78%); m.p. 81–83 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.54 (dd, J = 1.2 Hz and 4.8 Hz, 1H), 7.13–7.14 (m, 1H), 7.03 (dd, J = 3.6 Hz and 4.8 Hz, 1H), 5.38 (s, 2H), 4.10 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.42, 150.97, 136.35, 130.11, 127.90, 127.25, 127.18, 52.49, 43.46, 34.48.
Methyl 2-((4-(biphenyl-4-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (7i): White solid; 10.88 g (65%); m.p. 140–142 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.64–7.69 (m, 4H), 7.44–7.47 (m, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 5.26 (s, 2H), 4.11 (s, 2H), 3.64 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.43, 151.47, 139.99, 139.34, 133.56, 130.66, 128.91, 127.62, 127.42, 127.14, 126.64, 52.50, 47.84, 34.27.
Methyl 2-((5-bromo-4-(diphenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7j): Colorless oil; 9.54 g (57%); 1H-NMR (DMSO-d6, 400 MHz) δ: 7.32–7.40 (m, 6H), 7.21–7.25 (m, 4H), 6.93 (s, 1H), 3.95 (s, 2H), 3.55 (s, 3H).
Methyl 2-((4-(benzo[b]furan-5-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (7k): White solid; 10.40 g (68%); m.p. 89–91 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.01 (d, J = 2.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 0.8 Hz, 1H), 7.17 (dd, J = 1.6 Hz and 8.8 Hz, 1H), 6.96–6.97 (m, 1H), 5.30 (s, 2H), 4.09 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.44, 153.87, 151.44, 146.92, 130.60, 129.13, 127.60, 123.38, 119.87, 111.69, 106.73, 52.48, 48.24, 34.25.
Methyl 2-((5-bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7l): White solid; 10.85 g (65%); m.p. 120–122 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.37 (d, J = 8.0 Hz, 1H), 8.20–8.23 (m, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.86–7.94 (m, 2H), 6.70 (d, J = 7.6 Hz, 1H), 5.88 (s, 2H), 4.09 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.35, 151.95, 136.65, 132.94, 131.44, 131.26, 129.46, 129.26, 128.51, 125.12, 124.11, 121.74, 117.21, 109.24, 52.50, 46.07, 34.42.
Methyl 2-((5-bromo-4-(2-naphthylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7m): White solid; 9.41 g (60%); m.p. 91–93 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.88–7.96 (m, 3H), 7.64 (s, 1H), 7.51–7.55 (m, 2H), 7.34 (dd, J = 1.6 Hz and 8.4 Hz, 1H), 5.39 (s, 2H), 4.09 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.44, 151.57, 132.63, 132.42, 131.94, 130.74, 128.69, 127.74, 127.58, 126.64, 126.45, 125.63, 124.48, 52.46, 48.33, 34.30.
Methyl 2-((5-bromo-4-(quinoline-6-methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7n): White solid; 9.75 g (62%); m.p. 63–66 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.91 (dd, J = 1.6 Hz and 4.4 Hz, 1H), 8.36 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.67 (s, 1H), 7.60 (d, J = 2.0 Hz and 8.8 Hz, 1H), 7.53–7.56 (m, 1H), 5.44 (s, 2H), 4.09 (s, 2H), 3.61 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.45, 151.60, 150.97, 147.18, 135.98, 132.65, 130.80, 129.76, 128.08, 127.63, 125.79, 122.00, 52.48, 48.08, 34.39.
Ethyl 2-((4-((5-bromo-4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)-2-methylpropionate (7o): White foam; 11.94 g (65%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.38–8.40 (m, 1H), 8.20–8.22 (m, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.86–7.94 (m, 2H), 6.50 (d, J = 7.2 Hz, 1H), 5.89 (s, 2H), 4.07 (q, J = 7.2 Hz, 2H), 1.53 (s, 6H), 1.11 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.82, 149.65, 136.92, 132.91, 132.33, 131.37, 129.46, 129.05, 128.53, 125.12, 124.01, 121.42, 117.15, 109.21, 61.51, 53.89, 46.19, 25.83, 13.66.
Ethyl 1-((5-bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)cyclobutanecarboxylate (7p): White solid; 9.22 g (52%); m.p. 131–133 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.38 (d, J = 7.6 Hz, 1H), 8.20–8.23 (m, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.86–7.94 (m, 2H), 6.56 (d, J = 7.6 Hz, 1H), 5.87 (s, 2H), 4.06 (q, J = 7.1 Hz, 2H), 2.59–2.66 (m, 2H), 2.24–2.31 (m, 2H), 2.03–2.10 (m, 1H), 1.84–1.91 (m, 1H), 1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.06, 150.94, 137.53, 133.61, 132.67, 132.11, 130.19, 129.82, 129.27, 125.85, 124.70, 122.24, 117.87,109.98, 62.15, 55.09, 46.89, 32.58, 16.34, 14.42.
Methyl 2-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)acetate (7q): White solid; 8.33 g (50%); m.p. 105–107 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 7.6 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.85–7.93 (m, 2H), 7.31 (s, 1H), 6.49 (d, J = 7.6 Hz, 1H), 5.85 (s, 2H), 3.93 (s, 2H), 3.58 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.83, 141.67, 138.26, 133.01, 131.40, 130.25, 129.39, 129.20, 128.45, 125.11, 124.07, 121.33, 117.32, 108.87, 104.95, 52.34, 46.30, 35.11.
Ethyl 2-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (7r): White solid; 10.08 g (55%); m.p. 125–127 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (d, J = 7.6 Hz, 1H), 8.20 (d, J = 7.6 H, 1Hz), 8.07 (d, J = 7.6 Hz, 1H), 7.86–7.93 (m, 2H), 7.44 (s, 1H), 6.28 (d, J = 7.6 Hz, 1H), 5.89 (s, 2H), 4.05 (q, J = 7.1 Hz, 2H), 1.47 (s, 6H), 1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.28, 138.97, 138.57, 133.02, 131.33, 131.30, 129.42, 129.01, 128.50, 125.11, 123.97, 121.11, 117.28, 108.85, 106.69, 61.23, 53.31, 46.61, 25.56, 13.69.
Ethyl 1-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (7s): White solid; 9.78 g (52%); m.p. 102–105 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.42 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.86–7.93 (m, 2H), 7.40 (s, 1H), 6.34 (d, J = 7.2 Hz, 1H), 5.87 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 2.52–2.57 (m, 2H), 2.21–2.28 (m, 2H), 2.00–2.07 (m, 1H), 1.78–1.84 (m, 1H), 1.09 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.92, 139.49, 138.47, 132.99, 131.33, 131.15, 129.42, 129.02, 128.50, 125.11, 123.93, 121.18, 117.27, 108.87, 106.31, 61.14, 54.38, 46.54, 31.39, 15.36, 13.71.
Ethyl 2-((5-bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (7w): Oil; 8.72 g (56%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.46–7.47 (m, 1H), 7.24 (s, 1H), δ: 7.007–7.013 (m, 1H), 6.97–6.99 (m, 1H), 5.42 (s, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.48 (s, 6H), 1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.31, 138.40, 137.84, 130.86, 126.97, 126.71, 126.46, 105.57, 61.15, 53.31, 43.86, 25.47, 13.64.
Ethyl 1-((5-bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (7x): Colorless oil; 8.51 g (53%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.48 (dd, J = 0.8 Hz and 5.2 Hz, 1H), 7.20 (s, 1H), 7.03 (d, J = 2.8 Hz, 1H), 6.97–7.00 (m, 1H), 5.40 (s, 2H), 4.05 (q, J = 7.2 Hz, 2H), 2.51–2.59 (m, 2H), 2.24–2.31 (m, 2H), 2.06–2.13 (m, 1H), 1.79–1.87 (m, 1H), 1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.04, 138.40, 138.31, 130.73, 126.99, 126.80, 126.53, 105.17, 61.10, 54.52, 43.77, 31.26, 15.35, 13.71.
Methyl 2-((5-bromo-4-((5-chloro-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7ha): Colorless oil; 7.96 g (52%). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.05 (d, J = 3.6 Hz, 1H), 7.00 (d, J = 4.0 Hz, 1H), 5.34 (s, 2H), 4.11 (s, 2H), 3.63 (s, 3H).
Methyl 2-((5-bromo-4-((5-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hb): White solid; 8.40 g (58%); m.p. 57–59 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 6.93 (d, J = 3.6 Hz, 1H), 6.69–6.70 (m, 1H), 5.28 (s, 2H), 4.09 (s, 2H), 3.64 (s, 3H), 2.39 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.44, 150.91, 140.83, 133.82, 130.03, 127.97, 125.27, 52.49, 43.68, 34.45, 14.85.
Methyl 2-((5-bromo-4-((5-ethyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hc): Colorless oil; 9.03 g (60%). 1H-NMR (DMSO-d6, 400 MHz) δ: 6.95 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 3.2 Hz, 1H), 5.29 (s, 2H), 4.09 (s, 2H), 3.62 (s, 3H), 2.75 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.44, 150.95, 148.35, 133.46, 130.03, 127.86, 123.49, 52.50, 43.75, 34.51, 22.73, 15.60.
Methyl 2-((5-bromo-4-((3-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hd): White solid; 9.13 g (63%); m.p. 79–81 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.43 (d, J = 5.2 Hz, 1H), 6.88 (d, J = 5.2 Hz, 1H), 5.31 (s, 2H), 4.05 (s, 2H), 3.63 (s, 3H), 2.29 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.41, 151.00, 135.95, 130.20, 125.37, 52.47, 42.50, 34.49, 13.74.
Methyl 2-((4-((5-bromo-4-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7′he) White solid; 8.11 g (56%); m.p. 75–77 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.68 (s, 1H), 6.91 (s, 1H), 5.32 (s, 2H), 4.06 (s, 2H), 3.63 (s, 3H), 2.09 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.63, 147.94, 145.33, 137.37, 137.18, 129.84, 108.88, 52.41, 42.30, 34.59, 14.75.
Methyl 2-((5-bromo-4-((3-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hf): Colorless oil; 6.96 g (50%); 1H-NMR (DMSO-d6, 400 MHz) δ: 7.57–7.59 (m, 1H), 7.38 (d, J = 1.6 Hz, 1H), 7.01 (dd, J = 1.2 Hz and 5.2 Hz, 1H), 5.18 (s, 2H), 4.09 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.46, 151.12, 134.99, 130.27, 127.69, 126.47, 123.94, 52.49, 44.08, 34.27.
Methyl 2-((5-bromo-4-((5-phenyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hg): White solid; 9.00 g (53%); m.p. 78–80 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.59–7.61 (m, 2H), 7.38–7.41 (m, 3H), 7.31 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 5.40 (s, 2H), 4.12 (s, 2H), 3.63 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.43, 151.03, 144.36, 135.72, 133.05, 130.16, 129.21, 129.12, 127.98, 125.31, 123.49, 52.49, 43.70, 34.49.
Methyl 2-((4-((3-bromobenzo[b]thiophen-2-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7′hh): White solid; 10.36 g (65%); m.p. 94–96 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.77 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.47–7.56 (m, 2H), 5.60 (s, 2H), 4.04 (s, 2H), 3.60 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.28, 149.14, 146.39, 137.80, 137.74, 134.48, 127.01, 126.55, 123.96, 123.49, 108.30, 53.12, 43.52, 35.41.
Methyl 2-((5-bromo-4-((dibenzo[b,d]thiophen-4-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hi): White solid; 9.33 g (52%); m.p. 120–122 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.37–8.42 (m, 2H), 8.06–8.08 (m, 1H), 7.52–7.57 (m, 3H), 7.12 (d, J = 7.6 Hz, 1H), 5.49 (s, 2H), 4.04 (s, 2H), 3.60 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.39, 151.92, 138.04, 136.17, 135.98, 134.75, 131.25, 128.48, 127.49, 125.30, 125.24, 125.07, 123.00, 122.29, 121.97, 52.47, 47.59, 34.40.
3.2.9. General Procedure for the Synthesis of Brominated Esters 7he and 7hh
To a stirred solution of 7′he or 7′hh (20 mmol) in MeCN (100 mL) at room temperature was added NBS (3.56 g, 20 mmol), and the resulting mixture was stirred until completion of the reaction as indicated by TLC analysis (typically within 12h). The reaction mixture was poured into ice-water (200 mL), and the resulting mixture was extracted with CH2Cl2 (200 mL × 3). The combined extracts were washed successively with 5% aqueous Na2S2O3 (100 mL), saturated aqueous Na2CO3 (100 mL × 5) and 5% brine (100 mL), dried (Na2SO4) and evaporated on a rotary evaporator to give a residue, which was purified by column chromatography to afford the pure product 7he or 7hh.
Methyl 2-((5-bromo-4-((5-bromo-4-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7he): White solid; 5.92 g (62%); m.p. 91–93 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 6.91 (s, 1H), 5.29 (s, 2H), 4.11 (s, 2H), 3.63 (s, 3H), 2.09 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.40, 150.98, 137.21, 136.05, 130.12, 129.94, 109.04, 52.49, 43.48, 34.47, 14.73.
Methyl 2-((5-bromo-4-((3-bromobenzo[b]thiophen-2-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (7hh): White solid; 5.64 g (66%); m.p. 81–83 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.03 (d, J = 0.8 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.53–7.57 (m, 1H), 7.47–7.51 (m, 1H), 5.56 (s, 2H), 4.09 (s, 2H), 3.62 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.31, 151.42, 136.97, 136.83, 132.93, 130.58, 126.36, 125.90, 123.24, 122.78, 107.56, 52.48, 44.25, 34.51.
3.2.10 General Procedure for the Synthesis of Acids 8a–8x and 8ha–8hi
To a stirred solution of 6t–6v, 7a–7s, 7w–7x or 7ha–7hi (5 mmol) in MeOH (20 mL) at room temperature was added an aqueous solution of LiOH prepared by dissolving LiOH·H2O (0.42 g, 10 mmol) in a minimal amount of water, and the resulting mixture was stirred at room temperature until the reaction completed as indicated by TLC analysis (typically within 5 h). The reaction mixture was poured into ice-water (50 mL), and the resulting mixture was acidified to pH 4-5 (for 6t-6v, 7f and 7n) or 1–2 (for others) with concentrated hydrochloric acid before the extraction with CH2Cl2 (50 mL × 3). The combined extracts were washed with water (50 mL), dried (Na2SO4) and evaporated on a rotary evaporator to afford the crude product as a residue, which was purified by trituration with EtOAc/n-hexane to yield the pure product 8a–8x or 8ha–8hi.
2-((5-Bromo-4-ethyl-4H-1,2,4-triazol-3-yl)thio)acetic acid (8a): White solid; 0.98 g (78%); m.p. 136–138 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.95 (brs, 1H), 3.95 (s, 2H), 3.53 (s, 3H).
2-((5-Bromo-4-n-propyl-4H-1,2,4-triazol-3-yl)thio)acetic acid (8b): White solid; 0.10 g (75%); m.p. 143–145 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.94 (brs, 1H), 4.01 (s, 2H), 3.98 (q, J = 7.3 Hz, 2H), 1.25 (t, J = 7.2 Hz, 3H).
2-((5-Bromo-4-(cyclohexylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8c): White solid; 1.17 g (70%); m.p. 155–157 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.02 (brs, 1H), 4.03 (s, 2H), 3.76 (d, J = 7.6 Hz, 2H), 1.61–1.79 (m, 4H), 1.49–1.52 (m, 2H), 1.11–1.20 (m, 3H), 0.95–1.04 (m, 2H).
2-((4-(1-Adamantylmethyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetic acid (8d): White solid; 1.82 g (94%); m.p. 169–171 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.92 (brs, 1H), 4.00 (s, 2H), 3.66 (s, 2H), 1.95 (s, 3H), 1.63–1.66 (m, 3H), 1.54–1.58 (m, 3H), 1.52–1.53 (m, 6H).
2-((4-Benzyl-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetic acid (8e): White solid; 2.05 g (78%); m.p. 160–162 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.97 (brs, 1H), 7.31–7.40 (m, 3H), 7.16–7.18 (m, 2H), 5.20 (s, 2H), 4.01 (s, 2H).
2-((5-Bromo-4-(3-pyridinylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8f): White solid; 1.25 g (76%); m.p. 173 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 13.03 (brs, 1H), 8.54 (d, J = 4.0 Hz, 1H), 8.51 (d, J = 1.6 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.39–7.42 (m, 1H), 5.27 (s, 2H), 4.03 (s, 2H).
2-((5-Bromo-4-(2-furanylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8g): White solid; 1.24 g (78%); m.p. 113–116 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.97 (brs, 1H), 7.65 (s, 1H), 6.50 (d, J = 3.2 Hz, 1H), 6.45–6.46 (m, 1H), 5.21 (s, 2H), 4.00 (s, 2H).
2-((5-Bromo-4-(2-thiophenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8h): White solid; 1.40 g (84%); m.p. 113–115 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.98 (brs, 1H), 7.54 (dd, J = 1.2 Hz and 5.2 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 7.01–7.03 (m, 1H), 5.38 (s, 2H), 4.02 (s, 2H).
2-((4-(Biphenyl-4-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetic acid (8i): White solid; 1.44 g (71%); m.p. 178–179 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.01 (brs, 1H), 7.64–7.69 (m, 4H), 7.45 (t, J = 7.6 Hz, 2H), 7.36 (t, J = 7.2 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 5.25 (s, 2H), 4.02 (s, 2H).
2-((5-Bromo-4-(diphenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8j): White solid; 1.48 g (73%); m.p. 134–135 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.79 (brs, 1H), 7.32–7.40 (m, 6H), 7.23–7.25 (m, 4H), 6.93 (s, 1H), 3.88 (s, 2H).
2-((4-(Benzo[b]furan-5-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetic acid (8k): White solid; 1.51 g (82%); m.p. 171–173 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.97 (brs, 1H), 8.01 (d, J = 2.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.46 (s, 1H), 7.17–7.20 (m, 1H), 6.96 (d, J = 1.6 Hz, 1H), 5.29 (s, 2H), 4.02 (s, 2H).
2-((5-Bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8l): White solid; 1.61 g (80%); m.p. 170–172 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.93 (brs, 1H), 8.37 (d, J = 7.6 Hz, 1H), 8.20–8.22 (m, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.85–7.93 (m, 2H), 6.71 (d, J = 7.6 Hz, 1H), 5.87 (s, 2H), 4.01 (s, 2H).
2-((5-Bromo-4-(2-naphthylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8m): White solid; 1.48 g (78%); m.p. 172–174 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.97 (brs, 1H), 7.88–7.95 (m, 3H), 7.65 (s, 1H), 7.51–7.55 (m, 2H), 7.35 (dd, J = 1.6 Hz and 7.6 Hz, 1H), 5.38 (s, 2H), 4.03 (s, 2H).
2-((5-Bromo-4-(quinoline-6-methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8n): White solid; 1.37 g (72%); m.p. 203 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.98 (brs, 1H), 8.90–8.91 (m, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.69 (s, 1H), 7.60–7.63 (m, 1H), 7.54 (dd, J = 4.0 Hz and 8.4 Hz, 1H), 5.43 (s, 2H), 4.03 (s, 2H).
2-((4-((5-Bromo-4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)-2-methylpropionic acid (8o): White solid; 1.47 g (68%); m.p. 168–170 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.16 (brs, 1H), 8.35 (d, J = 7.6 Hz, 1H), 8.20–8.22 (m, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.86–7.94 (m, 2H), 6.53 (d, J = 7.6 Hz, 1H), 5.89 (s, 2H), 1.51 (s, 6H).
1-((5-Bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)cyclobutanecarboxylic acid (8p): White solid; 1.44 g (65%); m.p. 173–175 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.08 (brs, 1H), 8.34 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 7.6 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.86–7.94 (m, 2H), 6.60 (d, J = 7.6 Hz, 1H), 5.86 (s, 2H), 2.57–2.65 (m, 2H), 2.24–2.31 (m, 2H), 1.99–2.07 (m, 1H), 1.86–1.92 (m, 1H).
2-((5-Bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)acetic acid (8q): White solid; 1.45 g (72%); m.p. 180–182 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.84 (brs, 1H), 8.42 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 7.6 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.85–7.93 (m, 2H), 7.30 (s, 1H), 6.51 (d, J = 7.6 Hz, 1H), 5.85 (s, 2H), 3.86 (s, 2H).
2-((5-Bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionic acid (8r): White solid; 1.61 g (75%); m.p. 193–195 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.96 (brs, 1H), 8.39 (d, J = 7.6 Hz, 1H), 8.19–8.22 (m, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.86–7.93 (m, 2H), 7.43 (s, 1H), 6.33 (d, J = 7.6 Hz, 1H), 5.90 (s, 2H), 1.44 (s, 6H).
1-((5-Bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylic acid (8s): White solid; 1.50 g (68%); m.p. 210–213 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.96 (brs, 1H), 8.38 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.85–7.93 (m, 2H), 7.40 (s, 1H), 6.39 (d, J = 7.6 Hz, 1H), 5.88 (s, 2H), 2.53–2.56 (m, 2H), 2.19–2.26 (m, 2H), 1.95–2.02 (m, 1H), 1.79–1.85 (m, 1H).
2-((3-((4-Cyano-1-naphthyl)methyl)pyridin-4-yl)thio)acetic acid (8t): White solid; 1.30 g (78%); m.p. 246–249 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.10 (brs, 1H), 8.38 (d, J = 5.2 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.11 (s, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.4 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.36 (d, J = 5.2 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 4.56 (s, 2H), 4.01 (s, 2H).
2-((3-((4-Cyano-1-naphthyl)methyl)pyridin-4-yl)thio)-2-methylpropionic acid (8u) White soli; 1.30 g (72%); m.p. 175–177 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.11 (brs, 1H), 8.43 (d, J = 5.2 Hz, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.24 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 7.2 Hz, 1H), 7.83 (t, J = 7.6 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 5.2 Hz, 1H), 7.09 (d, J = 7.2 Hz, 1H), 4.64 (s, 2H), 1.50 (s, 6H).
1-((3-((4-Cyano-1-naphthyl)methyl)pyridin-4-yl)thio)cyclobutanecarboxylic acid (8v): White solid; 1.22 g (65%); m.p. 210–212 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.10 (brs, 1H), 8.34 (d, J = 5.6 Hz, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.10 (s, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.4 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.11 (d, J = 5.6 Hz, 1H), 4.54 (s, 2H), 2.78–2.86 (m, 2H), 2.18–2.25 (m, 2H), 2.07–2.16 (m, 1H), 1.92–2.00 (m, 1H).
2-((5-Bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionic acid (8w): White solid; 1.48 g (82%); m.p. 153–155 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.92 (brs, 1H), 7.46–7.47 (m, 1H), 7.22 (s, 1H), 7.03 (d, J = 3.2 Hz, 1H), 6.97–6.99 (m, 1H), 5.44 (s, 2H), 1.44 (s, 6H).
1-((5-Bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylic acid (8x): White solid; 1.40 g (75%); m.p. 164–166 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.96 (brs, 1H), 7.47–7.48 (m, 1H), 7.20 (s, 1H), 7.05 (d, J = 7.2 Hz, 1H), 6.99 (dd, J = 3.6 Hz and 4.8 Hz, 1H), 5.42 (s, 2H), 2.51–2.56 (m, 2H), 2.20–2.27 (m, 2H), 1.98–2.06 (m, 1H), 1.80–1.86 (m, 1H).
2-((5-Bromo-4-((5-chloro-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8ha): White solid; 1.25 g (68%); m.p. 142–143 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.95 (brs, 1H), 7.05 (d, J = 4.0 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 5.33 (s, 2H), 4.03 (s, 2H).
2-((5-Bromo-4-((5-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hb): White solid; 1.31 g (75%); m.p. 119–120 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.98 (brs, 1H), 6.93 (d, J = 3.2 Hz, 1H), 6.69–6.70 (m, 1H), 5.28 (s, 2H), 4.01 (s, 2H), 2.38 (s, 3H).
2-((5-Bromo-4-((5-ethyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hc): White solid; 1.41 g (78%); m.p. 119–120.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.98 (brs, 1H), 6.95 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 7.2 Hz, 1H), 5.29 (s, 2H), 4.02 (s, 2H), 2.75 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.4 Hz, 3H).
2-((5-Bromo-4-((3-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hd): White solid; 1.45 g (83%); m.p. 111–112 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.97 (brs, 1H), 7.42 (d, J = 4.8 Hz, 1H), 6.88 (d, J = 5.2 Hz, 1H), 5.30 (s, 2H), 3.98 (s, 2H), 2.29 (s, 3H).
2-((5-Bromo-4-((5-bromo-4-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8he): White solid; 1.82 g (85%); m.p. 144–145 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.99 (brs, 1H), 6.92 (s, 1H), 5.29 (s, 2H), 4.03 (s, 2H), 2.09 (s, 3H).
2-((5-Bromo-4-((3-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hf): White solid; 1.27 g (76%); m.p. 124–126 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.96 (brs, 1H), 7.56–7.58 (m, 1H), 7.39 (d, J = 2.0 Hz, 1H), 7.03 (dd, J = 0.8 Hz and 5.2 Hz, 1H), 5.17 (s, 2H), 4.01 (s, 2H).
2-((5-Bromo-4-((5-phenyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hg): White solid; 1.48 g (72%); m.p. 126–128 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 13.02 (brs, 1H), 7.59–7.61 (m, 2H), 7.38–7.41 (m, 3H), 7.31 (t, J = 7.2 Hz, 1H), 7.15 (d, J = 3.6 Hz, 1H), 5.39 (s, 2H), 4.04 (s, 2H).
2-((5-Bromo-4-((3-bromobenzo[b]thiophen-2-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hh): White solid; 1.81 g (78%); m.p. 153–155 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.98 (brs, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.53–7.57 (m, 1H), 7.47–7.51 (m, 1H), 5.55 (s, 2H), 4.02 (s, 2H).
2-((5-Bromo-4-((dibenzo[b,d]thiophen-4-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetic acid (8hi): White solid; 1.59 g (73%); m.p. 157 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.94 (brs, 1H), 8.37–8.42 (m, 2H), 8.06–8.08 (m, 1H), 7.52–7.57 (m, 3H), 7.13 (d, J = 7.2 Hz, 1H), 5.48 (s, 2H), 3.98 (s, 2H).
3.2.11 General Procedure for the Synthesis of Sodium Salts 1a–1x and 1ha–1hi
To a stirred mixture of 8a–8x or 8ha–8hi (accurately weighted to four decimal places, 2 mmol) in MeOH (10 mL) was added an aqueous solution of NaOH prepared by dissolving NaOH (0.0800 g, 2 mmol) in a minimal amount of water, and the resulting mixture was stirred at room temperature until a clear solution was obtained (typically within 1 h). The reaction mixture was filtered off and the filtrate was evaporated on a rotary evaporator to afford a residue, which was co-evaporated with CH2Cl2 (10 mL × 3) on a rotary evaporator and further dried in vacuo at room temperature to yield the pure product 1a–1x or 1ha–1hi.
Sodium 2-((5-bromo-4-ethyl-4H-1,2,4-triazol-3-yl)thio)acetate (1a): White solid; 0.55 g (100%); m.p. 203 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 3.62 (s, 2H), 3.50 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.38, 153.59, 129.80, 40.08, 31.71. ESI-HRMS [M − Na]–: (m/z) calcd. for C5H5BrN3O2S: 249.9291 (79Br), found: 249.9289; calcd. for C5H5BrN3O2S: 251.9271 (81Br), found: 251.9263. HPLC purity: 97.78%.
Sodium 2-((5-bromo-4-n-propyl-4H-1,2,4-triazol-3-yl)thio)acetate (1b): White solid; 0.58 g (100%); m.p. 106–108 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 3.94 (q, J = 7.2 Hz, 2H), 3.66 (s, 2H), 1.23 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.46, 153.07, 128.63, 40.29, 40.03, 14.47. ESI-HRMS [M − Na]–: (m/z) calcd. for C6H7BrN3O2S: 263.9448 (79Br), found: 263.9446; calcd. for C6H7BrN3O2S: 265.9427 (81Br), found: 265.9425. HPLC purity: 99.02%.
Sodium 2-((5-bromo-4-(cyclohexylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1c): White solid; 0.71 g (99%); m.p. 135 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 3.73 (d, J = 7.2 Hz, 2H), 3.68 (s, 2H), 1.61–1.77 (m, 4H), 1.48–1.51 (m, 2H), 1.13–1.17 (m, 3H), 0.98–1.03 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.41, 153.91, 129.38, 50.62, 40.10, 37.35, 29.88, 25.61, 25.04. ESI-HRMS [M − Na]–: (m/z) calcd. for C11H15BrN3O2S: 332.0074 (79Br), found: 332.0241; calcd. for C11H15BrN3O2S: 334.0053 (81Br), found: 334.0238. HPLC purity: 98.27%.
Sodium 2-((4-(1-adamantylmethyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (1d): White solid; 0.82 g (100%); m.p. 194 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 3.63 (s, 2H), 1.95 (s, 3H), 1.62–1.65 (m, 3H), 1.53–1.58 (m, 9H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.35, 155.05, 130.34, 56.18, 40.65, 40.36, 35.94, 35.46, 27.53. ESI-HRMS [M − Na]–: (m/z) calcd. for C15H19BrN3O2S: 384.0387 (79Br), found: 384.0393; calcd. for C15H19BrN3O2S: 386.0366 (81Br), found: 386.0375. HPLC purity: 98.41%.
Sodium 2-((4-benzyl-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (1e): White solid; 0.70 g (100%); m.p. 119–121 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.35–7.39 (m, 2H), 7.32 (d, J = 7.2 Hz, 1H), 7.13–7.15 (m, 2H), 5.17 (s, 2H), 3.60 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.26, 154.27, 134.87, 129.42, 128.81, 127.94, 126.71, 47.87, 40.61. ESI-HRMS [M − Na]–: (m/z) calcd. for C11H9BrN3O2S: 325.9604 (79Br), found: 325.9591; calcd. for C11H9BrN3O2S: 327.9584 (81Br), found: 327.9574. HPLC purity: 99.14%.
Sodium 2-((5-bromo-4-(3-pyridinylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1f): White solid; 0.68 g (97%); m.p. 109–111 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.53 (m, 1H), 8.48 (d, J = 1.6 Hz, 1H) 7.56 (d, J = 8.0 Hz, 1H), 7.39–7.42 (m, 1H), 5.25 (s, 2H), 3.65 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz), δ: 169.02, 154.06, 149.31, 148.38, 134.80, 130.69, 129.46, 123.90, 45.75, 40.33. ESI-HRMS [M − Na]–: (m/z) calcd. for C10H8BrN4O2S: 326.9557 (79Br), found: 326.9552; calcd. for C10H8BrN4O2S: 328.9536 (81Br), found: 328.9535. HPLC purity: 98.56%.
Sodium 2-((5-bromo-4-(2-furanylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1g): White solid; 0.68 g (100%); m.p. 108–111 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.64 (d, J = 1.2 Hz, 1H), 6.440–6.444 (m, 2H), 5.19 (s, 2H), 3.62 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.45, 153.85, 147.56, 143.71, 129.31, 110.80, 109.69, 41.66, 40.40. ESI-HRMS [M − Na]–: (m/z) calcd. for C9H7BrN3O3S: 315.9397 (79Br), found: 315.9392; calcd. for C9H7BrN3O3S: 317.9377 (81Br), found: 317.9370. HPLC purity: 97.68%.
Sodium 2-((5-bromo-4-(2-thiophenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1h): White solid; 0.71 g (100%); m.p. 122 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.51–7.52 (m, 1H), 7.11 (d, J = 2.8 Hz, 1H), 7.01 (dd, J = 3.6 Hz and 5.2 Hz, 1H), 5.36 (s, 2H), 3.65 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.00, 153.64, 136.88, 129.01, 127.71, 127.14, 127.04, 43.31, 40.67. ESI-HRMS [M − Na]–: (m/z) calcd. for C9H7BrN3O2S2: 331.9169 (79Br), found: 331.9167; calcd. for C9H7BrN3O2S2: 333.9148 (81Br), found: 333.9141. HPLC purity: 98.85%.
Sodium 2-((4-(biphenyl-4-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (1i): White solid; 0.85 g (100%); m.p. 199–201 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.64–7.68 (m, 4H), 7.45 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 5.22 (s, 2H), 3.63 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.61, 154.23, 139.85, 139.44, 134.00, 129.48, 128.91, 127.58, 127.36, 127.12, 126.65, 47.63, 40.49. ESI-HRMS [M − Na]–: (m/z) calcd. for C17H13BrN3O2S: 401.9917 (79Br), found: 401.9913; calcd. for C17H13BrN3O2S: 403.9897 (81Br), found: 403.9881. HPLC purity: 99.34%.
Sodium 2-((5-bromo-4-(diphenylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1j): White solid; 0.85 g (100%); m.p. 195 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.31–7.40 (m, 6H), 7.23–7.25 (m, 4H), 6.89 (s, 1H), 3.57 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.90, 162.42, 137.95, 129.96, 128.55, 128.18, 128.10, 65.42, 38.44. ESI-HRMS [M − Na]–: (m/z) calcd. for C17H13BrN3O2S: 401.9917 (79Br), found: 401.9863; calcd. for C17H13BrN3O2S: 403.9897 (81Br), found: 403.9845. HPLC purity: 98.86%.
Sodium 2-((4-(benzo[b]furan-5-methyl)-5-bromo-4H-1,2,4-triazol-3-yl)thio)acetate (1k): White solid; 0.78 g (100%); m.p. 92 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.00 (d, J = 2.0 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.42 (d, J = 0.8 Hz, 1H), 7.17 (dd, J = 1.6 Hz and 8.8 Hz, 1H), 6.965–6.970 (m, 1H), 5.26 (s, 2H), 3.64 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.51, 154.21, 153.79, 146.83, 129.59, 129.39, 127.56, 123.34, 119.71, 111.61, 106.76, 47.99, 40.49. ESI-HRMS [M − Na]–: (m/z) calcd. for C13H9BrN3O3S: 365.9553 (79Br), found: 365.9570; calcd. for C13H9BrN3O3S: 367.9533 (81Br), found: 367.9547. HPLC purity: 99.67%.
Sodium 2-((5-bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1l): White solid; 0.85 g (100%); m.p. 138 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.39 (d, J = 8.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.2 Hz, 1H), 7.85–7.93 (m, 2H), 6.64 (d, J = 7.6 Hz, 1H), 5.85 (s, 2H), 3.65 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.82, 154.57, 137.14, 133.01, 131.43, 130.14, 129.43, 129.25, 128.50, 125.12, 124.10, 121.51, 117.30, 109.04, 45.96, 40.54. ESI-HRMS [M − Na]–: (m/z) calcd. for C16H10BrN4O2S: 400.9713 (79Br), found: 400.9714; calcd. for C16H10BrN4O2S: 402.9693 (81Br), found: 402.9677. HPLC purity: 98.53%.
Sodium 2-((5-bromo-4-(2-naphthylmethyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1m): White solid; 0.78 g (98%); m.p. 118 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.87–7.94 (m, 3H), 7.60 (s, 1H), 7.50–7.54 (m, 2H), 7.32–7.34 (m, 1H), 5.36 (s, 2H), 3.69 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.36, 153.98, 132.68, 132.40, 132.39, 129.79, 128.69, 127.80, 127.62, 126.65, 126.43, 125.50, 124.56, 48.19, 39.82. ESI-HRMS [M − Na]–: (m/z) calcd. for C15H11BrN3O2S: 375.9761 (79Br), found: 375.9777; calcd. for C15H11BrN3O2S: 377.9740 (81Br), found: 377.9740. HPLC purity: 97.62%.
Sodium 2-((5-bromo-4-(quinoline-6-methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1n): White solid; 0.80 g (100%); m.p. 122 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.90 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.65 (s, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.53 (s, 1H), 5.40 (s, 2H), 3.67 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.72, 154.28, 150.90, 147.13, 136.04, 133.13, 129.70, 129.62, 128.10, 127.63, 125.59, 121.95, 47.86, 40.45. ESI-HRMS [M − Na]–: (m/z) calcd. for C14H10BrN4O2S: 376.9713 (79Br), found: 376.9702; calcd. for C14H10BrN4O2S: 378.9693 (81Br), found: 378.9676. HPLC purity: 99.14%.
Sodium 2-((4-((5-bromo-4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)-2-methylpropionate (1o): White solid; 0.91 g (100%); m.p. 143 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.33 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 7.2 Hz, 1H), 7.78 (t, J = 7.4 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 6.45 (d, J = 7.6 Hz, 1H), 5.91 (s, 2H), 1.48 (s, 6H). 13H-NMR (DMSO-d6, 100 MHz) δ: 174.54, 151.47, 137.46, 132.83, 131.80, 131.19, 129.06, 128.93, 128.21, 124.83, 123.80, 121.12, 117.23, 108.92, 56.64, 46.34, 26.83. ESI-HRMS [M − Na]–: (m/z) calcd. for C18H14BrN4O2S: 429.0026 (79Br), found: 429.0025; calcd. for C18H14BrN4O2S: 431.0006 (81Br), found: 431.0006. HPLC purity: 98.86%.
Sodium 1-((5-bromo-4-((4-cyano-1-naphthyl)methyl)-4H-1,2,4-triazol-3-yl)thio)cyclobutanecarboxylate (1p): White solid; 0.93 g (100%); m.p. 136 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.34 (d, J = 8.4 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.82–7.86 (m, 1H), 7.76–7.80 (m, 1H), 6.51 (d, J = 7.6 Hz, 1H), 5.94 (s, 2H), 2.43–2.49 (m, 2H), 2.22–2.29 (m, 2H), 1.82–1.85 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.74, 153.07, 137.73, 732.94, 131.34, 131.27, 129.37, 129.10, 128.47, 125.03, 124.13, 121.33, 117.28, 108.88, 60.10, 46.17, 33.07, 15.67. ESI-HRMS [M − Na]–: (m/z) calcd. for C19H14BrN4O2S: 441.0026 (79Br), found: 441.0029; calcd. for C19H14BrN4O2S: 443.0006 (81Br), found: 443.0008. HPLC purity: 99.30%.
Sodium 2-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)acetate (1q): White solid; 0.85 g (100%); m.p. 161 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.84–7.92 (m, 2H), 7.22 (s, 2H), 6.47 (d, J = 7.6 Hz, 1H), 5.87 (s, 2H), 3.53 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.55, 144.81, 138.85, 133.04, 131.38, 129.64, 129.32, 129.22, 128.38, 125.07, 124.08, 121.24, 117.40, 108.64, 103.57, 46.24, 41.22. ESI-HRMS [M − Na]–: (m/z) calcd. for C17H11BrN3O2S: 399.9761 (79Br), found: 399.9757; calcd. for C17H11BrN3O2S: 401.9740 (81Br), found: 401.9740. HPLC purity: 97.98%.
Sodium 2-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (1r): White solid; 0.90 g (100%); m.p. 243 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.45 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.83–7.91 (m, 2H), 7.30 (s, 1H), 6.31 (d, J = 7.6 Hz, 1H), 5.96 (s, 2H), 1.36 (s, 6H). 13C-NMR (DMSO-d6, 100 MHz) δ: 175.57, 143.52, 139.40, 132.95, 131.31, 130.19, 129.26, 129.12, 128.37, 125.00, 124.17, 121.16, 117.41, 108.50, 104.79, 59.03, 46.62, 28.10. ESI-HRMS [M − Na]–: (m/z) calcd. for C19H15BrN3O2S: 428.0074 (79Br), found: 428.0071; calcd. for C19H15BrN3O2S: 430.0053 (81Br), found: 430.0053. HPLC purity: 98.26%.
Sodium 1-((5-bromo-1-((4-cyano-1-naphthyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (1s): White solid; 0.93 g (100%); m.p. 285 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.41 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.82–7.91 (m, 2H), 7.33 (s, 1H), 6.35 (d, J = 7.6 Hz, 1H), 5.98 (s, 2H), 2.36–2.42 (m, 2H), 2.16–2.23 (m, 2H), 1.75–1.81 (m, 1H), 1.62–1.68 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 175.04, 143.20, 139.38, 132.98, 131.31, 130.38, 129.27, 129.11, 128.36, 125.00, 124.14, 121.17, 117.40, 108.48, 105.12, 59.84, 46.62, 32.83, 15.63. ESI-HRMS [M − Na]–: (m/z) calcd. for C20H15BrN3O2S: 440.0074 (79Br), found: 440.0070; calcd. for C20H15BrN3O2S: 442.0053 (81Br), found: 442.0052. HPLC purity: 99.15%.
Sodium 2-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)acetate (1t): White solid; 0.71 g (100%); m.p. 268 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.24–8.26 (m, 2H), 8.16 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 3.6 Hz, 1H), 7.95 (s, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 5.2 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 4.50 (s, 2H), 3.48 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.29, 150.24, 148.80, 147.62, 141.78, 133.01, 131.72, 131.18, 130.33, 128.87, 128.06, 125.52, 125.04, 124.91, 119.63, 117.74, 107.80, 37.60, 33.16. ESI-HRMS [M − Na]–: (m/z) calcd. for C19H13N2O2S: 333.0703, found: 333.0728. HPLC purity: 98.84%.
Sodium 2-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)-2-methylpropionate (1u): White solid; 0.77 g (100%); m.p. 273 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.23–8.25 (m, 2H), 8.16 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.98 (s, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.72–7.76 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 4.52 (s, 2H), 1.43 (s, 6H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.84, 149.42, 148.58, 146.99, 142.44, 133.00, 132.35, 131.70, 131.14, 128.86, 127.99, 125.62, 125.01, 123.85, 117.74, 107.70, 54.17, 33.40, 27.69. ESI-HRMS [M − Na]–: (m/z) calcd. for C21H17N2O2S: 361.1016, found: 361.1025. HPLC purity: 99.07%.
Sodium 1-((3-((4-cyano-1-naphthyl)methyl)pyridin-4-yl)thio)cyclobutanecarboxylate (1v): White solid; 0.79 g (100%); m.p. 296 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.24 (d, J = 8.0 Hz, 1H), 8.15–8.18 (m, 2H), 8.08 (d, J = 7.2 Hz, 1H), 7.91 (s, 1H), 7.82 (t, J = 7.4 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.60 (d, J = 5.6 Hz, 1H), 7.23 (d, J = 7.2 Hz, 1H), 4.46 (s, 2H), 2.69–2.77 (m, 2H), 1.93–2.02 (m, 3H), 1.79–1.86 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.51, 149.45, 148.94, 146.98, 141.93, 132.99, 131.70, 131.18, 130.38, 128.84, 127.99, 125.66, 125.01, 124.97, 121.42, 117.71, 107.77, 54.07, 33.21, 32.68, 16.08. ESI-HRMS [M − Na]–: (m/z) calcd. for C22H17N2O2S: 373.1016, found: 373.1003. HPLC purity: 98.34%.
Sodium 2-((5-bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)-2-methylpropionate (1w): White solid; 0.77 g (100%); m.p. 75 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.43 (d, J = 8.8 Hz, 1H), 7.11 (s, 1H), 6.95–6.98 (m, 2H), 5.49 (s, 2H), 1.33 (s, 6H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.91, 142.27, 140.00, 131.11, 127.92, 127.68, 126.85, 106.91, 58.53, 45.72, 27.90. ESI-HRMS [M − Na]–: (m/z) calcd. for C12H12BrN2O2S2: 358.9528 (79Br), found: 358.9526; calcd. for C12H12BrN2O2S2: 360.9509 (81Br), found: 360.9504. HPLC purity: 97.64%.
Sodium 1-((5-bromo-1-((2-thiophenyl)methyl)-1H-imidazol-2-yl)thio)cyclobutanecarboxylate (1x): White solid; 0.79 g (100%); m.p. 210 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.43–7.44 (m, 1H), 7.14 (s, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.95–6.97 (m, 1H), 5.49 (s, 2H), 2.34–2.41 (m, 2H), 2.10–2.17 (m, 2H), 1.73–1.79 (m, 1H), 1.61–1.67 (m, 1H). 13C-NMR (MeOH-d4, 100 MHz) δ: 180.25, 142.07, 140.02, 131.19, 128.03 127.70, 126.91, 107.11, 59.48, 45.72, 33.50, 15.97. ESI-HRMS [M − Na]–: (m/z) calcd. for C13H12BrN2O2S2: 370.9529 (79Br), found: 370.9527; calcd. for C13H12BrN2O2S2: 372.9509 (81Br), found: 372.9504. HPLC purity: 97.98%.
Sodium 2-((5-bromo-4-((5-chloro-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1ha): White solid; 0.78 g (100%); m.p. 97 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.03 (d, J = 3.6 Hz, 1H), 6.99 (d, J = 3.6 Hz, 1H), 5.32 (s, 2H), 3.75 (s, 2H). ESI-HRMS [M − Na]–: (m/z) calcd. for C9H6BrClN3O2S2: 365.8779 (79Br), found: 365.8788; calcd. for C9H6BrClN3O2S2: 367.8758 (81Br), found: 367.8765. HPLC purity: 96.26%.
Sodium 2-((5-bromo-4-((5-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hb): White solid; 0.74 g (100%); m.p. 157 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 6.90 (d, J = 3.2 Hz, 1H), 6.67–6.68 (m, 1H), 5.25 (s, 2H), 3.65 (s, 2H), 2.38 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.83, 153.46, 140.56, 134.32, 128.93, 127.74, 125.20, 43.51, 40.38, 14.86. ESI-HRMS [M − Na]–: (m/z) calcd. for C10H9BrN3O2S2: 345.9325 (79Br), found: 345.9321; calcd. for C10H9BrN3O2S2: 347.9305 (81Br), found: 347.9295. HPLC purity: 98.80%.
Sodium 2-((5-bromo-4-((5-ethyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hc): White solid; 0.77 g (100%); m.p. 178–180 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 6.92 (d, J = 3.2 Hz, 1H), 6.71 (d, J = 3.2 Hz, 1H), 5.26 (s, 2H), 3.66 (s, 2H), 2.74 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.15, 153.59, 148.05, 133.99, 128.95, 127.64, 123.45, 43.57, 40.58, 22.73, 15.66. ESI-HRMS [M − Na]–: (m/z) calcd. for C11H11BrN3O2S2: 359.9482 (79Br), found: 359.9479; calcd. for C11H11BrN3O2S2: 361.9461 (81Br), found: 361.9462. HPLC purity: 98.06%.
Sodium 2-((5-bromo-4-((3-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hd): White solid; 0.74 g (100%); m.p. 158 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.38 (d, J = 5.2 Hz, 1H), 6.86 (d, J = 5.2 Hz, 1H), 5.29 (s, 2H), 3.62 (s, 2H), 2.30 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.91, 153.65, 135.54, 130.86, 130.17, 129.12, 125.05, 42.37, 40.68, 13.80. ESI-HRMS [M − Na]–: (m/z) calcd. for C10H9BrN3O2S2 : 345.9325 (79Br), found: 345.9318; calcd. for C10H9BrN3O2S2 : 347.9305 (81Br), found: 347.9294. HPLC purity: 98.33%.
Sodium 2-((5-bromo-4-((5-bromo-4-methyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1he): White solid; 0.90 g (100%); m.p. 87 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 6.90 (s, 1H), 5.27 (s, 2H), 3.64 (s, 2H), 2.09 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.82, 153.62, 137.17, 136.60, 129.75, 128.98, 108.83, 43.35, 40.70, 14.77. ESI-HRMS [M − Na]–: (m/z) calcd. for C10H8Br2N3O2S2: 425.8410, found: 425.8399. HPLC purity: 97.71%.
Sodium 2-((5-bromo-4-((3-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hf): White solid; 0.71 g (100%); m.p. 124–126 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 7.55–7.57 (m, 1H), 7.34 (d, J = 1.6 Hz, 1H), 5.15 (s, 2H), 3.66 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.34, 153.69, 135.44, 129.23, 127.58, 126.57, 123.78, 43.91, 40.15. ESI-HRMS [M − Na]–: (m/z) calcd. for C9H7BrN3O2S2: 331.9169 (79Br), found: 331.9166; calcd. for C9H7BrN3O2S2: 333.9148 (81Br), found: 333.9140. HPLC purity: 99.40%.
Sodium 2-((5-bromo-4-((5-phenyl-2-thiophenyl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hg): White solid; 0.86 g (100%); m.p. 212 °C(dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 7.59–7.60 (m, 2H), 7.37–7.41 (m, 3H), 7.30 (t, J = 7.2 Hz, 1H), 7.12 (d, J = 3.6 Hz, 1H), 5.37 (s, 2H), 3.67 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 168.50, 153.77, 144.13, 136.31, 133.14, 129.12, 129.00, 127.92, 125.32, 123.45, 43.53, 40.85. ESI-HRMS [M − Na]–: (m/z) calcd. for C15H11BrN3O2S2: 407.9482 (79Br), found: 407.9498; calcd. for C15H11BrN3O2S2: 409.9461 (81Br), found: 409.9443. HPLC purity: 99.12%.
Sodium 2-((5-bromo-4-((3-bromobenzo[b]thiophen-2-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hh): White solid; 0.97 g (100%); m.p. 99 °C(dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.02 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.52–7.56 (m, 1H), 7.45–7.49 (m, 1H), 5.54 (s, 2H), 3.71 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.23, 153.82, 137.07, 136.75, 133.59, 129.58, 126.27, 125.90, 123.27, 122.72, 107.03, 44.21, 40.23. ESI-HRMS [M − Na]–: (m/z) calcd. for C13H8Br2N3O2S2: 461.8410, found: 461.8420. HPLC purity: 97.84%.
Sodium 2-((5-bromo-4-((dibenzo[b,d]thiophen-4-yl)methyl)-4H-1,2,4-triazol-3-yl)thio)acetate (1hi): White solid; 0.91 g (100%); m.p. 74 °C(dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.39–8.41 (m, 1H), 8.36 (d, J = 7.6 Hz, 1H), 8.05–8.08 (m, 1H), 7.51–7.57 (m, 3H), 7.01 (d, J = 7.6 Hz, 1H), 5.45 (s, 2H), 3.63 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 169.01, 154.56, 138.10, 135.94, 134.79, 130.13, 128.99, 127.48, 125.33, 125.07, 124.81, 123.02, 122.31, 121.77, 47.32, 40.37. ESI-HRMS [M − Na]–: (m/z) calcd. for C17H11BrN3O2S2: 431.9482 (79Br), found: 431.9640; calcd. for C17H11BrN3O2S2: 433.9461 (81Br), found: 433.9673. HPLC purity: 99.35%.



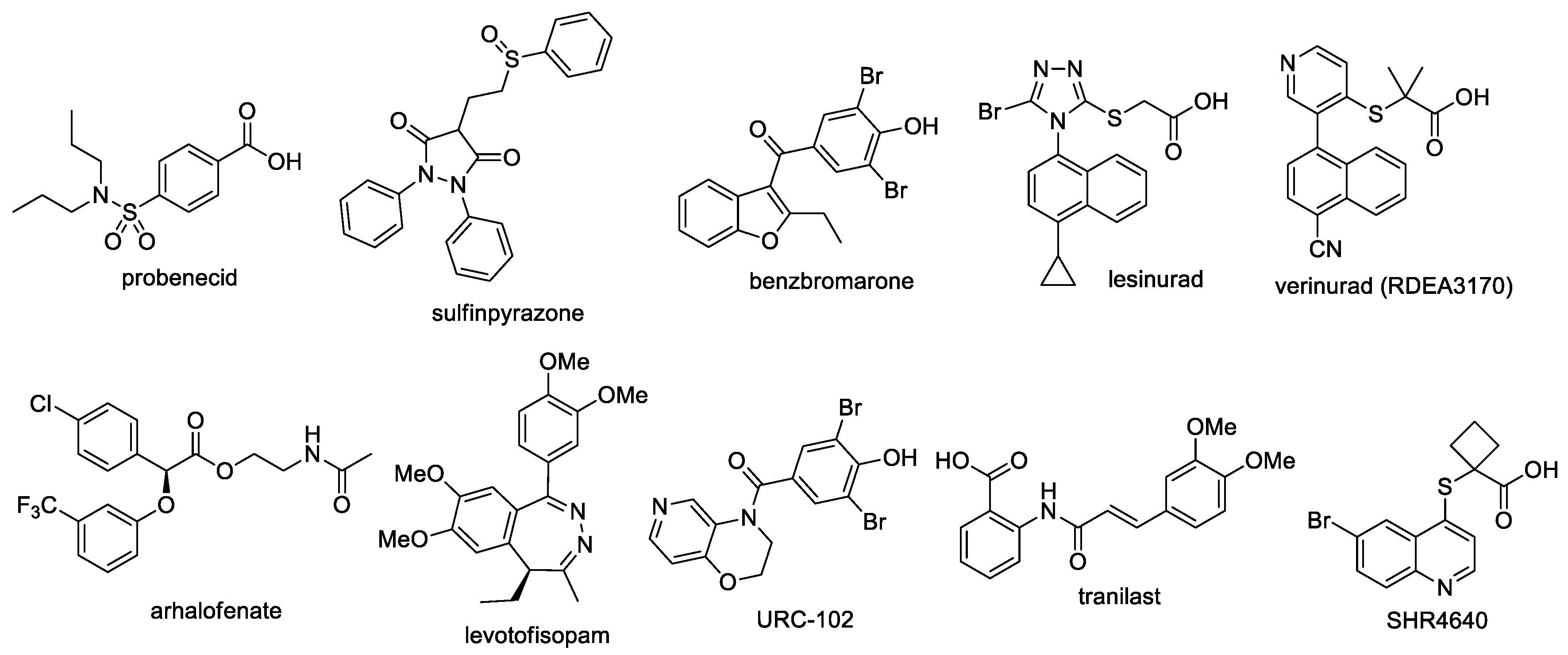
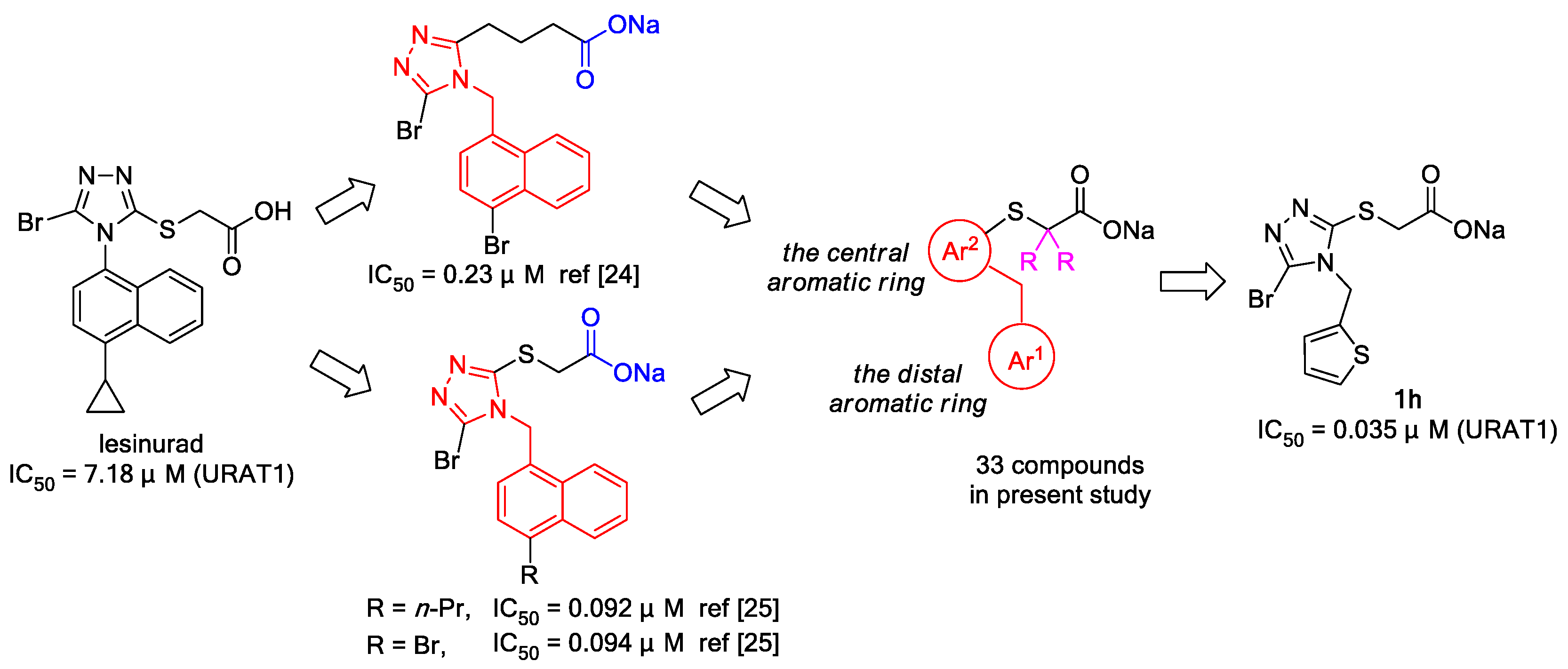
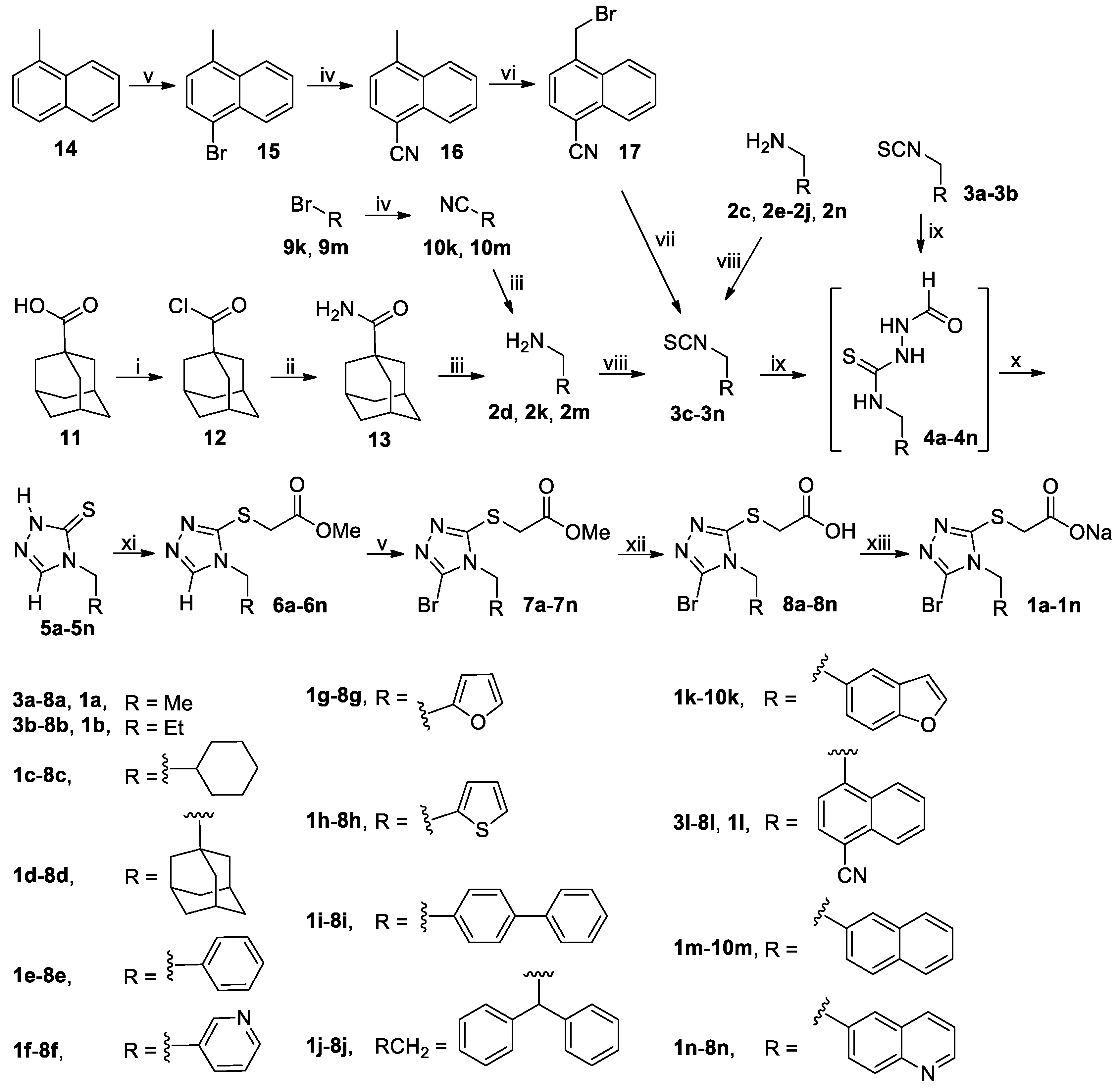
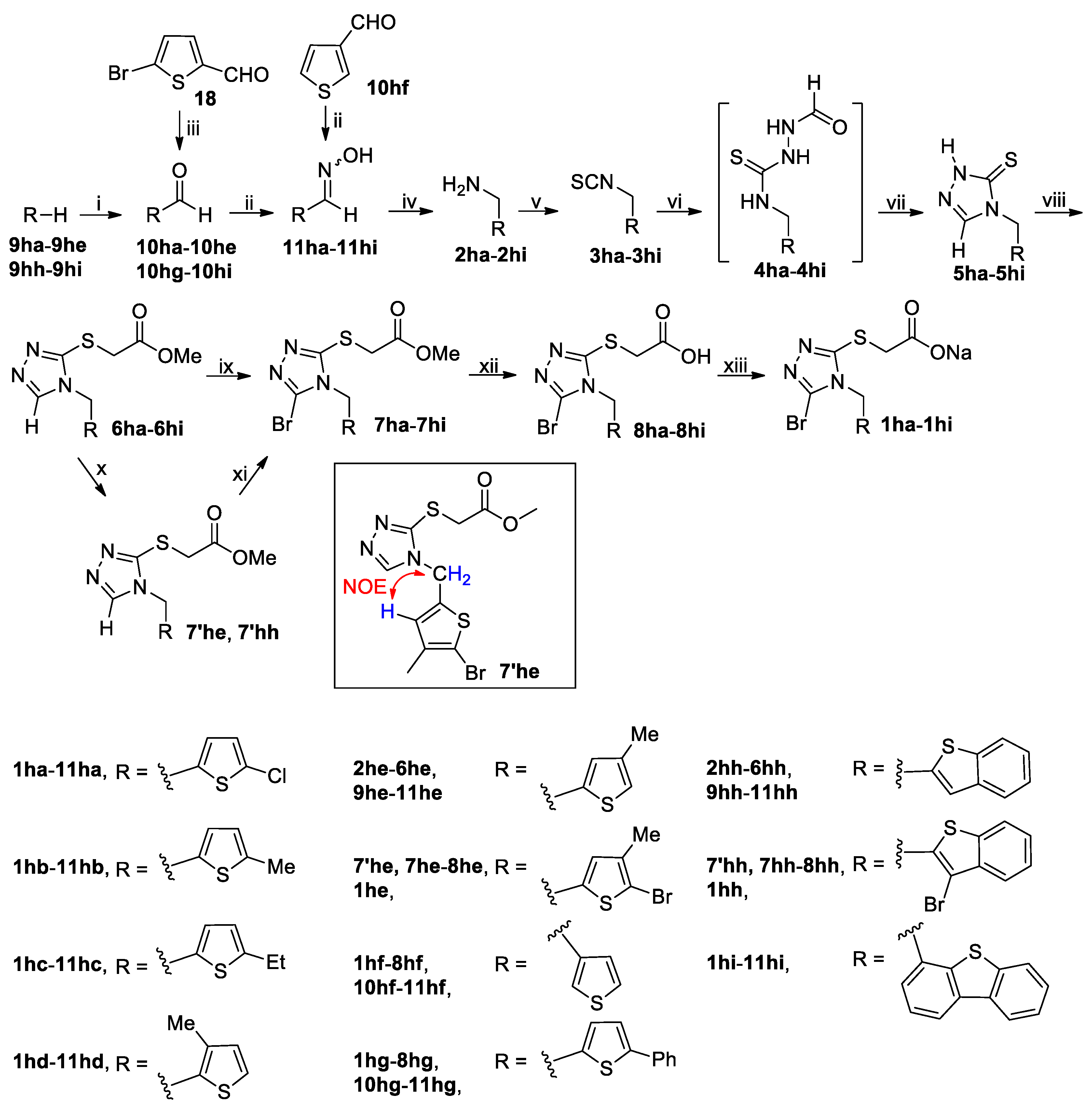
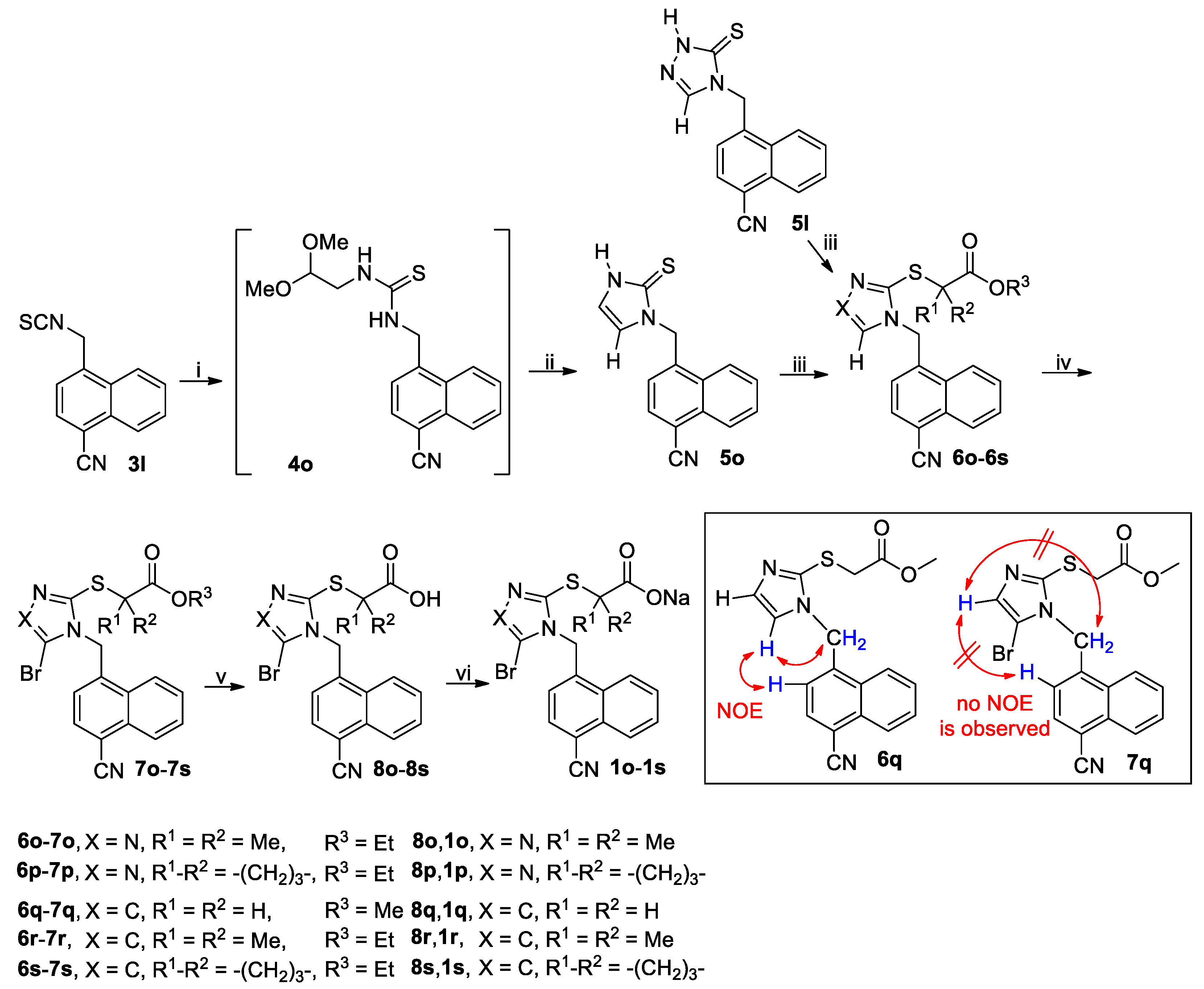
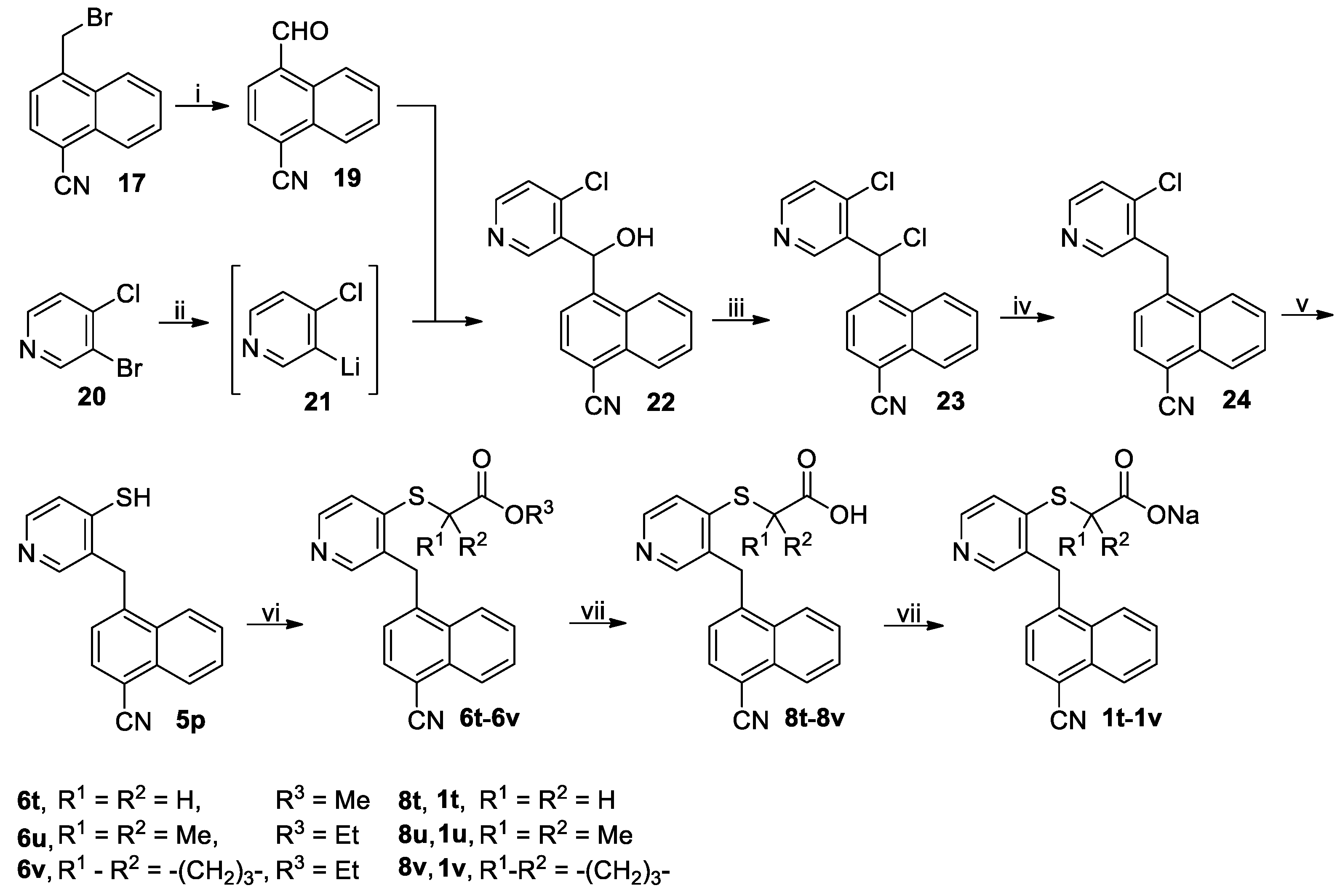
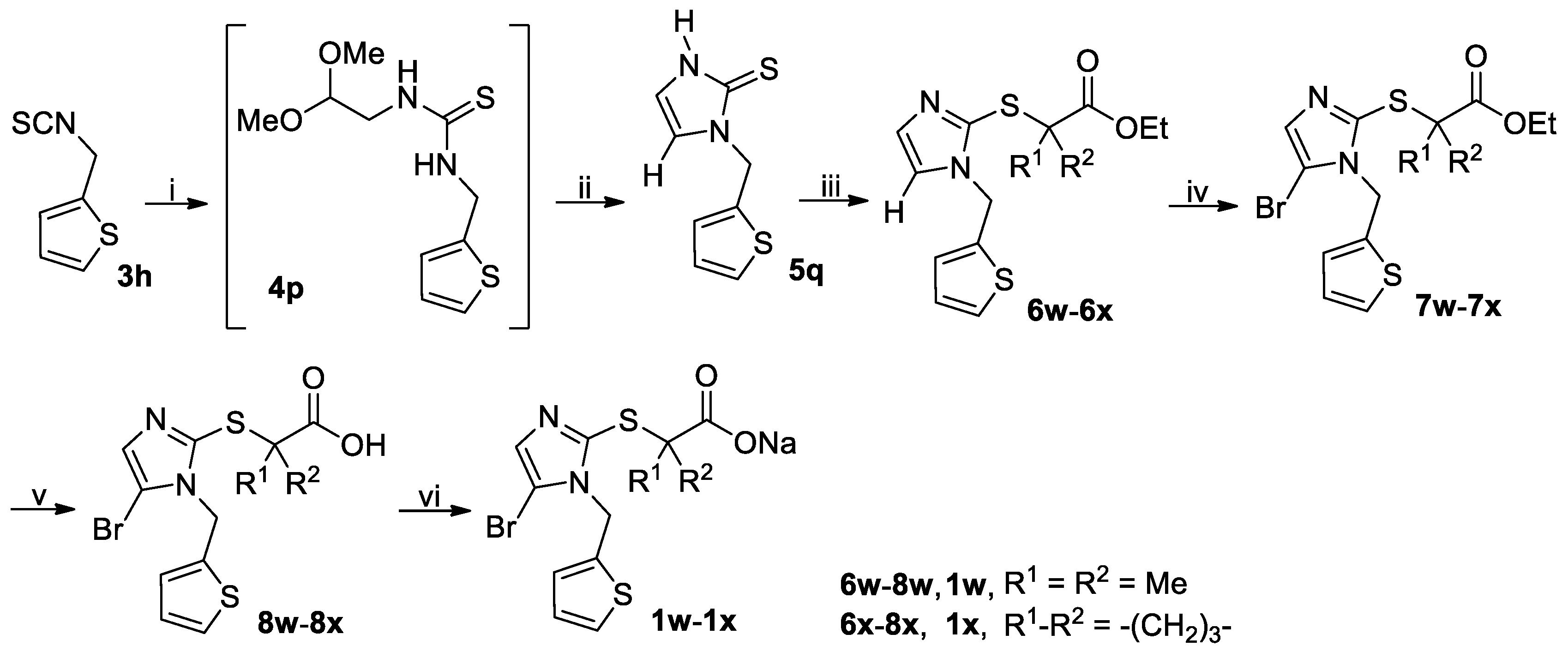

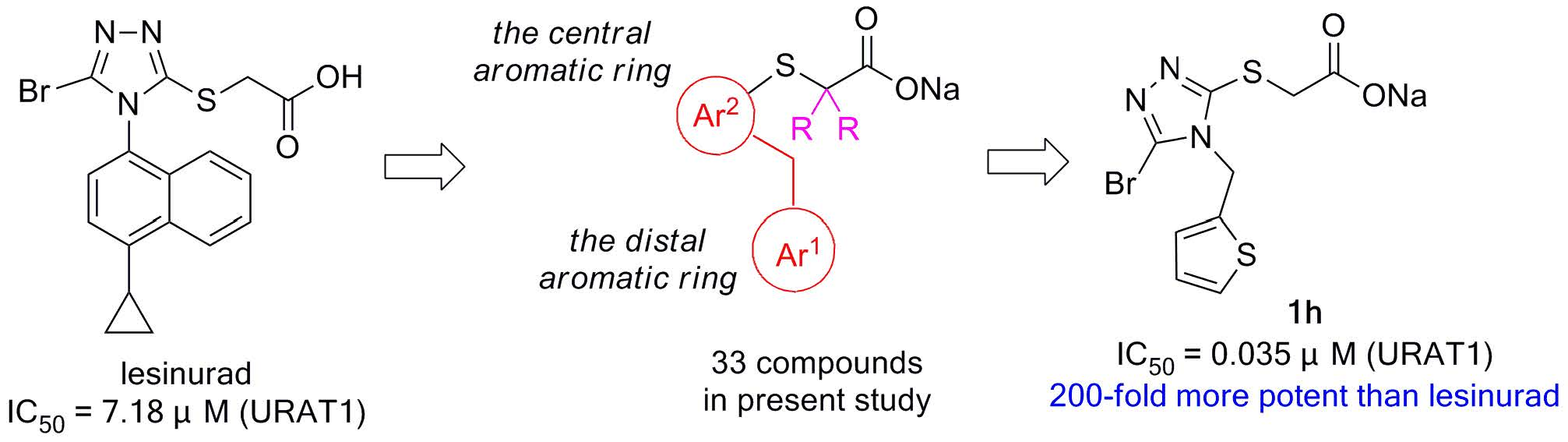{kind=link}
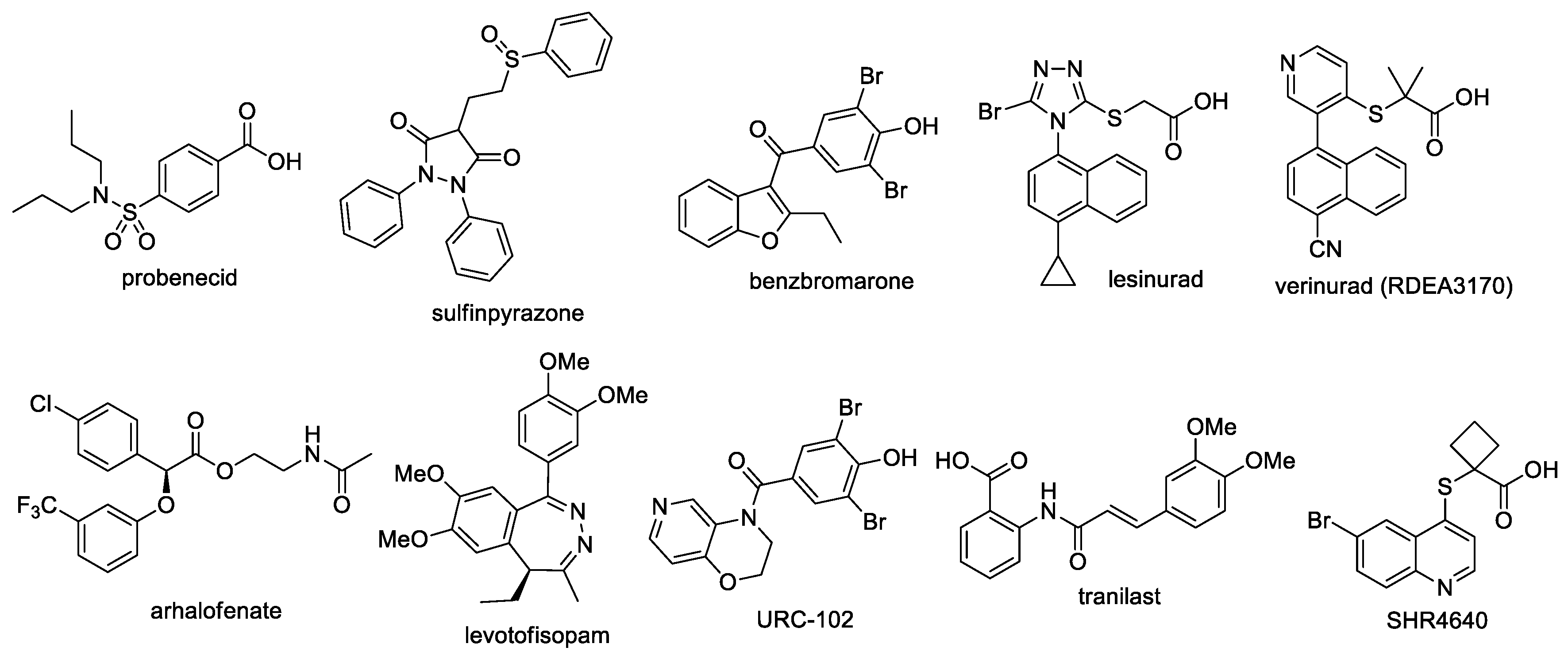{kind=link}
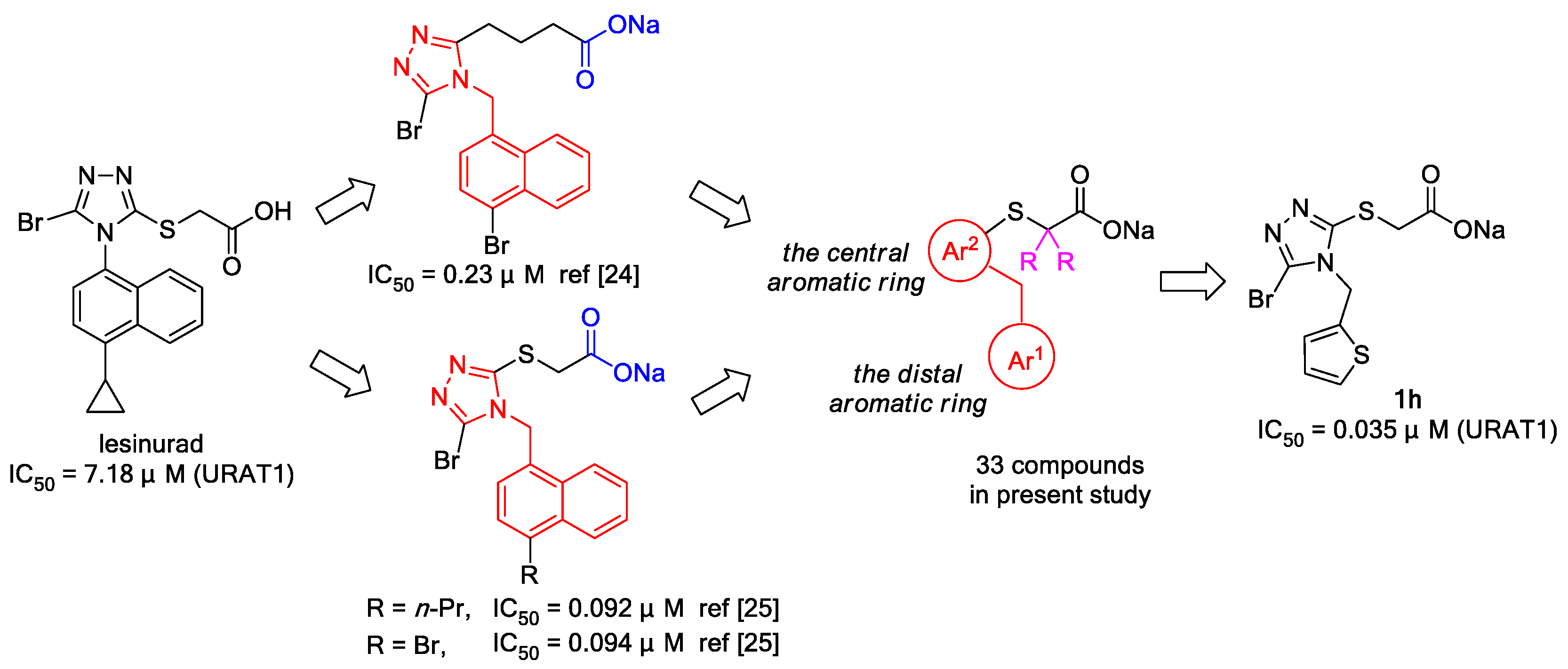{kind=link}
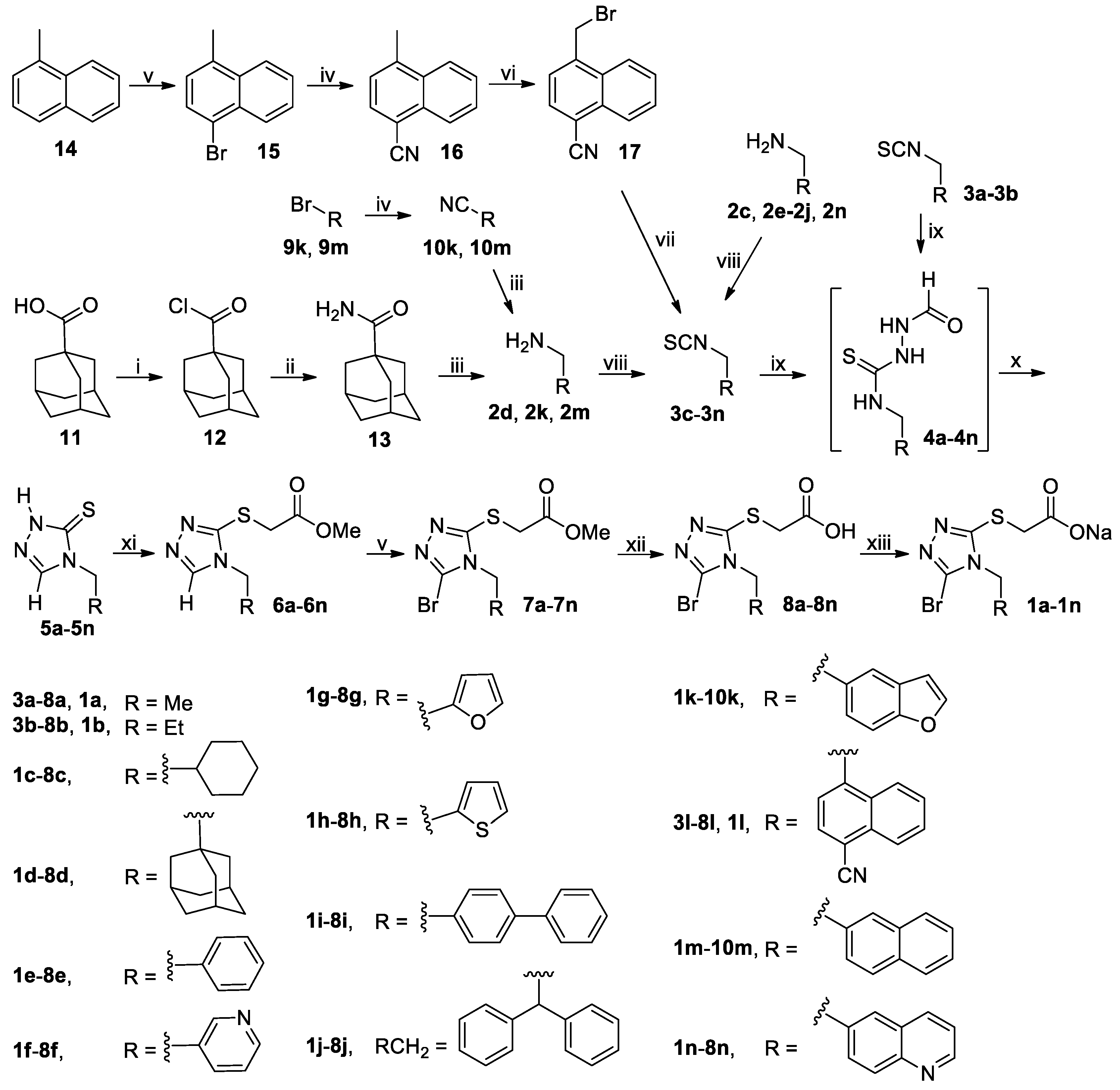{kind=link}
{kind=link}
{kind=link}
{kind=link}
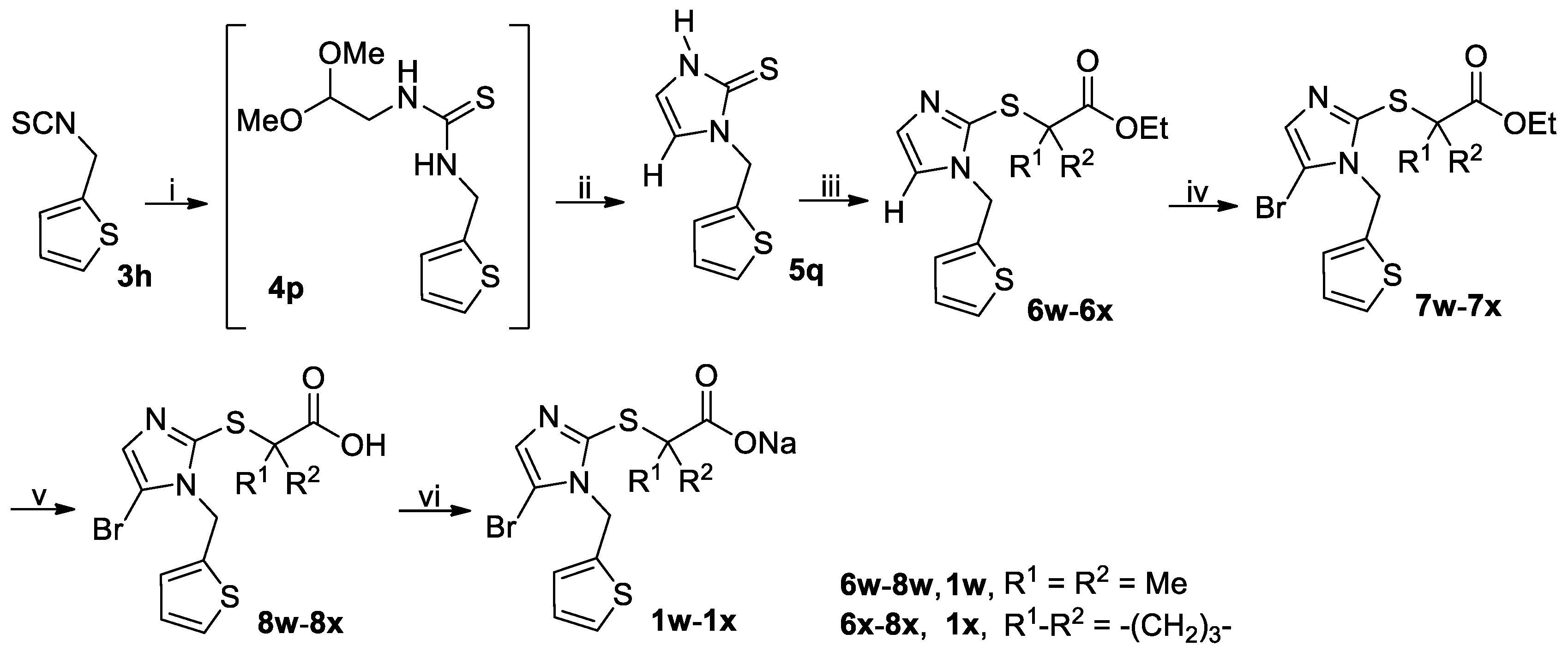{kind=link}





















