3.1. Chemistry
3.1.1. General Methods
Unless otherwise mentioned, all reactions were carried out under a nitrogen atmosphere with dry solvents under anhydrous conditions. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm Tsingdao silica gel plates (60F-254). Visualization was achieved using UV light, phosphomolybdic acid in ethanol, or potassium permanganate in water, each followed by heating. Tsingdao silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. NMR spectra were recorded with a 400 MHz (
1H: 400 MHz,
13C: 100 MHz) spectrometer. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br. = broad, m = multiplet), coupling constants, and integration. Purity testing was done by means of analytical HPLC on a Shimadzu LD-20A system with an ODS-C18 column (4.6 × 150 mm, 5 μm) eluted at 1–1.3 mL/min with Milli-Q water and CH
3CN. The purity of all tested compounds was ≥95% by HPLC (
Supplementary Materials).
3.1.2. Preparation of Cucurbitacin B (1)
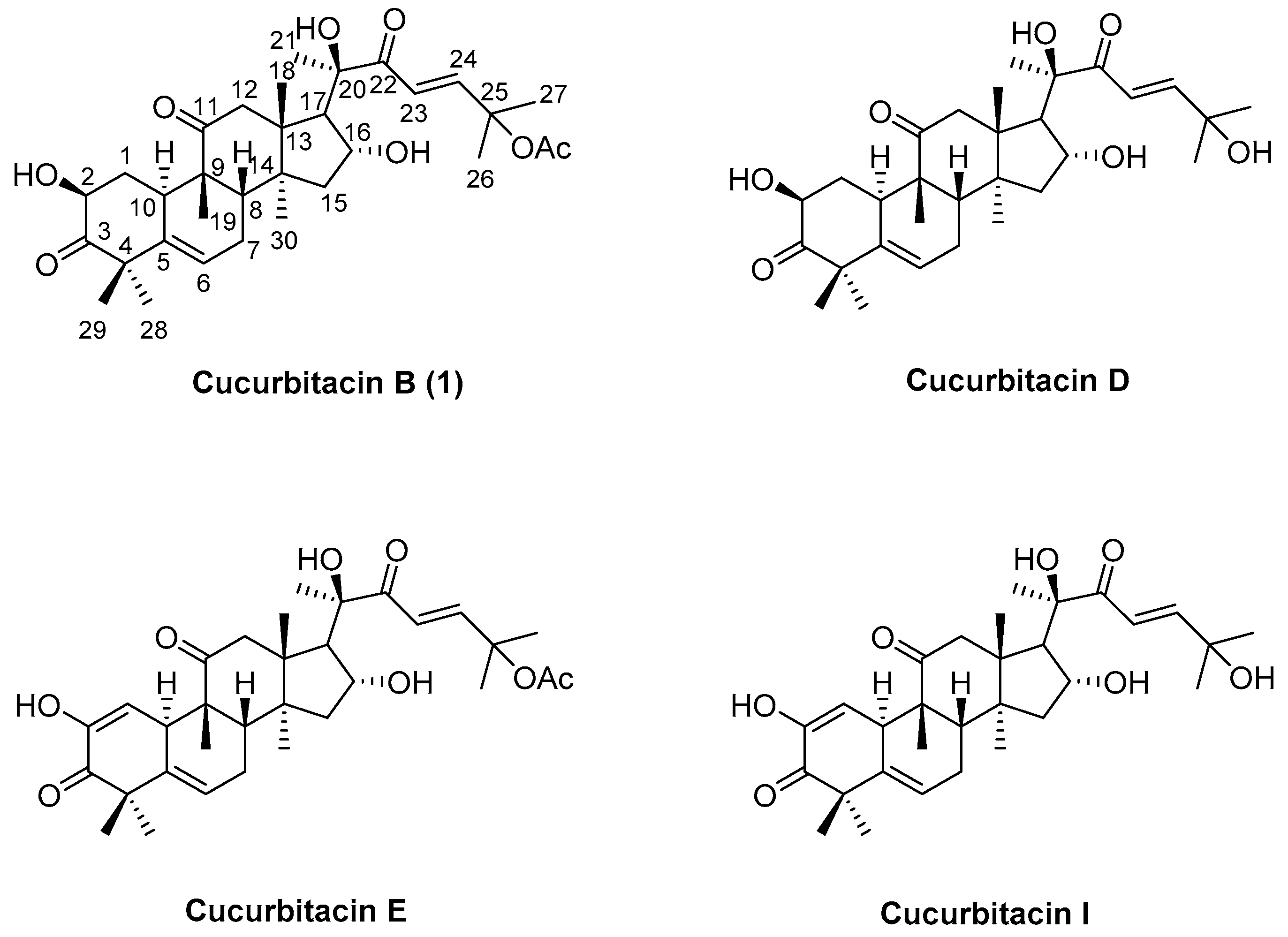
The starting material cucurbitacin B (1) was obtained from Pedicellus Melo in a yield of only 0.3%. (purity: 97%). 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 15.6 Hz, 1H), 6.46 (d, J = 15.6 Hz, 1H), 5.77 (d, J = 5.3 Hz, 1H), 4.47–4.37 (m, 1H), 4.33 (dd, J = 14.7, 7.4 Hz, 1H), 4.26 (s, 1H), 3.61 (d, J = 3.8 Hz, 1H), 3.23 (d, J = 14.5 Hz, 1H), 2.72 (d, J = 12.8 Hz, 1H), 2.66 (d, J = 14.6 Hz, 1H), 2.48 (d, J = 7.0 Hz, 1H), 2.39 (dd, J = 19.4, 7.9 Hz, 1H), 2.29 (ddd, J = 12.3, 5.7, 3.3 Hz, 1H), 2.07–1.92 (m, 6H), 1.85 (t, J = 10.2 Hz, 2H), 1.55 (s, 3H), 1.53 (s, 3H), 1.45–1.39 (m, 4H), 1.34 (s, 3H), 1.32 (s, 3H), 1.26 (s, 3H), 1.06 (s, 3H), 0.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.2, 212.3, 202.6, 170.3, 152.1, 140.5, 120.5, 120.4, 79.4, 78.3, 71.7, 71.3, 58.3, 50.8, 50.3, 48.7, 48.5, 48.2, 45.4, 42.5, 36.1, 33.8, 29.5, 26.5, 26.1, 24.0, 23.9, 22.1, 21.4, 20.2, 19.9, 18.9. HRMS (ESI) calcd. for C32H46NaO8 [M + Na]+ 581.3085, found 581.3088.
3.1.3. Procedure for the Synthesis of Compounds 2
(6R,E)-6-((2S,9R,13R,14S,16R)-2-((tert-butyldimethylsilyl)oxy)-16-Hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-6-hydroxy-2-methyl-5-oxohept-3-en-2-yl acetate (2). To a solution of cucurbitacin B (1) (200.0 mg, 0.36 mmol) and TBSCl (80.9 mg, 0.54 mmol) in dry DCM (5 mL) was added imidazole (48.7 mg, 0.72 mmol) at 0 °C. The mixture was stirred for 4 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was purified on a silica gel column to yield compound 2 (104 mg, 43.2%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 15.6 Hz, 1H), 6.45 (d, J = 15.6 Hz, 1H), 5.73 (d, J = 5.1 Hz, 1H), 4.45 (dd, J = 12.7, 5.7 Hz, 1H), 4.34 (dd, J = 14.3, 7.1 Hz, 1H), 4.25 (s, 1H), 3.25 (d, J = 14.5 Hz, 1H), 2.69 (t, J = 14.6 Hz, 2H), 2.49 (d, J = 7.0 Hz, 1H), 2.38 (dd, J = 19.4, 7.8 Hz, 1H), 2.10–2.02 (m, 1H), 2.01–1.91 (m, 5H), 1.86 (dd, J = 13.0, 9.3 Hz, 1H), 1.77 (d, J = 6.5 Hz, 1H), 1.54 (d, J = 7.2 Hz, 6H), 1.42 (s, 4H), 1.34 (s, 3H), 1.27 (s, 3H), 1.24 (s, 1H), 1.22 (s, 3H), 1.06 (s, 3H), 0.97 (s, 3H), 0.88 (s, 9H), 0.11 (s, 3H), −0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.5, 210.7, 202.6, 170.3, 151.9, 140.5, 120.5, 119.9, 79.4, 78.4, 73.8, 71.4, 58.3, 51.1, 50.8, 48.9, 48.7, 48.3, 45.5, 42.6, 36.2, 34.5, 29.4, 26.5, 26.1, 25.9, 24.1, 23.9, 22.1, 21.8, 20.2, 19.9, 19.0, 18.6, −4.4, −5.4. HRMS (ESI) calcd. for C38H60NaO8 [M + Na]+ 695.3950, found 695.3954.
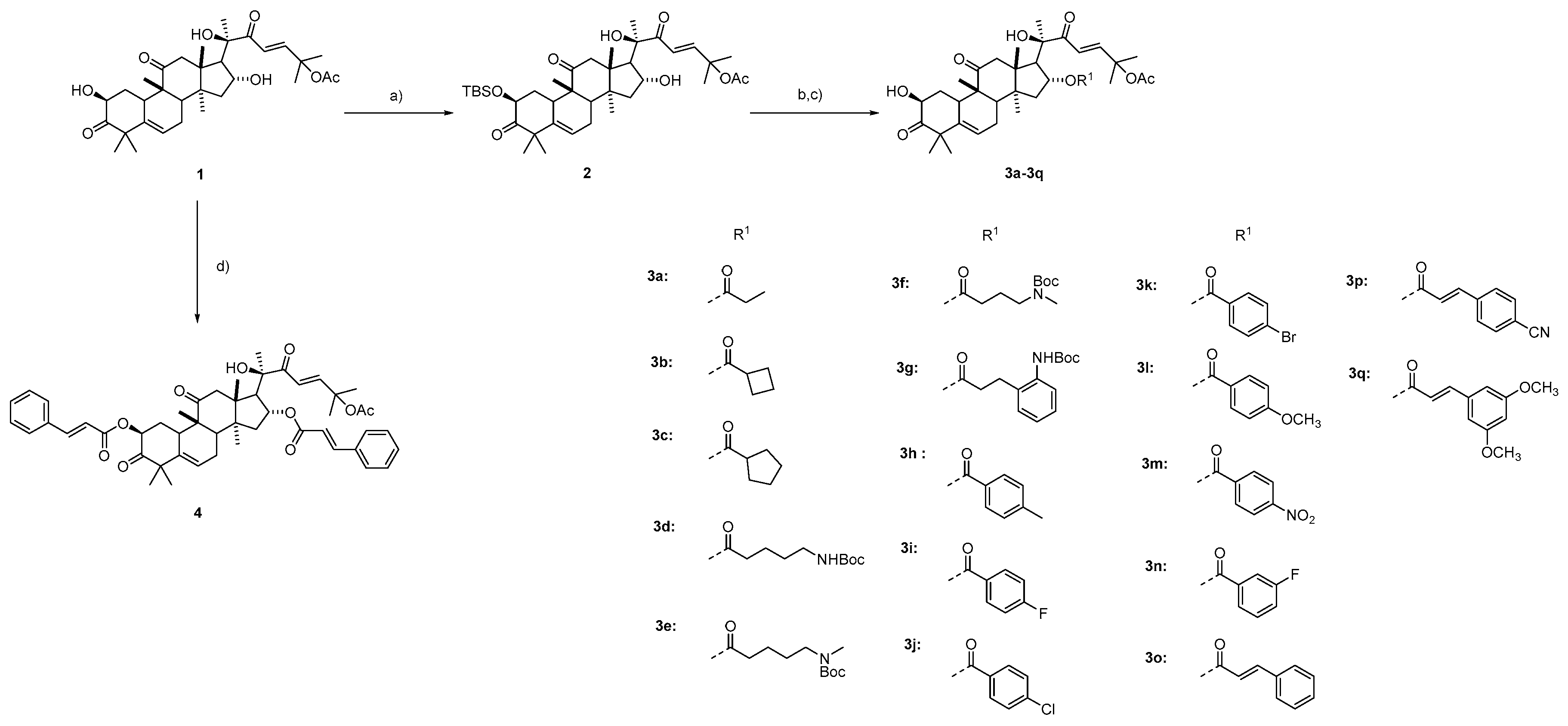
3.1.4. Procedure for the Synthesis of Compounds 3a–3q
To a solution of compound 2 (100 mg, 0.15 mmol), EDCI (85 mg, 0.45 mmol), DMAP (1.2 mg, 0.01 mmol), and the corresponding acid (0.45 mmol, 3 eq) in CH2Cl2 (2 mL) was added Et3N (62.5 μL, 0.45 mmol) at 0 °C. The mixture was stirred for 8 h at room temperature. The reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was simple purified to yield a white solid. The solid was used for the next step directly. The solid was dissolved in THF (2 mL). HOAc (38 μL, 0.65 mmol) and TBAF (169 mg, 0.65 mmol) were added to the mixture. The mixture was stirred at room temperature for 24 h, and then diluted with ethyl acetate (20 mL). The organic phase was washed with H2O (3 × 20 mL), dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel to obtain a white solid 3a–3q.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl propionate (3a) (65% yield in two steps). (purity: 97%) 1H NMR (400 MHz, CDCl3) δ 7.12 (dd, J = 15.5, 2.0 Hz, 1H), 6.40 (dd, J = 15.6, 1.8 Hz, 1H), 5.76 (s, 1H), 5.17 (t, J = 7.7 Hz, 1H), 4.49–4.35 (m, 1H), 4.29 (s, 1H), 3.60 (s, 1H), 3.24 (d, J = 14.5 Hz, 1H), 2.71 (dd, J = 20.2, 10.3 Hz, 3H), 2.46–2.24 (m, 2H), 2.09 (dt, J = 7.5, 5.6 Hz, 2H), 2.05–1.97 (m, 5H), 1.71–1.51 (m, 6H), 1.40 (s, 3H), 1.37 (s, 1H), 1.32 (s, 3H), 1.29–1.22 (m, 9H), 1.07 (s, 3H), 1.04 (dd, J = 7.6, 2.0 Hz, 2H), 1.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.8, 201.0, 173.9, 169.7, 152.7, 140.5, 120.5, 119.4, 79.3, 77.8, 73.6, 71.7, 54.3, 50.3, 50.1, 48.7, 48.5, 48.1, 43.3, 42.2, 36.1, 33.8, 29.5, 27.3, 26.6, 26.5, 23.9, 23.8, 22.0, 21.4, 20.2, 19.8, 18.9, 9.1. HRMS (ESI) calcd. for C35H50NaO9 [M + Na]+ 637.3347, found 637.3350.
(9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl cyclobutanecarboxylate (3b) (86% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 15.5 Hz, 1H), 6.41 (d, J = 15.5 Hz, 1H), 5.76 (s, 1H), 5.19 (t, J = 7.7 Hz, 1H), 4.40 (d, J = 8.1 Hz, 1H), 4.26 (s, 1H), 3.59 (s, 1H), 3.23 (d, J = 14.5 Hz, 1H), 2.99–2.83 (m, 1H), 2.71 (dd, J = 17.2, 10.4 Hz, 3H), 2.34 (dd, J = 38.6, 14.7 Hz, 2H), 2.22–2.12 (m, 2H), 2.08–1.96 (m, 7H), 1.93–1.83 (m, 2H), 1.58 (s, 6H), 1.40 (s, 3H), 1.32 (s, 3H), 1.26 (s, 9H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.8, 201.1, 174.9, 169.7, 152.4, 140.6, 120.5, 119.6, 79.3, 77.9, 73.6, 71.7, 54.3, 50.3, 50.2, 48.7, 48.5, 47.9, 43.5, 42.2, 37.8, 36.1, 33.8, 29.8, 29.5, 26.5, 25.1, 24.9, 23.9, 23.9, 22.0, 21.4, 20.2, 19.8, 18.8, 18.5. HRMS (ESI) calcd. for C37H52NaO9 [M + Na]+ 663.3504 found 663.3508.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl cyclopentanecarboxylate (3c) (52% yield in two steps). (purity: 98%). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 15.6 Hz, 1H), 6.42 (d, J = 15.6 Hz, 1H), 5.75 (dd, J = 3.7, 1.9 Hz, 1H), 5.16 (t, J = 7.8 Hz, 1H), 4.39 (ddd, J = 12.8, 5.8, 3.9 Hz, 1H), 4.26 (s, 1H), 3.59 (d, J = 3.9 Hz, 1H), 3.23 (d, J = 14.6 Hz, 1H), 2.71 (dd, J = 12.8, 11.3 Hz, 3H), 2.53–2.44 (m, 1H), 2.39 (dd, J = 19.5, 7.9 Hz, 1H), 2.28 (ddd, J = 12.5, 5.8, 3.4 Hz, 1H), 2.04–1.86 (m, 6H), 1.81–1.59 (m, 6H), 1.56 (d, J = 2.2 Hz, 6H), 1.51–1.48 (m, 2H), 1.39 (s, 3H), 1.31 (s, 3H), 1.26 (d, J = 3.1 Hz, 6H), 1.23 (s, 2H), 1.06 (s, 3H), 0.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.8, 201.0, 176.1, 169.7, 152.5, 140.5, 120.5, 119.6, 79.3, 77.9, 73.6, 71.7, 54.2, 50.3, 50.1, 48.7, 48.5, 47.9, 43.6, 43.5, 42.2, 30.1, 29.5, 29.5, 26.5, 26.4, 25.8, 25.7, 23.9, 23.8, 21.9, 21.4, 20.1, 19.8, 18.8. HRMS (ESI) calcd. for C38H54NaO9 [M + Na]+ 677.3660, found 677.3665.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 5-((tert-butoxycarbonyl)amino)pentanoate (3d) (67% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.13 (d, J = 15.6 Hz, 1H), 6.38 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.3 Hz, 1H), 5.17 (t, J = 8.0 Hz, 1H), 4.70 (s, 1H), 4.47–4.32 (m, 1H), 4.26 (s, 1H), 3.59 (d, J = 3.8 Hz, 1H), 3.23 (d, J = 14.6 Hz, 1H), 3.13–3.04 (m, 2H), 2.70 (dd, J = 19.5, 11.0 Hz, 3H), 2.39 (dd, J = 19.4, 7.7 Hz, 1H), 2.29 (ddd, J = 12.4, 5.7, 3.4 Hz, 1H), 2.10 (t, J = 7.2 Hz, 2H), 2.01 (s, 3H), 2.00–1.92 (m, 3H), 1.57 (s, 3H), 1.54 (s, 4H), 1.47–1.40 (m, 12H), 1.39 (s, 3H), 1.35 (s, 1H), 1.32 (s, 3H), 1.27 (d, J = 4.8 Hz, 6H), 1.23 (s, 1H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.9, 169.8, 156.1, 152.9, 140.5, 120.5, 119.4, 79.3, 77.8, 73.6, 71.7, 54.2, 50.3, 50.0, 48.7, 48.5, 48.1, 43.4, 42.2, 40.1, 36.1, 33.8, 33.3, 29.8, 29.5, 29.3, 28.5, 26.9, 26.4, 23.9, 23.8, 21.9, 21.8, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C42H63NNaO11 [M + Na]+ 780.4293, found 780.4296.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 5-((tert-butoxycarbonyl)(methyl)amino)pentanoate (3e) (66% yield in two steps). (purity: 96%). 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 15.6 Hz, 1H), 6.38 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.4 Hz, 1H), 5.18 (t, J = 7.9 Hz, 1H), 4.44–4.35 (m, 1H), 4.26 (s, 1H), 3.59 (d, J = 3.8 Hz, 1H), 3.28–3.12 (m, 3H), 2.80 (s, 3H), 2.69 (dd, J = 20.6, 11.0 Hz, 3H), 2.39 (dd, J = 19.5, 7.8 Hz, 1H), 2.28 (ddd, J = 12.3, 5.6, 3.4 Hz, 1H), 2.10 (s, 2H), 2.03–1.86 (m, 7H), 1.55 (d, J = 5.6 Hz, 6H), 1.51–1.45 (m, 4H), 1.42 (s, 9H), 1.39 (s, 3H), 1.31 (s, 3H), 1.27 (s, 3H), 1.25 (s, 3H), 1.23 (s, 1H), 1.06 (s, 3H), 0.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.7, 200.9, 172.9, 169.6, 155.9, 152.7, 140.6, 120.4, 119.4, 79.2, 77.8, 73.5, 71.7, 54.3, 50.3, 50.0, 48.7, 48.5, 48.1, 47.9, 43.4, 42.2, 36.1, 34.2, 33.8, 33.5, 29.8,29.4, 28.6, 26.7, 26.5, 23.9, 23.8, 21.9, 21.9, 21.4, 20.1, 19.8, 18.9. HRMS (ESI) calcd. for C43H65NNaO11 [M + Na]+ 794.4450, found 794.4453.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-((tert-butoxycarbonyl)(methyl)amino)butanoate (3f) (64% yield in two steps). (purity: 98%). 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 15.6 Hz, 1H), 6.39 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.3 Hz, 1H), 5.20 (t, J = 8.0 Hz, 1H), 4.44–4.35 (m, 1H), 4.25 (s, 1H), 3.59 (d, J = 3.7 Hz, 1H), 3.28–3.12 (m, 3H), 2.82 (s, 3H), 2.70 (dd, J = 18.3, 11.0 Hz, 3H), 2.40 (dd, J = 19.4, 7.5 Hz, 1H), 2.30 (ddd, J = 12.3, 5.5, 3.3 Hz, 1H), 2.08 (s, 2H), 2.01 (s, 3H), 1.99 (d, J = 5.7 Hz, 2H), 1.77–1.69 (m, 2H), 1.56 (d, J = 5.9 Hz, 6H), 1.43 (s, 9H), 1.40 (s, 3H), 1.32 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.24 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.7, 201.1, 172.6, 169.7, 155.9, 152.8, 140.6, 120.5, 119.4, 79.2, 77.8, 73.7, 71.7, 54.3, 50.34, 50.1, 48.7, 48.5, 48.1, 43.3, 42.2, 36.1, 34.4, 33.9, 30.9, 29.8, 29.5, 28.6, 26.5, 23.9, 23.8, 21.9, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C42H63NNaO11 [M + Na]+ 780.4293, found 780.4295.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 3-(2-((tert-butoxycarbonyl)amino)phenyl)propanoate (3g) (47% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 7.7 Hz, 1H), 7.20–7.04 (m, 4H), 7.00 (t, J = 7.3 Hz, 1H), 6.34 (d, J = 15.6 Hz, 1H), 5.74 (d, J = 3.5 Hz, 1H), 5.15 (t, J = 7.9 Hz, 1H), 4.38 (d, J = 9.1 Hz, 1H), 4.25 (s, 1H), 3.61 (s, 1H), 3.19 (d, J = 14.5 Hz, 1H), 2.81 (dt, J = 23.3, 7.6 Hz, 2H), 2.69 (d, J = 14.2 Hz, 2H), 2.61 (d, J = 7.3 Hz, 1H), 2.44 (t, J = 7.7 Hz, 2H), 2.36 (dd, J = 20.4, 7.5 Hz, 1H), 2.27 (d, J = 11.7 Hz, 1H), 1.95 (s, 3H), 1.94–1.78 (m, 3H), 1.53 (d, J = 9.1 Hz, 3H), 1.49 (s, 12H), 1.36 (s, 3H), 1.31 (s, 3H), 1.24 (s, 5H), 1.11 (s, 3H), 1.04 (s, 3H), 0.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.95, 211.70, 200.86, 173.16, 169.85, 153.82, 153.04, 140.37, 136.13, 129.31, 127.00, 124.28, 120.35, 119.09, 80.19, 79.15, 77.67, 74.02, 71.64, 65.61, 54.07, 50.28, 49.93, 48.55, 48.36, 47.92, 42.97, 42.07, 35.99, 34.12, 33.71, 29.36, 28.45, 27.06, 26.18, 25.59, 23.75, 23.57, 21.83, 21.34, 20.08, 19.69, 18.59. HRMS (ESI) calcd. for C46H63NNaO11 [M + Na]+ 828.4293, found 828.4296.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-methylbenzoate (3h) (79% yield in two steps). (purity: 97%). 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 7.9 Hz, 2H), 6.87 (d, J = 15.5 Hz, 1H), 6.51 (d, J = 15.5 Hz, 1H), 5.74 (d, J = 4.2 Hz, 1H), 5.54 (t, J = 7.8 Hz, 1H), 4.42 (d, J = 10.8 Hz, 1H), 4.19 (s, 1H), 3.61 (d, J = 2.2 Hz, 1H), 3.29 (d, J = 14.5 Hz, 1H), 2.89 (d, J = 7.2 Hz, 1H), 2.74 (d, J = 18.2 Hz, 2H), 2.47 – 2.31 (m, 4H), 2.10 (dd, J = 13.6, 9.1 Hz, 1H), 2.03 (d, J = 12.0 Hz, 5H), 1.46 (s, 6H), 1.40 (s, 3H), 1.37 (s, 3H), 1.31 (s, 3H), 1.25 (d, J = 8.9 Hz, 6H), 1.09 (s, 3H), 1.06 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.9, 201.3, 169.8, 165.9, 152.5, 143.8, 140.5, 129.7, 129.1, 127.4, 120.5, 119.5, 79.5, 78.2, 74.1, 71.7, 54.8, 50.4, 48.7, 48.5, 48.0, 43.6, 42.3, 36.1, 33.8, 32.0, 29.8, 29.5, 26.6, 25.8, 24.2, 23.9, 22.8, 22.1, 21.8, 21.4, 20.2, 19.8, 18.8. HRMS (ESI) calcd. for C40H52NaO9 [M + Na]+ 699.3504, found 699.3508.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-fluorobenzoate (3i) (81% yield in two steps). (purity: 98%). 1H NMR (400 MHz, CDCl3) δ 7.96–7.82 (m, 2H), 7.05 (t, J = 8.4 Hz, 2H), 6.90 (d, J = 15.6 Hz, 1H), 6.48 (d, J = 15.6 Hz, 1H), 5.74 (d, J = 3.6 Hz, 1H), 5.55 (t, J = 7.8 Hz, 1H), 4.42 (dd, J = 12.7, 5.7 Hz, 1H), 4.14 (s, 1H), 3.61 (s, 1H), 3.29 (d, J = 14.6 Hz, 1H), 2.87 (d, J = 7.3 Hz, 1H), 2.78 (d, J = 14.7 Hz, 1H), 2.73 (d, J = 12.4 Hz, 1H), 2.41 (dd, J = 18.9, 6.5 Hz, 1H), 2.35–2.27 (m, 1H), 2.19–2.07 (m, 1H), 2.03 (d, J = 10.1 Hz, 4H), 1.97–1.86 (m, 1H), 1.46 (s, 6H), 1.43 (s, 3H), 1.36 (s, 3H), 1.31 (s, 3H), 1.26 (s, 3H), 1.24 (s, 2H), 1.09 (s, 3H), 1.06 (s, 3H). 19F NMR (376 MHz, CDCl3) δ −105.30–−105.40 (m). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.3, 169.7, 165.9 (d, J = 254.2 Hz), 164.9, 152.8, 140.5, 132.2 (d, J = 9.2 Hz), 131.1, 128.9, 126.3 (d, J = 2.7 Hz), 120.4, 119.3, 115.6 (d, J = 22.0 Hz), 79.4, 78.1, 74.5, 71.7, 54.6, 50.4, 50.3, 48.7, 48.5, 48.0, 43.6, 42.2, 36.1, 33.8, 29.4, 26.7, 26.1, 24.1, 23.9, 22.1, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C39H49FNaO9 [M + Na]+ 703.3253, found 703.3258.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-chlorobenzoate (3j) (69% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 6.88 (d, J = 15.6 Hz, 1H), 6.48 (d, J = 15.6 Hz, 1H), 5.73 (s, 1H), 5.55 (t, J = 7.8 Hz, 1H), 4.41 (dd, J = 12.7, 5.7 Hz, 1H), 3.28 (d, J = 14.5 Hz, 1H), 2.87 (d, J = 7.2 Hz, 1H), 2.81–2.67 (m, 2H), 2.50–2.26 (m, 2H), 2.16–2.06 (m, 1H), 2.02 (d, J = 10.9 Hz, 4H), 1.97–1.86 (m, 2H), 1.45 (s, 6H), 1.42 (s, 3H), 1.35 (s, 3H), 1.30 (s, 3H), 1.25 (s, 3H), 1.24 (s, 3H), 1.08 (s, 3H), 1.05 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.7, 201.3, 169.7, 165.0, 152.8, 140.5, 139.6, 131.1, 128.8, 120.4, 119.4, 79.4, 78.1, 74.6, 71.7, 54.6, 50.3, 50.3, 48.6, 48.4, 48.0, 43.5, 42.2, 36.1, 33.8, 29.4, 26.6, 26.1, 24.1, 23.9, 22.0, 21.3, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C39H49ClNaO9 [M + Na]+ 719.2957, found 719.2962.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-bromobenzoate (3k) (64% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 15.6 Hz, 1H), 6.47 (d, J = 15.6 Hz, 1H), 5.74 (d, J = 5.4 Hz, 1H), 5.55 (t, J = 7.9 Hz, 1H), 4.42 (dd, J = 12.9, 5.9 Hz, 1H), 4.13 (d, J = 7.0 Hz, 1H), 3.29 (d, J = 14.7 Hz, 1H), 2.87 (d, J = 7.4 Hz, 1H), 2.76 (dd, J = 21.3, 13.9 Hz, 2H), 2.47–2.28 (m, 2H), 2.16–2.07 (m, 1H), 2.06–2.01 (m, 4H), 1.95 (d, J = 3.4 Hz, 1H), 1.46 (d, J = 4.2 Hz, 6H), 1.44 (s, 3H), 1.36 (s, 3H), 1.31 (s, 3H), 1.27 (s, 3H), 1.24 (s, 3H), 1.09 (s, 3H), 1.06 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.2, 169.8, 165.2, 152.9, 140.5, 131.8, 131.2, 128.9, 128.3, 120.4, 119.3, 79.4, 78.1, 74.7, 71.7, 54.6, 50.4, 50.3, 48.7, 48.5, 48.0, 43.5, 42.2, 36.1, 33.8, 29.4, 26.7, 26.1, 24.1, 23.9, 22.1, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C39H49BrNaO9 [M + Na]+ 763.2452, found 763.2458.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-methoxybenzoate (3l) (78% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.8 Hz, 2H), 6.87 (t, J = 12.0 Hz, 3H), 6.51 (d, J = 15.6 Hz, 1H), 5.74 (d, J = 5.4 Hz, 1H), 5.54 (t, J = 7.9 Hz, 1H), 4.42 (dd, J = 7.2, 3.2 Hz, 1H), 4.15 (s, 1H), 3.83 (s, 3H), 3.60 (d, J = 3.4 Hz, 1H), 3.29 (d, J = 14.6 Hz, 1H), 2.88 (d, J = 7.4 Hz, 1H), 2.78 (d, J = 14.7 Hz, 1H), 2.47–2.28 (m, 2H), 2.14–2.03 (m, 2H), 2.02 (s, 3H), 1.97–1.88 (m, 1H), 1.46 (d, J = 5.3 Hz, 6H), 1.42 (s, 3H), 1.37 (s, 3H), 1.32 (d, J = 5.5 Hz, 3H), 1.27 (d, J = 3.5 Hz, 3H), 1.25 (s, 3H), 1.09 (s, 3H), 1.06 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.9, 201.4, 169.8, 165.6, 163.6, 152.5, 140.6, 131.7, 122.6, 120.5, 119.6, 113.7, 79.5, 78.3, 74.0, 71.8, 55.6, 54.9, 50.4, 48.8, 48.5, 48.0, 43.6, 42.3, 36.1, 33.9, 29.8, 29.5, 26.7, 25.9, 24.3, 22.8, 22.1, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C40H52BrNaO10 [M + Na]+ 715.3453, found 715.3458.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 4-nitrobenzoate (3m) (64% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 8.7 Hz, 2H), 8.05 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 15.6 Hz, 1H), 6.47 (d, J = 15.6 Hz, 1H), 5.74 (s, 1H), 5.61 (t, J = 7.9 Hz, 1H), 4.42 (d, J = 10.8 Hz, 1H), 4.12 (s, 1H), 3.60 (d, J = 2.8 Hz, 1H), 3.29 (d, J = 14.6 Hz, 1H), 2.90 (d, J = 7.4 Hz, 1H), 2.80 (d, J = 14.6 Hz, 1H), 2.73 (d, J = 12.8 Hz, 1H), 2.52 – 2.27 (m, 2H), 2.16 (dd, J = 14.1, 9.0 Hz, 1H), 2.07–1.98 (m, 4H), 1.48 (s, 3H), 1.47 (s, 3H), 1.37 (s, 3H), 1.31 (s, 3H), 1.27 (s, 3H), 1.25 (s, 6H), 1.10 (s, 3H), 1.08 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.5, 201.1, 169.8, 164.2, 153.4, 150.7, 140.7, 135.4, 130.9, 128.9, 123.7, 120.4, 119.2, 79.3, 77.9, 75.5, 71.7, 54.3, 50.4, 50.3, 48.6, 48.5, 48.1, 43.6, 42.2, 36.1, 33.9, 29.8, 29.5, 26.6, 24.0, 23.9, 22.0, 21.4, 20.2, 19.7, 19.0. HRMS (ESI) calcd. for C39H49NNaO11 [M + Na]+ 730.3198, found 730.3210.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl 3-fluorobenzoate (3n) (80% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 7.3 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.42–7.33 (m, 1H), 7.22 (d, J = 7.4 Hz, 1H), 6.89 (d, J = 15.5 Hz, 1H), 6.49 (d, J = 15.6 Hz, 1H), 5.74 (s, 1H), 5.57 (t, J = 7.5 Hz, 1H), 4.42 (d, J = 7.2 Hz, 1H), 4.19 (s, 1H), 3.60 (s, 1H), 3.30 (d, J = 14.4 Hz, 1H), 2.89 (d, J = 7.0 Hz, 1H), 2.85–2.70 (m, 2H), 2.41 (d, J = 19.3 Hz, 1H), 2.31 (s, 1H), 2.12 (t, J = 11.3 Hz, 1H), 2.03 (d, J = 15.4 Hz, 3H), 1.93 (d, J = 15.3 Hz, 1H), 1.46 (s, 6H), 1.42 (s, 3H), 1.37 (s, 3H), 1.31 (s, 3H), 1.26 (d, J = 10.4 Hz, 6H), 1.08 (d, J = 10.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.6, 201.3, 169.7, 164.7, 162.5 (d, J = 247.4 Hz), 152.9, 140.6, 132.2 (d, J = 7.1 Hz), 130.2 (d, J = 7.8 Hz), 125.6 (d, J = 2.7 Hz), 120.4, 120.2 (d, J = 21.3 Hz), 119.4, 116.4 (d, J = 22.7 Hz), 79.4, 78.1, 74.8, 71.7, 54.6, 50.4, 48.7, 48.5, 48.1, 43.6, 42.3, 36.1, 33.9, 29.5, 26.7, 25.7, 24.1, 23.1, 22.1, 21.4, 20.2, 19.8, 18.9. 19F NMR (376 MHz, CDCl3) δ−112.09 (dd, J = 13.9, 8.1 Hz). HRMS (ESI) calcd. for C39H49FNaO9 [M + Na]+ 703.3253, found 703.3258.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl cinnamate (3o) (81% yield in two steps). (purity: 97%). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 15.9 Hz, 1H), 7.52 (s, 2H), 7.35 (s, 3H), 7.07 (d, J = 15.5 Hz, 1H), 6.45 (d, J = 15.5 Hz, 1H), 6.32 (d, J = 16.0 Hz, 1H), 5.76 (s, 1H), 5.37 (t, J = 7.5 Hz, 1H), 4.41 (d, J = 7.7 Hz, 1H), 4.29 (s, 1H), 3.61 (s, 1H), 3.28 (d, J = 14.5 Hz, 1H), 2.85–2.66 (m, 3H), 2.48–2.26 (m, 2H), 2.03 (s, 2H), 1.97 (s, 3H), 1.52 (d, J = 11.6 Hz, 6H), 1.43 (s, 3H), 1.34 (s, 3H), 1.32 (s, 3H), 1.27 (s, 3H), 1.25 (s, 3H), 1.09 (s, 3H), 1.04 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 213.0, 211.8, 200.9, 169.7, 166.5, 152.8, 145.4, 140.5, 134.5, 130.4, 128.9, 128.3, 120.5, 119.3, 117.8, 79.2, 77.9, 73.7, 71.7, 54.5, 50.3, 50.2, 48.7, 48.5, 48.1, 43.4, 42.3, 36.1, 33.9, 29.8, 29.4, 26.8, 26.5, 23.9, 21.9, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C41H52NaO9 [M + Na]+ 711.3504, found 711.3507.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl (E)-3-(4-cyanophenyl)acrylate (3p) (85% yield in two steps). (purity: 97%). 1H NMR (400 MHz, CDCl3) δ 7.65 (dd, J = 17.0, 8.1 Hz, 4H), 7.58 (d, J = 15.9 Hz, 1H), 7.11 (d, J = 15.5 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 3.9 Hz, 1H), 5.36 (t, J = 8.1 Hz, 1H), 4.41 (dd, J = 12.4, 5.3 Hz, 1H), 4.29 (s, 1H), 3.61 (s, 1H), 3.27 (d, J = 14.6 Hz, 1H), 2.75 (dd, J = 19.3, 10.9 Hz, 3H), 2.48 – 2.27 (m, 2H), 2.02 (d, J = 8.0 Hz, 2H), 1.98 (s, 3H), 1.56 (s, 3H), 1.48 (s, 3H), 1.43 (s, 3H), 1.32 (d, J = 5.5 Hz, 6H), 1.26 (s, 3H), 1.24 (s, 3H), 1.09 (s, 3H), 1.05 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.7, 200.8, 169.7, 165.9, 153.1, 142.9, 140.6, 138.9, 132.6, 128.8, 121.6, 120.4, 119.1, 113.4, 79.2, 77.8, 74.2, 71.7, 54.2, 50.3, 50.1, 48.7, 48.5, 48.1, 43.4, 42.2, 36.1, 33.9, 29.8, 29.4, 27.3, 26.5, 23.9, 23.8, 21.9, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C42H51NNaO9 [M + Na]+ 736.3456, found 736.3458.
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl (E)-3-(3,5-dimethoxyphenyl)acrylate (3q) (82% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 15.9 Hz, 1H), 7.07 (d, J = 15.6 Hz, 1H), 6.66 (d, J = 2.2 Hz, 2H), 6.45 (t, J = 9.2 Hz, 2H), 6.28 (d, J = 15.9 Hz, 1H), 5.75 (d, J = 5.5 Hz, 1H), 5.36 (t, J = 7.8 Hz, 1H), 4.41 (dd, J = 12.9, 6.0 Hz, 1H), 3.78 (s, 7H), 3.27 (d, J = 14.7 Hz, 1H), 2.82 – 2.68 (m, 3H), 2.48 – 2.26 (m, 2H), 2.11 – 1.94 (m, 6H), 1.53 (d, J = 3.2 Hz, 6H), 1.43 (s, 3H), 1.34 (s, 3H), 1.31 (s, 3H), 1.26 (s, 3H), 1.24 (s, 3H), 1.08 (s, 3H), 1.04 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.9, 200.9, 169.8, 166.4, 161.1, 152.9, 145.5, 140.5, 136.3, 120.5, 119.3, 118.2, 106.4, 102.5, 79.2, 77.9, 73.8, 71.7, 55.5, 54.4, 50.3, 50.2, 48.7, 48.5, 48.1, 43.4, 42.2, 36.1, 33.8, 29.8, 29.4, 26.8, 26.7, 23.9, 21.9, 21.4, 20.2, 19.8, 18.8. HRMS (ESI) calcd. for C43H56NaO11 [M + Na]+ 771.3715, found 771.3718.
3.1.5. Procedure for the Synthesis of Compounds 4
(2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthrene-2,16-diyl (2E,2′E)-bis(3-phenylacrylate) (4). To a solution of compound 1 (100.0 mg, 0.18 mmol), EDCI (171.6 mg, 0.9 mmol), DMAP (1.2 mg, 0.01 mmol), and cinnamic acid (133.4 mg, 0.9 mmol) in CH2Cl2 (2 mL) was added Et3N (125 μL, 0.9 mmol) at 0 °C. The mixture was stirred for 8 h at room temperature. The reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was purified on a silica gel column to yield compound 4 (112mg, yield: 76%) as a white solid. (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 16.0 Hz, 1H), 7.61 (d, J = 15.9 Hz, 1H), 7.52 (s, 4H), 7.37 (d, J = 8.6 Hz, 6H), 7.08 (d, J = 15.5 Hz, 1H), 6.49 (t, J = 15.0 Hz, 2H), 6.34 (d, J = 16.0 Hz, 1H), 5.79 (s, 1H), 5.64 (dd, J = 13.2, 4.5 Hz, 1H), 5.38 (t, J = 7.5 Hz, 1H), 3.31 (d, J = 14.5 Hz, 1H), 2.90 (d, J = 11.9 Hz, 1H), 2.79 (t, J = 10.5 Hz, 2H), 2.43 (d, J = 19.1 Hz, 1H), 2.23 (d, J = 11.6 Hz, 1H), 2.07 (t, J = 10.6 Hz, 2H), 1.99 (s, 3H), 1.54 (s, 3H), 1.52 (s, 3H), 1.45 (s, 3H), 1.37 (d, J = 4.4 Hz, 5H), 1.30 (s, 3H), 1.25 (s, 5H), 1.13 (s, 3H), 1.06 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.1, 205.8, 200.9, 169.7, 166.5, 166.0, 152.9, 145.9, 145.4, 139.9, 134.5, 134.4, 130.6, 130.4, 129.0, 128.9, 128.3, 120.6, 119.2, 117.8, 117.4, 79.3, 77.9, 73.7, 73.5, 54.4, 51.4, 50.2, 48.8, 48.6, 48.1, 43.4, 42.3, 34.5, 32.2, 32.0, 29.5, 28.9, 26.7, 26.6, 23.9, 22.8, 21.9, 21.5, 20.1, 19.9, 18.8. HRMS (ESI) calcd. for C50H58NaO10 [M + Na]+ 841.3922, found 841.3928.
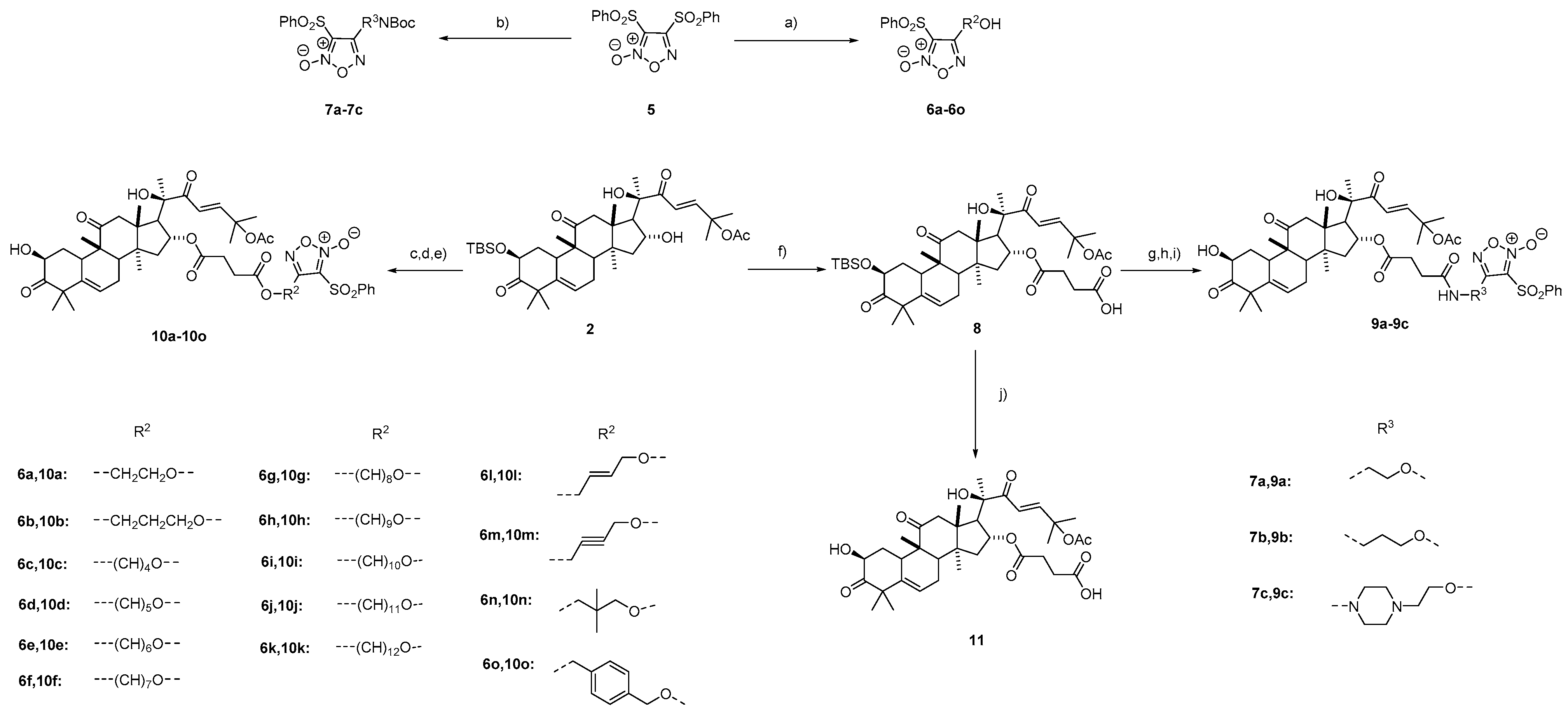
3.1.6. Procedure for the Synthesis of Compounds 6a–6o and 7a–7c
Compounds
6a–
6o and
7a–
7c were synthesized according to the procedure previously reported [
42,
43,
44,
46].
3.1.7. Procedure for the Synthesis of Compound 8
4-(((2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-((tert-butyldimethylsilyl)oxy)-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoic acid (8). To a solution of compound 2 (50.0 mg, 0.074 mmol) in dry DCM (2 mL) was added succinic anhydride (37.4 mg, 0.37 mmol) and DMAP (10.0 mg, 0.08 mmol). The mixture was stirred for 4 h at room temperature. The reaction was quenched with 1N HCl, the pH was adjusted to 2–3, and extraction was performed with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was purified on a silica gel column to yield compound 8 (49.0 mg, yield: 86%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 6.88 (d, J = 15.7 Hz, 1H), 6.73 (d, J = 15.8 Hz, 1H), 5.65 (s, 1H), 5.32 (s, 2H), 4.72 (d, J = 11.9 Hz, 1H), 3.47 (d, J = 14.2 Hz, 1H), 3.02 (d, J = 11.6 Hz, 1H), 2.60 (d, J = 6.2 Hz, 1H), 2.44 (s, 1H), 2.39–2.17 (m, 5H), 1.95 (s, 3H), 1.84 (dd, J = 26.6, 10.6 Hz, 4H), 1.46 (s, 6H), 1.26 (s, 3H), 1.20 (s, 7H), 1.12 (d, J = 15.3 Hz, 4H), 0.88 (s, 3H), 0.83 (s, 12H), 0.02 (s, 3H), −0.05 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 212.3, 210.7, 203.5, 173.4, 171.3, 169.4, 150.3, 140.5, 120.4, 118.8, 79.3, 78.3, 73.5, 73.0, 55.1, 50.6, 49.8, 48.7, 47.7, 47.3, 42.7, 41.6, 35.9, 32.5, 29.0, 28.9, 28.7, 26.3, 26.2, 25.8, 24.7, 23.4, 21.7, 21.5, 19.8, 19.3, 18.1, 18.0, −4.5, −5.2. HRMS (ESI) calcd. for C42H64NaO11Si [M + Na]+ 795.4110, found 795.4115.
3.1.8. General Procedure for the Synthesis of Compounds 9a–9c
Compounds 7a–7c (1.5 mmol) were dissolved in dry DCM. TFA (20% in DCM) was dropwise added into the reaction mixture at 0 °C, then the mixture was allowed to warm at 20 °C for 2 h. The solvent was removed by reduced pressure. The residue was purified by flash chromatography to obtain the crude amine as a colored oily matter.
Compound 8 (154 mg, 0.2 mmol) and HATU (95.1 mg, 0.25 mmol) were dissolved in dry DMF (4 mL), then DIPEA (44 μL, 0.25 mmol) was added. The mixture was stirred at 30 °C for 1 h, then the corresponding amine (0.6 mmol, 3 eq.) was added. The reaction was stirred for 2 h at this temperature. Then, the reaction was quenched with saturated aqueous NH4Cl and extracted with EA (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was purified on a silica gel column (PE/EA = 3:1–1:1) to give compound 7 as a yellow solid, which was simple purified to yield a white solid. The solid was used for the next step directly. The solid was dissolved in THF (2 mL). To the mixture was added HOAc (38 μL, 0.65 mmol) and TBAF (169 mg, 0.65 mmol). The mixture was stirred at room temperature for 24 h, and then diluted with ethyl acetate (20 mL). The organic phase was washed with H2O (3 × 20 mL), dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel to obtain a white solid 9a–9c.
3-((l1-oxidanyl)(oxo)(phenyl)-l5-sulfanyl)-4-(2-(4-(((9R,13R,14S,16R)-17-((R,E)-6-acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-Hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanamido)ethoxy)-1,2,5-oxadiazole 2-oxide (9a) (67% yield in three steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.13 – 8.02 (m, 2H), 7.78 (t, J = 7.5 Hz, 1H), 7.65 (t, J = 7.8 Hz, 2H), 7.16 (d, J = 15.6 Hz, 1H), 6.57 (t, J = 5.7 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.4 Hz, 1H), 5.18 (t, J = 7.9 Hz, 1H), 4.52 (t, J = 5.8 Hz, 2H), 4.46–4.34 (m, 1H), 4.25 (s, 1H), 3.64–3.42 (m, 3H), 3.24 (d, J = 14.6 Hz, 1H), 2.71 (dd, J = 17.2, 7.2 Hz, 3H), 2.56–2.27 (m, 6H), 2.16–2.05 (m, 2H), 2.02 (s, 3H), 2.00–1.87 (m, 3H), 1.56 (d, J = 7.8 Hz, 6H), 1.40 (s, 3H), 1.33 (s, 3H), 1.26 (s, 3H), 1.24 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.8, 201.2, 172.5, 172.0, 170.1, 158.9, 153.1, 140.4, 137.8, 136.0, 129.9, 128.7, 120.5, 119.3, 110.6, 79.4, 77.8, 73.9, 71.7, 70.7, 54.2, 50.4, 50.1, 48.7, 48.5, 48.1, 43.1, 42.2, 37.1, 36.1, 33.8, 30.8, 29.4, 28.6, 26.9, 26.5, 23.8, 23.7, 22.1, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C46H59N3NaO15S [M + Na]+ 948.3559, found 948.3562.
4-(3-(4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanamido)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9b) (73% yield in three steps) (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.11–8.01 (m, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.64 (t, J = 7.9 Hz, 2H), 7.18 (d, J = 15.6 Hz, 1H), 6.65 (t, J = 5.9 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.5 Hz, 1H), 5.17 (t, J = 7.9 Hz, 1H), 4.50 (t, J = 5.1 Hz, 2H), 4.45–4.35 (m, 1H), 4.25 (s, 1H), 3.79–3.66 (m, 2H), 3.61 (d, J = 3.8 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.70 (t, J = 11.3 Hz, 3H), 2.57–2.24 (m, 6H), 2.03 (s, 3H), 1.99–1.80 (m, 4H), 1.58 (s, 3H), 1.54 (s, 3H), 1.40 (s, 3H), 1.32 (s, 3H), 1.28 (s, 3H), 1.24 (d, J = 4.4 Hz, 6H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.8, 201.3, 172.3, 172.2, 170.3, 159.1, 153.3, 140.4, 137.9, 135.9, 129.9, 128.7, 120.5, 119.3, 110.7, 79.4, 77.8, 74.0, 71.7, 70.5, 54.0, 50.4, 50.1, 48.6, 48.5, 48.1, 43.1, 42.2, 38.4, 36.1, 33.8, 31.0, 29.8, 29.4, 29.4, 27.2, 26.4, 23.8, 23.6, 22.1, 21.4, 20.1, 19.8, 18.9. HRMS (ESI) calcd. for C47H61N3NaO15S [M + Na]+ 962.3716, found 962.3720.
4-(2-(4-(4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)piperazin-1-yl)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9c) (44% yield in three steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.6 Hz, 2H), 7.76 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.9 Hz, 2H), 7.16 (d, J = 15.6 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.4 Hz, 1H), 5.26–5.12 (m, 1H), 4.55 (t, J = 5.3 Hz, 2H), 4.40 (dd, J = 12.8, 5.9 Hz, 1H), 4.29 (s, 1H), 3.68–3.52 (m, 3H), 3.47 (t, J = 8.3 Hz, 2H), 3.25 (d, J = 14.6 Hz, 1H), 2.88 (t, J = 5.3 Hz, 2H), 2.79 (s, 1H), 2.71 (dd, J = 11.0, 7.4 Hz, 3H), 2.63 (dt, J = 9.5, 6.1 Hz, 2H), 2.54 (dd, J = 9.3, 4.9 Hz, 2H), 2.50–2.25 (m, 5H), 2.01 (s, 3H), 1.98 (d, J = 9.7 Hz, 2H), 1.57 (d, J = 1.3 Hz, 6H), 1.41 (s, 3H), 1.31 (d, J = 7.5 Hz, 6H), 1.26 (s, 3H), 1.24 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.9, 201.2, 172.7, 169.8, 169.7, 159.0, 153.0, 140.4, 138.1, 135.8, 129.8, 128.6, 120.5, 119.4, 110.6, 79.2, 77.8, 73.9, 71.7, 69.4, 56.2, 54.3, 53.5, 53.2, 50.4, 50.1, 48.7, 48.5, 48.1, 45.3, 43.1, 42.2, 41.8, 38.7, 36.1, 33.8, 29.4, 28.9, 27.8, 26.7, 26.6, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C50H67N4O15S [M + H]+ 995.4318, found 995.4322.
3.1.9. General Procedure for the Synthesis of Compounds 10a–10o
To a solution of 6a–6o (0.5 mmol) in dry DCM (20 mL) was added succinic anhydride (75 mg, 0.75 mmol) and DMAP (61 mg, 0.5 mmol). The mixture was stirred for 4 h at room temperature. The reaction was quenched with 1N HCl, the pH was adjusted to 2–3 and extraction was performed with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was purified on a silica gel column to obtain the corresponding acid in a yield of 86–96%.
To a solution of compound 2 (135 mg, 0.2 mmol), EDCI (96 mg, 0.5 mmol), DMAP (1.2 mg, 0.01 mmol), and the corresponding acid (0.4 mmol, 2 eq.) in CH2Cl2 (2 mL) was added Et3N (69.5 μL, 0.5 mmol) at 0 °C. The mixture was stirred for 8 h at room temperature. The reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated brine, dried over Na2SO4, and concentrated to give an oily crude product, which was simple purified to yield a white solid. The solid was used for the next step directly. The solid was dissolved in THF (2 mL). HOAc (38 μL, 0.65 mmol) and TBAF (169 mg, 0.65 mmol) were added to the mixture. The mixture was stirred at room temperature for 24 h, and then diluted with ethyl acetate (20 mL). The organic phase was washed with H2O (3 × 20 mL), dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel to obtain a white solid 10a–10o.
4-(2-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10a) (81% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.7 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.63 (t, J = 7.8 Hz, 2H), 7.18 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.2 Hz, 1H), 5.19 (s, 1H), 4.67–4.56 (m, 2H), 4.55–4.46 (m, 2H), 4.45–4.35 (m, 1H), 4.28 (s, 1H), 3.60 (d, J = 3.7 Hz, 1H), 3.25 (d, J = 14.6 Hz, 1H), 2.78–2.66 (m, 3H), 2.65–2.25 (m, 6H), 2.05–1.85 (m, 6H), 1.58 (s, 3H), 1.55 (s, 3H), 1.40 (s, 3H), 1.32 (s, 3H), 1.29 (s, 3H), 1.25 (s, 3H), 1.24 (s, 2H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.8, 201.1, 172.2, 171.7, 169.8, 158.8, 153.2, 140.5, 138.0, 135.9, 129.9, 128.7, 120.5, 119.2, 110.5, 79.2, 77.8, 74.1, 71.7, 68.9, 61.5, 54.2, 50.3, 50.1, 48.6, 48.5, 48.1, 43.1, 42.2, 36.1, 33.8, 29.4, 28.8, 28.6, 26.9, 26.3, 23.8, 23.7, 22.0, 21.4, 20.1, 19.8, 18.9. HRMS (ESI) calcd. for C46H58N2NaO16S [M + Na]+ 949.3399, found, 949.3402.
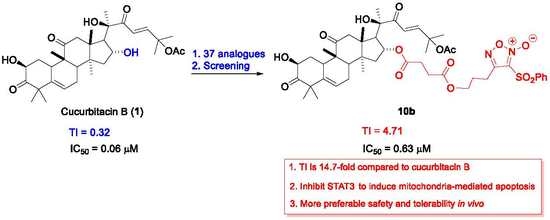
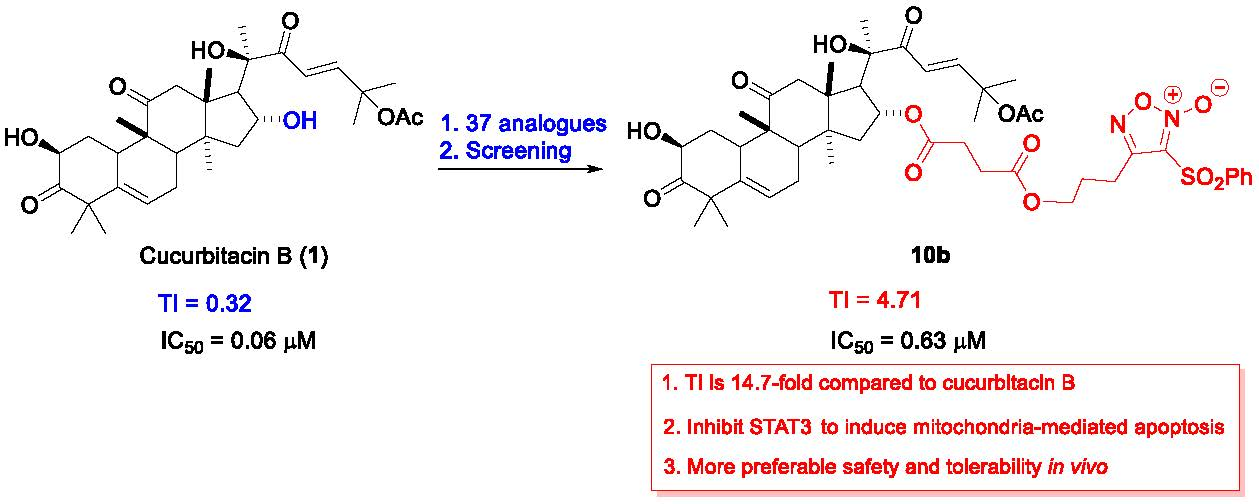
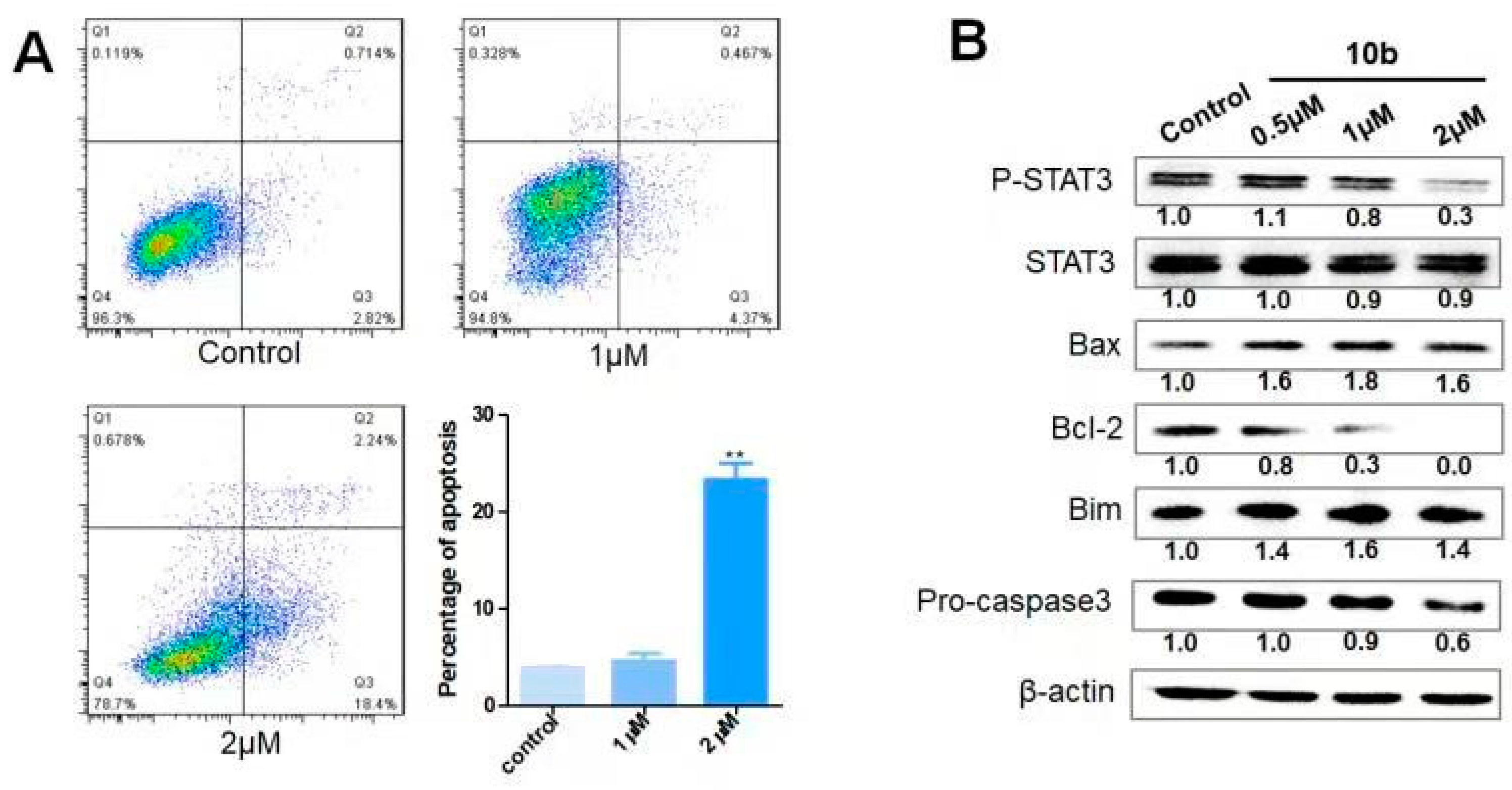
4-(3-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10b) (74% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.0 Hz, 2H), 7.76 (t, J = 7.4 Hz, 1H), 7.63 (t, J = 7.7 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 6.39 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.0 Hz, 1H), 5.18 (t, J = 8.0 Hz, 1H), 4.51 (t, J = 6.0 Hz, 2H), 4.45 – 4.36 (m, 1H), 4.36 – 4.21 (m, 3H), 3.60 (d, J = 3.7 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.71 (dd, J = 15.6, 11.0 Hz, 3H), 2.65–2.34 (m, 5H), 2.30 (dd, J = 9.0, 5.7 Hz, 1H), 2.21 (p, J = 5.9 Hz, 2H), 2.01 (s, 3H), 2.00–1.86 (m, 3H), 1.58 (s, 3H), 1.55 (s, 3H), 1.45–1.33 (m, 4H), 1.32 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.24 (s, 1H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.7, 201.0, 172.3, 171.8, 169.8, 159.0, 153.1, 140.5, 138.1, 135.8, 129.8, 128.7, 120.5, 119.3, 110.6, 79.2, 77.8, 74.1, 71.7, 68.1, 60.6, 54.2, 50.3, 50.0, 48.6, 48.5, 48.1, 43.1, 42.2, 36.1, 33.8, 29.4, 28.9, 28.7, 28.1, 26.9, 26.4, 23.8, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C47H60N2NaO16S [M + Na]+ 963.3556, found 963.3560.
4-(4-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)butoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10c) (78% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.09–8.00 (m, 2H), 7.76 (t, J = 7.5 Hz, 1H), 7.63 (t, J = 7.9 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.5 Hz, 1H), 5.19 (t, J = 7.9 Hz, 1H), 4.45 (t, J = 6.3 Hz, 2H), 4.42–4.36 (m, 1H), 4.27 (s, 1H), 4.23–4.11 (m, 2H), 3.60 (d, J = 3.9 Hz, 1H), 3.25 (d, J = 14.6 Hz, 1H), 2.78–2.65 (m, 3H), 2.65–2.24 (m, 6H), 2.01 (s, 3H), 2.00–1.93 (m, 4H), 1.82 (dt, J = 13.0, 6.4 Hz, 2H), 1.67 (s, 1H), 1.58 (s, 3H), 1.55 (s, 3H), 1.41 (s, 3H), 1.37 (d, J = 14.5 Hz, 1H), 1.32 (s, 3H), 1.29 (s, 3H), 1.26 (s, 3H), 1.22 (d, J = 13.6 Hz, 1H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.4, 171.8, 169.8, 159.0, 153.1, 140.5, 138.1, 135.8, 129.8, 128.7, 120.5, 119.3, 110.6, 79.2, 77.8, 74.1, 71.7, 71.0, 64.0, 54.2, 50.3, 50.1, 48.6, 48.5, 48.1, 43.1, 42.2, 36.1, 33.8, 29.4, 28.9, 28.7, 26.9, 26.4, 25.3, 25.1, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C48H62N2NaO16S [M + Na]+ 977.3712, found 977.3718.
4-((5-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)pentyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10d) (82% yield in two steps). (purity: 98%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.9 Hz, 2H), 7.76 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.7 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 4.7 Hz, 1H), 5.19 (s, 1H), 4.42 (t, J = 6.3 Hz, 3H), 4.26 (s, 1H), 4.12 (dd, J = 9.8, 6.2 Hz, 2H), 3.59 (d, J = 3.6 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.80–2.66 (m, 3H), 2.64–2.25 (m, 6H), 2.09–1.85 (m, 8H), 1.76–1.61 (m, 3H), 1.58 (s, 3H), 1.55 (d, J = 7.0 Hz, 4H), 1.40 (d, J = 6.5 Hz, 3H), 1.32 (s, 3H), 1.29 (s, 3H), 1.25 (d, J = 7.4 Hz, 5H), 1.07 (s, H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.5, 171.8, 169.7, 159.1, 153.0, 140.5, 138.2, 135.8, 129.8, 128.7, 120.5, 119.3, 110.6, 79.2, 77.8, 74.1, 71.7, 71.4, 64.4, 54.3, 50.3, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.1, 33.8, 29.5, 29.0, 28.8, 28.2, 28.2, 26.8, 26.4, 23.9, 23.7, 22.3, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (MALDI) calcd. for C49H64N2NaO16S [M + Na]+ 991.3869, found 991.3875.
4-((6-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)hexyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10e) (72% yield in two steps). (purity: 96%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.7 Hz, 2H), 7.76 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.8 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.75 (s, 1H), 5.19 (t, J = 7.9 Hz, 1H), 4.42 (t, J = 6.4 Hz, 3H), 4.28 (s, 1H), 4.08 (dt, J = 10.7, 6.3 Hz, 2H), 3.60 (d, J = 3.7 Hz, 1H), 3.25 (d, J = 14.5 Hz, 1H), 2.71 (dd, J = 15.1, 11.0 Hz, 3H), 2.64 – 2.26 (m, 6H), 2.06–1.93 (m, 6H), 1.91–1.85 (m, 2H), 1.71–1.62 (m, 2H), 1.58 (s, 3H), 1.56 (s, 3H), 1.50–1.39 (m, 7H), 1.33 (s, 3H), 1.29 (s, 3H), 1.26 (s, 3H), 1.24 (s, 2H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.5, 171.8, 169.7, 159.2, 153.1, 140.5, 138.2, 135.8, 129.8, 128.7, 120.5, 119.3, 110.6, 79.2, 77.8, 74.0, 71.7, 71.5, 64.6, 54.3, 50.4, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.1, 33.8, 29.5, 29.0, 28.8, 28.6, 28.4, 26.8, 26.4, 25.6, 25.4, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C50H66N2NaO16S [M + Na]+ 1005.4025, found 1005.4028.
4-((7-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)heptyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10f) (76% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.9 Hz, 2H), 7.76 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.8 Hz, 2H), 7.16 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.3 Hz, 1H), 5.19 (t, J = 7.9 Hz, 1H), 4.40 (t, J = 6.5 Hz, 3H), 4.27 (s, 1H), 4.07 (td, J = 6.6, 2.0 Hz, 2H), 3.59 (d, J = 3.8 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.71 (dd, J = 16.0, 7.8 Hz, 3H), 2.63–2.24 (m, 6H), 2.04–1.82 (m, 8H), 1.67–1.60 (m, 2H), 1.58 (s, 3H), 1.55 (s, 3H), 1.44 (dd, J = 8.3, 5.8 Hz, 2H), 1.40 (s, 3H), 1.35 (s, 4H), 1.32 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.24 (s, 2H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.5, 171.8, 169.7, 159.2, 153.1, 140.5, 138.2, 135.8, 129.8, 128.6, 120.5, 119.3, 110.6, 79.2, 77.8, 74.0, 71.7, 71.6, 64.8, 54.2, 50.3, 50.1, 48.6, 48.5, 48.1, 43.1, 42.2, 36.1, 33.8, 29.4, 29.0, 28.8, 28.8, 28.6, 28.4, 26.8, 26.4, 25.9, 25.6, 23.8, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (MALDI) calcd. for C51H68N2NaO16S [M + Na]+ 1019.4182, found 1019.4185.
4-((8-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)octyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10g) (66% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.04 (dd, J = 8.5, 1.2 Hz, 2H), 7.79–7.72 (m, 1H), 7.61 (dd, J = 10.8, 5.0 Hz, 2H), 7.16 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.75 (d, J = 5.6 Hz, 1H), 5.19 (t, J = 7.8 Hz, 1H), 4.40 (t, J = 6.5 Hz, 3H), 4.26 (s, 1H), 4.06 (td, J = 6.7, 1.7 Hz, 2H), 3.58 (d, J = 3.7 Hz, 1H), 3.24 (d, J = 14.7 Hz, 1H), 2.71 (dd, J = 15.6, 7.5 Hz, 3H), 2.60–2.34 (m, 5H), 2.30 (ddd, J = 9.1, 6.7, 3.4 Hz, 1H), 2.04–1.92 (m, 6H), 1.90–1.81 (m, 2H), 1.66–1.60 (m, 2H), 1.57 (d, J = 8.1 Hz, 6H), 1.45 (dd, J = 9.4, 5.9 Hz, 2H), 1.40 (s, 3H), 1.39 (s, 1H), 1.35 (d, J = 1.4 Hz, 6H), 1.32 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.24 (s, 1H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.9, 211.6, 200.9, 172.3, 171.7, 169.6, 159.1, 152.9, 140.4, 138.1, 135.6, 129.6, 128.5, 120.3, 119.2, 110.5, 79.1, 77.7, 73.9, 71.6, 71.6, 64.8, 54.2, 50.2, 50.0, 48.5, 48.4, 48.0, 43.1, 42.1, 36.0, 33.7, 29.3, 29.1, 29.0, 28.9, 28.7, 28.6, 28.4, 26.7, 26.3, 25.8, 25.5, 23.8, 23.6, 21.8, 21.3, 20.0, 19.7, 18.8. HRMS (MALDI) calcd. for C52H70N2NaO16S [M + Na]+ 1033.4338, found 1033.4342.
4-((9-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)nonyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10h) (69% yield in two steps). (purity: 97%). 1H NMR (400 MHz, CDCl3) δ 8.05 (dd, J = 8.4, 1.1 Hz, 2H), 7.76 (dd, J = 10.7, 4.3 Hz, 1H), 7.62 (t, J = 7.9 Hz, 2H), 7.16 (d, J = 15.6 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.6 Hz, 1H), 5.20 (t, J = 7.8 Hz, 1H), 4.41 (t, J = 6.6 Hz, 3H), 4.27 (s, 1H), 4.08 (dt, J = 6.9, 6.5 Hz, 2H), 3.70 (s, 1H), 3.59 (d, J = 3.9 Hz, 1H), 3.25 (d, J = 14.7 Hz, 1H), 2.72 (dd, J = 15.2, 11.0 Hz, 3H), 2.61–2.35 (m, 5H), 2.35–2.27 (m, 1H), 2.04–1.93 (m, 6H), 1.92–1.81 (m, 2H), 1.61 (d, J = 7.5 Hz, 3H), 1.59 (s, 3H), 1.56 (s, 3H), 1.44 (d, J = 7.4 Hz, 2H), 1.41 (s, 3H), 1.33 (s, 8H), 1.29 (s, 3H), 1.27 (s, 3H), 1.25 (s, 3H), 1.08 (s, 3H), 1.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.7, 201.0, 172.5, 171.8, 169.8, 159.1, 153.1, 140.5, 138.2, 135.7, 129.8, 128.7, 120.5, 119.3, 110.6, 100.1, 79.2, 77.8, 74.0, 71.7, 65.0, 54.3, 50.4, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.1, 33.8, 29.8, 29.5, 29.3, 29.2, 29.0, 28.8, 28.7, 28.5, 26.8, 26.4, 26.0, 25.7, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (MALDI) calcd. for C53H72N2NaO16S [M + Na]+ 1047.4495, found 1047.4498.
4-((10-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)decyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10i) (71% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.08–7.98 (m, 2H), 7.74 (t, J = 7.5 Hz, 1H), 7.60 (t, J = 7.9 Hz, 2H), 7.14 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.74 (d, J = 5.5 Hz, 1H), 5.18 (t, J = 7.9 Hz, 1H), 4.44–4.35 (m, 3H), 4.27 (d, J = 7.8 Hz, 1H), 4.10–3.99 (m, 2H), 3.58 (d, J = 3.9 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.76–2.66 (m, 3H), 2.60–2.23 (m, 6H), 2.04–1.90 (m, 6H), 1.90–1.79 (m, 2H), 1.65–1.58 (m, 2H), 1.56 (s, 3H), 1.55 (s, 3H), 1.38–1.44 (m, 6H), 1.31 (s, 13H), 1.27 (s, 3H), 1.25 (s, 3H), 1.23 (s, 1H), 1.05 (s, 3H), 0.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.4, 171.7, 169.7, 159.1, 153.0, 140.5, 138.2, 135.7, 129.7, 128.6, 120.4, 119.3, 110.6, 79.2, 77.8, 74.0, 71.7, 71.7, 64.9, 54.2, 50.3, 50.0, 48.6, 48.5, 48.1, 43.1, 42.2, 36.0, 33.8, 29.5, 29.4, 29.3, 29.1, 29.0, 28.8, 28.7, 28.5, 26.7, 26.4, 25.9, 25.6, 23.8, 23.7, 21.9, 21.3, 20.1, 19.8, 18.8. HRMS (MALDI) calcd. for C54H74N2NaO16S [M + Na]+ 1061.4651, found 1061.4655.
4-((11-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)undecyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10j) (75% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.1 Hz, 2H), 7.75 (t, J = 7.3 Hz, 1H), 7.61 (t, J = 7.5 Hz, 2H), 7.16 (d, J = 15.5 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 4.0 Hz, 1H), 5.20 (t, J = 8.0 Hz, 1H), 4.41 (t, J = 6.5 Hz, 3H), 4.27 (s, 1H), 4.05 (t, J = 6.7 Hz, 2H), 3.59 (s, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.71 (dd, J = 14.1, 11.1 Hz, 3H), 2.65–2.26 (m, 6H), 2.05–1.92 (m, 6H), 1.92–1.79 (m, 3H), 1.61 (d, J = 6.1 Hz, 1H), 1.58 (s, 3H), 1.56 (s, 3H), 1.48–1.38 (m, 6H), 1.28 (dd, J = 21.2, 12.1 Hz, 22H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.5, 171.8, 169.7, 159.2, 153.0, 140.6, 138.3, 135.7, 129.8, 128.7, 120.5, 119.3, 110.6, 79.2, 77.8, 74.0, 71.8, 71.7, 65.0, 54.3, 50.4, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.1, 33.8, 29.8, 29.6, 29.6, 29.5, 29.4, 29.2, 29.0, 28.9, 28.7, 28.5, 26.8, 26.4, 26.0, 25.7, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (MALDI) calcd. for C55H76N2NaO16S [M + Na]+ 1075.4808, found 1075.4812.
4-((12-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)dodecyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10k) (73% yield in two steps). (purity: 96%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.5 Hz, 2H), 7.75 (t, J = 7.5 Hz, 1H), 7.61 (t, J = 7.8 Hz, 2H), 7.15 (d, J = 15.6 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.3 Hz, 1H), 5.20 (t, J = 7.9 Hz, 1H), 4.40 (t, J = 6.6 Hz, 3H), 4.26 (s, 1H), 4.05 (t, J = 6.8 Hz, 2H), 3.58 (d, J = 3.6 Hz, 1H), 3.24 (d, J = 14.7 Hz, 1H), 2.79–2.65 (m, 3H), 2.60–2.34 (m, 5H), 2.30 (ddd, J = 12.3, 6.1, 3.2 Hz, 1H), 2.05–1.92 (m, 6H), 1.91–1.81 (m, 2H), 1.61 (d, J = 6.2 Hz, 2H), 1.57 (d, J = 7.9 Hz, 6H), 1.46–1.37 (m, 6H), 1.32 (s, 6H), 1.28 (s, 12H), 1.26 (s, 3H), 1.24 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.1, 172.4, 171.8, 169.7, 159.2, 153.0, 140.6, 138.3, 135.7, 129.7, 128.7, 120.5, 119.4, 110.6, 79.2, 77.8, 74.0, 71.8, 71.7, 65.0, 54.3, 50.3, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.1, 33.9, 29.8, 29.7, 29.6, 29.6, 29.5, 29.4, 29.2, 29.0, 28.9, 28.7, 28.5, 26.8, 26.4, 26.0, 25.7, 23.9, 23.8, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (MALDI) calcd. for C56H78N2NaO16S [M + Na]+ 1089.4964, found 1089.4968.
4-(((E)-4-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)but-2-en-1-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10l) (61% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 7.5 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.63 (t, J = 7.8 Hz, 2H), 7.18 (d, J = 15.6 Hz, 1H), 6.39 (d, J = 15.6 Hz, 1H), 5.91 (t, J = 5.5 Hz, 2H), 5.76 (d, J = 5.4 Hz, 1H), 5.20 (t, J = 8.0 Hz, 1H), 5.05 (d, J = 5.0 Hz, 2H), 4.74 (d, J = 4.9 Hz, 2H), 4.46 – 4.36 (m, 1H), 4.28 (s, 1H), 3.60 (d, J = 3.8 Hz, 1H), 3.25 (d, J = 14.7 Hz, 1H), 2.72 (dd, J = 19.1, 10.9 Hz, 3H), 2.66–2.27 (m, 7H), 2.04–1.97 (m, 4H), 1.58 (s, 3H), 1.55 (s, 3H), 1.41 (s, 3H), 1.33 (s, 3H), 1.29 (s, 3H), 1.26 (s, 3H), 1.25 (s, 3H), 1.08 (s, 3H), 1.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.7, 201.0, 172.1, 171.8, 169.8, 153.2, 140.5, 135.9, 130.4, 129.9, 128.7, 125.9, 120.5, 119.2, 79.2, 77.8, 74.1, 71.7, 66.7, 60.3, 54.2, 50.4, 50.1, 48.6, 48.5, 48.2, 43.2, 42.2, 36.1, 33.8, 29.8, 29.5, 28.9, 28.7, 26.9, 26.4, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C48H60N2NaO16S [M+Na]+ 975.3556, found 975.3560.
4-((4-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)but-2-yn-1-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10m) (77% yield in two steps). (purity: 99%). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.5 Hz, 2H), 7.77 (t, J = 7.3 Hz, 1H), 7.64 (t, J = 7.6 Hz, 2H), 7.18 (d, J = 14.9 Hz, 1H), 6.40 (d, J = 15.4 Hz, 1H), 5.77 (s, 1H), 5.21 (t, J = 7.5 Hz, 1H), 5.10 (s, 2H), 4.79–4.67 (m, 2H), 4.41 (d, J = 12.7 Hz, 1H), 4.27 (s, 1H), 3.59 (s, 1H), 3.25 (d, J = 14.8 Hz, 1H), 2.74–2.67 (m, 3H), 2.65–2.54 (m, 2H), 2.43 (d, J = 4.5 Hz, 2H), 2.40–2.27 (m, 2H), 2.01 (d, J = 9.7 Hz, 4H), 1.59 (s, 3H), 1.56 (s, 3H), 1.41 (s, 3H), 1.33 (s, 3H), 1.29 (s, 3H), 1.27 (s, 3H), 1.25 (s, 4H), 1.08 (s, 3H), 1.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.0, 211.7, 201.1, 171.6, 169.8, 158.0, 153.2, 140.6, 137.8, 135.9, 129.9, 128.8, 120.5, 119.3, 100.0, 84.0, 79.2, 78.8, 77.8, 74.2, 71.8, 58.7, 54.3, 52.2, 50.4, 50.1, 48.7, 48.5, 48.2, 43.2, 42.2, 36.1, 33.9, 29.8, 29.5, 28.8, 28.7, 27.0, 26.4, 23.9, 23.8, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C48H58N2NaO16S [M + Na]+ 973.3399, found 973.3402.
4-(3-((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)-2,2-dimethylpropoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10n) (71% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.9 Hz, 2H), 7.75 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.7 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 6.39 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 4.3 Hz, 1H), 5.15 (t, J = 8.0 Hz, 1H), 4.45–4.35 (m, 1H), 4.26 (s, 1H), 4.22–4.14 (m, 2H), 4.09 (d, J = 10.9 Hz, 1H), 3.98 (d, J = 11.0 Hz, 1H), 3.60 (d, J = 3.5 Hz, 1H), 3.24 (d, J = 14.6 Hz, 1H), 2.76–2.26 (m, 9H), 2.04–1.85 (m, 6H), 1.56 (d, J = 11.9 Hz, 6H), 1.40 (s, 3H), 1.32 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.24 (s, 2H), 1.09 (s, 6H), 1.07 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.7, 201.1, 172.2, 171.8, 169.8, 159.3, 153.1, 140.5, 138.3, 135.8, 129.9, 128.7, 120.6, 119.3, 110.6, 79.2, 77.8, 75.8, 74.1, 71.8, 68.8, 54.3, 50.4, 50.1, 48.7, 48.5, 48.2, 43.1, 42.2, 35.6, 33.9, 30.5, 29.5, 28.9, 28.8, 27.0, 26.4, 23.7, 22.0, 21.7, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C49H64N2NaO16S [M + Na]+ 991.3869, found 991.3873.
4-((4-(((4-(((9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoyl)oxy)methyl)benzyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (10o) (63% yield in two steps). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.07 – 7.98 (m, 2H), 7.74 (d, J = 7.5 Hz, 1H), 7.59 (t, J = 7.9 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 15.6 Hz, 1H), 7.17 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.76 (d, J = 5.5 Hz, 1H), 5.44 (s, 2H), 5.21 (t, J = 7.9 Hz, 1H), 5.15 (s, 2H), 4.45–4.36 (m, 1H), 4.28 (s, 1H), 3.60 (d, J = 3.9 Hz, 1H), 3.25 (d, J = 14.6 Hz, 1H), 2.78–2.25 (m, 10H), 2.05–1.86 (m, 6H), 1.57 (s, 3H), 1.54 (s, 3H), 1.41 (s, 3H), 1.33 (s, 3H), 1.29 (s, 3H), 1.25 (d, J = 6.5 Hz, 4H), 1.07 (s, 3H), 1.01 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 213.0, 211.7, 201.0, 172.2, 171.8, 169.8, 158.7, 153.2, 140.5, 138.1, 137.0, 135.8, 133.9, 129.8, 128.7, 128.6, 128.5, 120.5, 119.3, 110.6, 79.2, 77.8, 74.1, 72.3, 71.7, 66.0, 54.3, 50.3, 50.1, 48.6, 48.5, 48.1, 43.2, 42.2, 36.1, 33.8, 29.5, 29.0, 28.8, 26.9, 26.4, 23.9, 23.7, 22.0, 21.4, 20.2, 19.8, 18.9. HRMS (ESI) calcd. for C52H62N2NaO16S [M + Na]+ 1025.3712, found 1025.3718.
3.1.10. Procedure for the Synthesis of Compound 11
4-(((2S,9R,13R,14S,16R)-17-((R,E)-6-Acetoxy-2-hydroxy-6-methyl-3-oxohept-4-en-2-yl)-2-hydroxy-4,4,9,13,14-pentamethyl-3,11-dioxo-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-16-yl)oxy)-4-oxobutanoic acid (11). Compound 8 (77 mg, 0.1 mmol) was dissolved in THF (5 mL). HOAc (28 μL, 0.5 mmol) and TBAF (131 mg, 0.5 mmol) were added to the mixture. The mixture was stirred at room temperature for 24 h, and then diluted with ethyl acetate (20 mL). The organic phase was washed with H2O (3 × 20 mL), dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel to obtain a white solid 11 (52 mg, 79% yield). (purity: 95%). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 15.6 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.77 (d, J = 5.5 Hz, 1H), 5.22 (t, J = 7.8 Hz, 1H), 4.41 (dd, J = 12.9, 6.0 Hz, 1H), 3.25 (d, J = 14.6 Hz, 1H), 2.77–2.67 (m, 3H), 2.58 (dd, J = 12.2, 6.5 Hz, 2H), 2.43 (t, J = 6.7 Hz, 2H), 2.38 (d, J = 7.8 Hz, 1H), 2.31 (ddd, J = 12.5, 5.8, 3.4 Hz, 1H), 2.04–1.93 (m, 6H), 1.59 (s, 3H), 1.55 (s, 3H), 1.41 (s, 3H), 1.39 (s, 1H), 1.33 (s, 3H), 1.28 (d, J = 4.5 Hz, 6H), 1.25 (s, 3H), 1.08 (s, 3H), 1.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 213.1, 211.8, 201.1, 176.9, 171.7, 170.0, 153.1, 140.5, 120.5, 119.3, 79.4, 77.9, 74.3, 71.8, 54.3, 50.4, 50.1, 48.7, 48.5, 48.1, 43.2, 42.2, 36.09, 33.9, 29.5, 28.9, 28.8, 27.0, 26.3, 23.9, 23.8, 21.9, 21.4, 20.2, 19.9, 18.8. HRMS (ESI) calcd. for C36H50NaO11 [M + Na]+ 681.3245, found 681.3250.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}