Synthesis, Biological Evaluation and Low-Toxic Formulation Development of Glycosylated Paclitaxel Prodrugs

 ,
,
Abstract
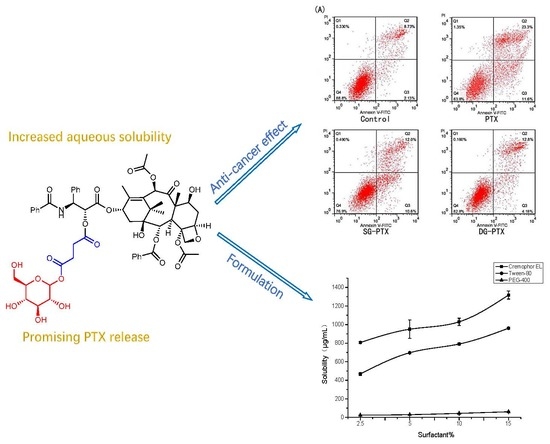
:
1. Introduction
2. Results
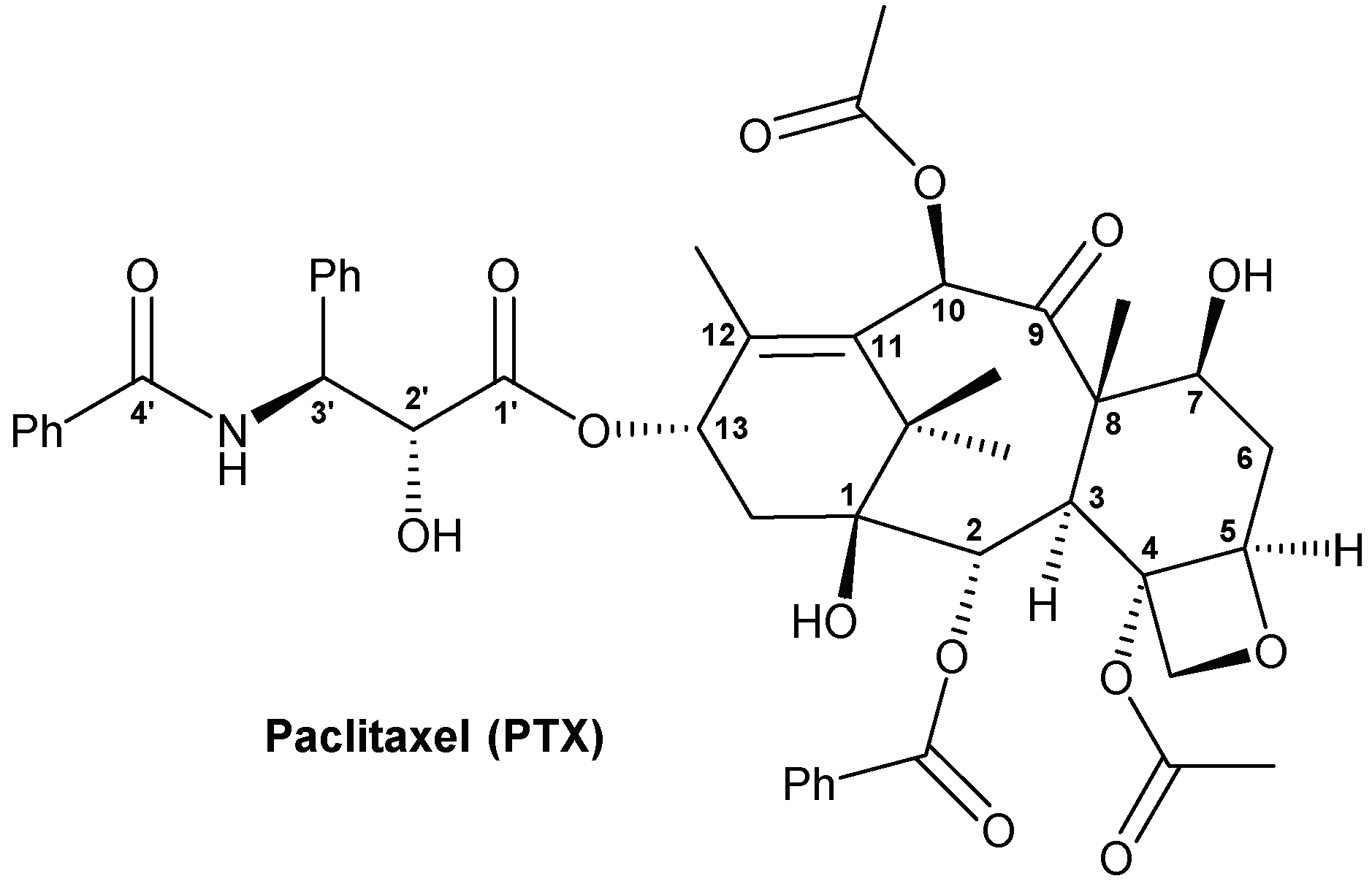
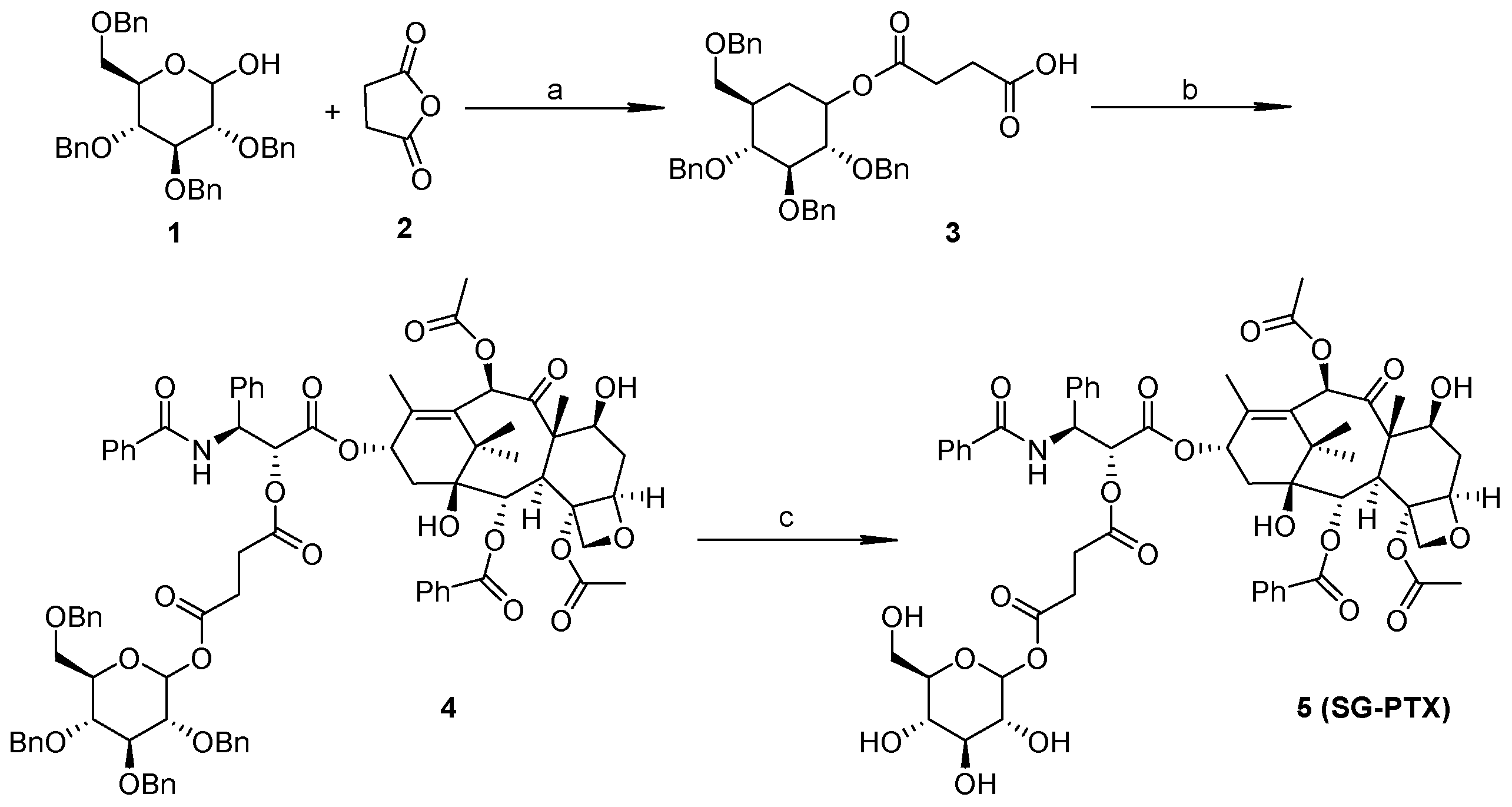
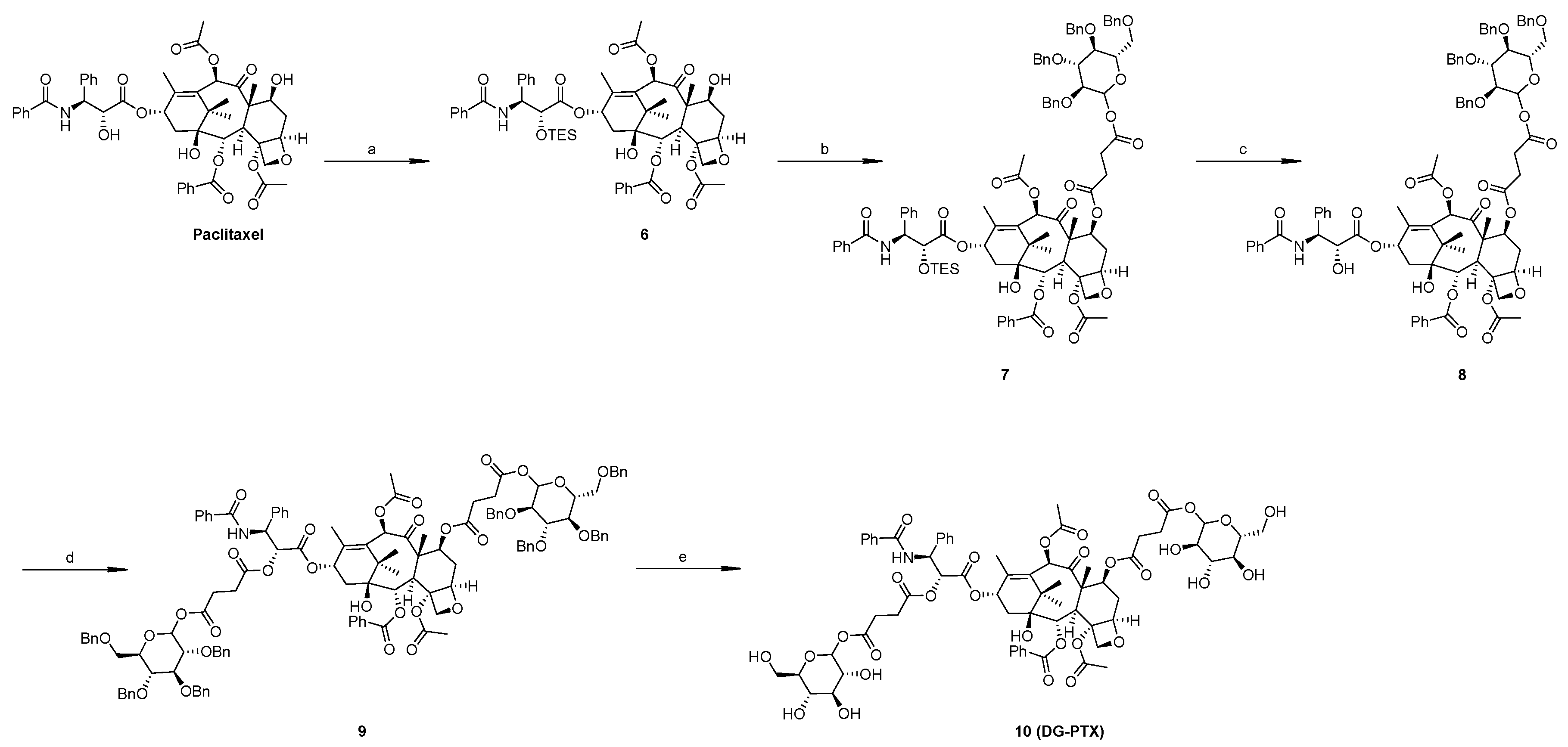
2.1. Synthesis of SG-PTX and DG-PTX
2.2. Solubility Assays of SG-PTX and DG-PTX
2.3. Determination of Partition Coefficient (Log P) of SG-PTX and DG-PTX
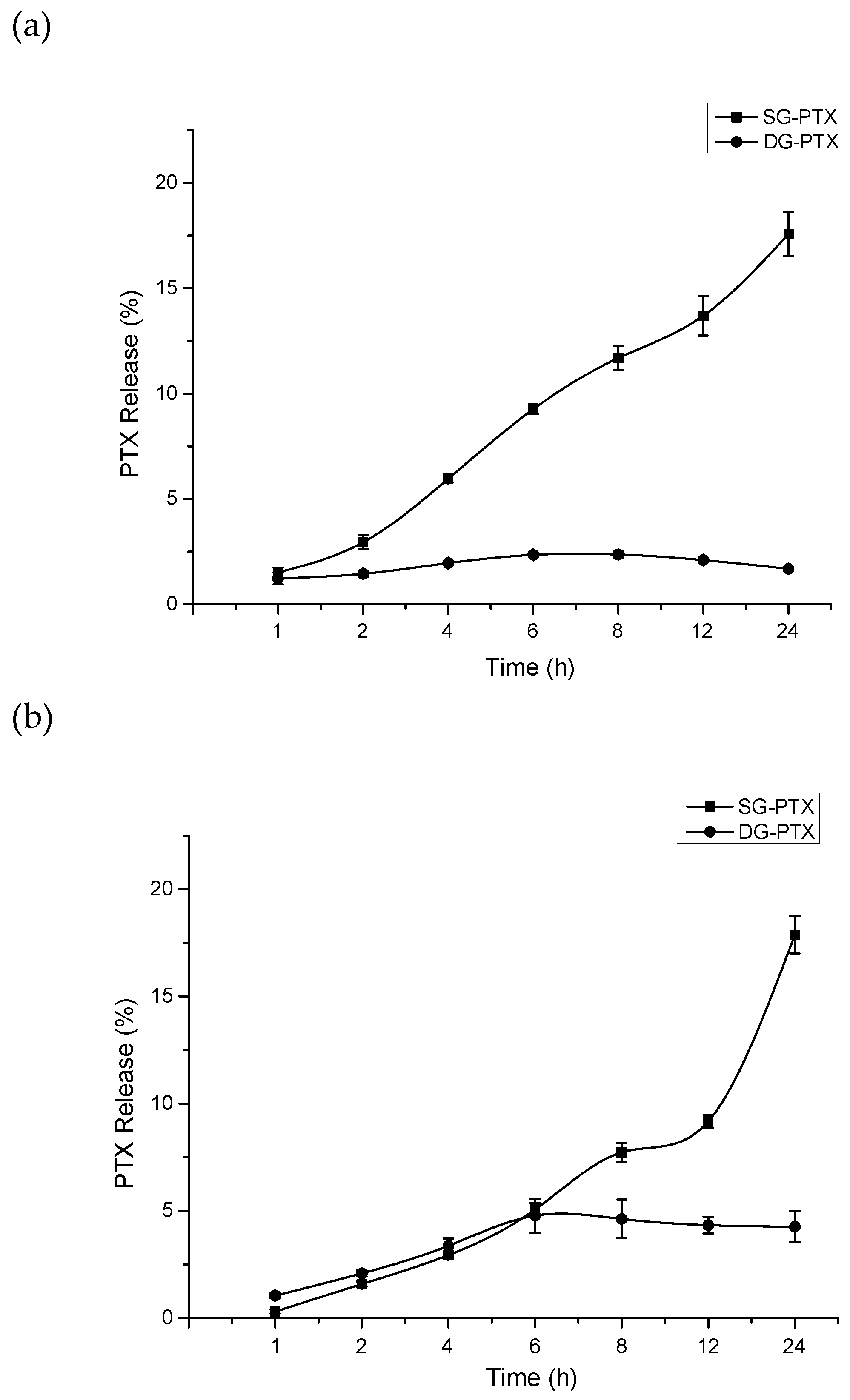
2.4. Parent Drug Release Assays of SG-PTX and DG-PTX
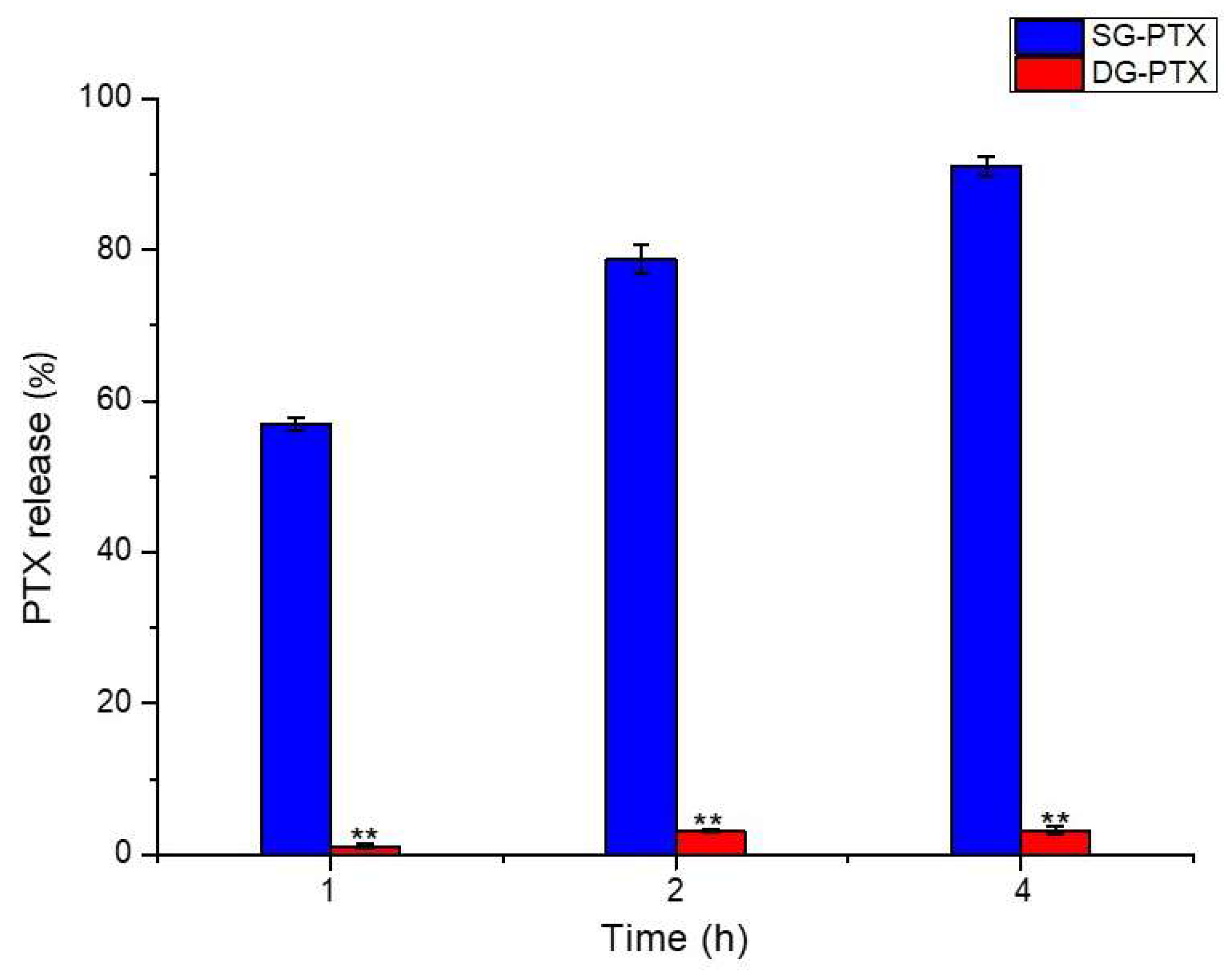
2.5. Enzymatic Hydrolysis of SG-PTX and DG-PTX
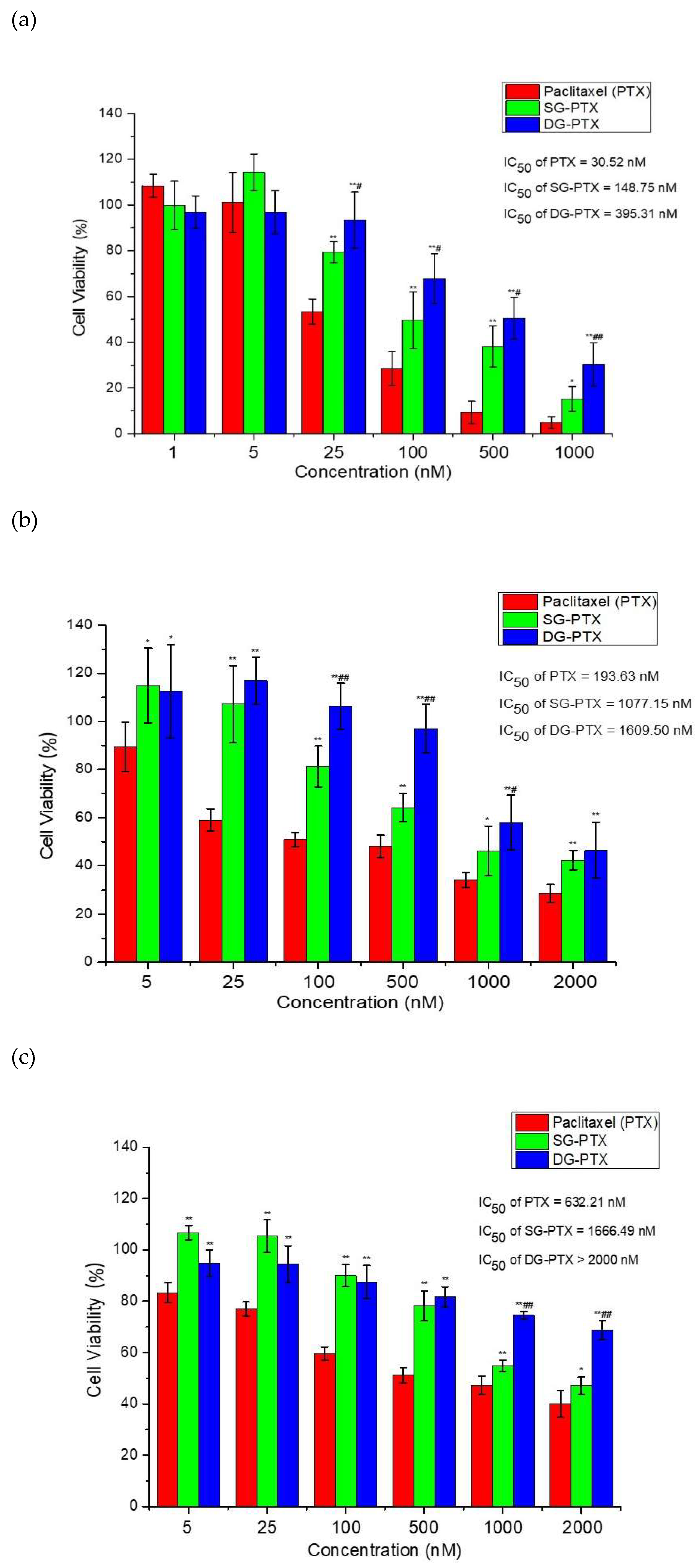
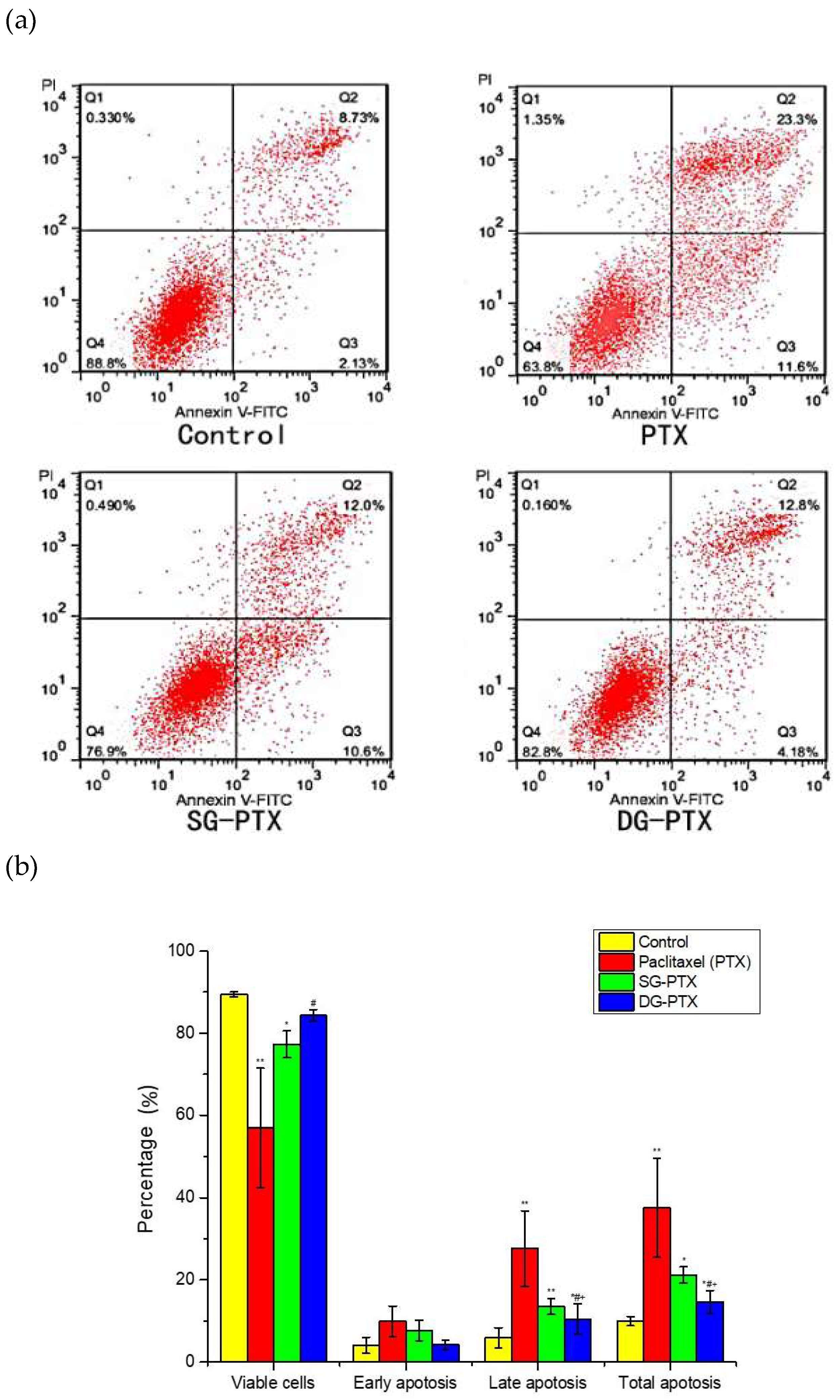
2.6. In Vitro Anti-Cancer Effect Evaluations of SG-PTX and DG-PTX
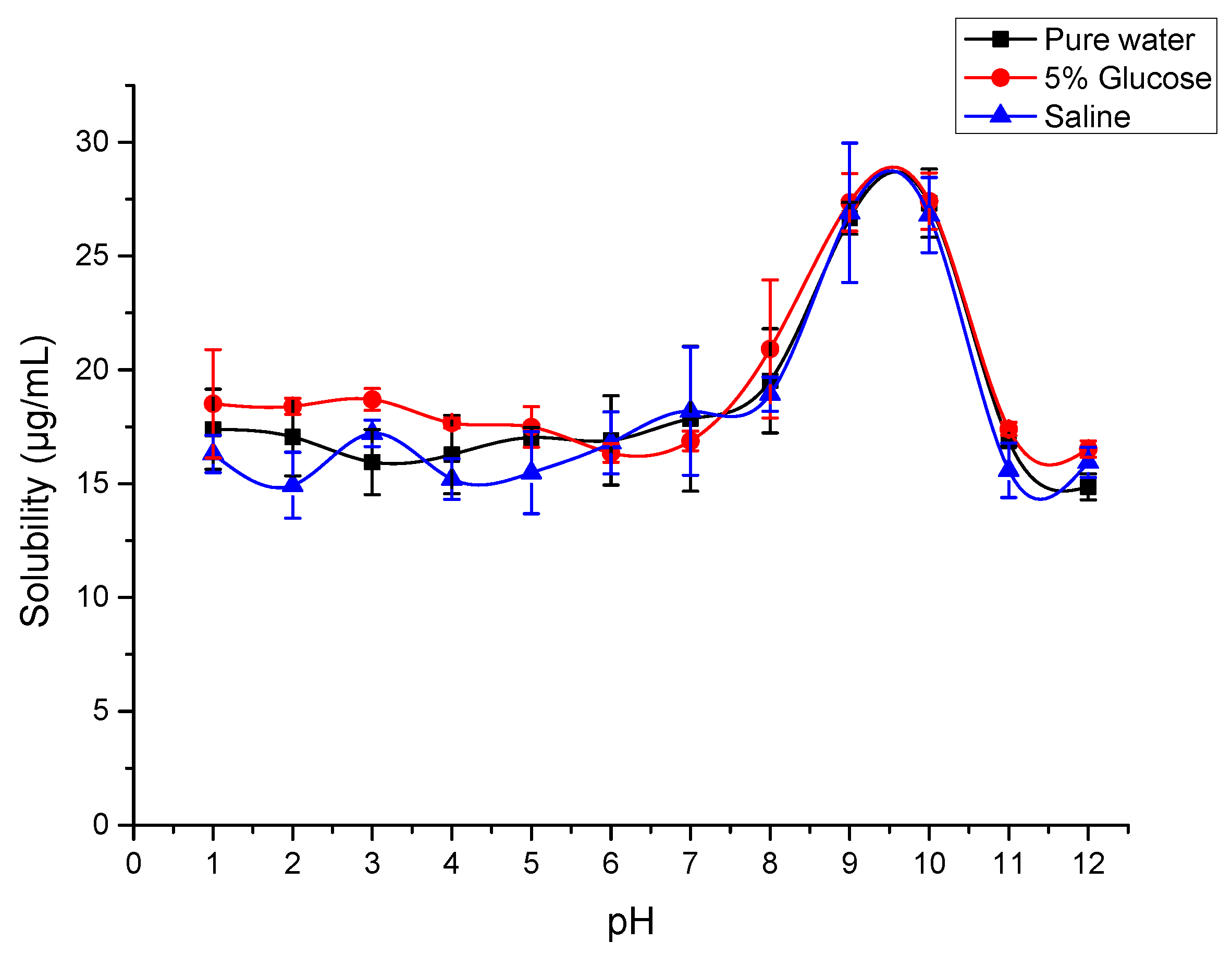
2.7. Solubility of SG-PTX at Different pH Conditions
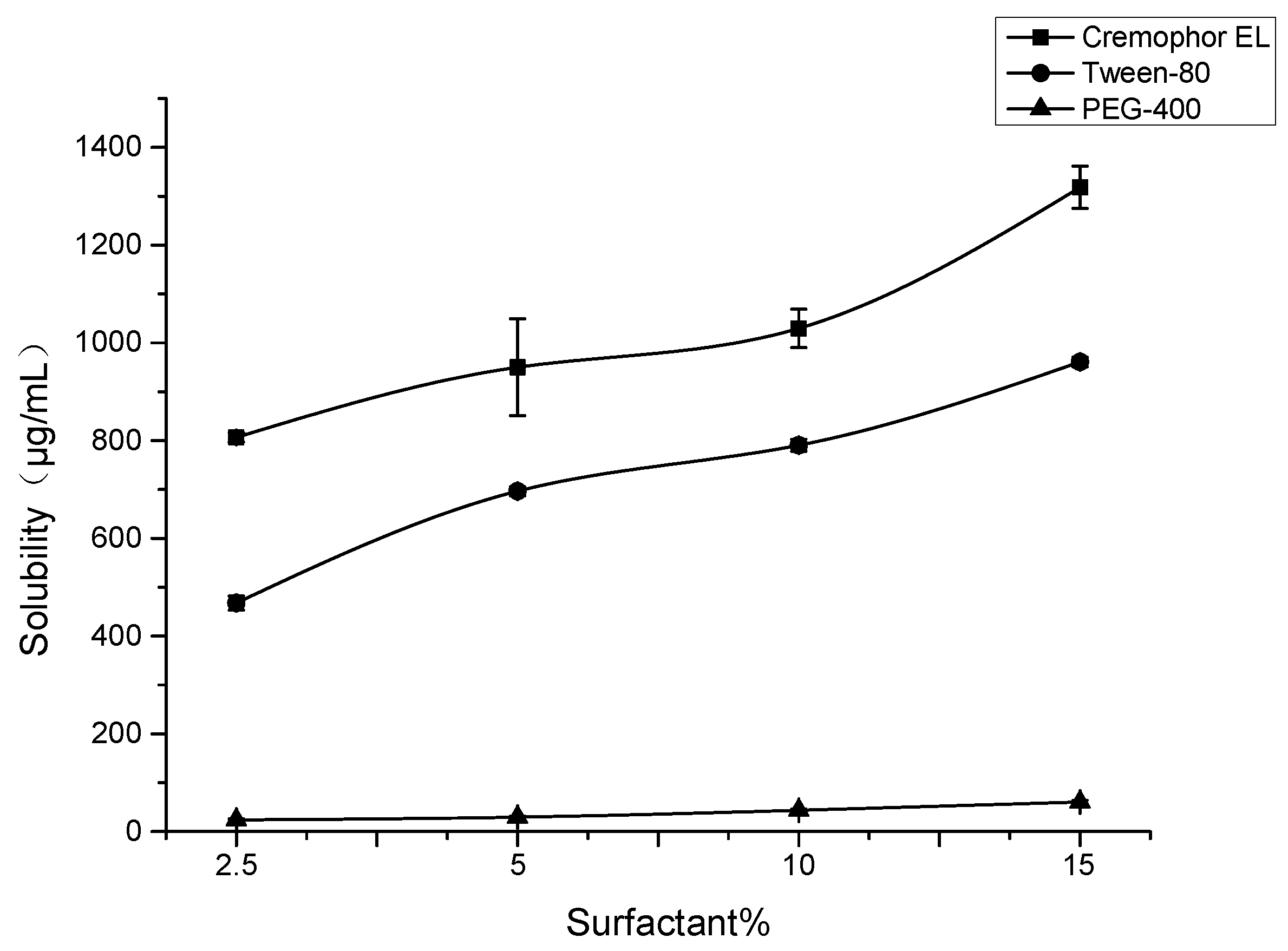
2.8. Formulation Development of SG-PTX
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthesis of SG-PTX
4.3. Synthesis of DG-PTX
4.4. HPLC Determination Assays
4.5. Solubility Measurements
4.6. Determination of Log P
4.7. Parent Drug Release in Serum
4.8. Enzymatic Hydrolysis
4.9. Cell Culture
4.10. MTT Assays
4.11. Annexin V-FITC/PI Assays
4.12. Solubility of SG-PTX at Various pH Conditions
4.13. Solubility of SG-PTX in Various Formulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- De Weger, V.A.; Beijnen, J.H.; Schellens, J.H. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel-a review. Anticancer Drugs 2014, 25, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Dai, W.; Guy, R.K. Chemistry and biology of TAXOL. Ange. Chem. Int. Ed. 1994, 33, 15–44. [Google Scholar] [CrossRef]
- Chu, Q.; Vincent, M.; Logan, D.; Mackay, J.A.; Evans, W.K. Taxanes as first-line therapy for advanced non-small cell lung cancer: A systematic review and practice guideline. Lung Cancer 2005, 50, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Petrelli, F.; Coinu, A.; Raspagliesi, F.; Barni, S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol. Oncol. 2014, 133, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H. Progress in the knowledge and treatment of advanced pancreatic cancer: From benchside to bedside. Cancer Treat Rev. 2014, 40, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Ellis, A.G.; Webster, L.K. Inhibition of paclitaxel elimination in the isolated perfused rat liver by Cremophor EL. Cancer Chemother. Pharmacol. 1999, 43, 13–18. [Google Scholar] [CrossRef]
- Reddy, L.H.; Bazile, D. Drug delivery design for intravenous route with integrated physicochemistry, pharmacokinetics and pharmacodynamics: Illustration with the case of taxane therapeutics. Adv. Drug Deliv. Rev. 2014, 71, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Van Zuylen, L.; Verweij, J.; Sparreboom, A. Role of formulation vehicles in taxane pharmacology. Invest. New Drugs 2001, 19, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Chen, K.; Wang, C.; Jiang, X.; Dong, H.; Gong, Z.; Liu, K. Design, synthesis, and evaluation of water-soluble morpholino-decorated paclitaxel prodrugs with remarkably decreased toxicity. Bioorg. Med. Chem. Lett. 2016, 26, 3598–3602. [Google Scholar] [CrossRef] [PubMed]
- Tavakolifard, S.; Biazar, E.; Pourshamsian, K.; Moslemin, M.H. Synthesis and evaluation of single-wall carbon nanotube-paclitaxel-folic acid conjugate as an anti-cancer targeting agent. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Loira-Pastoriza, C.; Patil, H.P.; Ucakar, B.; Muccioli, G.G.; Bosquillon, C.; Vanbever, R. PEGylation of paclitaxel largely improves its safety and anti-tumor efficacy following pulmonary delivery in a mouse model of lung carcinoma. J. Control Release 2016, 239, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trant, J.F.; McEachran, M.J.; Sran, I.; Turowec, B.A.; de Bruyn, J.R.; Gillies, E.R. Covalent Polyisobutylene-Paclitaxel Conjugates for Controlled Release from Potential Vascular Stent Coatings. ACS Appl. Mater. Interfaces 2015, 7, 14506–14517. [Google Scholar] [CrossRef]
- Wang, W.; Li, M.; Zhang, Z.; Cui, C.; Zhou, J.; Yin, L.; Lv, H. Design, synthesis and evaluation of multi-functional tLyP-1-hyaluronic acid-paclitaxel conjugate endowed with broad anticancer scope. Carbohydr. Polym. 2017, 156, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Tungpradit, R.; Sinchaikul, S.; An, F.M.; Liu, D.Z.; Phutrakul, S.; Chen, S.T. Targeting the delivery of glycan-based paclitaxel prodrugs to cancer cells via glucose transporters. J. Med. Chem. 2008, 51, 7428–7441. [Google Scholar] [CrossRef]
- Juan, T.Y.; Roffler, S.R.; Hou, H.S.; Huang, S.M.; Chen, K.C.; Leu, Y.L.; Prijovich, Z.M.; Yu, C.P.; Wu, C.C.; Sun, G.H.; et al. Antiangiogenesis targeting tumor microenvironment synergizes glucuronide prodrug antitumor activity. Clin. Cancer Res. 2009, 15, 4600–4611. [Google Scholar] [CrossRef]
- Cheng, T.C.; Roffler, S.R.; Tzou, S.C.; Chuang, K.H.; Su, Y.C.; Chuang, C.H.; Kao, C.H.; Chen, C.S.; Harn, I.H.; Liu, K.Y.; et al. An activity-based near-infrared glucuronide trapping probe for imaging beta-glucuronidase expression in deep tissues. J. Am. Chem. Soc. 2012, 134, 3103–3110. [Google Scholar] [CrossRef]
- Mikuni, K.; Nakanishi, K.; Hara, K.; Hara, K.; Iwatani, W.; Amano, T.; Nakamura, K.; Tsuchiya, Y.; Okumoto, H.; Mandai, T. In Vivo Antitumor Activity of Novel Water-Soluble Taxoids. Biol. Pharm. Bull. 2008, 31, 1155–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Chen, L.; Wang, X.; Liu, X.; You, Q.; Xi, W.; Gao, L.; Chen, G.; Chen, Y.L.; Xiong, B.; et al. Direct glycosylation of bioactive small molecules with glycosyl iodide and strained olefin as acid scavenger. J. Org. Chem. 2014, 79, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Kubota, N. Chemo-enzymatic synthesis of ester-linked docetaxel-monosaccharide conjugates as water-soluble prodrugs. Molecules 2011, 16, 6769–6777. [Google Scholar] [CrossRef] [PubMed]
- Shigehiro, T.; Zhai, W.; Vaidyanath, A.; Masuda, J.; Mizutani, A.; Kasai, T.; Murakami, H.; Hamada, H.; Salomon, D.S.; Mikuni, K.; et al. Evaluation of glycosylated docetaxel-encapsulated liposomes prepared by remote loading under solubility gradient. J. Microencapsul. 2016, 33, 172–182. [Google Scholar] [CrossRef]
- Mandai, T.; Okumoto, H.; Oshitari, T.; Nakanishi, K.; Mikuni, K.; Hara, K.-J.; Hara, K.-Z.; Iwatani, W.; Amano, T.; Nakamura, K.; et al. Synthesis and biological evaluation of water soluble taxoids bearing sugar moieties. Heterocycles 2001, 54, 561–566. [Google Scholar] [CrossRef]
- Bharate, S.S.; Vishwakarma, R.A. Thermodynamic equilibrium solubility measurements in simulated fluids by 96-well plate method in early drug discovery. Bioorg. Med. Chem. Lett. 2015, 25, 1561–1567. [Google Scholar] [CrossRef]
- Compound Report Card of Paclitaxel. Available online: https://www.ebi.ac.uk/chembl/compound/inspect/CHEMBL428647 (accessed on 5 July 2018).
- Kumar, V.; Bharate, S.S.; Vishwakarma, R.A. Modulating lipophilicity of rohitukine via prodrug approach: Preparation, characterization, and in vitro enzymatic hydrolysis in biorelevant media. Eur. J. Pharm. Sci. 2016, 92, 203–211. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. Lipophilicity. In Drug-Like Properties: Concepts, Structure Design and Methods From ADME to Toxicity Optimization; Elsevier’s Science & Technology Rights: Oxford, UK, 2008; pp. 43–47. ISBN 978-0-12-369520-8. [Google Scholar]
- Mathew, A.E.; Mejillano, M.R.; Nath, J.P.; Himes, R.H.; Stella, V.J. Synthesis and evaluation of some water-soluble prodrugs and derivatives of taxol with antitumor activity. J. Med. Chem. 1992, 35, 145–151. [Google Scholar] [CrossRef]
- Vyas, D.M.; Wong, H.; Crosswell, A.R.; Casazza, A.M.; Knipe, J.O. Synthesis and antitumor evaluation of water soluble taxol phosphates. Bioorg. Med. Chem. 1993, 3, 1357–1360. [Google Scholar] [CrossRef]
- Fu, Q.; Wang, Y.; Ma, Y.; Zhang, D.; Fallon, J.K.; Yang, X.; Liu, D.; He, Z.; Liu, F. Programmed hydrolysis in designing paclitaxel prodrug for nanocarrier assembly. Sci. Rep. 2015, 5, 12023. [Google Scholar] [CrossRef]
- Zhou, Z.; Yan, J.; Sun, T.; Wang, X.; Xie, Z. Nanoprodrug of retinoic acid-modified paclitaxel. Org. Biomol. Chem. 2017, 15, 9611–9615. [Google Scholar] [CrossRef]
- Luo, C.; Sun, J.; Liu, D.; Sun, B.; Miao, L.; Musetti, S.; Li, J.; Han, X.; Du, Y.; Li, L.; et al. Self-assembled redox dual-responsive prodrug-nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for cancer chemotherapy. Nano Lett. 2016, 16, 5401–5408. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Taxol (Paclitaxel) Injection Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf (accessed on 14 September 2018).
- Khan, K.M.; Rahim, F.; Halim, S.A.; Taha, M.; Khan, M.; Perveen, S.; Zaheer Ul, H.; Mesaik, M.A.; Iqbal Choudhary, M. Synthesis of novel inhibitors of beta-glucuronidase based on benzothiazole skeleton and study of their binding affinity by molecular docking. Bioorg. Med. Chem. 2011, 19, 4286–4294. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Jin, Y.-H. Intestinal bacterial β-glucuronidase activity of patients with colon cancer. Arch. Pharm. Res. 2001, 24, 564–567. [Google Scholar] [CrossRef]
- Graaf, M.D.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. Beta-Glucuronidase-Mediated Drug Release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar] [CrossRef]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Islam, M.R.; Tomatsu, S.; Shah, G.N.; Grubb, J.H.; Jain, S.; Sly, W.S. Active site residues of human β-glucuronidase. J. Biol. Chem. 1999, 274, 23451–23455. [Google Scholar] [CrossRef]
- Zitomersky, N.L.; Coyne, M.J.; Comstock, L.E. Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect Immun. 2011, 79, 2012–2020. [Google Scholar] [CrossRef]
- Pellock, S.J.; Walton, W.G.; Biernat, K.A.; Torres-Rivera, D.; Creekmore, B.C.; Xu, Y.; Liu, J.; Tripathy, A.; Stewart, L.J.; Redinbo, M.R. Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biolog. Chem. 2018. [Google Scholar] [CrossRef]
- Hou, J.; Sun, E.; Sun, C.; Wang, J.; Yang, L.; Jia, X.B.; Zhang, Z.H. Improved oral bioavailability and anticancer efficacy on breast cancer of paclitaxel via Novel Soluplus((R))-Solutol((R)) HS15 binary mixed micelles system. Int. J. Pharm. 2016, 512, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Bardelmeijer, H.A.; Ouwehand, M.; Malingre, M.M.; Schellens, J.; Beijnen, J.H.; van Tellingen, O. Entrapment by Cremophor EL decreases the absorption of paclitaxel from the gut. Cancer Chemother. Pharmacol. 2002, 49, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Borkar, N.; Li, B.; Holm, R.; Hakansson, A.E.; Mullertz, A.; Yang, M.; Mu, H. Lipophilic prodrugs of apomorphine I: Preparation, characterisation, and in vitro enzymatic hydrolysis in biorelevant media. Eur. J. Pharm. Biopharm. 2015, 89, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Graffner-Nordberg, M.; Sjödin, K.; Tunek, A.; Hallberg, A. Synthesis and enzymatic hydrolysis of esters, constituting simple models of soft drugs. Chem. Pharm. Bull. 1998, 46, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Hong, L.; Yao, T.; Yang, X.; He, Q.; Yang, B.; Hu, Y. Synthesis and evaluation of water-soluble docetaxel prodrugs-docetaxel esters of malic acid. Bioorg. Med. Chem. 2007, 15, 6323–6330. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Lin, S.W.; Dai, W.; Lu, J.K.; Yang, T.Y.; Xiang, Y.; Zhang, Y.; Li, R.T.; Zhang, Q. Novel cathepsin B-sensitive paclitaxel conjugate: Higher water solubility, better efficacy and lower toxicity. J. Control. Release 2012, 160, 618–629. [Google Scholar] [CrossRef]
- Singh, R.; Kumar, V.; Bharate, S.S.; Vishwakarma, R.A. Synthesis, pH dependent, plasma and enzymatic stability of bergenin prodrugs for potential use against rheumatoid arthritis. Bioorg. Med. Chem. 2017, 25, 5513–5521. [Google Scholar] [CrossRef]
- Mohammed, M.O.; Hussain, K.S.; Haj, N.Q. Preparation and bioactivity assessment of chitosan-1-acetic acid-5-flurouracil conjugates as cancer prodrugs. Molecules 2017, 22, 1629. [Google Scholar] [CrossRef]
- Duan, Z.; Chen, C.; Qin, J.; Liu, Q.; Wang, Q.; Xu, X.; Wang, J. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017, 24, 752–764. [Google Scholar] [CrossRef]
Sample Availability: Compound-3 to compound-10 are available from the authors. Please refer to Materials and Methods for the sources of compound-1, compound-2 and other reagents |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pure Water | 5% Glucose | Saline |
|---|---|---|---|
| SG-PTX | 19.93 ± 4.07 | 17.85 ± 0.28 | 17.83 ± 0.09 |
| DG-PTX | 38.97 ± 5.66 | 34.78 ± 4.22 | 36.72 ± 2.05 |
| PTX | <1 | <1 | <1 |
| Compound | Log P |
|---|---|
| SG-PTX | 2.77 ± 0.16 |
| DG-PTX | 2.12 ± 0.08 |
| PTX | 3.97 [27] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Y.; Zhang, Y.; Luo, Z.; Zhan, R.; Xu, H.; Chen, W.; Huang, H. Synthesis, Biological Evaluation and Low-Toxic Formulation Development of Glycosylated Paclitaxel Prodrugs. Molecules 2018, 23, 3211. https://doi.org/10.3390/molecules23123211
Mao Y, Zhang Y, Luo Z, Zhan R, Xu H, Chen W, Huang H. Synthesis, Biological Evaluation and Low-Toxic Formulation Development of Glycosylated Paclitaxel Prodrugs. Molecules. 2018; 23(12):3211. https://doi.org/10.3390/molecules23123211
Chicago/Turabian StyleMao, Yukang, Yili Zhang, Zheng Luo, Ruoting Zhan, Hui Xu, Weiwen Chen, and Huicai Huang. 2018. "Synthesis, Biological Evaluation and Low-Toxic Formulation Development of Glycosylated Paclitaxel Prodrugs" Molecules 23, no. 12: 3211. https://doi.org/10.3390/molecules23123211