Complete Chloroplast Genome Sequence of Malus hupehensis: Genome Structure, Comparative Analysis, and Phylogenetic Relationships

Abstract
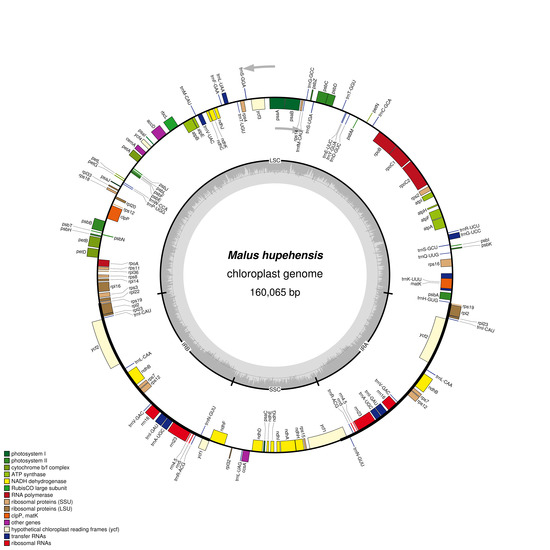
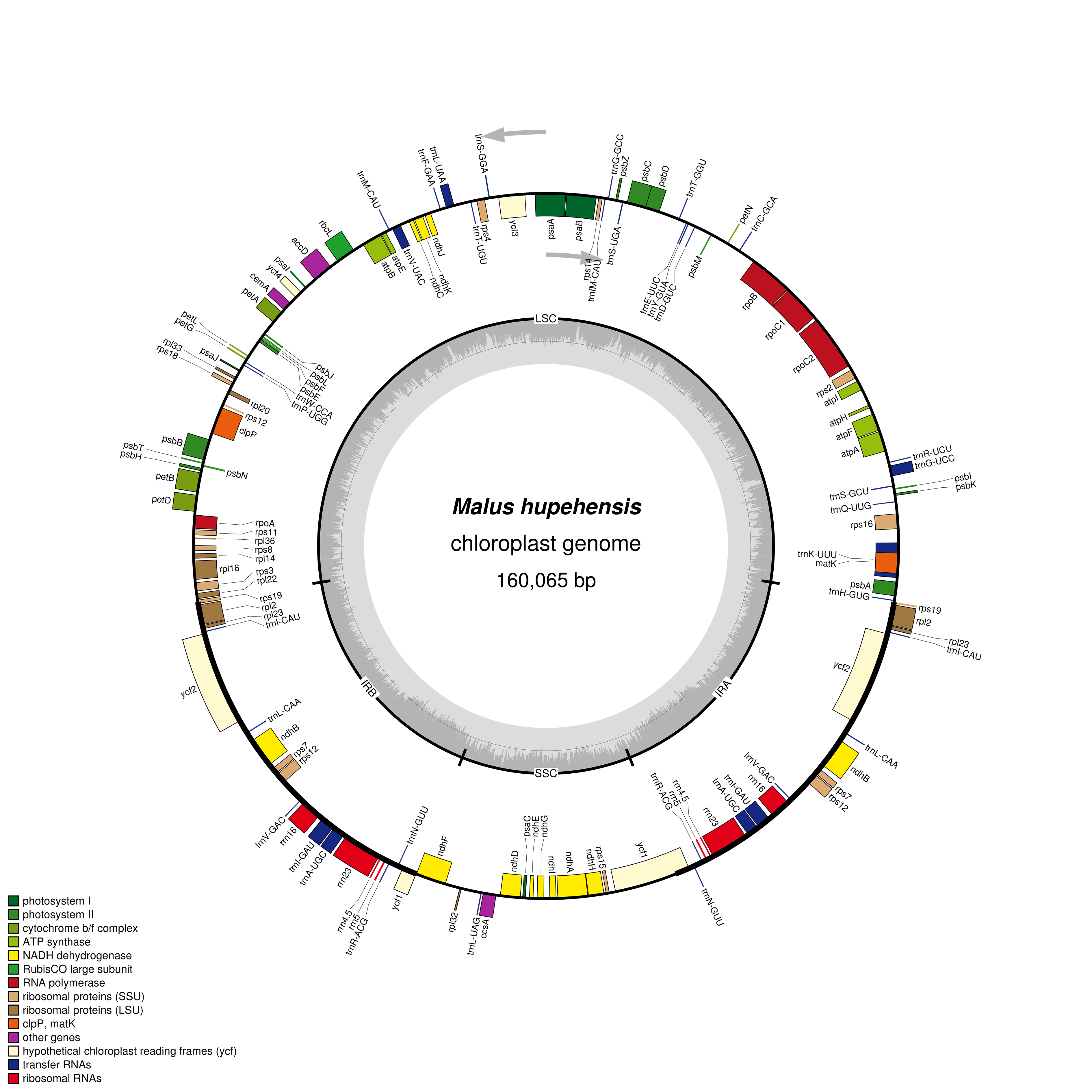
:
1. Introduction
2. Results and Discussions
2.1. Chloroplast Genome Features of M. hupehensis
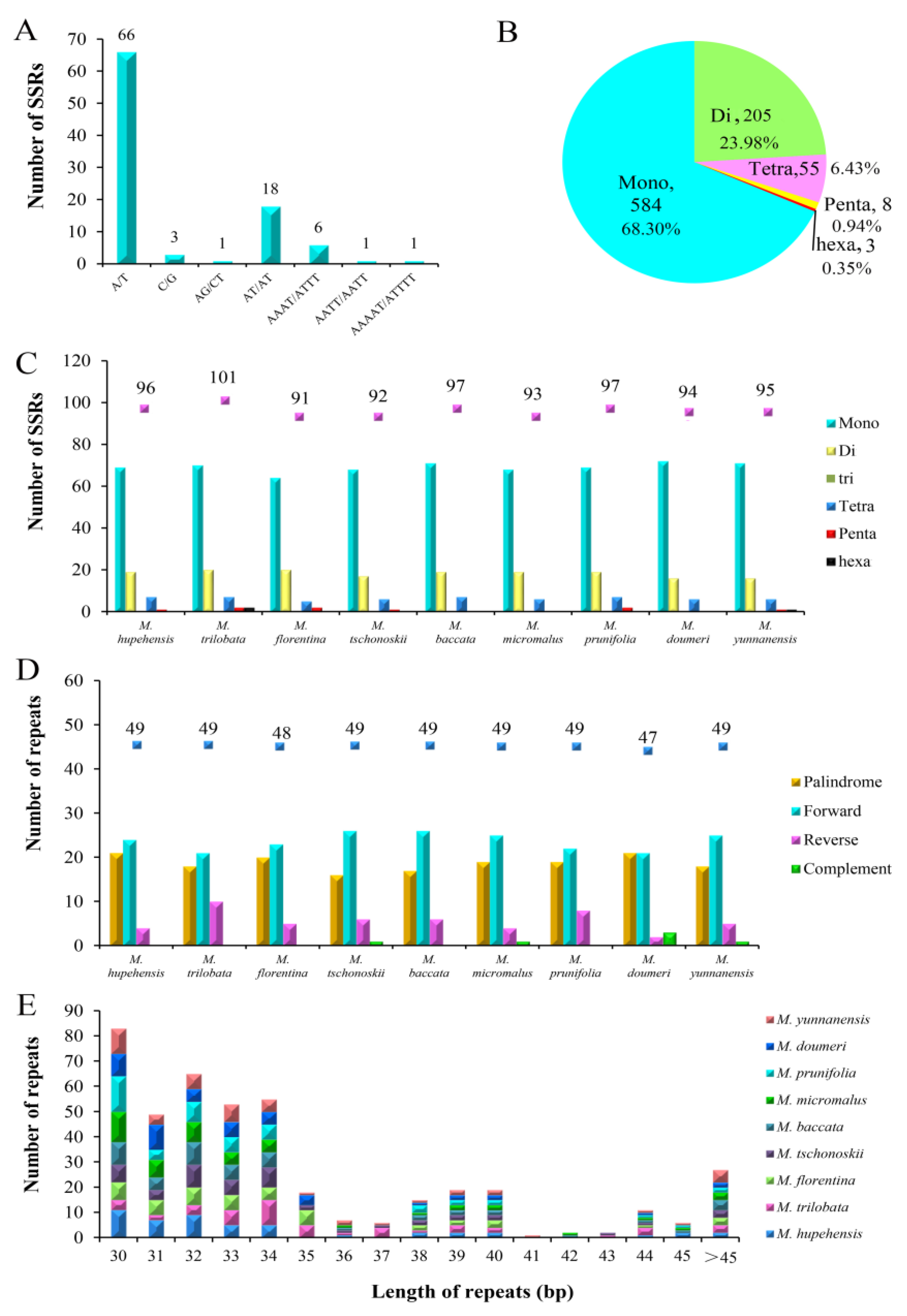
2.2. SSR and Long-Repeat Analysis
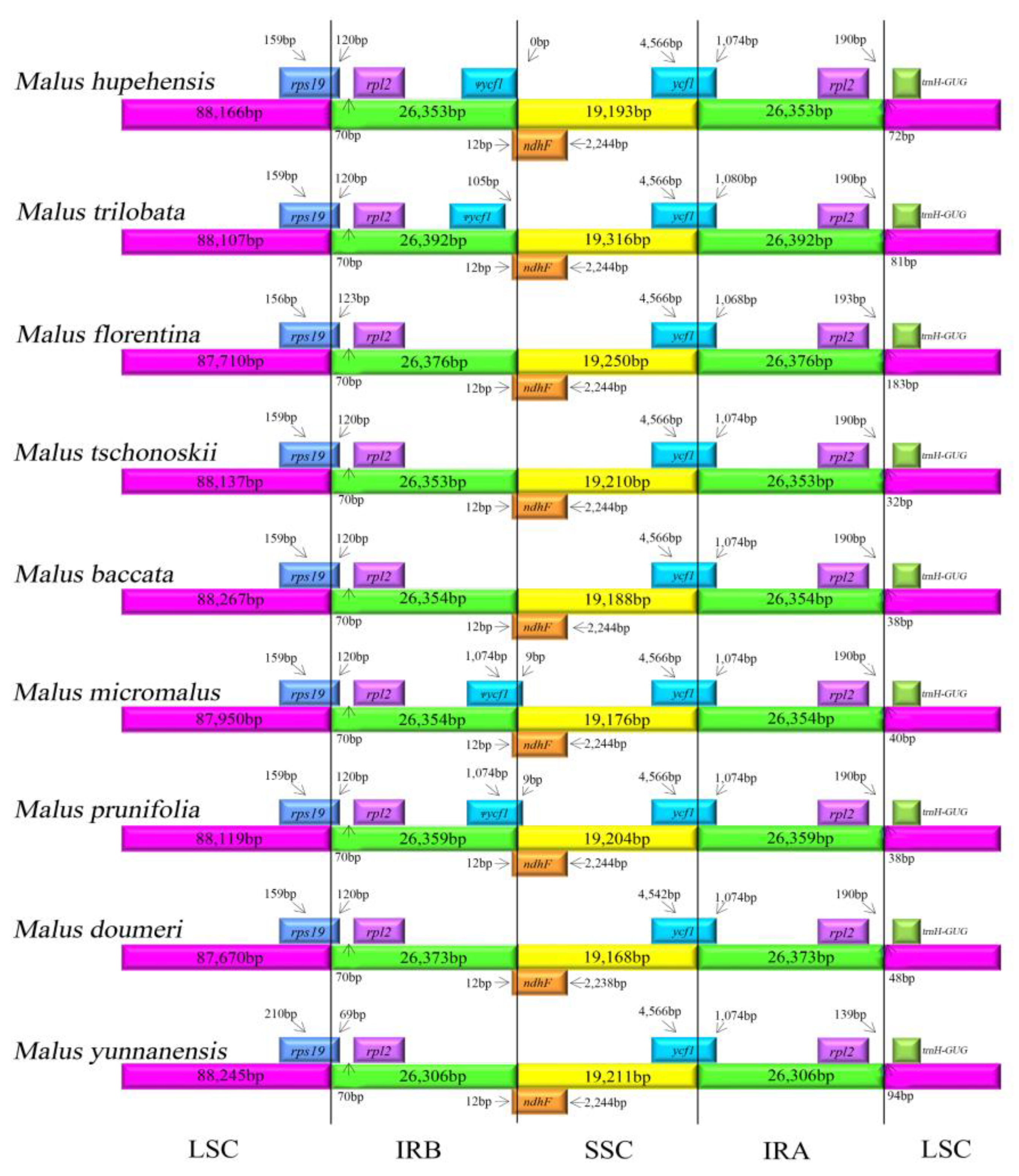
2.3. IR Contraction and Expansion
2.4. Comparative Chloroplast Genomic Analysis
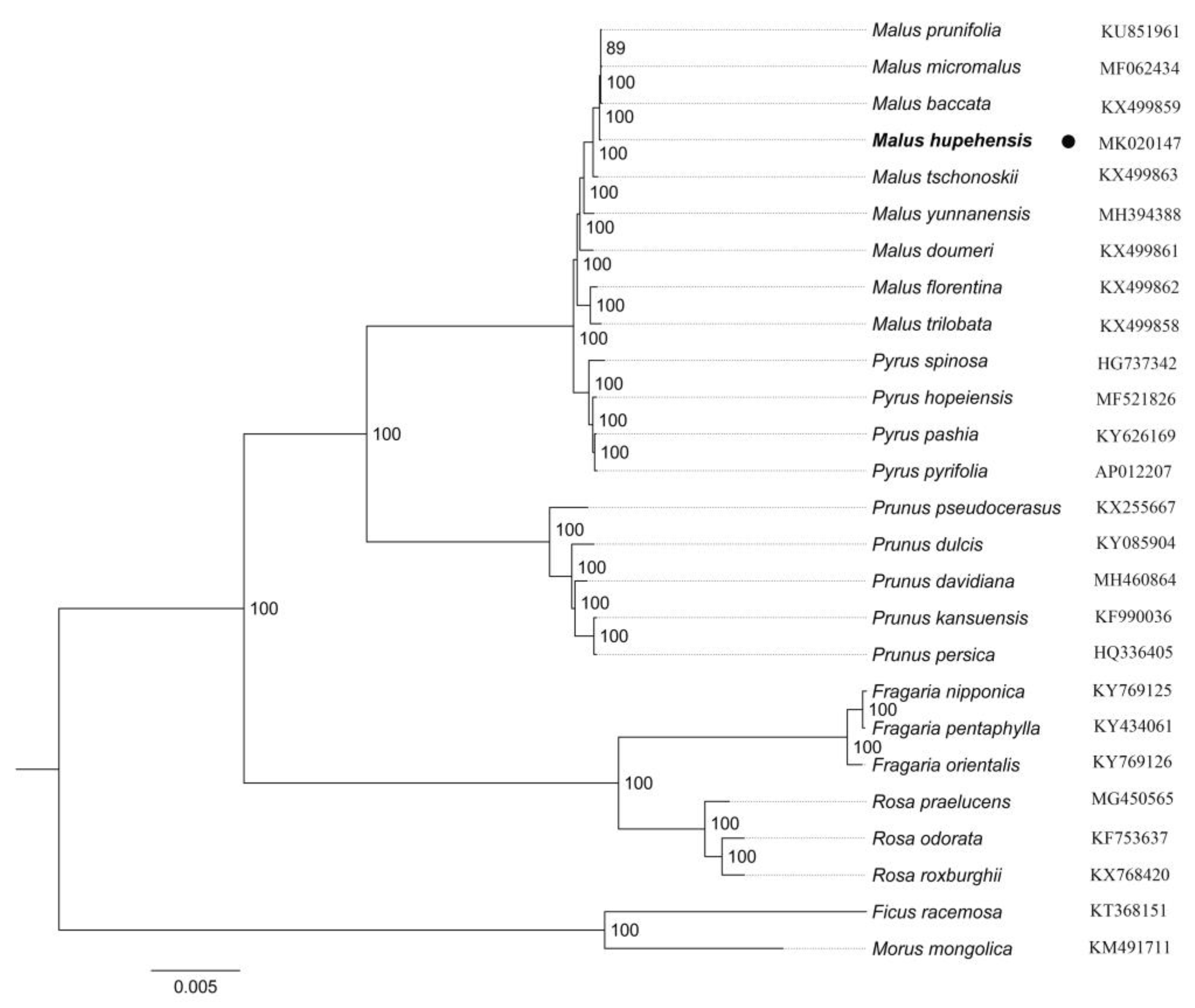
2.5. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Materials and DNA Sequencing
3.2. Genome Assembly and Genome Annotation
3.3. Sequence Analysis
3.4. Comparative Genome Analysis
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| cp | Chloroplast |
| PCGs | Protein-coding genes |
| tRNAs | Transfer RNA genes |
| rRNAs | Ribosomal RNA genes |
| IRs | Inverted repeat regions |
| LSC | Large single copy |
| SSC | Small single copy |
| SSR | Simple sequence repeat |
| ML | Maximum likelihood |
References
- Chaney, L.; Mangelson, R.; Ramaraj, T.; Jellen, E.N.; Maughan, P.J. The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Appl. Plant Sci. 2016, 4, 1600063. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Cohill, P.R.; Kumar, S.; Dufourmantel, N. Chloroplast Genetic Engineering. In Molecular Biology and Biotechnology of Plant Organelles; Springer: Dordrecht, The Netherlands, 2004; pp. 443–490. [Google Scholar] [CrossRef]
- Smith, D.R. Mutation rates in plastid genomes: They are lower than you might think. Genom. Biol. Evol. 2015, 7, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cui, L.; Feng, K.; Deng, P.; Du, X.; Wan, F.; Song, W.; Nie, X. Comparative analysis of asteraceae chloroplast genomes: Structural organization, RNA editing and evolution. Plant. Mol. Biol. Rep. 2015, 33, 1526–1538. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Ge, L.; Xing, G.; Wang, M.; Song, W.; Nie, X. Identification and analysis of RNA editing sites in the chloroplast transcripts of Aegilops tauschii L. Genes 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes-applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed]
- Bock, R.; Khan, M.S. Taming plastids for a green future. Trends Biotechnol. 2004, 22, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, X.; Zhao, X.; Wang, S.; Zhang, T.; Li, C.; Liu, H.; Tong, W.; Guo, Y. Complete chloroplast genome sequence of Broomcorn Millet (Panicum miliaceum L.) and comparative analysis with other Panicoideae species. Agronomy 2018, 8, 159. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Ku, T.C.; Spongberg, S.A. Malus (Rosaceae). In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2003; Volume 9, pp. 179–189. ISBN 9787030130440. [Google Scholar]
- Zhi-Qin, Z. The apple genetic resources in China: The wild species and their distributions, informative characteristics and utilisation. Genet. Res. Crop. Evol. 1999, 46, 599–609. [Google Scholar] [CrossRef]
- Cui, L.; Xing, M.; Xu, L.; Wang, J.; Zhang, X.; Ma, C.; Kang, W. Antithrombotic components of Malus halliana Koehne flowers. Food. Chem. Toxicol. 2018, 119, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.A.N.; Rodrigues, S.; Carcel, J.A.; Garcia-Perez, J.V. Ultrasound-assisted air-drying of apple (Malus domestica L.) and its effects on the vitamin of the dried product. Food Bioprocess Technol. 2015, 8, 1503–1511. [Google Scholar] [CrossRef]
- Fang, L.; Meng, W.; Min, W. Phenolic compounds and antioxidant activities of flowers, leaves and fruits of five crabapple cultivars (Malus Mill. species). Sci. Hortic. 2018, 235, 460–467. [Google Scholar] [CrossRef]
- Huang, S.; Liu, H.F.; Meng, N.; Li, B.; Wang, J.L. Hypolipidemic and antioxidant effects of Malus toringoides (Rehd.) Hughes leaves in high-fat-diet-induced hyperlipidemic rats. J. Med. Food 2017, 20, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, F.Z.; Wang, K.; Wang, D.J.; Gong, X.; Liu, L.J.; Richards, C.M.; Henk, A.D.; Volk, G.M. Genetic diversity of Malus cultivars and wild relatives in the Chinese National Repository of Apple Germplasm Resources. Tree Genet. Genomes 2015, 11. [Google Scholar] [CrossRef]
- Kumar, C.; Singh, S.K.; Pramanick, K.K.; Verma, M.K.; Srivastav, M.; Singh, R.; Bharadwaj, C.; Naga, K.C. Morphological and biochemical diversity among the Malus species including indigenous Himalayan wild apples. Sci. Hortic. 2018, 233, 204–219. [Google Scholar] [CrossRef]
- Zhang, W.-X.; Zhao, M.-M.; Fan, J.-J.; Zhou, T.; Chen, Y.-X.; Cao, F.-L. Study on relationship between pollen exine ornamentation pattern and germplasm evolution in flowering crabapple. Sci Rep. 2017, 7, 39759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, B.L.; Zhong, G.-Y.; Brown, S.K. Genetic diversity of dihydrochalcone content in Malus germplasm. Genet. Res. Crop. Evol. 2018, 65, 1485–1502. [Google Scholar] [CrossRef]
- Robinson, J.P.; Harris, S.A.; Juniper, B.E. Taxonomy of the genus Malus Mill. (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh. Plant. Syst. Evol. 2001, 226, 35–58. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Zhou, T.Y.; Xiao, P.G. Xinhua Materia Medica Outline. Jiangsu Institute of Botany; Shanghai Science and Technology Press: Shanghai, China, 1990; Volume 3, pp. 103–104. ISBN 7-5323-1421-9. [Google Scholar]
- State Administration of Traditional Chinese Medicine “Chinese Materia Medica Committee”. Zhong Hua Ben Cao (Chinese Materia Medica); Shanghai Science and Technology Press: Shanghai, China, 1999; Volume 4, pp. 158–159. ISBN 7-5323-5106-8. [Google Scholar]
- Chen, Y.L.; Tan, Z.X.; Peng, Y. Traditional uses and modern research of leaves of Malus hupehensis (Pamp.) rehd. Modern. Chin. Med. 2017, 10, 1505–1510. [Google Scholar] [CrossRef]
- Wen, C.; Wang, D.; Li, X.; Huang, T.; Huang, C.; Hu, K. Targeted isolation and identification of bioactive compounds lowering cholesterol in the crude extracts of crabapples using UPLC-DAD-MS-SPE/NMR based on pharmacology-guided PLS-DA. J. Pharm. Biomed. Anal. 2018, 150, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Chen, Y.-Y.; Jiao, Q.-Y.; Khan, A.; Shan, J.; Cao, G.-D.; Li, F.; Zhang, C.; Lou, H.-X. Polyphenolic compounds from Malus hupehensis and their free radical scavenging effects. Nat. Prod. Res. 2018, 32, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Q.; Zhu, X.-F.; Wang, X.-N.; Shen, T.; Xiang, F.; Lou, H.-X. Flavonoids from Malus hupehensis and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Phytochemistry 2013, 87, 119–125. [Google Scholar] [CrossRef] [PubMed]
- You-Qin, D.; Tian-Yan, F.; Gai-Gai, D.; Ying, L.; Jun-Zhi, W. Inhibitory effect of total flavonoids of Malus hupehensis on hepatic fibrosis induced by Schistosoma japonicum in mice. Chin. J. Schi. Cont. 2011, 23, 551–554. [Google Scholar] [CrossRef]
- Chun-Nian, H.E.; Peng, Y.; Xiao, W.; Li-Jia, X.U.; Xiao, P.G. The prevention and treatment of cancer with non-camellia tea from China. Modern. Chin. Med. 2013, 16, 13–17. [Google Scholar] [CrossRef]
- Zhang, J.G.; Hu, H.J.; Xu, Y.H.; Luo, Z.R. Germplasm identification and genetic relationship of some Malus hupehensis (Pamp.) Rehd. accessions. J. Huazhong Agric. Univ. 2009, 6, 736–740. [Google Scholar] [CrossRef]
- Jansen, R.K.; Saski, C.; Lee, S.-B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Waqas, M.; Kang, S.-M.; Yun, B.-W.; Lee, I.-J. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Sci. Rep. 2017, 7, 7556. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikanai, T.; Shimizu, K.; Ueda, K.; Nishimura, Y.; Kuroiwa, T.; Hashimoto, T. The chloroplast clpP gene, encoding a proteolytic subunit of ATP-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol. 2001, 42, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Sy Nguyen, D.; Sai, T.Z.T.; Nawaz, G.; Lee, K.; Kang, H. Abiotic stresses affect differently the intron splicing and expression of chloroplast genes in coffee plants (Coffea arabica) and rice (Oryza sativa). Plant Physiol. 2016, 201, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Dong, B.; Xu, L.; Tembrock, L.R.; Zheng, S.; Wu, Z. The complete chloroplast genome of Heimia myrtifolia and comparative analysis within Myrtales. Molecules 2018, 23, 846. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, P.; Palme, A.; Morales, L.O.; Lannenpaa, M.; Keinanen, M.; Sopanen, T.; Lascoux, M. Phylogenetic relationships of Betula species (Betulaceae) based on nuclear ADH and chloroplast matK sequences. Am. J. Bot. 2004, 91, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.X.; Gadagkar, S.R.; Potter, D.; Xu, D.X.; Zhang, M.; Li, Z.Y. Phylogeny of Spiraea (Rosaceae) based on plastid and nuclear molecular data: Implications for morphological character evolution and systematics. Perspect. Plant Ecol. Evol. Syst. 2018, 34, 109–119. [Google Scholar] [CrossRef]
- Johnson, L.A.; Soltis, D.E. matK DNA sequences and phylogenetic reconstruction in Saxifragaceae s. str. Syst. Bot. 1994, 19, 143–156. [Google Scholar] [CrossRef]
- Steele, K.P.; Vilgalys, R. Phylogenetic analyses of polemoniaceae using nucleotide sequences of the plastid gene matK. Syst. Bot. 1994, 19, 126–142. [Google Scholar] [CrossRef]
- Plunkett, G.M.; Soltis, D.E.; Soltis, P.S. Clarification of the relationship between Apiaceae and Araliaceae based on matK and rbcL sequence data. Am. J. Bot. 1997, 84, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; He, R.; Zhang, H.Y.; Huang, Y.B.; Tian, M.L.; Zhang, J.J. Analysis of synonymous codon usage in Zea mays. Mol. Biol. Rep. 2010, 37, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Xing, H.X.; Yuan, Y.C.; Wang, X.L.; Saeed, M.; Tao, J.C.; Feng, W.; Zhang, G.H.; Song, X.L.; Sun, X.Z. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.B.; Yin, J.L.; Guo, H.Y.; Zhang, Y.Y.; Xiao, W.; Sun, C.; Wu, J.Y.; Qu, X.B.; Yu, J.; Wang, X.M.; et al. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front. Plant Sci. 2015, 5, 696. [Google Scholar] [CrossRef] [PubMed]
- Gichira, A.W. The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): Structural comparative analysis, gene content and microsatellite detection. PeerJ 2017, 5, e2846. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, J.F.; Zhang, H.; Cai, B.H.; Gao, Z.H.; Qiao, Y.S.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria X ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3931. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.Y.; Zhang, Y.H.; Yan, H.J.; Qiu, X.Q.; Wang, Q.G.; Li, S.B.; Zhang, S.D. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium x hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Isono, K.; Kojima, T.; Endo, A.; Hanaoka, M.; Shiina, T.; Terachi, T.; Utsugi, S.; Murata, M.; Mori, N.; et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol. Genet. Genom. 2002, 266, 740–746. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S.L. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid. DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, K.; Szczecinska, M.; Sawicki, J. Evaluation of 11 single-locus and seven multilocus DNA barcodes in Lamium. L. (Lamiaceae). Mol. Ecol. Resour. 2014, 14, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Gyulai, G.; Hidvegi, N.; Kerti, B.; Al Hemaid, F.M.A.; Pandey, A.K.; Lee, J.K. The changing epitome of species identification DNA barcoding. Saudi J. Biol. Sci. 2014, 21, 204–231. [Google Scholar] [CrossRef]
- Niu, Z.; Xue, Q.; Zhu, S.; Sun, J.; Liu, W.; Ding, X. The complete plastome sequences of four orchid species: Insights into the evolution of the Orchidaceae and the utility of plastomic mutational hotspots. Front. Plant. Sci. 2017, 8, 715. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.S.; Fu, P.C.; Zhou, X.J.; Cheng, Y.W.; Zhang, F.Q.; Chen, S.L.; Gao, Q.B. The complete plastome sequences of seven species in Gentianasect. Kudoa (Gentianaceae): Insights into plastid gene loss and molecular evolution. Front. Plant Sci. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.Z.; Huang, C.H.; Hu, Y.; Wen, J.; Li, S.S.; Yi, T.S.; Chen, H.Y.; Xiang, J.; Ma, H. Evolution of Rosaceae Fruit Types Based on Nuclear Phylogeny in the Context of Geological Times and Genome Duplication. Mol. Biol. Evol. 2017, 34, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Yang, J.Y.; Kim, S.C. Complete chloroplast genome of Ulleung Island endemic flowering cherry, Prunus takesimensis (Rosaceae), in Korea. Mitochondrial DNA Part B Resour. 2018, 3, 274–275. [Google Scholar] [CrossRef]
- Terakami, S.; Matsumura, Y.; Kurita, K.; Kanamori, H.; Katayose, Y.; Yamamoto, T.; Katayama, H. Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet. Genomes 2012, 8, 841–854. [Google Scholar] [CrossRef]
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.E.; Morgan, D.R.; Kerr, M.; Robertson, K.R.; Arsenault, M.; Dickinson, T.A.; et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.Y.Y.; Donoghue, M.J. Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol. Phylogenet. Evol. 2012, 63, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucl. Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kaya, T.; Balta, F.; Sensoy, S. Fruit quality parameters and molecular analysis of apple germplasm resources from Van Lake Basin, Turkey. Turk. J. Agric. For. 2015, 39, 864–875. [Google Scholar] [CrossRef]
- Potts, S.M.; Han, Y.P.; Khan, M.A.; Kushad, M.M.; Rayburn, A.L.; Korban, S.S. Genetic diversity and characterization of a core collection of Malus germplasm using simple sequence repeats (SSRs). Plant Mol. Biol. Rep. 2012, 30, 827–837. [Google Scholar] [CrossRef]
- Potts, S.M.; Kushad, M.M.; Korban, S.S. Genetic diversity and classification of Malus germplasm using simple sequence repeats (SSRs). Hortscience 2009, 44, 1173. [Google Scholar]
- Linden, L.; Iwarsson, M. Identification of weeping crabapple cultivars by microsatellite DNA markers and morphological traits. Sci. Hortic. 2014, 179, 221–226. [Google Scholar] [CrossRef]
- Guo, Q.G.; Yu, Y.; He, Q.; Li, X.L.; Liang, G.L. AFLP analysis of four wild Malus Mill. Acta Hortic. 2007, 16, 131–135. [Google Scholar] [CrossRef]
- Xu, R.X.; Hu, D.C.; Chen, Z.Y.; Zhang, P.; Jiang, X.M.; Tang, G.G. SRAP analysis on genetic relationships of genotypes in the genus Malus Mill. Biotechnol. Biotechnol. Equip. 2014, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.Z.; Gao, Y.; Wang, D.J.; Gong, X.; Liu, L.J. The natural distribution, diversity and utilization of wild in China. J. Plant Genet. Resour. 2013, 14, 1013–1019. [Google Scholar] [CrossRef]
Sample Availability: Samples of M. hupehensis are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Characteristics | M. hupehensis | M. trilobata | M. florentina | M. tschonoskii | M. baccata | M. micromalus | M. prunifolia | M. doumeri | M. yunnanensis |
|---|---|---|---|---|---|---|---|---|---|
| Accession number | MK020147 | KX499858 | KX499862 | KX499863 | KX499859 | MF062434 | KU851961 | KX499861 | MH394388 |
| Genome size (bp) | 160,065 | 160,207 | 159,712 | 160,053 | 160,163 | 159,834 | 160,041 | 159,584 | 160,068 |
| LSC length (bp) | 88,166 | 88,107 | 87,710 | 88,137 | 88,267 | 87,950 | 88,119 | 87,670 | 88,245 |
| SSC length (bp) | 19,193 | 19,316 | 19,250 | 19,210 | 19,188 | 19,176 | 19,204 | 19,168 | 19,211 |
| IR length (bp) | 26,353 | 26,392 | 26,376 | 26,353 | 26,354 | 26,354 | 26,359 | 26,373 | 26,306 |
| No. of different genes | 112 | 110 | 110 | 110 | 109 | 111 | 111 | 110 | 112 |
| No. of different protein-coding genes | 78 | 76 | 77 | 77 | 76 | 77 | 77 | 77 | 78 |
| No. of different tRNA genes | 30 | 30 | 29 | 29 | 29 | 30 | 30 | 29 | 30 |
| No. of different rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| % GC content in LSC | 34.2 | 34.2 | 34.3 | 34.2 | 34.2 | 34.3 | 34.2 | 34.4 | 34.2 |
| % GC content in SSC | 30.4 | 30.3 | 30.4 | 30.4 | 30.4 | 30.4 | 30.4 | 30.4 | 30.4 |
| % GC content in IR | 42.7 | 42.6 | 42.6 | 42.7 | 42.7 | 42.7 | 42.7 | 42.6 | 42.7 |
| % GC content of genome | 36.6 | 36.5 | 36.6 | 36.5 | 36.5 | 36.6 | 36.6 | 36.6 | 36.5 |
| Group of Genes | Gene Name |
|---|---|
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1#, rpoC2 |
| tRNA genes | trnA-UGC# (×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC #, trnG-UCC, trnH-GUG, trnI-CAU (×2), trnI-GAU # (×2), trnK-UUU #, trnL-CAA (×2), trnL-UAA #, trnL-UAG, trnfM-CAU, trnM-CAU, trnN-GUU (×2), trnP-GGG, trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC #, trnW-CCA, trnY-GUA |
| Ribosomal small subunit | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 # (×2), rps14, rps15, rps16 #, rps18, rps19 (×2) |
| Ribosomal large subunit | rpl2# (×2), rpl14, rpl16 #, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 |
| rRNA genes | rrn16 (×2), rrn23 (×2), rrn4.5 (×2), rrn5 (×2) |
| ATP synthase | atpA, atpB, atpE, atpF#, atpH, atpI |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| NADH dehydrogenase | ndhA#, ndhB# (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Cytochrome b/f complex | petA, petB#, petD #, petG, petL, petN |
| Large subunit of rubisco | rbcL |
| Maturase | matK |
| Subunit of acetyl-CoA carboxylase | accD |
| Envelope membrane protein | cemA |
| Protease | clpP## |
| c-type cytochrome synthesis | ccsA |
| Conserved open reading frames | ycf1 (×2), ycf2 (×2), ycf3 ##, ycf4 |
| Gene | Location | ExonI (bp) | IntronI (bp) | ExonII (bp) | IntronII (bp) | ExonIII (bp) |
|---|---|---|---|---|---|---|
| trnK-UUU | LSC | 37 | 2497 | 35 | ||
| trnG-UCC | LSC | 23 | 698 | 48 | ||
| trnL-UAA | LSC | 37 | 514 | 50 | ||
| trnV-UAC | LSC | 39 | 592 | 37 | ||
| trnI-GAU | IR | 42 | 943 | 35 | ||
| trnA-UGC | IR | 38 | 807 | 35 | ||
| rps12 * | LSC | 114 | - | 232 | 541 | 26 |
| rps16 | LSC | 40 | 864 | 221 | ||
| rpl16 | LSC | 9 | 983 | 399 | ||
| rpl2 | IR | 390 | 686 | 435 | ||
| rpoC1 | LSC | 435 | 741 | 1611 | ||
| ndhA | SSC | 552 | 1134 | 540 | ||
| ndhB | IR | 777 | 669 | 756 | ||
| ycf3 | SSC | 126 | 708 | 228 | 744 | 153 |
| petB | LSC | 6 | 797 | 642 | ||
| atpF | LSC | 144 | 737 | 411 | ||
| clpP | LSC | 71 | 826 | 292 | 627 | 228 |
| petD | LSC | 8 | 724 | 475 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Rong, C.; Qin, L.; Mo, C.; Fan, L.; Yan, J.; Zhang, M. Complete Chloroplast Genome Sequence of Malus hupehensis: Genome Structure, Comparative Analysis, and Phylogenetic Relationships. Molecules 2018, 23, 2917. https://doi.org/10.3390/molecules23112917
Zhang X, Rong C, Qin L, Mo C, Fan L, Yan J, Zhang M. Complete Chloroplast Genome Sequence of Malus hupehensis: Genome Structure, Comparative Analysis, and Phylogenetic Relationships. Molecules. 2018; 23(11):2917. https://doi.org/10.3390/molecules23112917
Chicago/Turabian StyleZhang, Xin, Chunxiao Rong, Ling Qin, Chuanyuan Mo, Lu Fan, Jie Yan, and Manrang Zhang. 2018. "Complete Chloroplast Genome Sequence of Malus hupehensis: Genome Structure, Comparative Analysis, and Phylogenetic Relationships" Molecules 23, no. 11: 2917. https://doi.org/10.3390/molecules23112917