Chemical Constituents of the Ethyl Acetate Extract from Diaphragma juglandis Fructus and Their Inhibitory Activity on Nitric Oxide Production In Vitro

Abstract
:
1. Introduction
2. Results and Discussion
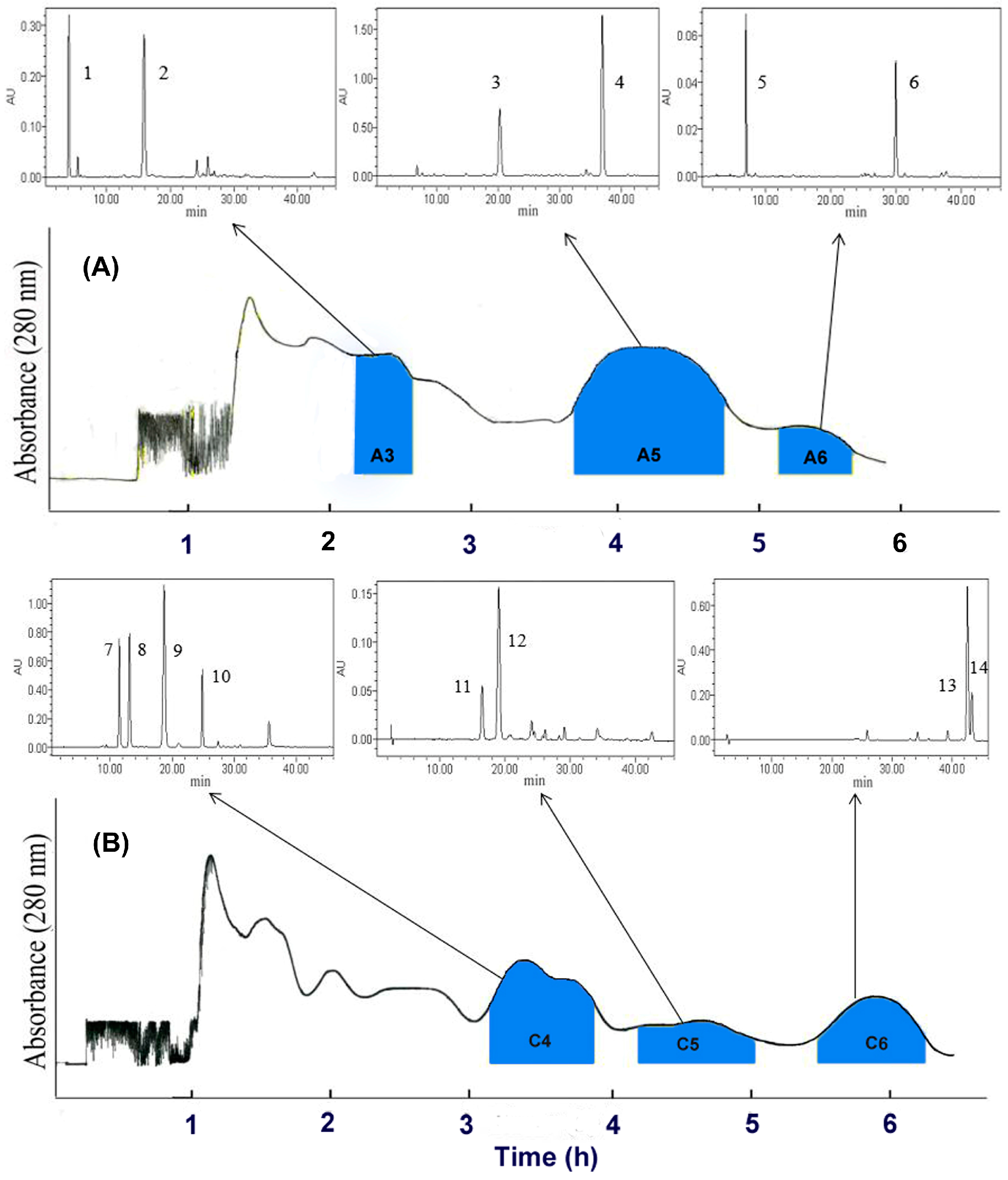
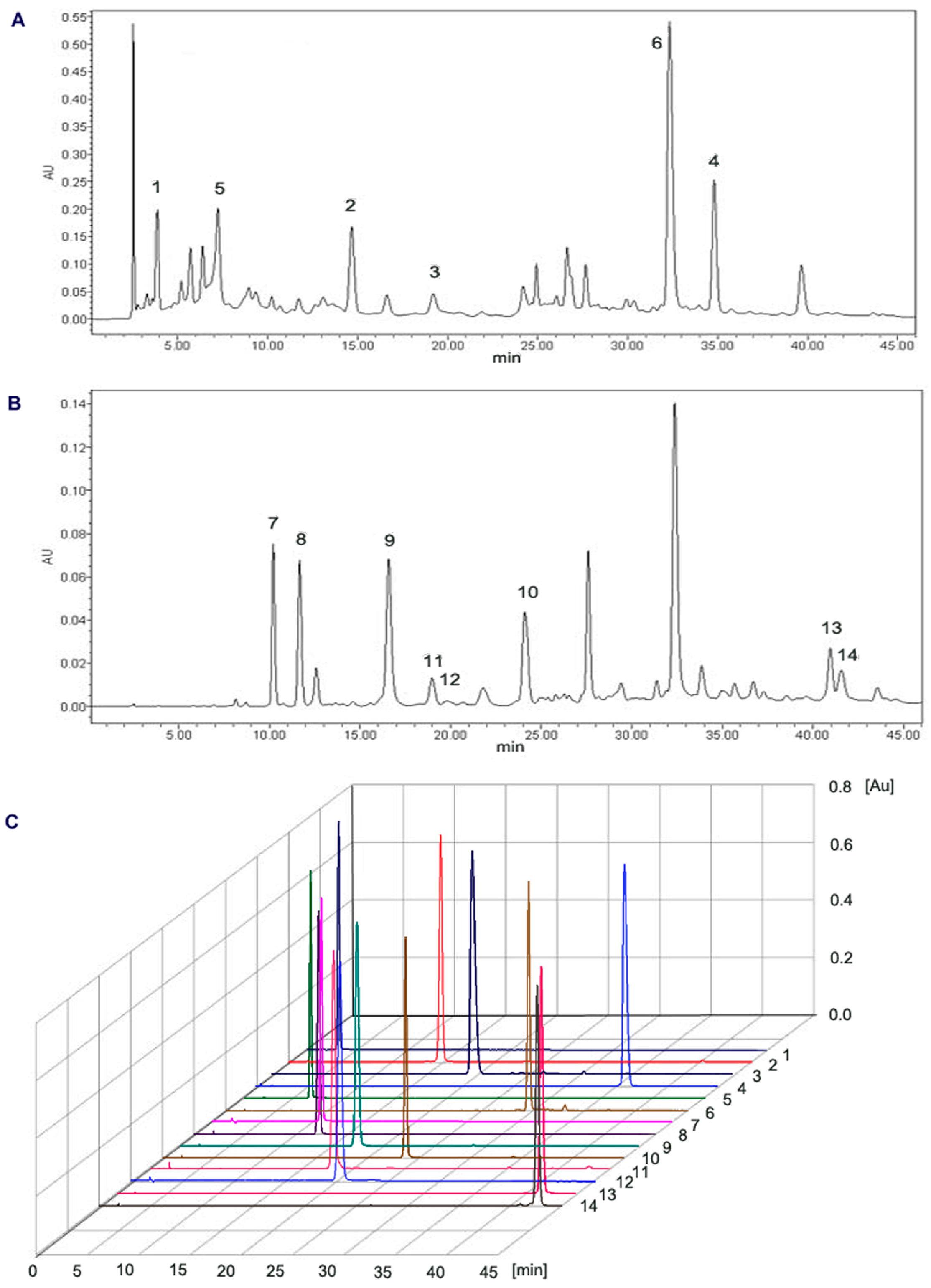
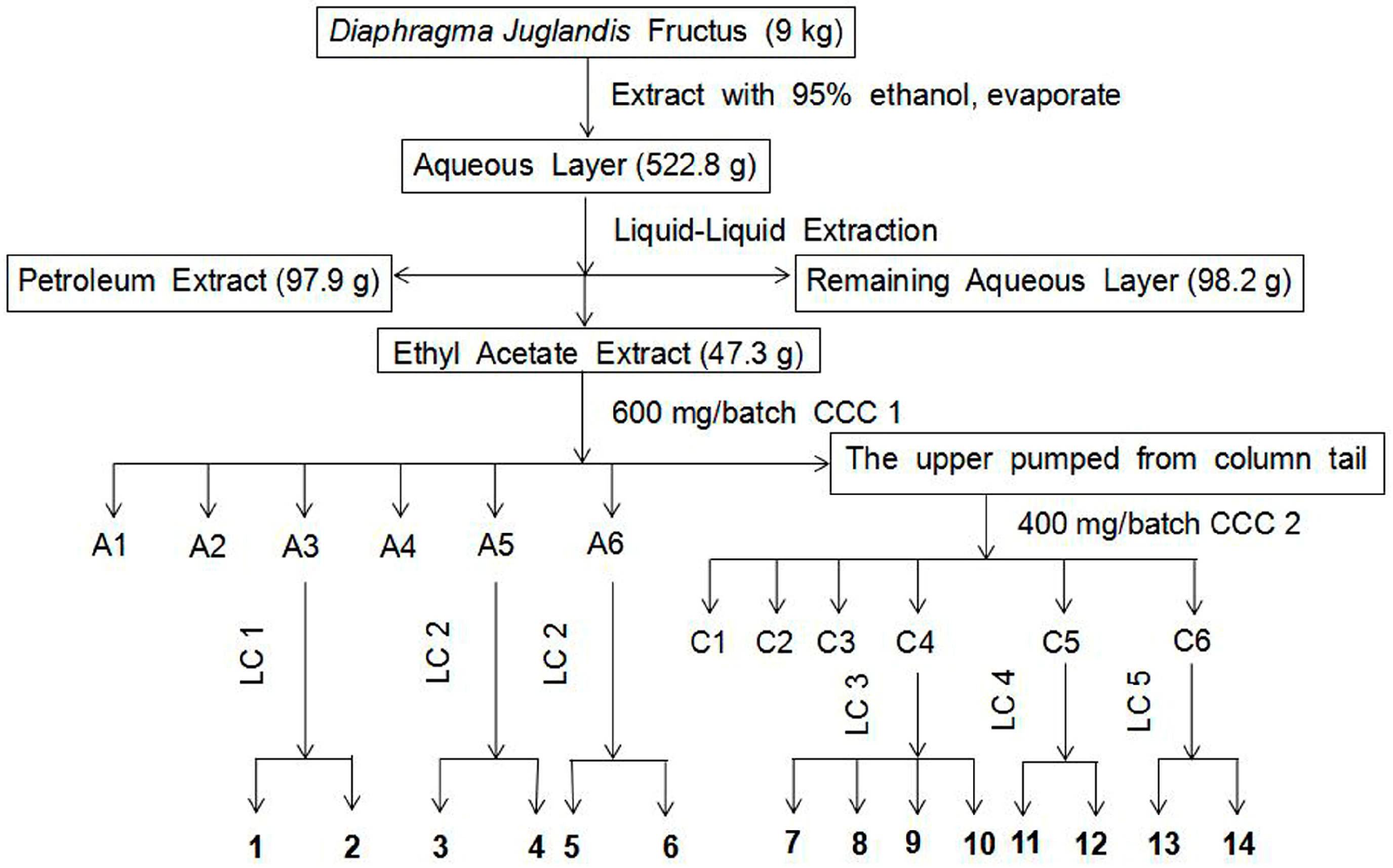
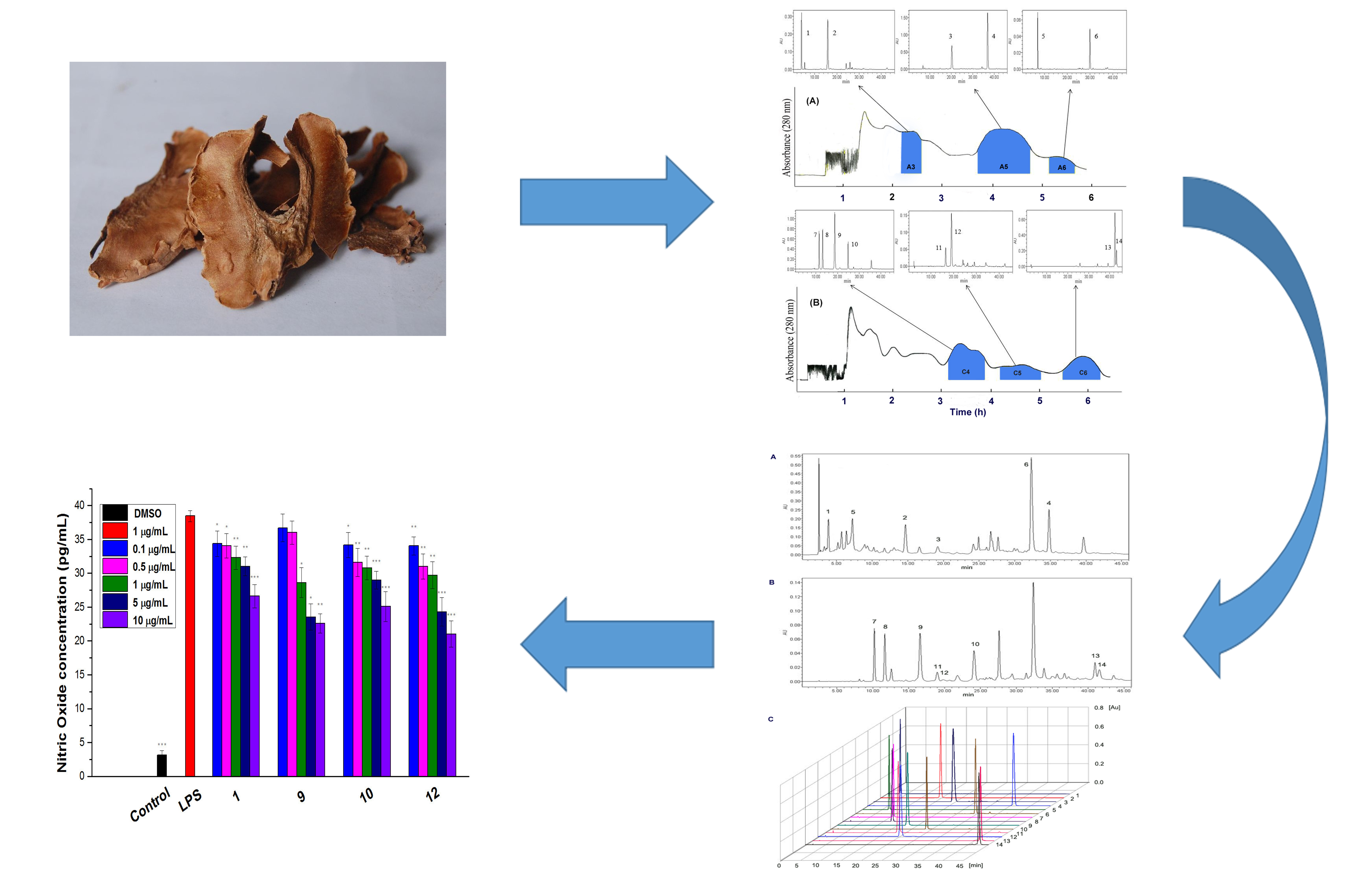
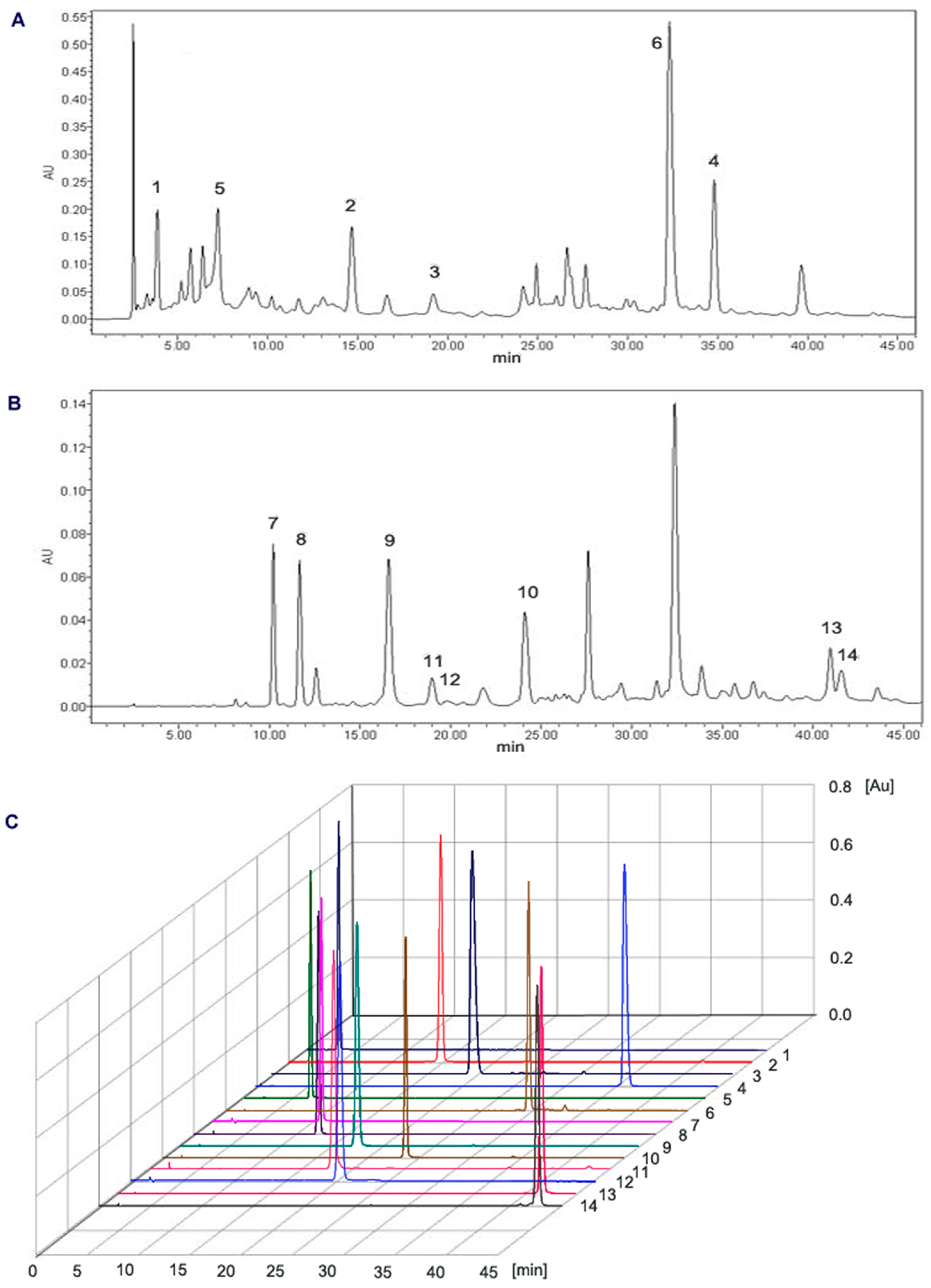
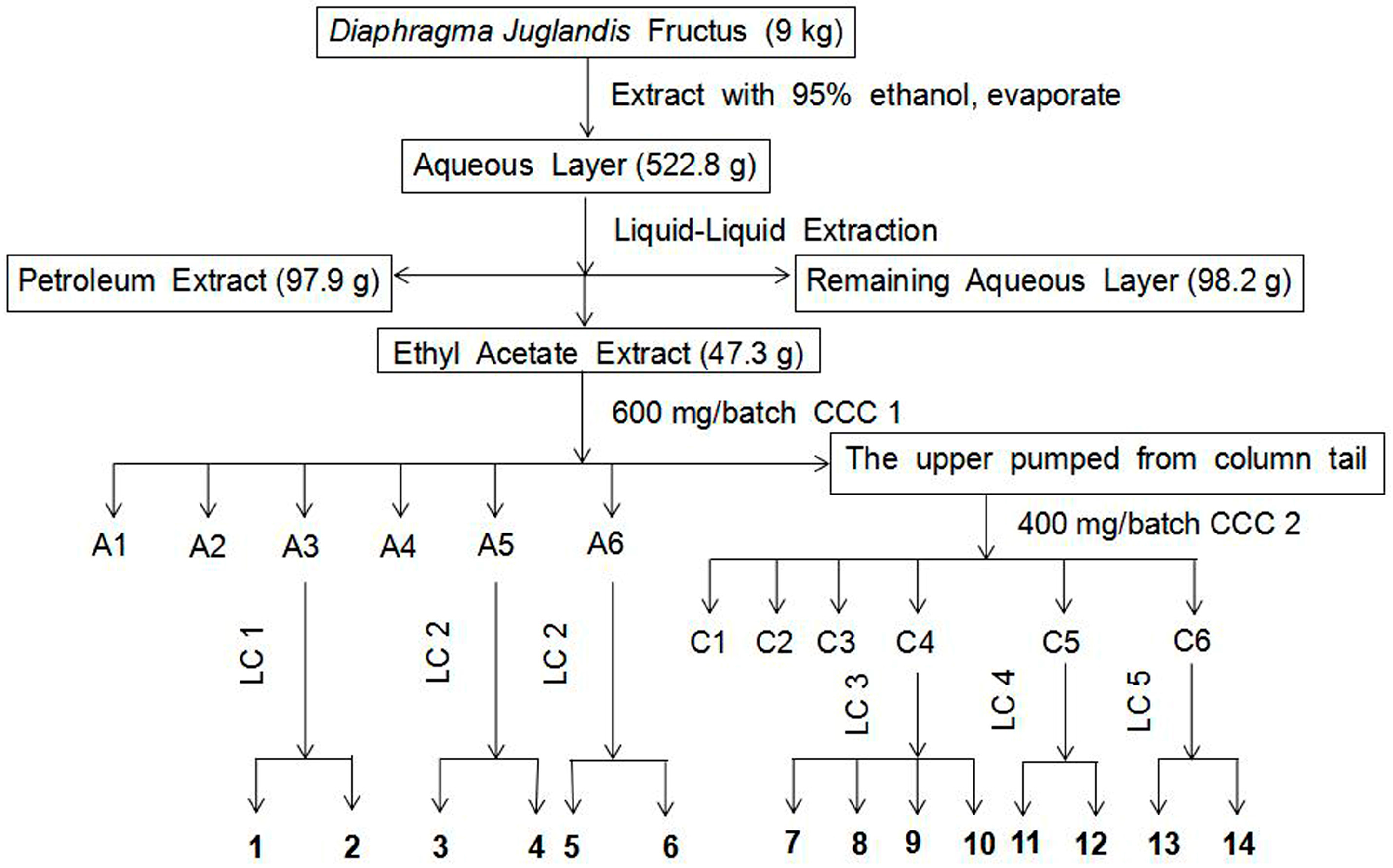
2.1. Separation and Purification of Compounds
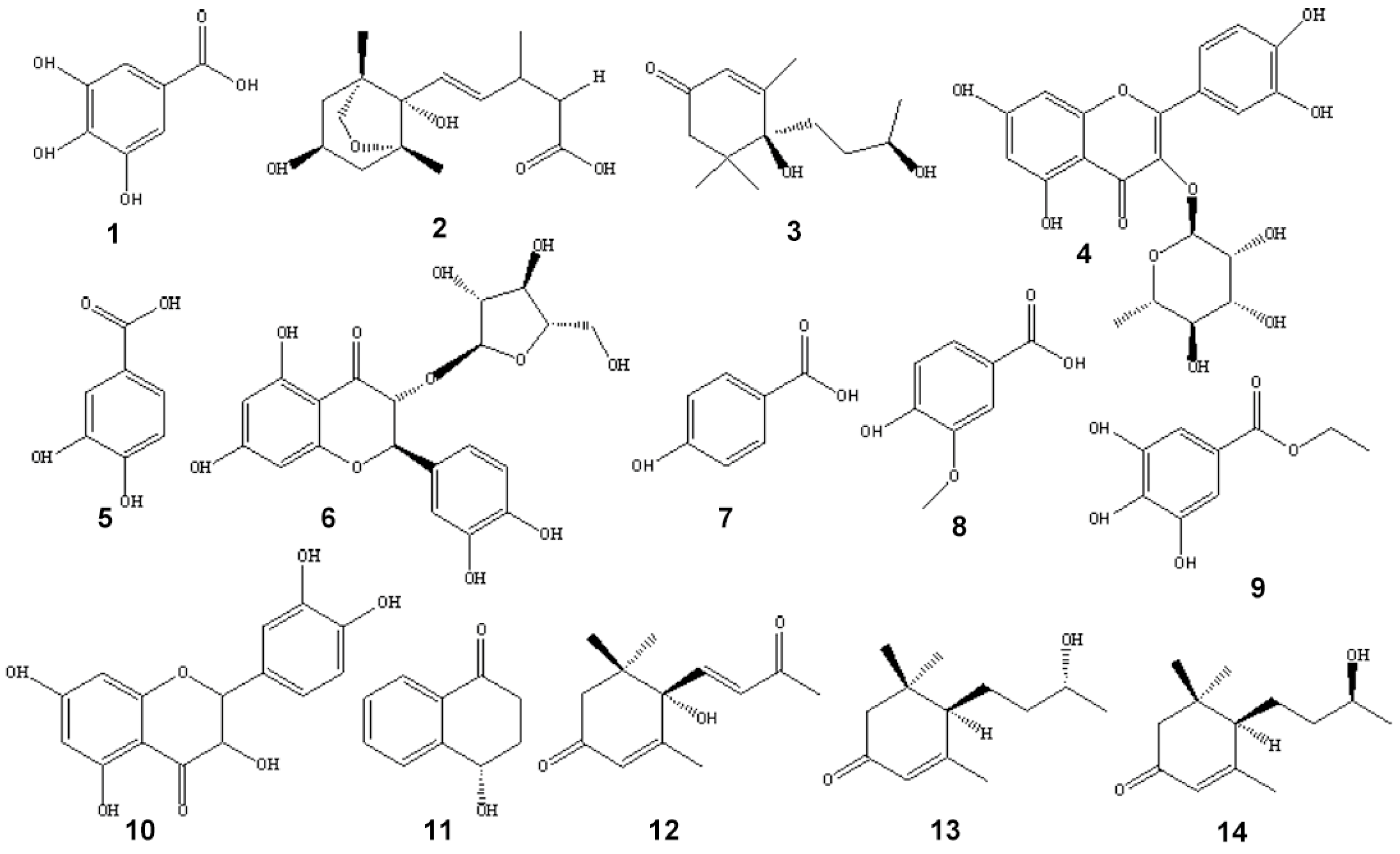
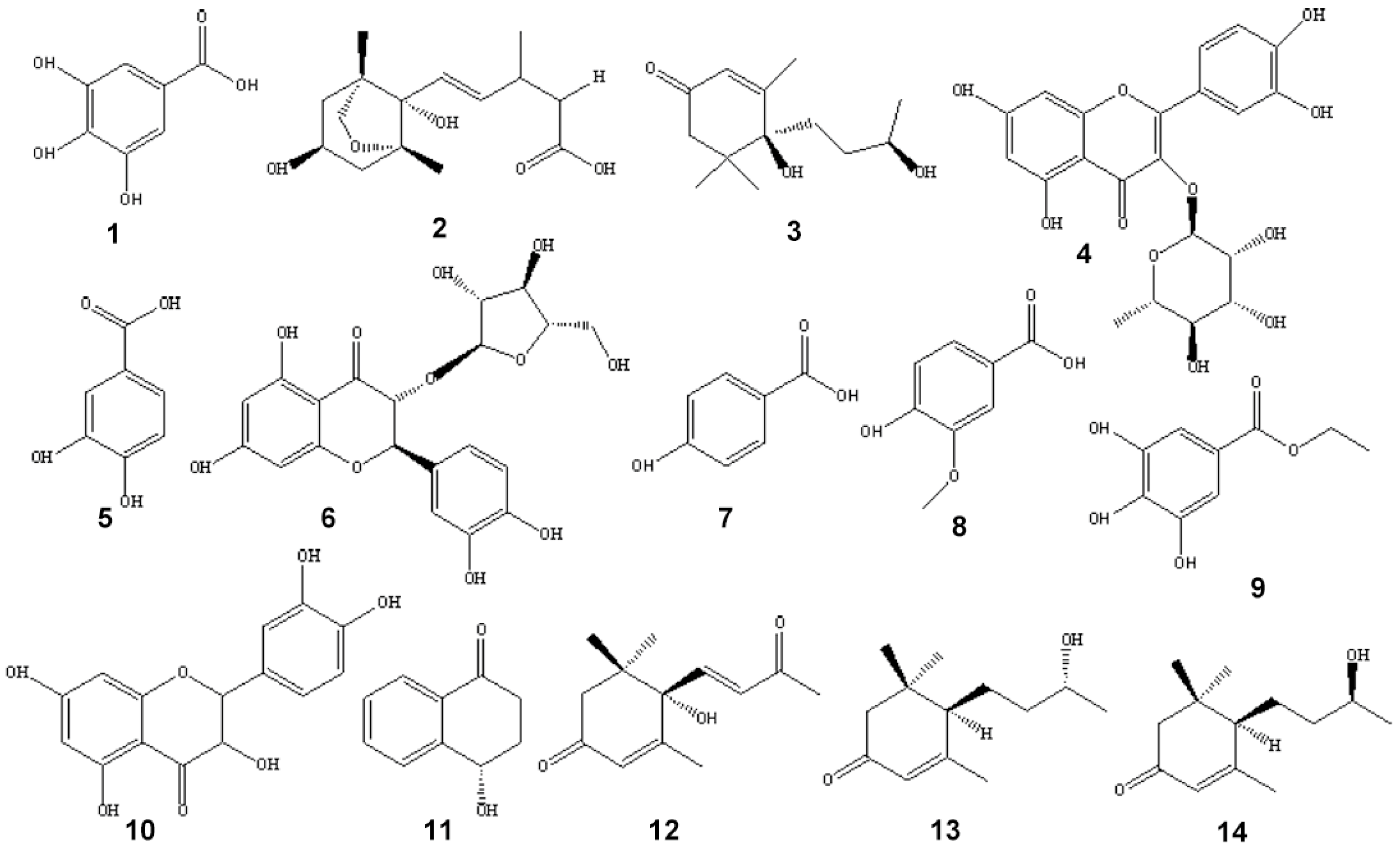
2.2. Structural Identification
2.3. The Anti-Inflammatory Activity of Isolated Compounds Based on the NO Production in LPS-Stimulated RAW 264.7 Cells
3. Materials and Methods
3.1. Materials and Reagents
3.2. Apparatus
3.3. Preparation of the Crude Extracts
3.4. Separation and Purification of the Ethyl Acetate Extract
3.4.1. Solvent Selection and HSCCC Separation
3.4.2. Fractionation of the Ethyl Acetate Extract
3.4.3. Purification by Pre-HPLC
3.5. Analysis and Identification of Isolated Pure Compounds
3.6. Anti-Inflammatory Activity Assay against NO Production
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Banel, D.K.; Hu, F.B. Effects of walnut consumption on blood lipids and other cardiovascular risk factors: A meta-analysis and systematic review. Am. J. Clin. Nutr. 2009, 90, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Sun, Q.; Manson, J.E.; Willett, W.C.; Hu, F.B. Walnut consumption is associated with lower risk of type 2 diabetes in women. J. Nutr. 2013, 143, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.A.; Vasu, V.T.; Gohil, K.; Kim, H.; Khan, I.H.; Cross, C.E.; Yokoyama, W. A high-fat diet containing whole walnuts (Juglans regia L.) reduces tumour size and growth along with plasma insulin-like growth factor 1 in the transgenic adenocarcinoma of the mouse prostate model. Br. J. Nutr. 2012, 108, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Qi, Y.; Zhu, C.; Wang, Q. Purification and identification of antioxidant peptides from Chinese cherry (Prunus pseudocerasus Lindl.) seeds. J. Funct. Foods 2015, 19, 394–403. [Google Scholar] [CrossRef]
- Prasad, R.B.N. Walnuts and pecans. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press Inc.: Oxford, UK, 2003; pp. 6071–6079. [Google Scholar]
- Xiang, L.; Wang, Y.; Yi, X.; Wang, X.; He, X. Chemical constituent and antioxidant activity of the husk of Chinese hickory. J. Funct. Foods 2016, 23, 378–388. [Google Scholar] [CrossRef]
- Pereira, J.A.; Oliveira, I.; Sousa, A.; Valento, P.; Andrade, P.B.; Ferreira, I.C.F.R.; Ferreres, F.; Bento, A.; Seabra, R.; Estevinho, L. Walnut (Juglans regia L.) leaves: phenolic compounds, antimicrobial activity and antioxidant potential of different cultivars. Food Chem. Toxicol. 2007, 45, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Ferreira, I.C.F.R.; Calhelha, R.; Andrade, P.B.; Valent, P.; Seabra, R.; Estevinho, L.; Bento, A.; Pereira, J.A. Phenolics and antimicrobial activity of traditional stoned table olives “alcaparra”. Bioorg. Med. Chem. 2006, 14, 8533–8538. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.P.; Laires, A.; Gaspar, J.; Leaoa, D.; Oliveira, J.S.; Rueff, J. Genotoxicity of instant coffee: Possible involvement of phenolic compounds. Mutat. Res.-Rev. Mutat. 1999, 442, 43–51. [Google Scholar] [CrossRef]
- Rocha-Guzmán, N.E.; Herzog, A.; González-Laredo, R.F.; Ibarra-Pérez, F.J.; Zambrano, G.; Gallegos-Infante, J.A. Antioxidant and antimutagenic activity of phenolic compounds in three different colour groups of common bean cultivars (Phaseolus vulgaris). Food Chem. 2007, 103, 521–527. [Google Scholar] [CrossRef]
- Pietta, P.; Gardana, C.; Pietta, A. Flavonoids in herbs. In Flavonoids in Health and Disease; Packer, L., Cadenas, E., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2003; pp. 43–71. [Google Scholar]
- Meng, Q.; Li, Y.; Xiao, T.; Zhang, L.; Xu, D. Antioxidant and antibacterial activities of polysaccharides isolated and purified from Diaphragma juglandis fructus. Int. J. Biol. Macromol. 2017, 105, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E. The role of cytokines, chemokines, and adhesion molecules in the pathogenesis of idiopathic inflammatory myopathies. Curr. Rheumatol. Rep. 2000, 2, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Yoon, W.J.; Kim, K.N.; Ahn, G.N.; Kang, S.M.; Kang, D.H.; Affan, A.; Oh, C.; Jung, W.K.; Jeon, Y.J. Evaluation of anti-inflammatory effect of fucoxanthin isolated from brown algae in lipopolysaccharide-stimulated RAW 264.7 macrophages. Food Chem. Toxicol. 2010, 48, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.C.; Kuo, C.D. Pro-inammatory effects of commercial alpha-lactalbumin on RAW 264.7 macrophages is due to endotoxin contamination. Food Chem. Toxicol. 2010, 48, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.J.; Wang, M.L.; Wang, D.J.; Duan, W.J.; Wang, X.; Zheng, C.C. Preparative separation of alkaloids from Nelumbo nucifera leaves by conventional and pH-zone-refining counter-current chromatography. J. Chromatogr. B 2010, 878, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Ya, Q.K.; Chen, J.Y.; Liu, B.M. A new flavonol glycoside from Baeckea Frutescens L. Acta Pharm. Sin. 2008, 43, 1032–1035. [Google Scholar]
- Abdelhady, M.I.S.; Kamal, A.M.; Tawfik, N.F.; Abdelkhalik, S.M. Polyphenolic constituents of the methanolic extract of Callistemon viridiflorous leaves and its antimicrobial activity. Pharmacogn. J. 2012, 4, 47–53. [Google Scholar] [CrossRef]
- Ye, G.; Ma, C.H.; Huang, X.Y.; Li, Z.X.; Huang, C.G. Components of Sophora alopecuroides seeds. Chem. Nat. Compd. 2009, 45, 545–546. [Google Scholar] [CrossRef]
- Quan, L.; Chen, C.J.; Xiang, S.; Li, Z.; Chen, H.J.; Gao, K. Chemical constituents from Aphanamixis grandifolia. Chem. Pharm. Bull. 2010, 58, 1431–1435. [Google Scholar]
- Li, Y.L.; Li, J.; Wang, N.L.; Yao, X.S. Flavonoids and a new polyacetylene from Bidens parviflora Willd. Molecules 2008, 13, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, N.H.; Salim, F.; Ahmad, R. Chemical constituents of Malaysian U. cordata var. ferruginea and their in vitro α-glucosidase inhibitory activities. Molecules 2016, 21, 525. [Google Scholar]
- Lee, J.C. Flavonoid fraction purified from Rhus verniciflua Stokes actively inhibits cell growth via induction of apoptosis in mouse tumorigenic hepatocytes. Nat. Prod. Sci. 2004, 29, 2032–2034. [Google Scholar]
- Hernandez, C.L.C.; Villaseñor, I.M.; Joseph, E.; Tolliday, N. Isolation and evaluation of antimitotic activity of phenolic compounds from Pouteria campechiana Baehni. Philipp. J. Sci. 2008, 137, 1–10. [Google Scholar]
- Zhao, H.X.; Bai, H.; Jing, Y.C.; Li, W.; Yin, S.J.; Zhou, H.L. A pair of taxifolin-3-O-arabinofuranoside isomers from Juglans regia L. Nat. Prod. Res. 2017, 31, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, Q.Y.; Huang, S.Z.; Guo, Z.K.; Dai, H.F.; Zhao, Y.X. Chemical constituents from Ganoderma hainanense. J. Trop. Subtrop. Bot. 2014, 22, 190–194. [Google Scholar]
- Ren, Q.; Chen, W.; Zhao, H.; Wu, Z.; Zhang, H. Organic acids from Capparis spinosa fruit. Chem. Nat. Compd. 2012, 48, 868–869. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sang, U.C.; Lee, J.H.; Dong, U.L.; Kang, R.L. A new phenylpropane glycoside from the rhizome of Sparganium stoloniferum. Arch. Pharm. Res. 2010, 33, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, S.; Suphapong, S.; Worachartcheewan, A.; Lawung, R.; Ruchirawat, S.; Prachayasittikul, V. Bioactive metabolites from Spilanthes acmella Murr. Molecules 2009, 14, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.; Lu, Y.; Ping, G.; Shao, Q.; Wei, Y. Isolation of four phenolic compounds from Mangifera indica L. flowers by using normal phase combined with elution extrusion two-step high speed counter-current chromatography. J. Chromatogr. B 2017, 1046, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Mao, Y.; Wang, N.L.; Yao, X.S. A new phloroglucinol diglycoside derivative from Hypericum japonicum Thunb. Molecules 2008, 13, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Yang, B.Y.; Jiang, Y.Q.; Liu, Z.X.; Liu, Y.X.; Wang, X.L.; Kuang, H.X. Studies on cytotoxic activity against HepG-2 cells of naphthoquinones from green walnut husks of Juglans mandshurica Maxim. Molecules 2015, 20, 15572–15588. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lee, S.; Park, S.Y. A new monoterpene from the flower buds of Buddleja officinalis. Nat. Prod. Sci. 2013, 19, 355–359. [Google Scholar]
- Jing, W.G. Studies on Chemical Constituents and Bioactivity of the Rapeseed Meal (Brassica napus L.). Ph.D. Thesis, China Academy of Chinese Medical Sciences, Beijing, China, 2014. [Google Scholar]
- Pan, L.; Acuña, U.M.; Li, J.; Jena, N.; Ninh, T.N.; Pannell, C.M.; Chai, H. Bioactive flavaglines and other constituents isolated from Aglaia perviridis. J. Nat. Prod. 2013, 76, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.G.; Wu, G.Z.; Fang, X.; Tian, X.H.; Dong, H.Y.; Xu, X.K.; Li, H.L.; Xie, N.; Zhang, W.D.; Shen, Y.H. Chlorajaponols A–F, sesquiterpenoids from Chloranthus japonicus and their in vitro anti-inflammatory and anti-tumor activities. Fitoterapia 2017, 119, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Song, X.; Wang, D.; Wang, X.; Wang, X. Five new chromone glycosides from Scindapsus officinalis (Roxb.) Schott. Fitoterapia 2017, 122, 101–106. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the lignans and lignan glycosides compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | NO Production Relative to LPS Group (Mean ± SD) | |
|---|---|---|
| Control (DMSO) | 0.23 ± 0.03 | |
| LPS (1 μg/mL) | 1 ± 0 | |
| Compounds (50 μM) | 1 | 0.59 ± 0.05 |
| 2 | 0.97 ± 0.07 | |
| 3 | 0.93 ± 0.07 | |
| 4 | 0.95 ± 0.06 | |
| 5 | 0.63 ± 0.09 | |
| 6 | 0.99 ± 0.08 | |
| 7 | 0.61 ± 0.1 | |
| 8 | 0.81 ± 0.04 | |
| 9 | 0.57 ± 0.04 | |
| 10 | 0.92 ± 0.03 | |
| 11 | 0.72 ± 0.03 | |
| 12 | 0.62 ± 0.05 | |
| 13 | 0.98 ± 0.05 | |
| 14 | 0.86 ± 0.07 | |
| Group | NO Concentration (μM) (Mean ± SD) | ||
|---|---|---|---|
| Control (DMSO) | 3.13 ± 0.67 | ||
| LPS (1 μg/mL) | 38.47 ± 0.82 | ||
| Compounds | 1 | 0.5 μM | 28.05 ± 1.87 |
| 2.5 μM | 27.78 ± 1.79 | ||
| 5 μM | 26.28 ± 1.72 | ||
| 25 μM | 25.20 ± 1.41 | ||
| 50 μM | 21.45 ± 1.71 | ||
| 9 | 0.5 μM | 40.16 ± 2.05 | |
| 2.5 μM | 35.60 ± 1.73 | ||
| 5 μM | 28.25 ± 2.22 | ||
| 25 μM | 23.23 ± 1.93 | ||
| 50 μM | 22.3 ± 1.44 | ||
| 12 | 0.5 μM | 37.01 ± 1.34 | |
| 2.5 μM | 33.69 ± 1.86 | ||
| 5 μM | 32.31 ± 2.01 | ||
| 25 μM | 26.47 ± 2.10 | ||
| 50 μM | 22.97 ± 1.95 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Mu, Y.; Dong, H.; Yan, H.; Hao, C.; Wang, X.; Zhang, L. Chemical Constituents of the Ethyl Acetate Extract from Diaphragma juglandis Fructus and Their Inhibitory Activity on Nitric Oxide Production In Vitro. Molecules 2018, 23, 72. https://doi.org/10.3390/molecules23010072
Wang D, Mu Y, Dong H, Yan H, Hao C, Wang X, Zhang L. Chemical Constituents of the Ethyl Acetate Extract from Diaphragma juglandis Fructus and Their Inhibitory Activity on Nitric Oxide Production In Vitro. Molecules. 2018; 23(1):72. https://doi.org/10.3390/molecules23010072
Chicago/Turabian StyleWang, Dan, Yan Mu, Hongjing Dong, Huijiao Yan, Cui Hao, Xiao Wang, and Lisi Zhang. 2018. "Chemical Constituents of the Ethyl Acetate Extract from Diaphragma juglandis Fructus and Their Inhibitory Activity on Nitric Oxide Production In Vitro" Molecules 23, no. 1: 72. https://doi.org/10.3390/molecules23010072