Identification of Inhibitors Targeting Ferredoxin-NADP+ Reductase from the Xanthomonas citri subsp. citri Phytopathogenic Bacteria

 , ,
, , 
Abstract
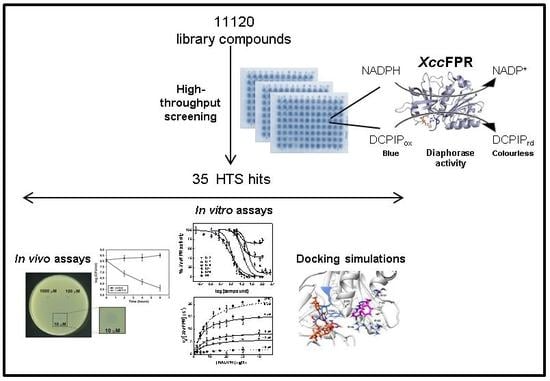
:
1. Introduction
2. Results and Discussion
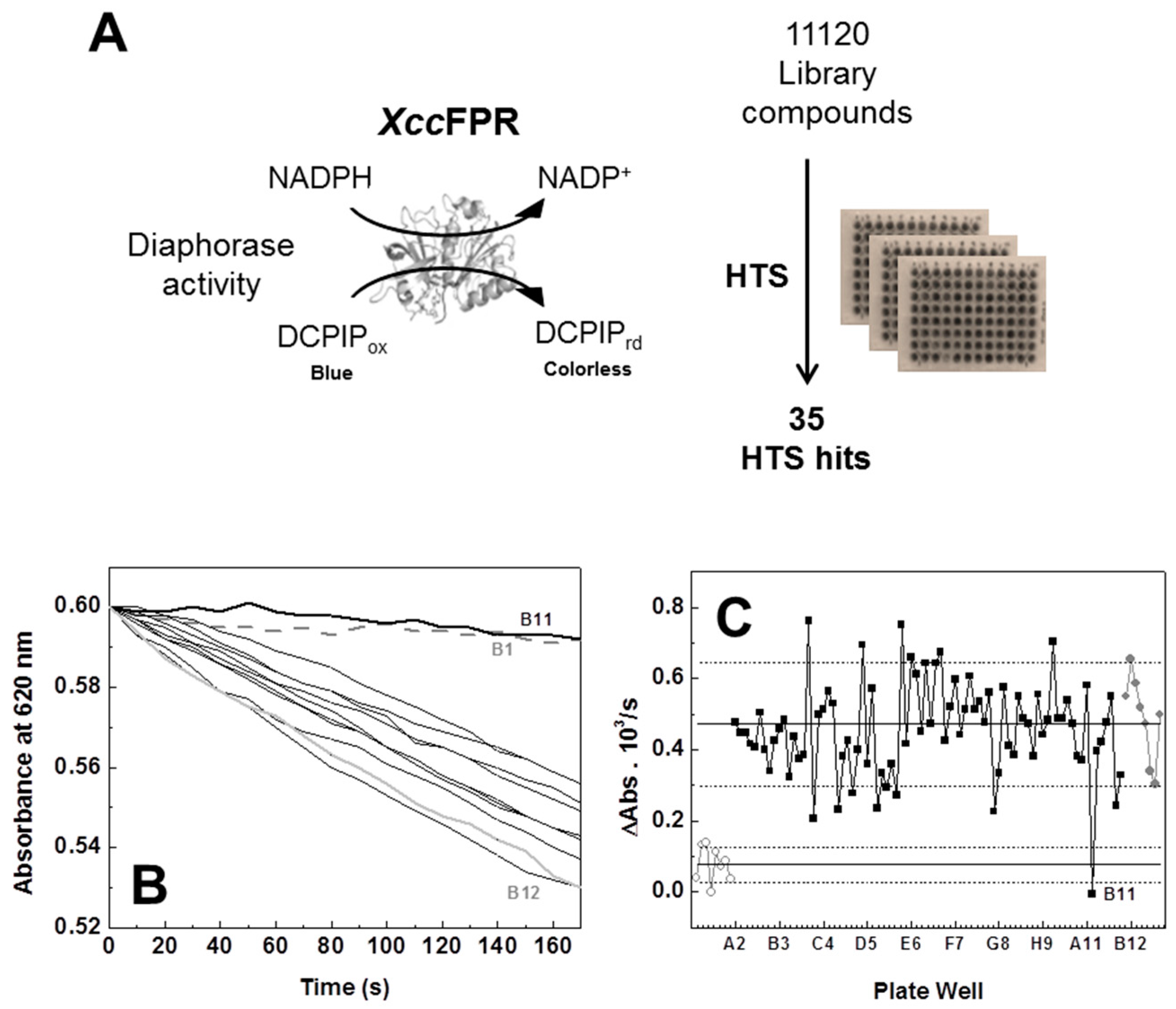
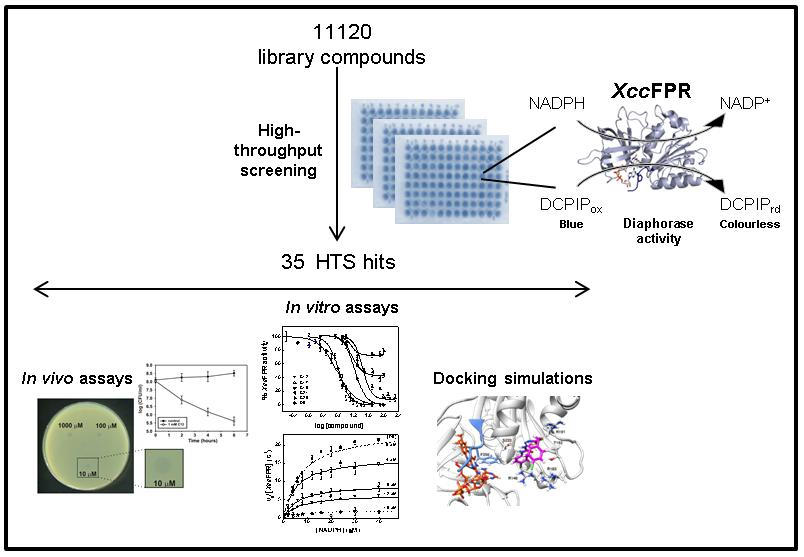
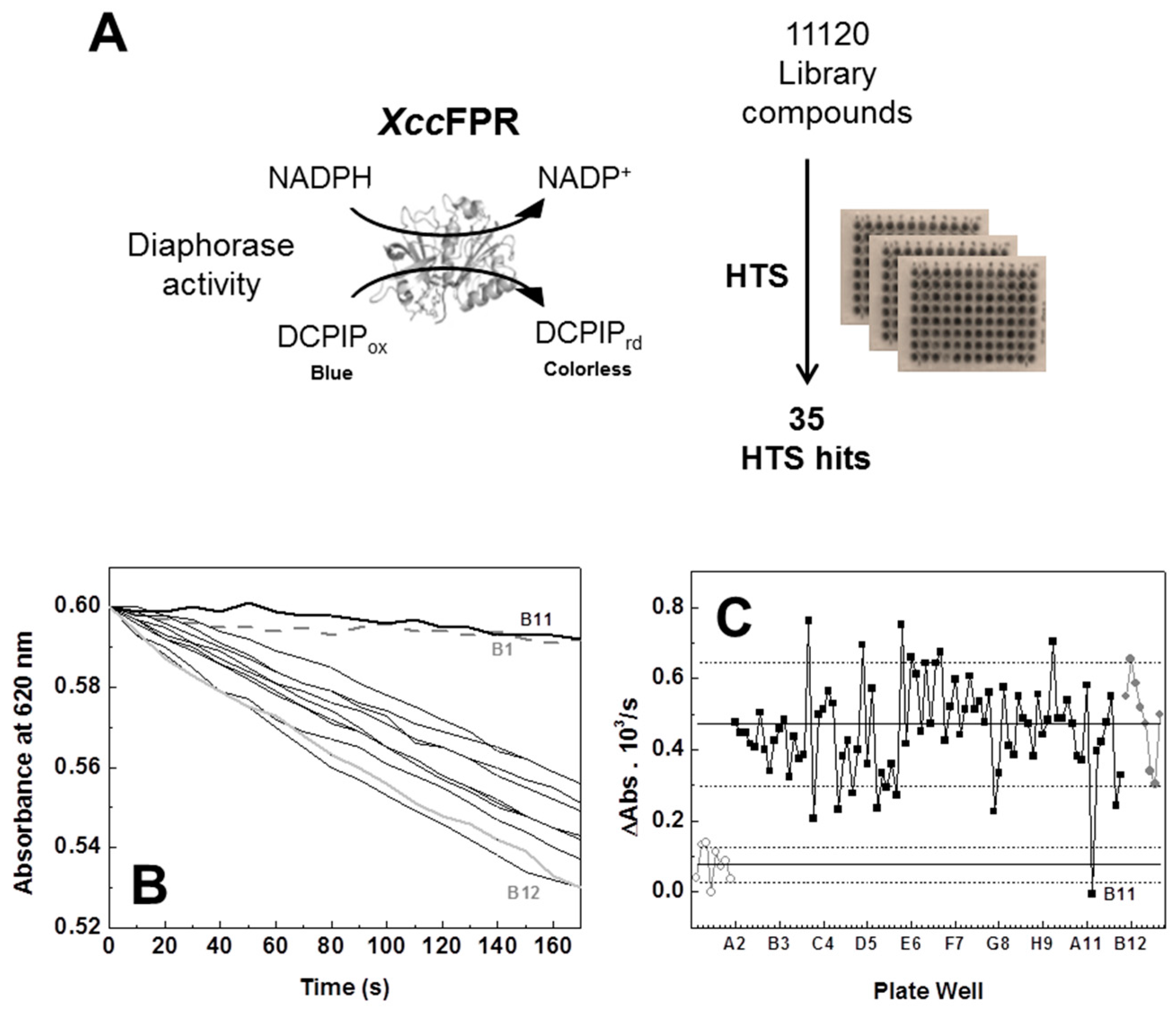
2.1. An XccFPR Activity Based High-Throughput Screening to Discover Xcc Antimicrobials
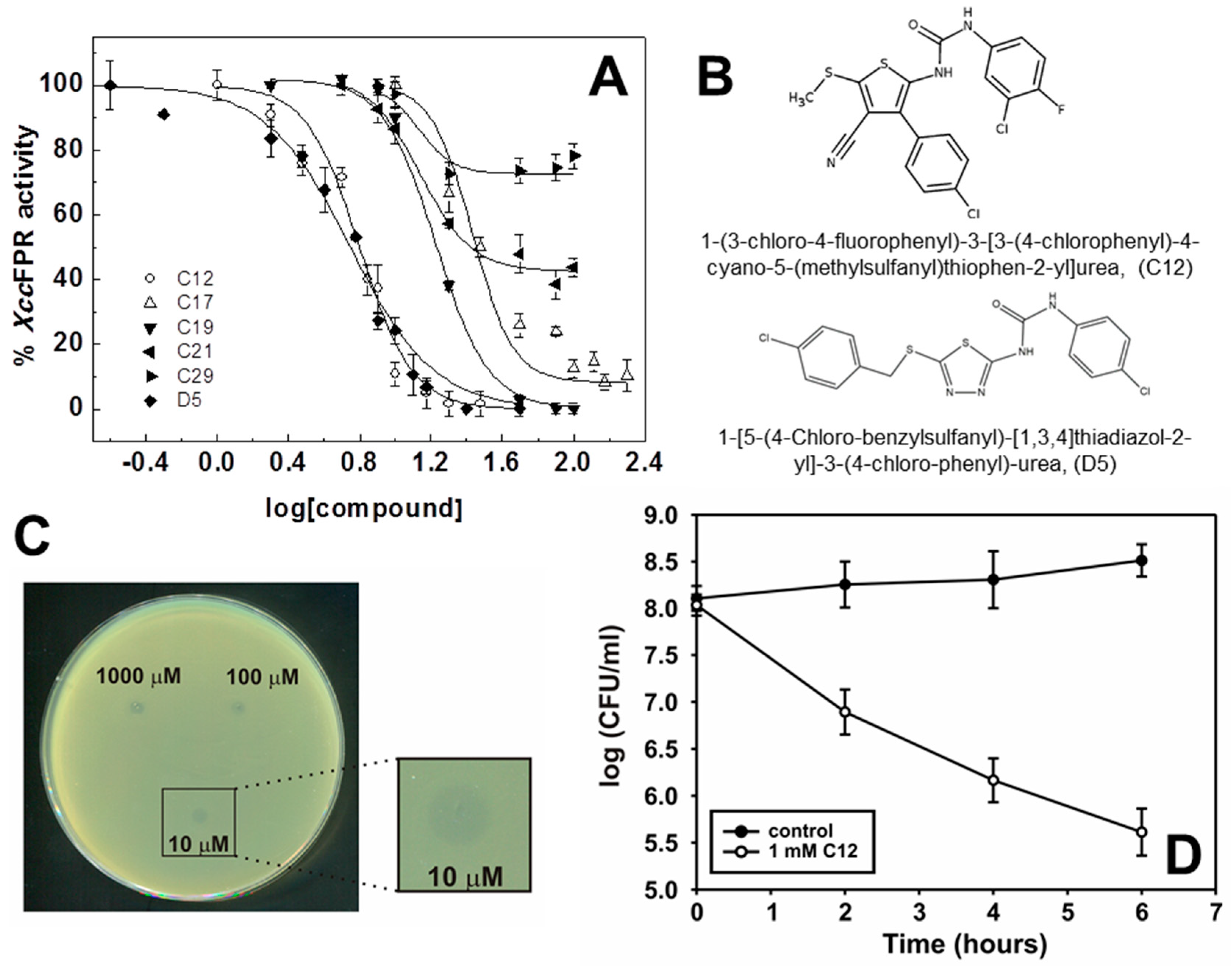
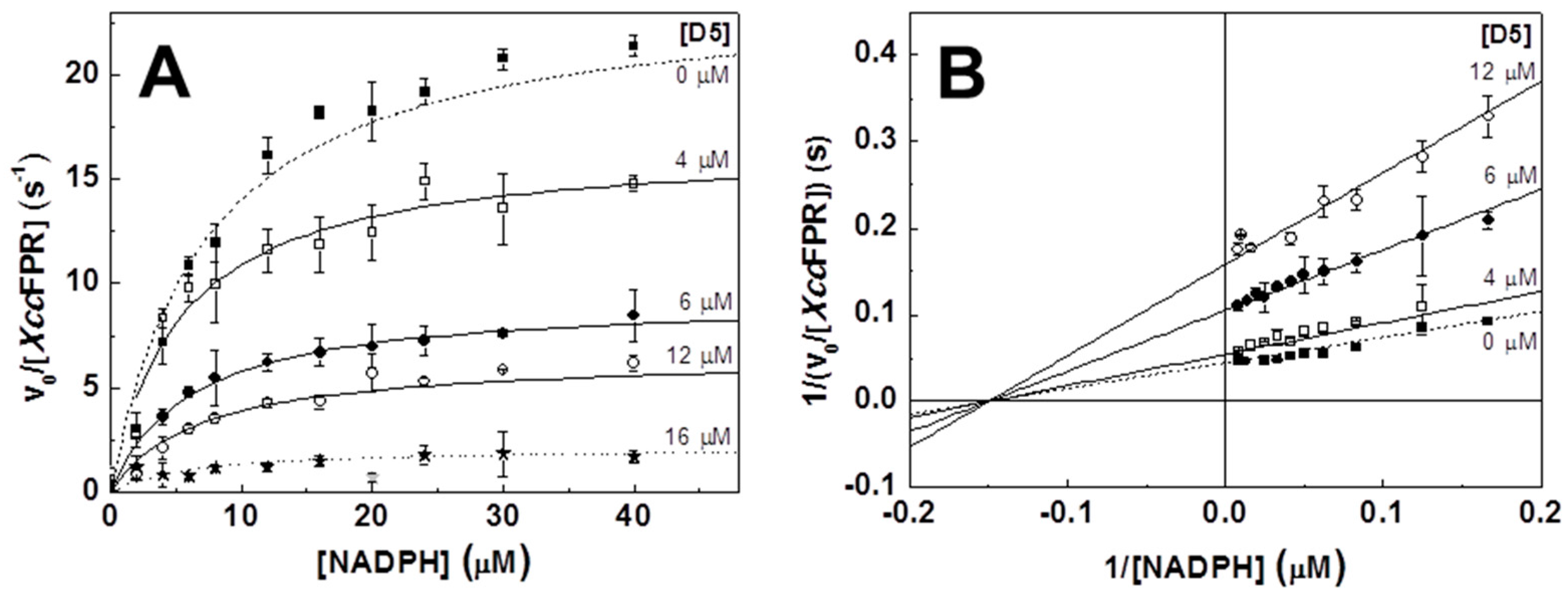
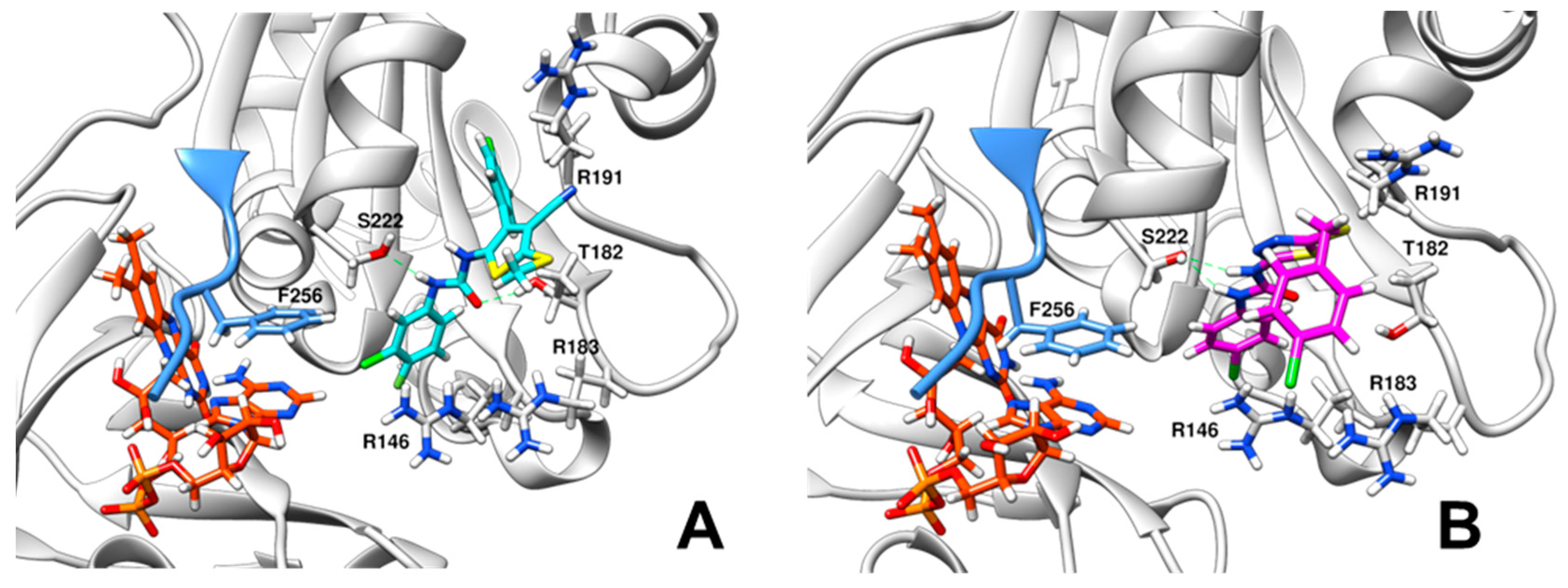
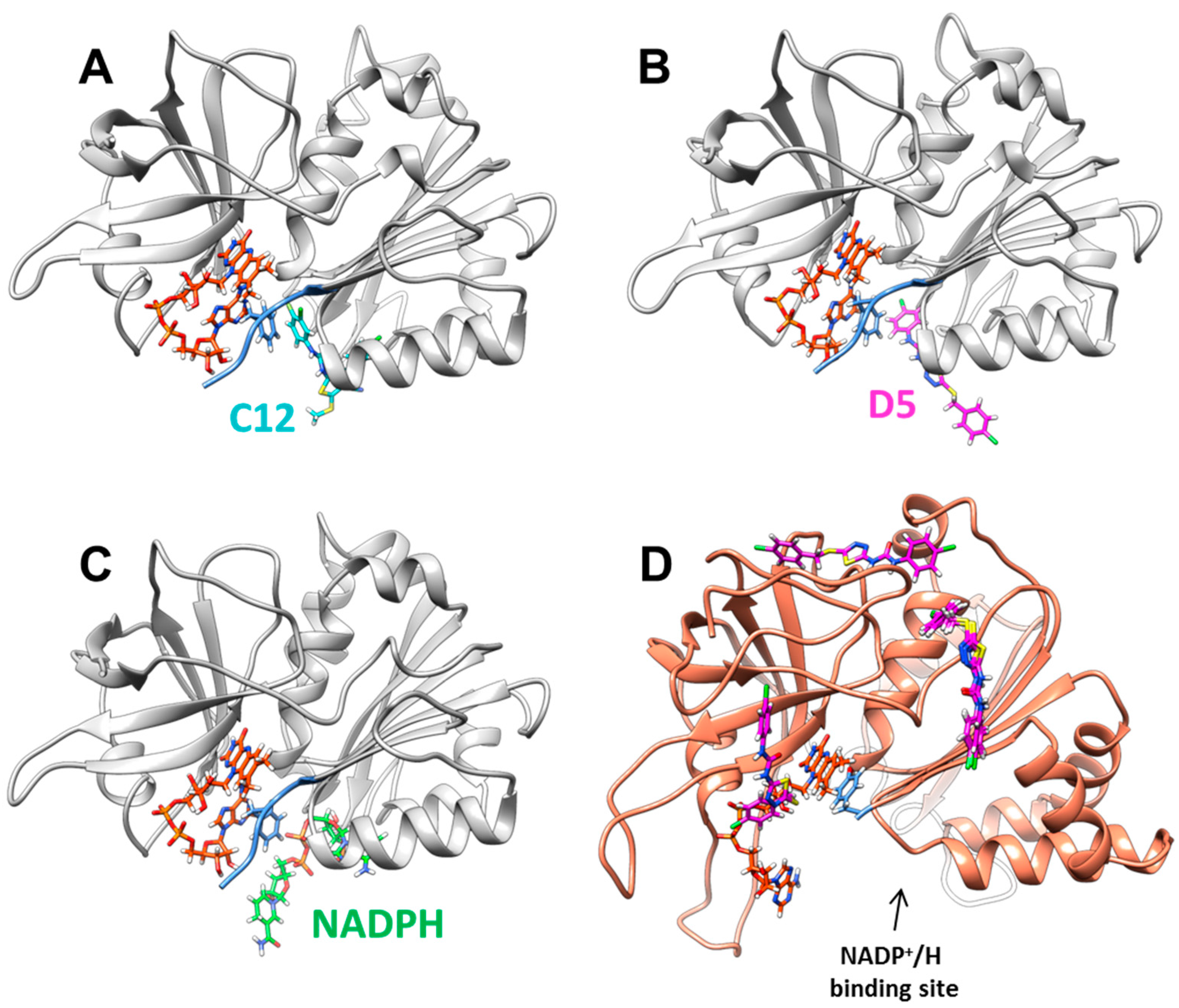
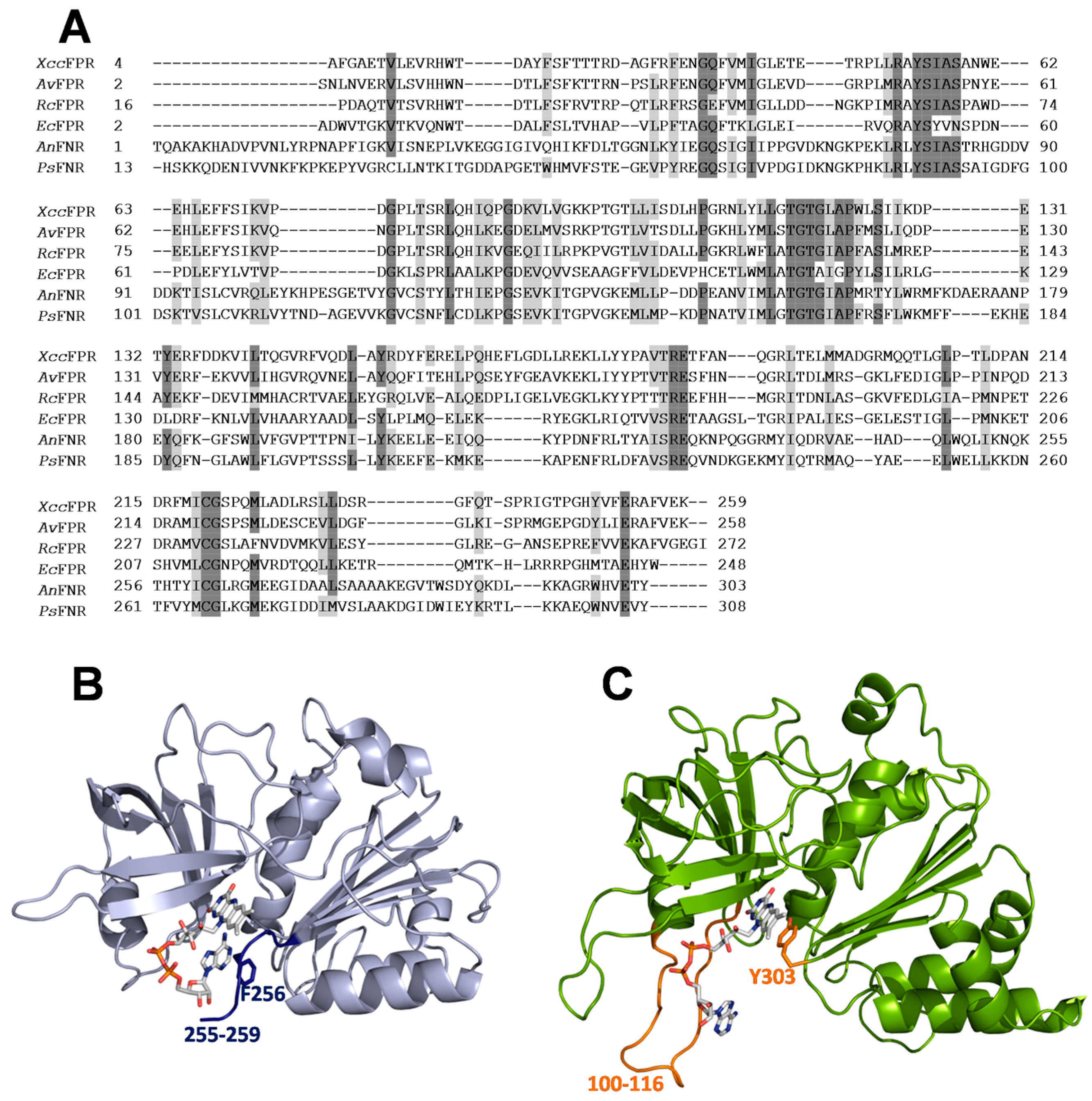
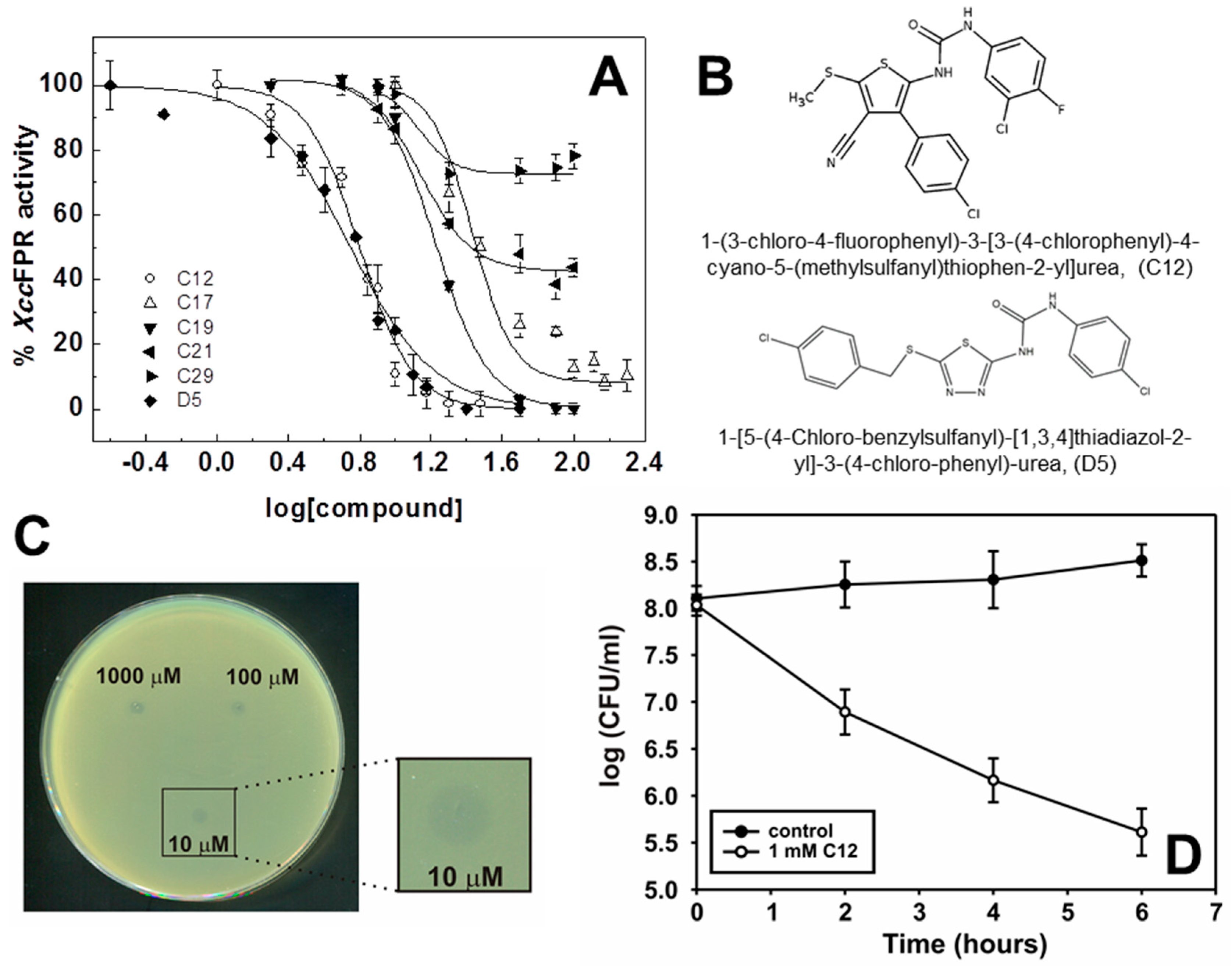
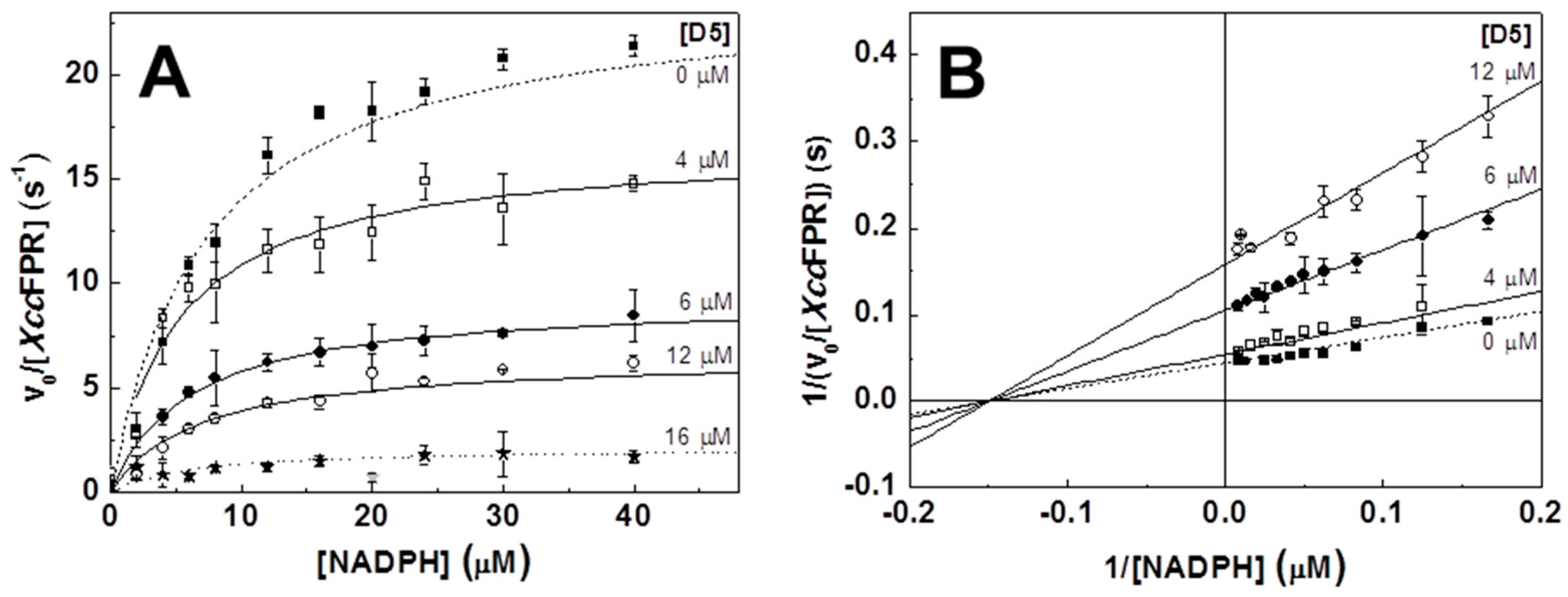
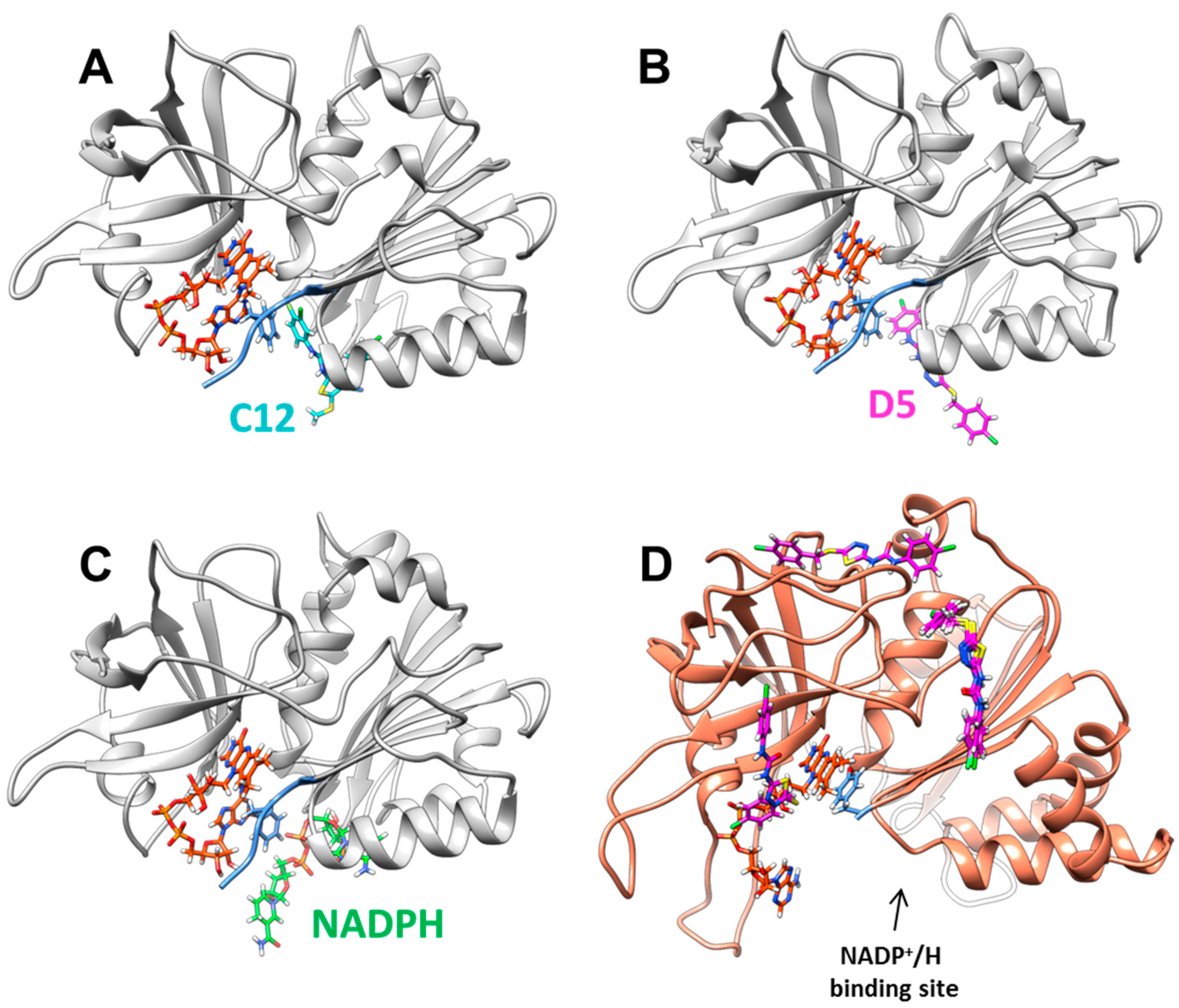
2.2. Inhibition and Binding Mechanisms of XccFPR Inhibitors
3. Materials and Methods
3.1. Biological Material and Chemicals
3.2. High-Throughput Screening of Chemical Libraries against XccFPR
3.3. Cell Viability Assays
3.4. Analysis of the Effect of Compounds on the Thermal Stability of XccFPR
3.5. Steady-State Kinetic Parameters of XccFPR and Inhibitory Effect of Selected Hits
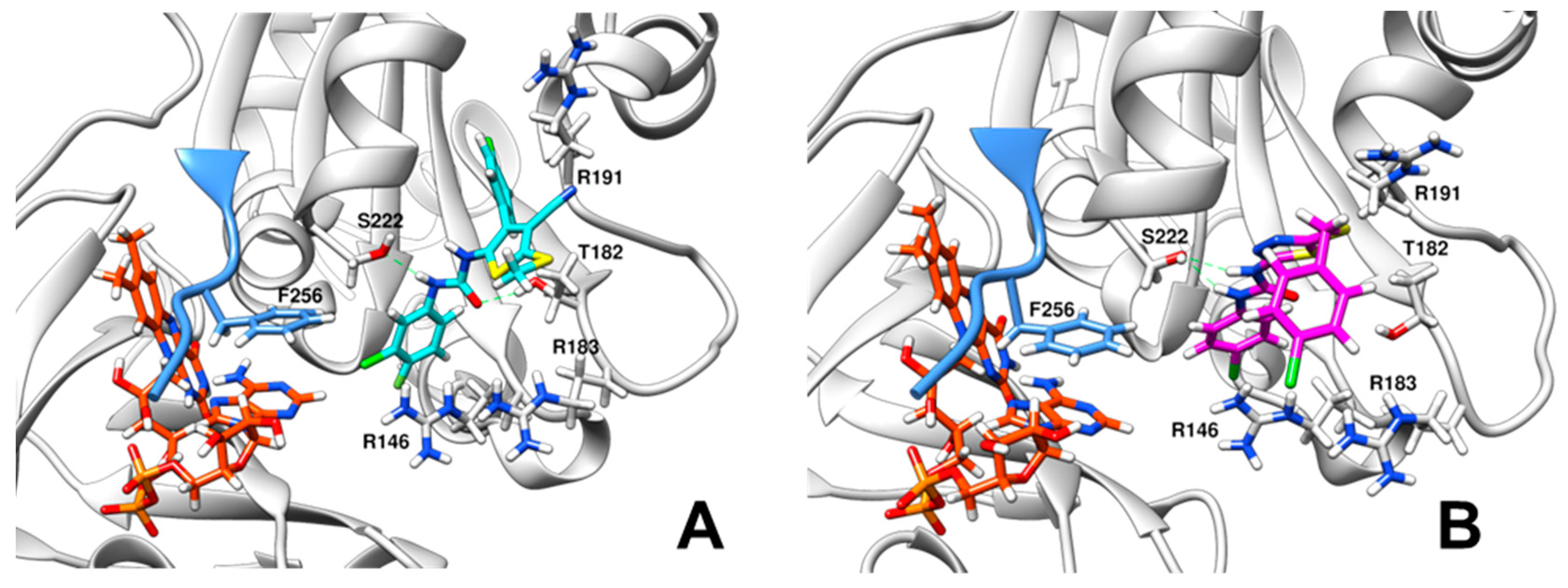
3.6. Docking of Compounds C12 and D5 to XccFPR
3.7. Data Analysis and Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Graham, J.H.; Gottwald, T.R.; Cubero, J.; Achor, D.S. Xanthomonas axonopodis pv. citri: Factors affecting successful eradication of citrus canker. Mol. Plant Pathol. 2004, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brunings, A.M.; Gabriel, D.W. Xanthomonas citri: Breaking the surface. Mol. Plant Pathol. 2003, 4, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, R.; Russi, P.; Mara, P.; Mara, H.; Peyrou, M.; de León, I.P.; Gaggero, C. Xanthomonas axonopodis pv. citri enters the VBNC state after copper treatment and retains its virulence. FEMS Microbiol. Lett. 2009, 298, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Paget, M.S. Bacterial redox sensors. Nat. Rev. Microbiol. 2004, 2, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.K.; Andrio, E.; Marino, D.; Pauly, N.; Dunand, C. Reactive oxygen species during plant-microorganism early interactions. J. Integr. Plant Biol. 2010, 52, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.A.; Jones, J.D.; Dangl, J.L. Reactive oxygen species signaling in response to pathogens. Plant Physiol. 2006, 141, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Aliverti, A.; Pandini, V.; Pennati, A.; de Rosa, M.; Zanetti, G. Structural and functional diversity of ferredoxin-NADP+ reductases. Arch. Biochem. Biophys. 2008, 474, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, A.K.; Ceccarelli, E.A.; Carrillo, N. Plant-type ferredoxin-NADP+ reductases: A basal structural framework and a multiplicity of functions. FASEB J. 1997, 11, 133–140. [Google Scholar] [PubMed]
- Krapp, A.R.; Rodriguez, R.E.; Poli, H.O.; Paladini, D.H.; Palatnik, J.F.; Carrillo, N. The flavoenzyme ferredoxin (flavodoxin)-NADP(H) reductase modulates NADP(H) homeostasis during the soxRS response of Escherichia coli. J. Bacteriol. 2002, 184, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Peña-Llopis, S.; Lee, Y.; Demple, B. Regulation of superoxide stress in Pseudomonas putida KT2440 is different from the SoxR paradigm in Escherichia coli. Biochem. Biophys. Res. Commun. 2006, 341, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Tondo, M.L.; Musumeci, M.A.; Delprato, M.L.; Ceccarelli, E.A.; Orellano, E.G. Structural-functional characterization and physiological significance of ferredoxin-NADP+ reductase from Xanthomonas axonopodis pv. citri. PLoS ONE 2011, 6, e27124. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Azqueta, A.; Catalano-Dupuy, D.L.; Lopez-Rivero, A.; Tondo, M.L.; Orellano, E.G.; Ceccarelli, E.A.; Medina, M. Dynamics of the active site architecture in plant-type ferredoxin-NADP(+) reductases catalytic complexes. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Razquin, P.; Fillat, M.F.; Schmitz, S.; Stricker, O.; Bohme, H.; Gómez-Moreno, C.; Peleato, M.L. Expression of ferredoxin-NADP+ reductase in heterocysts from Anabaena sp. Biochem. J. 1996, 316 Pt 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Medina, M. Structural and mechanistic aspects of flavoproteins: Photosynthetic electron transfer from photosystem I to NADP(+). FEBS J. 2009, 276, 3942–3958. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Gomez-Moreno, C. Interaction of ferredoxin-NADP(+) reductase with its substrates: Optimal interaction for efficient electron transfer. Photosynth. Res. 2004, 79, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Nogues, I.; Perez-Dorado, I.; Frago, S.; Bittel, C.; Mayhew, S.; Gomez-Moreno, C.; Hermoso, J.; Medina, M.; Cortez, N.; Carrillo, N. The ferredoxin-NADP(H) reductase from Rhodobacter capsulatus: Molecular structure and catalytic mechanism. Biochemistry 2005, 44, 11730–11740. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, E.A.; Arakaki, A.K.; Cortez, N.; Carrillo, N. Functional plasticity and catalytic efficiency in plant and bacterial ferredoxin-NADP(H) reductases. Biochim. Biophys. Acta 2004, 1698, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tondo, M.L.; Hurtado-Guerrero, R.; Ceccarelli, E.A.; Medina, M.; Orellano, E.G.; Martinez-Julvez, M. Crystal Structure of the FAD-Containing Ferredoxin-NADP(+) Reductase from the Plant Pathogen Xanthomonas axonopodis pv. citri. Biomed. Res. Int. 2013, 2013, 906572. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, A.; Pérez-Dorado, I.; Goñi, G.; Medina, M.; Hermoso, J.A.; Carrillo, N.; Cortez, N. Coenzyme binding and hydride transfer in Rhodobacter capsulatus ferredoxin/flavodoxin NADP(H) oxidoreductase. Biochim. Biophys. Acta 2009, 1794, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zeng, Y.; Han, H.; Weeratunga, S.; Morgan, B.N.; Moenne-Loccoz, P.; Schonbrunn, E.; Rivera, M. Biochemical and structural characterization of Pseudomonas aeruginosa Bfd and FPR: Ferredoxin NADP+ reductase and not ferredoxin is the redox partner of heme oxygenase under iron-starvation conditions. Biochemistry 2007, 46, 12198–12211. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, A.; Sánchez-Azqueta, A.; Maya, C.M.; Velázquez-Campoy, A.; Hermoso, J.A.; Medina, M.; Cortez, N. The C-terminal extension of bacterial flavodoxin-reductases: Involvement in the hydride transfer mechanism from the coenzyme. Biochim. Biophys. Acta 2014, 1837, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Kita, Y.; Timasheff, S.N. Protein precipitation and denaturation by dimethyl sulfoxide. Biophys. Chem. 2007, 131, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nissink, J.W.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the assay: Chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Martinez-Julvez, M.; Hurley, J.K.; Tollin, G.; Gomez-Moreno, C. Involvement of glutamic acid 301 in the catalytic mechanism of ferredoxin-NADP(+) reductase from Anabaena PCC 7119. Biochemistry 1998, 37, 2715–2728. [Google Scholar] [CrossRef] [PubMed]
- Tondo, M.L.; Ottado, J.; Orellano, E.G. Expression, purification and characterization of the ferredoxin-NADP(H) reductase from the phytophathogen Xanthomonas axonopodis pv. citri. In Flavins and Flavoproteins; Frago, S., Medina, M., Gomez-Moreno, C., Eds.; Prensas Universitarias de Zaragoza: Zaragoza, Spain, 2008; p. 255. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–25. [Google Scholar] [CrossRef]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Orru, R.; Bonivento, D.; Chiarelli, L.R.; Mattevi, A. ThermoFAD, a Thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J. 2009, 276, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Velázquez-Campoy, A.; Martínez-Júlvez, M.; Neira, J.L.; Pérez-Dorado, I.; Hermoso, J.; Jimenez, P.; Lanas, A.; Hoffman, P.S.; Sancho, J. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem. Biol. 2009, 4, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newslett. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HTS Hit | IC50 (µM) | ICmax (µM) | % Inhibition at ICmax | HTS Hit | IC50 (µM) | ICmax (µM) | % Inhibition at ICmax |
|---|---|---|---|---|---|---|---|
| 1 | 110 ± 1 | 130 ± 1 | 84 ± 4 | 19 | 17 ± 1 | 80 ± 1 | 100 ± 1 |
| 2 | 27 ± 1 | 300 ± 1 | 95 ± 3 | 20 | 53 ± 2 | 100 ± 1 | 100 ± 1 |
| 3 | 190 ± 1 | 300 ± 3 | 31 ± 8 | 21 | 14 ± 1 | 80 ± 1 | 62 ± 7 |
| 4 | 36 ± 1 | 80 ± 1 | 86 ± 1 | 22 | 58 ± 2 | 150 ± 1 | 100 ± 4 |
| 5 | 46 ± 1 | 150 ± 1 | 98 ± 1 | 23 | >604 | >1000 | 73 ± 3 |
| 6 | >605 | >1000 | ~70 | 24 | 35 ± 1 | 120 ± 1 | 43 ± 4 |
| 7 | 85 ± 16 | 450 ± 1 | 97 ± 7 | 25 | 91 ± 1 | >500 | ~91 |
| 8 | 79 ± 1 | 250 ± 1 | 88 ± 9 | 26 | 33 ± 2 | 120 ± 1 | 94 ± 1 |
| 9 | 147 ± 1 | 250 ± 1 | 60 ± 4 | 27 | 89 ± 1 | 120 ± 1 | 92 ± 3 |
| 10 | 98 ± 1 | 150 ± 1 | 57 ± 7 | 28 | >652 | >1500 | 81 ± 2 |
| 11 | 78 ± 1 | 140 ± 1 | 92 ± 5 | 29 | 11 ± 2 | 50 ± 1 | 27 ± 2 |
| 12 | 7.7 ± 1.0 | 45 ± 1 | 100 ± 2 | 30 | 29 ± 1 | 80 ± 1 | 92 ± 2 |
| 13 | 95 ± 1 | 180 ± 1 | 80 ± 3 | 31 | 90 ± 1 | 150 ± 3 | 85 ± 1 |
| 14 | 127 ± 1 | 200 ± 1 | 85 ± 4 | 32 | >2720 | >4000 | ~56 |
| 15 | 30 ± 1 | 150 ± 1 | 93 ± 4 | 33 | >1920 | >2800 | ~53 |
| 16 | 58 ± 1 | 150 ± 1 | 79 ± 6 | 34 | 210 ± 1 | >1500 | ~87 |
| 17 | 27 ± 2 | 150 ± 1 | 91 ± 4 | 35 | 63 ± 1 | 100 ± 2 | 100 ± 1 |
| 18 | 24 ± 1 | 150 ± 1 | 100 ± 1 |
| C12 Derived Compound | IC50 (µM) | ICmax (µM) | % Inhibition at ICmax |
|---|---|---|---|
| D1 | 76 ± 1 | 200 ± 1 | 98 ± 1 |
| D2 | 26 ± 1 | 175 ± 1 | 100 ± 1 |
| D3 | >5000 | - | - |
| D4 | 113 ± 1 | 400 ± 1 | 78 ± 2 |
| D5 | 6.6 ± 1.1 | 23 ± 2 | 100 ± 2 |
| D6 | >1500 | - | - |
| D7 | 151 ± 1 | 220 ± 1 | 84 ± 1 |
| D8 | 38 ± 1 | 150 ± 1 | 100 ± 1 |
| Inhibitors | IC50 (µM) | ICmax (µM) | % Inhibition at ICmax |
|---|---|---|---|
| D2 | 27 ± 1 | 100 ± 2 | 99 ± 11 |
| D5 | 102 ± 1 | >300 | ~47 |
| D8 | 122 ± 1 | >350 | ~73 |
| Ki (μM) | Inhibition Mechanism | |
|---|---|---|
| C12 | 4.2 ± 0.9 | Non-competitive |
| D2 | 28 ± 2 | Uncompetitive |
| D5 | 4.3 ± 1.1 | Non-competitive |
| D8 | 62 ± 19 | Non-competitive |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Júlvez, M.; Goñi, G.; Pérez-Amigot, D.; Laplaza, R.; Ionescu, I.A.; Petrocelli, S.; Tondo, M.L.; Sancho, J.; Orellano, E.G.; Medina, M. Identification of Inhibitors Targeting Ferredoxin-NADP+ Reductase from the Xanthomonas citri subsp. citri Phytopathogenic Bacteria. Molecules 2018, 23, 29. https://doi.org/10.3390/molecules23010029
Martínez-Júlvez M, Goñi G, Pérez-Amigot D, Laplaza R, Ionescu IA, Petrocelli S, Tondo ML, Sancho J, Orellano EG, Medina M. Identification of Inhibitors Targeting Ferredoxin-NADP+ Reductase from the Xanthomonas citri subsp. citri Phytopathogenic Bacteria. Molecules. 2018; 23(1):29. https://doi.org/10.3390/molecules23010029
Chicago/Turabian StyleMartínez-Júlvez, Marta, Guillermina Goñi, Daniel Pérez-Amigot, Rubén Laplaza, Irina Alexandra Ionescu, Silvana Petrocelli, María Laura Tondo, Javier Sancho, Elena G. Orellano, and Milagros Medina. 2018. "Identification of Inhibitors Targeting Ferredoxin-NADP+ Reductase from the Xanthomonas citri subsp. citri Phytopathogenic Bacteria" Molecules 23, no. 1: 29. https://doi.org/10.3390/molecules23010029