Synthesis and Detailed Examination of Spectral Properties of (S) and (R)-Higenamine 4′-O-β-d-Glucoside and HPLC Analytical Conditions to Distinguish the Diastereomers

Abstract
:1. Introduction
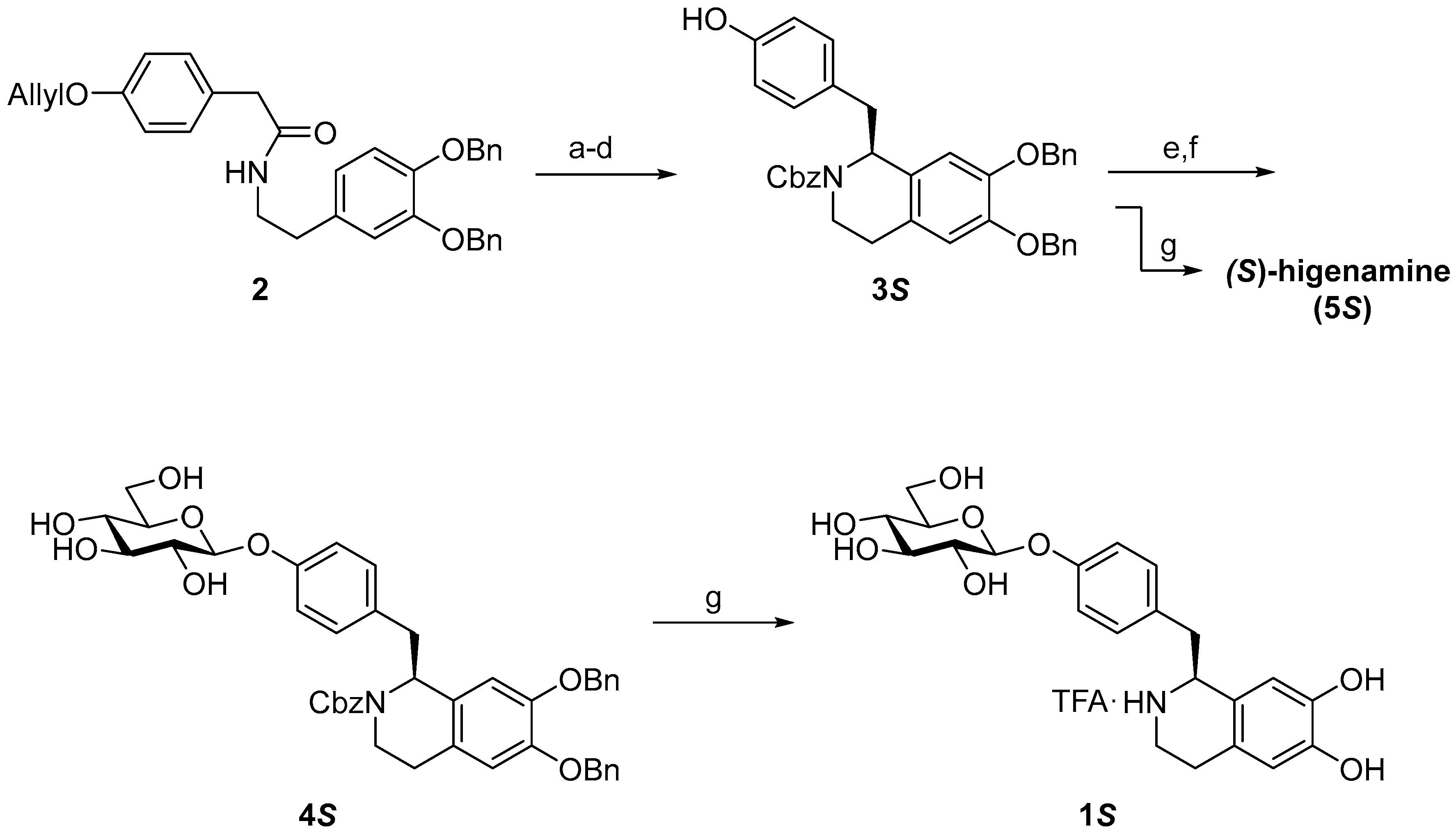
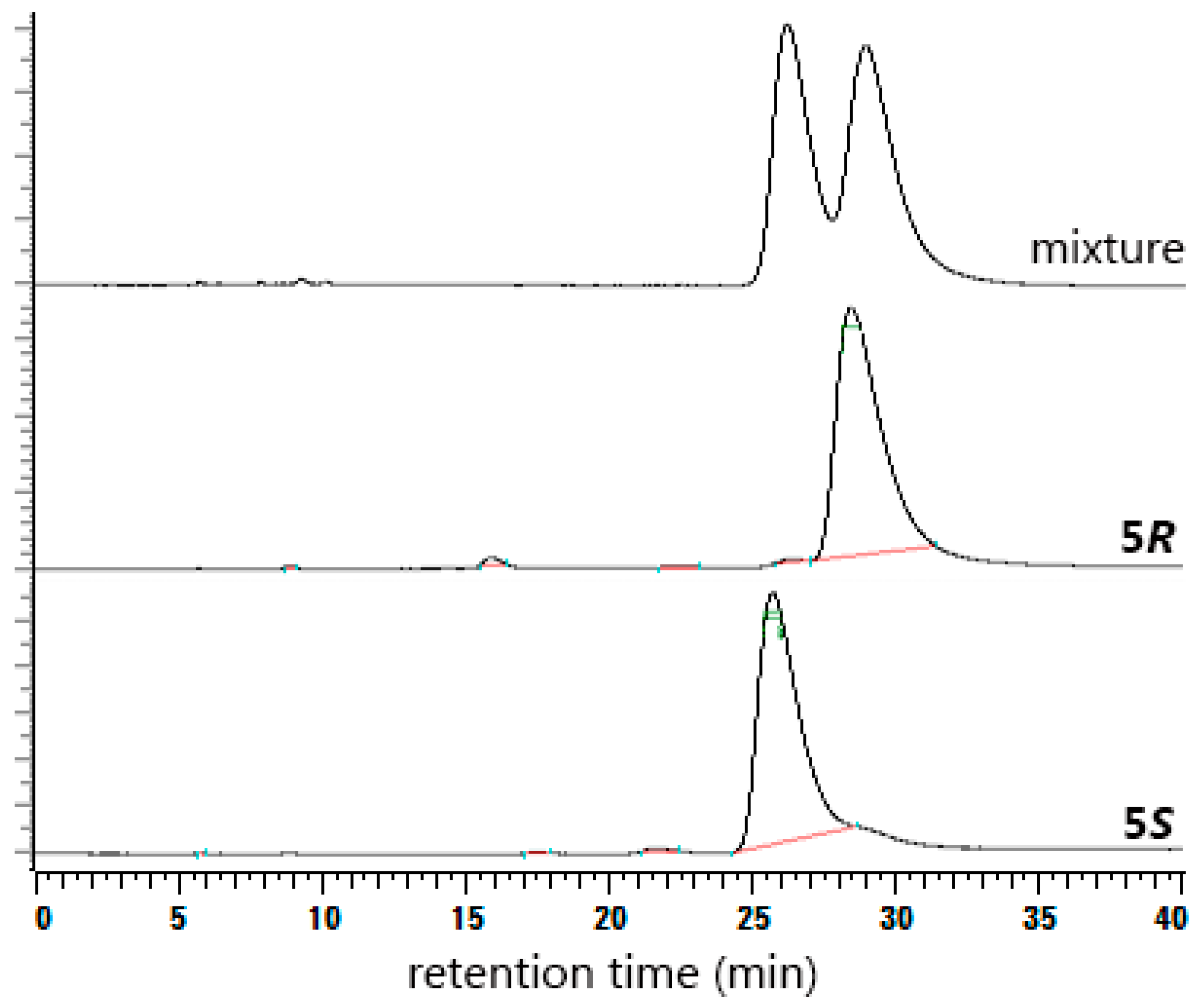
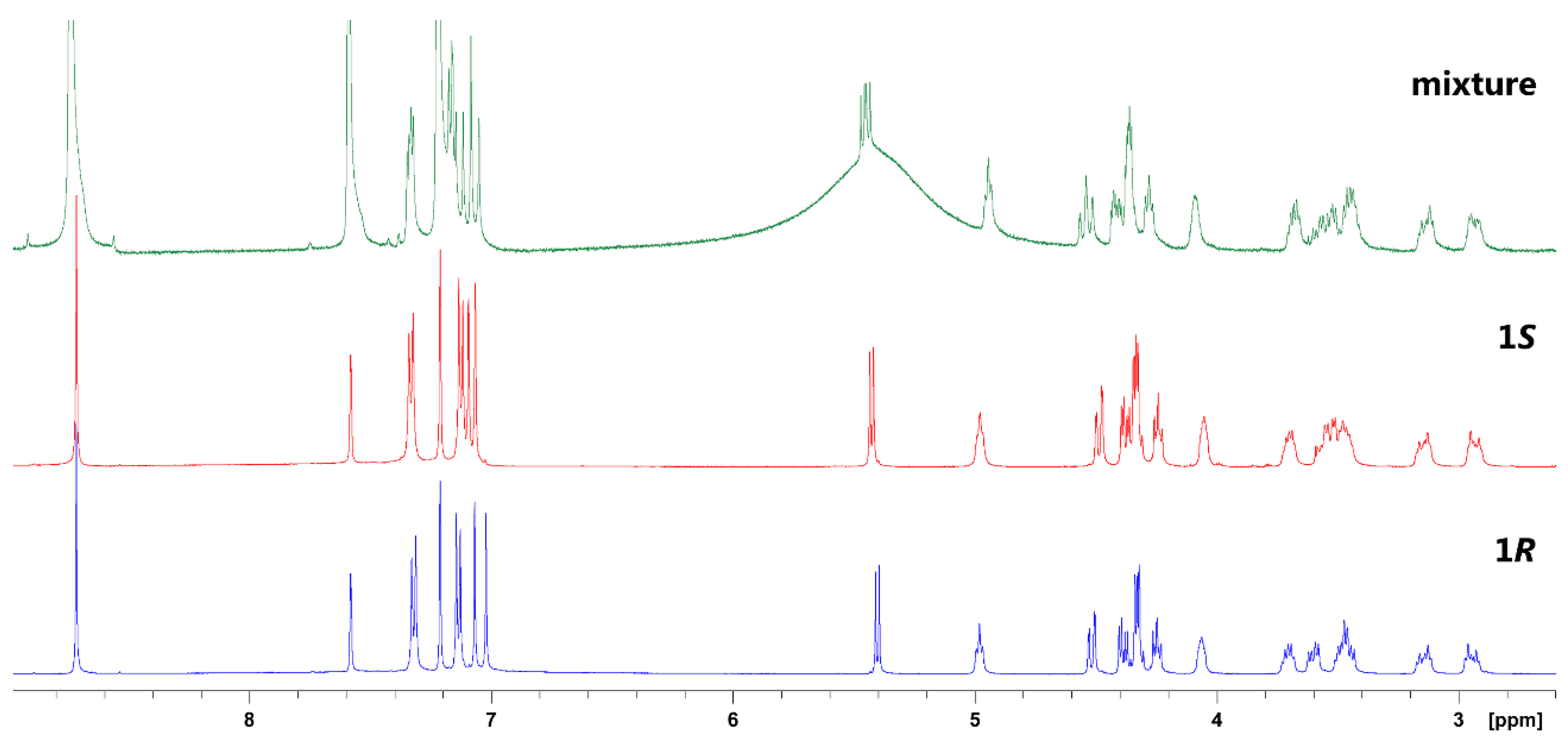
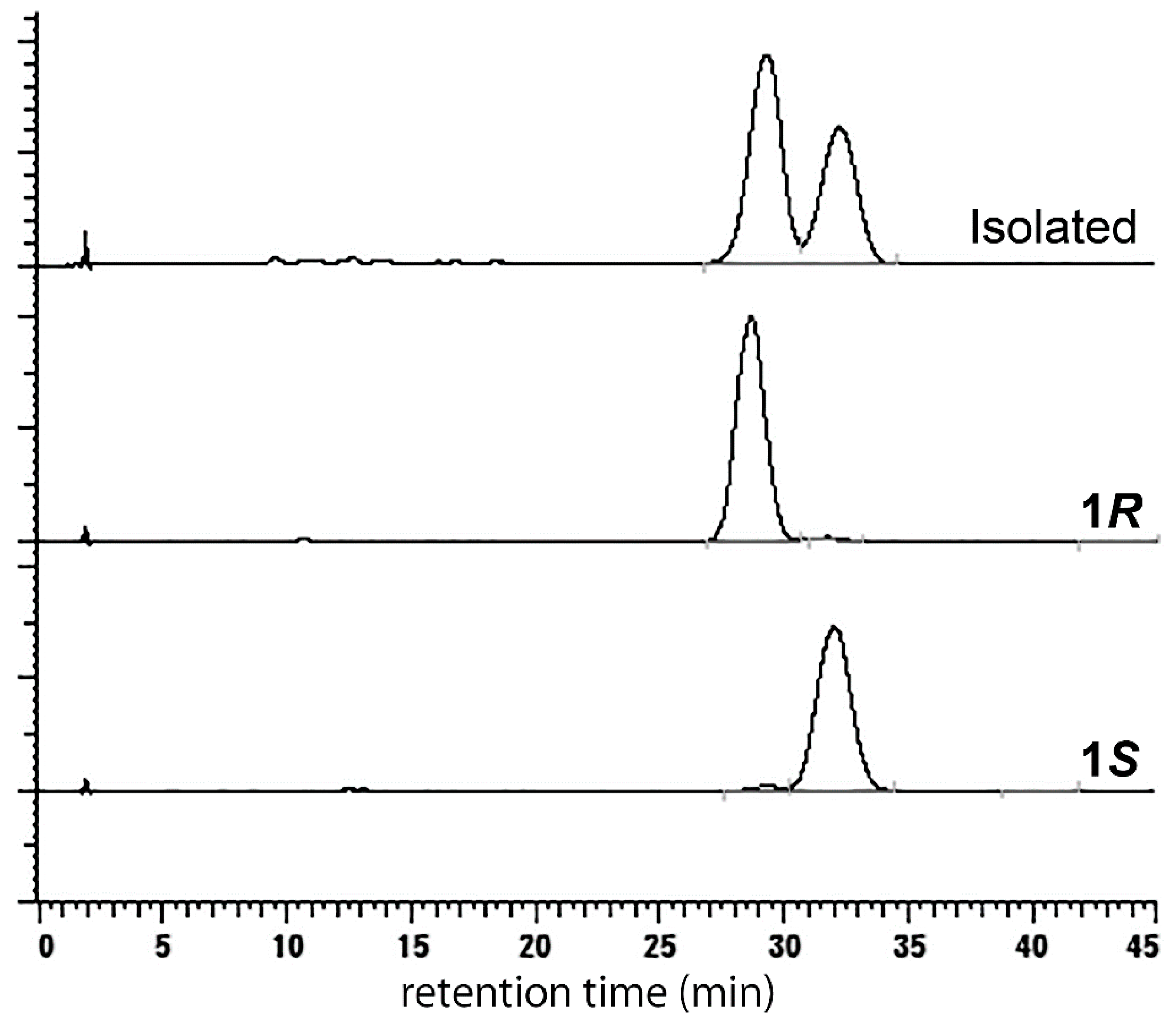
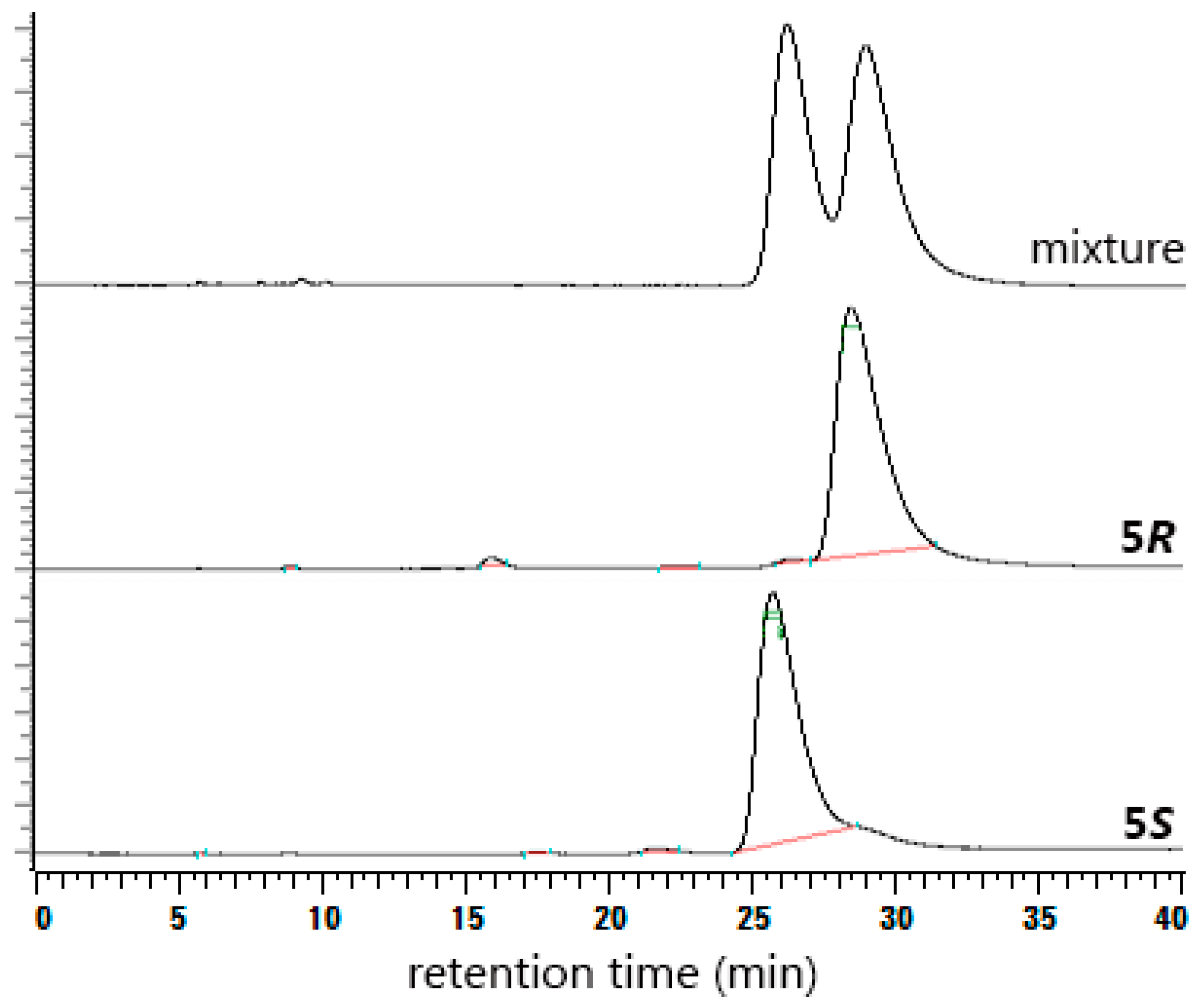
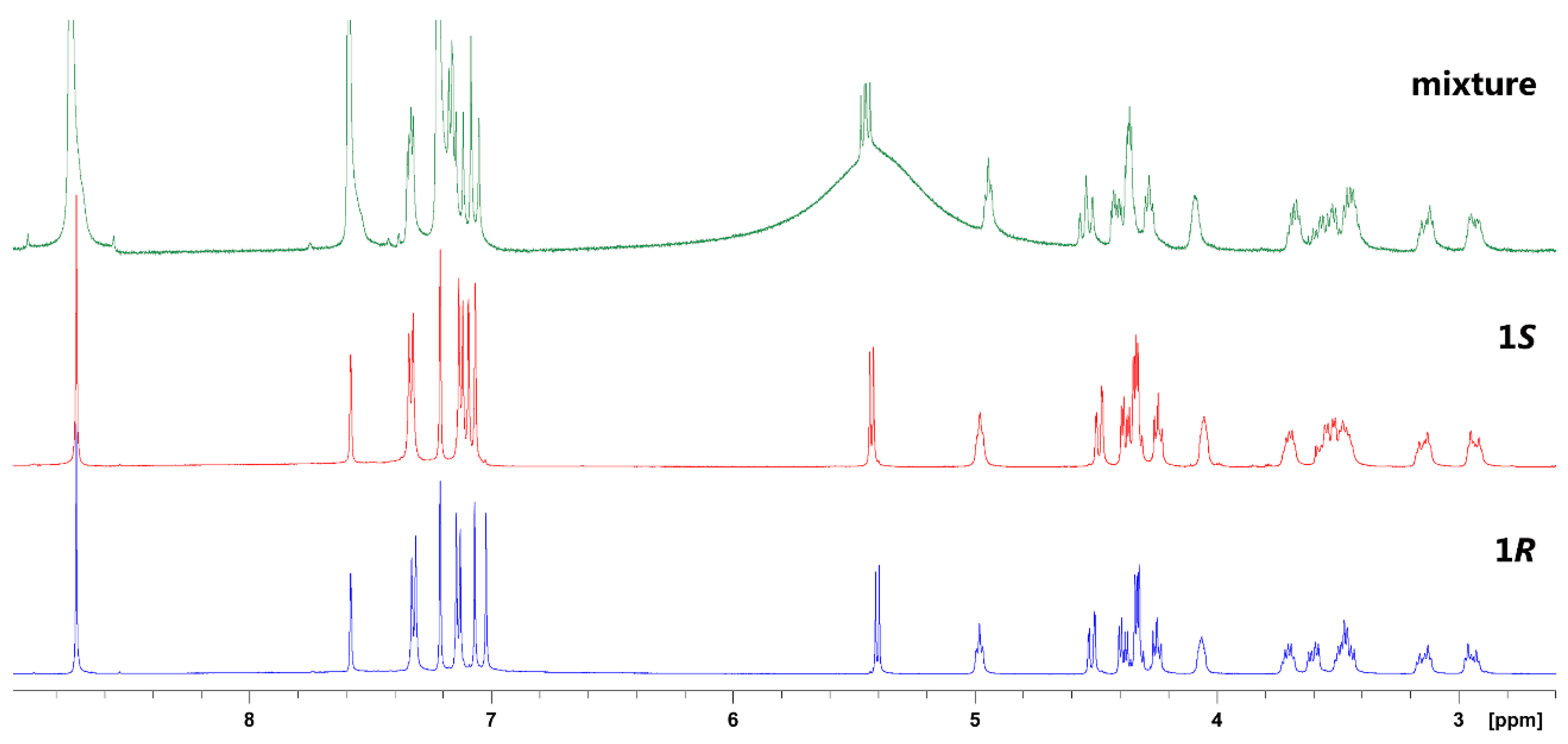
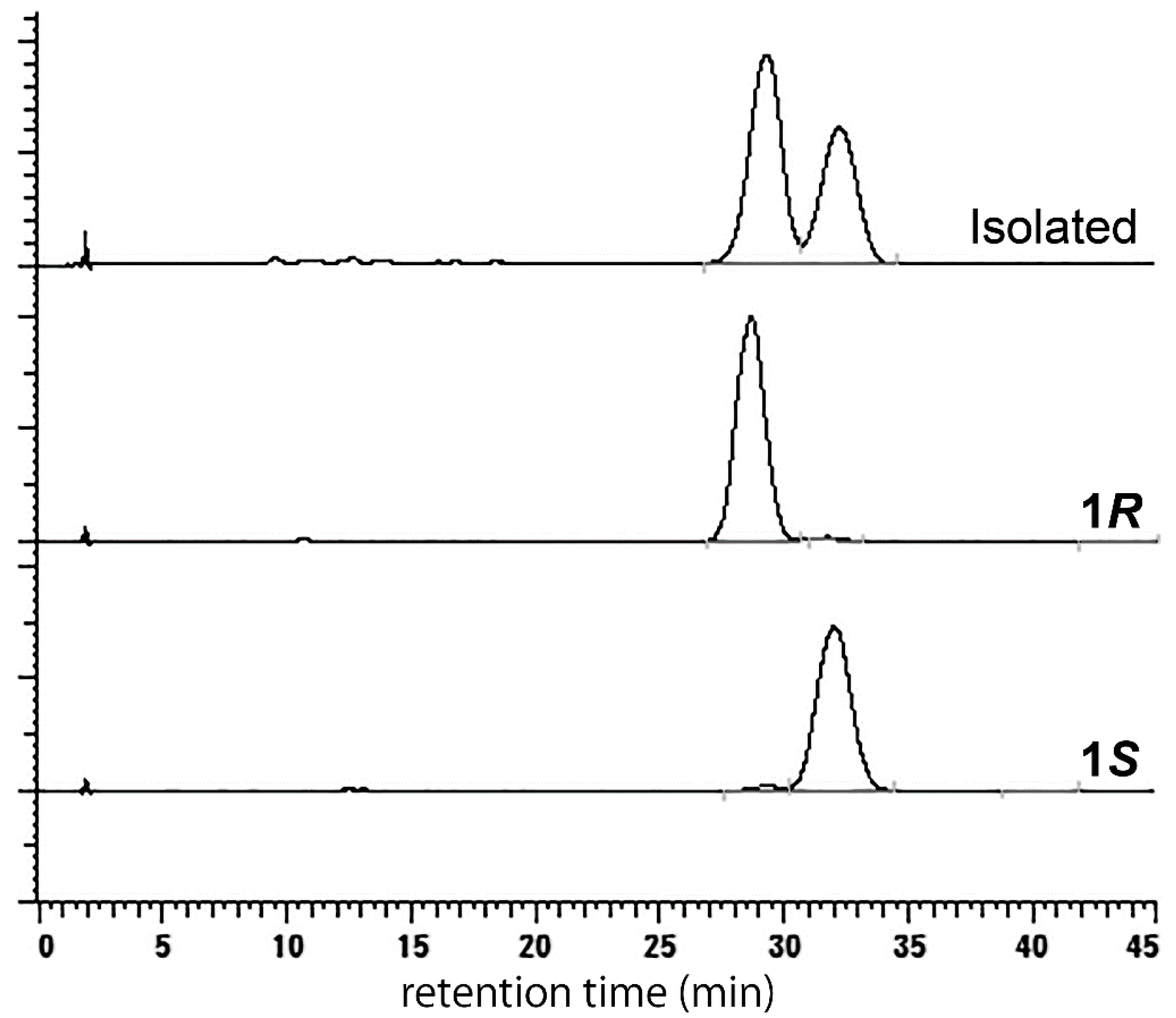
2. Results and Discussion
3. Materials and Methods
3.1. General
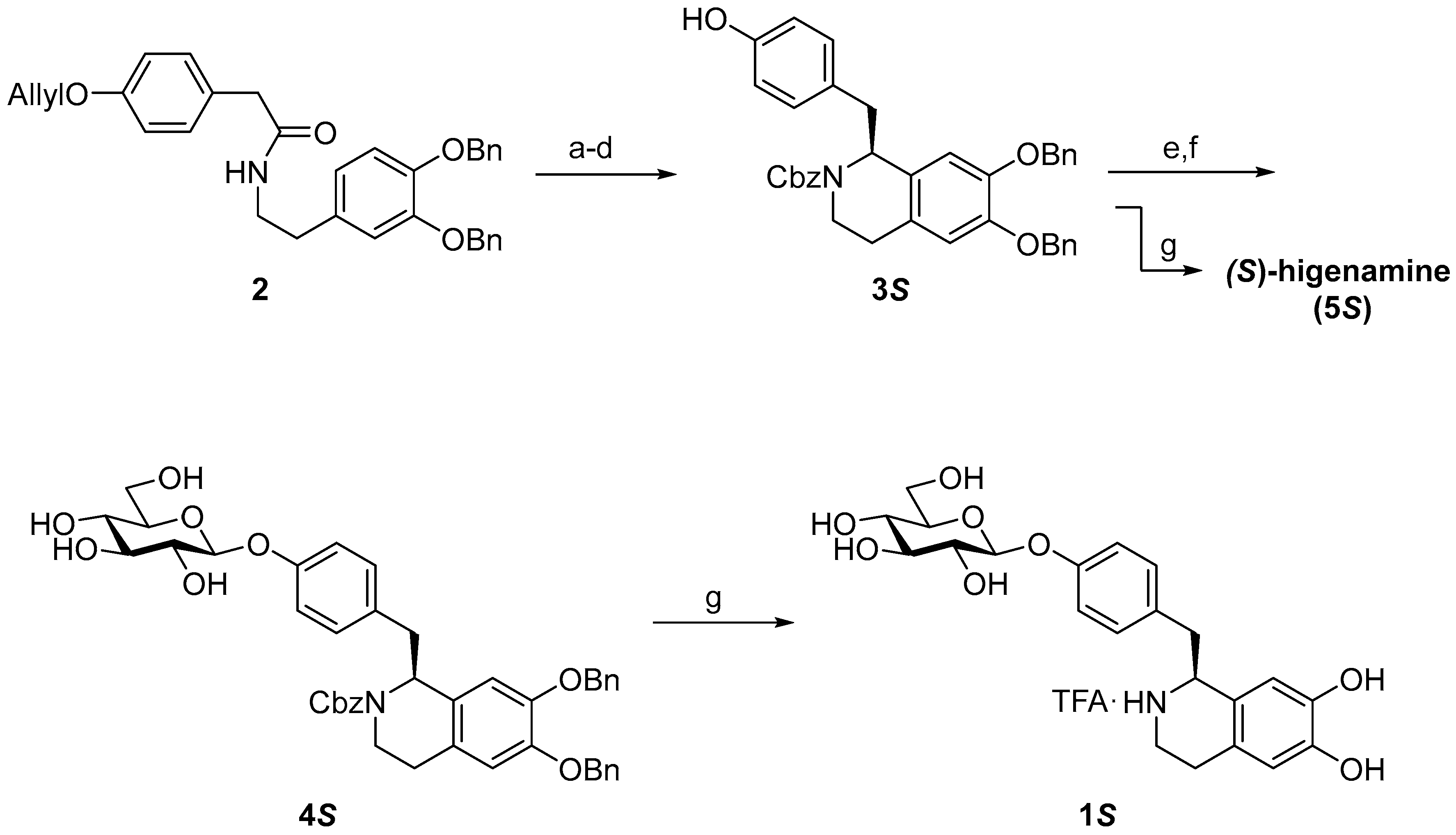
3.2. Synthesis
3.2.1. (S)-N-Cbz-6,7-di-O-benzylhigenamine (3S)
3.2.2. (S)-N-Cbz-6,7-di-O-benzylhigenamine 4′-O-β-d-glucoside (4S)
3.2.3. (S)-higenamine 4′-O-β-d-glucoside (1S)
3.2.4. (S)-Higenamine (5S)
3.2.5. (R)-Higenamine 4′-O-β-d-glucoside (1R)
3.2.6. (R)-Higenamine (5R)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bai, G.; Yang, Y.; Shi, Q.; Liu, Z.; Zhang, Q.; Zhu, Y.Y. Identification of higenamine in Radix Aconiti Lateralis Preparata as a beta2-adrenergic receptor agonist. Acta Pharmacol. Sin. 2008, 29, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Mukherjee, D.; Maji, A.K.; Rai, S.; Heinrich, M. The sacred lotus (Nelumbo nucifera)—Phytochemical and therapeutic profile. J. Pharm. Pharmacol. 2009, 61, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Lin, M. Benzylisoquinoline alkaloids from Gnetum parvifolium. J. Nat. Prod. 1999, 62, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Praman, S.; Mulvany, M.J.; Williams, D.E.; Andersen, R.J.; Jansakul, C. Crude extract and purified components isolated from the stems of Tinospora crispa exhibit positive inotropic effects on the isolated left atrium of rats. J. Ethnopharmacol. 2013, 149, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, V.; Singh, A.; Chandra, P.; Negi, M.P.S.; Kumar, N.; Kumar, B. Analysis of phytochemical variations in dioecious Tinospora cordifolia stems using HPLC/QTOF MS/MS and UPLC/QqQLIT-MS/MS. Phytochem. Anal. 2016, 27, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama, M.; Ueki, T.; Yasuda, Y.; Kikuchi, H.; Akaishi, T.; Okumura, H.; Abe, K. β2-Adrenoceptor-mediated tracheal relaxation induced by Higenamine from Nandina domestica thunberg. Planta Med. 2009, 75, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.P.; Zhang, Y.S.; Zhou, Q.M.; Xiong, J.; Dong, Y.R.; Yan, C. Higenamine protects ischemia/reperfusion induced cardiac injury and myocyte apoptosis through activation of β2-AR/PI3K/AKT signaling pathway. Pharmacol. Res. 2016, 104, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Chen, J.; Wu, Y.; Zhang, Y.; Xu, Y. Protective effect of higenamine ameliorates collagen-induced arthritis through heme oxygenase-1 and PI3K/Akt/Nrf-2 signaling pathways. Exp. Ther. Med. 2016, 12, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, M.; Wang, Y.; Wu, J.; Li, J. Higenamine promotes M2 macrophage activation and reduces Hmgb1 production through HO-1 induction in a murine model of spinal cord injury. Int. Immunopharmacol. 2014, 23, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lian, Z.; Peng, X.; Li, Z.; Zhu, H. Applications of Higenamine in pharmacology and medicine. J. Ethnopharmacol. 2017, 196, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-R.; Schriefer, J.M.; Gunnels, T.A.; Harvey, I.C.; Bloomer, R.J. Acute oral intake of a higenamine-based dietary supplement increases circulating free fatty acids and energy expenditure in human subjects. Lipids Health Dis. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Tying the knot: Occurrence and possible significance of gene fusions in plant metabolism and beyond. J. Exp. Bot. 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ghirga, F.; Quaglio, D.; Ghirga, P.; Berardozzi, S.; Zappia, G.; Botta, B.; Mori, M.; D’acquarica, I. Occurrence of enantioselectivity in nature: The case of (S)-norcoclaurine. Chirality 2016, 28, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, T.; Yokota, M. Studies on cardiac principle of aconite root. Chem. Pharm. Bull. 1976, 24, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Kashiwada, Y.; Aoshima, A.; Ikeshiro, Y.; Chen, Y.P.; Furukawa, H.; Itoigawa, M.; Fujioka, T.; Mihashi, K.; Cosentino, L.M.; Morris-Natschke, S.L.; et al. Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids. Bioorg. Med. Chem. 2005, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, H.; Ohkuma, H.; Kawaguchi, H.; Hsu, H.-Y.; Chen, Y.-P. Isolation of 1-(p-hydroxybenzyl)-6, 7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (demethylcoclaurine), an active alkaloid from Nelumbo nucifera. Chem. Pharm. Bull. 1970, 18, 2564–2568. [Google Scholar] [CrossRef]
- Kato, E.; Inagaki, Y.; Kawabata, J. Higenamine 4′-O-β-d-glucoside in the lotus plumule induces glucose uptake of L6 cells through β2-adrenergic receptor. Bioorg. Med. Chem. 2015, 23, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-P.; Lu, T.-N.; Hsu, N.-Y.; Chang, C.-C. Absolute stereochemical assignment of SCH 71450, a selective dopamine D4 receptor antagonist, through enantioselective epimer synthesis. Eur. J. Org. Chem. 2013, 2013, 2898–2905. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. The catalytic potential of Coptis japonica NCS2 revealed—Development and utilisation of a fluorescamine-based assay ETI. Adv. Synth. Catal. 2012, 354, 2997–3008. [Google Scholar] [CrossRef]
- Pyo, M.K.; Lee, D.-H.; Kim, D.-H.; Lee, J.-H.; Moon, J.-C.; Chang, K.C.; Yun-Choi, H.S. Enantioselective synthesis of (R)-(+)- and (S)-(−)-higenamine and their analogues with effects on platelet aggregation and experimental animal model of disseminated intravascular coagulation. Bioorg. Med. Chem. Lett. 2008, 18, 4110–4114. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Dai, P.; Ladislaw, C.; Patel, M.G.; Puar, M.S.; Pachter, J.A. D4 dopamine receptor-selective compounds from the Chinese plant Phoebe chekiangensis. Bioorg. Med. Chem. Lett. 1997, 7, 1207–1212. [Google Scholar] [CrossRef]
- Xu, C.; Xu, K.; Gu, H.; Zheng, R.; Liu, H.; Zhang, X.; Guo, Z.; Xu, B. Dopamine as a robust anchor to immobilize functional molecules on the iron oxide shell of magnetic nanoparticles. J. Am. Chem. Soc. 2004, 126, 9938–9939. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4S, 4R, 1S and 1R are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Synthetic 1R | Synthetic 1S | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 4.64 (dd, J = 5.7, 8.8 Hz) | 56.78 | 4.61 (dd, J = 5.7, 9.1 Hz) | 56.75 |
| 3 | 3.46–3.52 (m) | 39.93 | 3.47 (td, J = 6.3, 12.6 Hz) | 39.90 |
| 3.28 (td, J = 6.3, 13.2 Hz) | 3.27 (td, J = 6.3, 12.6 Hz) | |||
| 4 | 2.96–3.00 (m) | 24.69 | 2.95–3.00 (m) | 24.67 |
| 2.91 (td, J = 6.3, 17.3 Hz) | 2.90 (td, J = 6.3, 17.3 Hz) | |||
| 5 | 6.74 (s) | 116.49 | 6.73 (s) | 116.48 |
| 6 or 7 | - | 143.53 | - | 143.51 |
| 6 or 7 | - | 144.77 | - | 144.76 |
| 8 | 6.66 (s) | 114.53 | 6.64 (s) | 114.50 |
| 9 or 10 | - | 123.84 | - | 123.79 |
| 9 or 10 | - | 124.47 | - | 124.45 |
| 1′ | - | 130.30 | - | 130.27 |
| 2′, 6′ | 7.26 (2H, d, J = 8.5 Hz) | 131.61 | 7.25 (2H, d, J = 8.6 Hz) | 131.58 |
| 3′, 5′ | 7.13 (2H, d, J = 8.5 Hz) | 117.72 | 7.12 (2H, d, J = 8.6 Hz) | 117.74 |
| 4′ | - | 156.74 | - | 156.75 |
| 7′ | 3.00 (dd, J = 8.8, 14.5 Hz) | 39.13 | 2.97 (dd, J = 9.1, 14.5 Hz) | 39.14 |
| 3.40 (dd, J = 5.7, 14.5 Hz) | 3.36 (dd, J = 5.7, 14.5 Hz) | |||
| Glc-1 | 5.12 (d, J = 7.6 Hz) | 100.78 | 5.09 (d, J = 7.3 Hz) | 100.84 |
| Glc-2 | 3.58 (dd, J = 7.6, 9.5 Hz) | 73.59 | 3.58 (dd, J = 7.3, 9.5 Hz) | 73.60 |
| Glc-3 | 3.50 (dd, J = 8.6, 9.5 Hz) | 76.25 | 3.50 (dd, J = 9.1, 9.5 Hz) | 76.26 |
| Glc-4 | 3.61 (dd, J = 8.6, 9.5 Hz) | 70.11 | 3.61 (dd, J = 8.2, 9.1 Hz) | 70.12 |
| Glc-5 | 3.63 (ddd, J = 1.9, 5.7, 9.5 Hz) | 76.82 | 3.60–3.63 (m) | 76.81 |
| Glc-6 | 3.93 (dd, J = 1.9, 12.5 Hz) | 61.23 | 3.93 (dd, J = 2.2, 12.6 Hz) | 61.26 |
| 3.75 (dd, J = 5.7, 12.5 Hz) | 3.76 (dd, J = 5.7, 12.6 Hz) | |||
| No. | Synthetic 1R | Synthetic 1S | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 4.61 (dd, J = 6.1, 8.4 Hz) | 57.78 | 4.59 (dd, J = 5.8, 8.2 Hz) | 57.75 |
| 3 | 3.42–3.48 (m) | 40.87 | 3.42–3.48 (m) | 40.82 |
| 3.25 (td, J = 6.2, 13.0 Hz) | 3.25 (td, J = 6.4, 13.0 Hz) | |||
| 4 | 2.98 (td, J = 6.2, 17.3 Hz) | 25.69 | 2.98 (td, J = 6.4, 17.3 Hz) | 25.64 |
| 2.90 (td, J = 6.2, 17.3 Hz) | 2.90 (td, J = 6.4, 17.3 Hz) | |||
| 5 | 6.62 (s) | 116.21 | 6.62 (s) | 116.21 |
| 6 or 7 | 158.69 | 158.61 | ||
| 6 or 7 | 146.91 | 146.82 | ||
| 8 | 6.58 (s) | 114.25 | 6.56 (s) | 114.27 |
| 9 or 10 | 123.65 | 123.70 | ||
| 9 or 10 | 123.60 | 123.60 | ||
| 1′ | 130.29 | 130.32 | ||
| 2′, 6′ | 7.23 (2H, d, J = 8.6 Hz) | 131.64 | 7.23 (2H, d, J = 8.6 Hz) | 131.66 |
| 3′, 5′ | 7.11 (2H, d, J = 8.6 Hz) | 118.43 | 7.11 (2H, d, J = 8.6 Hz) | 118.37 |
| 4′ | 158.69 | 158.61 | ||
| 7′ | 3.35–3.42 (m) | 40.43 | 3.35–3.42 (m) | 40.40 |
| 3.03 (dd, J = 8.4, 14.5 Hz) | 3.04 (dd, J = 8.2, 14.4 Hz) | |||
| Glc-1 | 4.90 (d, J = 7.6 Hz) | 102.30 | 4.90 (d, J = 7.7 Hz) | 102.27 |
| Glc-2,3,5 | 3.42–3.48 (3H, m) | 78.20 | 3.42–3.48 (3H, m) | 78.12 |
| 78.01 | 77.94 | |||
| 74.87 | 74.85 | |||
| Glc-4 | 3.35–3.42 (m) | 71.43 | 3.35–3.42 (m) | 71.38 |
| Glc-6 | 3.89 (dd, J = 2.2, 12.0 Hz) | 62.59 | 3.89 (dd, J = 2.3, 12.0 Hz) | 62.53 |
| 3.68 (dd, J = 5.8, 12.0 Hz) | 3.68 (dd, J = 5.8, 12.0 Hz) | |||
| No. | Synthetic 1R | Synthetic 1S | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 5.00 (t, J = 5.7 Hz) | 57.11 | 5.00 (t, J = 6.5 Hz) | 57.05 |
| 3 | 3.73 (ddd, J = 6.0, 6.5, 12.3 Hz) | 40.19 | 3.73 (ddd, J = 6.0, 6.3, 12.5 Hz) | 40.00 |
| 3.46–3.62 (m) | 3.51 (ddd, J = 6.0, 6.3, 12.5 Hz) | |||
| 4 | 3.17 (ddd, J = 6.5, 6.5, 14.0 Hz) | 25.91 | 3.17 (ddd, J = 6.3, 6.3, 17.0 Hz) | 25.81 |
| 2.96 (ddd, J = 6.0, 6.0, 14.0 Hz) | 2.97 (ddd, J = 6.0, 6.0, 17.0 Hz) | |||
| 5 | 7.12 (s) | 116.98 | 7.09 (s) | 116.98 |
| 6 | - | 146.69 | - | 146.62 |
| 7 | - | 147.79 | - | 147.82 |
| 8 | 7.09 (s) | 115.04 | 7.05 (s) | 115.00 |
| 9 | - | 124.36 | - | 124.36 |
| 10 | - | 124.20 | - | 123.64 |
| 1′ | - | 130.61 | - | 130.62 |
| 2′, 6′ | 7.36 (2H, d, J = 8.4 Hz) | 131.74 | 7.34 (2H, d, J = 8.5 Hz) | 131.75 |
| 3′, 5′ | 7.15 (2H, d, J = 8.4 Hz) | 117.56 | 7.16 (2H, d, J = 8.5 Hz) | 117.53 |
| 4′ | - | 158.21 | - | 158.23 |
| 7′ | 3.46–3.62 (2H, m) | 40.40 | 3.63 (dd, J = 6.5, 14.2 Hz) | 40.48 |
| - | 3.48 (dd, J = 6.5, 14.2 Hz) | |||
| Glc-1 | 5.45 (d, J = 7.9 Hz) | 102.44 | 5.43 (d, J = 7.7 Hz) | 102.53 |
| Glc-2 | 4.27 (dd, J = 7.9, 8.8 Hz) | 75.27 | 4.27 (dd, J = 7.7, 9.1 Hz) | 75.27 |
| Glc-3, 4 | 4.33–4.37 (2H, m) | 78.83 | 4.33–4.38 (2H, m) | 78.86 |
| - | 71.62 | - | 71.64 | |
| Glc-5 | 4.06–4.09 (m) | 79.18 | 4.07–4.10 (m) | 79.25 |
| Glc-6 | 4.51 (dd, J = 2.2, 12.0 Hz) | 62.71 | 4.54 (dd, J = 2.2, 12.0 Hz) | 62.74 |
| 4.40 (dd, J = 5.0, 12.0 Hz) | 4.41 (dd, J = 5.0, 12.0 Hz) | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, E.; Iwata, R.; Kawabata, J. Synthesis and Detailed Examination of Spectral Properties of (S) and (R)-Higenamine 4′-O-β-d-Glucoside and HPLC Analytical Conditions to Distinguish the Diastereomers. Molecules 2017, 22, 1450. https://doi.org/10.3390/molecules22091450
Kato E, Iwata R, Kawabata J. Synthesis and Detailed Examination of Spectral Properties of (S) and (R)-Higenamine 4′-O-β-d-Glucoside and HPLC Analytical Conditions to Distinguish the Diastereomers. Molecules. 2017; 22(9):1450. https://doi.org/10.3390/molecules22091450
Chicago/Turabian StyleKato, Eisuke, Ryohei Iwata, and Jun Kawabata. 2017. "Synthesis and Detailed Examination of Spectral Properties of (S) and (R)-Higenamine 4′-O-β-d-Glucoside and HPLC Analytical Conditions to Distinguish the Diastereomers" Molecules 22, no. 9: 1450. https://doi.org/10.3390/molecules22091450