Pharmacological and Toxicological Screening of Novel Benzimidazole-Morpholine Derivatives as Dual-Acting Inhibitors

 , ,
, ,
Abstract
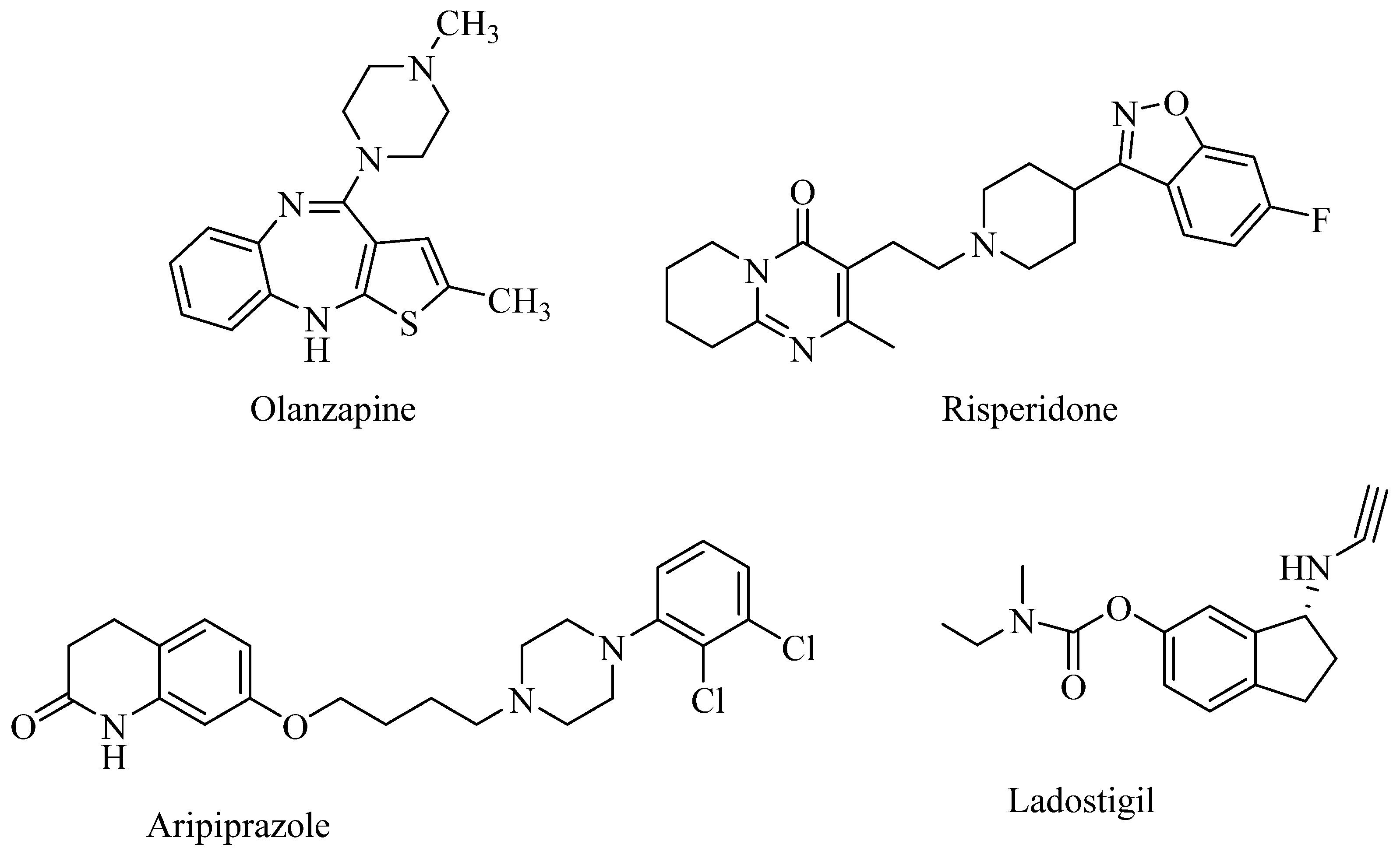
:1. Introduction
2. Results and Discussion
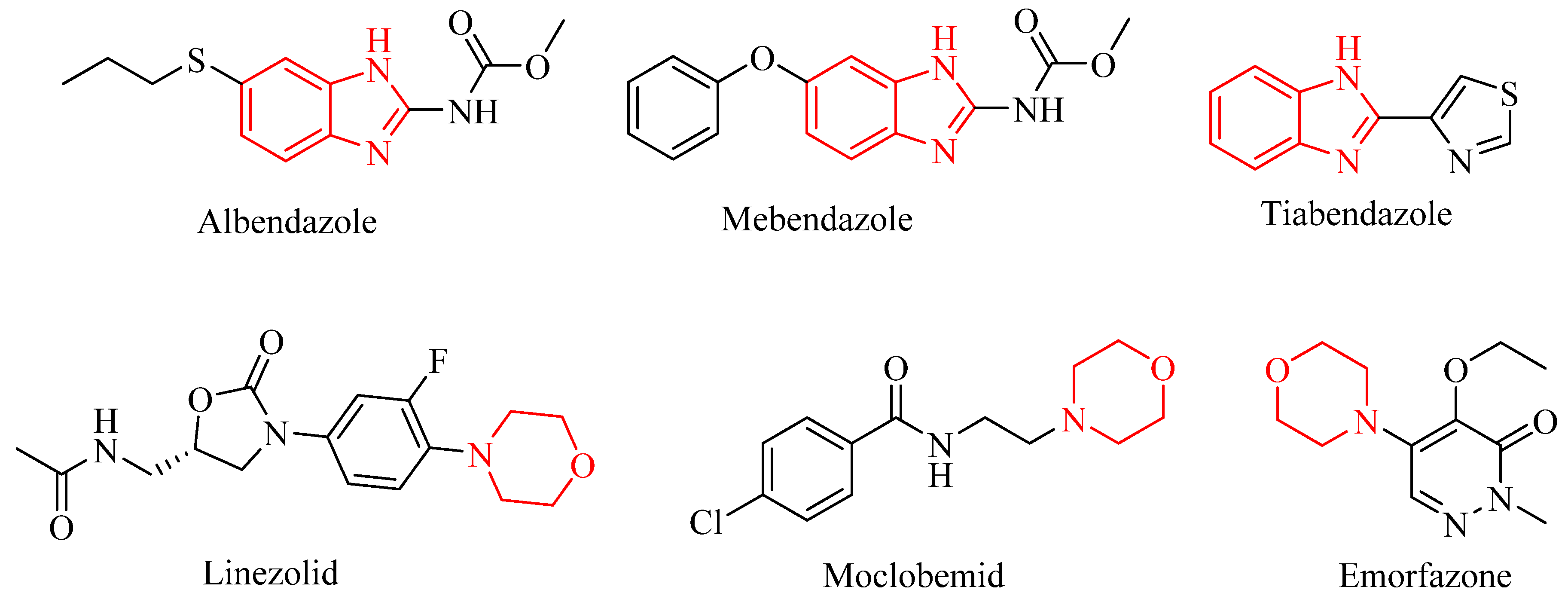
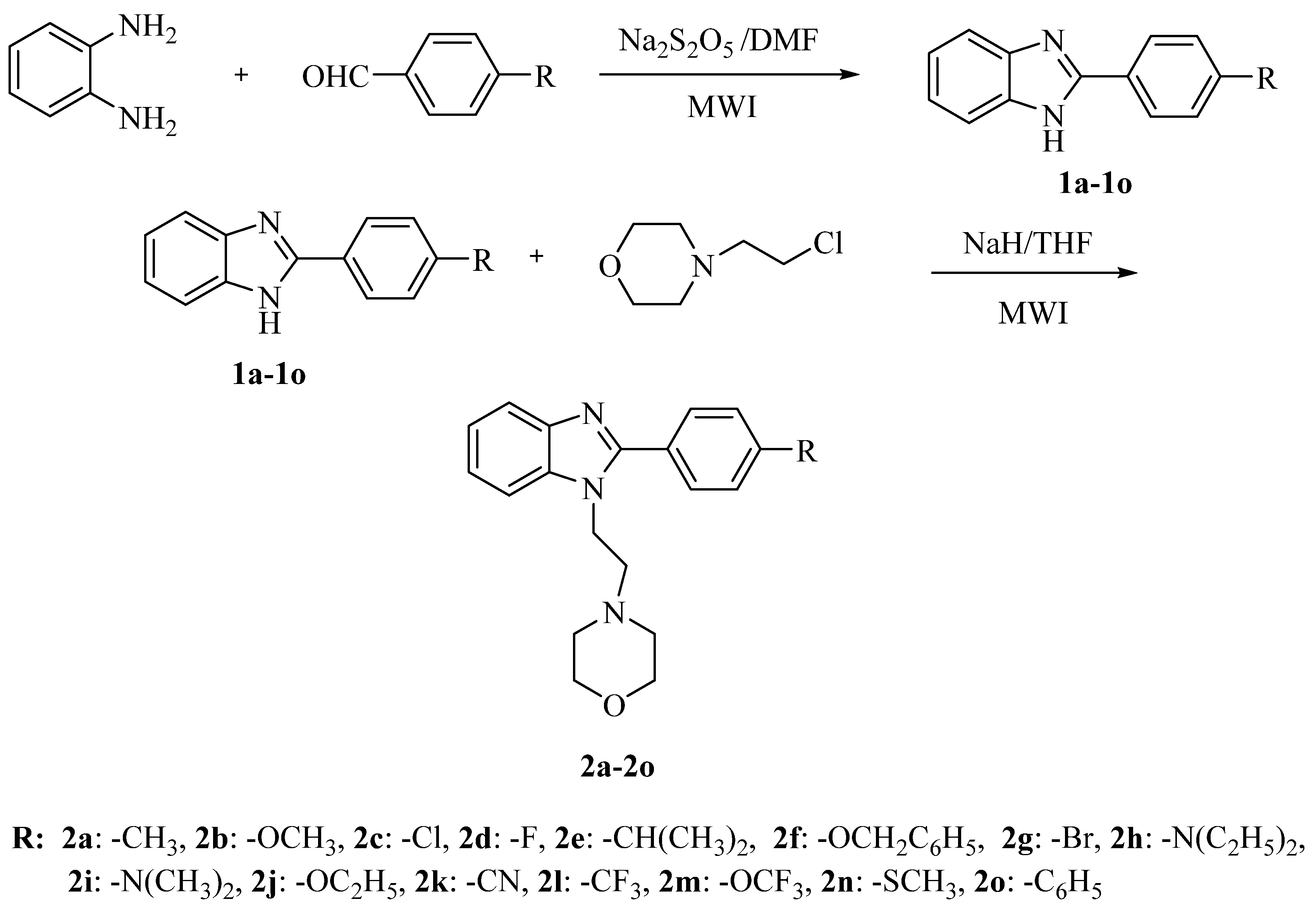
2.1. Chemistry
2.2. Biological Activity Screening
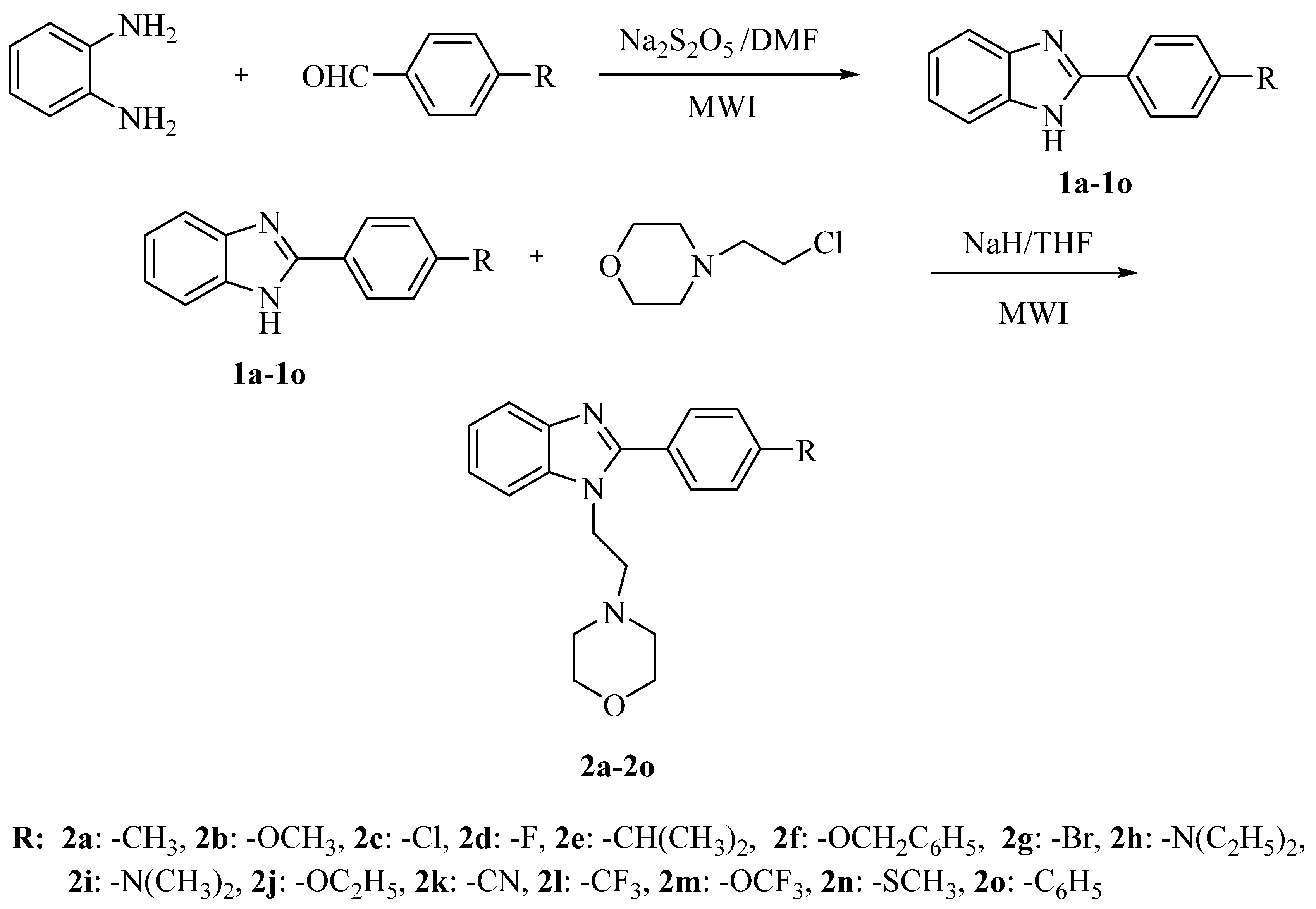
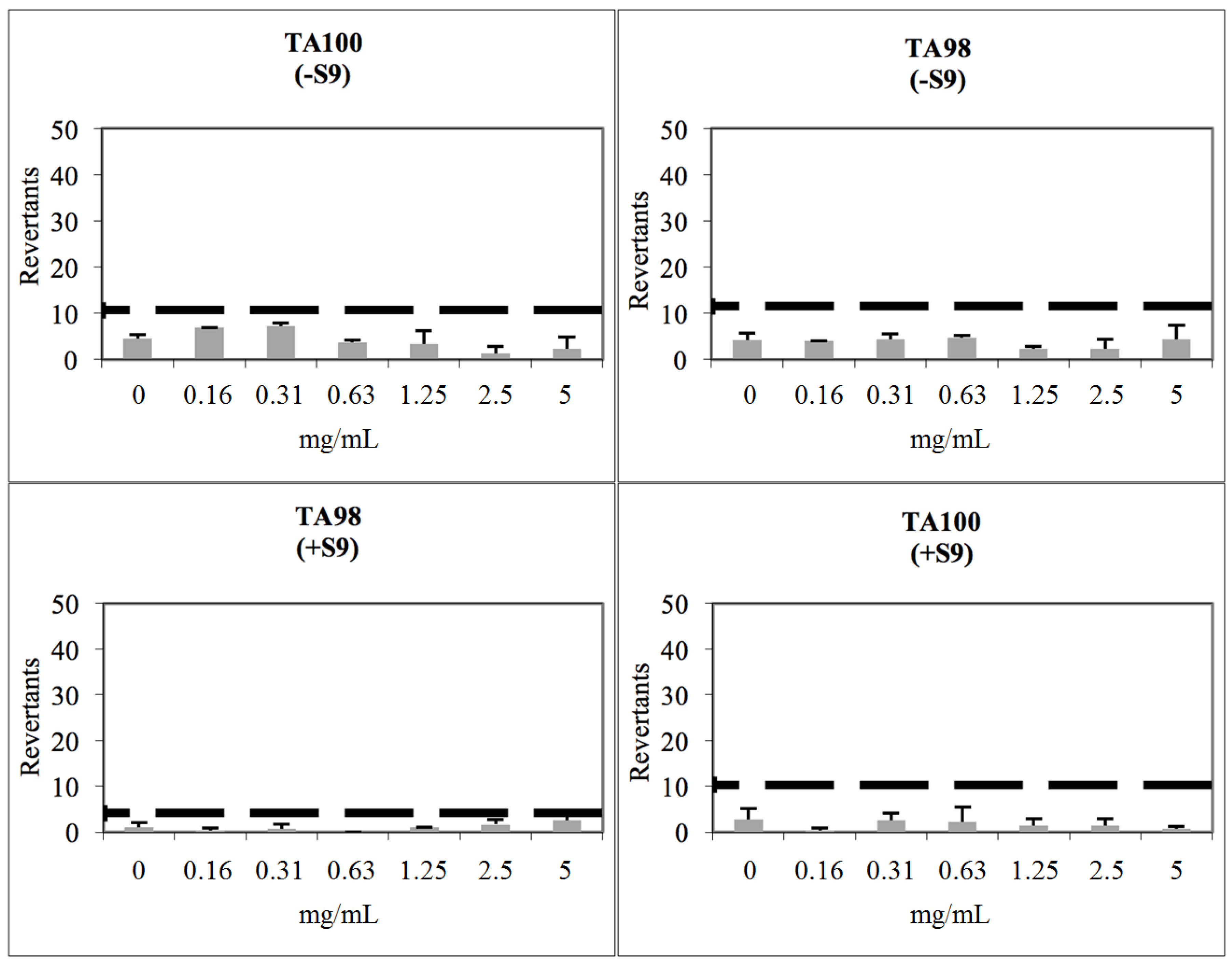
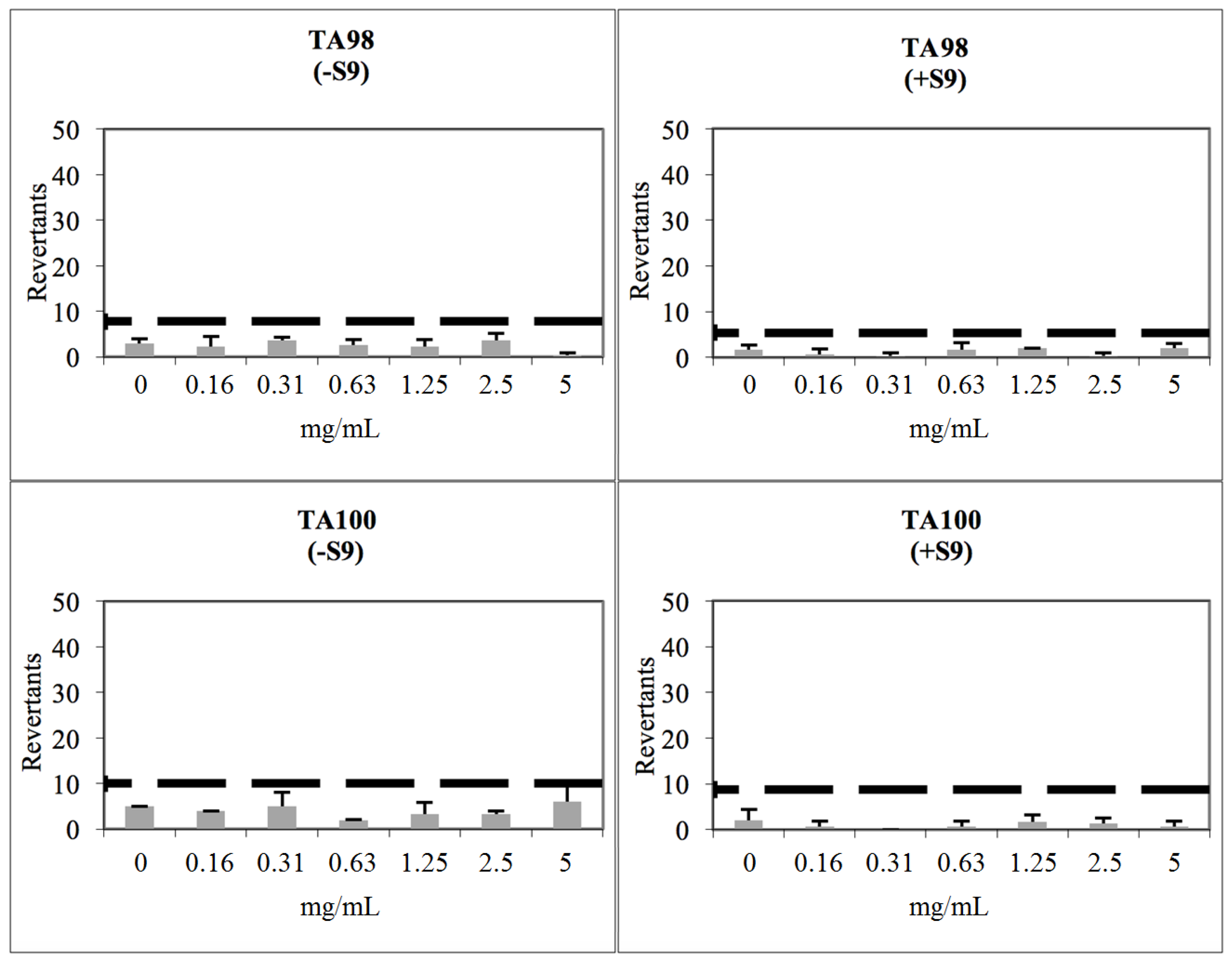
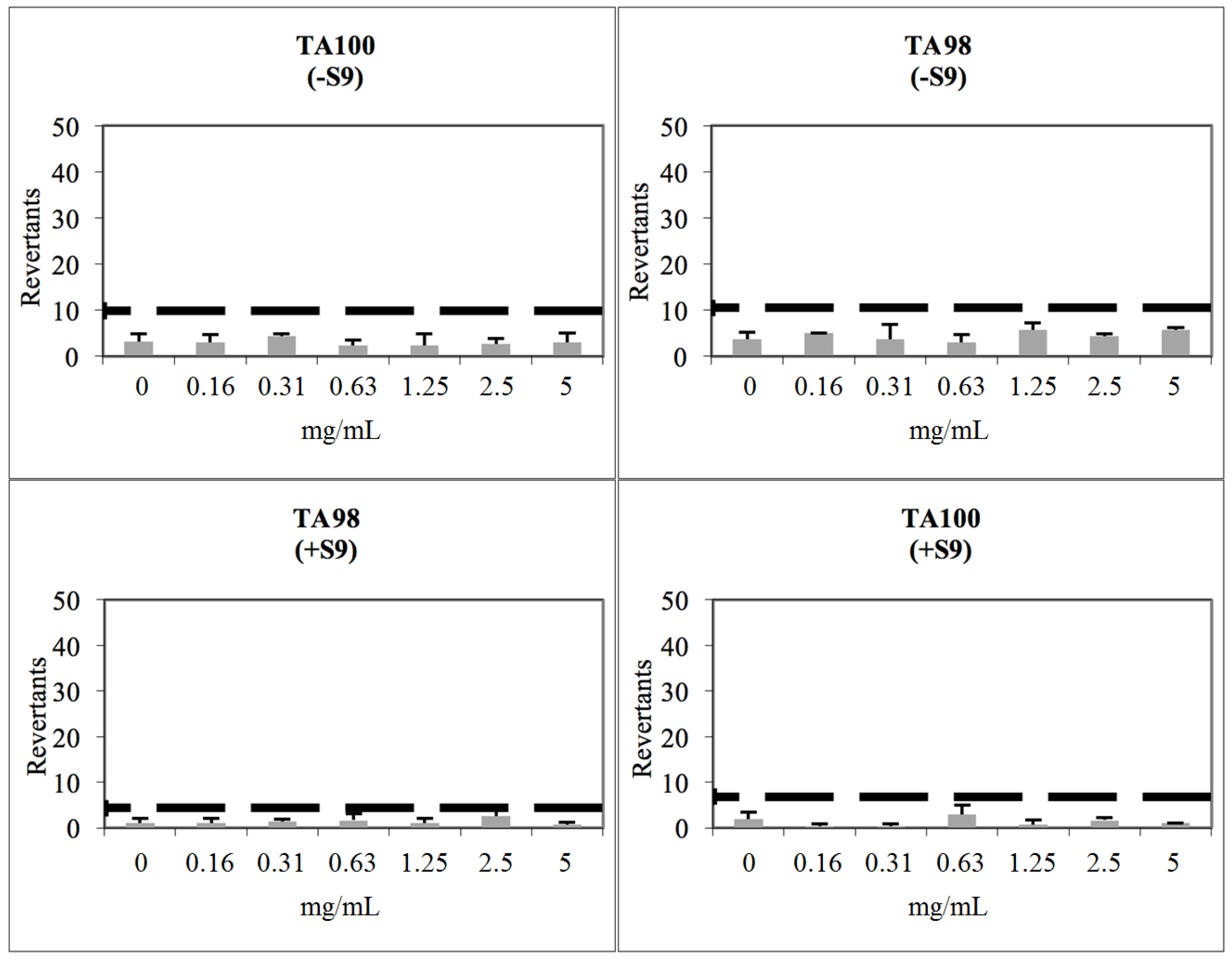
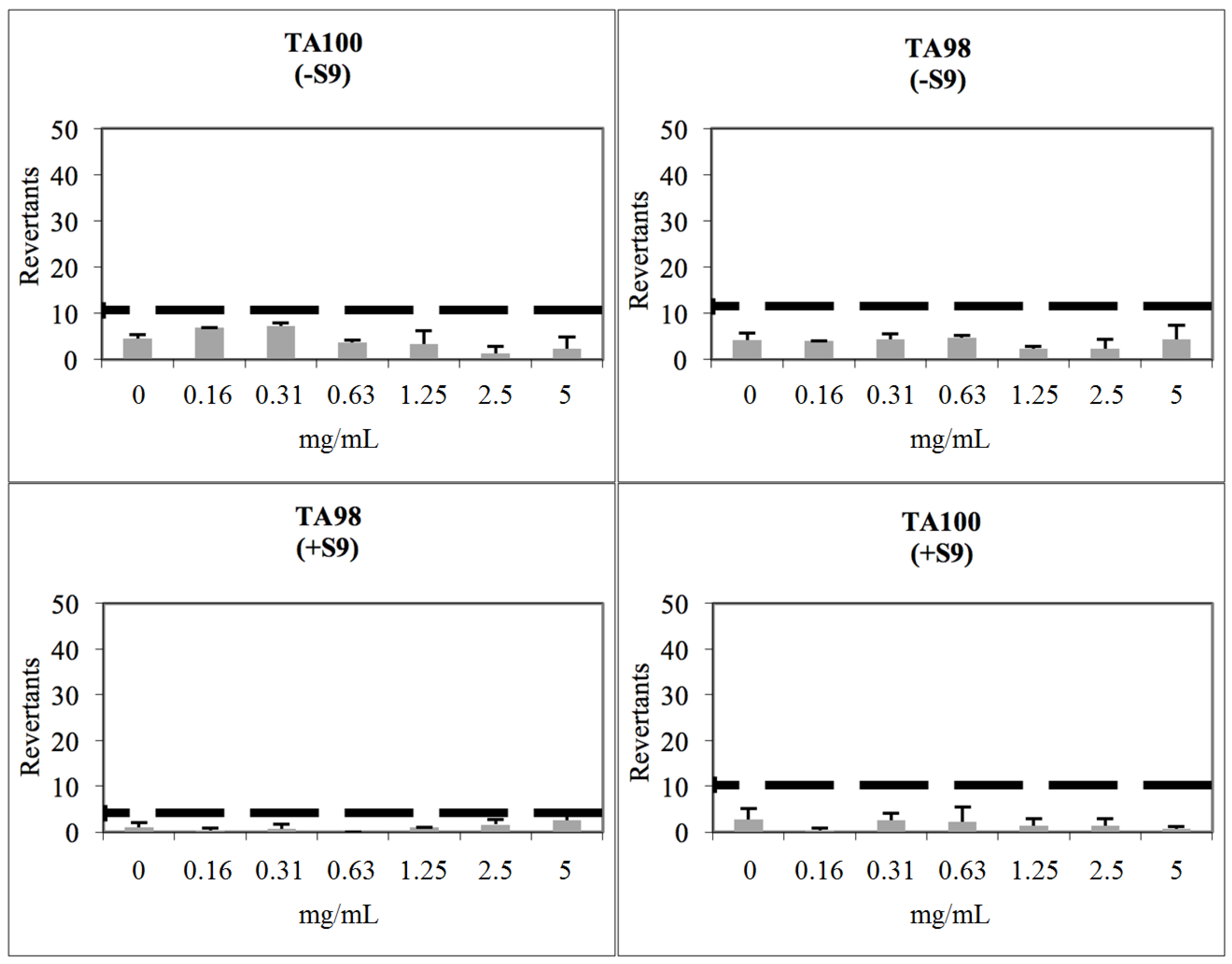
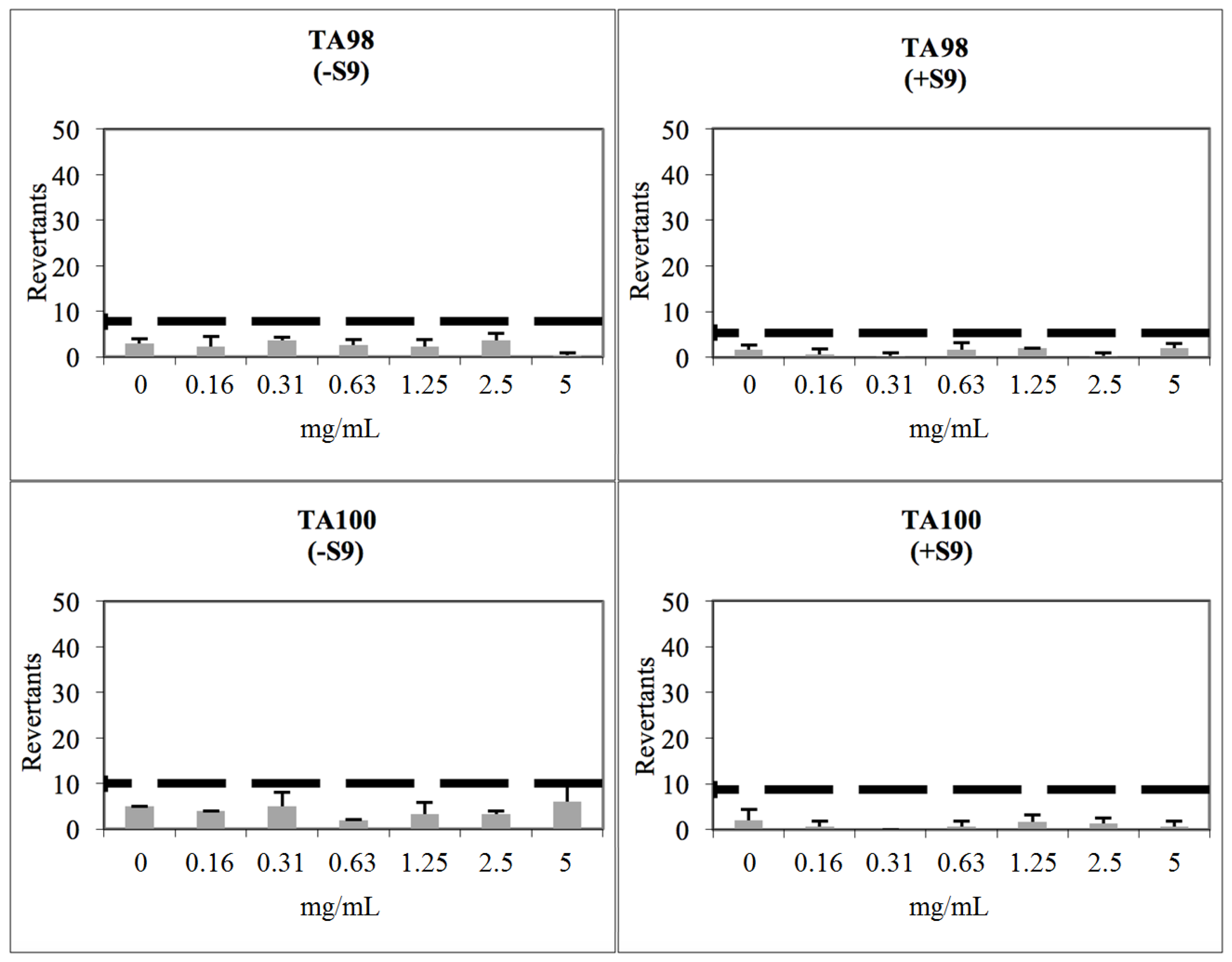
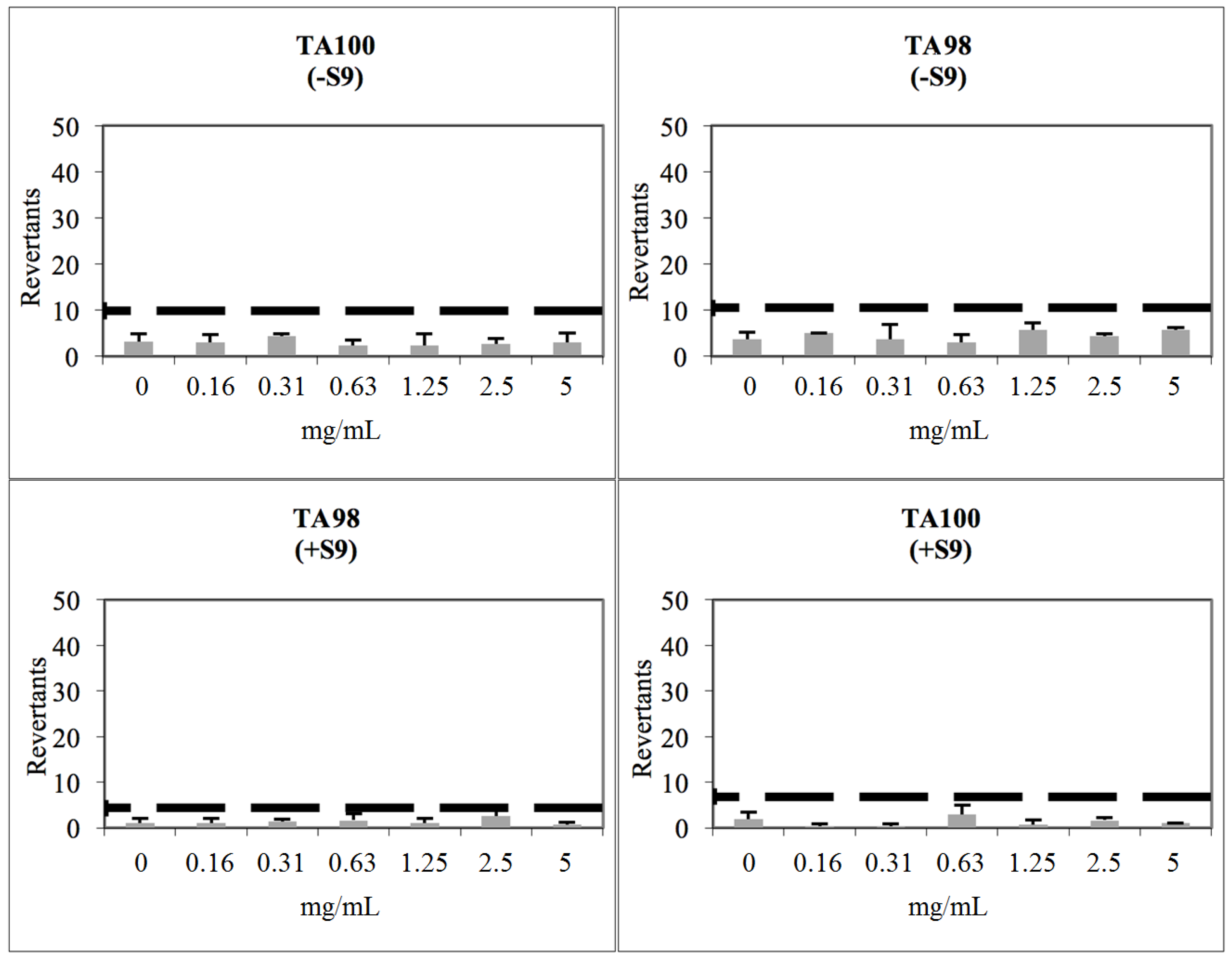
2.3. Toxicological Studies
2.4. Prediction of ADME and Drug Likeness
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Microwave-Assisted Synthesis of 2-Substituted-1H-benzimidazole Derivatives 1a–1o
3.2.2. Microwave-Assisted Synthesis of 2-Substituted-1-[2-(morpholin-4-yl)ethyl]-1H-benzimidazole Derivatives 2a–2o
3.3. Antimicrobial Assay
3.4. COX-1 and COX-2 Inhibition Assays
3.5. AChE Inhibition Assay
3.6. MAO Inhibition Assay
3.7. Determination of Cytotoxicity
3.8. Determination of Genotoxicity
3.9. Prediction of ADME Properties and Drug Likeness
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kimura, I. Medical benefits of using natural compounds and their derivatives having multiple pharmacological actions. Yakugaku Zasshi 2006, 126, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Manssour Fraga, C.A.; Barreiro, E.J. New insights for multifactorial disease therapy: The challenge of the symbiotic drugs. Curr. Drug Ther. 2008, 3, 1–13. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory resolution: New opportunities for drug discovery. Nat. Rev. Drug Discov. 2004, 3, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Guo, Y.; Wang, Z.; Huang, L.; Li, X. Dual functional cholinesterase and MAO inhibitors for the treatment of Alzheimer’s disease: Synthesis, pharmacological analysis and molecular modeling of homoisoflavonoid derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Dual inhibitors against topoisomerases and histone deacetylases. J. Cancer Prev. 2015, 20, 85. [Google Scholar] [CrossRef] [PubMed]
- Özkay, Y.; Tunalı, Y.; Karaca, H.; Işıkdağ, İ. Antimicrobial activity of a new combination system of benzimidazole and various azoles. Arch. Pharm. 2011, 344, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Subramanian, M.R.; Chinnaiyan, S.K. Synthesis, characterisation and evaluation of N-mannich bases of 2-substituted Benzimidazole derivatives. J. Young Pharm. 2013, 5, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Kuldipsinh, P.B.; Stoyanka, N.; Ivanov, I.; Manjunath, D.G. Novel research strategies of benzimidazole derivatives: A review. Mini Rev. Med. Chem. 2013, 13, 1421–1447. [Google Scholar]
- Qu, Z.; Li, F.; Xing, C.; Jiang, L. Synthesis and antifungal activity of novel benzimidazol-2-ylcyanoketone oxime ethers containing morpholine moiety. Chin. J. Org. Chem. 2015, 10, 028. [Google Scholar] [CrossRef]
- Vyas, M.; Swarnkar, N.; Mehta, S.; Joshi, H.; Ameta, R.; Punjabi, P.B. Microwave assisted synthesis and biological assay of 2-substituted [(morpholin-4-yl/4-methylpiperazin-1-yl)methyl]-1H-benzimidazoles using acidic alúmina as solid suport. Afinidad 2008, 65, 537. [Google Scholar]
- Shanmugapandiyan, P.; Denshing, K.S.; Ilavarasan, R.; Anbalagan, N.; Nirmal, R. Synthesis and biological activity of 2-(thiazolidin-4-one)phenyl]-1H-phenylbenzimidazoles and 2-[4-(azetidin-2-one)-3-chloro-4-phenyl]-1H-phenyl benzimidazoles. IJPSDR 2010, 2, 115–119. [Google Scholar]
- Eisa, H.M.; Barghash, A.E.M.; Badr, S.M.; Farahat, A.A. Synthesis and antimicrobial activity of certain benzimidazole and fused benzimidazole derivatives. Indian J. Chem. 2010, 49, 1515. [Google Scholar]
- Tunçbilek, M.; Kiper, T.; Altanlar, N. Synthesis and in vitro antimicrobial activity of some novel substituted benzimidazole derivatives having potent activity against MRSA. Eur. J. Med. Chem. 2009, 44, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.N.; Chaturvedi, S.C.; Kale, D.L.; Kakde, R.B.; Dhiker, S.B. Synthesis and antimicrobial activity of some benzimidazole derivatives. Cont. J. Pharm. Sci. 2008, 2, 44–48. [Google Scholar]
- Gill, C.; Jadhav, G.; Shaikh, M.; Kale, R.; Ghawalkar, A.; Nagargoje, D.; Shiradkar, M. Clubbed [1,2,3] triazoles by fluorine benzimidazole: A novel approach to H37Rv inhibitors as a potential treatment for tuberculosis. Bioorg. Med. Chem. Lett. 2008, 18, 6244–6247. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Han, X.; Zhou, Z. New substituted benzimidazole derivatives: A patent review (2013–2014). Expert. Opin. Ther. Pat. 2015, 25, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Naim, M.J.; Alam, O.; Alam, M.J.; Alam, P.; Shrivastava, N. A review on pharmacological profile of morpholine derivatives. Int. J. Pharmacol. Pharm. Sci. 2015, 3, 40–51. [Google Scholar]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Nadri, H.; Moghadam, F.H.; Edraki, N.; Khan, M.I.; Amini, M. Synthesis, docking study and neuroprotective effects of some novel pyrano [3, 2-c] chromene derivatives bearing morpholine/phenylpiperazine moiety. Bioorg. Med. Chem. 2017, 25, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Polshettiwar, V.; Varma, R.S. Greener and expeditious synthesis of bioactive heterocycles using microwave irradiation. Pure Appl. Chem. 2008, 80, 777–790. [Google Scholar] [CrossRef]
- Yoon, Y.K.; Ali, M.A.; Wei, A.C.; Choon, T.S.; Khaw, K.Y.; Murugaiyah, V.; Osman, H.; Masand, V.H. Synthesis, characterization, and molecular docking analysis of novel benzimidazole derivatives as cholinesterase inhibitors. Bioorg. Chem. 2013, 49, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Coban, G.; Carlino, L.; Tarikogullari, A.H.; Parlar, S.; Sarıkaya, G.; Alptüzün, V.; Alpan, A.S.; Güneş, H.S.; Erciyas, E. 1H-benzimidazole derivatives as butyrylcholinesterase inhibitors: Synthesis and molecular modeling studies. Med. Chem. Res. 2016, 25, 2005–2014. [Google Scholar] [CrossRef]
- Menteşe, E.; Ülker, S.; Kahveci, B. Synthesis and study of α-glucosidase inhibitory, antimicrobial and antioxidant activities of some benzimidazole derivatives containing triazole, thiadiazole, oxadiazole, and morpholine rings. Chem. Heterocycl. Compd. 2015, 50, 1671–1682. [Google Scholar] [CrossRef]
- Zhou, S.; Li, F.; Zhang, P.; Jiang, L. Synthesis and antifungal activity of novel 1-(1H-benzoimidazol-1-yl) propan-2-one oxime-ethers containing the morpholine moiety. Res. Chem. Intermed. 2013, 39, 1735–1743. [Google Scholar] [CrossRef]
- Zhang, P.; Wan, F.; Li, Y.; Li, C.; Jiang, L. Synthesis and antibacterial activity of novel ethyl 2-alkoxyimino-2-benzimidazol-2-yl acetates bearing a morpholine group. Res. Chem. Intermed. 2015, 41, 3349–3357. [Google Scholar] [CrossRef]
- Achar, K.C.; Hosamani, K.M.; Seetharamareddy, H.R. In Vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- El-Nezhawy, A.O.; Biuomy, A.R.; Hassan, F.S.; Ismaiel, A.K.; Omar, H.A. Design, synthesis and pharmacological evaluation of omeprazole-like agents with anti-inflammatory activity. Bioorg. Med. Chem. 2013, 21, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Jesudason, E.P.; Sridhar, S.K.; Malar, E.P.; Shanmugapandiyan, P.; Inayathullah, M.; Arul, V.; Selvaraj, D.; Jayakumar, R. Synthesis, pharmacological screening, quantum chemical and in vitro permeability studies of N-Mannich bases of benzimidazoles through bovine cornea. Eur. J. Med. Chem. 2009, 44, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Gaba, M.; Singh, D.; Singh, S.; Sharma, V.; Gaba, P. Synthesis and pharmacological evaluation of novel 5-substituted-1-(phenylsulfonyl)-2-methylbenzimidazole derivatives as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2010, 45, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.A. Synthesis, characterization and biological evaluation of 1,2-disubstituted benzimidazole derivatives using Mannich bases. J. Chem. 2010, 7, 222–226. [Google Scholar]
- Mathew, B.; Suresh, J.; Anbazhagan, S. Development of novel (1-H) benzimidazole bearing pyrimidine-trione based MAO-A inhibitors: Synthesis, docking studies and antidepressant activity. J. Saudi Chem. Soc. 2016, 20 (Suppl. 1), S132–S139. [Google Scholar] [CrossRef]
- Van den Berg, D.; Zoellner, K.R.; Ogunrombi, M.O.; Malan, S.F.; Terre’Blanche, G.; Castagnoli, N.; Bergh, J.J.; Petzer, J.B. Inhibition of monoamine oxidase B by selected benzimidazole and caffeine analogues. Bioorg. Med. Chem. 2007, 15, 3692–3702. [Google Scholar] [CrossRef] [PubMed]
- Petzer, J.P.; Steyn, S.; Castagnoli, K.P.; Chen, J.F.; Schwarzschild, M.A.; Van der Schyf, C.J.; Castagnoli, N. Inhibition of monoamine oxidase B by selective adenosine A 2A receptor antagonists. Bioorg. Med. Chem. 2003, 11, 1299–1310. [Google Scholar] [CrossRef]
- Grandi, T.; Sparatore, F.; Gnerre, C.; Crivori, P.; Carrupt, P.A.; Testa, B. Monoamine oxidase inhibitory properties of some benzazoles: Structure-; Activity relationships. AAPS J. 1999, 1, 1–4. [Google Scholar] [CrossRef]
- Shukla, J.S.; Saxena, S.; Rastogi, R. Synthesis of some newer, 1-heterocyclic amino/iminomethyl-2-substituted benzimidazoles as a potent CNS, anticonvulsant and monoamine oxidase inhibitory agents. Curr. Sci. 1982, 51, 820–822. [Google Scholar]
- Avramova, P.; Danchev, N.; Buyukliev, R.; Bogoslovova, T. Synthesis, toxicological, and pharmacological assessment of derivatives of 2-aryl-4-(3-arylpropyl) morpholines. Arch. Pharm. 1998, 331, 342–346. [Google Scholar] [CrossRef]
- Ghanbarpour, A.; Hadizadeh, F.; Piri, F.; Rashidi-Ranjbar, P. Synthesis, conformational analysis and antidepressant activity of moclobemide new analogues. Pharm. Acta Helv. 1997, 72, 119–122. [Google Scholar] [CrossRef]
- Avramova, P.D.; Danchev, N.D.; Buyukliev, R.T. Synthesis, toxicological and pharmacological assessment of esters of carbonic and carbamic acids with 2-aryl-4-hydroxyethylmorpholines. Eur. J. Med. Chem. 1996, 31, 909–914. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Abdel-Aziem, T. Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Ashour, H.M.; Ghany, Y.S.A.; Bekhit, A.E.D.A.; Baraka, A. Synthesis and biological evaluation of some thiazolyl and thiadiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Eur. J. Med. Chem. 2008, 43, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Yanez, M.; Vina, D. Dual inhibitors of monoamine oxidase and cholinesterase for the treatment of Alzheimer disease. Curr. Top. Med. Chem. 2013, 3, 1692–1706. [Google Scholar] [CrossRef]
- Secci, D.; Bolasco, A.; D’Ascenzio, M.; della Sala, F.; Yáñez, M.; Carradori, S. Conventional and microwave-assisted synthesis of benzimidazole derivatives and their in vitro inhibition of human cyclooxygenase. J. Heterocycl. Chem. 2012, 49, 1187–1195. [Google Scholar] [CrossRef]
- Sakram, B.; Rambabu, S.; Ashok, K.; Sonyanaik, B.; Ravi, D. Microwave assisted aqueous phase synthesis of benzothiazoles and benzimidazoles in the presence of Ag2O. Russ. J. Gen. Chem. 2016, 86, 2737–2743. [Google Scholar] [CrossRef]
- Alp, A.S.; Kılcıgil, G.; Özdamar, E.D.; Çoban, T.; Eke, B. Synthesis and evaluation of antioxidant activities of novel 1,3,4-oxadiazole and imine containing 1H-benzimidazoles. Turk. J. Chem. 2015, 39, 42–53. [Google Scholar] [CrossRef]
- Kathirvelan, D.; Yuvaraj, P.; Babu, K.; Nagarajan, A.S.; Reddy, B.S. A green synthesis of benzimidazoles. Indian J. Chem. 2013, 52, 1152–1156. [Google Scholar]
- Hasanpour, M.; Eshghi, H.; Bakavoli, M.; Mirzaeia, M. A novel ionic liquid based on imidazolium cation as an efficient and reusable catalyst for the one-pot synthesis of benzoxazoles, benzthiazoles, benzimidazoles and 2-arylsubstituted benzimidazoles. J. Chin. Chem. Soc. 2015, 62, 412–419. [Google Scholar] [CrossRef]
- Alapati, M.L.P.R.; Abburi, S.R.; Mukkamala, S.B.; Krishnaji Rao, M. Simple and efficient one-pot synthesis of 2-substituted benzimidazoles from θ-diaminoarene and aryl aldehydes. Synth. Commun. 2015, 45, 2436–2443. [Google Scholar] [CrossRef]
- Vinodkumar, R.; Rajagopal, K.; Devanna, N. Synthesis of highly functionalized 2-(substituted biphenyl) benzimidzoles via suzuki-miyaura cross coupling reaction. J. Heterocycl. Chem. 2007, 44, 1521–1523. [Google Scholar] [CrossRef]
- Fathima, N.; Krishnamurthy, M.S.; Begum, N.S. 2-[4-(Trifluoromethoxy) phenyl]-1H-benzimidazole. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, 264. [Google Scholar] [CrossRef] [PubMed]
- BioVision Inc. COX-1 Inhibitor Screening Kit Guide (Fluorometric); Catalog K548-100; BioVision: Milpitas, CA, USA; Available online: http://www.biovision.com/documentation/datasheets/K548.pdf (accessed on 17 August 2017).
- BioVision Inc. COX-2 Inhibitor Screening Kit Guide (Fluorometric); Catalog K547-100; BioVision: Milpitas, CA, USA; Available online: http://www.biovision.com/documentation/datasheets/K547.pdf (accessed on 17 August 2017).
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Matsumoto, T.; Suzuki, O.; Furuta, T.; Asai, M.; Kurokawa, Y.; Nimura, Y.; Katsumata, Y.; Takahashi, I. A sensitive fluorometric assay for serum monoamine oxidase with kynuramine as substrate. Clin. Biochem. 1985, 18, 126–129. [Google Scholar] [CrossRef]
- Flückiger-Isler, S.; Kamber, M. Direct comparison of the Ames microplate format (MPF) test in liquid medium with the standard Ames pre-incubation assay on agar plates by use of equivocal to weakly positive test compounds. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2012, 747, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Calculation of Molecular Properties and Bioactivity Score Software, Molinspiration Cheminformatics. Available online: http://www.molinspiration.com/cgi-bin/properties (accessed on 17 August 2017).
- Drug-Likeness and Molecular Property Prediction Software, Molsoft LLC. Available online: http://molsoft.com/mprop/ (accessed on 17 August 2017).
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard-Ninth ed.; CLSI document M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST). EUCAST definitive Document EDef 7.1: Method for the Determination of Broth Dilution MICs of Antifungal Agents for Fermentative Yeasts. Clin. Microbiol. Infect. 2008, 14, 398–405. [Google Scholar]
- Can, Ö.D.; Osmaniye, D.; Demir Özkay, Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2017, 131, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Yurttaş, L.; Özkay, Y.; Akalın-Çiftçi, G.; Ulusoylar-Yıldırım, Ş. Synthesis and anticancer activity evaluation of N-[4-(2-methylthiazol-4-yl)phenyl] acetamide derivatives containing (benz) azole moiety. J. Enzym. Inhib. Med. Chem. 2014, 29, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Gheewala, N.; Suthar, A.; Shah, A. In Vitro cytotoxicity activity of Solanum nigrum extract against Hela cell line and Vero cell line. Int. J. Pharm. Pharm. Sci. 2009, 1, 38–46. [Google Scholar]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, C.V.; Sundarajan, K.; Gupta, A.; Srikanth, H.S.; Edwin, J.; Agarwal, A. Evaluation of the genotoxic potential of standardized extract of Glycyrrhiza glabra (GutGard™). Regul. Toxicol. Pharmacol. 2011, 61, 373–380. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1a–1o and 2a–2o are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A | B | C | D | E | F | G | H | I | J | K |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2b | >1 | >1 | >1 | 0.0156 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2c | >1 | >1 | >1 | 0.125 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2d | >1 | >1 | >1 | 0.500 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2e | >1 | >1 | >1 | 0.250 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2f | >1 | >1 | >1 | 0.500 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2g | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2h | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2i | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2j | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2k | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2l | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2m | >1 | >1 | >1 | 0.125 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2n | >1 | >1 | >1 | 0.500 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| 2o | >1 | >1 | >1 | 0.500 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| Ref-1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | 0.0039 | 0.0312 | 0.0019 | 0.0625 |
| Ref-2 | 0.0312 | >1 | 0.0156 | 0.250 | 0.0625 | 0.250 | 0.0312 | >1 | >1 | >1 | >1 |
| Comp. | COX 1 Inhibition % | COX 2 Inhibition % | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | IC50 µM | 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | IC50 µM | |
| 2a | 47.39 ± 1.08 | 40.12 ± 0.92 | 35.68 ± 0.97 | 30.46 ± 0.83 | 28.81 ± 0.79 | 56.08 | 43.72 ± 1.07 | 40.32 ± 1.10 | 38.26 ± 0.92 | 36.20 ± 0.87 | 32.44 ± 0.91 | 58.66 |
| 2b | 59.37 ± 1.24 | 42.75 ± 1.02 | 37.08 ± 0.92 | 26.77 ± 0.87 | 19.56 ± 0.76 | 8.096 | 57.85 ± 1.28 | 43.70 ± 1.12 | 38.12 ± 0.97 | 27.45 ± 0.87 | 21.35 ± 0.74 | 8.369 |
| 2f | 45.38 ± 0.99 | 42.08 ± 0.87 | 34.23 ± 0.76 | 32.68 ± 0.78 | 29.88 ± 0.68 | 43.23 | 46.98 ± 1.13 | 40.22 ± 1.24 | 35.62 ± 0.91 | 31.46 ± 0.84 | 28.74 ± 0.63 | 44.92 |
| 2h | 46.29 ± 1.09 | 40.88 ± 0.99 | 35.62 ± 0.81 | 30.77 ± 0.93 | 24.23 ± 0.72 | 42.97 | 47.54 ± 1.08 | 42.60 ± 1.16 | 31.75 ± 0.99 | 28.35 ± 0.82 | 23.04 ± 0.68 | 51.19 |
| 2j | 51.41 ± 1.15 | 44.45 ± 1.07 | 37.66 ± 0.76 | 27.46 ± 0.82 | 23.44 ± 0.61 | 14.18 | 52.54 ± 1.35 | 43.40 ± 1.27 | 33.98 ± 1.06 | 26.85 ± 0.86 | 20.54 ± 0.76 | 13.09 |
| 2m | 47.36 ± 1.16 | 42.02 ± 0.88 | 36.65 ± 0.92 | 26.19 ± 0.79 | 20.01 ± 0.62 | 21.94 | 46.11 ± 1.13 | 41.23 ± 1.05 | 34.48 ± 0.97 | 28.69 ± 0.83 | 22.30 ± 0.52 | 22.66 |
| Ref-1 | 74.67 ± 1.28 | 45.08 ± 1.16 | 28.23 ± 0.91 | 22.66 ± 0.82 | 14.76 ± 0.60 | 2.435 | 69.51 ± 1.28 | 36.38 ± 0.91 | 29.42 ± 0.84 | 24.03 ± 0.76 | 17.08 ± 0.58 | 5.327 |
| Ref-2 | 68.39 ± 1.26 | 41.91 ± 0.98 | 29.05 ± 0.92 | 20.37 ± 0.81 | 18.84 ± 0.63 | 3.810 | 78.69 ± 1.19 | 48.28 ± 1.09 | 32.97 ± 0.96 | 21.30 ± 0.88 | 14.12 ± 0.61 | 1.683 |
| Comp. | Cytotoxicity NIH3T3 Cells IC50 (µM) | Concentration (mg/mL) for Ames Test | REVERTANTS Fold Increase (over Baseline) | |||
|---|---|---|---|---|---|---|
| TA98 | TA100 | |||||
| S9+ | S9− | S9+ | S9− | |||
| 2b | 316 | 0.156 | 0.16 | 0.69 | 0.06 | 1.31 *** |
| 0.3125 | 0.32 | 0.75 | 0.52 | 1.37 *** | ||
| 0.625 | 0.00 | 0.81 | 0.45 | 0.69 | ||
| 1.25 | 0.48 | 0.40 | 0.26 | 0.62 | ||
| 2.5 | 0.80 | 0.40 | 0.26 | 0.25 *** | ||
| 5 | 1.27 | 0.75 | 0.13 | 0.44 * | ||
| 2j | 316 | 0.156 | 0.25 | 0.60 | 0.15 | 0.80 |
| 0.3125 | 0.12 * | 0.94 | 0.00 | 1.00 | ||
| 0.625 | 0.62 | 0.68 | 0.15 | 0.40 | ||
| 1.25 | 0.74 | 0.60 | 0.38 | 0.67 | ||
| 2.5 | 0.12 | 0.94 | 0.31 | 0.67 *** | ||
| 5 | 0.74 * | 0.09 *** | 0.15 | 1.20 | ||
| 2m | 100 | 0.156 | 0.46 | 0.95 | 0.10 | 0.61 |
| 0.3125 | 0.62 | 0.70 | 0.10 | 0.88 | ||
| 0.625 | 0.77 | 0.57 | 0.88 | 0.47 | ||
| 1.25 | 0.46 | 1.08 * | 0.19 | 0.47 | ||
| 2.5 | 1.23 | 0.82 | 0.49 | 0.54 | ||
| 5 | 0.31 | 1.08 * | 0.29 | 0.61 | ||
| Compound | MW | logP | tPSA | nON | nOHNH | Volume | Vio | DLS |
|---|---|---|---|---|---|---|---|---|
| 2a | 321.42 | 3.94 | 30.30 | 4 | 0 | 308.95 | 0 | 0.74 |
| 2b | 337.42 | 3.54 | 39.53 | 5 | 0 | 317.94 | 0 | 1.12 |
| 2c | 341.84 | 4.17 | 30.30 | 4 | 0 | 305.93 | 0 | 1.21 |
| 2d | 325.39 | 3.65 | 30.30 | 4 | 0 | 297.32 | 0 | 1.10 |
| 2e | 349.48 | 5.00 | 30.30 | 4 | 0 | 342.34 | 1 | 1.15 |
| 2f | 375.39 | 4.38 | 30.30 | 4 | 0 | 323.69 | 0 | 0.69 |
| 2g | 386.29 | 4.30 | 30.30 | 4 | 0 | 310.28 | 0 | 0.89 |
| 2h | 378.52 | 4.34 | 33.54 | 5 | 0 | 371.90 | 0 | 0.61 |
| 2i | 350.47 | 3.59 | 33.54 | 5 | 0 | 338.30 | 0 | 0.59 |
| 2j | 351.45 | 3.92 | 39.53 | 5 | 0 | 334.74 | 0 | 1.01 |
| 2k | 332.41 | 3.24 | 54.09 | 5 | 0 | 309.25 | 0 | 0.63 |
| 2l | 413.52 | 5.14 | 39.53 | 5 | 0 | 389.59 | 1 | 0.92 |
| 2m | 391.39 | 4.46 | 39.53 | 5 | 0 | 332.67 | 0 | 0.88 |
| 2n | 353.49 | 3.92 | 30.30 | 4 | 0 | 327.08 | 0 | 0.92 |
| 2o | 383.50 | 5.28 | 30.30 | 4 | 0 | 363.80 | 1 | 0.68 |
| Donepezil | 379.50 | 4.10 | 38.78 | 4 | 0 | 367.89 | 0 | 1.76 |
| Moclobemide | 268.74 | 1.69 | 41.57 | 4 | 0 | 240.70 | 0 | 1.36 |
| Selegiline | 187.29 | 2.64 | 3.24 | 1 | 0 | 202.64 | 0 | 1.03 |
| Chloramphenicol | 323.13 | 0.73 | 115.38 | 7 | 3 | 249.16 | 0 | 0.89 |
| Ketoconazole | 531.44 | 3.77 | 69.08 | 8 | 0 | 452.47 | 1 | 1.32 |
| Ibuprofen | 206.28 | 3.46 | 37.30 | 2 | 1 | 211.19 | 0 | 0.96 |
| Nimesulide | 308.31 | 2.81 | 101.23 | 7 | 1 | 248.17 | 0 | −1.40 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Can, N.Ö.; Çevik, U.A.; Sağlık, B.N.; Özkay, Y.; Atlı, Ö.; Baysal, M.; Özkay, Ü.D.; Can, Ö.D. Pharmacological and Toxicological Screening of Novel Benzimidazole-Morpholine Derivatives as Dual-Acting Inhibitors. Molecules 2017, 22, 1374. https://doi.org/10.3390/molecules22081374
Can NÖ, Çevik UA, Sağlık BN, Özkay Y, Atlı Ö, Baysal M, Özkay ÜD, Can ÖD. Pharmacological and Toxicological Screening of Novel Benzimidazole-Morpholine Derivatives as Dual-Acting Inhibitors. Molecules. 2017; 22(8):1374. https://doi.org/10.3390/molecules22081374
Chicago/Turabian StyleCan, Nafiz Öncü, Ulviye Acar Çevik, Begüm Nurpelin Sağlık, Yusuf Özkay, Özlem Atlı, Merve Baysal, Ümide Demir Özkay, and Özgür Devrim Can. 2017. "Pharmacological and Toxicological Screening of Novel Benzimidazole-Morpholine Derivatives as Dual-Acting Inhibitors" Molecules 22, no. 8: 1374. https://doi.org/10.3390/molecules22081374