β-Formyl- and β-Vinylporphyrins: Magic Building Blocks for Novel Porphyrin Derivatives †

 ,
,  , , and
, , and
Abstract
:
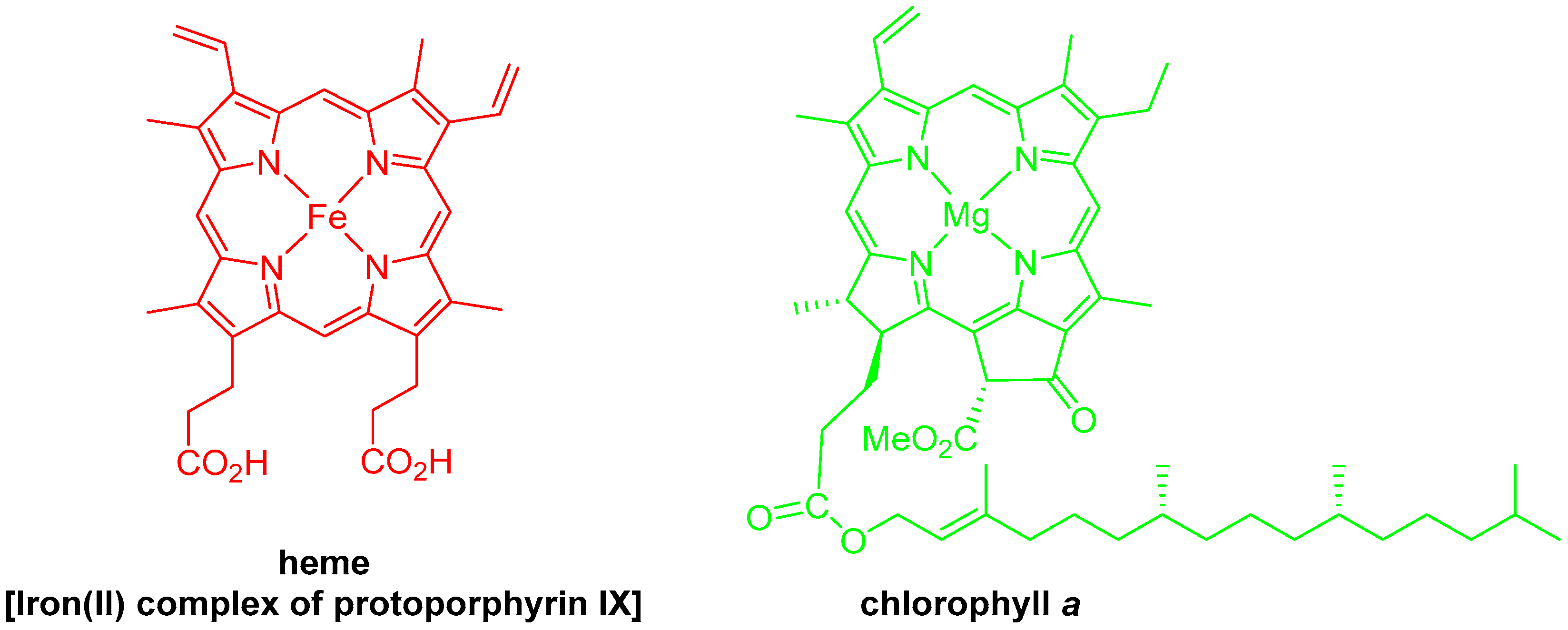
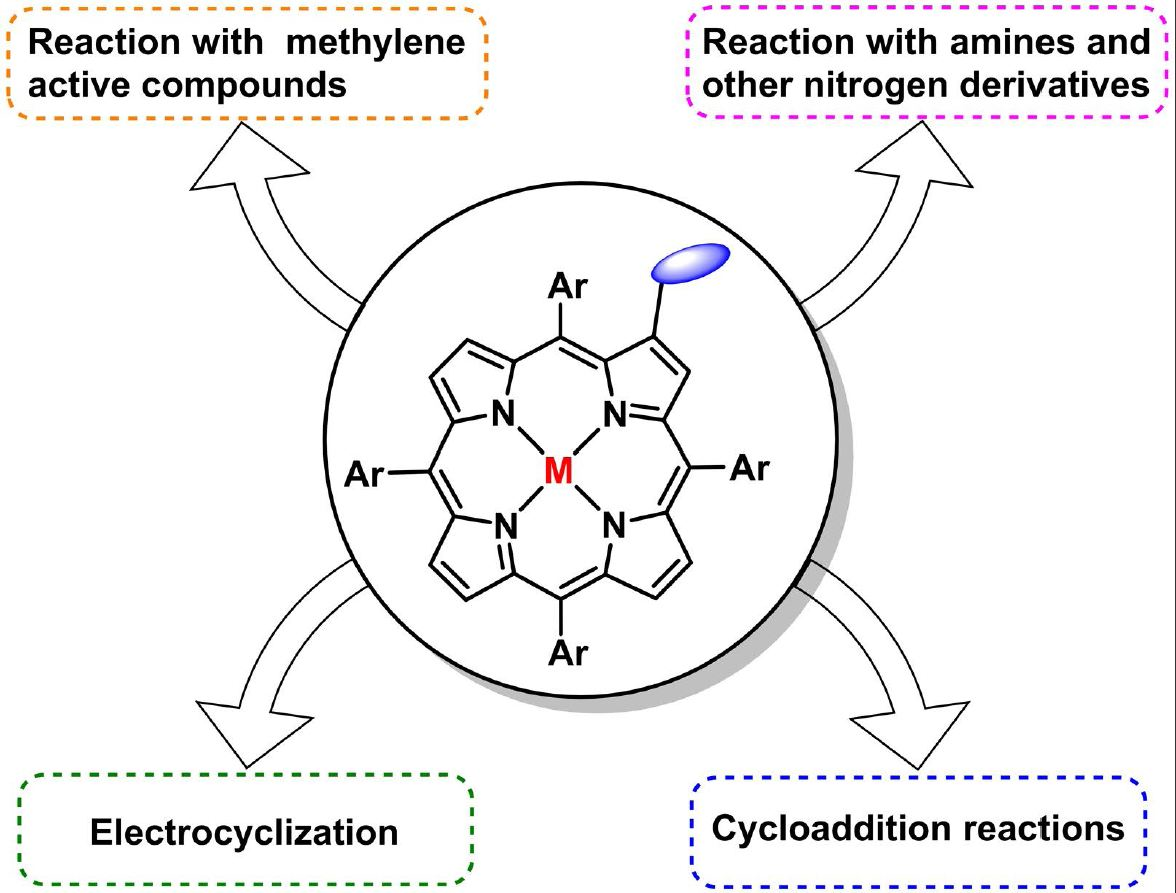
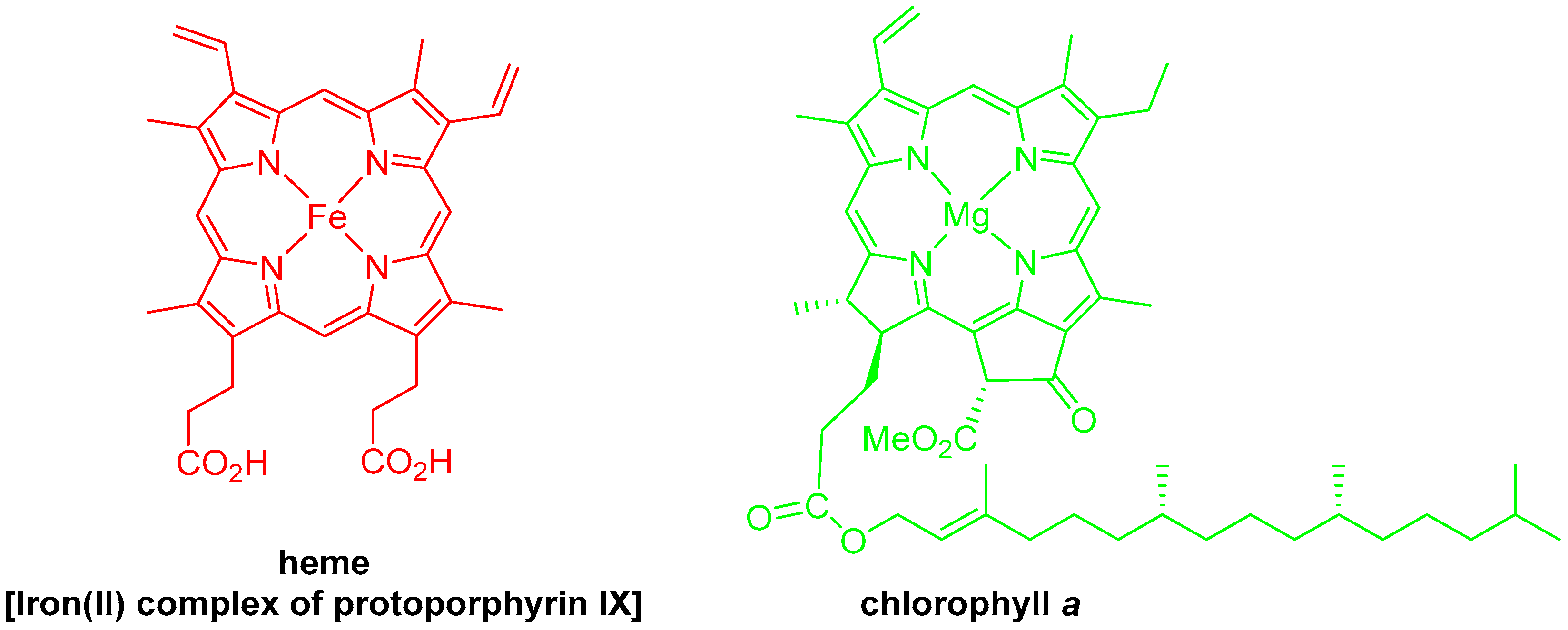
1. Introduction
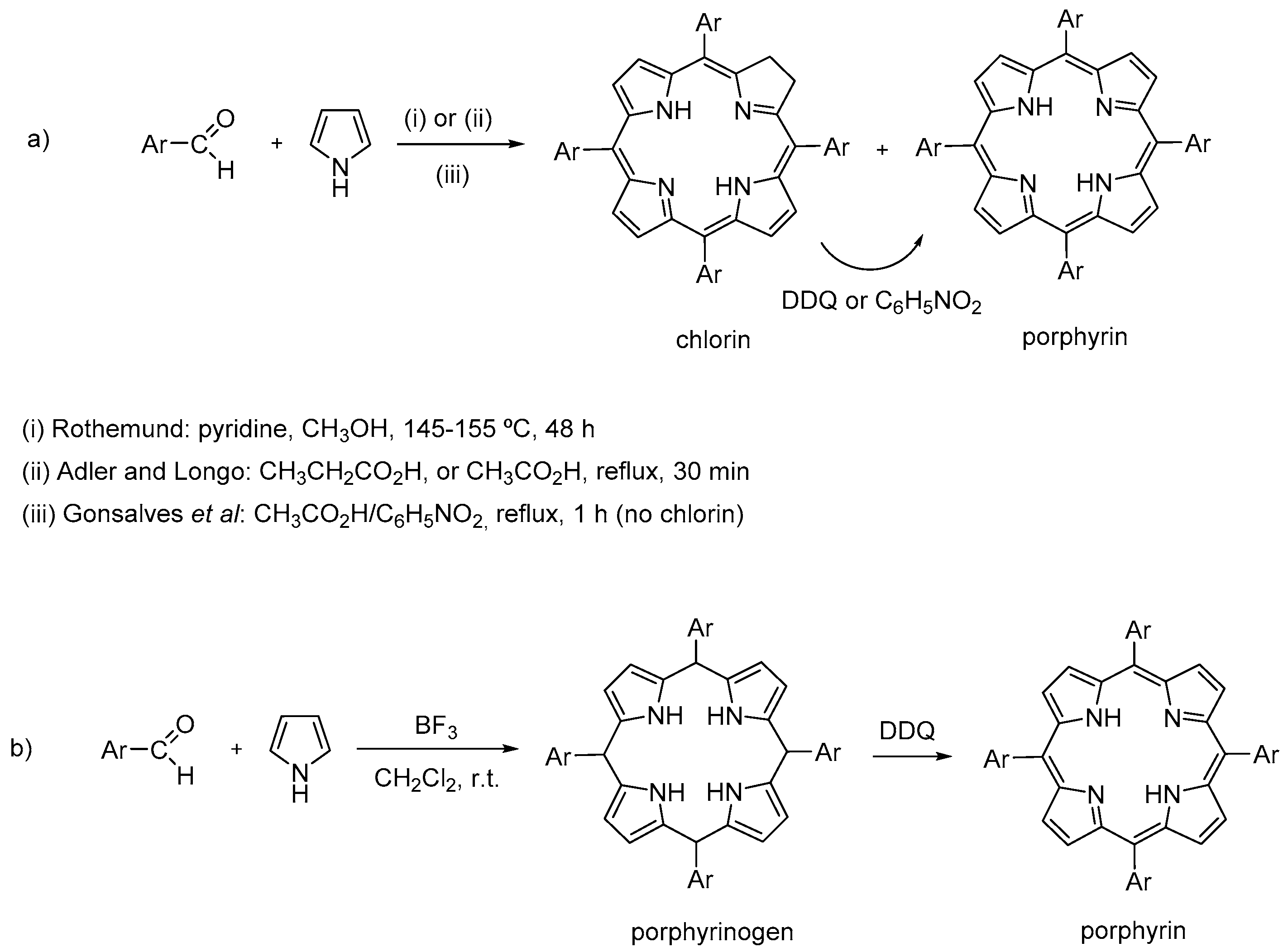
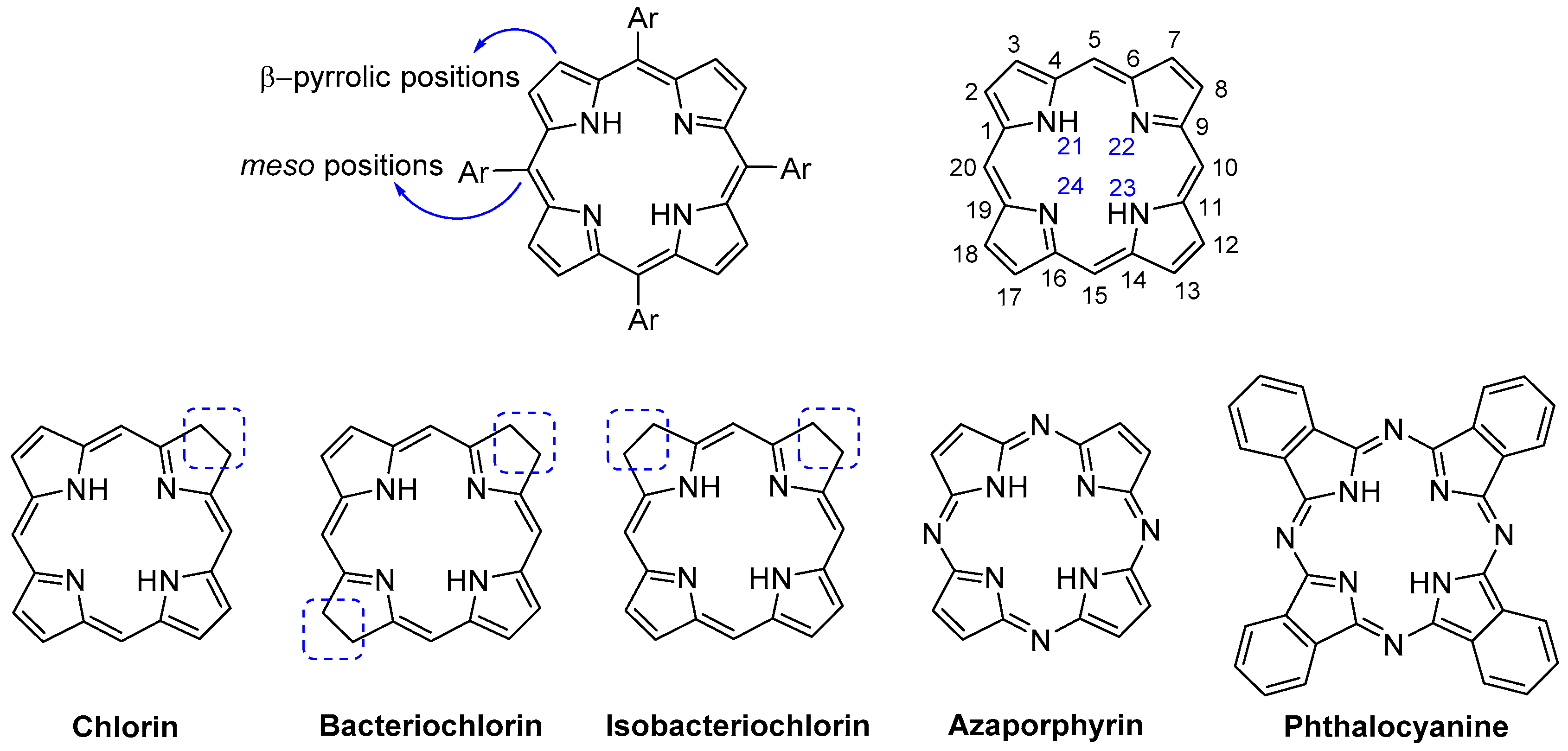
2. General Considerations about the Synthesis and Properties of meso-Tetraarylporphyrins
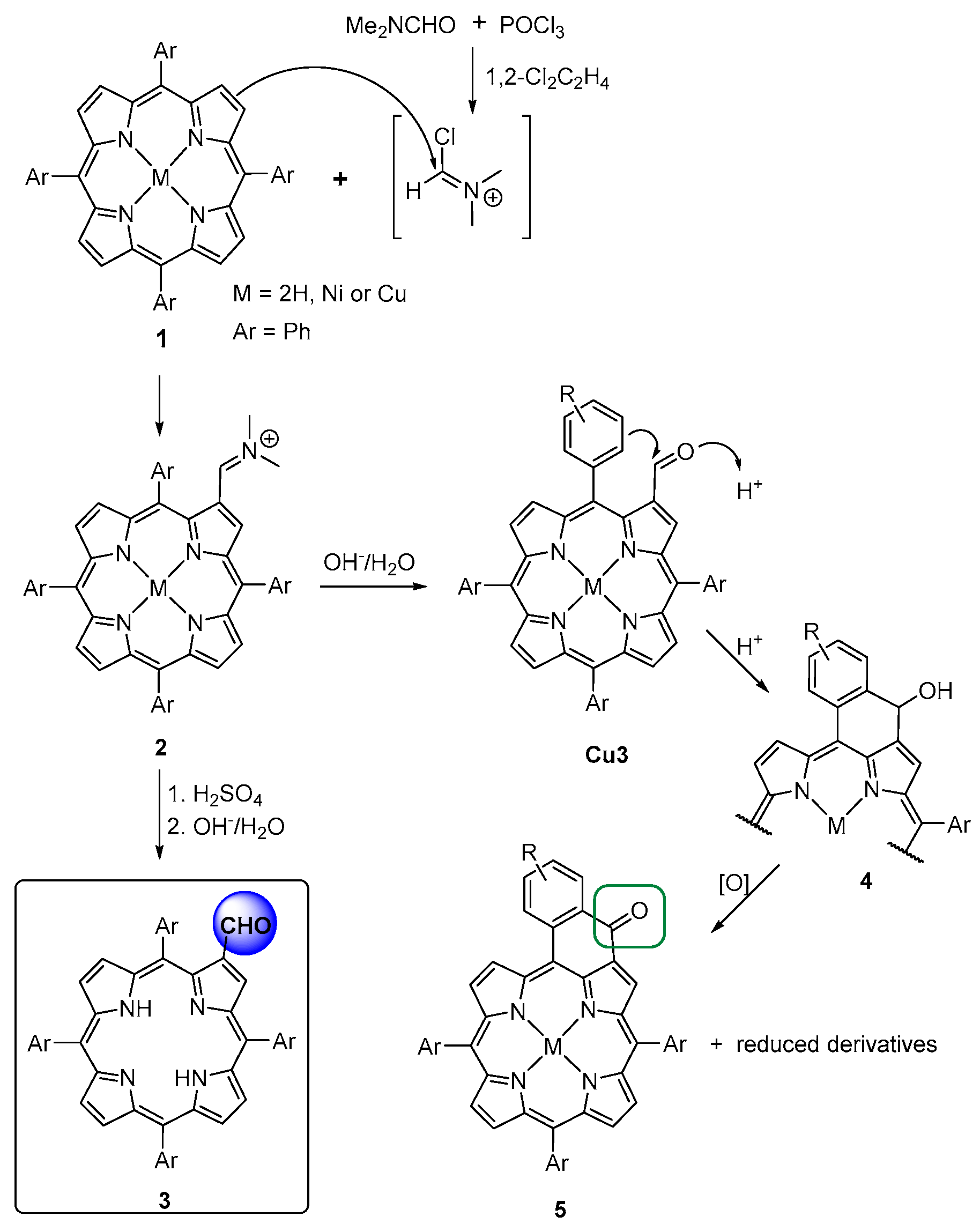
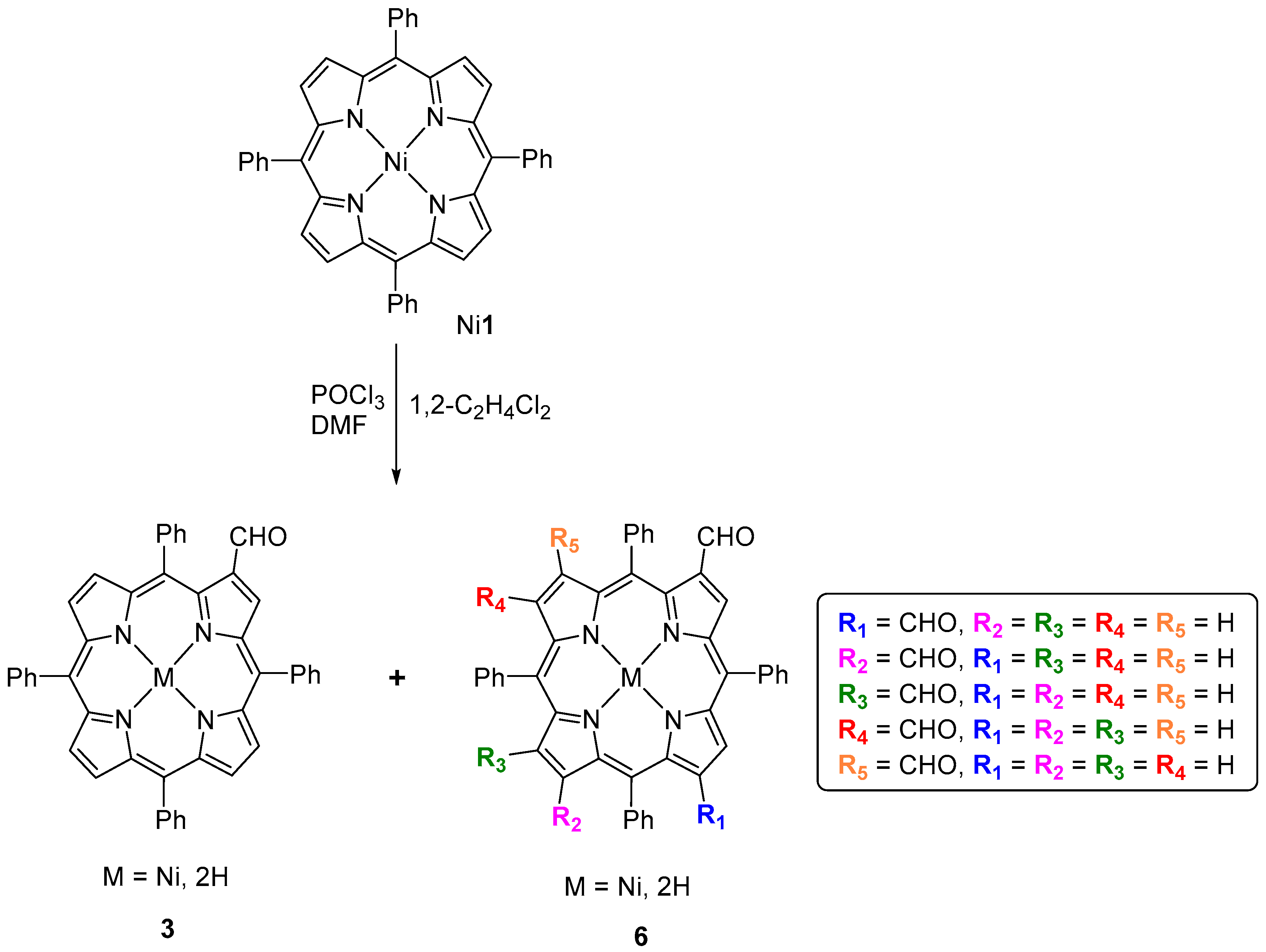
3. β-Formylation of meso-Tetraarylporphyrins
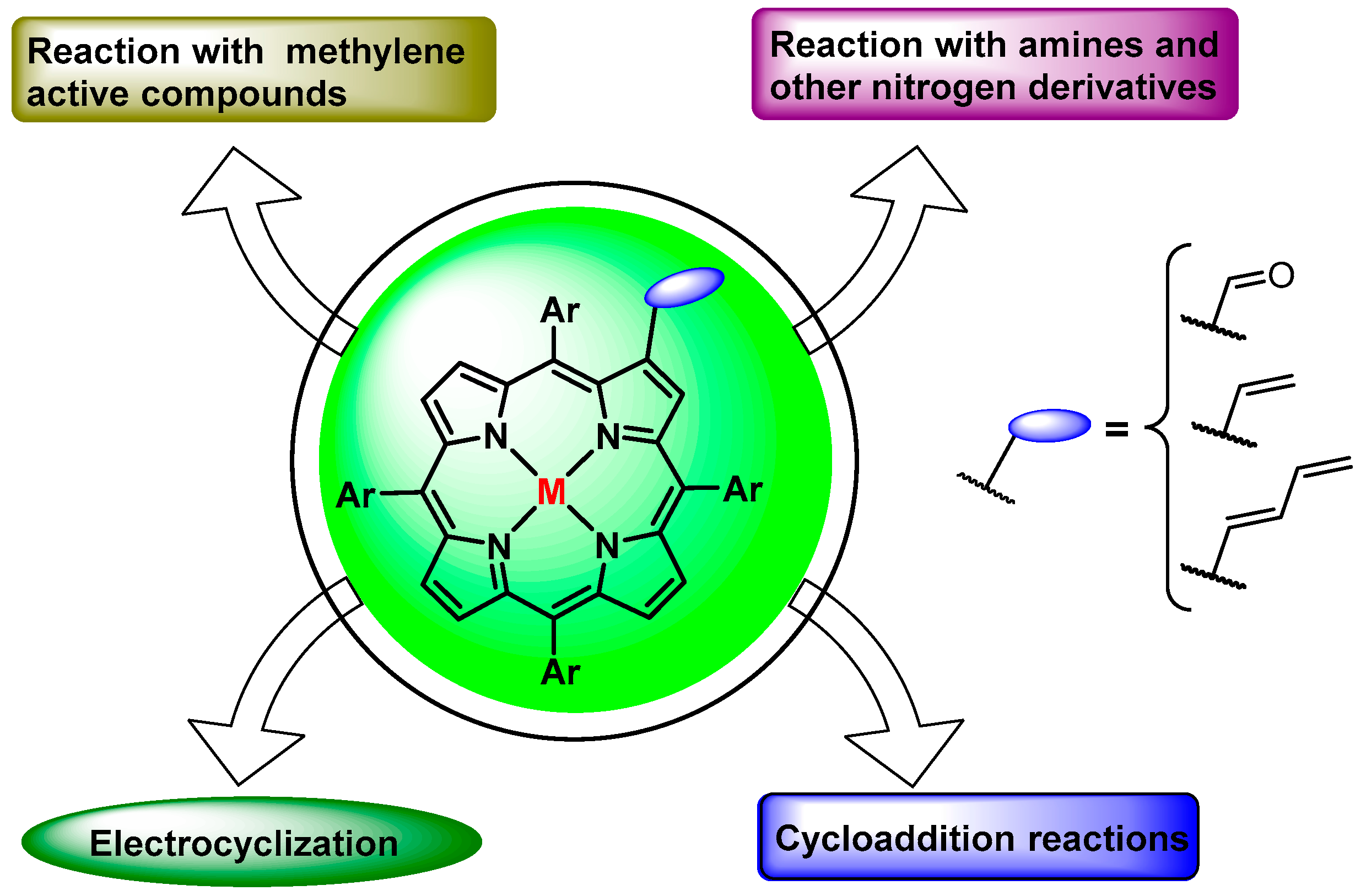
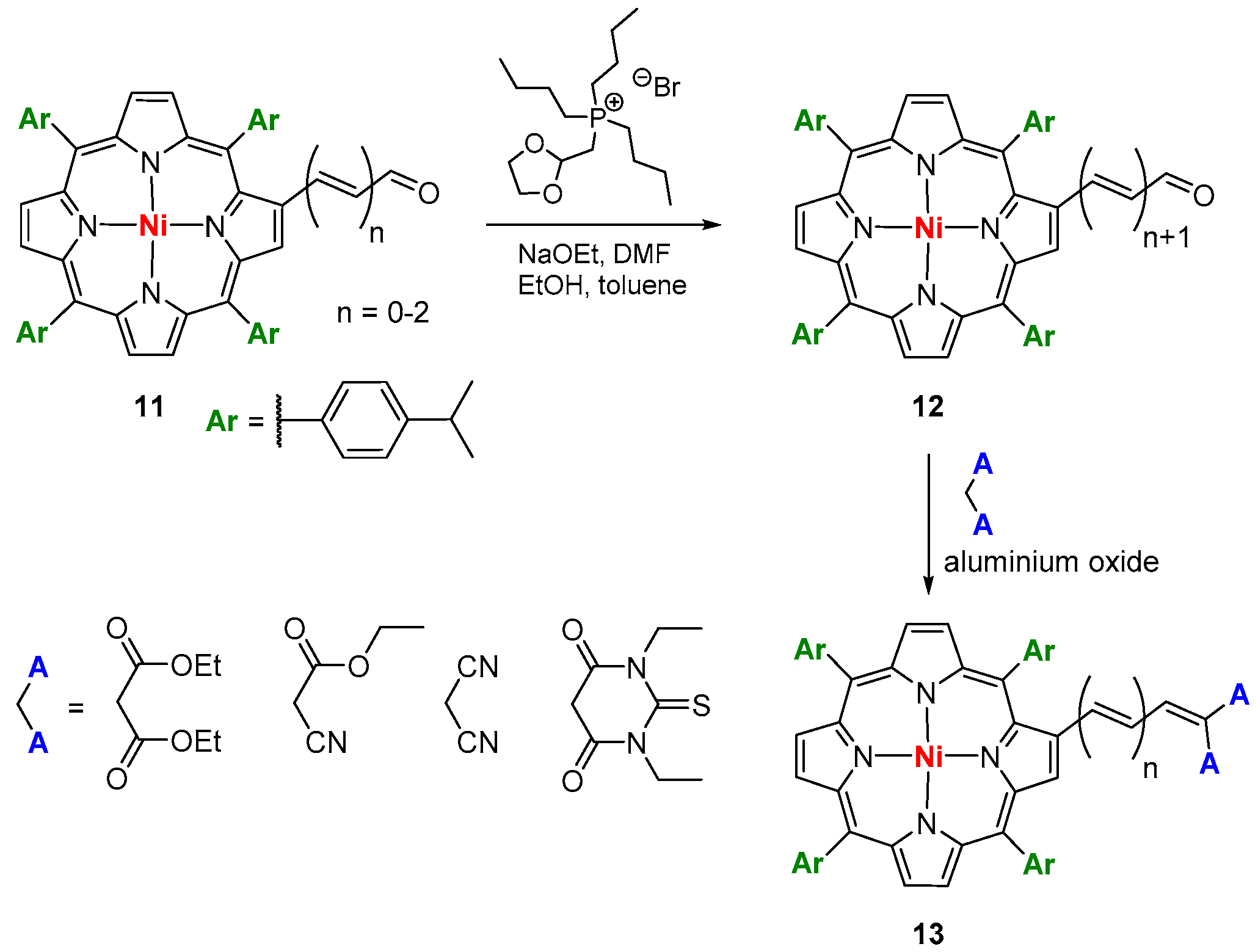
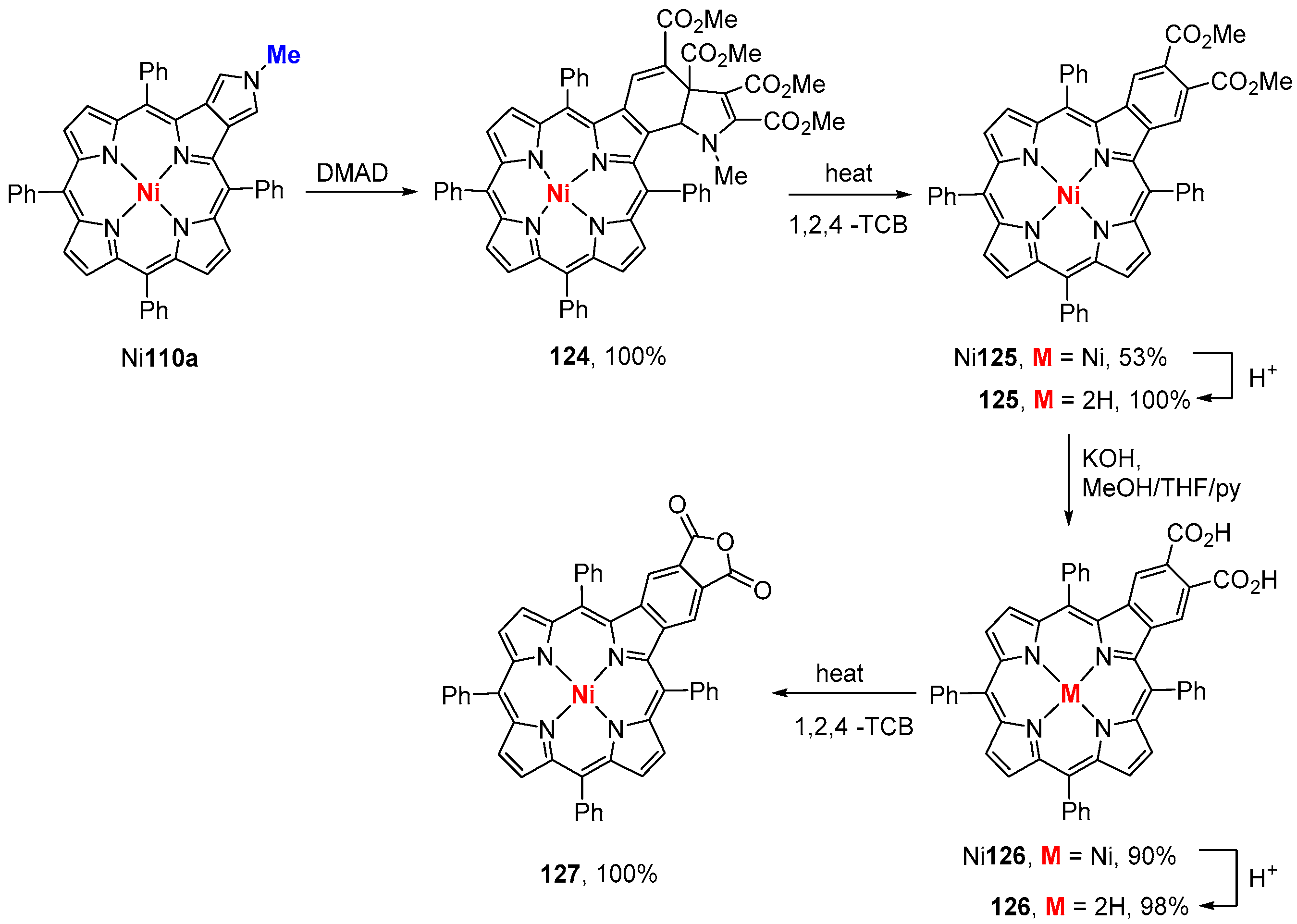
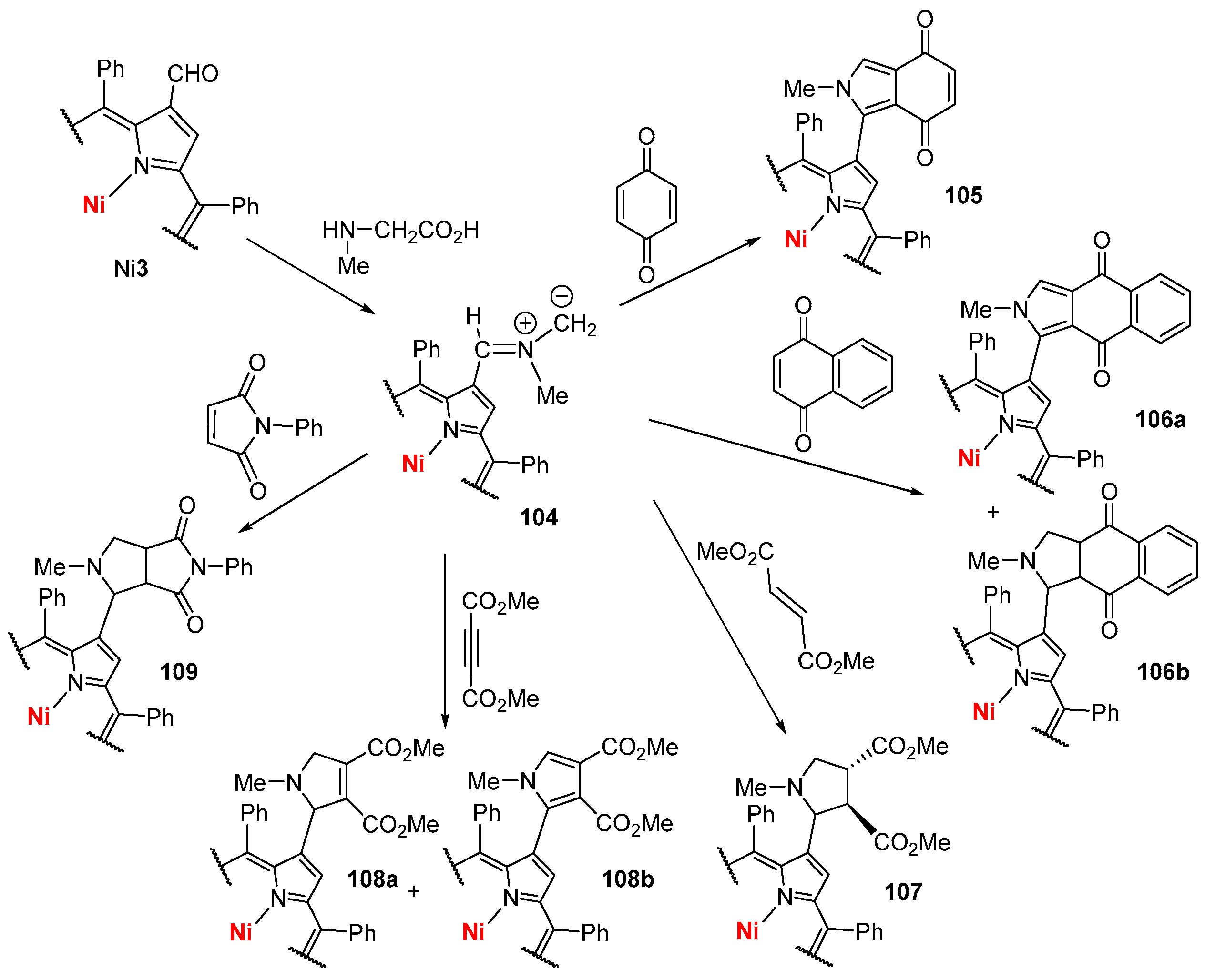
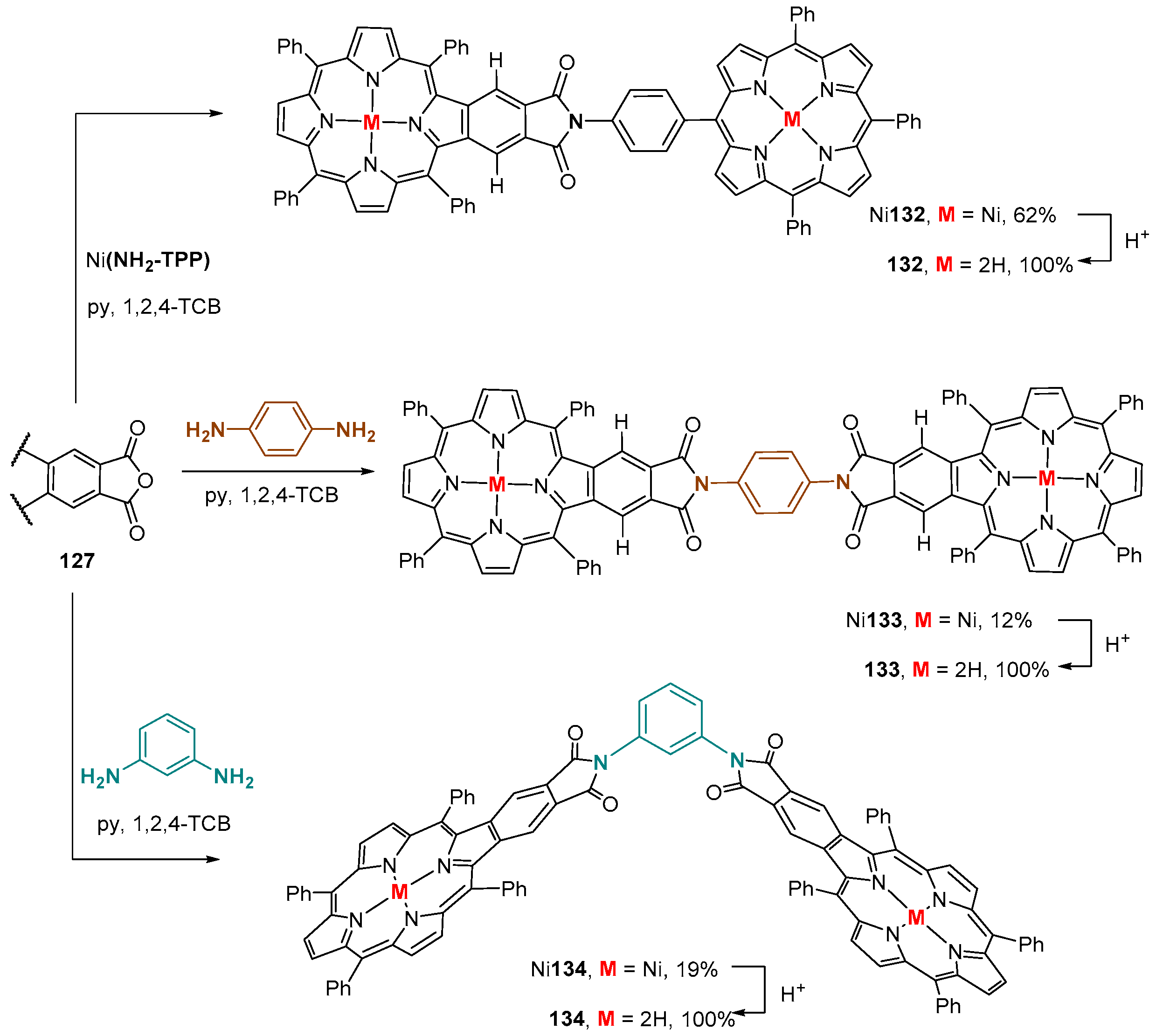
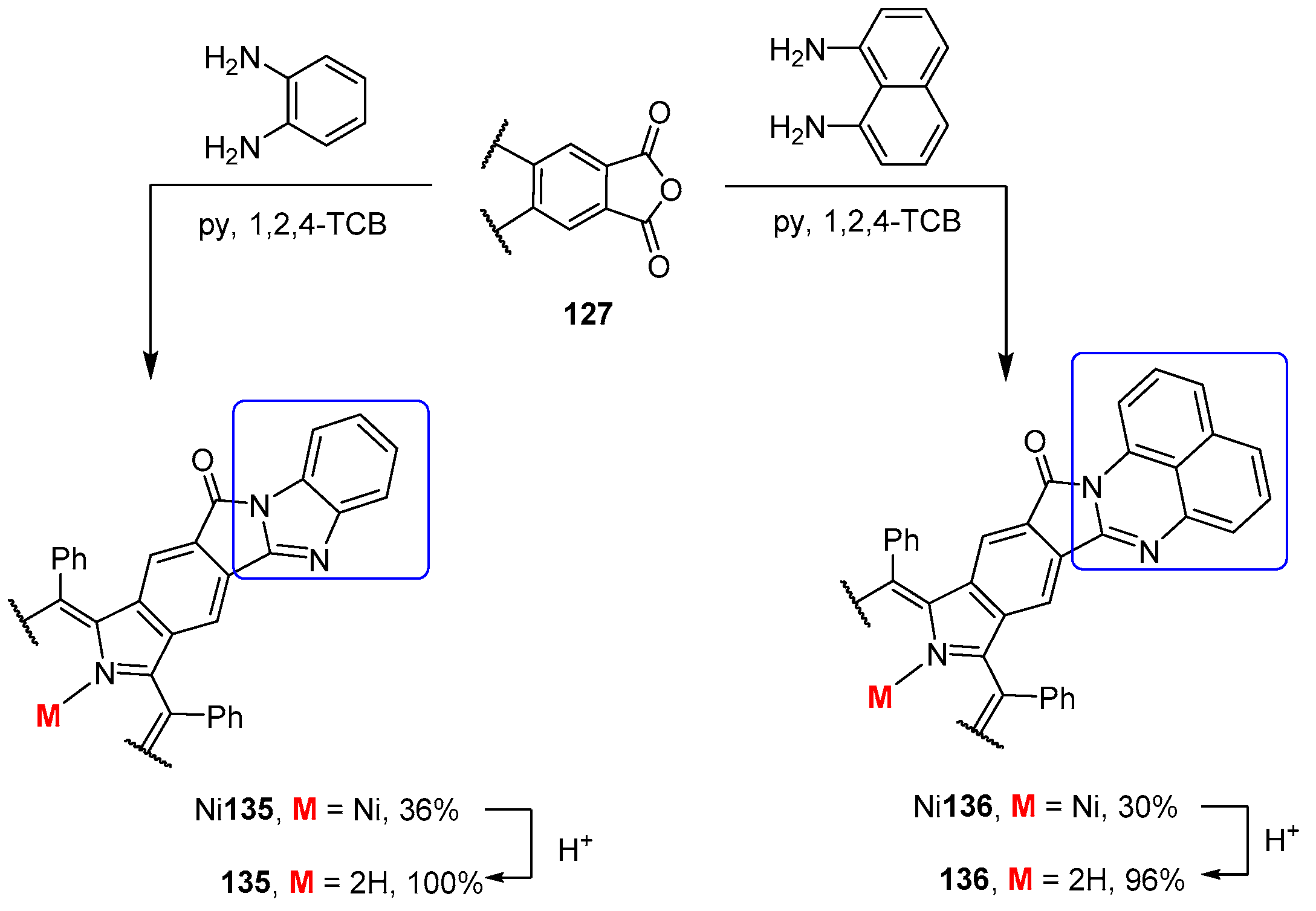
4. Reactivity of β-Formyl-meso-tetraarylporphyrins with Methylene Active Compounds
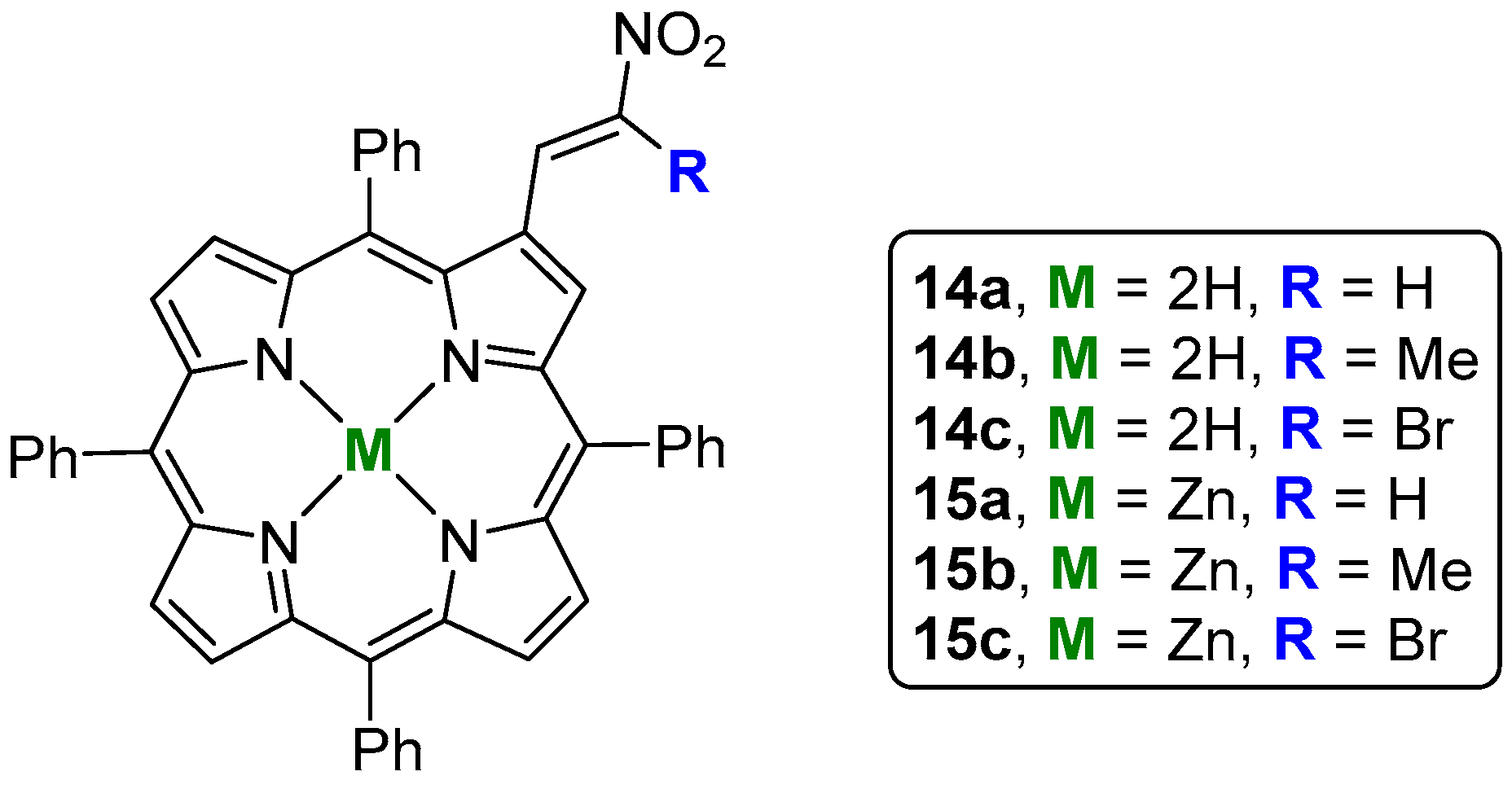
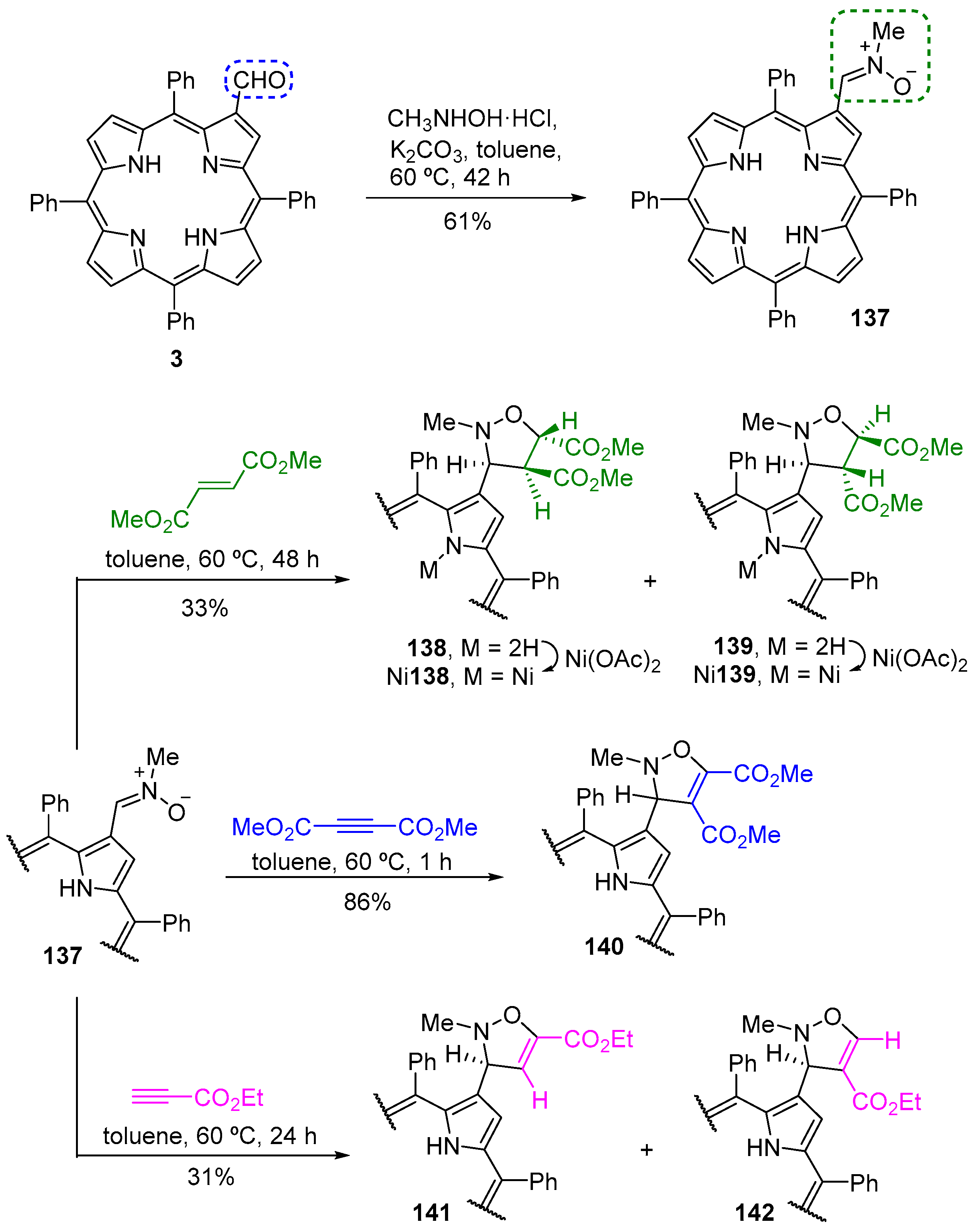
5. Reactivity of β-Formyl-meso-tetraarylporphyrins with Wittig Reagents
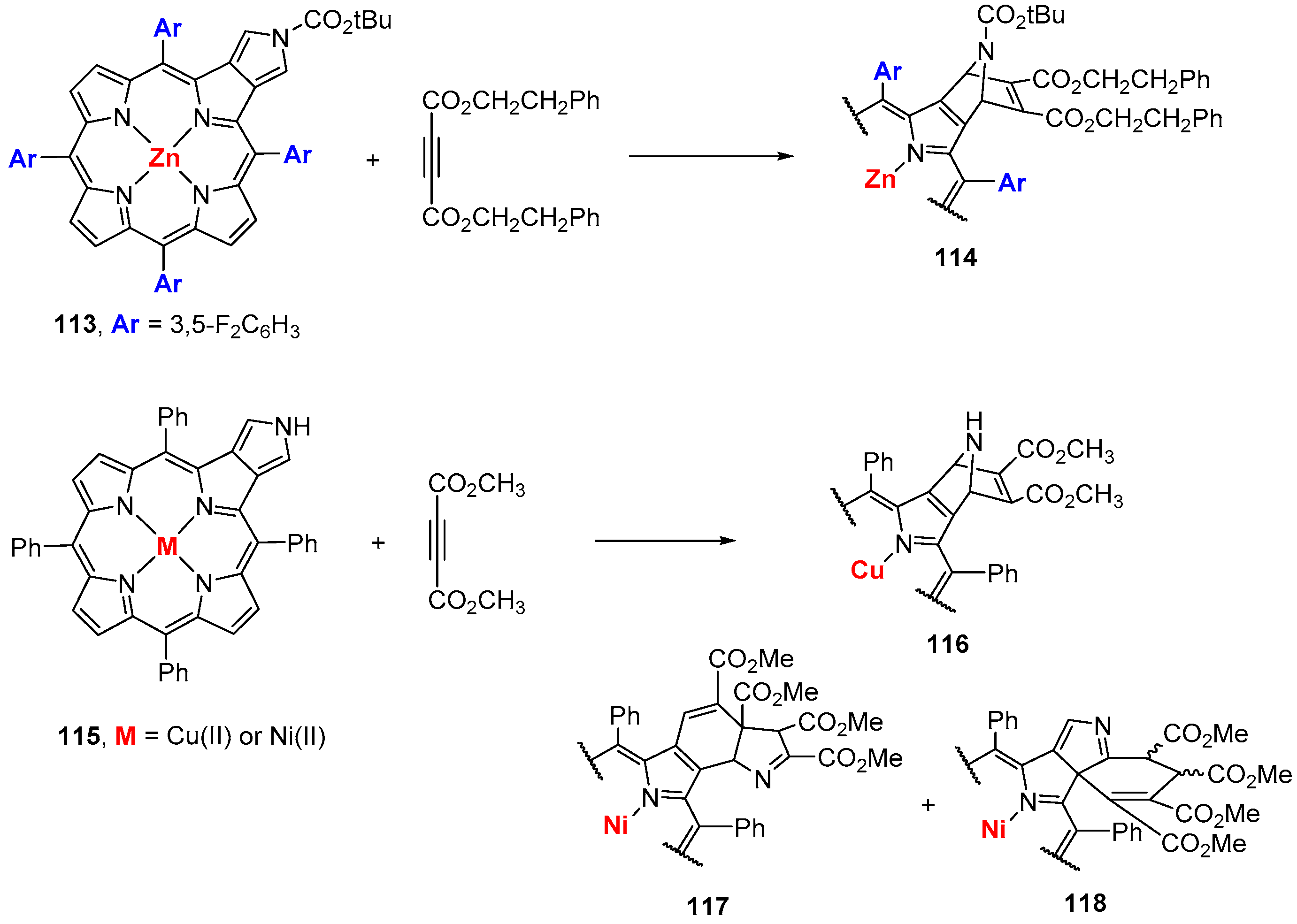
5.1. β-Vinylporphyrins as 4π Components in Cycloaddition Reactions
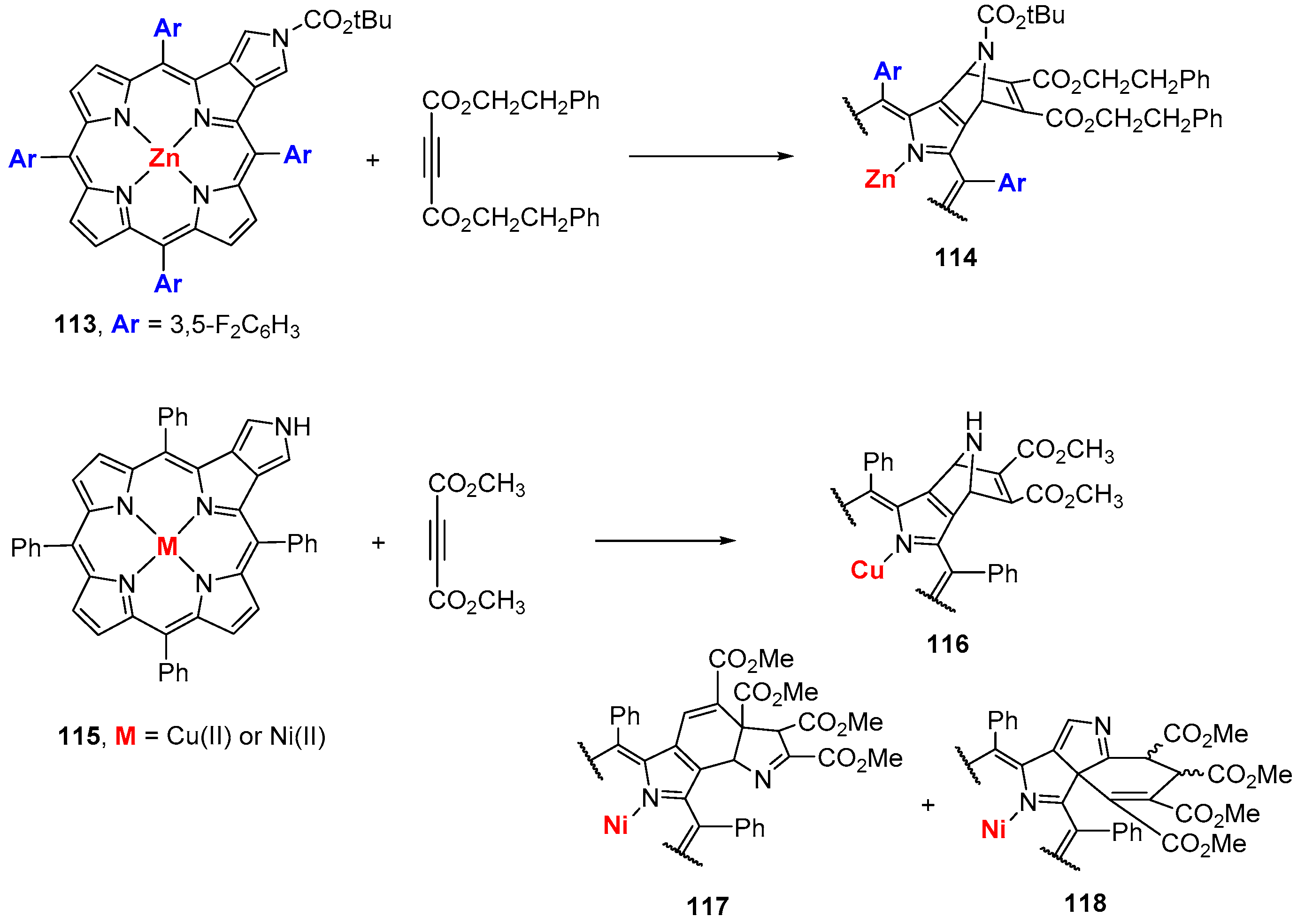
5.2. β-Vinylporphyrins As 2π Component in Cycloaddition Reactions
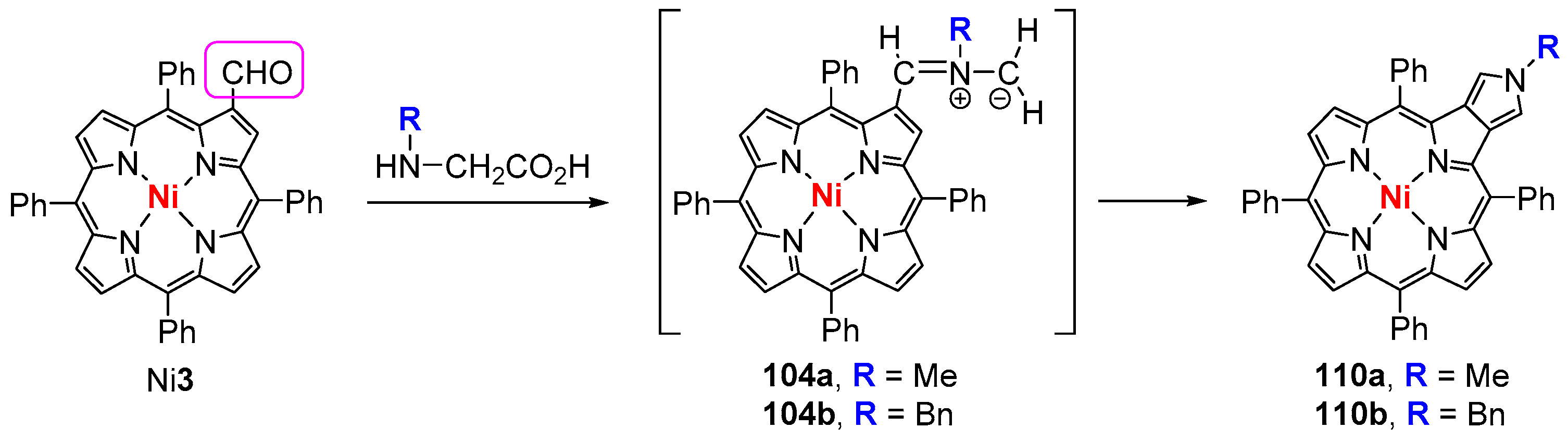
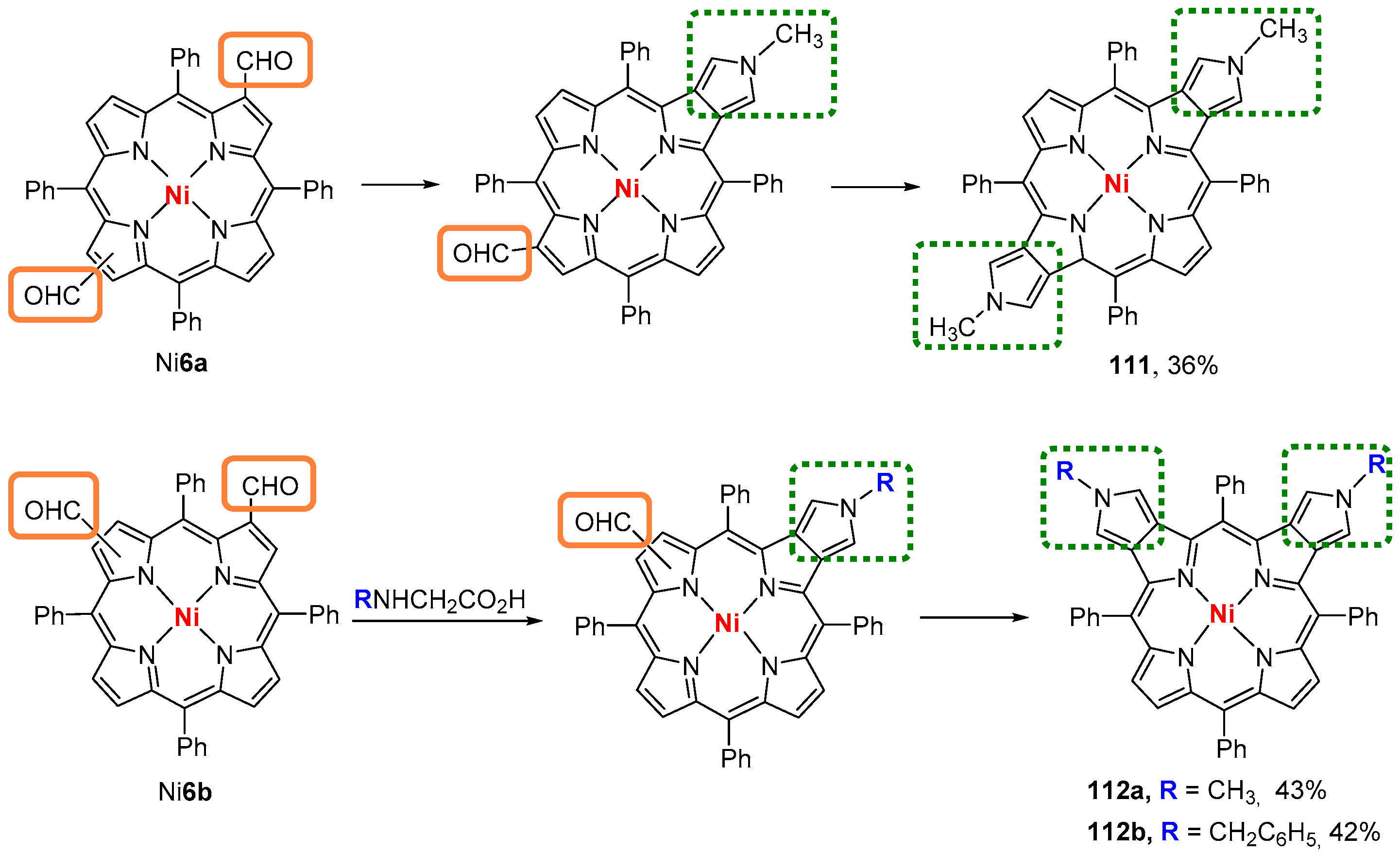
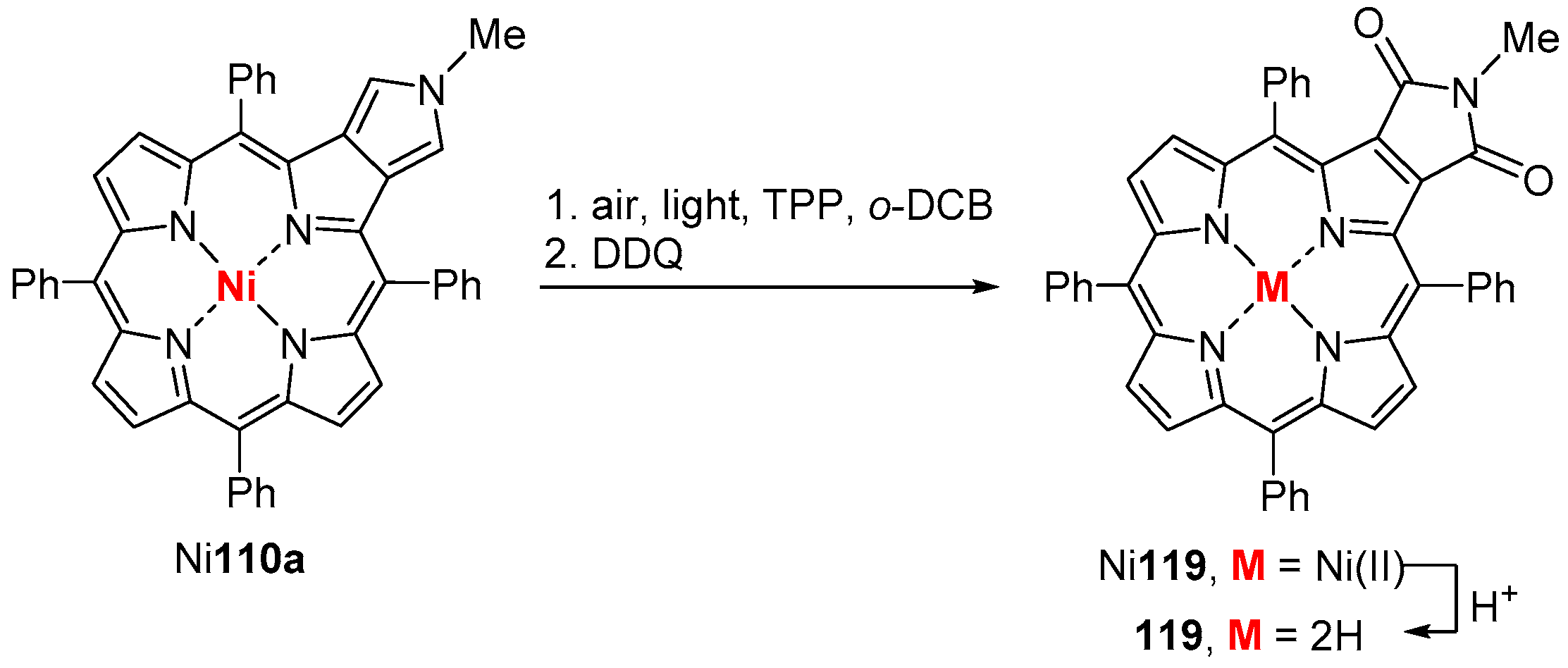
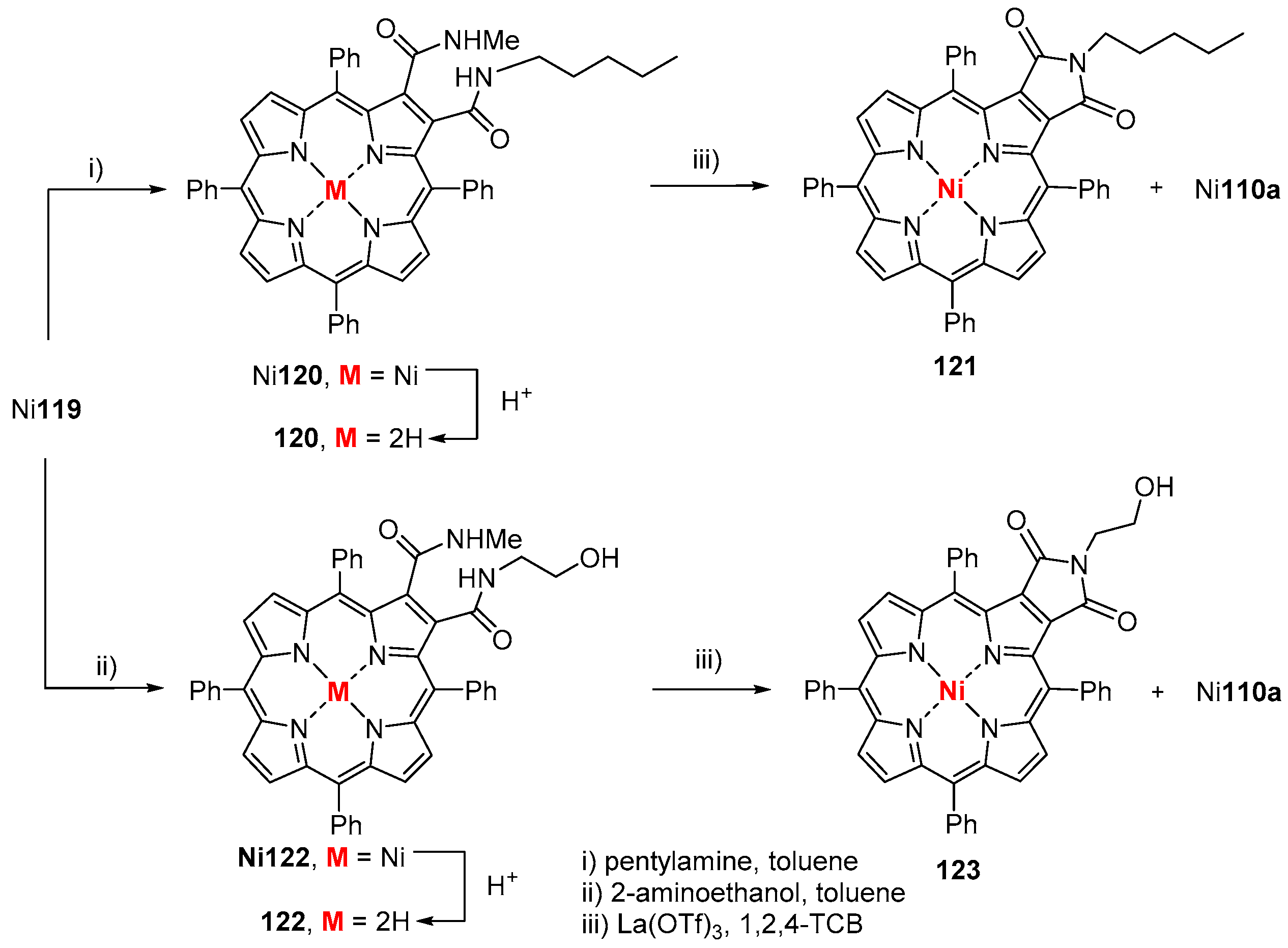
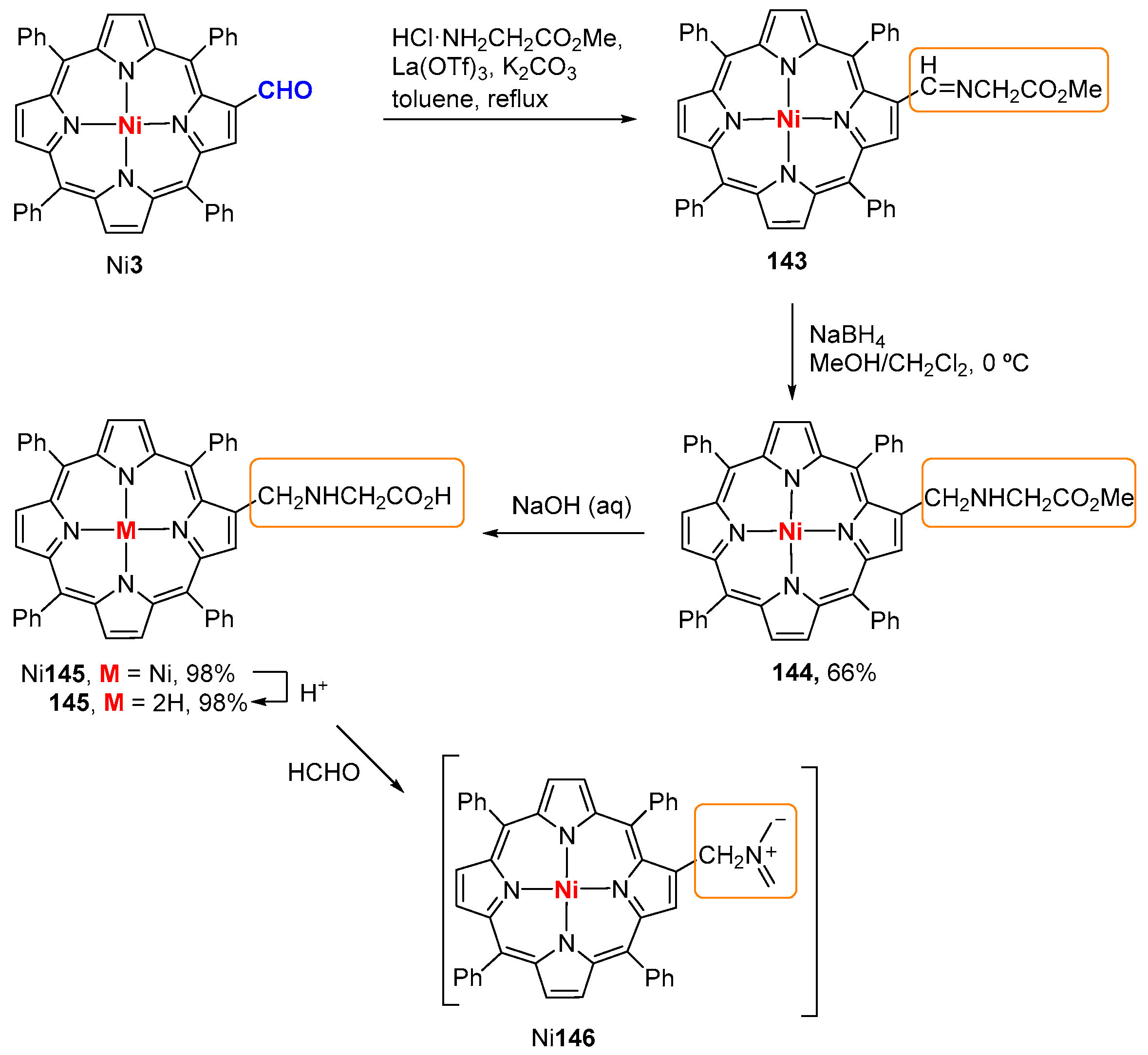
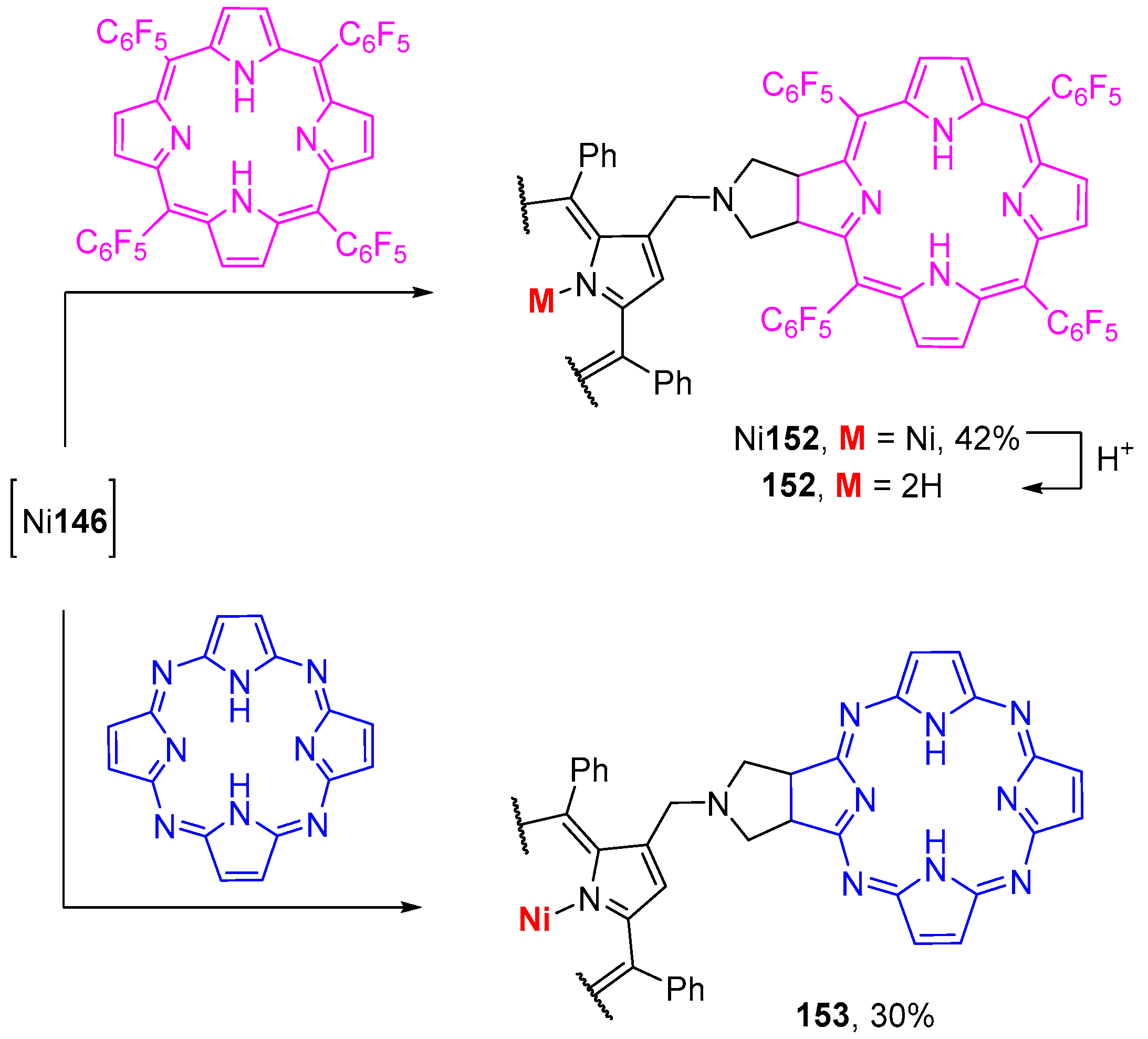
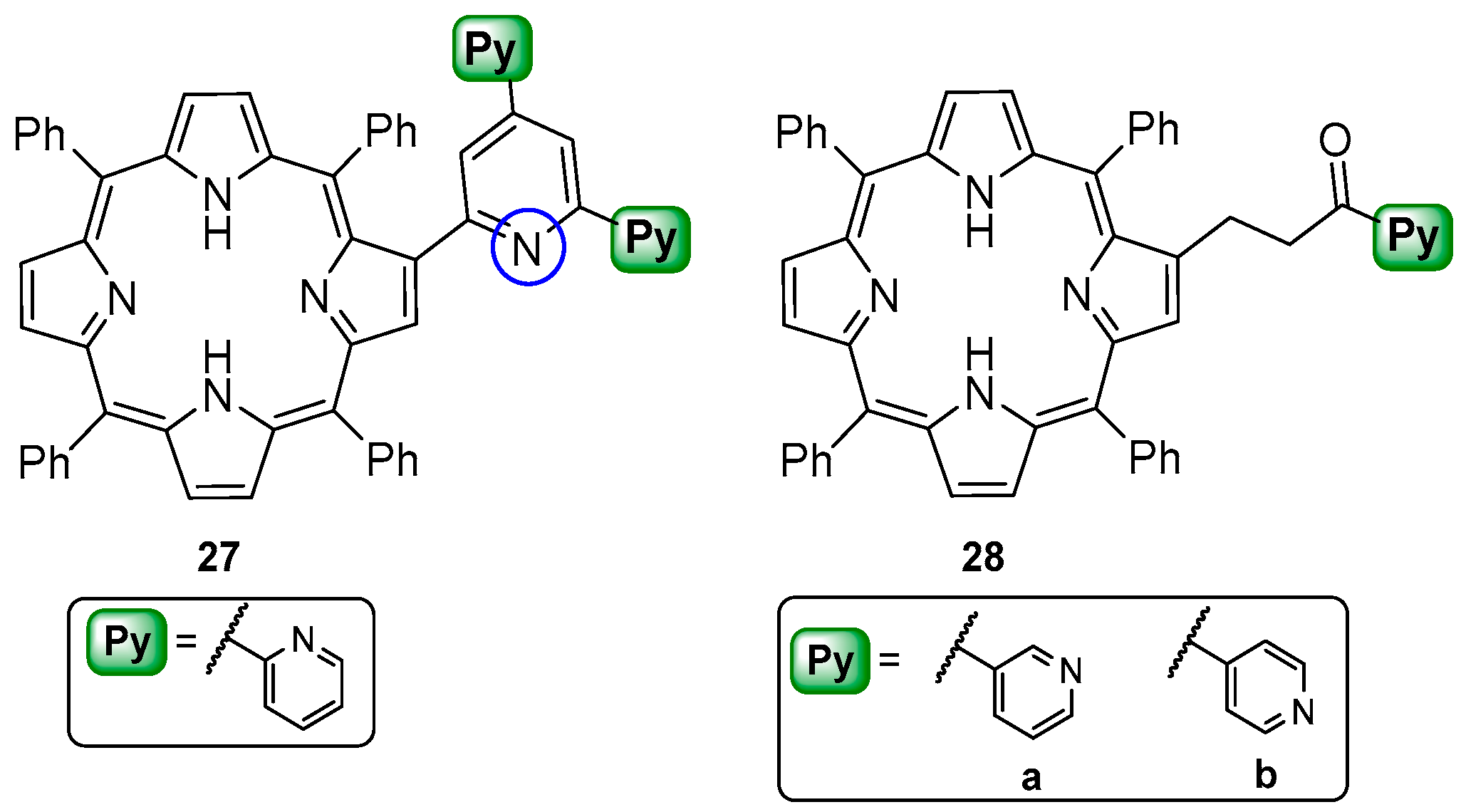
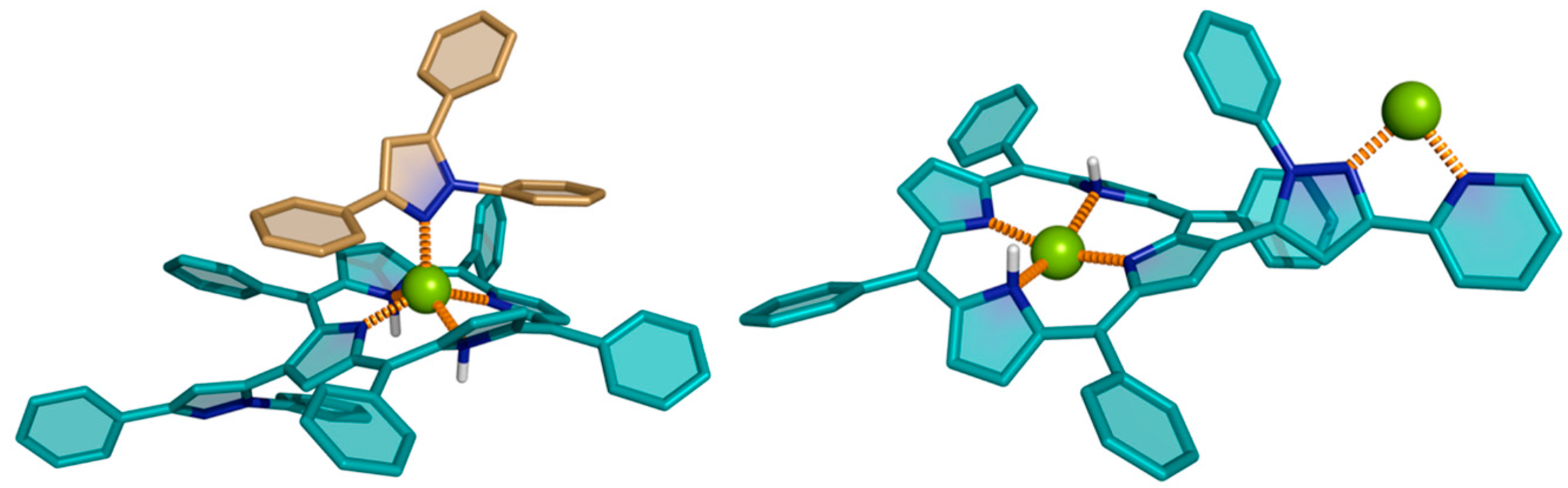
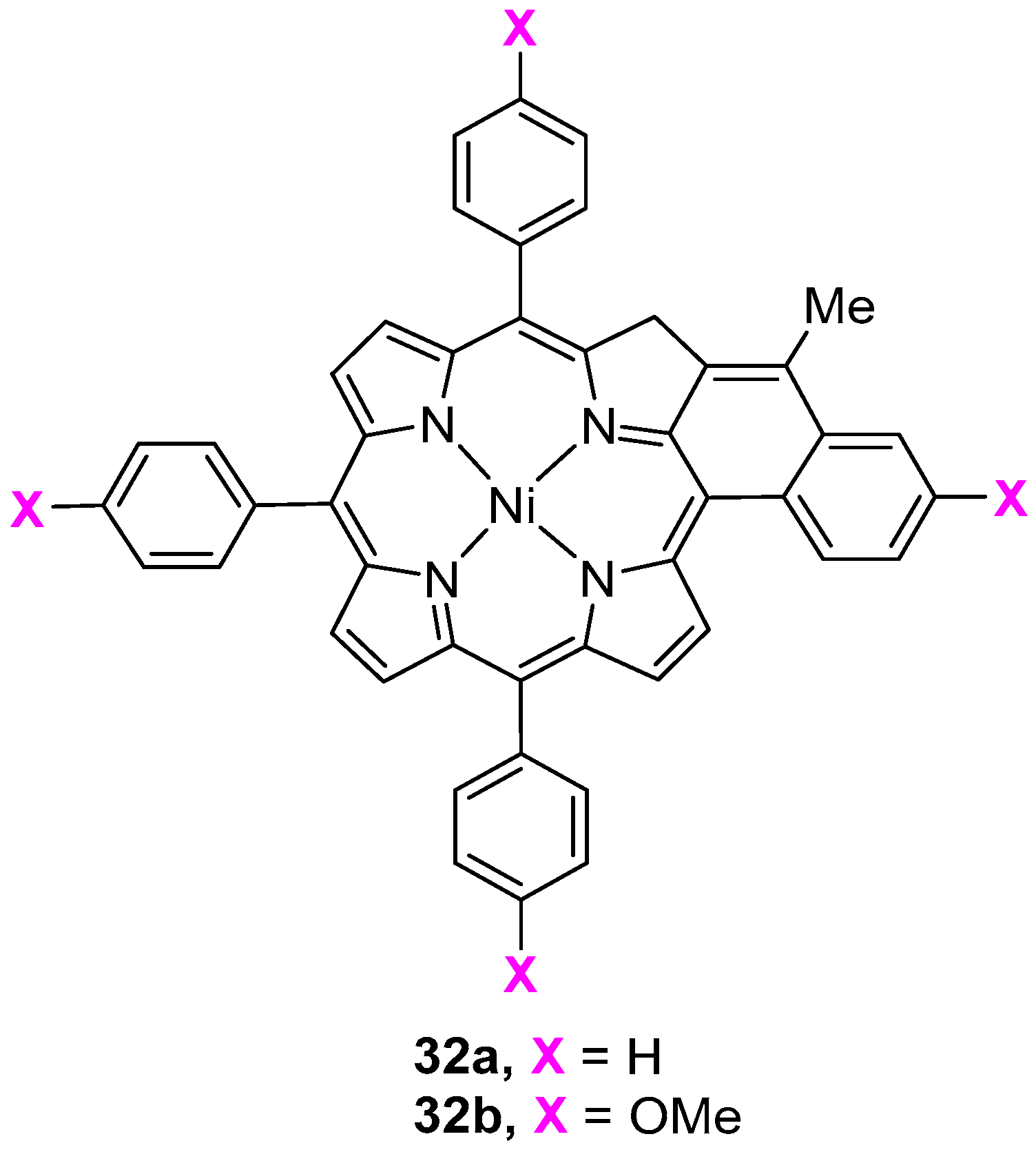
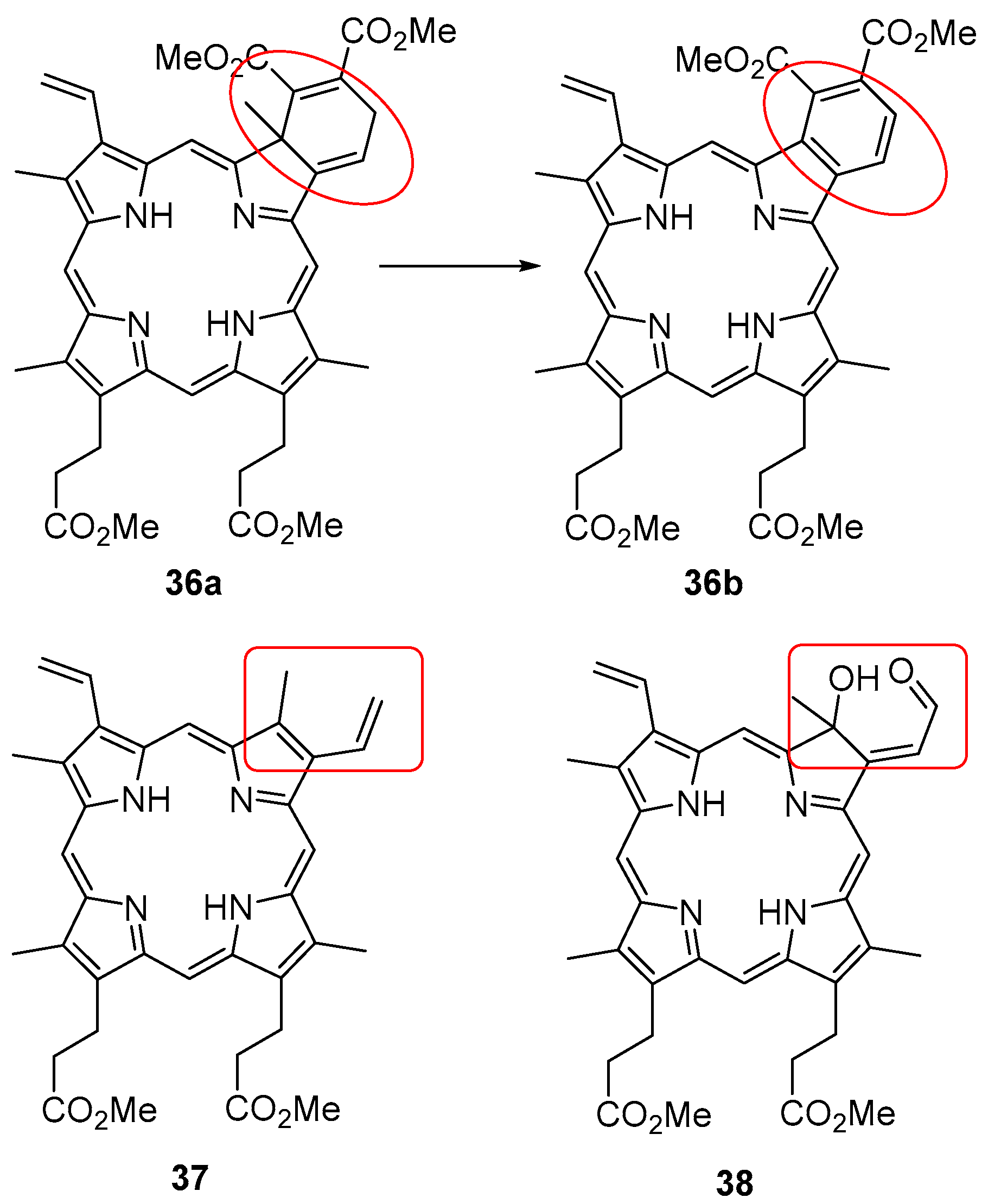
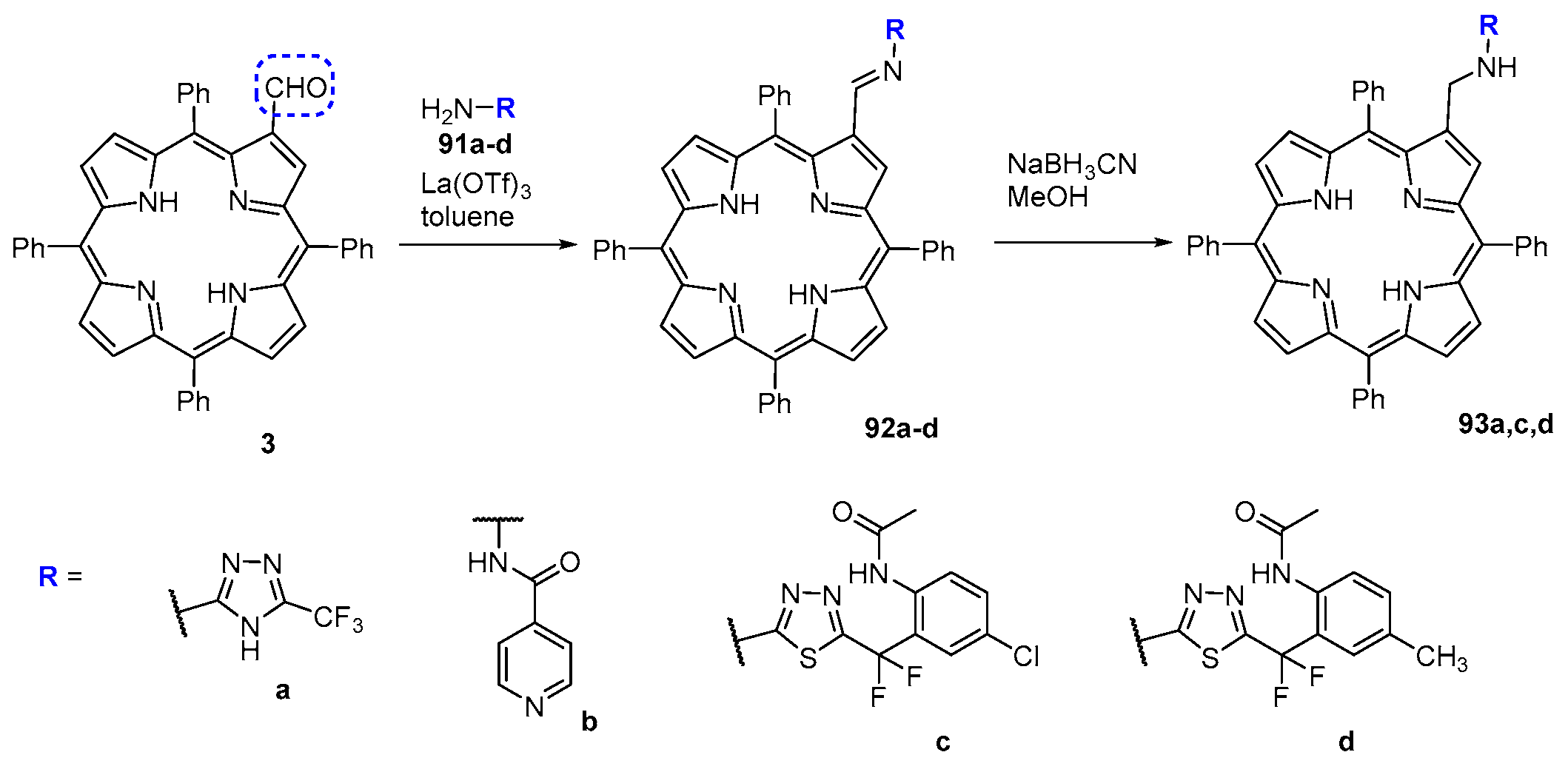
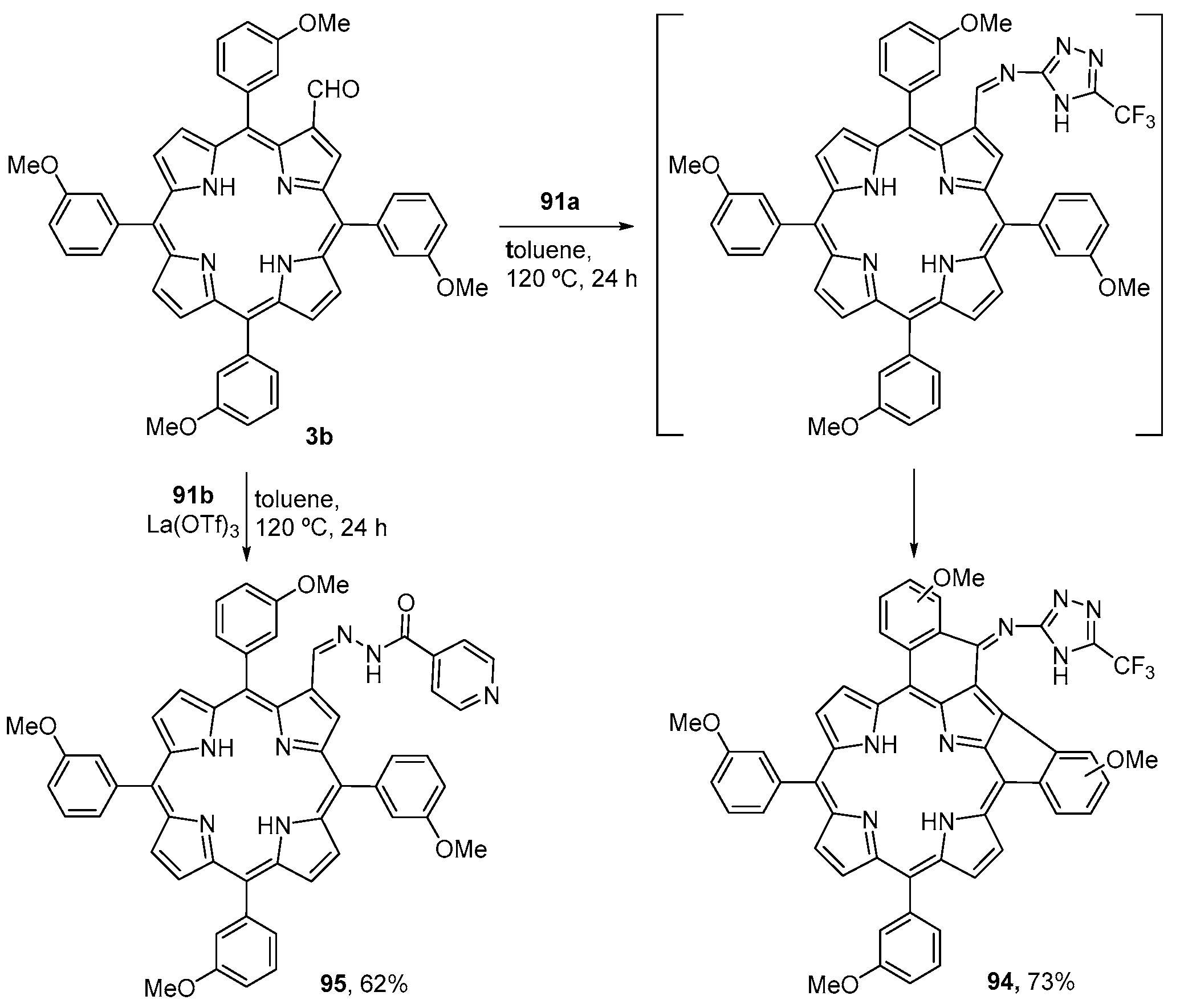
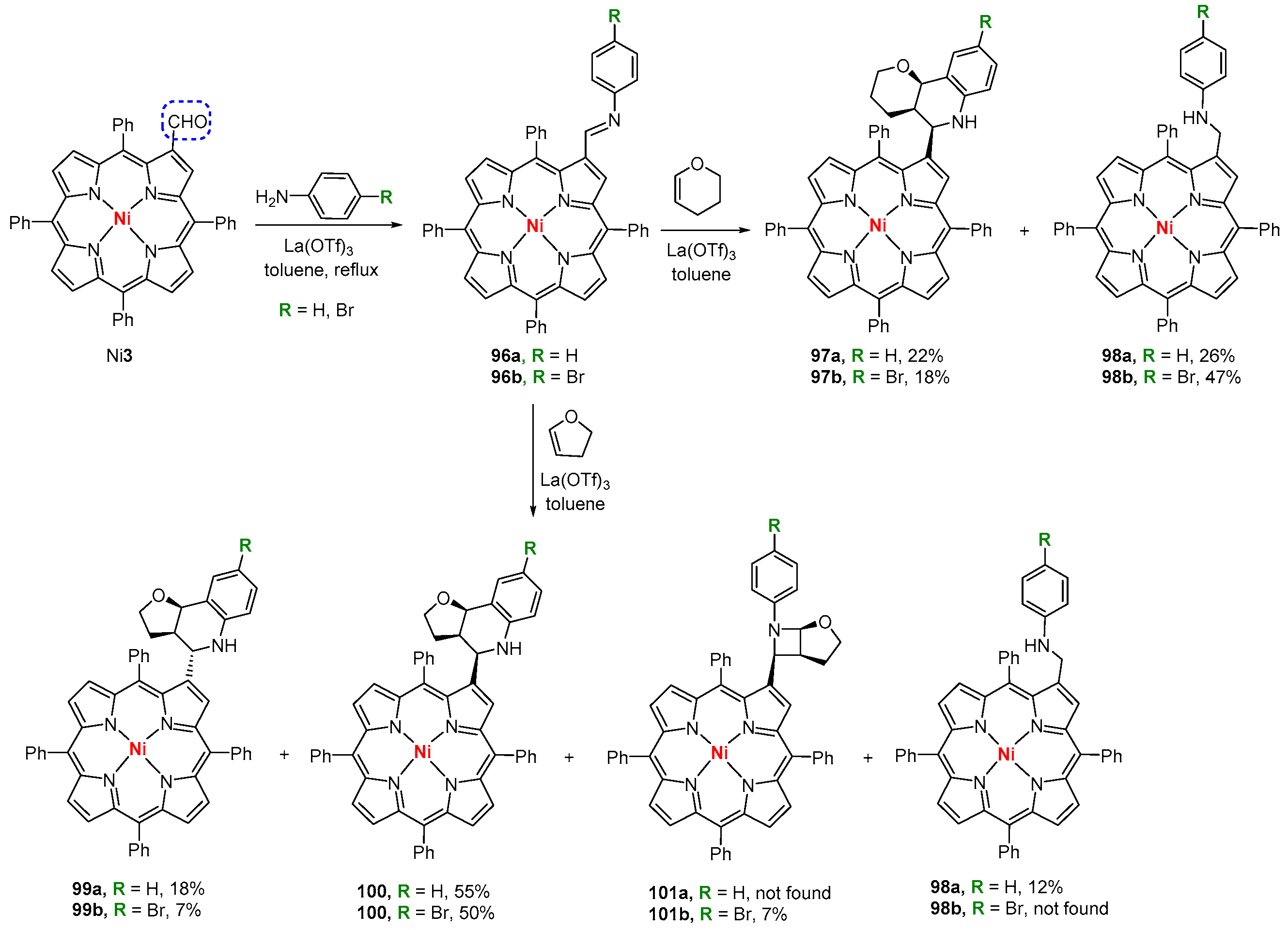
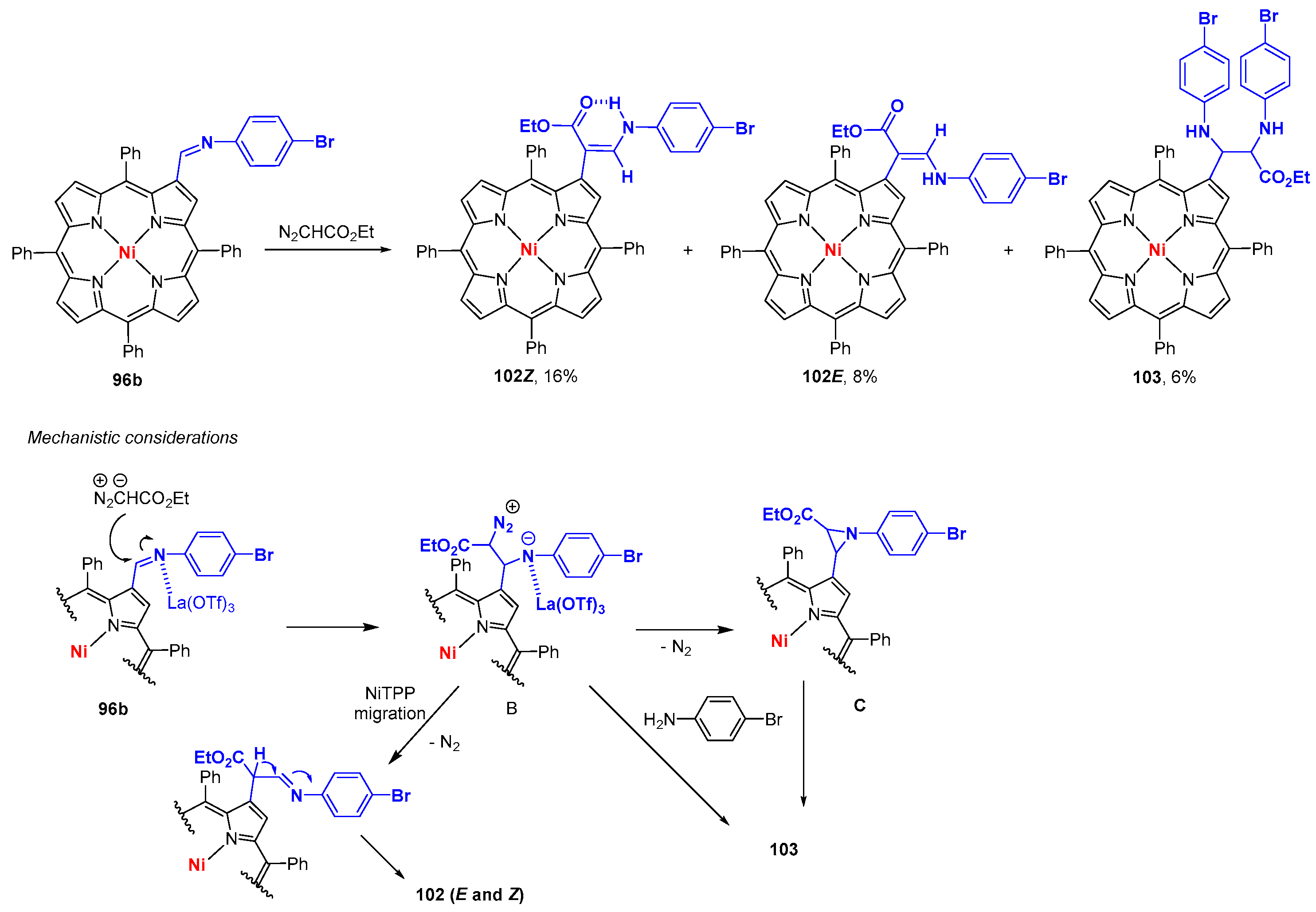
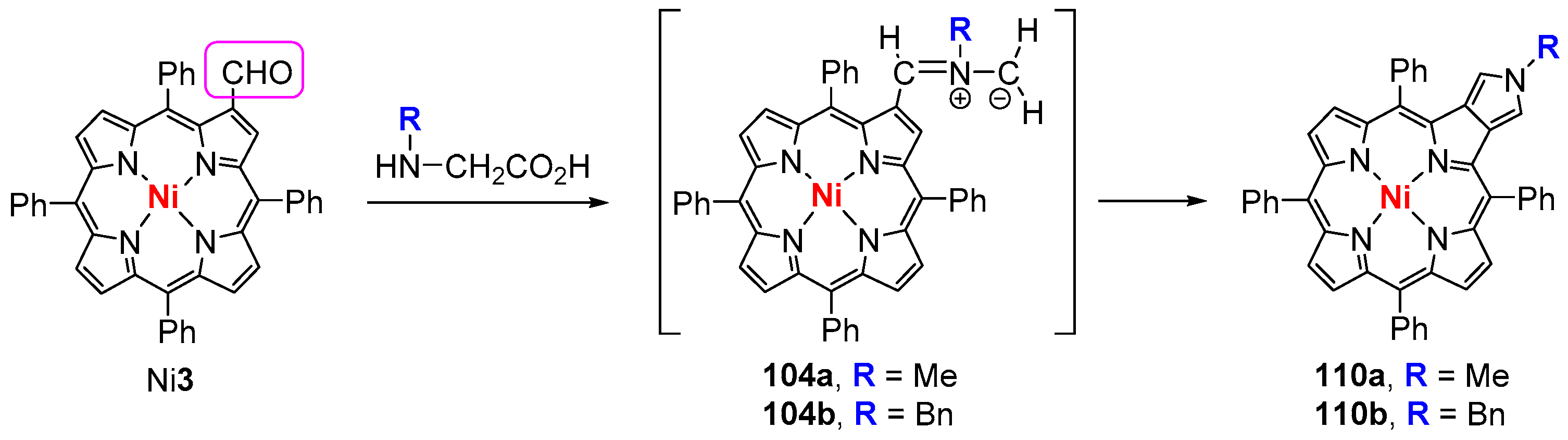
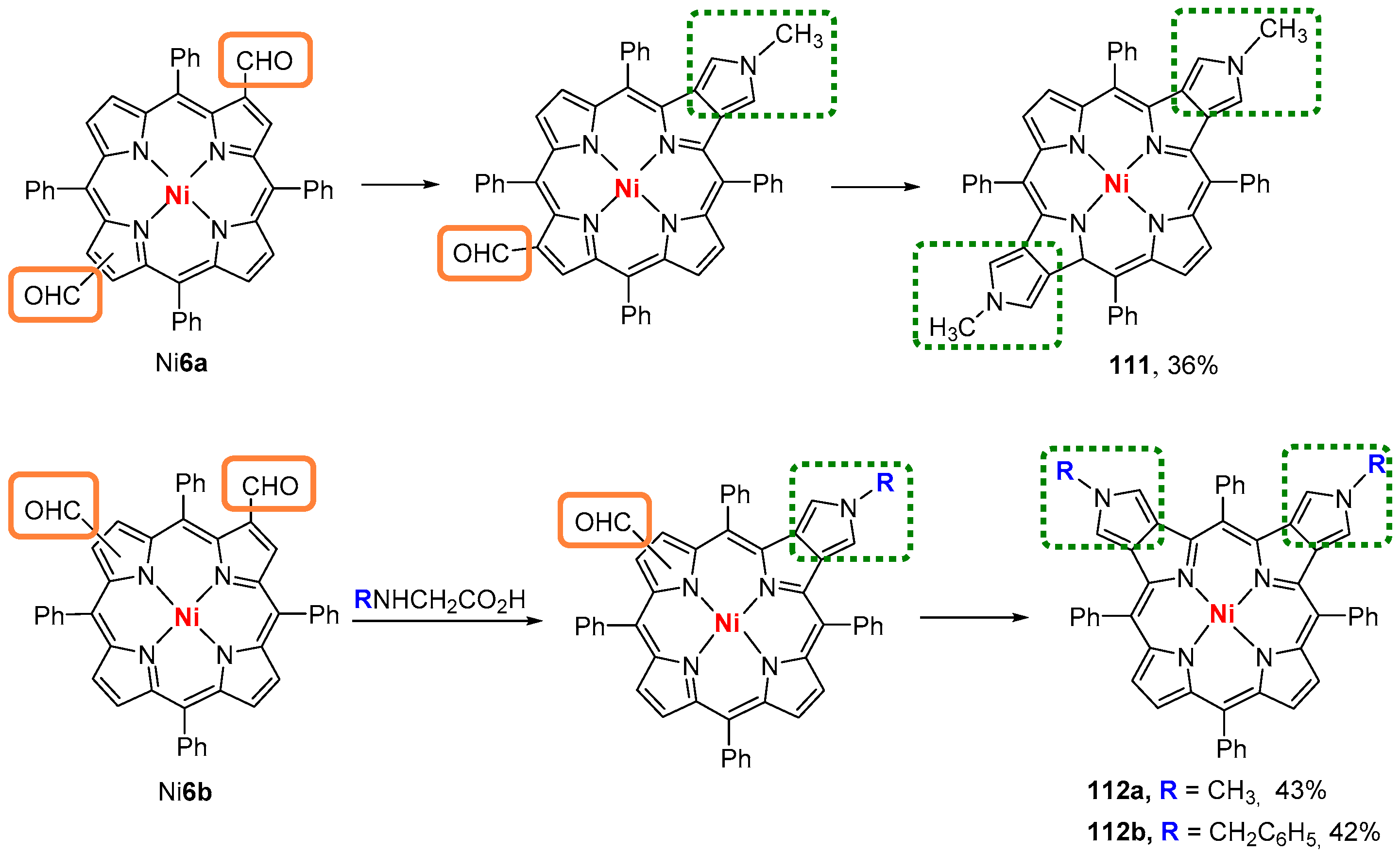
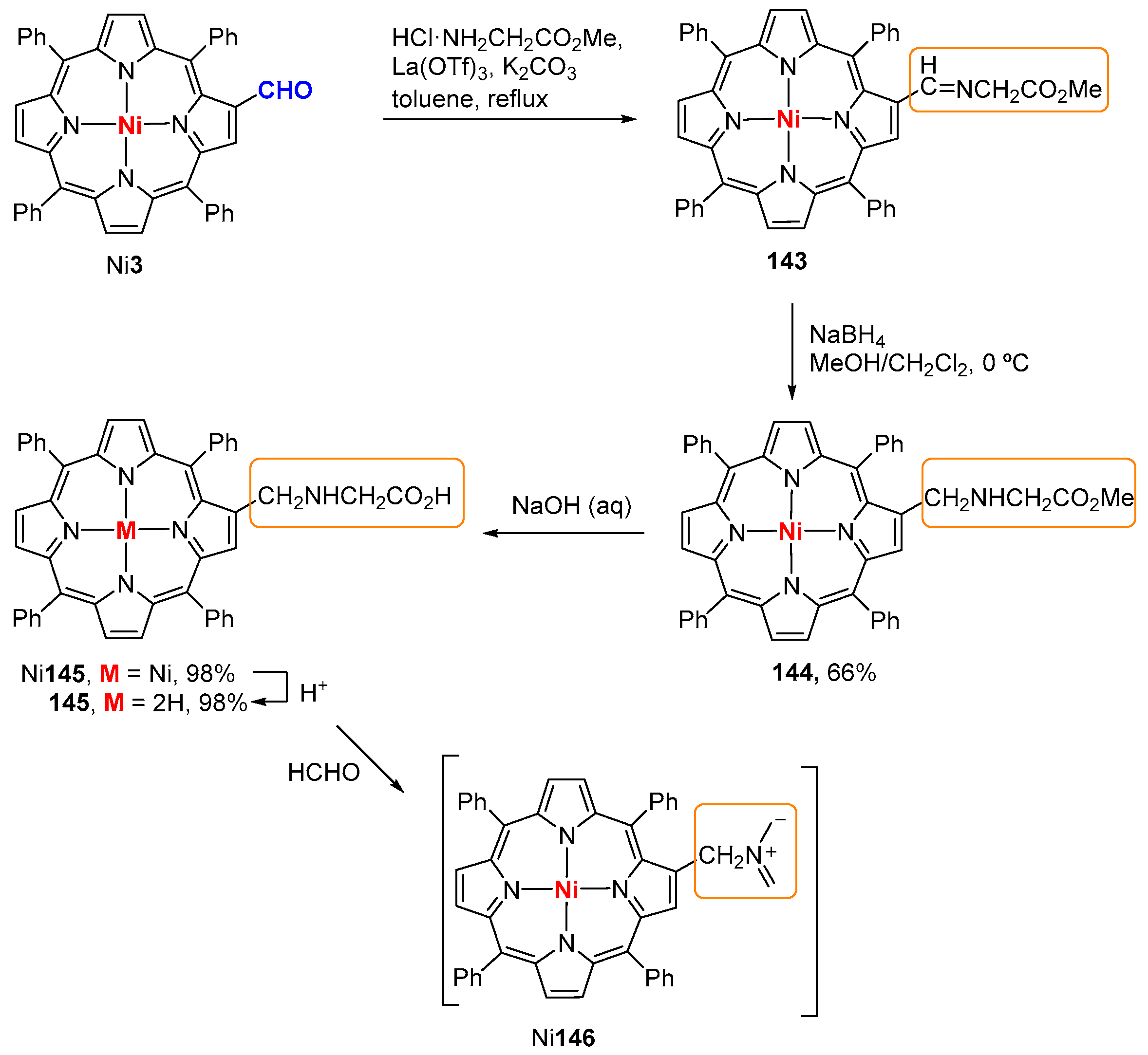
6. Reaction of β-Formyl-meso-tetraarylporphyrins with Amines and Other Nitrogen Derivatives
7. Final Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,2,4-TCB | 1,2,4-trichlorobenzene |
| AcOH | acetic acid |
| AOT | sodium 1,4-bis(2-ethylhexyl)sulfosuccinate |
| DBSA | p-dodecylbenzenesulfonic acid |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DFT | density functional theory |
| DMAD | dimethyl acetylenedicarboxylate |
| DMF | dimethylformamide |
| DPBF | 1,3-diphenylisobenzofuran |
| ESI-MS | electrospray ionisation mass spectrometry |
| ESI-MS/MS | electrospray ionization tandem mass spectrometry |
| GC | gas chromatography |
| ISC | intersystem crossing |
| IUPAC | International Union of Pure and Applied Chemistry |
| MALDI-TOF-MS | matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| min | minute |
| MS | mass spectrometry |
| MW | microwave |
| NiTPP | (5,10,15,20-tetraphenylporphyrinato)nickel(II) |
| NMR | nuclear magnetic resonance |
| o-DCB | 1,2-dichlorobenzene |
| PDT | photodynamic therapy |
| PMMA | poly(methylmethacrylate) |
| TCNE | tetracyanoethylene |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TPP | 5,10,15,20-tetraphenylporphyrin |
| UV-vis | ultraviolet-visible |
| β-CHOTPP | 2-formyl-5,10,15,20-tetrapenhylporphyrin |
| β-CHOTPP-m-MeOTPP | 2-formyl-5,10,15,20-tetrakis(3-methoxyphenyl)porphyrin |
| β-vinylTPP | 2-vinyl-5,10,15,20- tetraphenylporphyrin |
References
- García-Sánchez, M.A.; Rojas-González, F.; Menchaca-Campos, E.C.; Tello-Solís, S.R.; Quiroz-Segoviano, R.I.Y.; Diaz-Alejo, L.A.; Salas-Bañales, E.; Campero, A. Crossed and linked histories of tetrapyrrolic macrocycles and their use for engineering pores within sol-gel matrices. Molecules 2013, 18, 588–653. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Zeile, K. Synthese des hämatoporphyrins, protoporphyrins und hämins. Ann. Chem. 1929, 468, 98–116. [Google Scholar] [CrossRef]
- Sheldon, R.A. (Ed.) Metalloporphyrins in Catalytic Oxidations; Marcel Dekker: New York, NY, USA, 1994; p. 3. [Google Scholar]
- Costas, M. Selective C–H oxidation catalysed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Current issues in molecular catalysis illustrated by iron porphyrins as catalysts of the CO2-to-CO electrochemical conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Neves, C.M.B.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cocco, G.; Sanjust, E. Immobilized lignin peroxidase-like metalloporphyrins as reusable catalysts in oxidative bleaching of industrial dyes. Molecules 2016, 21, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagaki, S.; Mantovani, K.M.; Machado, G.S.; Castro, K.A.D.F.; Wypych, F. Recent advances in solid catalysts obtained by metalloporphyrins immobilization on layered anionic exchangers: A short review and some new catalytic results. Molecules 2016, 21, 291. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, S.L.H.; Silva, A.M.N.; Medforth, C.J.; Freire, C. Iron(III) fluorinated porphyrins: Greener chemistry from synthesis to oxidative catalysis reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular electronic components of functional devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-T.; Bassani, D.M. Energy transfer in supramolecular materials for new applications in photonics and electronics. NPG Asia Mater. 2014, 6, e116. [Google Scholar] [CrossRef]
- Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Porphyrinoids for chemical sensor applications. Chem. Rev. 2017, 117, 2517–2583. [Google Scholar] [CrossRef] [PubMed]
- Auwärter, W.; Écija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Zhua, W.; Xie, Y. Fluorescent and colorimetric ion probes based on conjugated oligopyrroles. Chem. Soc. Rev. 2015, 44, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Zhou, W.; Lu, J.; Lu, C. Luminescent films for chemo-and biosensing. Chem. Soc. Rev. 2015, 44, 6981–7009. [Google Scholar] [CrossRef] [PubMed]
- Figueira, F.; Rodrigues, J.M.M.; Farinha, A.A.S.; Cavaleiro, J.A.S.; Tomé, J.P.C. Synthesis and anion binding properties of porphyrins and related compounds. J. Porphyr. Phthalocyanines 2016, 20, 951–965. [Google Scholar] [CrossRef]
- Ding, Y.; Zhu, W.-H.; Xie, Y. Development of ion chemosensors based on porphyrin analogues. Chem. Rev. 2017, 117, 2203–2256. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-L.; Diau, E.W.-G. Porphyrin-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Urbani, M.; Grätzel, M.; Nazeeruddin, M.K.; Torres, T. Meso-substituted porphyrins for dye-sensitized solar cells. Chem. Rev. 2014, 114, 12330–12396. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.V.M.; Cerqueira, A.F.R.; Moura, N.M.M.; Iglesias, B.A.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Lima, M.J.C.; da Cunha, A.F. β-(p-Carboxyaminophenyl)porphyrin derivatives: New dyes for TiO2 dye-sensitized solar cells. J. Nanopart. Res. 2014, 16, 2647. [Google Scholar] [CrossRef]
- Birel1, Ö.; Nadeem, S.; Duman, H. Porphyrin-based Dye-Sensitized Solar Cells (DSSCs): A review. J. Fluoresc. 2017, 27, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Patra, A. Nanoscale strategies for light harvesting. Chem. Rev. 2017, 117, 712–757. [Google Scholar] [CrossRef] [PubMed]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Chaparro, R.E.; Sasaki, T.; Izutsu, M.; Pearlstein, R.D.; Tovmasyan, A.; Warner, D.S. Metalloporphyrins as therapeutic catalytic oxidoreductants in central nervous system disorders. Antioxid. Redox Signal. 2014, 20, 2437–2464. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cunha, Â.; Nadais, H.; Almeida, A. Potential applications of porphyrins in photodynamic inactivation beyond the medical scope. J. Photochem. Photobiol. C Photochem. Rev. 2015, 22, 34–37. [Google Scholar] [CrossRef]
- Zhou, Y.M.; Liang, X.L.; Dai, Z.F. Porphyrin-loaded nanoparticles for cancer theranostics. Nanoscale 2016, 8, 12394–12405. [Google Scholar] [CrossRef] [PubMed]
- Calvete, M.J.F.; Pinto, S.M.A.; Pereira, M.M.; Geraldes, C.F.G.C. Metal coordinated pyrrole-based macrocycles as contrast agents for magnetic resonance imaging technologies: Synthesis and applications. Coord. Chem. Rev. 2017, 333, 82–107. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, W.; Sun, J.; Guo, S. Triplet photosensitizers: From molecular design to applications. Chem. Soc. Rev. 2013, 42, 5323–5351. [Google Scholar] [CrossRef] [PubMed]
- Xodo, L.E.; Cogoi, S.; Rapozzi, V. Photosensitizers binding to nucleic acids as anticancer agents. Future Med. Chem. 2016, 8, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carter, K.A.; Miranda, D.; Lovell, J.F. Chemophototherapy: An emerging treatment option for solid tumors. Adv. Sci. 2017, 4, 1600106. [Google Scholar] [CrossRef] [PubMed]
- Grin, M.A.; Mironov, A.F. Chemical transformations of bacteriochlorophyll a and its medical applications. Russ. Chem. Bull. 2016, 65, 333–349. [Google Scholar] [CrossRef]
- Grin, M.A.; Mironov, A.F.; Shtil, A.A. Bacteriochlorophyll a and its derivatives: Chemistry and perspectives for cancer therapy. Anti-Cancer Agent Med. Chem. 2008, 8, 683–697. [Google Scholar] [CrossRef]
- Marciel, M.; Teles, L.; Moreira, B.; Pacheco, M.; Lourenço, L.M.O.; Neves, M.G.P.M.S.; Tomé, J.P.C.; Faustino, M.A.F.; Almeida, A. An effective and potentially safe blood disinfection protocol using tetrapyrrolic photosensitizers. Future Med. Chem. 2016, 9, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Luksiene, Z. New approach to inactivation of harmful and pathogenic microorganisms by photosensitization. Food Technol. Biotechnol. 2005, 43, 411–418. [Google Scholar]
- Felgenträger, A.; Maisch, T.; Späth, A.; Schröder, J.A.; Bäumler, W. Singlet oxygen generation in porphyrin-doped polymeric surface coating enables antimicrobial effects on Staphylococcus aureus. Phys. Chem. Chem. Phys. 2014, 16, 20598–20607. [Google Scholar] [CrossRef] [PubMed]
- Jemli, M.; Alouini, Z.; Sabbahi, S.; Gueddari, M. Destruction of fecal bacteria in wastewater by three photosensitizers. J. Environ. Monit. 2002, 4, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Gomes, A.T.P.C.; Fernandes, S.C.D.; Prata, A.C.B.; Almeida, M.A.; Cunha, M.A.; Tomé, J.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; et al. Photoinactivation of bacteria in wastewater by porphyrins: Bacterial β-galactosidase activity and leucine-uptake as methods to monitor the process. J. Photochem. Photobiol. B 2007, 88, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Alves, E.; Costa, L.; Tomé, J.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Almeida, A.; Cunha, A.; et al. Functional cationic nanomagnet−porphyrin hybrids for the photoinactivation of microorganisms. ACS Nano 2010, 4, 7133–7140. [Google Scholar] [CrossRef] [PubMed]
- Arrojado, C.; Pereira, C.; Tomé, J.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Cunha, A.; Calado, R.; Gomes, N.C.M.; et al. Applicability of photodynamic antimicrobial chemotherapy as an alternative to inactivate fish pathogenic bacteria in aquaculture systems. Photochem. Photobiol. Sci. 2011, 10, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Coppellottu, O.; Fabris, C.; Soncin, M.; Magaraggia, M.; Camerin, M.; Jori, G.; Guidolin, L. Porphyrin photosensitised processes in the prevention and treatment of water-and vector-borne diseases. Curr. Med. Chem. 2012, 19, 808–819. [Google Scholar] [CrossRef]
- Almeida, J.; Tomé, J.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Cunha, A.; Costa, L.; Faustino, M.A.F.; Almeida, A. Photodynamic inactivation of multidrug-resistant bacteria in hospital wastewaters: Influence of residual antibiotics. Photochem. Photobiol. Sci. 2014, 13, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Behbahani, A.; Eghbali, H.; Ardjmand, M.; Noufal, M.M.M.; Williamson, H.C.; Sayar, O. A novel bio-compatible sorbent based on carbon nanostructure modified by porphyrin for heavy metal separation from industrial wastewaters. J. Environ. Chem. Eng. 2016, 4, 398–404. [Google Scholar] [CrossRef]
- Jaquinod, L. Functionalization of 5,10,15,20-tetra-substituted porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Boston, MA, USA, 2000; Volume 1, pp. 201–237. [Google Scholar]
- Vicente, M.G.H. Reactivity and functionalization of β-substituted porphyrins and chlorins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Boston, MA, USA, 2000; Volume 1, Chapter 4; pp. 149–200. [Google Scholar]
- Tokuji, S.; Awane, H.; Yorimitsu, H.; Osuka, A. Direct arylation of meso-formyl porphyrin. Chem. Eur. J. 2013, 19, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Vicente, M.G.H.; Smith, K. Syntheses and functionalizations of porphyrin macrocycles. Curr. Org. Synth. 2014, 11, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Moss, G.P. Nomenclature of tetrapyrroles (Recommendations 1986). Pure Appl. Chem. 1987, 59, 779–832. [Google Scholar] [CrossRef]
- Rothemund, P. Formation of porphyrins from pyrrole and aldehydes. J. Am. Chem. Soc. 1935, 57, 2010–2011. [Google Scholar] [CrossRef]
- Rothemund, P. A new porphyrin synthesis. The synthesis of porphin. J. Am. Chem. Soc. 1936, 58, 625–627. [Google Scholar] [CrossRef]
- Rothemund, P. Porphyrin studies. III. The structure of the porphine ring system. J. Am. Chem. Soc. 1939, 61, 2912–2915. [Google Scholar] [CrossRef]
- Aronoff, S.; Calvin, M. The porphyrin-like products of the reaction of pyrrole with benzaldehyde. J. Org. Chem. 1943, 8, 205–223. [Google Scholar] [CrossRef]
- Calvin, M.; Ball, R.H.; Aronoff, S. α,β,γ,δ-Tetraphenylchlorin. J. Am. Chem. Soc. 1943, 65, 2259. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A simplified synthesis for meso-tetraphenylporphin. J. Org. Chem. 1967, 32, 476. [Google Scholar] [CrossRef]
- Barnett, G.H.; Hudson, M.F.; Smith, K.M. Concerning meso-tetraphenylporphyrin purification. J. Chem. Soc. Perkin Trans. 1 1975, 0, 1401–1403. [Google Scholar] [CrossRef]
- Gonsalves, A.; Varejão, J.M.; Pereira, M.M. Some new aspects related to the synthesis of meso-substituted porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Gonsalves, A.D.A.R.; Pereira, M.M. A new look into the Rothemund meso-tetraalkyl and tetraarylporphyrins synthesis. J. Heterocycl. Chem. 1985, 22, 931–933. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Hsu, H.C.; Schreiman, I.C. Synthesis of tetraphenylporphyrins under very mild conditions. Tetrahedron Lett. 1986, 27, 4969–4970. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler–Longo reactions revisited: Synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthesis of meso-substituted porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Boston, MA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Sharghi, H.; Nejad, A.H. Phosphorus pentachloride (PCl5) mediated synthesis of tetraarylporphyrins. Helv. Chim. Acta 2003, 86, 408–414. [Google Scholar] [CrossRef]
- Sharghi, H.; Nejad, A.H. Novel synthesis of meso-tetraarylporphyrins using CF3SO2Cl under aerobic oxidation. Tetrahedron 2004, 60, 1863–1868. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Tomé, A.C.; Neves, M.G.P.M.S. Meso-tetraarylporphyrin derivatives: New synthetic methodologies. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2010; Volume 2, Chapter 9; pp. 193–294. [Google Scholar]
- Chauhan, S.; Sahoo, B.; Srinivas, K. Microwave-assisted synthesis of 5,10,15,20-tetraaryl porphyrins. Synth. Commun. 2001, 31, 33–37. [Google Scholar] [CrossRef]
- De Paula, R.; Faustino, M.A.; Pinto, D.C.; Neves, M.G.; Cavaleiro, J.A. Kinetic study of meso-tetraphenylporphyrin synthesis under microwave irradiation. J. Heterocycl. Chem. 2008, 45, 453–459. [Google Scholar] [CrossRef]
- Liu, M.O.; Tai, C.-H.; Wang, W.-Y.; Chen, J.-R.; Hu, A.T.; Wei, T.-H. Microwave-assisted synthesis and reverse saturable absorption of phthalocyanines and porphyrins. J. Organomet. Chem. 2004, 689, 1078–1084. [Google Scholar] [CrossRef]
- Nascimento, B.F.; Pineiro, M.; Rocha Gonsalves, A.M.d.A.; Ramos Silva, M.; Matos Beja, A.; Paixão, J.A. Microwave-assisted synthesis of porphyrins and metalloporphyrins: A rapid and efficient synthetic method. J. Porphyr. Phthalocyanines 2007, 11, 77–84. [Google Scholar] [CrossRef]
- Henriques, C.A.; Pinto, S.; Aquino, G.L.; Pineiro, M.; Calvete, M.J.; Pereira, M.M. Ecofriendly porphyrin synthesis by using water under microwave irradiation. Chem. Sustain. Chem. 2014, 7, 2821–2824. [Google Scholar] [CrossRef] [PubMed]
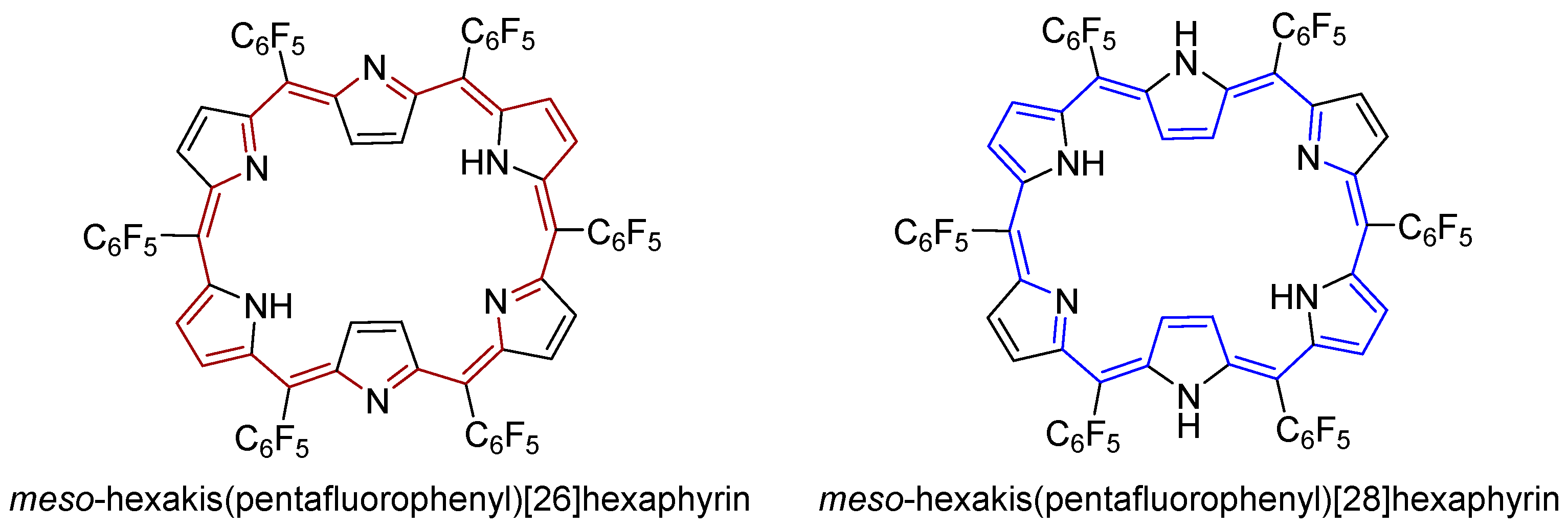
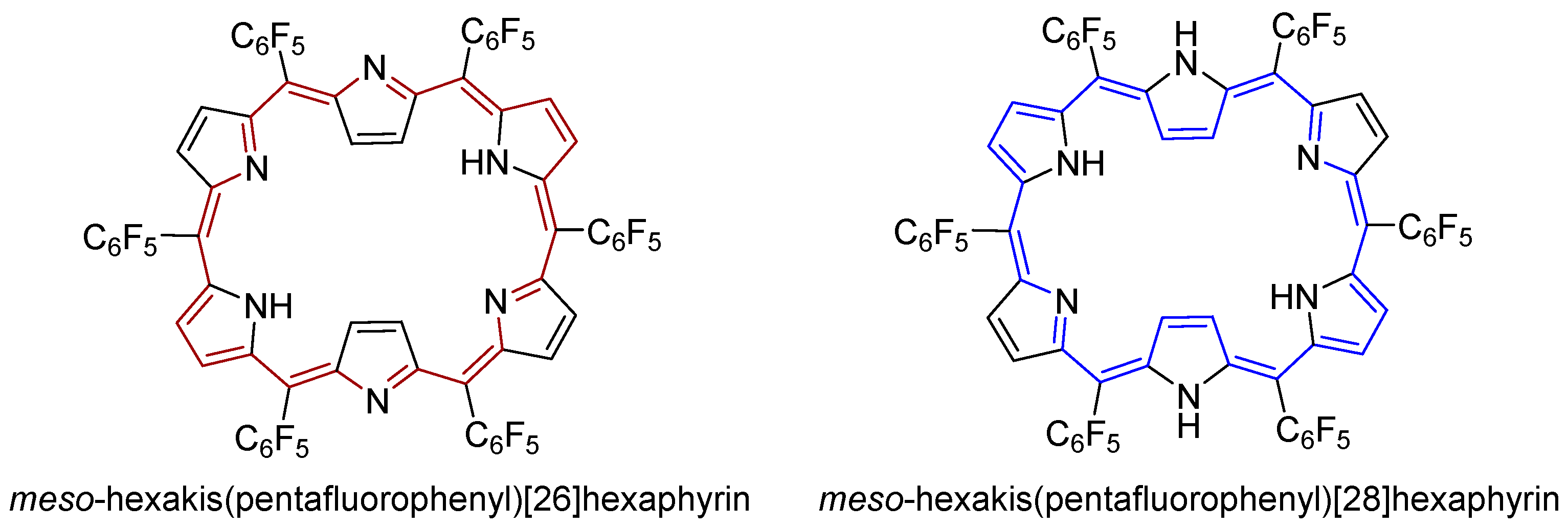
- Neves, M.G.P.M.S.; Martins, R.M.; Tomé, A.C.; Silvestre, A.J.D.; Silva, A.M.S.; Félix, V.; Drew, M.G.B.; Cavaleiro, J.A.S. Meso-substituted expanded porphyrins: New and stable hexaphyrins. Chem. Commun. 1999, 0, 385–386. [Google Scholar] [CrossRef]
- Tanaka, T.; Osuka, A. Chemistry of meso-aryl-substituted expanded porphyrins: Aromaticity and molecular twist. Chem. Rev. 2017, 117, 2584–2640. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.D.; Costa, J.I.T.; Tomé, A.C. Porphyrin macrocycle modification: Pyrrole ring-contracted or-expanded porphyrinoids. Molecules 2016, 21, 320. [Google Scholar] [CrossRef] [PubMed]
- Inhoffen, H.H.; Fuhrhop, J.-H.; Voigt, H.; Brockmann, H., Jr. Zur weiteren Kenntnis des Chlorophylls und des Hämins, VI. Formylierung der meso-kohlenstoffatome von alkyl-substituierten porphyrinen. Justus Liebigs Ann. Chem. 1966, 695, 133–143. [Google Scholar] [CrossRef]
- Johnson, A.W.; Oldfield, D. Meso-Substitution products of ætioporphyrin I. J. Chem. Soc. C 1966, 0, 794–798. [Google Scholar] [CrossRef]
- Inhoffen, H.H.; Buchler, J.W.; Thomas, R. Zur weiteren kenntnis des chlorophylls und des hamins, XXV. 3, 4,7,8-Tetrahydro-octaäthylporphin (“Bacterio-octaäthylchlorin”). Tetrahedron. Lett. 1969, 10, 1141–1144. [Google Scholar] [CrossRef]
- Callot, H.J. Nouvelles voies d’accès aux vinylporphyrines. Tetrahedron 1973, 29, 899–901. [Google Scholar] [CrossRef]
- Callot, H.J. Wittig reaction on some formylporphyrins-example of extremely facile deformylation. Bull. Soc. Chim. Fr. 1973, 12, 3413–3416. [Google Scholar]
- Callot, H.J.; Castro, B.; Selve, C. Porphyrines synthetic ues porteuses de chaines laterales peptidiques. I Atropoisomérie d’amides cis(méso-tétraphénylporphyrinyl)-3-propénoïques. Tetrahedron. Lett. 1978, 32, 2877–2880. [Google Scholar] [CrossRef]
- Momenteau, M.; Loock, B.; Bisagni, E.; Rougee, M. Five-coordinate iron(II) porphyrins derived from meso-α,β,γ,δ-tetraphenylporphin: Synthesis, characterization, and coordinating properties. Can. J. Chem. 1979, 57, 1804–1813. [Google Scholar] [CrossRef]
- Barloy, L.; Dolphin, D.; Dupré, D.; Wijesekera, T.P. Anomalous double cyclization reactions of β-formylporphyrins. J. Org. Chem. 1994, 59, 7976–7985. [Google Scholar] [CrossRef]
- Balakumar, A.; Mutukumaran, K.; Lindsey, J.S. A New route to meso-formyl porphyrins. J. Org. Chem. 2004, 69, 5112–5115. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, G.V.; Yashunskii, D.V.; Moskovkin, A.S. Porphyrins. 35. Unusual course of the Vilsmeir reaction when formylating the platinum complex of octaethylporphyrin (revision of the known reaction). Chem. Heterocycl. Compd. 1997, 33, 271–275. [Google Scholar] [CrossRef]
- Henrick, K.; Owston, P.G.; Peters, R.; Tasker, P.A.; Dell, A. Cyclisation involving a meso-phenyl substituent of a metalloporphyrin: X-ray structure of 5,10,15-triphenyl{22-oxo-benzo[2324]cyclohexa[a,b]porphinato(2-)} copper(II). Inorg. Chim. Acta 1980, 45, L161–L163. [Google Scholar] [CrossRef]
- Callot, H.J.; Schaeffer, E.; Cromer, R.; Metz, F. Unexpected routes to naphthoporphyrin derivatives. Tetrahedron 1990, 46, 5253–5262. [Google Scholar] [CrossRef]
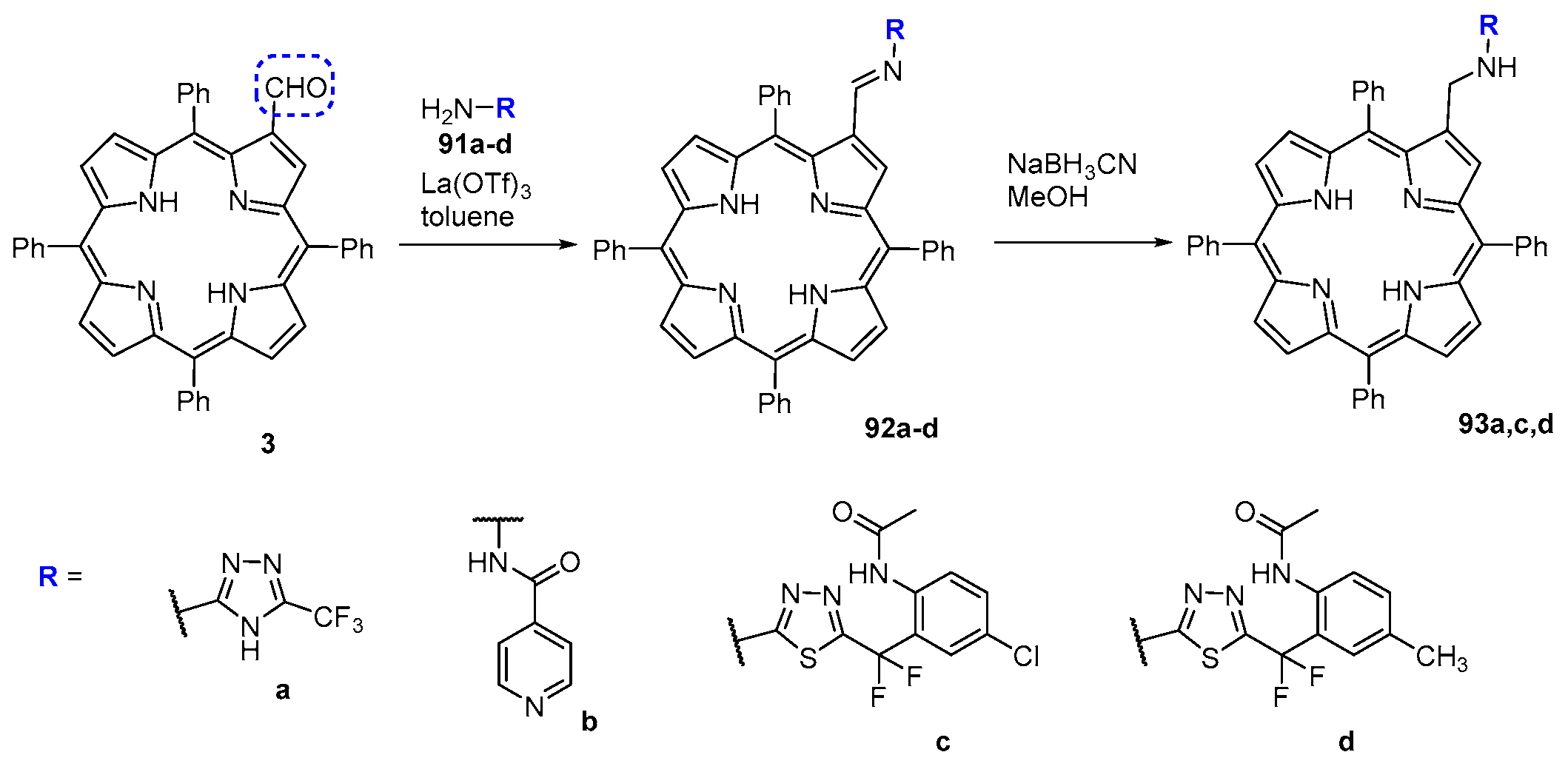
- Bonfantini, E.E.; Burrell, A.K.; Campbell, W.M.; Crossley, M.J.; Gosper, J.J.; Harding, M.M.; Officer, D.L.; Reid, D.C.W. Efficient synthesis of free-base 2-formyl-5,10,15,20-tetraarylporphyrins, their reduction and conversion to [(porphyrin-2-yl)methyl]phosphonium salts. J. Porphyrins. Phthalocyanines 2002, 6, 708–719. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Maravin, G.B. Porphyrins. 14. Synthesis and properties of 1-substituted derivatives of 5,10,15,20-tetraphenylporphyrin. Chem. Heterocycl. Compd. 1982, 18, 50–55. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Faustino, M.A.F.; Silva, T.M.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S. NMR characterisation of five isomeric β,β´-diformyl-meso-tetraphenylporphyrins. J. Chem. Soc. Perkin Trans. 1 2002, 1, 1774–1777. [Google Scholar] [CrossRef]
- Mironov, A.F.; Grin, M.A.; Moskalchuk, T.V.; Shashkov, A.S.; Lokshin, B.V. Synthesis and unusual spectroscopic properties of diformyltetraarylporphyrins. Mendeleev Commun. 2002, 12, 204–205. [Google Scholar] [CrossRef]
- Mironov, A.F.; Moskalchuk, T.V.; Shashkov, A.S. Formylation reaction in series of meso-tetraaryl substituted porphyrins and chlorins. Russ. J. Bioorg. Chem. 2004, 30, 261–267. [Google Scholar] [CrossRef]
- Yaseen, M.; Ali, M.; NajeebUllah, M.; Ali Munawar, M.; Khokhara, I. Microwave-assisted synthesis, metallation, and Duff formylation of porphyrins. J. Heterocycl. Chem. 2009, 46, 251–255. [Google Scholar] [CrossRef]
- Moura, N.M.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Duarte, A.C.; Cavaleiro, J.A.S. Vilsmeier-Haack formylation of Cu(II) and Ni(II) porphyrin complexes under microwaves irradiation. J. Porphyrins. Phthalocyanines 2011, 15, 652–658. [Google Scholar] [CrossRef]
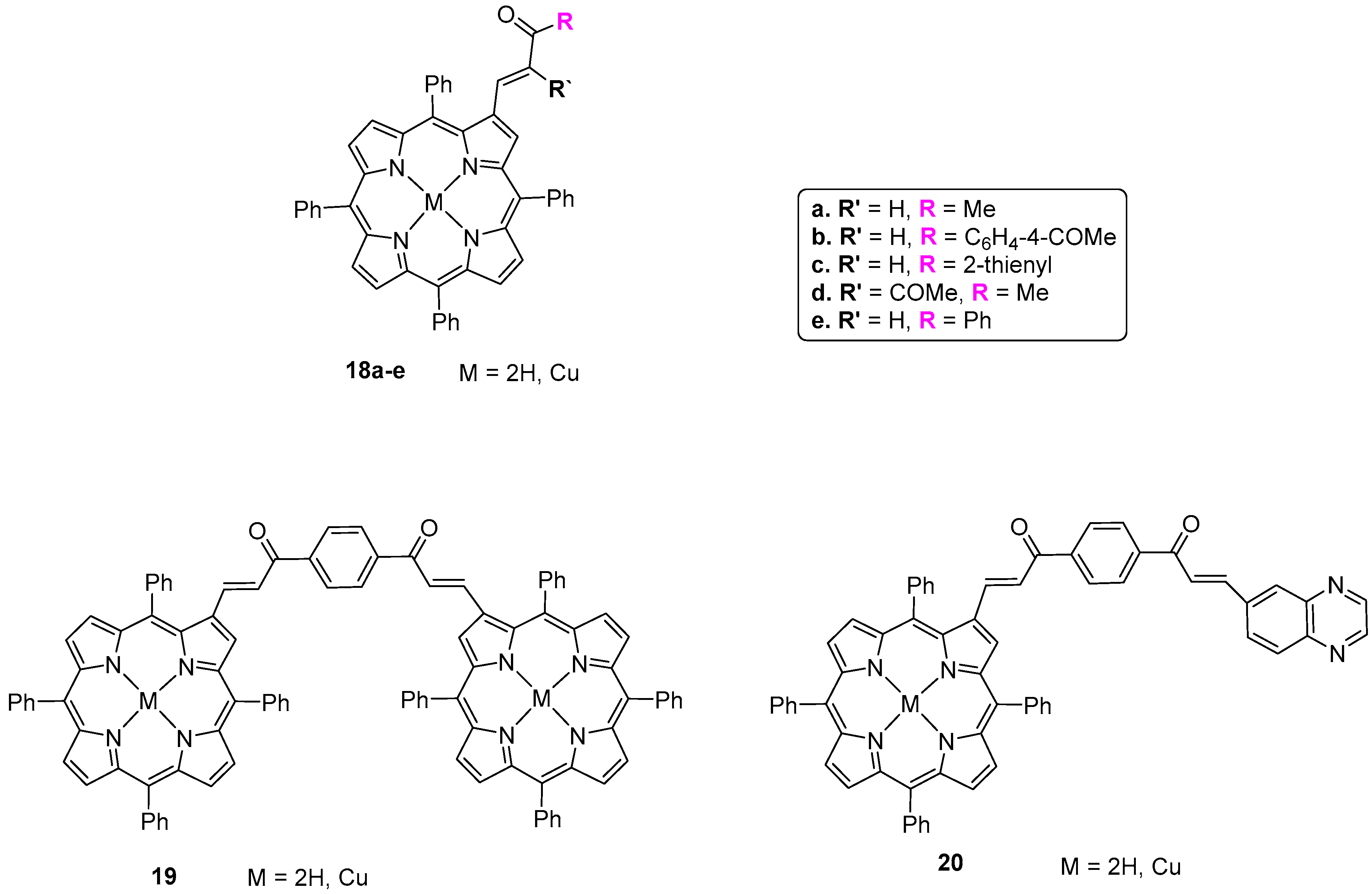
- Rish, I.G.; Pshezhetskii, V.S.; Askarov, K.A.; Ponomarev, G.V. Porphyrins. 20. Interaction of 2-formyl-5,10,15,20-tetraphenylporphyrin with CH acids. Chem. Heterocycl. Compd. 1985, 21, 777–781. [Google Scholar] [CrossRef]
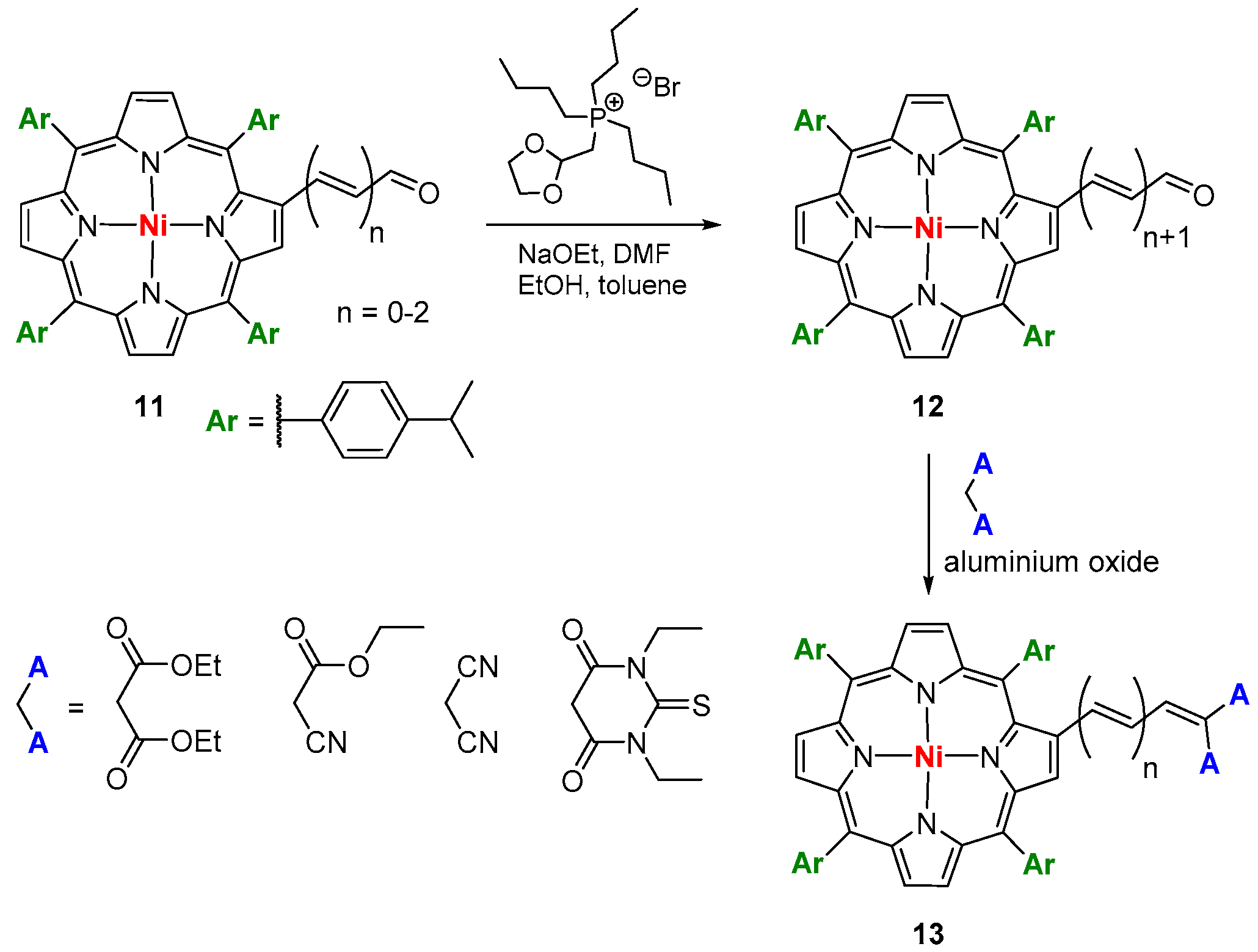
- Chen, C.-T.; Yeh, H.-C.; Zhang, X.; Yu, J. Olefin-mediated interaction observed for nickel tetraphenylporphyrins with an acceptor substituted on the β-carbon. Org. Lett. 1999, 1, 1767–1770. [Google Scholar] [CrossRef]
- Yeha, H.-C.; Chen, C.-T.; Yu, J.; Tsaic, P.-C.; Wang, J.-K. Conformation and π-conjugation of olefin-bridged acceptor on the pyrrole β-carbon of nickel tetraphenylporphyrins: Implicit evidence from linear and nonlinear optical properties. J. Porphyrins. Phthalocyanines 2007, 11, 857–873. [Google Scholar] [CrossRef]
- Ramos, C.I.V.; Moura, N.M.M.; Santos, S.M.F.; Faustino, M.A.; Tomé, J.P.C.; Amado, F.M.L.; Neves, M.G.P.M.S. An insight into the gas-phase fragmentations of potential molecular sensors with porphyrin-chalcone structures. Int. J. Mass Spectrom. 2015, 392, 164–172. [Google Scholar] [CrossRef]
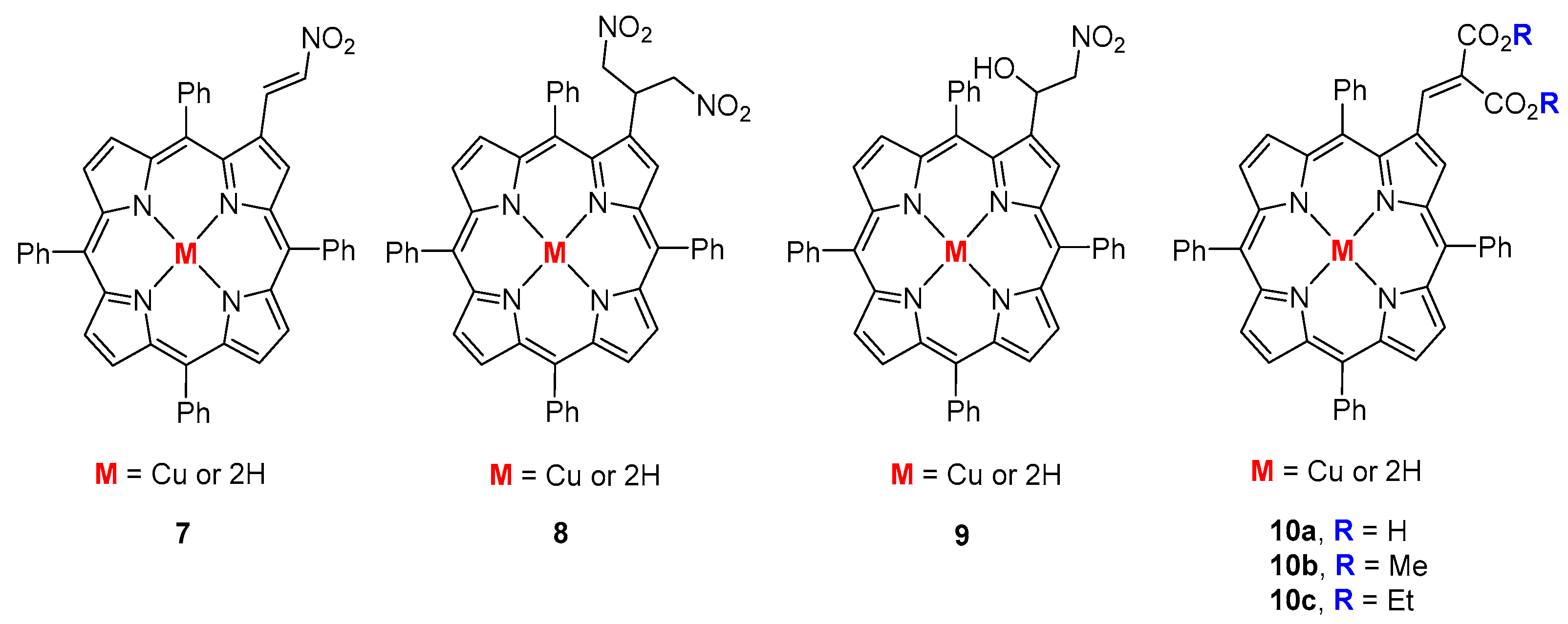
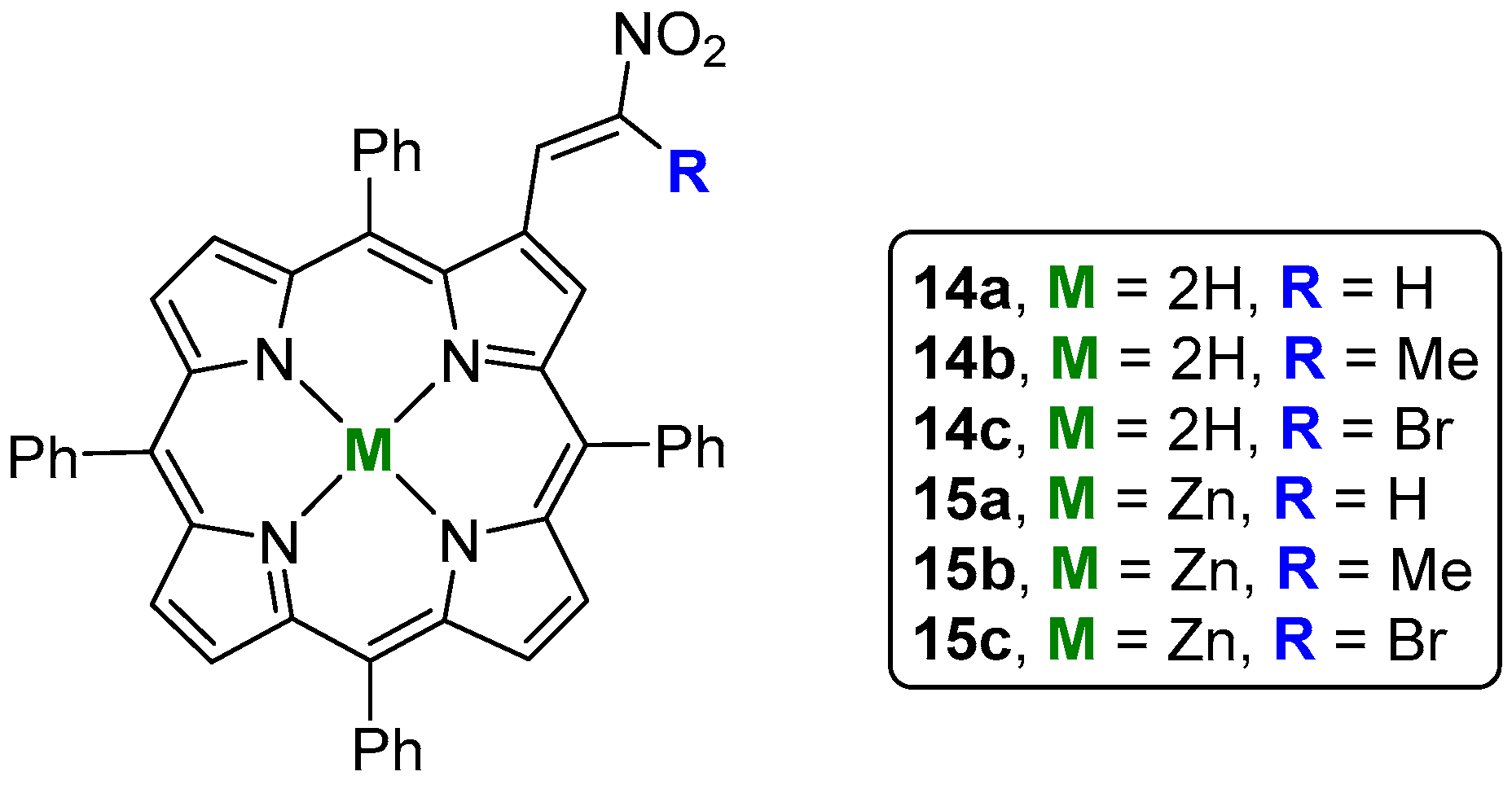
- Silva, E.M.P.; Domingues, P.; Tomé, J.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Dauzonne, D.; Silva, A.M.S.; Cavaleiro, J.A.S.; Ferrer-Correia, A.J.; et al. Electrospray tandem mass spectrometry of β-nitroalkenyl meso-tetraphenylporphyrins. Eur. J. Mass Spectrom. 2008, 14, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Giribabu, L.; Kumar, C.V.; Reddy, P.Y. Porphyrin-rhodanine dyads for dye sensitized solar cells. J. Porphyrins. Phthalocyanines 2006, 10, 1007–1016. [Google Scholar] [CrossRef]
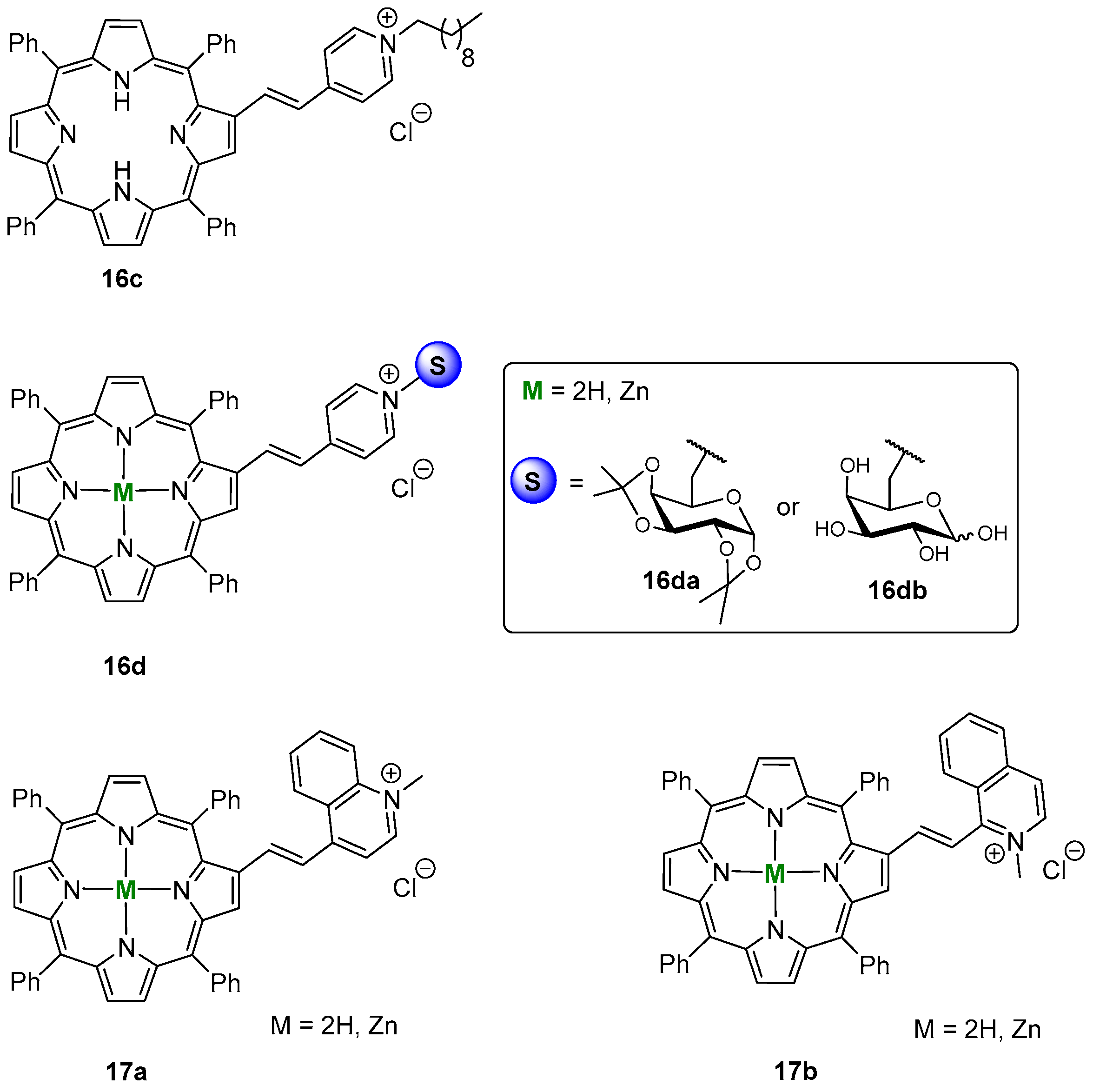
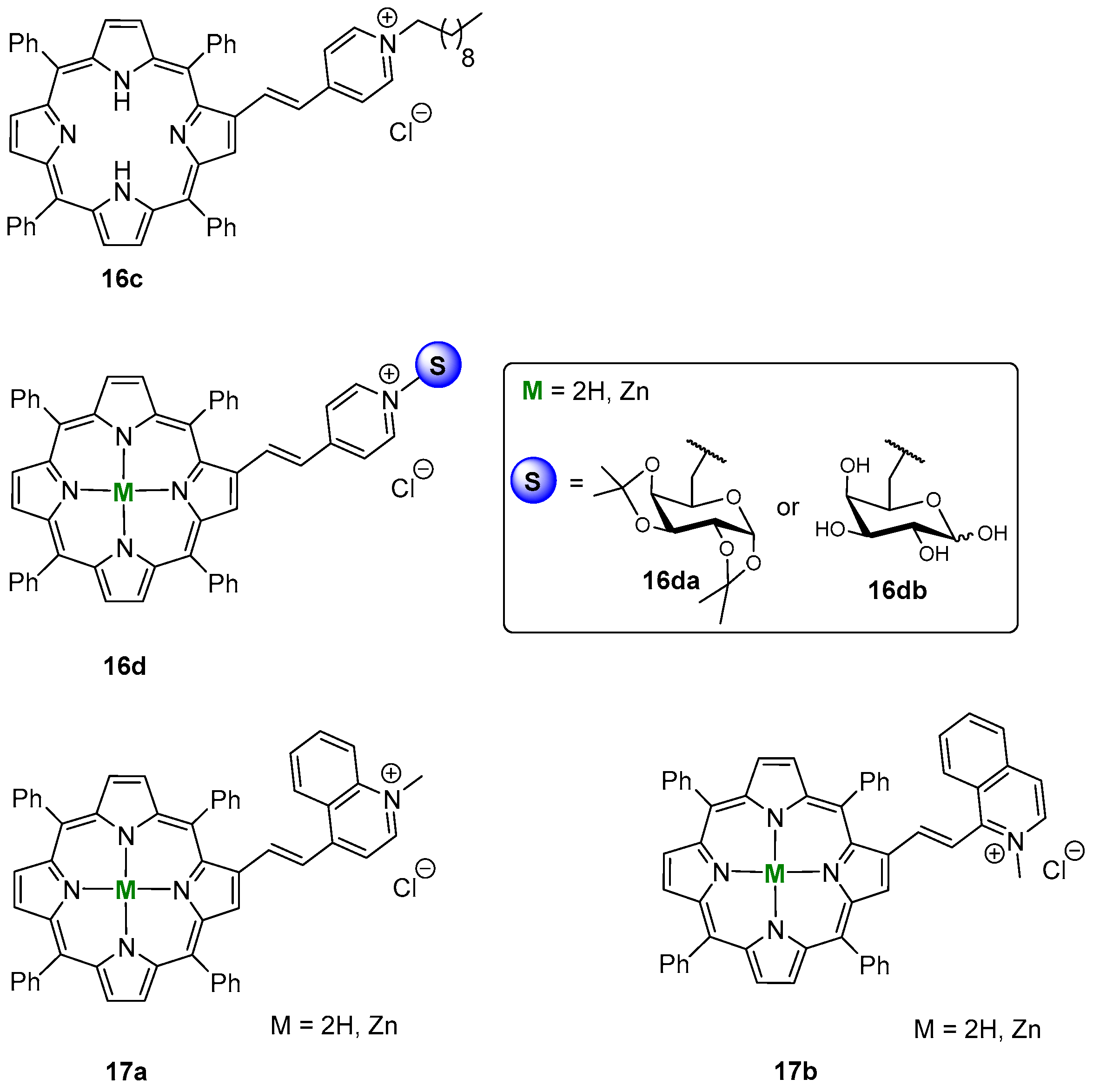
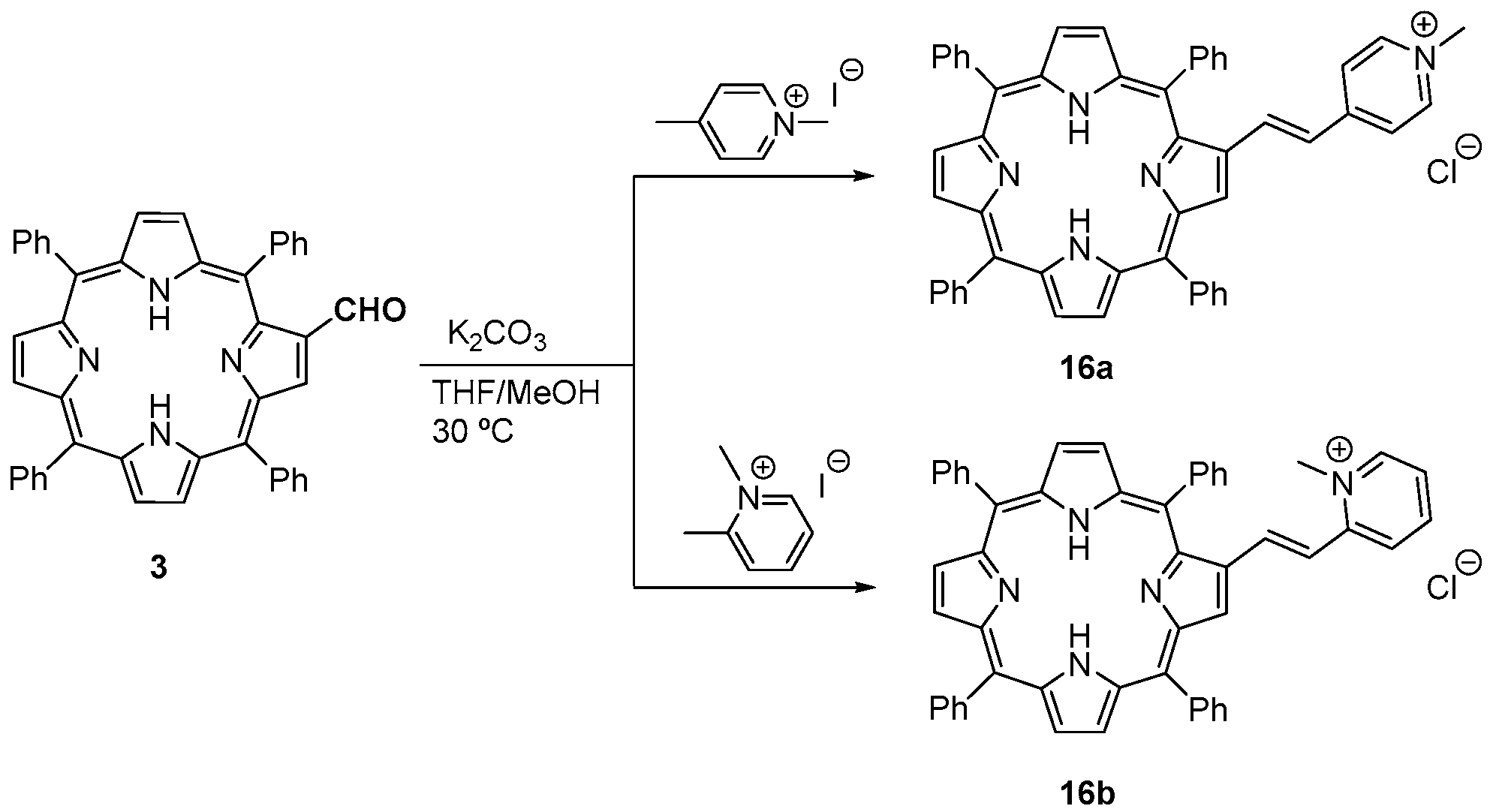
- Silva, E.M.P.; Giuntini, F.; Faustino, M.A.F.; Tomé, J.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Santana-Marques, M.G.; Ferrer-Correia, A.J.; Cavaleiro, J.A.S.; et al. Synthesis of cationic β-vinyl substituted meso-tetraphenylporphyrins and their in vitro activity against herpes simplex virus type. Bioorg. Med. Chem. Lett. 2005, 15, 3333–3337. [Google Scholar] [CrossRef] [PubMed]
- Burrel, A.K.; Officer, D.L.; Reid, D.C.W.; Wild, K.Y. Controlling the structure of supramolecular porphyrin arrays. Angew. Chem. Int. Ed. Engl. 1998, 37, 114–117. [Google Scholar] [CrossRef]
- Burrel, A.K.; Officer, D.L. Functionalizing porphyrins via Wittig reactions: A building block approach. Synlett 1998, 12, 1297–1307. [Google Scholar] [CrossRef]
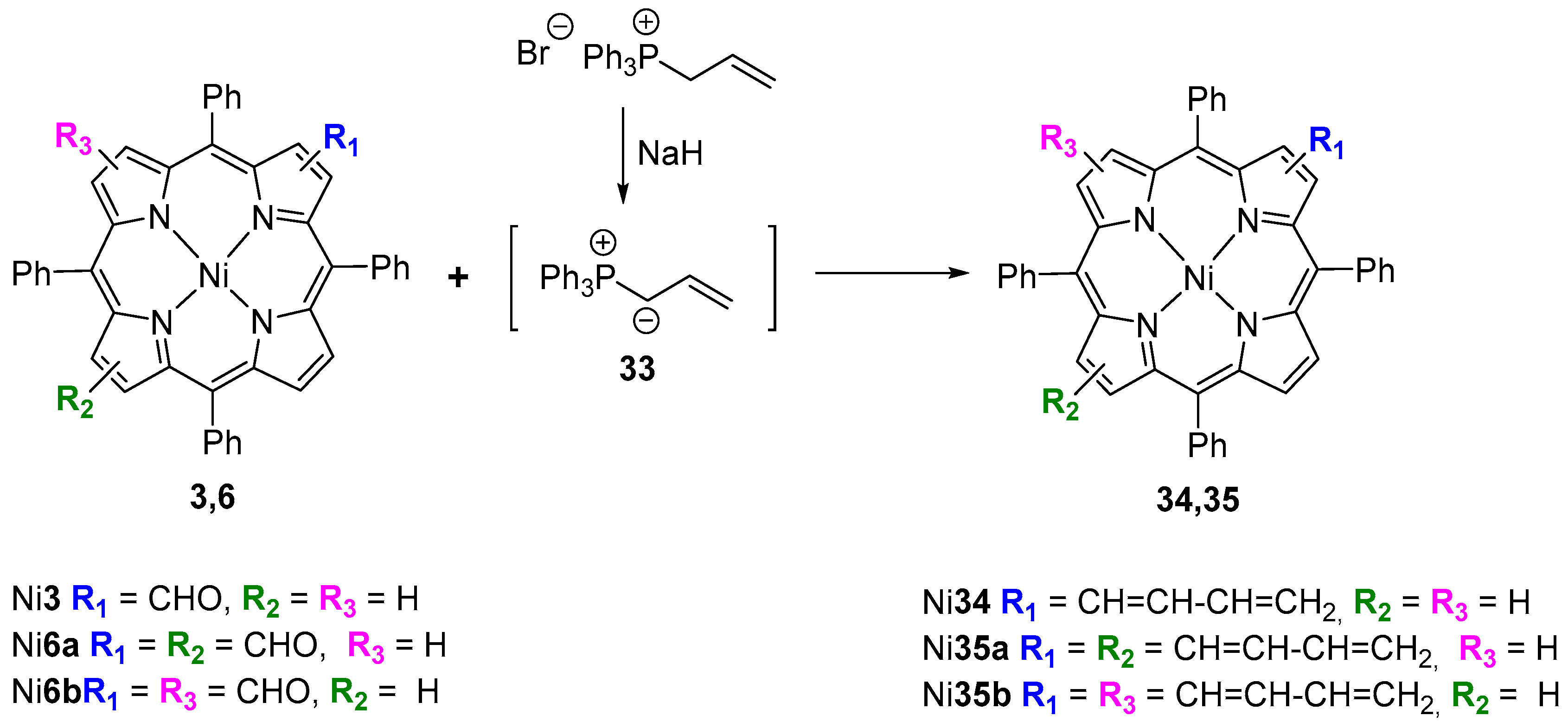
- Bonfantini, E.E.; Officer, D.L. The synthesis of butadiene-bridged porphyrin dimers and styryl porphyrins using a porphyrin-derived Wittig reagent. Tetrahedron Lett. 1993, 34, 8531–8534. [Google Scholar] [CrossRef]
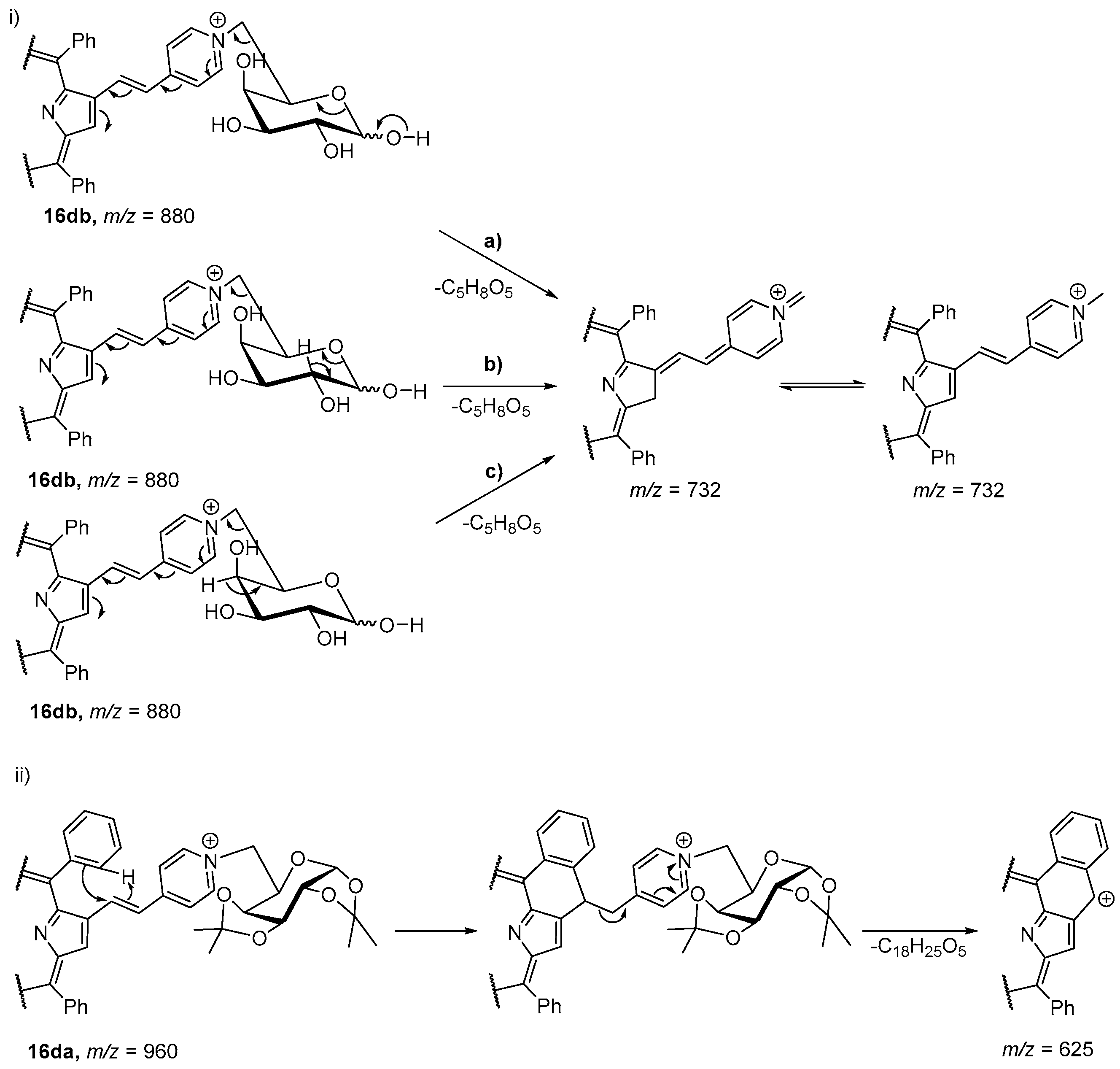
- Izquierdo, R.A.; Barros, C.M.; Santana-Marques, M.G.; Ferrer-Correia, A.J.; Silva, E.M.P.; Giuntini, F.; Faustino, M.A.F.; Tomé, J.P.C.; Tomé, A.C.; Silva, A.M.S.; et al. Characterization of isomeric cationic porphyrins with β-pyrrolic substituents by electrospray mass spectrometry: The singular behavior of a potential virus photoinactivator. J. Am. Soc. Mass Spectrom. 2007, 18, 218–225. [Google Scholar] [CrossRef] [PubMed]
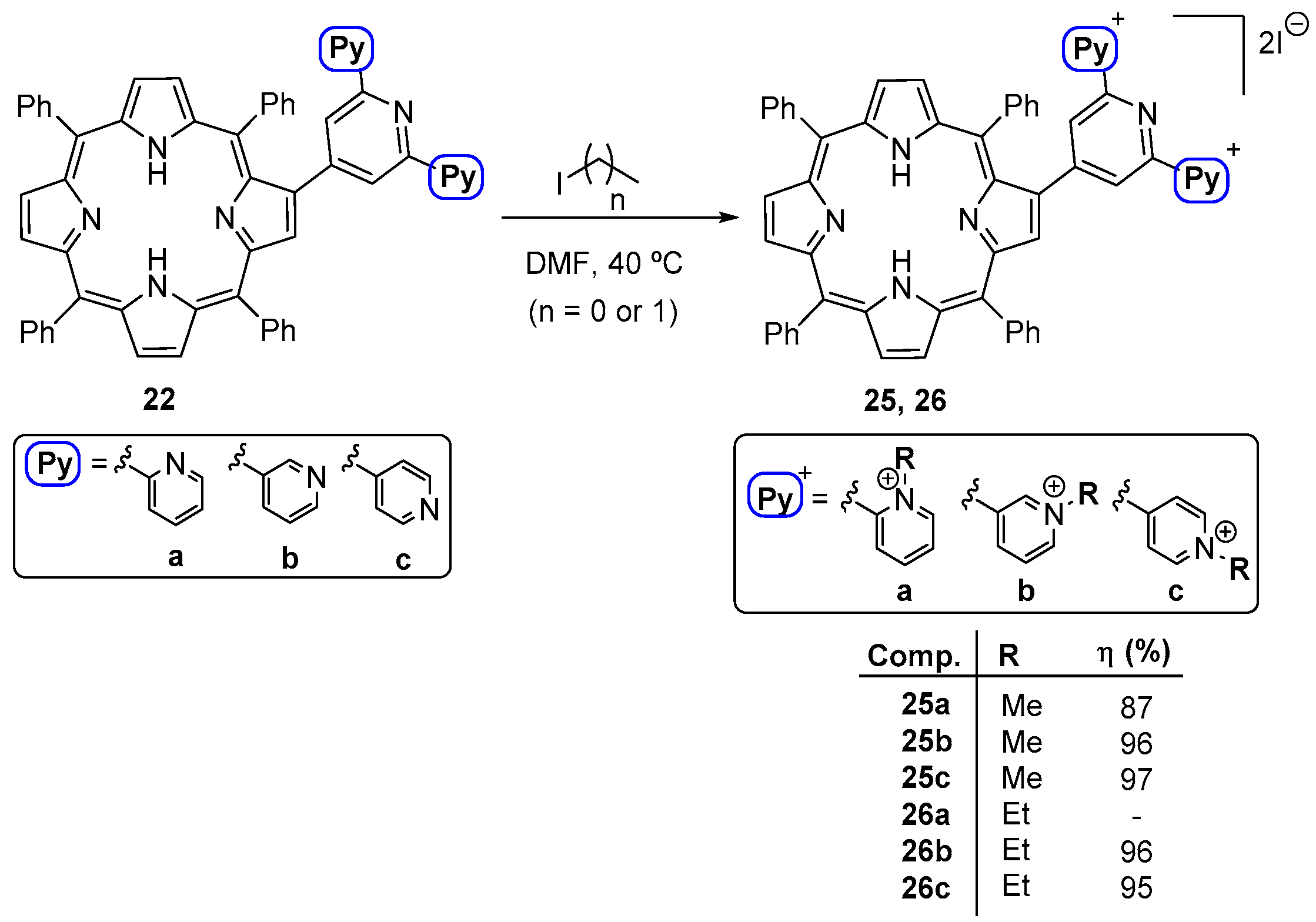
- Serra, V.V.; Andrade, S.M.; Silva, E.M.P.; Silva, A.M.S.; Neves, M.G.P.M.S.; Costa, S.M.B. Structural effects of the β-vinyl linker in pyridinium porphyrins: Spectroscopic studies in organic solvents and AOT reverse micelles. J. Phys. Chem. B 2013, 117, 15023–15032. [Google Scholar] [CrossRef]
- Silva, E.M.P.; Ramos, C.I.V.; Pereira, P.M.R.; Giuntini, F.; Faustino, M.A.F.; Tomé, J.P.C.; Tomé, A.C.; Silva, A.M.S.; Santana-Marques, M.G.; Neves, M.G.P.M.S.; et al. Cationic β-vinyl substituted meso-tetraphenylporphyrins: Synthesis and non-covalent interactions with a short poly(dGdC) duplex. J. Porphyrins. Phthalocyanines 2012, 16, 101–113. [Google Scholar] [CrossRef]
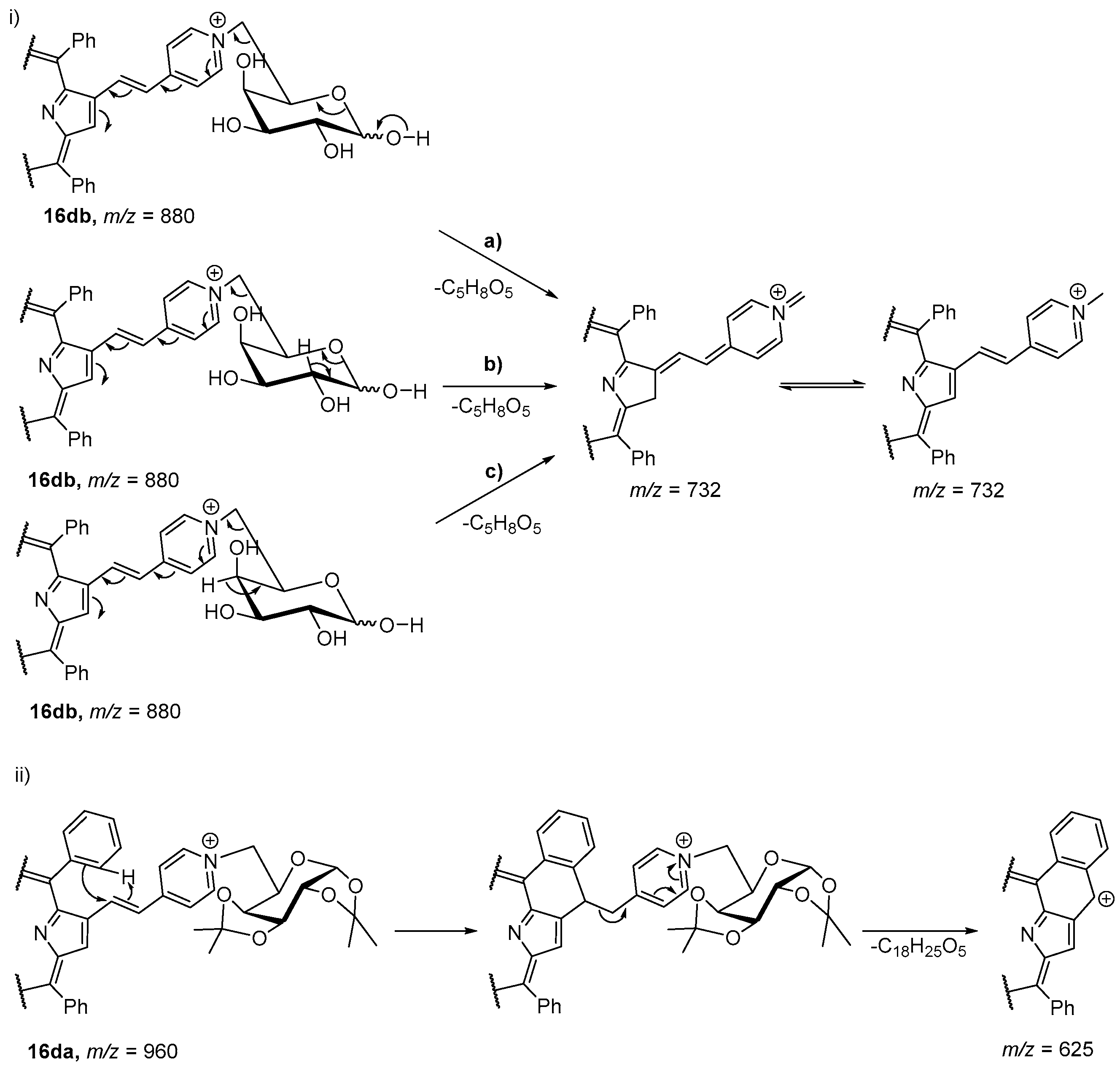
- Silva, E.M.P.; Serra, V.V.; Ribeiro, A.O.; Tomé, J.P.C.; Domingues, P.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Ferrer-Correia, A.J.; et al. Characterization of cationic glycoporphyrins by electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 3605–3611. [Google Scholar] [CrossRef] [PubMed]
- Ishkov, Y.V.; Zhilina, Z.I.; Barday, L.P. The interaction of formylporphyrins with weak CH-acids. J. Porphyrins. Phthalocyanines 2003, 7, 761–765. [Google Scholar] [CrossRef]
- Ishkov, Y.V.; Zhilina, Z.I.; Barday, L.P.; Vodzinskii, S.V. Porphyrins and their derivatives: XXIII. Reaction of formylporphyrins with weak CH acids. Russ. J. Org. Chem. 2004, 40, 434–437. [Google Scholar] [CrossRef]
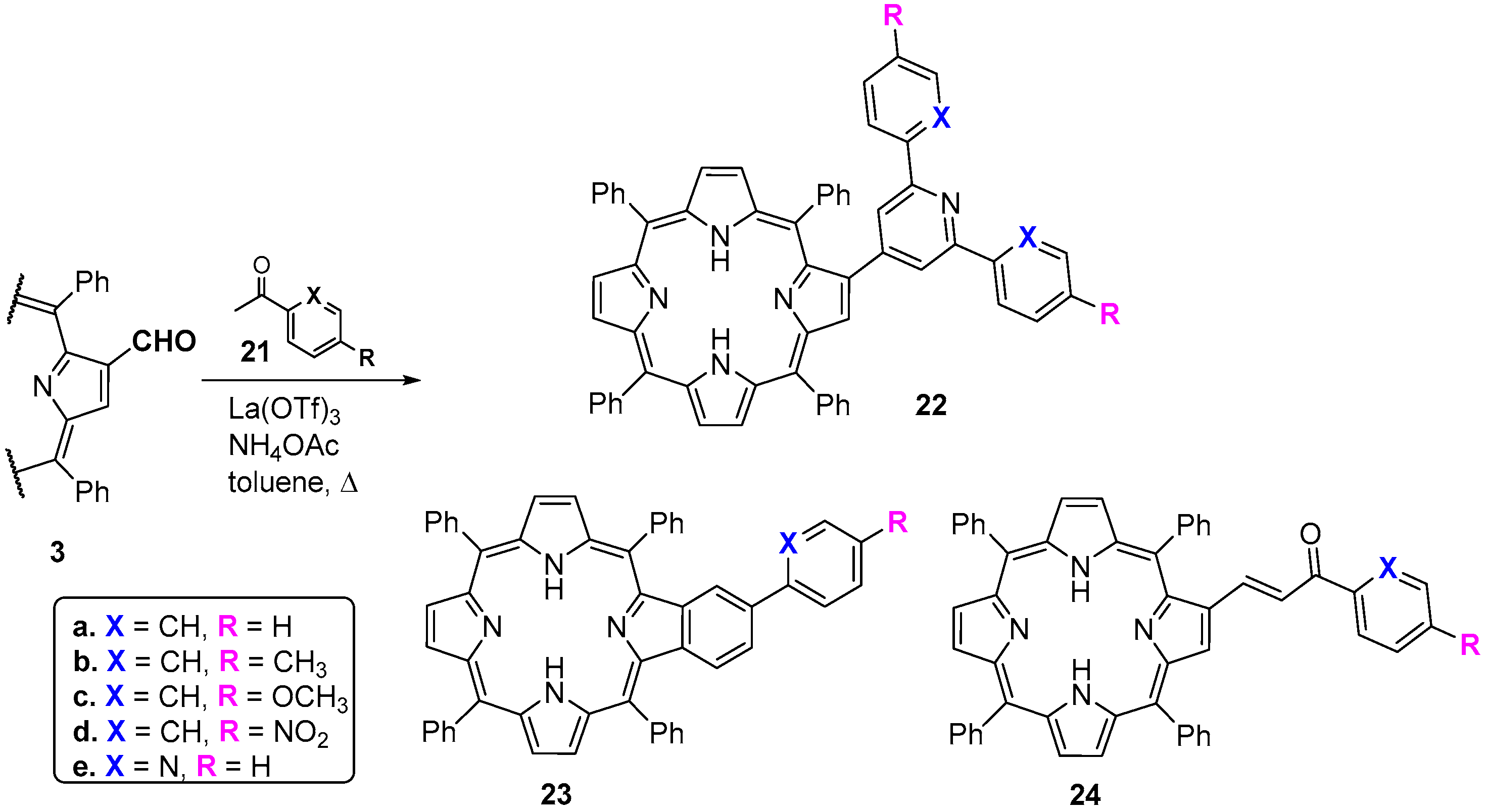
- Moura, N.M.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Paz, F.A.A.; Silva, A.M.S.; Tomé, A.C.; Cavaleiro, J.A.S. A new synthetic approach to benzoporphyrins and Kröhnke type porphyrin-2-ylpyridines. Chem. Commun. 2012, 48, 6142–6144. [Google Scholar] [CrossRef] [PubMed]
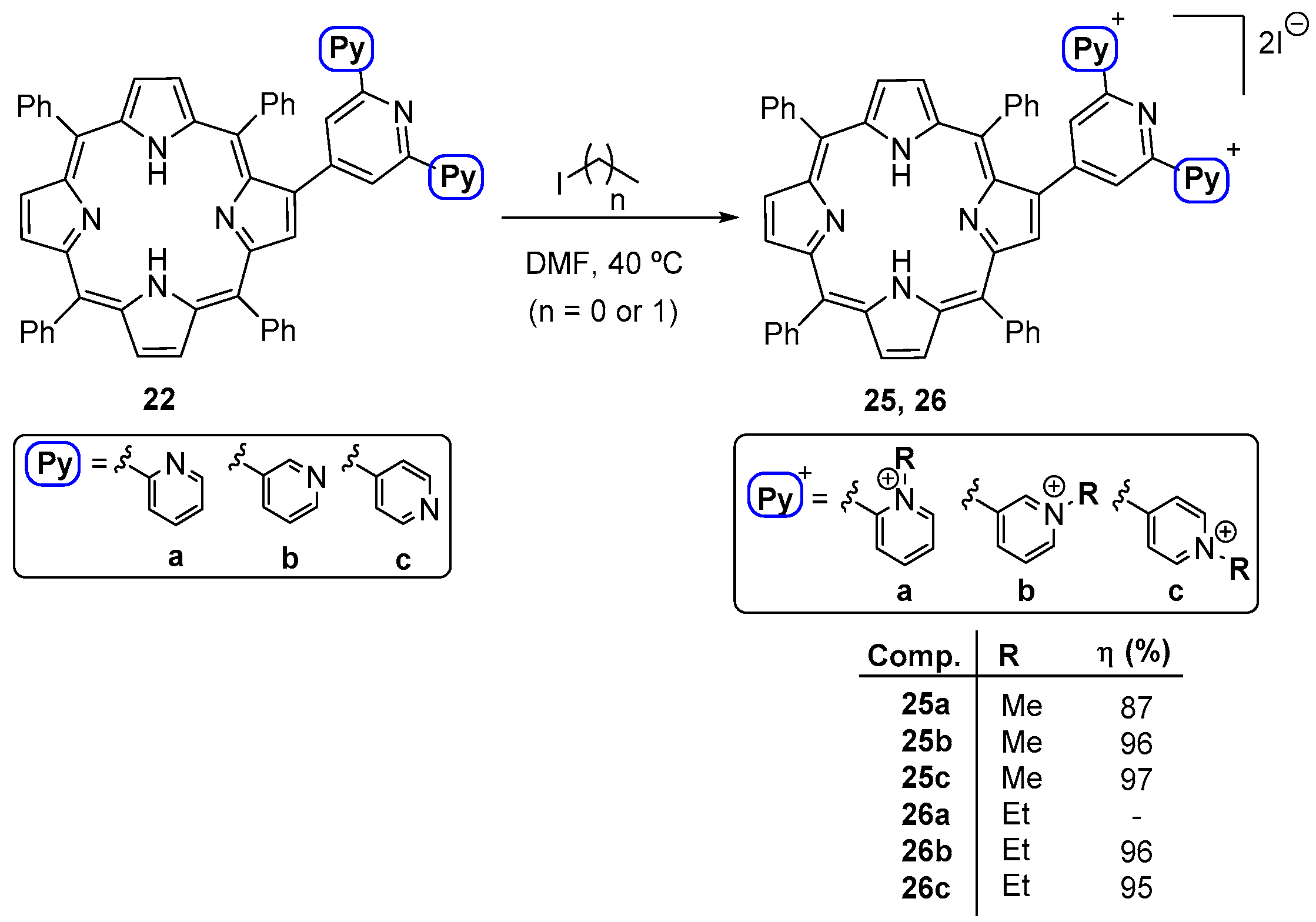
- Moura, N.M.M.; Ramos, C.I.V.; Linhares, I.; Santos, S.M.; Faustino, M.A.F.; Almeida, A.; Cavaleiro, J.A.S.; Amado, F.M.L.; Lodeiro, C.; Neves, M.G.P.M.S. Synthesis, characterization and biological evaluation of cationic porphyrin-terpyridine derivatives. RSC Adv. 2016, 6, 110674–110685. [Google Scholar] [CrossRef]
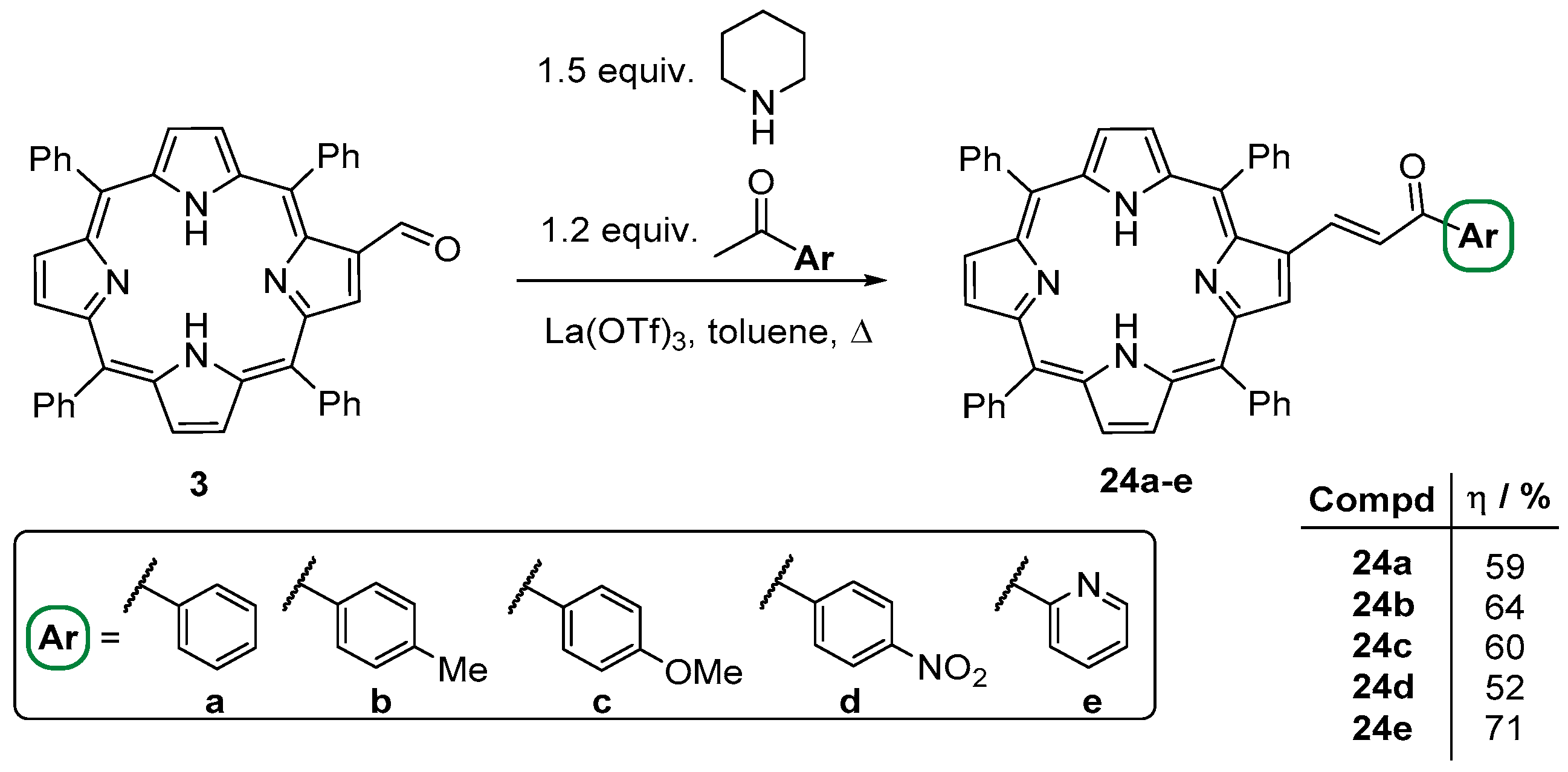
- Moura, N.M.M.; Nuñez, C.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S.; Capelo, J.L.; Lodeiro, C. Preparation and ion recognition features of porphyrin-chalcone type compounds as efficient red-fluorescent materials. J. Mater. Chem. C 2014, 2, 4772–4782. [Google Scholar] [CrossRef]
- Fleischer, E.B.; Dixon, F. Definitive evidence for the existence of the “sitting-atop” porphyrin complexes in nonaqueous solutions. Bioinorg. Chem. 1977, 7, 129–139. [Google Scholar] [CrossRef]
- Callot, H.J.; Chevrier, B.; Weiss, R. Sitting-atop porphyrin complexes. The structure of the bischloromercury(II) complex of N-tosylaminooctaethylporphyrin. J. Am. Chem. Soc. 1979, 101, 7729–7730. [Google Scholar] [CrossRef]
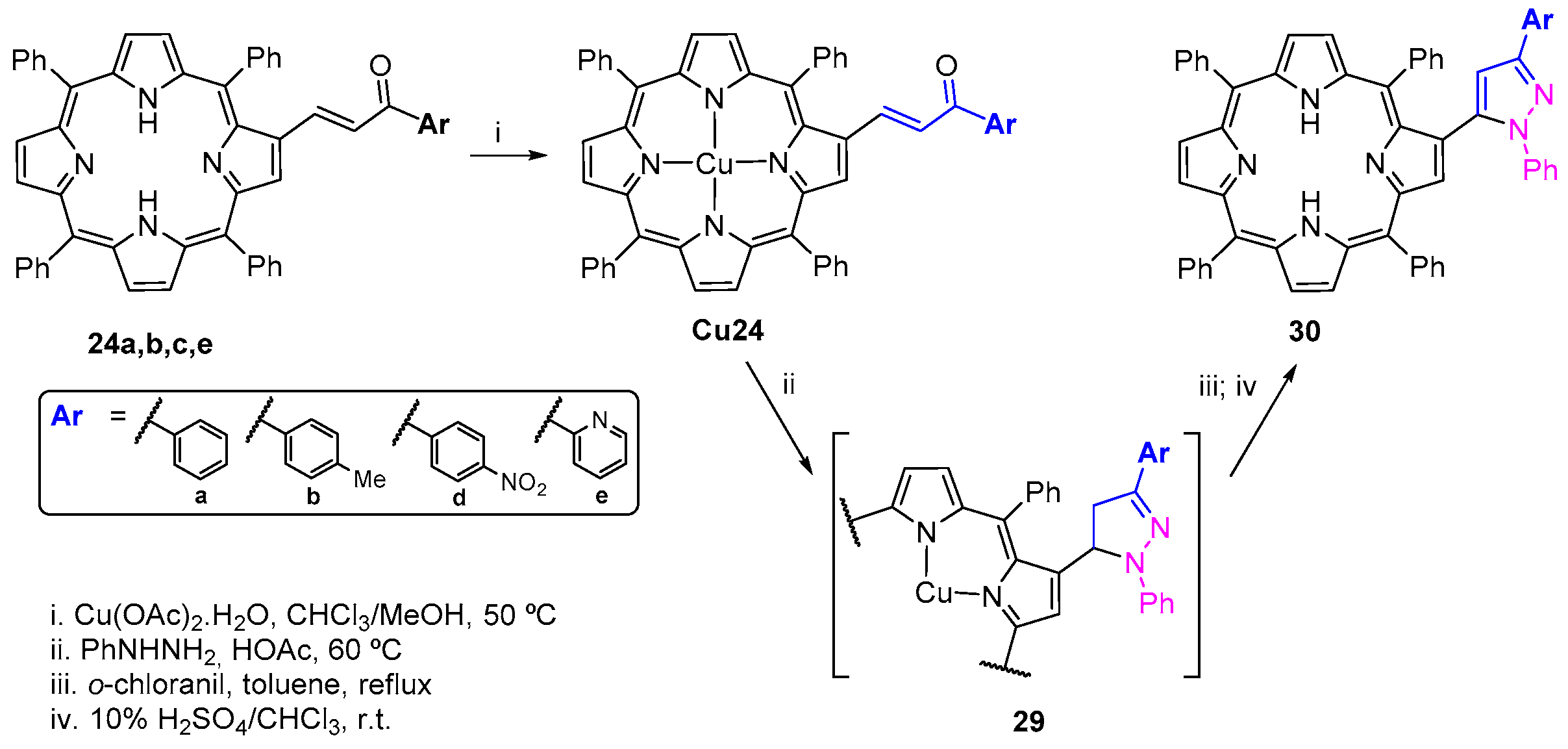
- Moura, N.M.M.; Nuñez, C.; Santos, S.M.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S.; Capelo, J.L.; Lodeiro, C. Synthesis, spectroscopy studies, and theoretical calculations of new fluorescent probes based on pyrazole containing porphyrins for Zn(II), Cd(II), and Hg(II) optical detection. Inorg. Chem. 2014, 53, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.P.; Nitschinsk, L.J. Porphyrin dimers linked by conjugated butadiynes. Tetrahedron 1992, 48, 8781–8792. [Google Scholar] [CrossRef]
- Gosper, J.J.; Ali, M. A conformationally constrained conjugated porphyrin dimer. J. Chem. Soc. Chem. Commun. 1994, 1707–1708. [Google Scholar] [CrossRef]
- Arnold, D.P.; Gaete-Holmes, R.; Johnson, A.W.; Smith, A.R.P.; Williams, G.A. Wittig condensation products from nickel meso-formyl-octaethyl-porphyrin and -aetioporphyrin I and some cyclisation reactions. J. Chem. Soc. Perkin Trans. 1 1978, 1660–1670. [Google Scholar] [CrossRef]
- Burrell, A.K.; Officer, D.L.; Plieger, P.G.; Reid, D.C.W. Synthetic routes to multiporphyrin arrays. Chem. Rev. 2001, 101, 2751–2796. [Google Scholar] [CrossRef] [PubMed]
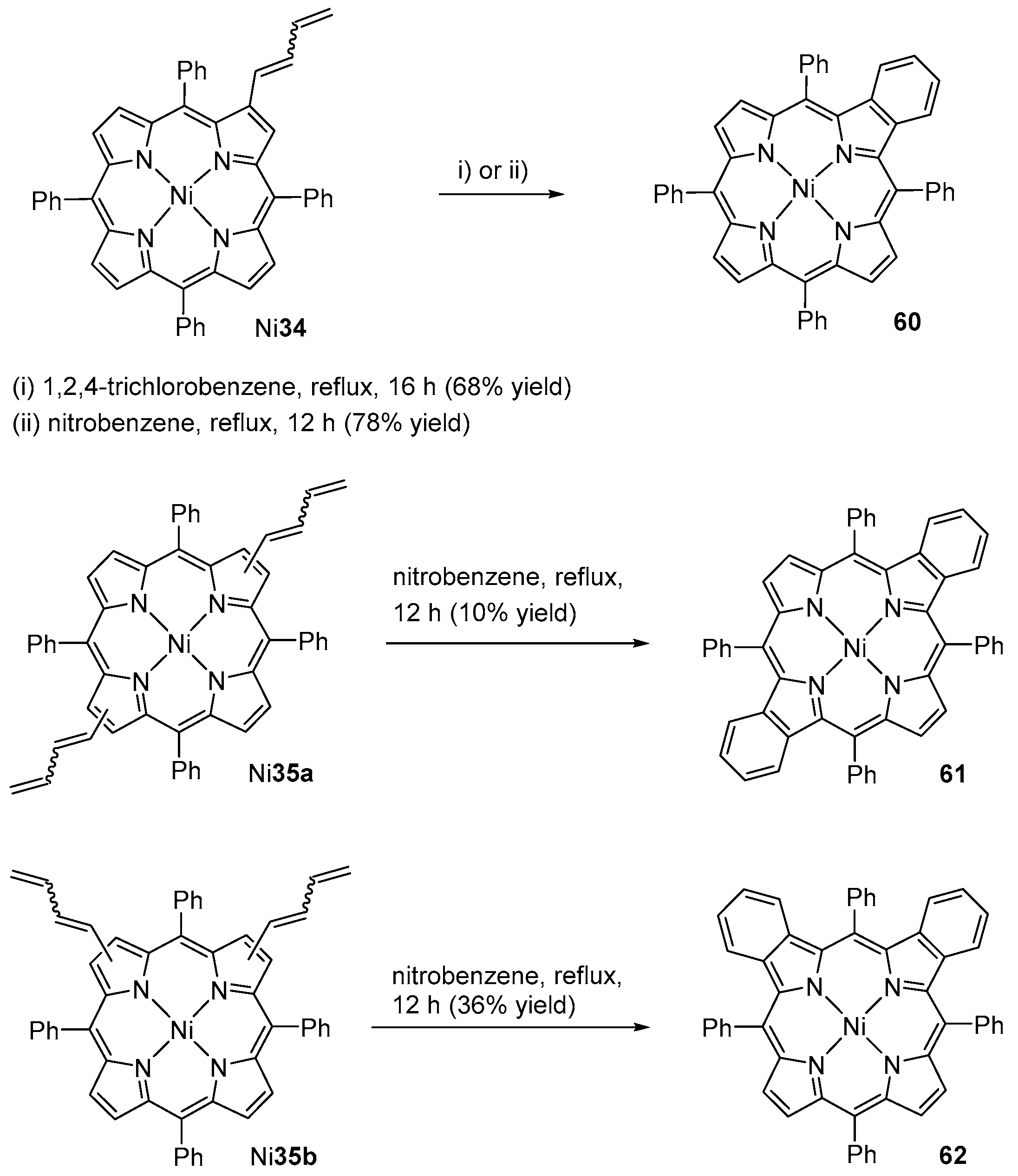
- Faustino, M.A.; Neves, M.G.P.M.S.; Vicente, M.G.H.; Silva, A.M.S.; Cavaleiro, J.A.S. New naphthochlorins from the intramolecular cyclization of β-vinyl-meso-tetraarylporphyrins. Tetrahedron Lett. 1995, 36, 5977–5978. [Google Scholar] [CrossRef]
- Silva, A.M.G.; de Oliveira, K.T.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S.; Brandão, P.; Felix, V. Chemical transformations of mono- and bis(buta-1,3-dien-1-yl)porphyrins: A new synthetic approach to mono- and dibenzoporphyrins. Eur. J. Org. Chem. 2008, 2008, 704–712. [Google Scholar] [CrossRef]
- Grigg, R.; Johnson, A.W.; Sweeney, A. Diels–Alder additions to protoporphyrin IX dimethyl ester. J. Chem. Soc. Chem. Commun. 1968, 0, 697. [Google Scholar] [CrossRef]
- Callot, H.J.; Johnson, A.W.; Sweeney, A. Additions to porphins involving the formation of new carbon-carbon bonds. J. Chem. Soc. Perkin Trans. 1 1973, 0, 1424–1427. [Google Scholar] [CrossRef]
- DiNello, R.K.; Dolphin, D. Reactions of protoporphyrin with tetracyanoethylene. J. Org. Chem. 1980, 45, 5196–5204. [Google Scholar] [CrossRef]
- Morgan, A.R.; Pangka, V.S.; Dolphin, D. Ready syntheses of benzoporphyrins via Diels–Alder reactions with protoporphyrin IX. J. Chem. Soc. Chem. Commun. 1984, 0, 1047–1048. [Google Scholar] [CrossRef]
- Pangka, V.S.; Morgan, A.R.; Dolphin, A. The Diels–Alder reactions of protoporphyrin IX dimethyl ester with electron-deficient alkynes. J. Org. Chem. 1986, 51, 1094–1100. [Google Scholar] [CrossRef]
- Yon-Hin, P.; Wijesekera, T.P.; Dolphin, D. Transformation of a monovinylporphyrin to benzoporphyrins via Diels–Alder adducts. Tetrahedron Lett. 1989, 30, 6135–6138. [Google Scholar] [CrossRef]
- Inhoffen, H.H.; Brockmann, H.; Bliesener, K.-M. Zur weiteren Kenntnis des Chlorophylls und des Hamins, XXX. Photoprotoporphyrine und ihre Umwandlung in Spirographis-sowie Isospirographis-porphyrin. Liebigs. Ann. Chem. 1969, 730, 173–185. [Google Scholar] [CrossRef]
- De Oliveira, K.T.; Silva, A.M.S.; Tomé, A.C.; Neves, M.G.P.M.S.; Neri, C.R.; Garcia, V.S.; Serra, O.A.; Iamamoto, Y.; Cavaleiro, J.A.S. Synthesis of new amphiphilic chlorin derivatives from protoporphyrin-IX dimethyl ester. Tetrahedron 2008, 64, 8709–8715. [Google Scholar] [CrossRef]
- Petrilli, R.; Praça, F.S.G.; Carollo, A.R.H.; Medina, W.S.G.; de Oliveira, K.T.; Fantini, M.C.A.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Serra, O.A.; Iamamoto, Y.; et al. Nanoparticles of lyotropic liquid crystals: A novel strategy for the topical delivery of a chlorin derivative for photodynamic therapy of skin cancer. Curr. Nanosci. 2013, 9, 434–441. [Google Scholar] [CrossRef]
- Morgan, A.R.; Kohli, D.H. Diels-Alder reactions of protoporphyrin ix dimethyl ester. Tetrahedron Lett. 1995, 36, 7603–7606. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Neves, M.G.P.M.S.; Tomé, A.C. Cycloaddition reactions of porphyrins. ARKIVOC 2003, 2003, 107–130. [Google Scholar] [CrossRef]
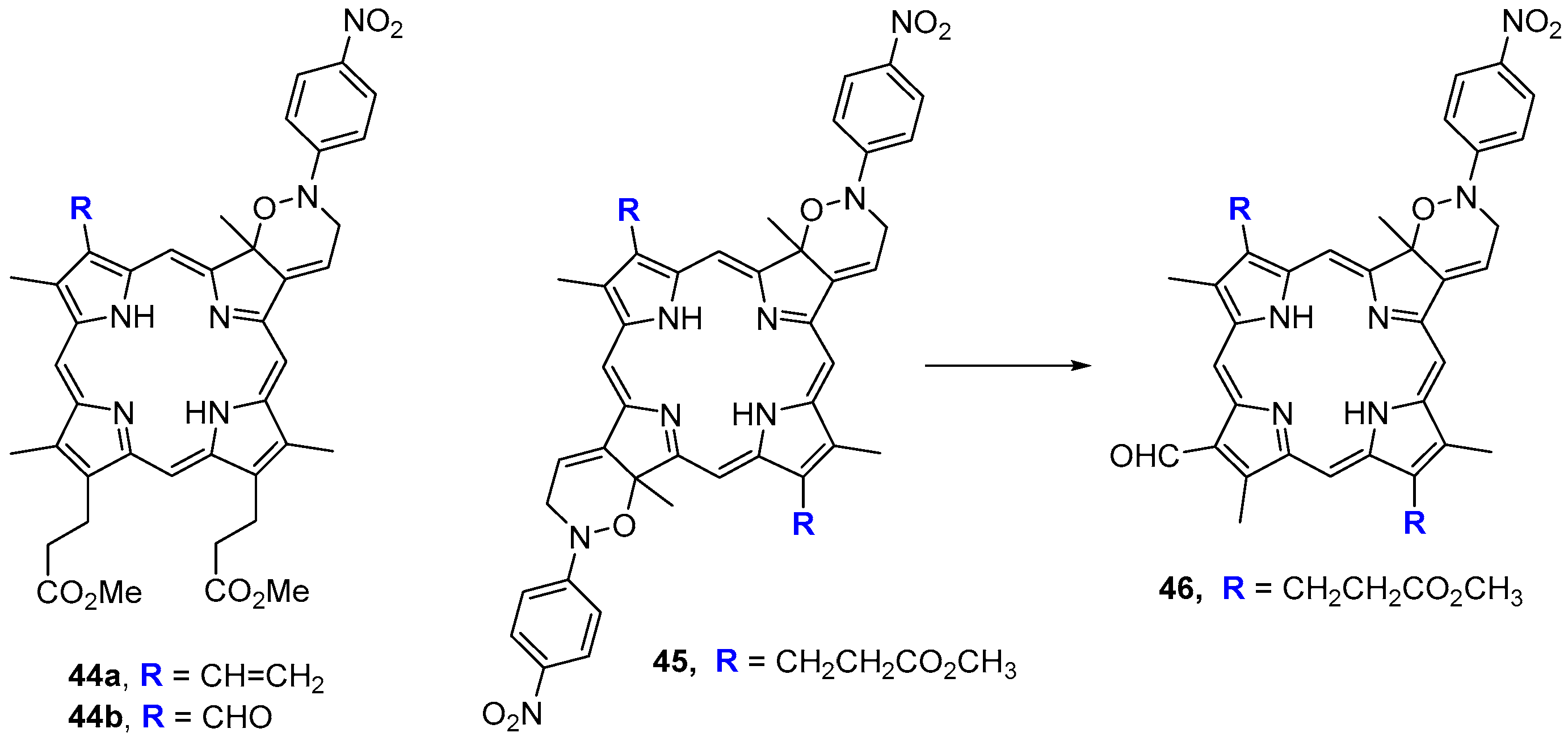
- Cavaleiro, J.A.S.; Jackson, A.H.; Neves, M.G.P.M.S.; Rao, K.R.N. Diels–Alder reactions of protoporphyrin dimethyl esters with nitrosobenzenes; a novel degradation to formyl porphyrins. J. Chem. Soc. Chem. Commun. 1985, 0, 776–777. [Google Scholar] [CrossRef]
- Faustino, M.A.F.; Neves, M.G.P.M.S.; Vicente, M.G.H.; Silva, A.M.S.; Cavaleiro, J.A.S. Diels–Alder reactions of Ni(II) β-vinyl-meso-tetraarylporphyrins. Tetrahedron Lett. 1996, 37, 3569–3570. [Google Scholar] [CrossRef]
- Faustino, M.A.F.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. A new meso-tetraphenylchlorin and meso-tetraphenylbenzoporphyrin from Diels–Alder reactions of Ni(II) β-vinyl meso-tetraphenylporphyrin. Chimia 1997, 51, 472. [Google Scholar]
- Matsumoto, K.; Kimura, S. Diels–Alder reaction of Ni(II) β-vinyl-meso-tetraphenylporphyrin with dimethyl acetylenedicarboxylate and N-phenyl maleimide. Heterocycl. Commun. 2000, 6, 31–34. [Google Scholar] [CrossRef]
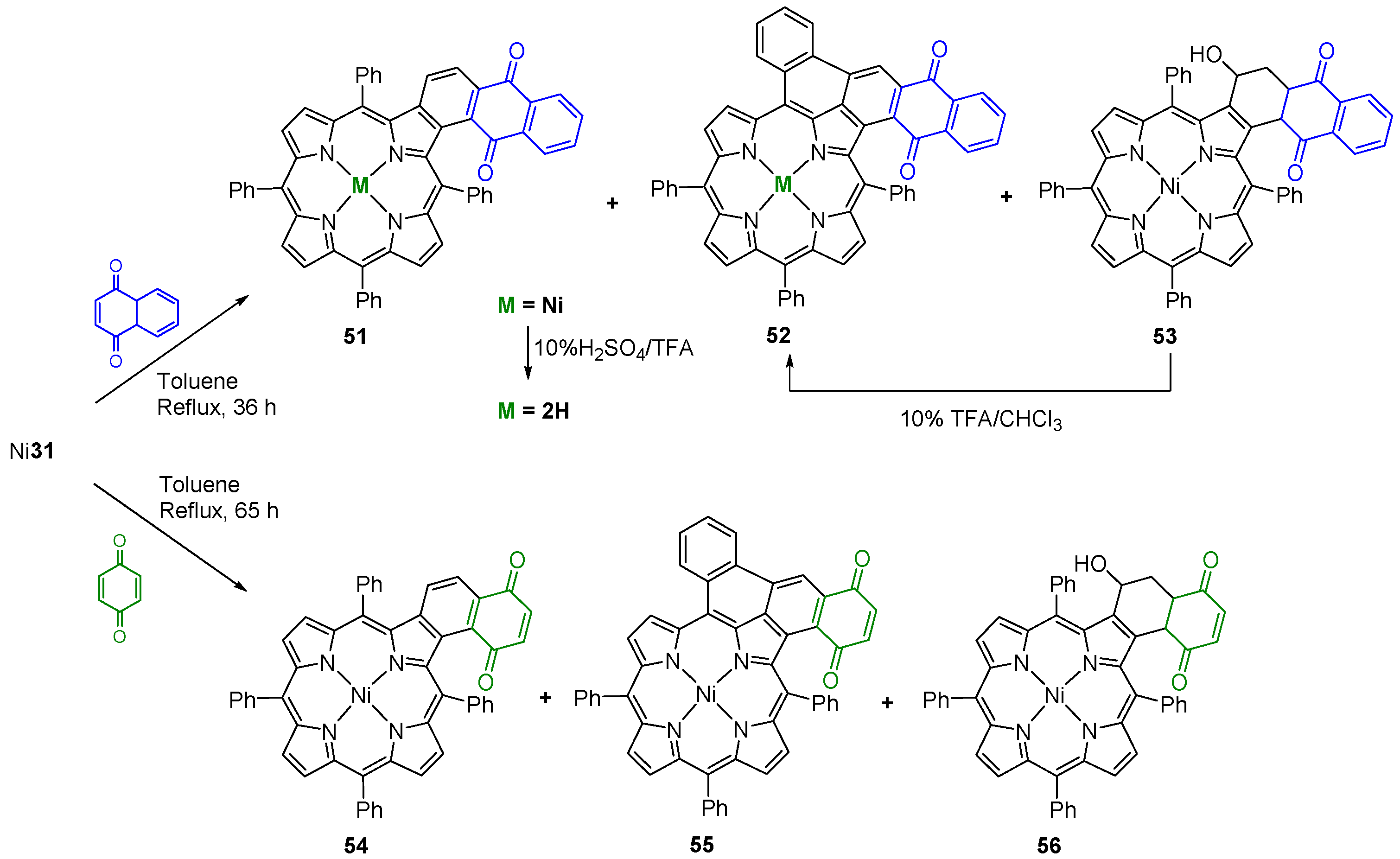
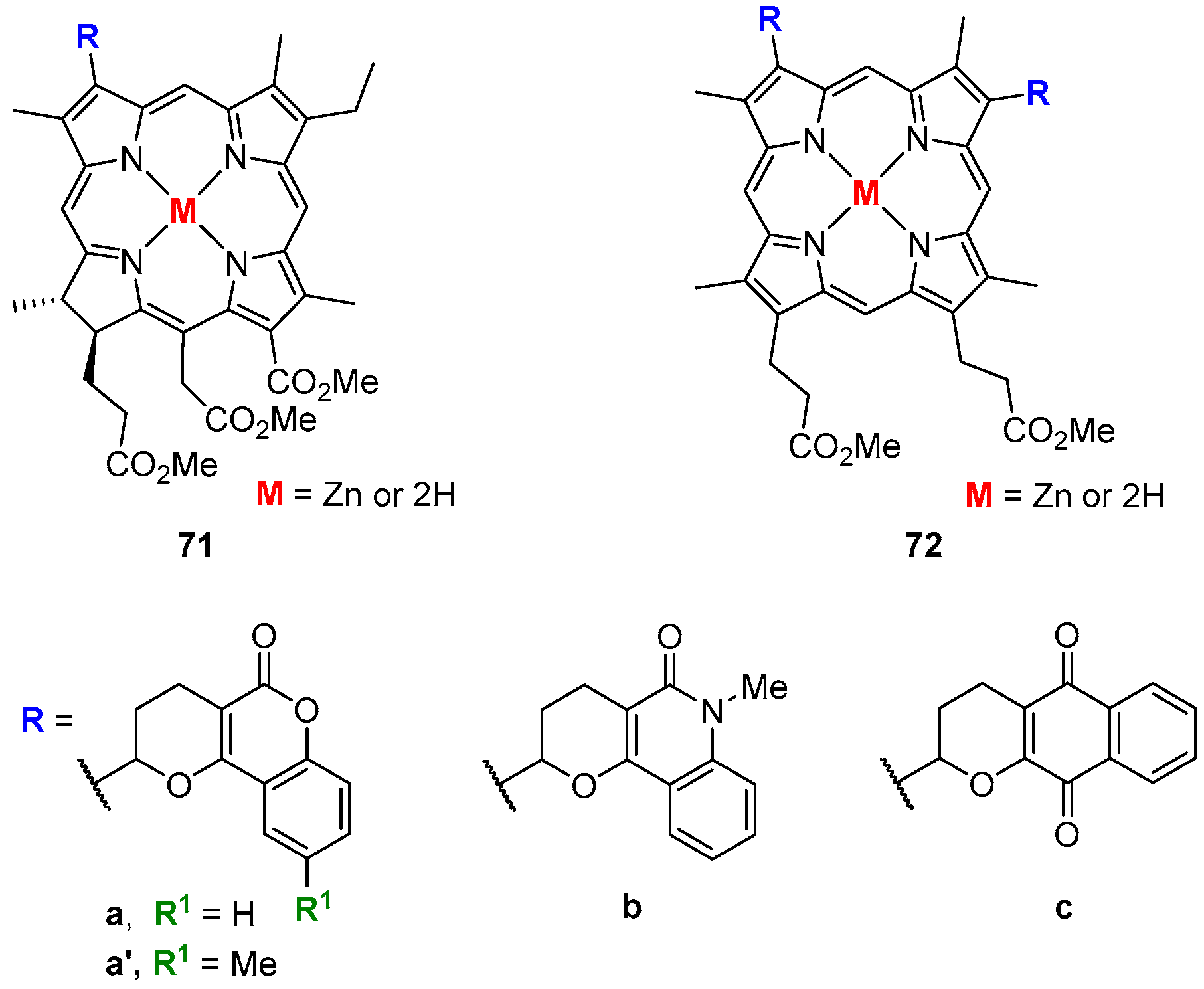
- Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S. Diels–Alder reactions of beta-vinyl-meso-tetraphenylporphyrin with quinones. ARKIVOC 2005, 9, 332–343. [Google Scholar]
- Matsumoto, K.; Kimura, S.; Morishita, T.; Misumi, Y.; Hayashi, N. Diels–Alder reaction of Ni(II) β-vinyl-meso-tetraphenylporphyrin: A general method for synthesis of functionalized porphyrins. Synlett 2000, 2000, 233–235. [Google Scholar] [CrossRef]
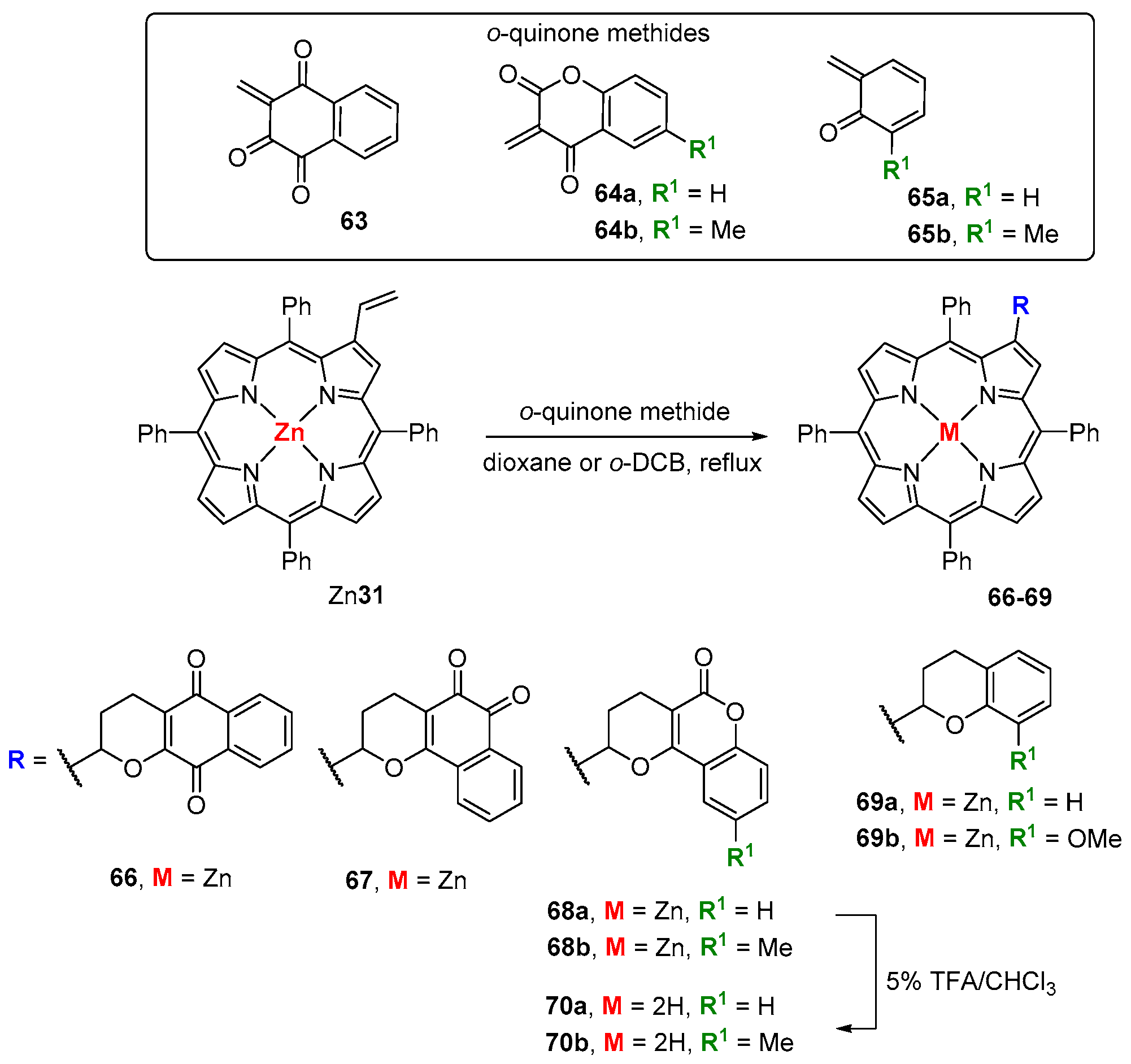
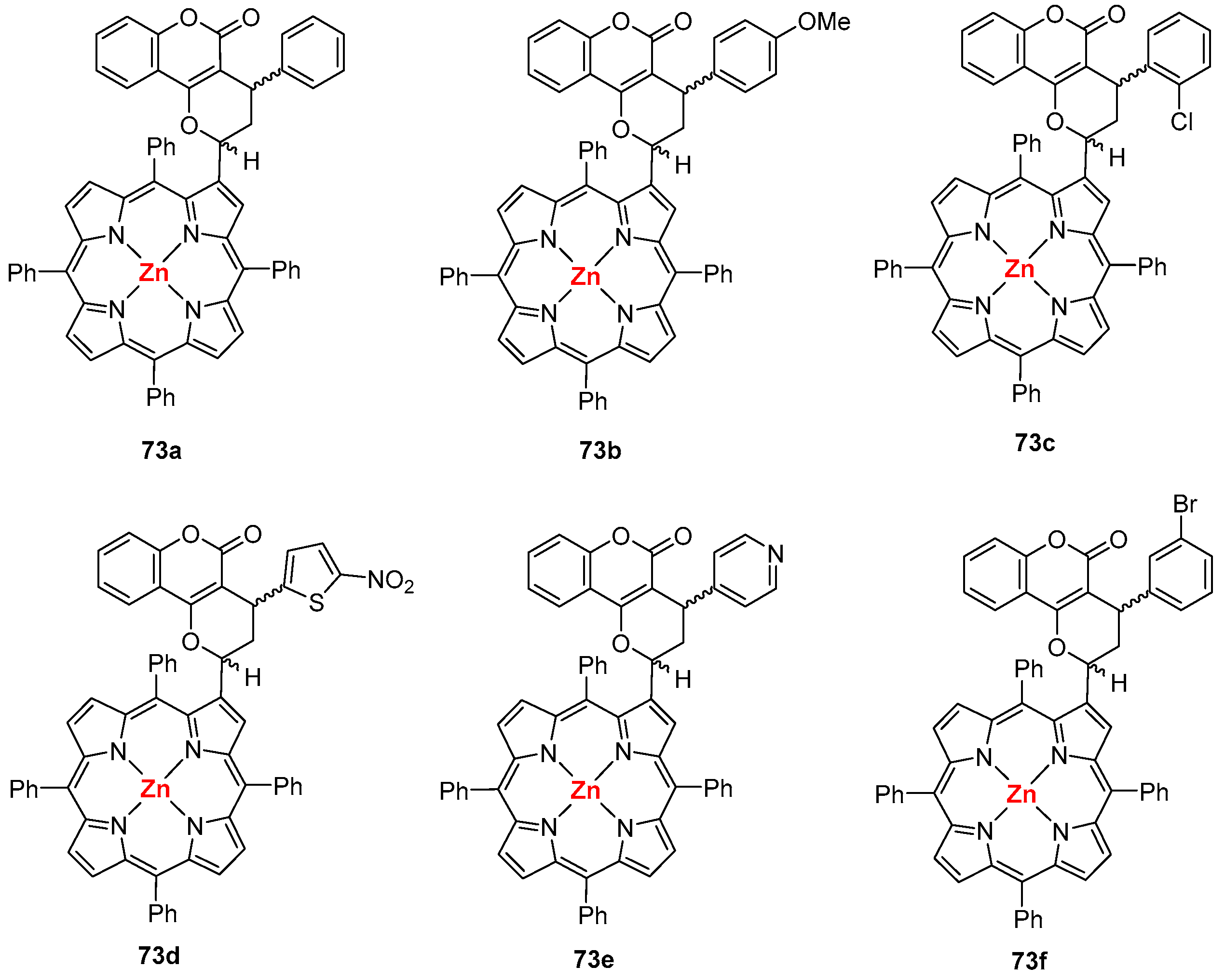
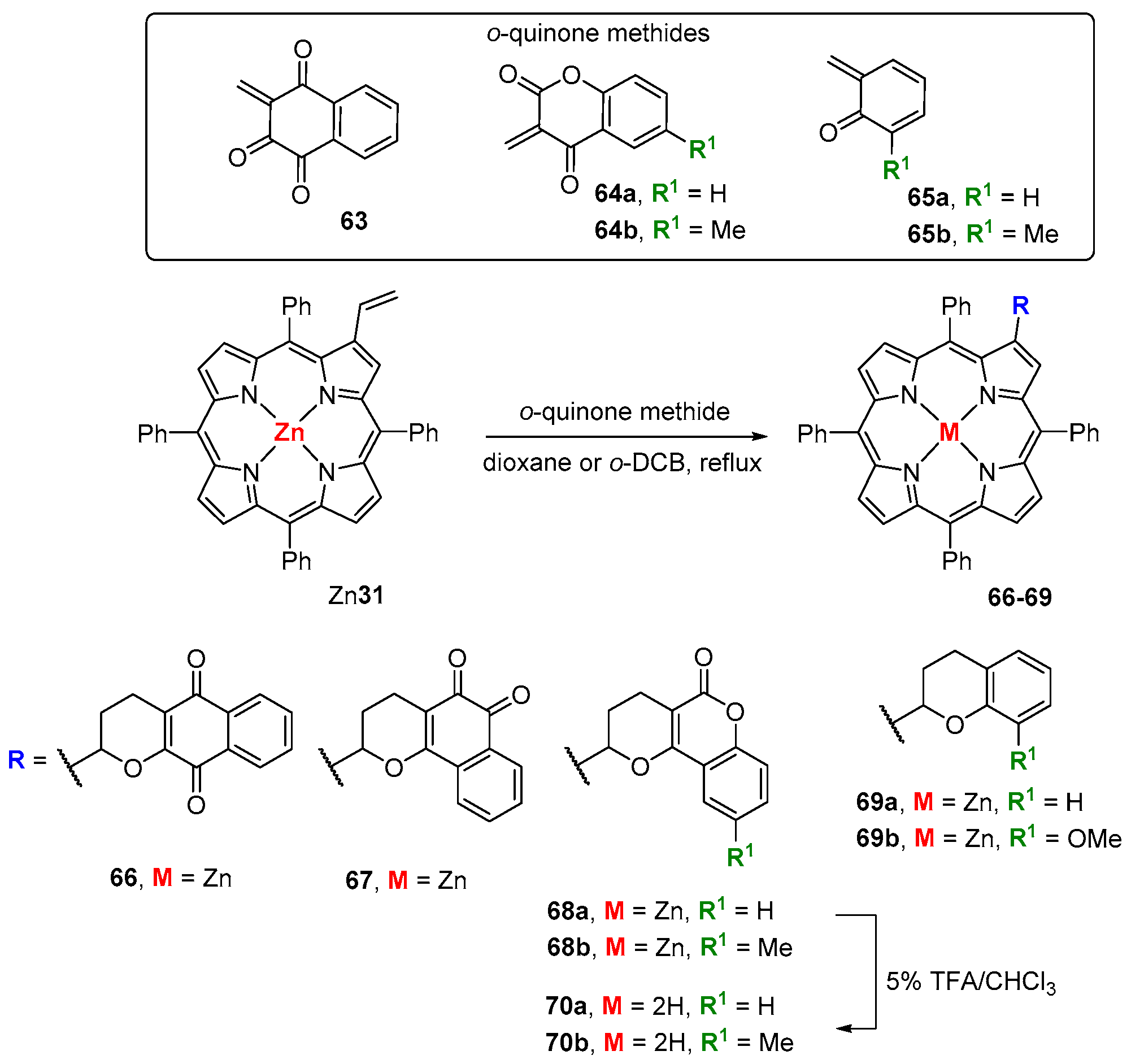
- Menezes, J.C.J.M.D.S.; Gomes, A.T.P.C.; Silva, A.M.S.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; da Silva, F.C.; Ferreira, V.F.; Cavaleiro, J.A.S. Reaction of β-vinyl-meso-tetraphenylporphyrin with o-quinone methines. Synlett 2011, 13, 1841–1844. [Google Scholar] [CrossRef]
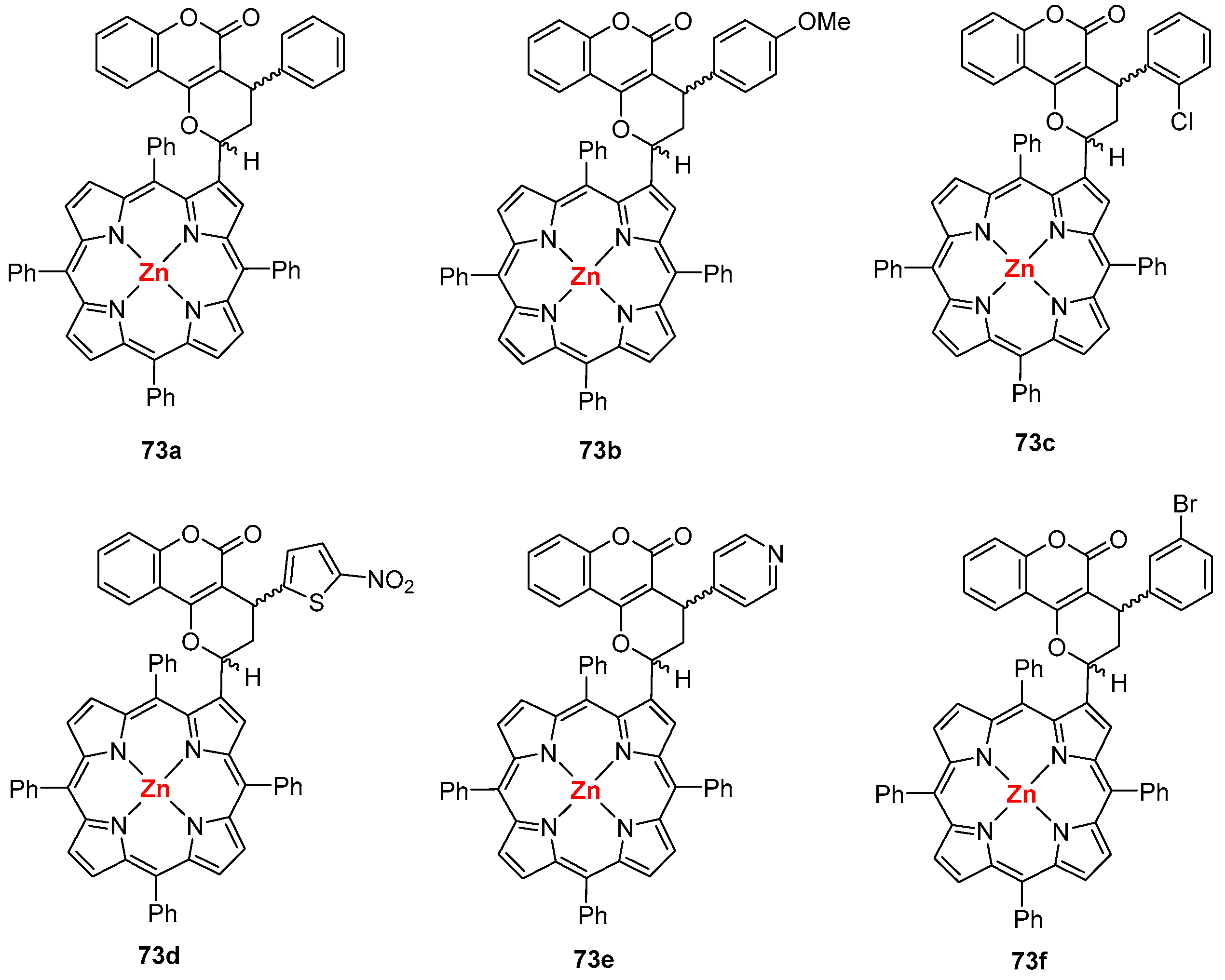
- Santos, C.I.M.; Oliveira, E.; Menezes, J.C.J.M.D.S.; Barata, J.F.B.; Faustino, M.A.F.; Ferreira, V.F.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S.; Lodeiro, C. New coumarin-corrole and -porphyrin conjugate multifunctional probes for anionic or cationic interactions: Synthesis, spectroscopy, and solid supported studies. Tetrahedron 2014, 70, 3361–3370. [Google Scholar] [CrossRef]
- Santos, C.I.M.; Oliveira, E.; Santos, H.M.; Menezes, J.C.J.M.D.S.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Capelo, J.L.; Neves, M.G.P.M.S.; Lodeiro, C. Untangling interactions of a zinc(II) complex containing a coumarin-porphyrin unit with alkaloids in water solutions: A photophysical study. Photochem. Photobiol. Sci. 2015, 14, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Menezes, J.C.J.M.D.S.; Faustino, M.A.F.; de Oliveira, K.T.; Uliana, M.P.; Ferreira, V.F.; Hackbarth, S.; Röder, B.; Tasso, T.T.; Furuyama, T.; Kobayashi, N.; et al. Synthesis of new chlorin e6 trimethyl and protoporphyrin IX dimethyl ester derivatives and their photophysical and electrochemical characterizations. Chem. Eur. J. 2014, 20, 13644–13655. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.F.C.; Gomes, A.T.P.C.; Silva, V.L.M.; Silva, A.M.S.; Neves, M.G.P.M.S.; da Silva, F.C.; Ferreira, V.F.; Cavaleiro, J.A.S. Ohmic heating assisted synthesis of coumarinyl porphyrin derivatives. RSC Adv. 2015, 5, 66192–66199. [Google Scholar] [CrossRef]
- Cerqueira, A.F.R.; Almodôvar, V.A.S.; Neves, M.G.P.M.S.; Tomé, A.C. Coumarin-tetrapyrrolic macrocycle conjugates: Synthesis and applications. Molecules 2017, 22, 994. [Google Scholar] [CrossRef] [PubMed]
- Silva Vera, L.M.; Santos Luis, M.N.B.F.; Silva Artur, M.S. Ohmic heating: An emerging concept in organic synthesis. Chem. Eur. J. 2017, 23, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
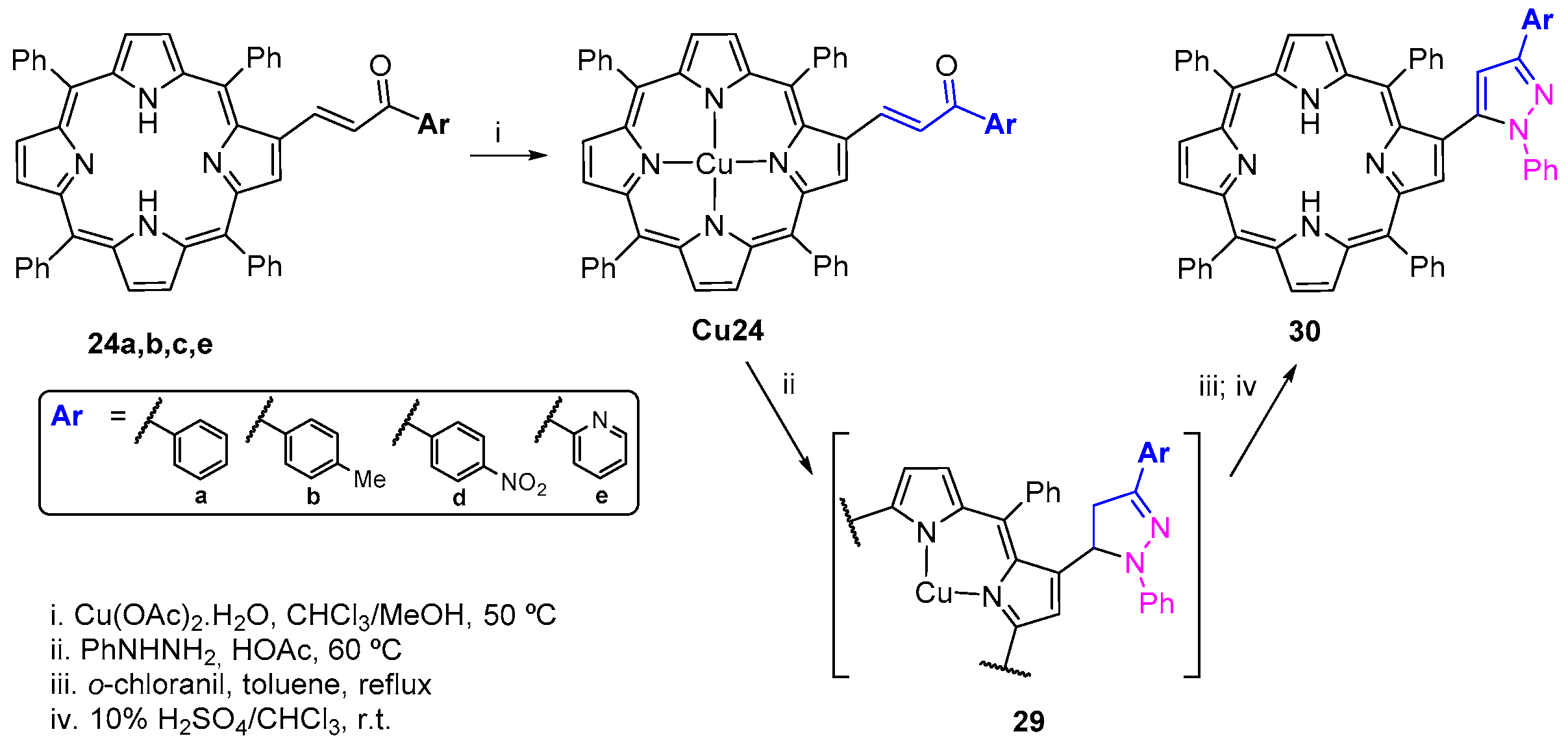
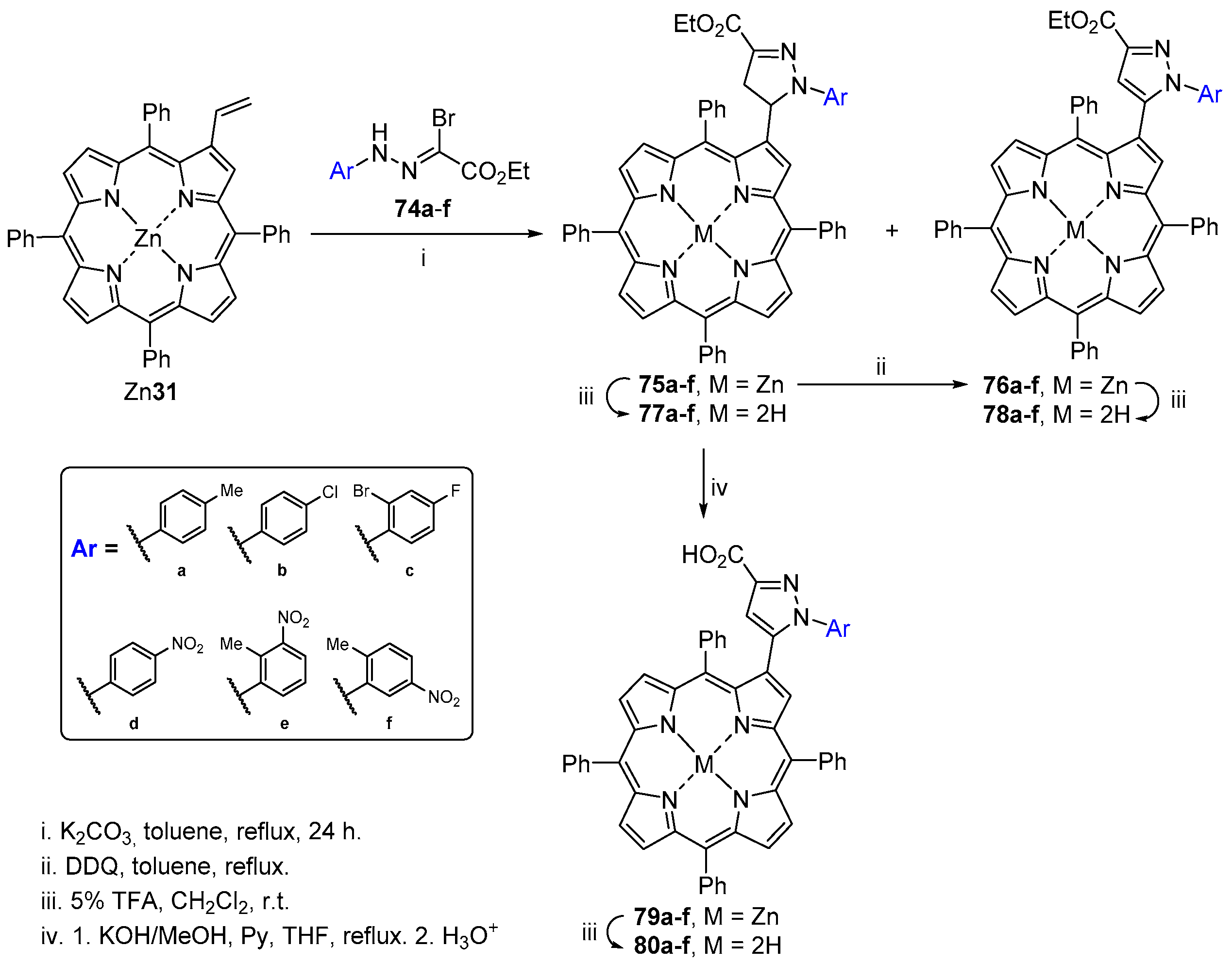
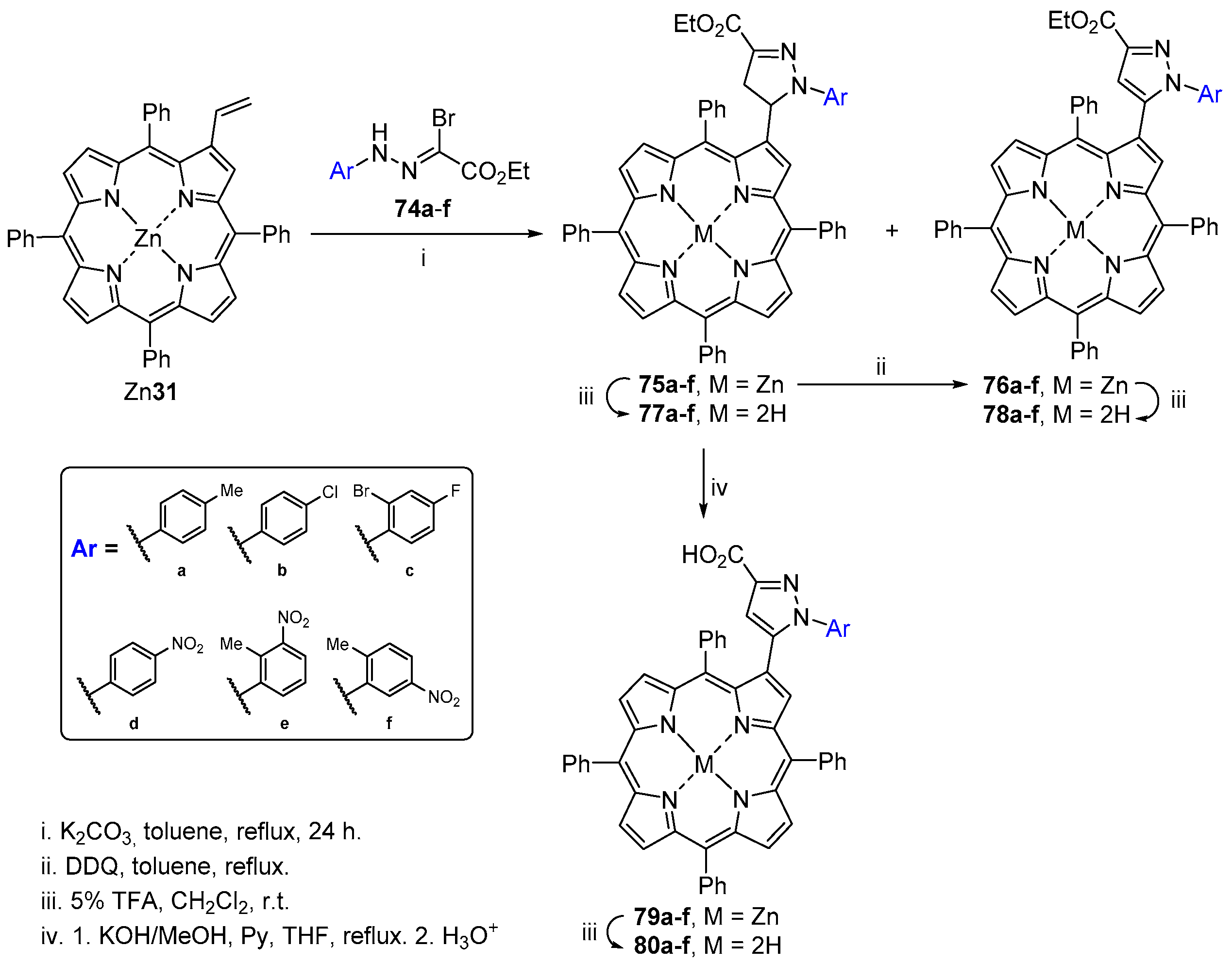
- Moura, N.M.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Rakib, E.M.; Hannioui, A.; Mojahidi, S.; Hackbarth, S.; Röder, B.; Paz, F.A.A.; et al. Novel pyrazoline and pyrazole porphyrin derivatives: Synthesis and photophysical properties. Tetrahedron 2012, 68, 8181–8193. [Google Scholar] [CrossRef]
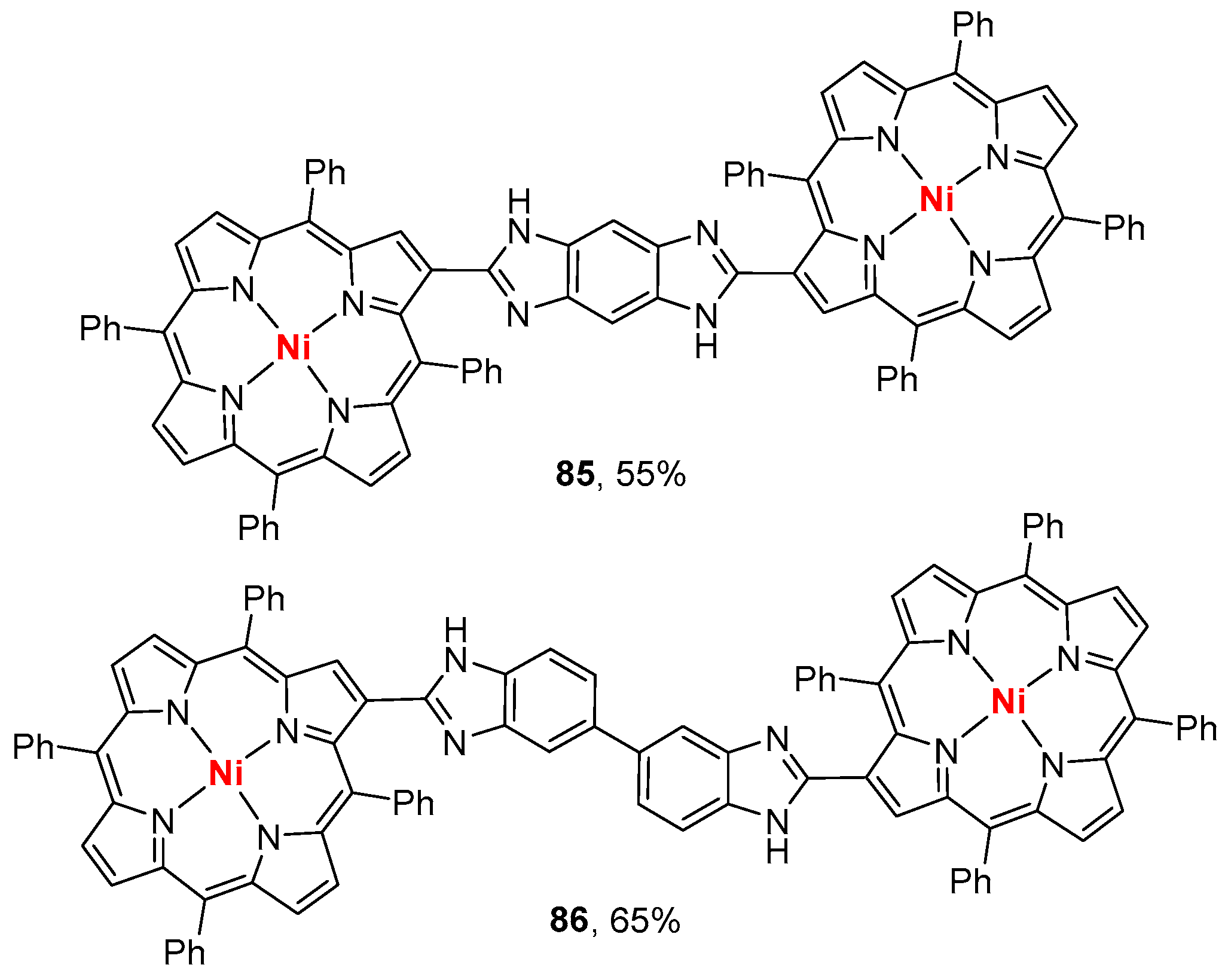
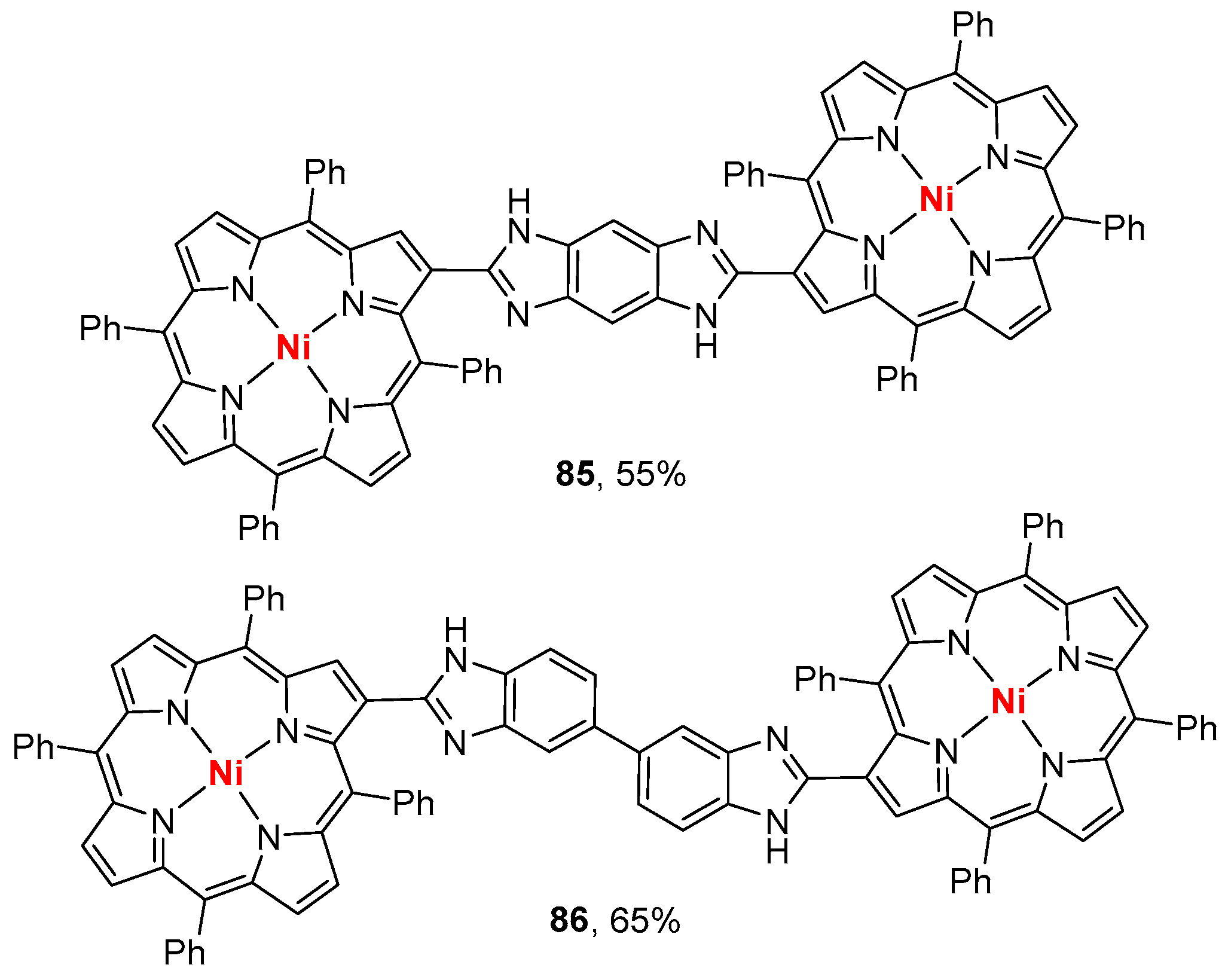
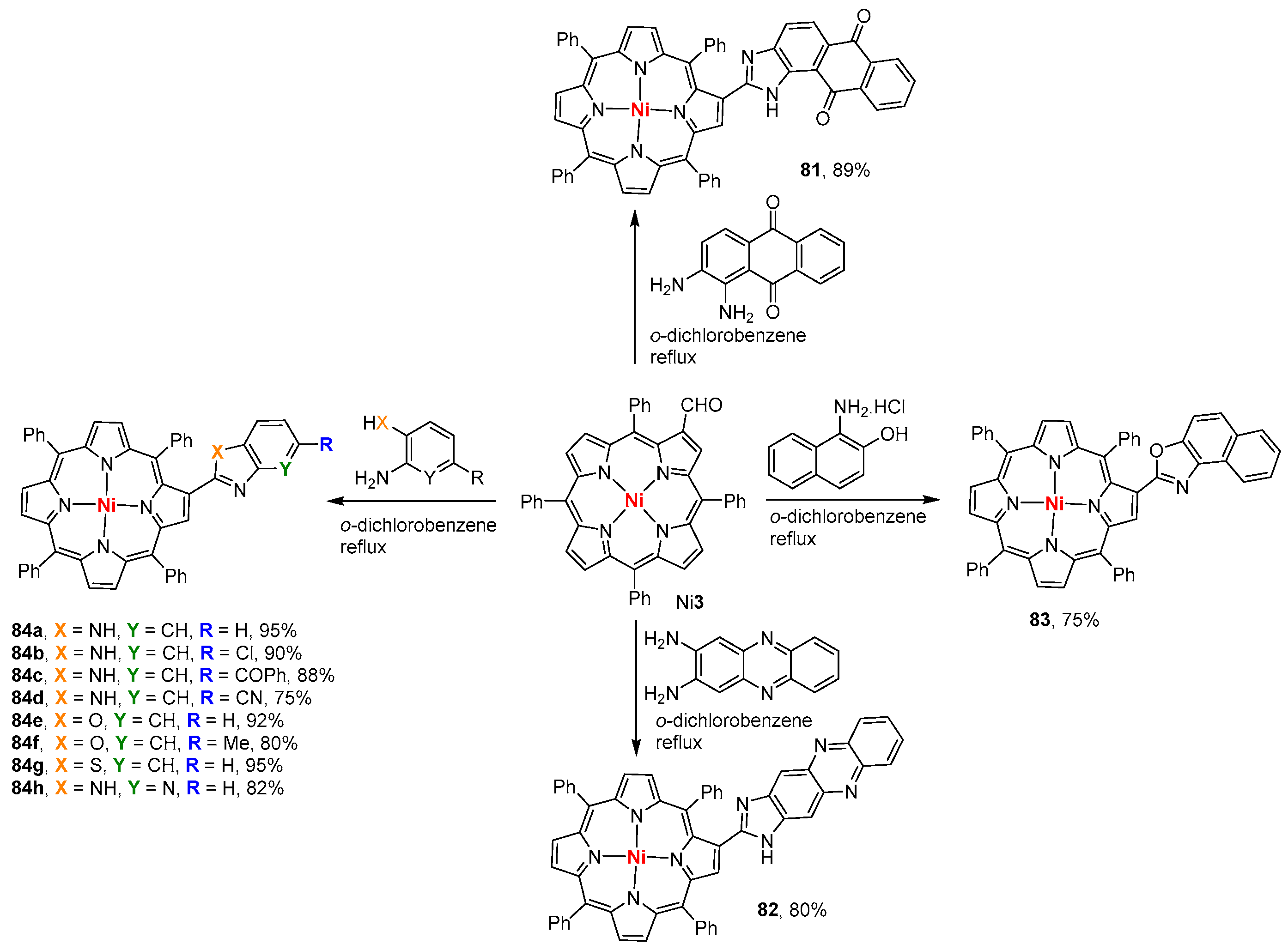
- Sharma, S.; Nath, M. An efficient synthetic approach to novel nickel(II) 2-benzazolo-5,10,15,20-tetraphenylporphyrins. J. Heterocycl. Chem. 2012, 49, 88–92. [Google Scholar] [CrossRef]
- Tekuri, C.; Singh, D.K.; Nath, M. Synthesis, characterization and optical properties of β-substituted pyrrolo- and indolo[1,2-a]quinoxalinoporphyrins. Dyes Pigment. 2016, 132, 194–203. [Google Scholar] [CrossRef]
- Giribabu, L.; Reeta, P.S.; Kanaparthi, R.K.; Srikanth, M.; Soujanya, Y. Bis(porphyrin)-anthraquinone triads: Synthesis, spectroscopy, and photochemistry. J. Phys. Chem. A 2013, 117, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Bastos, M.M.; Gomes, A.T.P.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Santos-Filho, O.A.; Boechat, N.; Cavaleiro, J.A.S. Synthesis of β-substituted porphyrin derivatives containing heterocyclic moieties as potential photosensitizers against cutaneous Leishmaniasis. Eur. J. Org. Chem. 2013, 1485–1493. [Google Scholar] [CrossRef]
- Alonso, C.M.A.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S. β-Imino-meso-tetraphenylporphyrin derivatives in hetero-Diels–Alder reactions. Eur. J. Org. Chem. 2004, 3233–3239. [Google Scholar] [CrossRef]
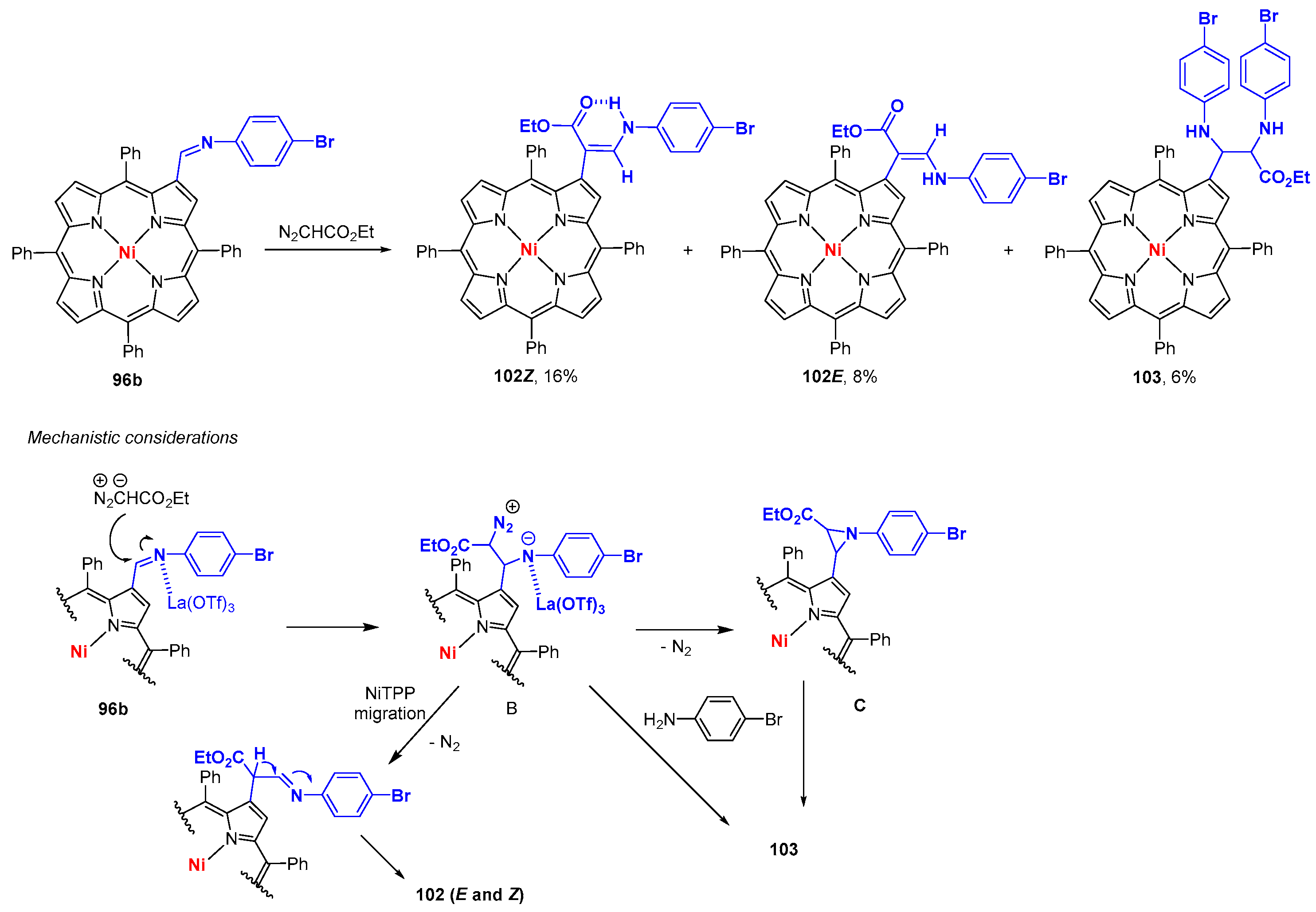
- Alonso, C.M.A.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S. Beta-imino-meso-tetraphenylporphyrin derivatives: Reactivity towards ethyl diazoacetate. Jordan J. Chem. 2006, 2, 95–107. [Google Scholar]
- Drovetskaya, T.; Reed, C.A.; Boyd, P. A fullerene-porphyrin conjugate. Tetrahedron Lett. 1995, 36, 7971–7974. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. Synthesis of new β-substituted meso-tetraphenylporphyrins via 1,3-dipolar cycloaddition reactions. 1. J. Org. Chem. 2002, 67, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.G.; Faustino, M.A.F.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. A novel approach to the synthesis of mono- and dipyrroloporphyrins. J. Chem. Soc. Perkin Trans. 1 2001, 0, 2752–2753. [Google Scholar] [CrossRef]
- Jaquinod, L.; Gros, C.; Olmstead, M.M.; Antolovich, M.; Smith, K.M. First syntheses of fused pyrroloporphyrins. Chem. Commun. 1996, 12, 1475–1476. [Google Scholar] [CrossRef]
- Knapp, S.; Vasudevan, J.; Emge, T.J.; Arison, B.H.; Potenza, J.A.; Schugar, H.J. A tethered porphyrin dimer with π overlap of a single pyrrole ring. Angew. Chem. Int. Ed. 1998, 37, 2368–2370. [Google Scholar] [CrossRef]
- Vicente, M.G.H.; Jaquinod, L.; Khoury, R.G.; Madrona, A.Y.; Smith, K.M. Synthesis and chemistry of new benzoporphyrins. Tetrahedron Lett. 1999, 40, 8763–8766. [Google Scholar] [CrossRef]
- Liu, W.; Fronczek, F.R.; Vicente, M.G.H.; Smith, K.M. Diels–Alder reactions of pyrrolo[3,4-b]porphyrins. Tetrahedron Lett. 2005, 46, 7321–7324. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.; Neves, M.G.P.M.S.; Tomé, A.C.; Paz, F.A.A.; Silva, A.M.S.; Cavaleiro, J.A.S. 1,3-dioxopyrrolo[3,4-b]porphyrins: Synthesis and chemistry. Org. Lett. 2011, 13, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Santos, S.M.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Rocha, J.; Cavaleiro, J.A.S. Meso-tetraphenylbenzoporphyrin-2(2),2(3)-dicarboxylic anhydride: A platform to benzoporphyrin derivatives. J. Org. Chem. 2013, 78, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.F.F.; Barata, J.F.B.; Silva, A.M.G.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Cavaleiro, J.A.S. Reactivity of tetrapyrrolyl nitrones towards dipolarophiles bearing electron-withdrawing groups. Tetrahedron Lett. 2015, 56, 2878–2881. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Lacerda, P.S.S.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S.; Makarova, E.A.; Lukyanets, E.A. Porphyrins in 1,3-dipolar cycloaddition reactions. Synthesis of new porphyrin-chlorin and porphyrin-tetraazachlorin dyads. J. Org. Chem. 2006, 71, 8352–8356. [Google Scholar] [CrossRef] [PubMed]
- Enes, R.F.; Cid, J.-J.; Hausmann, A.; Trukhina, O.; Gouloumis, A.; Vázquez, P.; Cavaleiro, J.A.S.; Tomé, A.C.; Guldi, D.M.; Torres, T. Synthesis and photophysical properties of fullerene-phthalocyanine-porphyrin triads and pentads. Chem. Eur. J. 2012, 18, 1727–1736. [Google Scholar] [CrossRef] [PubMed]







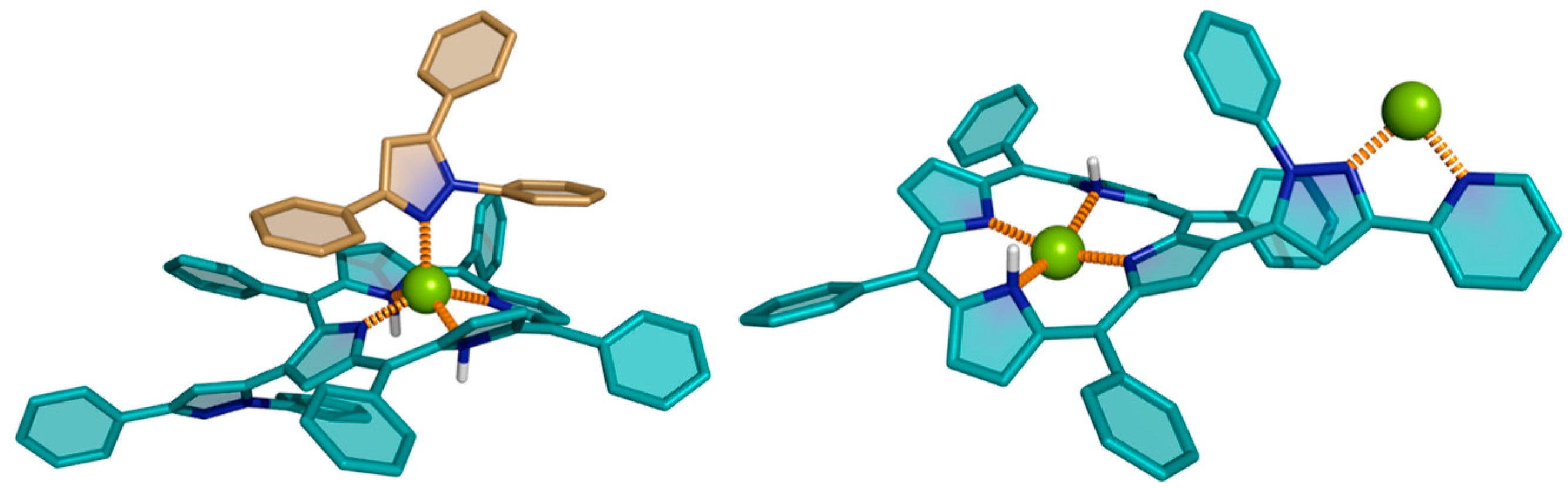

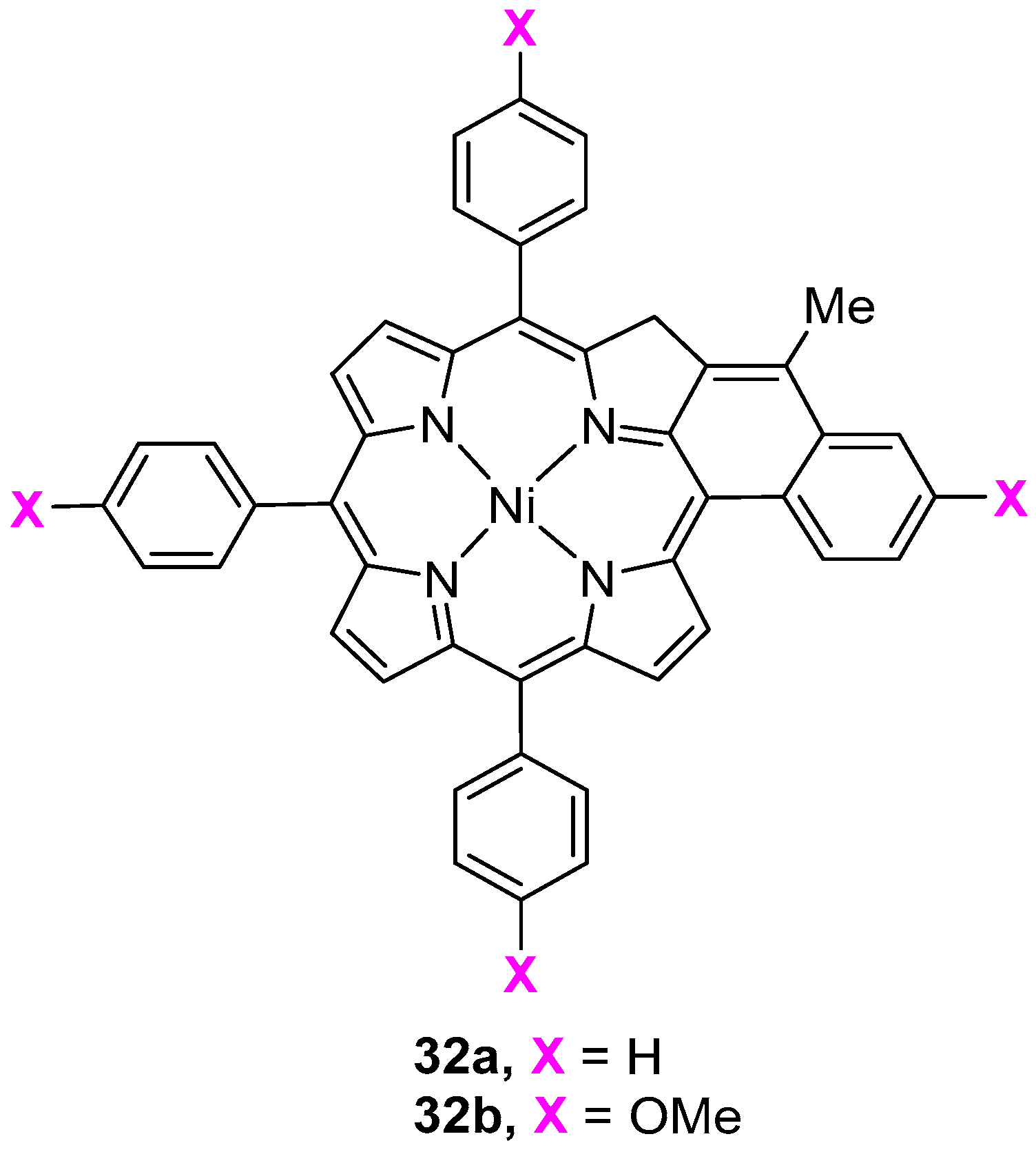
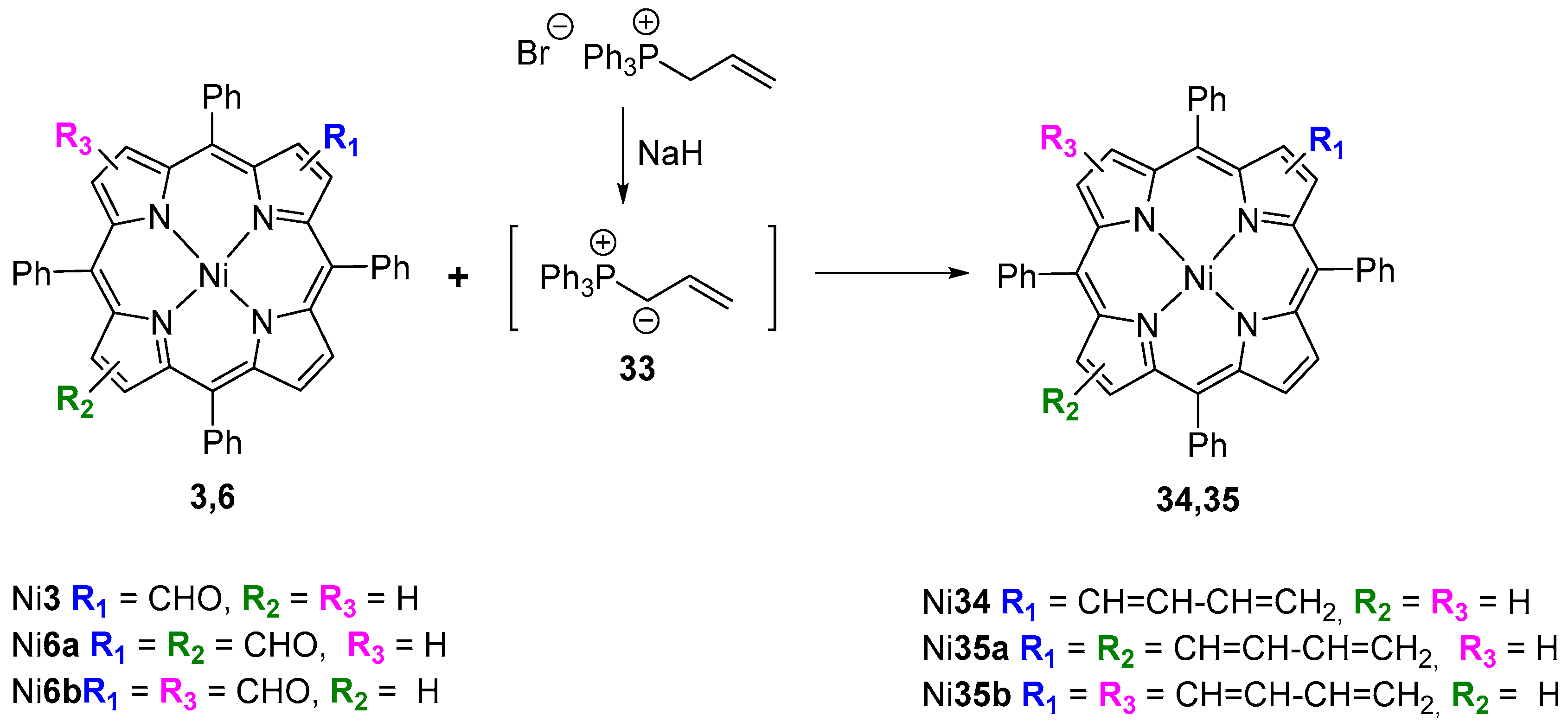
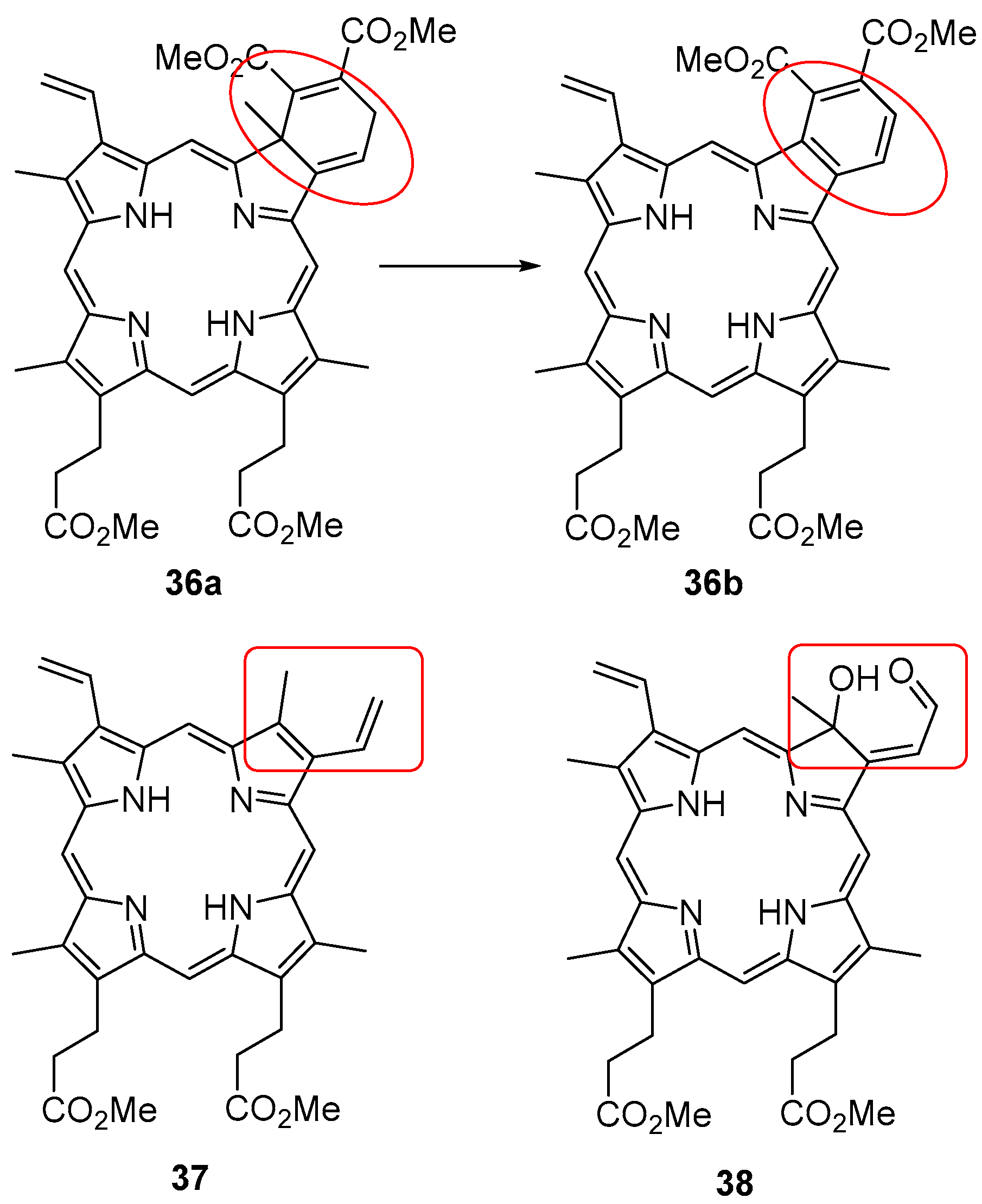

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
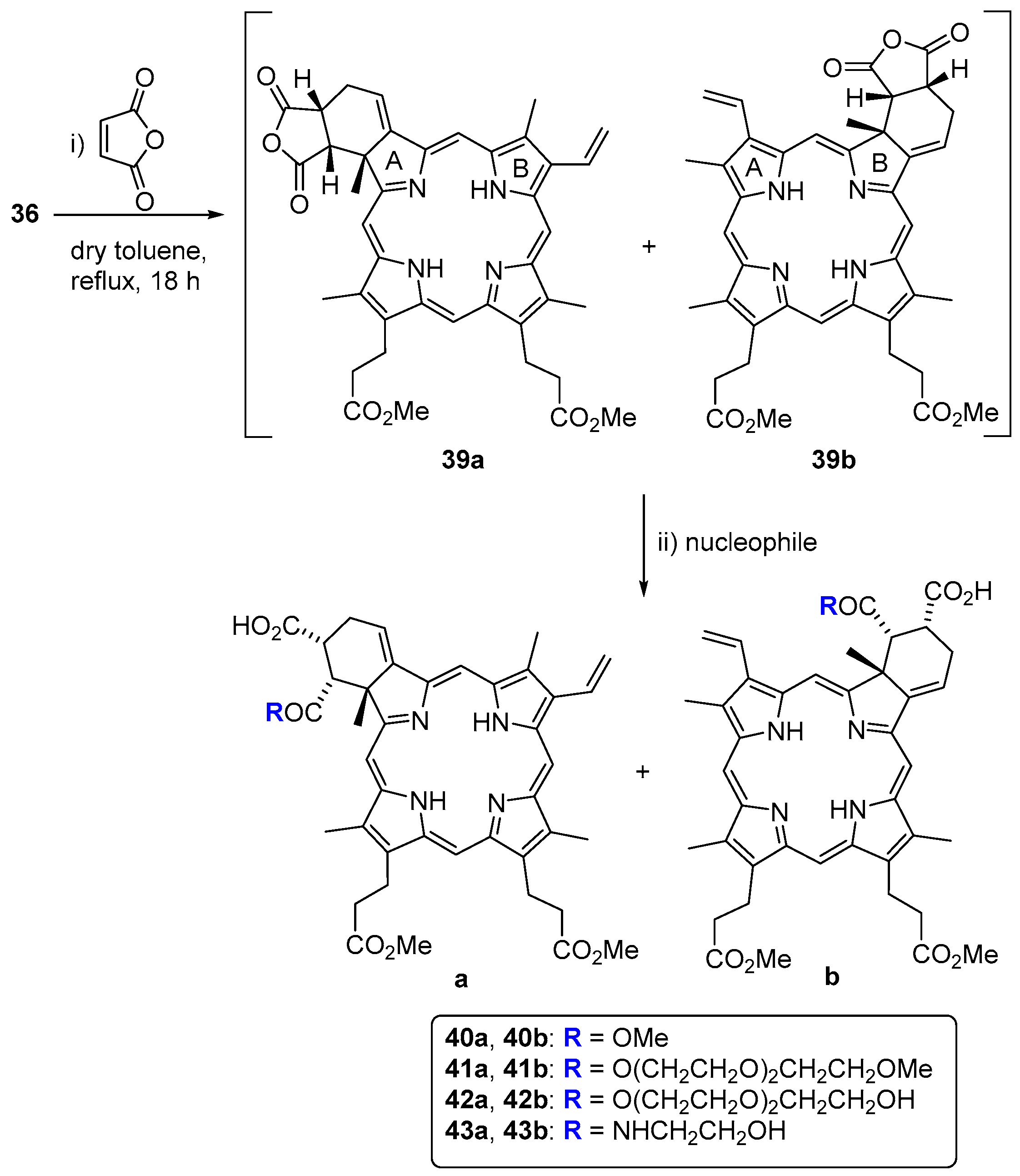{kind=link}
{kind=link}
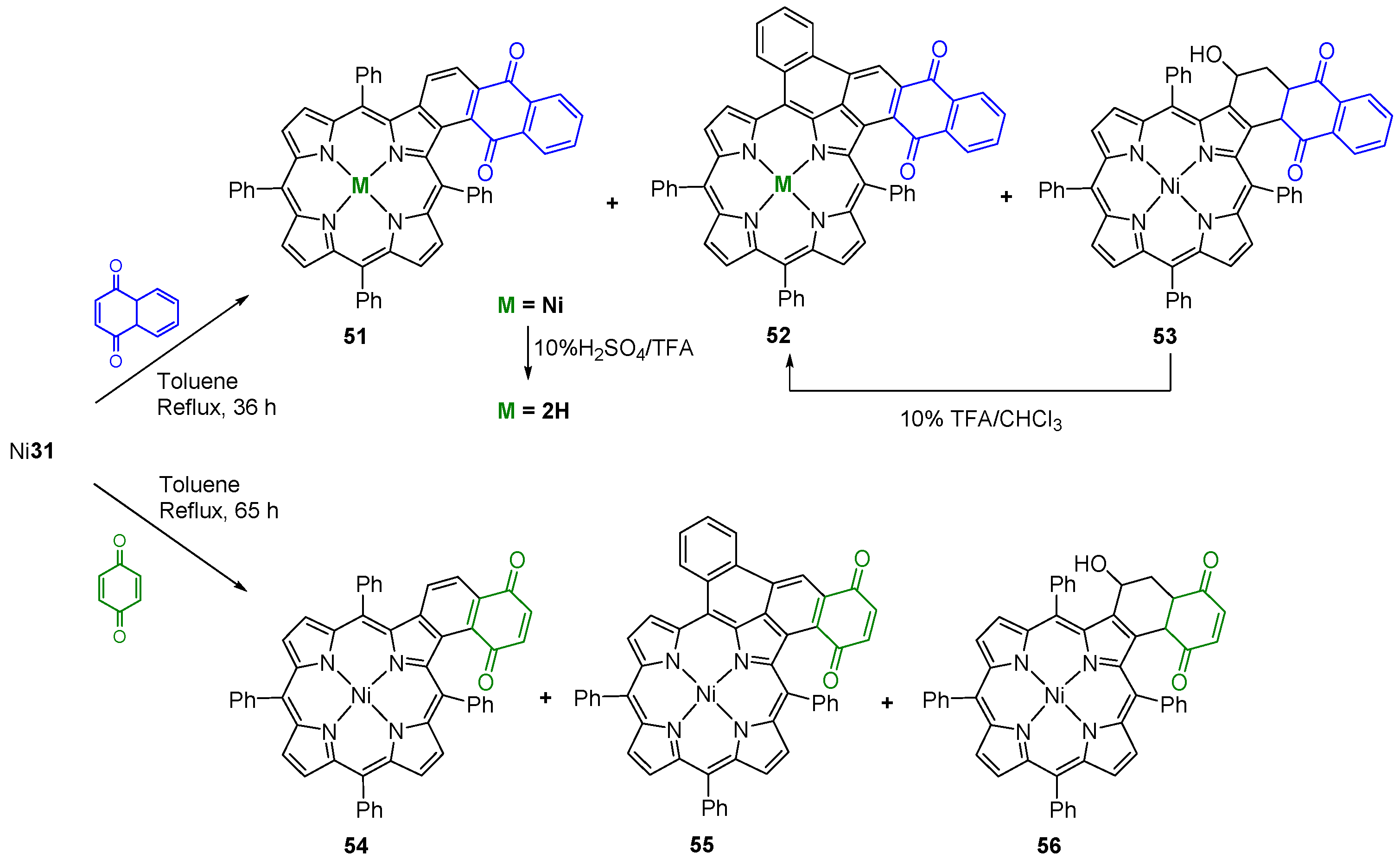{kind=link}
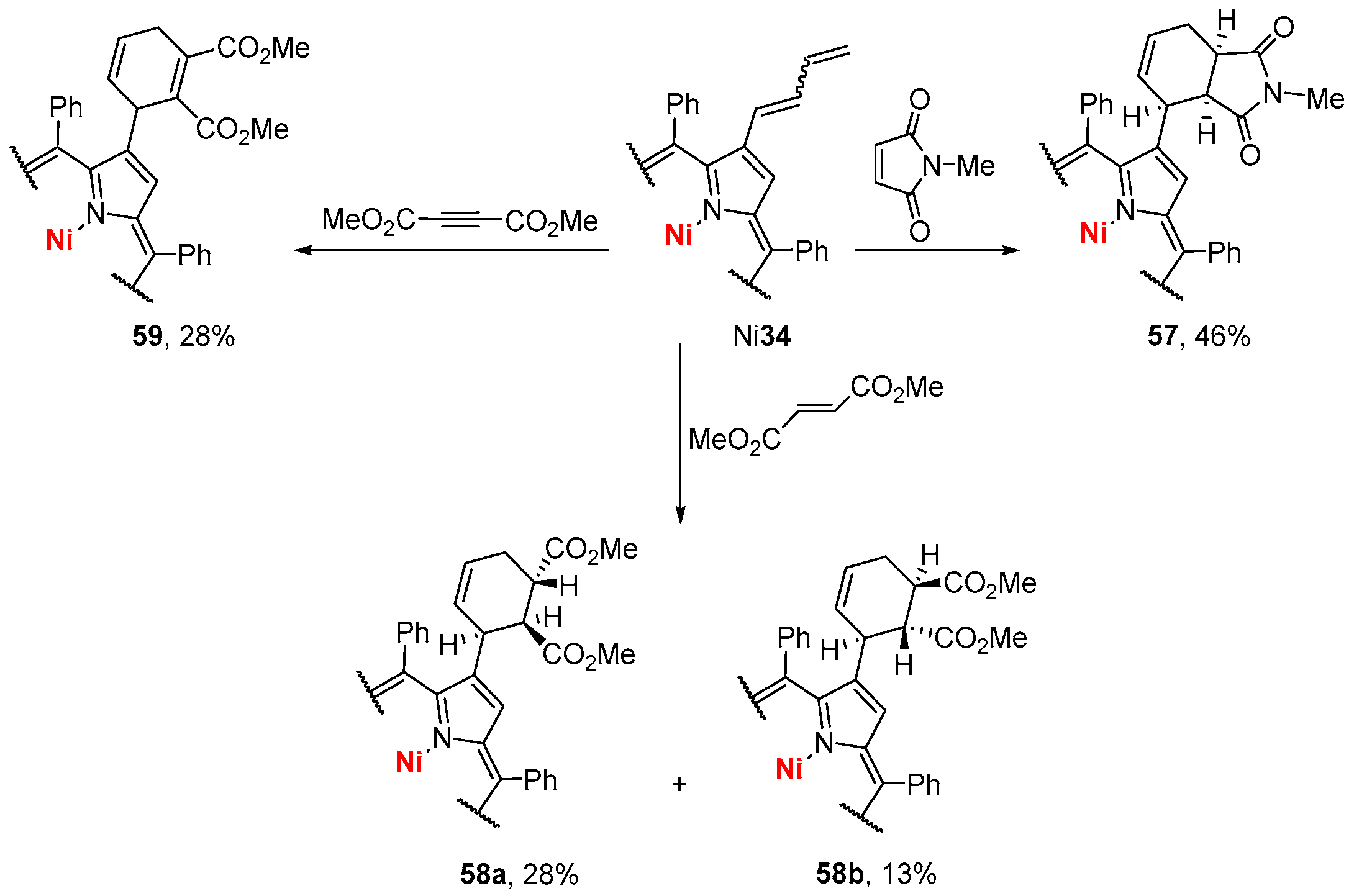{kind=link}
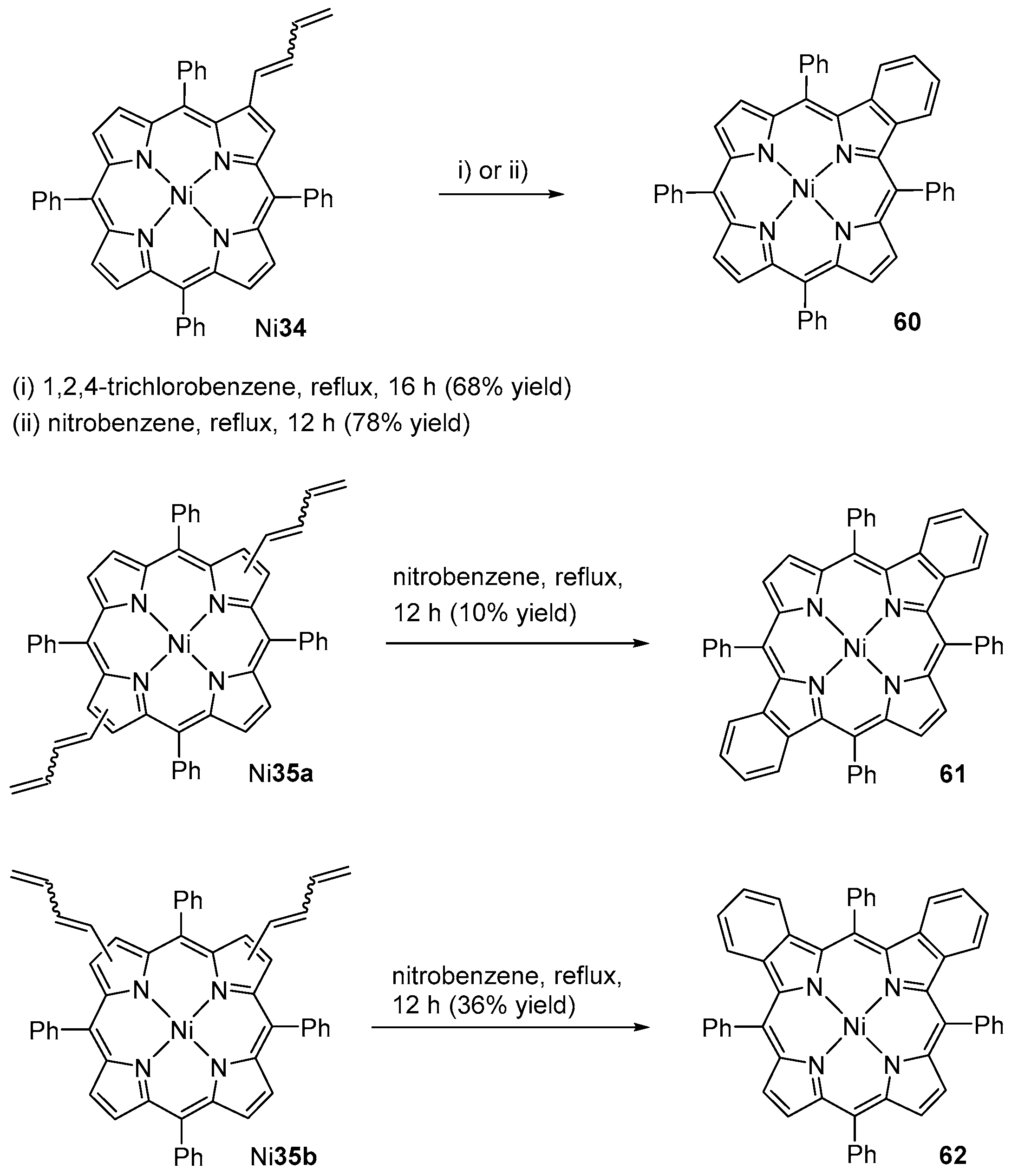{kind=link}
{kind=link}
{kind=link}
{kind=link}
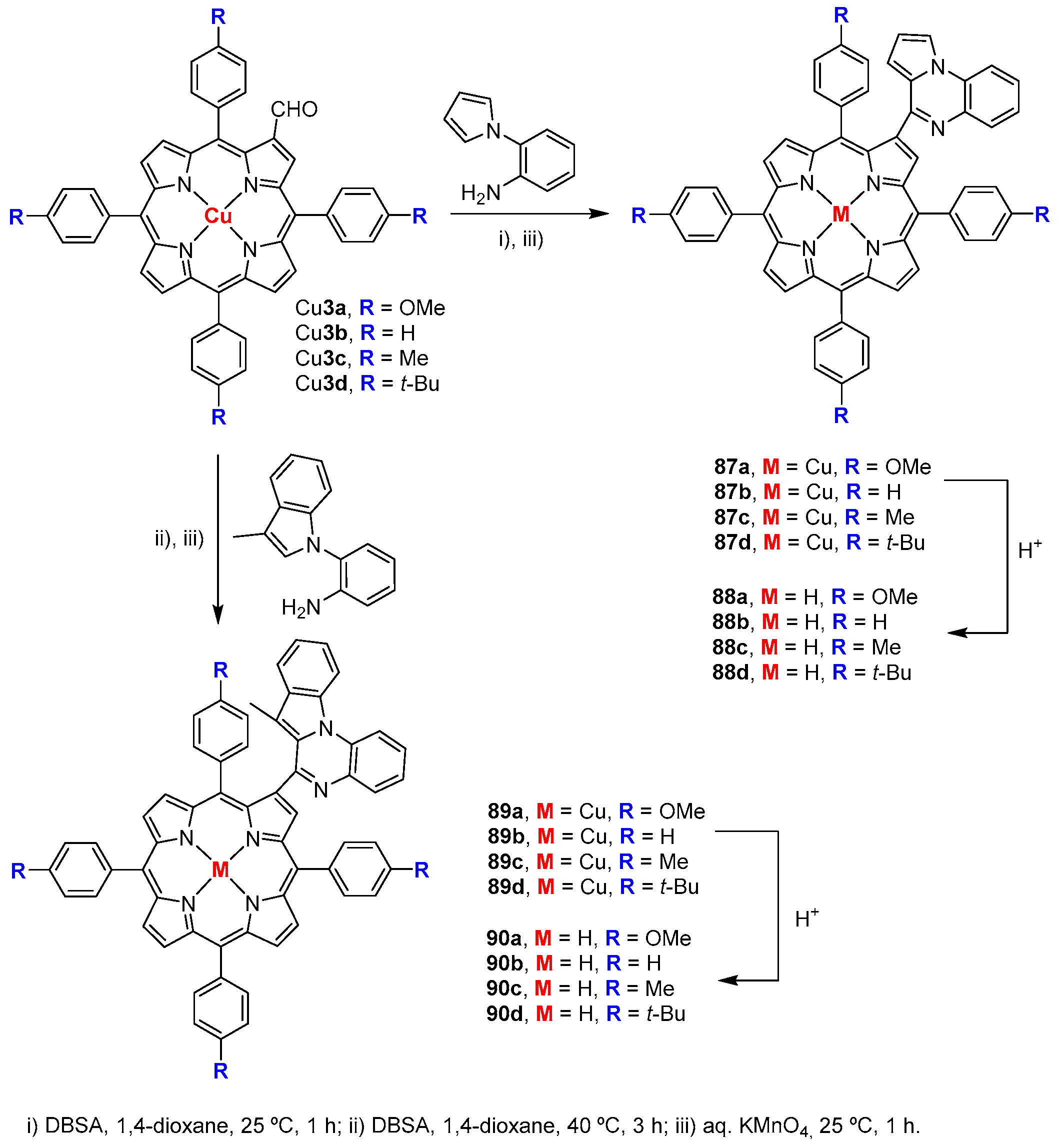{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
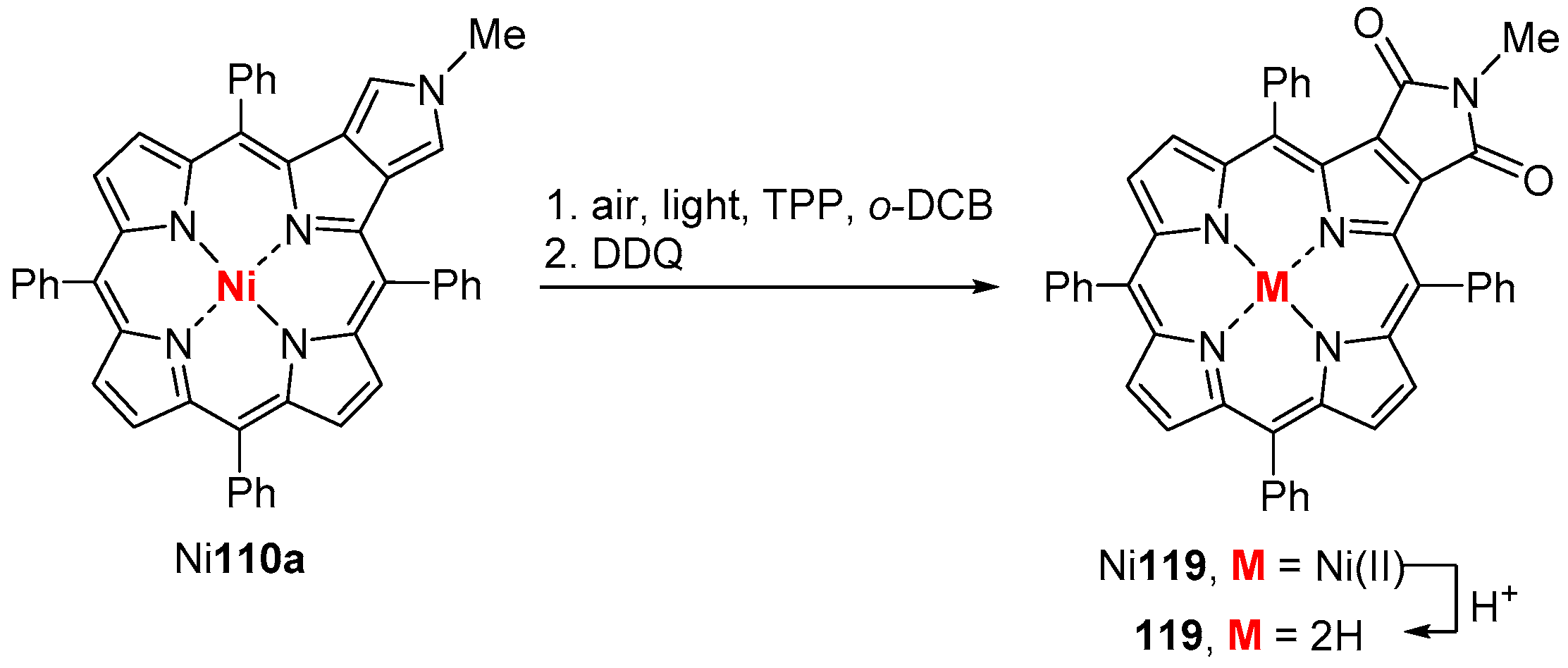{kind=link}
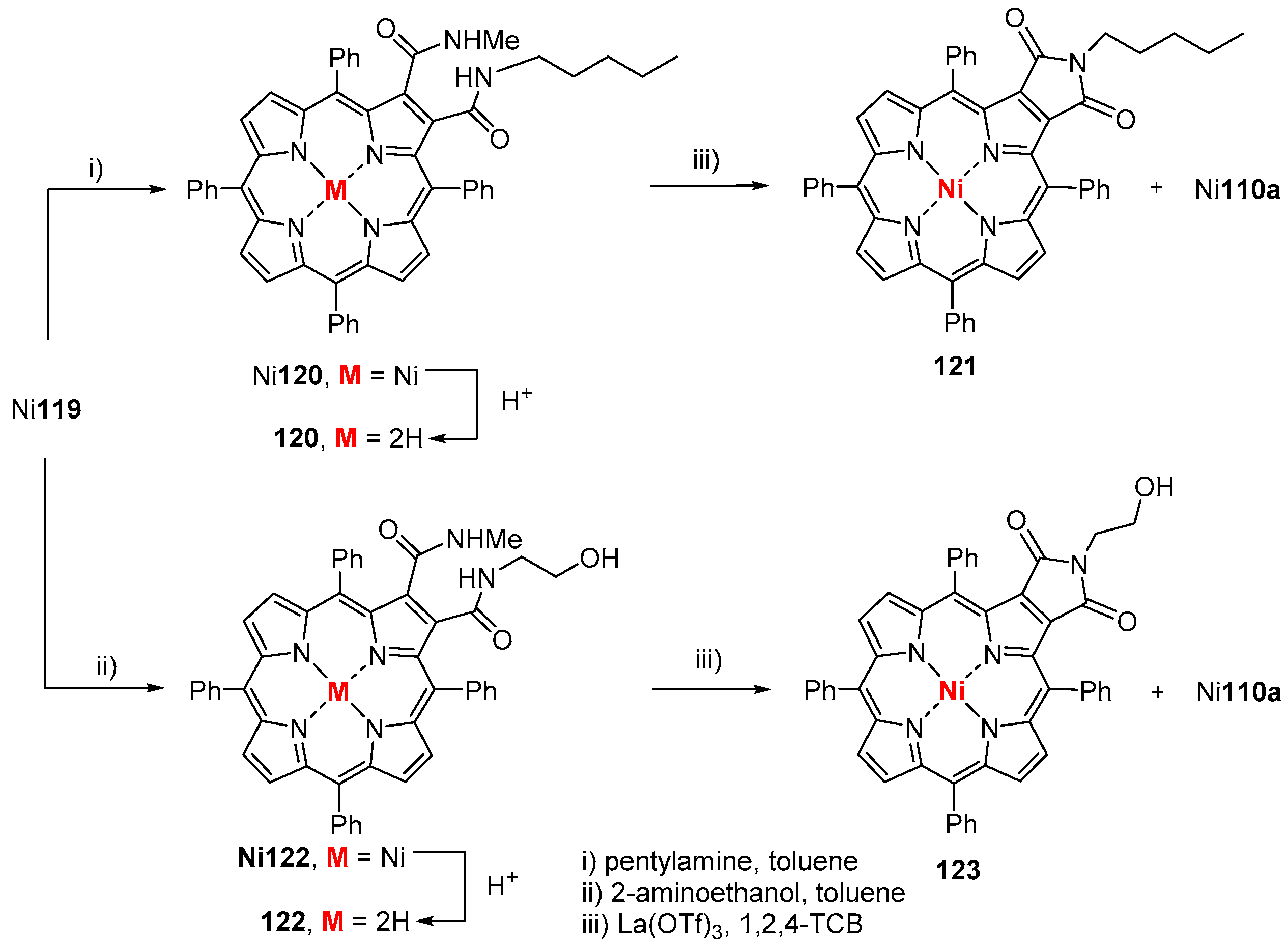{kind=link}
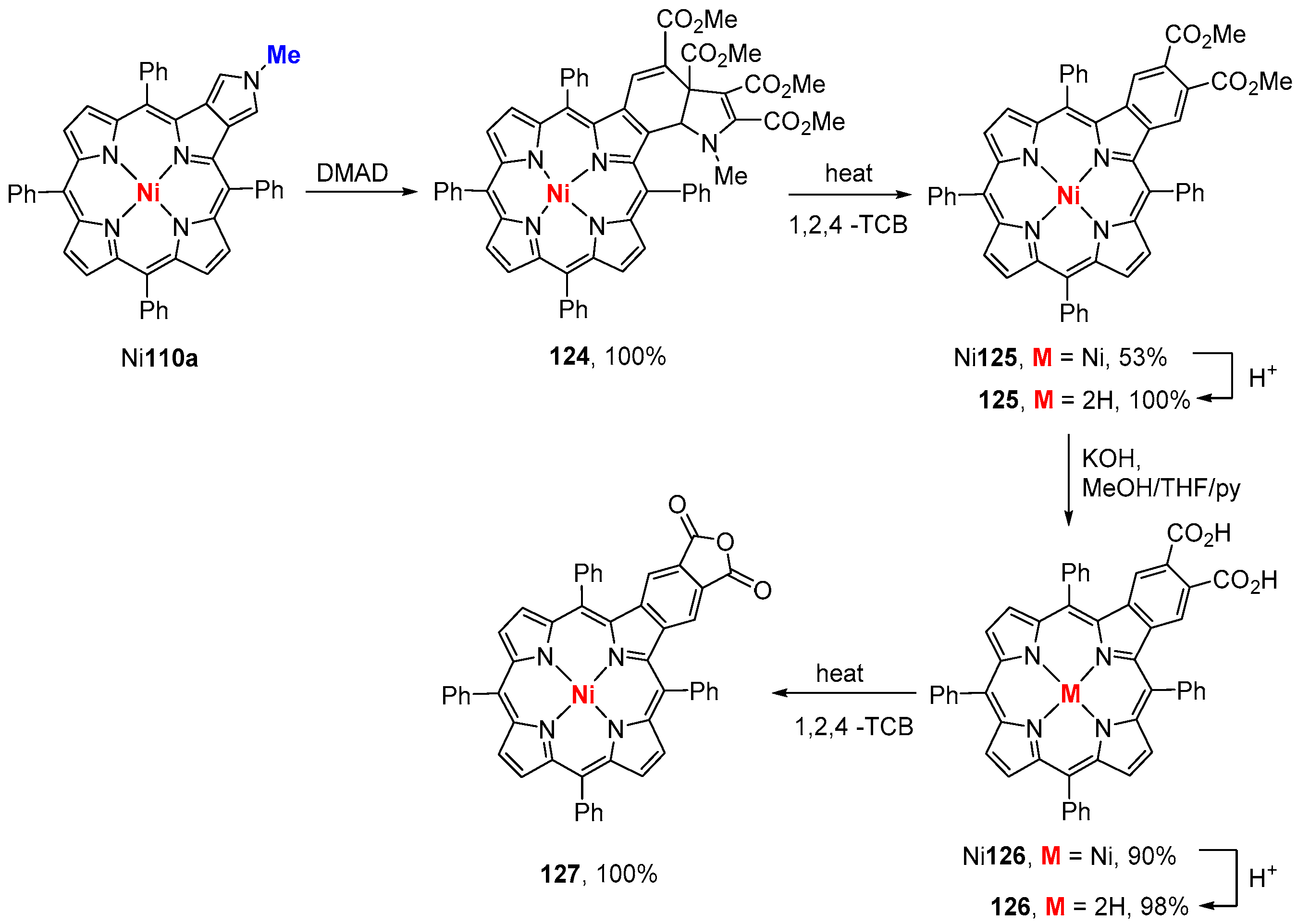{kind=link}
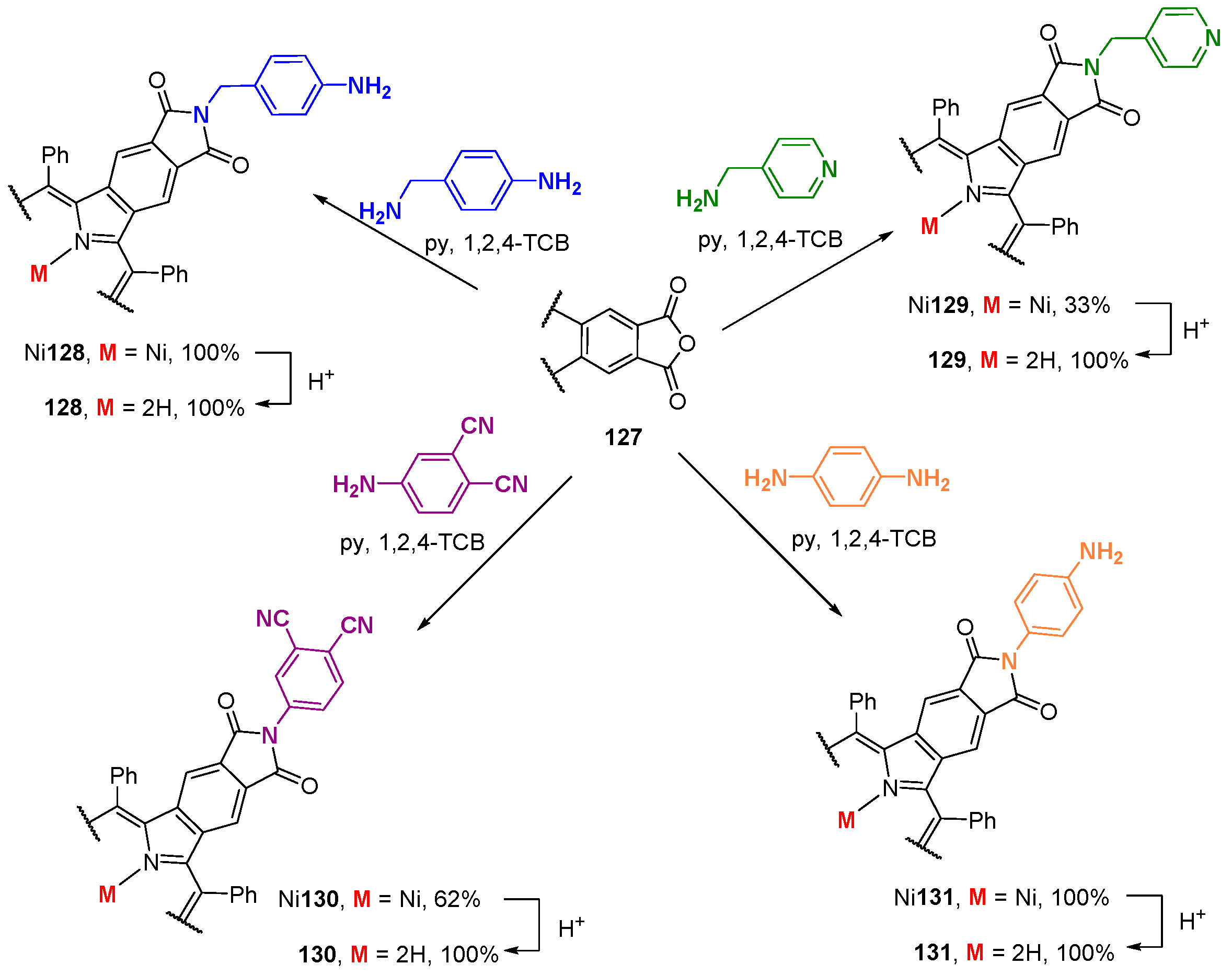{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
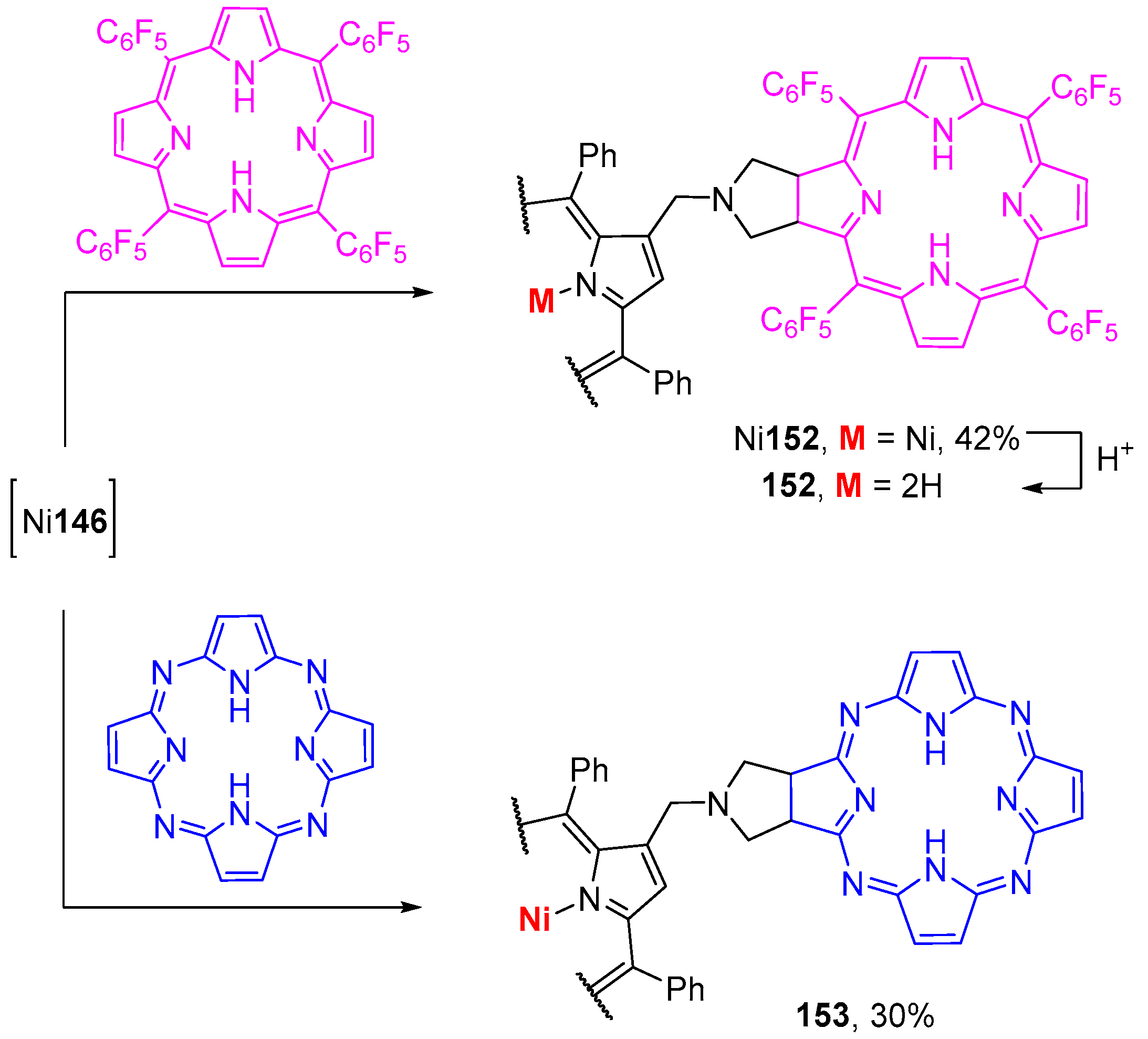{kind=link}
| Ar | M | MW Power (W)/Time (min) | Yield of Ni3 or Cu3 Under classical Heating Conditions (%) | Yield of Ni3 or Cu3 Under MW Heating Condition (%) |
|---|---|---|---|---|
 | Ni | 500/30 | 86 | 74 |
 | 57 | 50 | ||
 | 41 | 36 | ||
 | 51 | a | ||
 | 43 | a | ||
 | Cu | 160/15 | 95 | 90 |
 | 68 | a | ||
 | 88 | 41 | ||
 | 83 | 19 | ||
 | 87 | 26 |
| Entry | Ketone 21 | Time (h) | 22 (% Yield) | 23 (% Yield) | 24 (% Yield) |
|---|---|---|---|---|---|
| 1 a | a | 2 | 68 | 14 | - |
| 2 | b | 4 | 61 | 22 | - |
| 3 | c | 5.5 | 10 | 6 | 46 |
| 4 b | c | 2.5 | 55 | 26 | 6 |
| 5 | d | 8 | 19 | 8 | 71 |
| 6 b | d | 8 | 58 | 15 | 9 |
| 7 | e | 3 | 23 | 19 | 40 |
| 8 b | e | 3 | 45 | 29 | 10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerqueira, A.F.R.; Moura, N.M.M.; Serra, V.V.; Faustino, M.A.F.; Tomé, A.C.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S. β-Formyl- and β-Vinylporphyrins: Magic Building Blocks for Novel Porphyrin Derivatives. Molecules 2017, 22, 1269. https://doi.org/10.3390/molecules22081269
Cerqueira AFR, Moura NMM, Serra VV, Faustino MAF, Tomé AC, Cavaleiro JAS, Neves MGPMS. β-Formyl- and β-Vinylporphyrins: Magic Building Blocks for Novel Porphyrin Derivatives. Molecules. 2017; 22(8):1269. https://doi.org/10.3390/molecules22081269
Chicago/Turabian StyleCerqueira, Ana F. R., Nuno M. M. Moura, Vanda Vaz Serra, M. Amparo F. Faustino, Augusto C. Tomé, José A. S. Cavaleiro, and M. Graça P. M. S. Neves. 2017. "β-Formyl- and β-Vinylporphyrins: Magic Building Blocks for Novel Porphyrin Derivatives" Molecules 22, no. 8: 1269. https://doi.org/10.3390/molecules22081269