Effects of K11R and G31P Mutations on the Structure and Biological Activities of CXCL8: Solution Structure of Human CXCL8(3-72)K11R/G31P


Abstract
:
1. Introduction
2. Results and Discussion
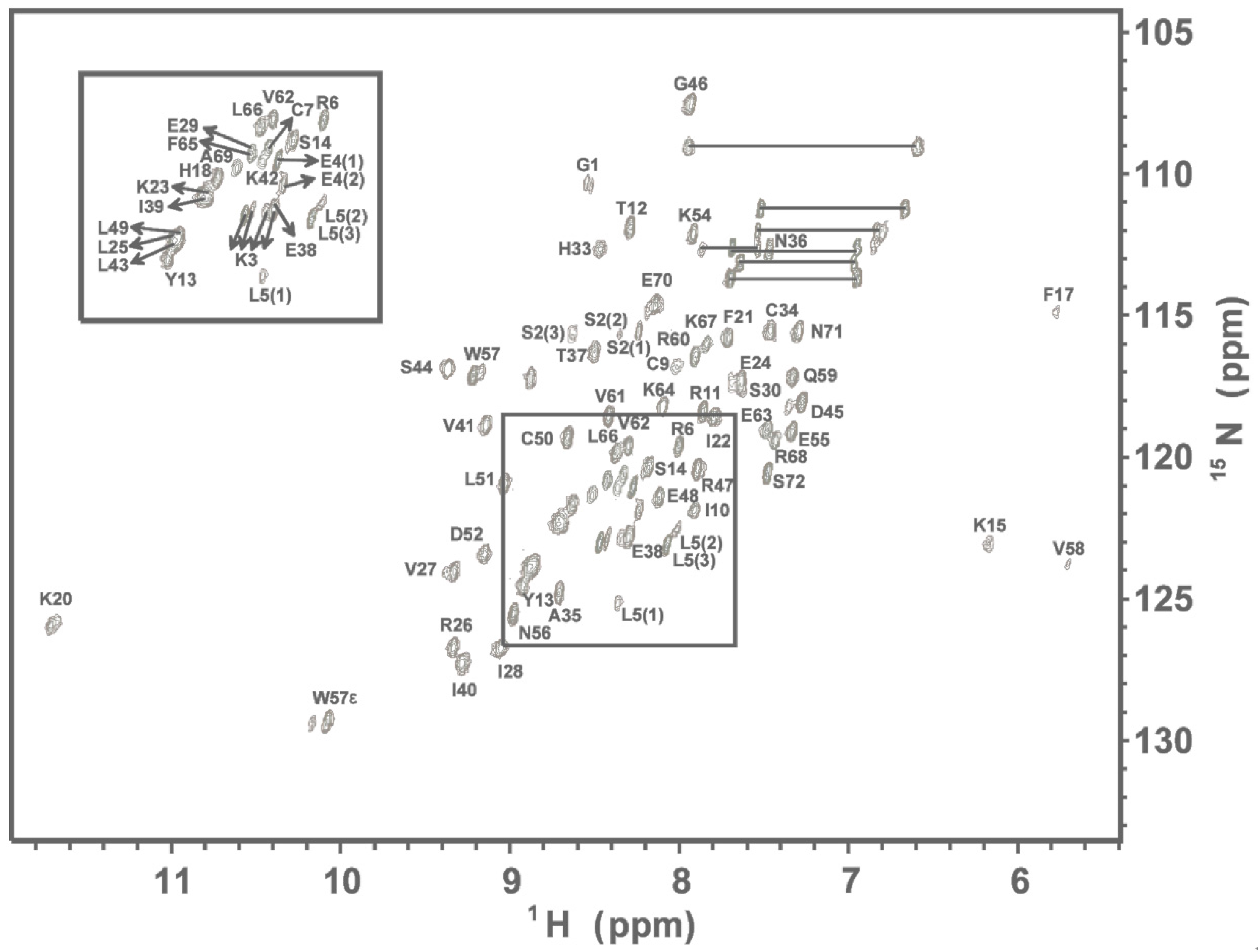
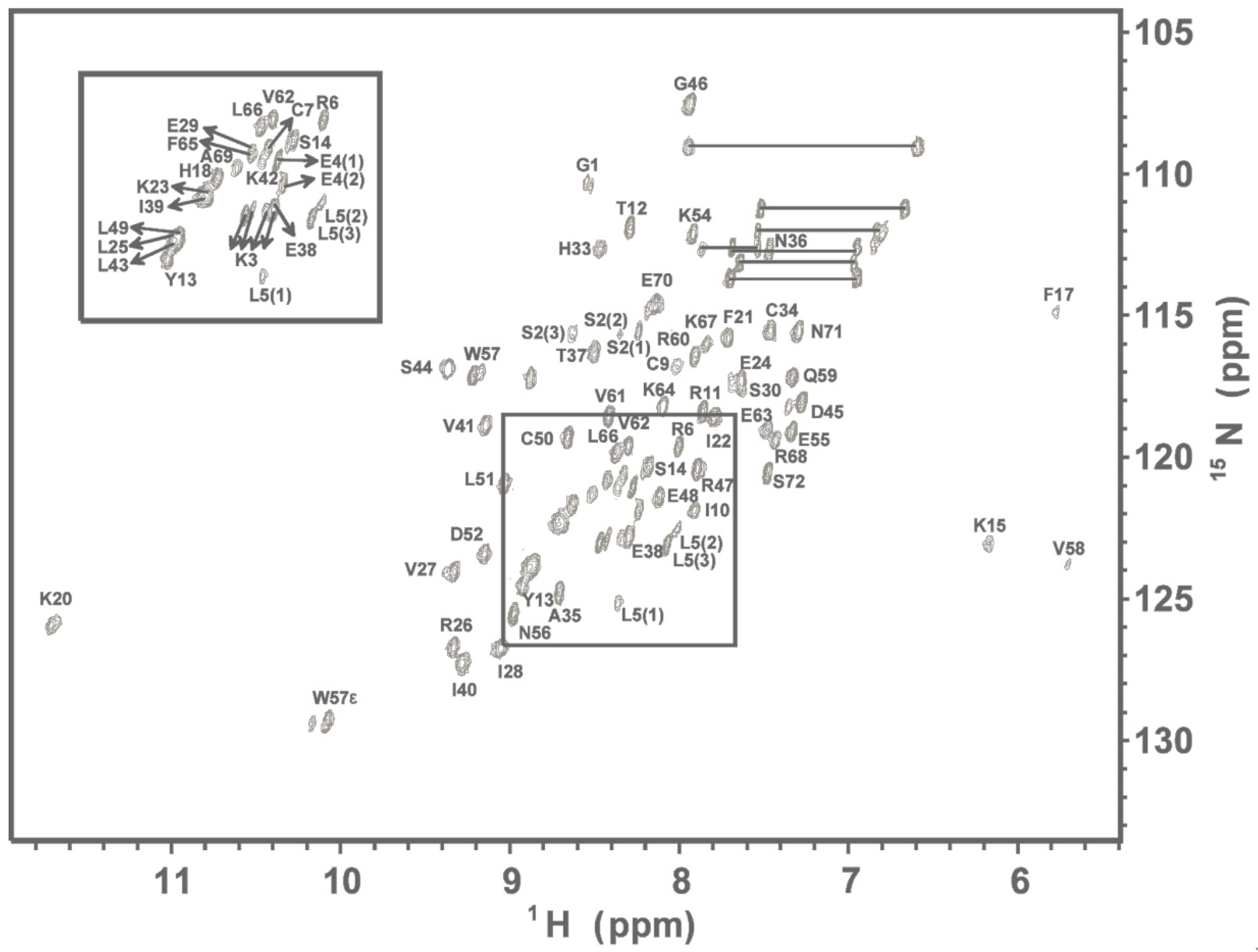
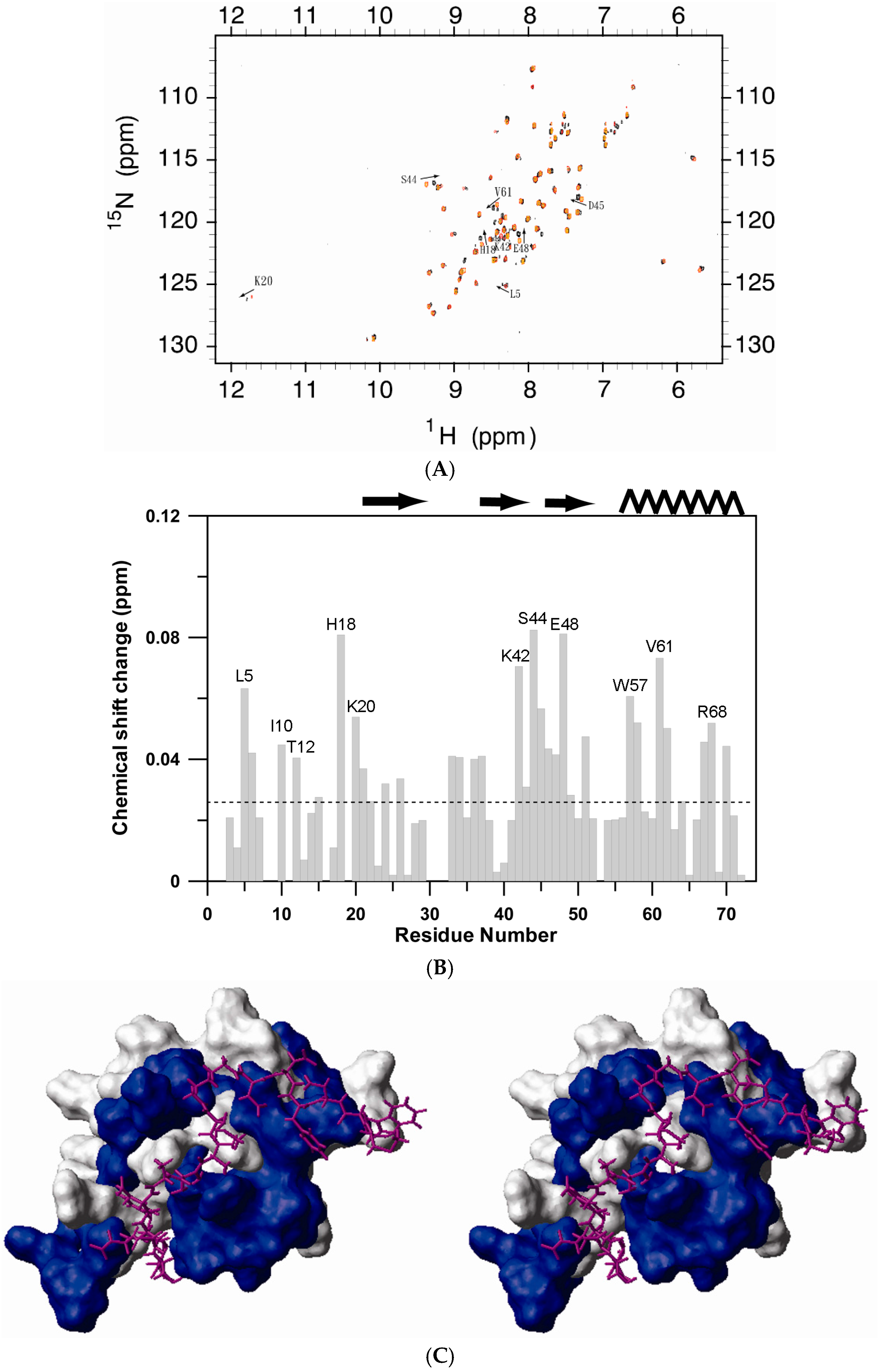
2.1. NMR Spectroscopy
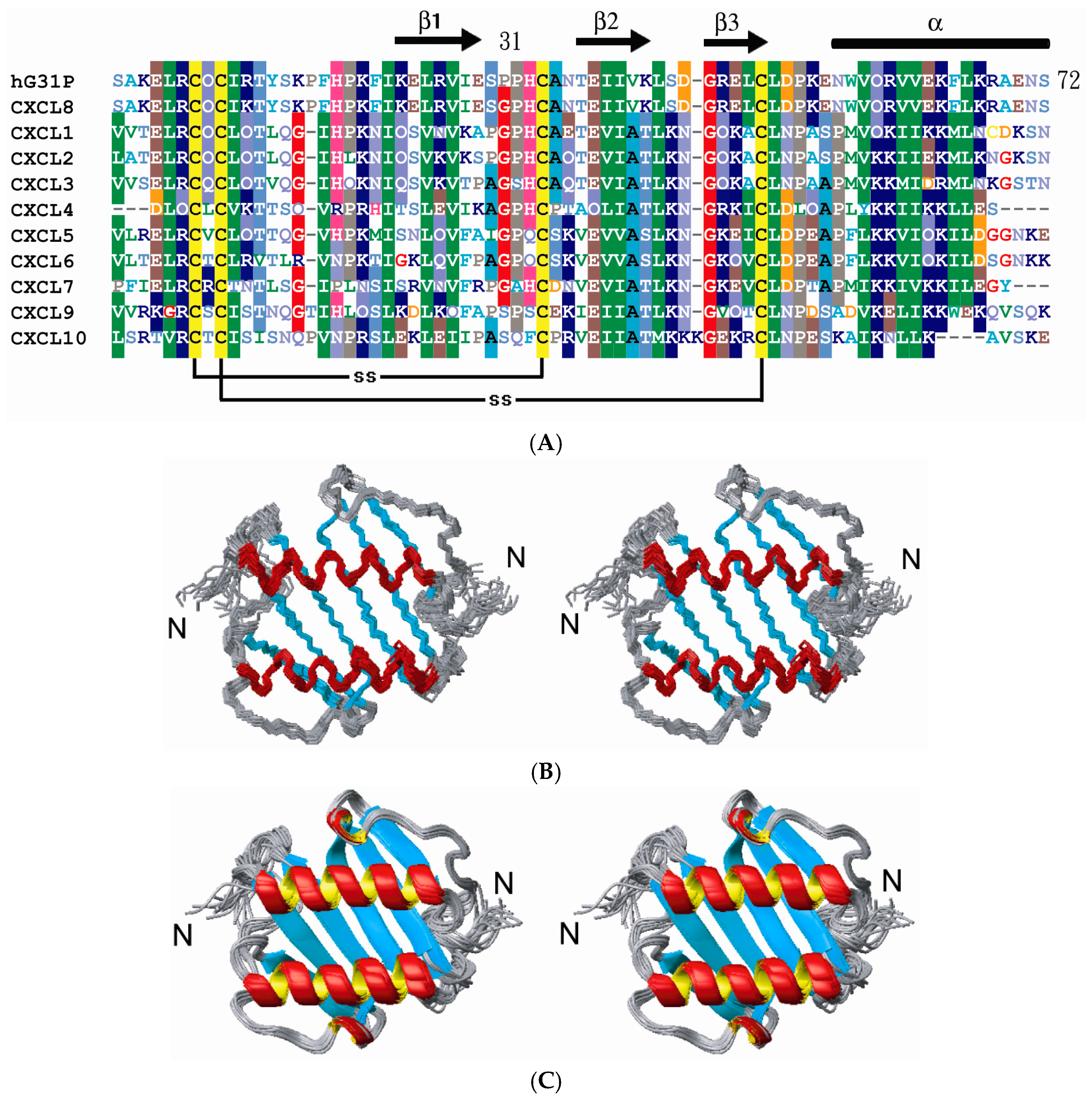
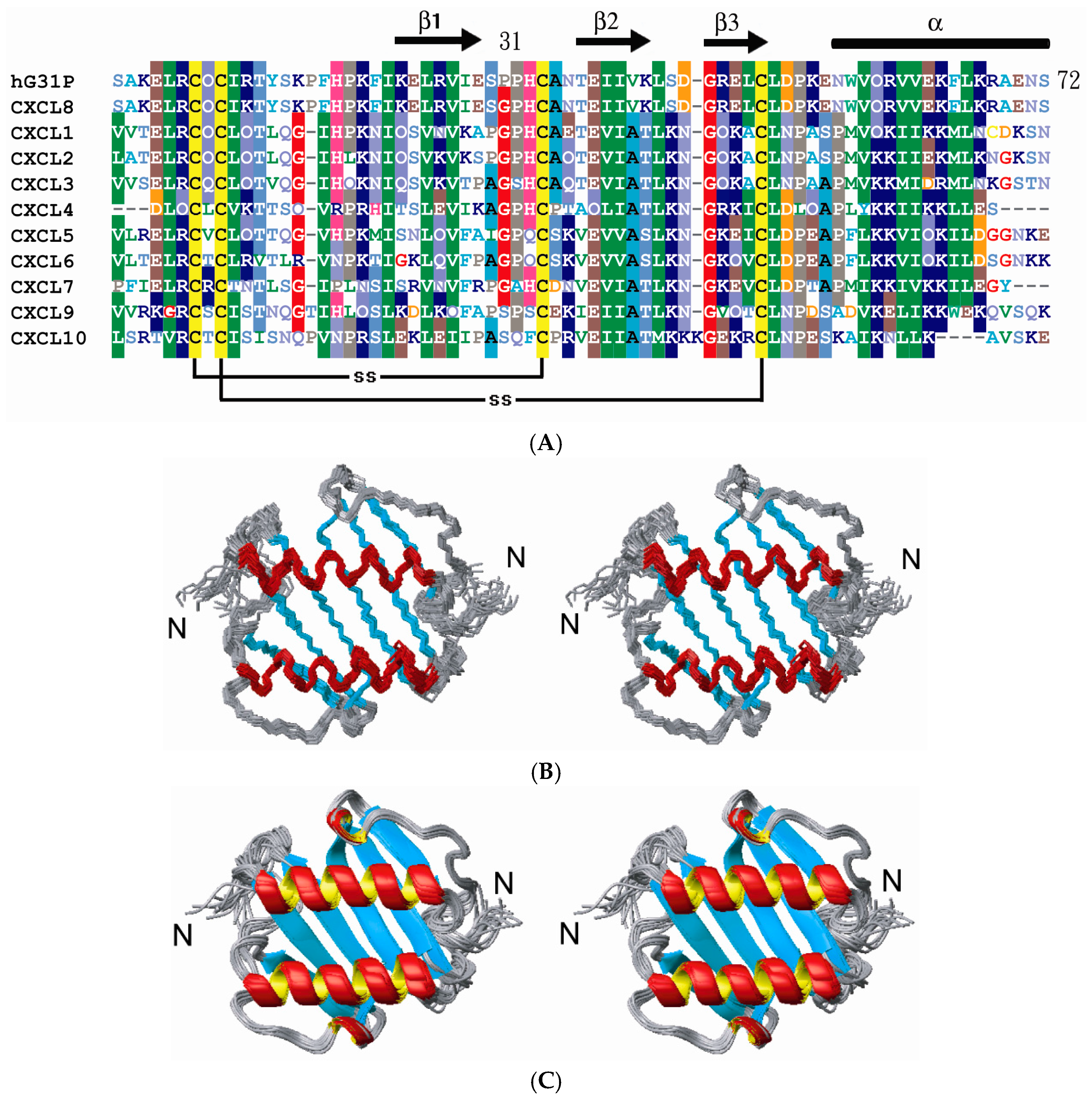
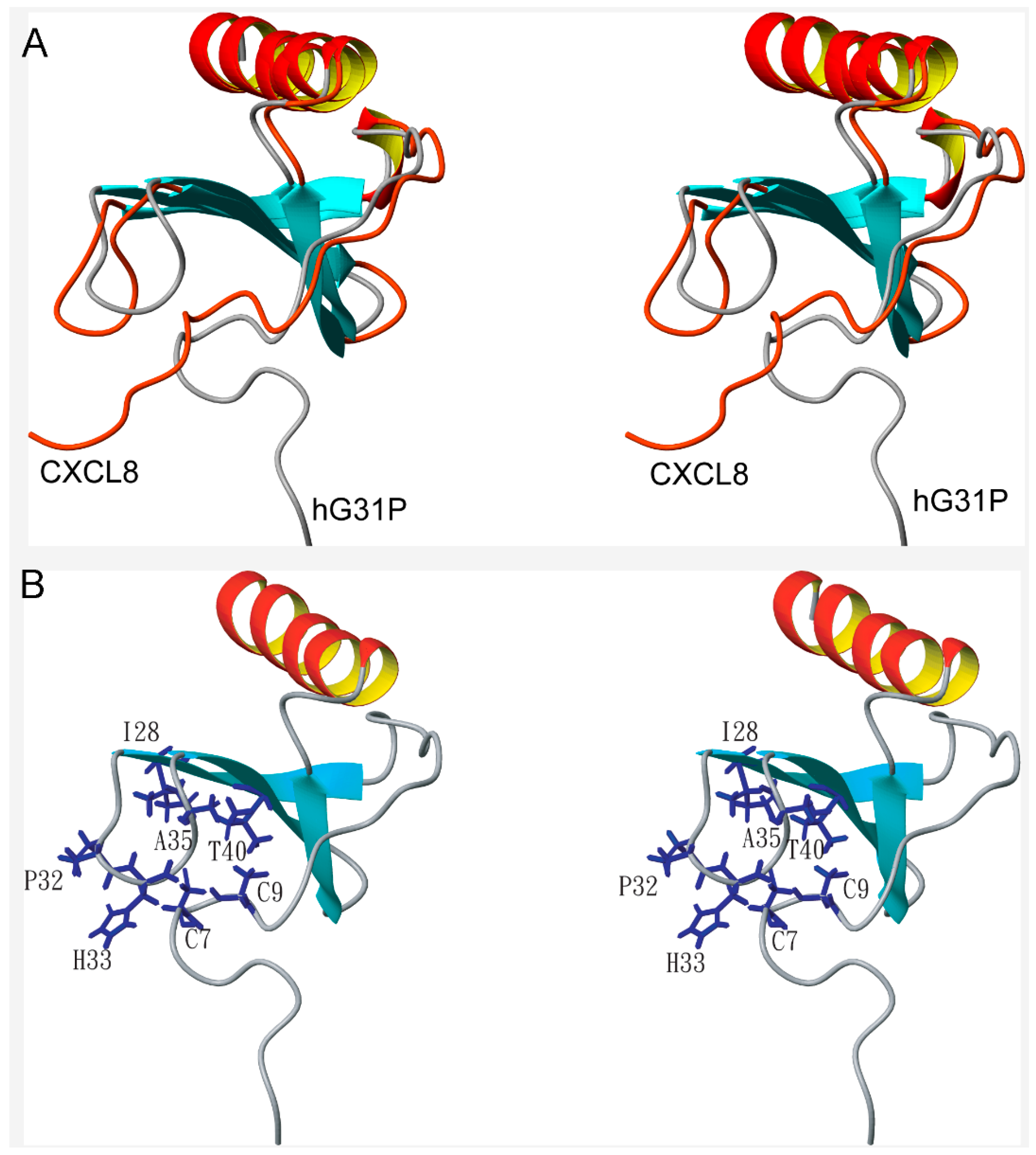
2.2. Structure Comparison with CXCL8
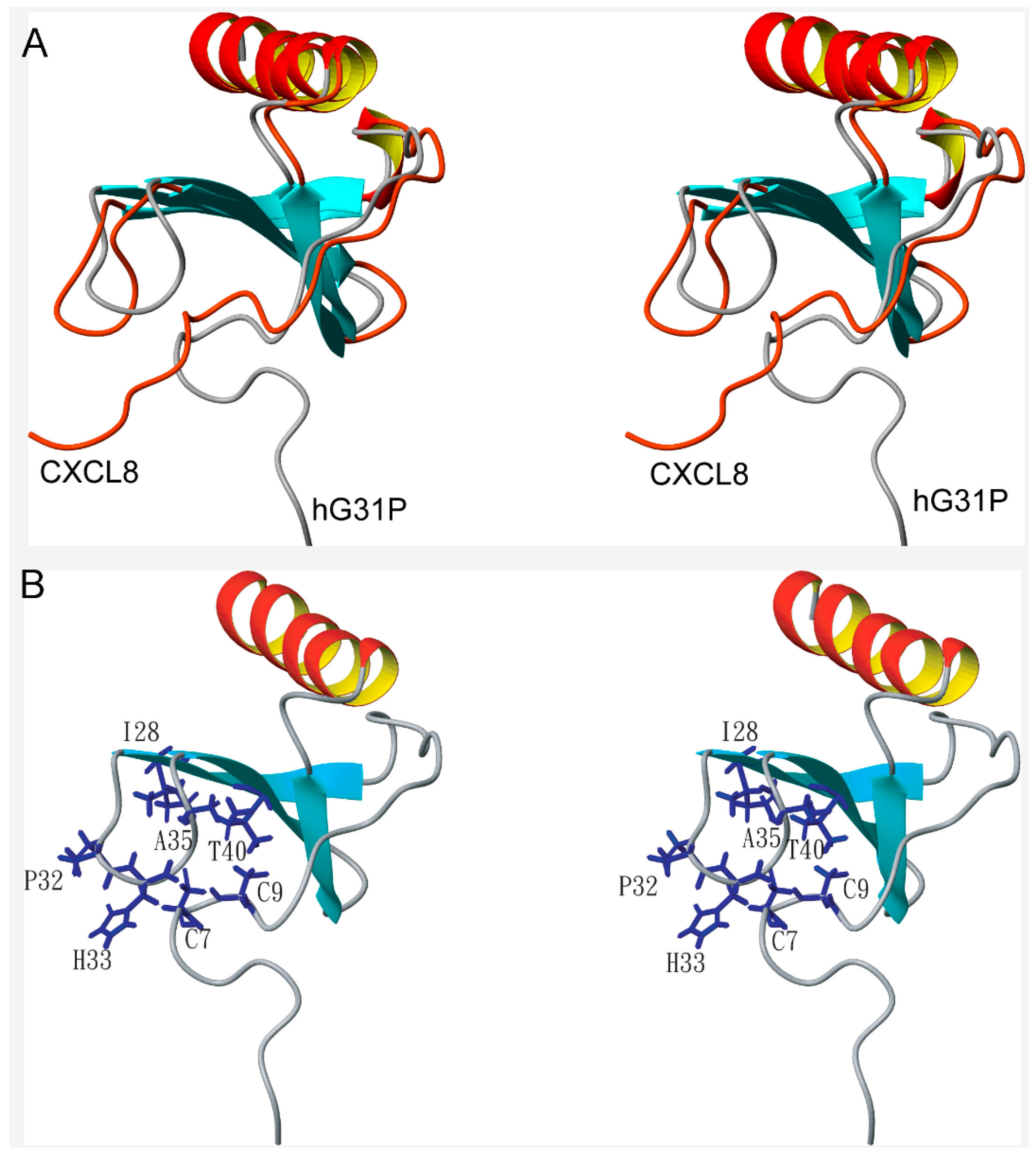
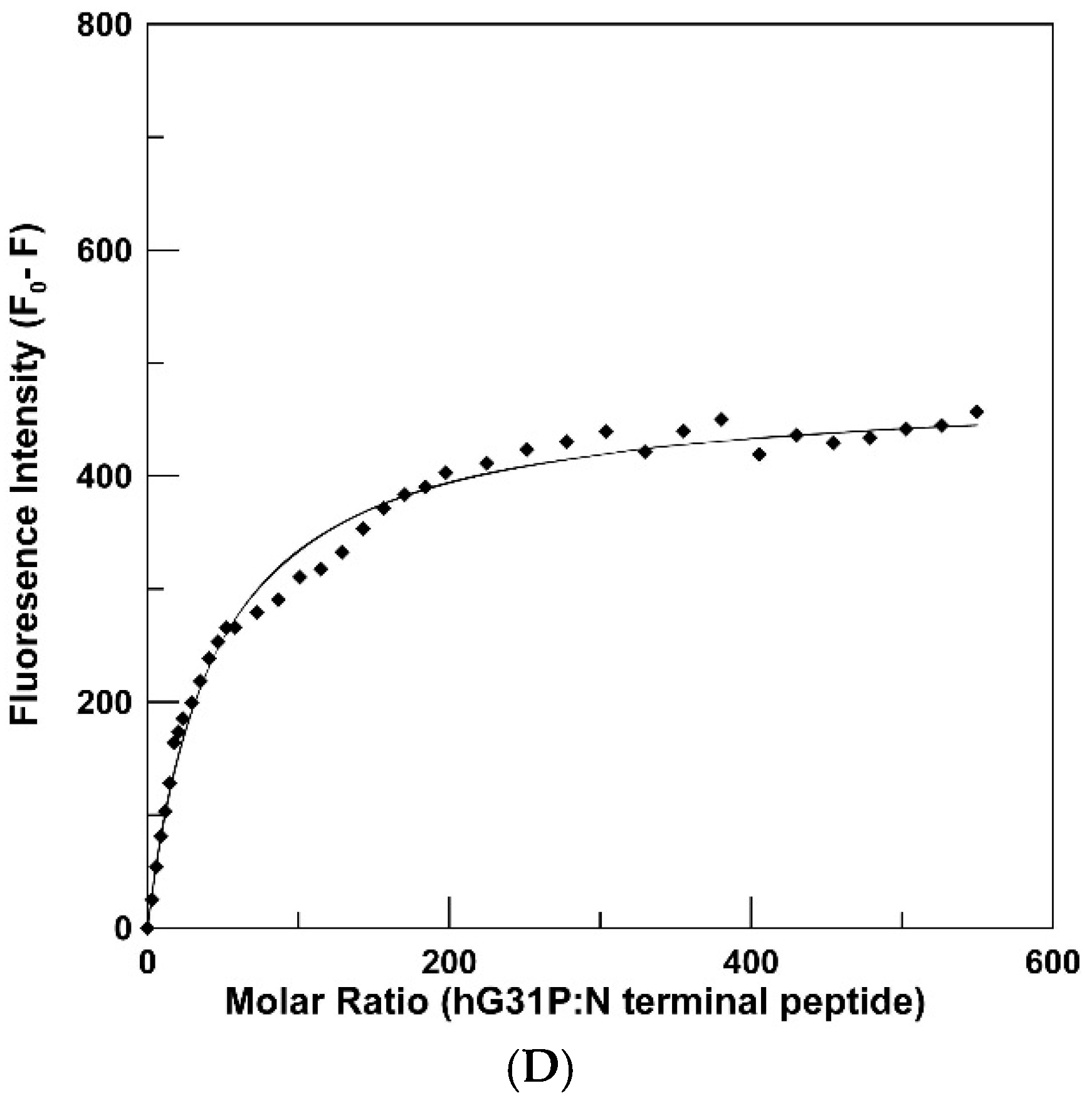
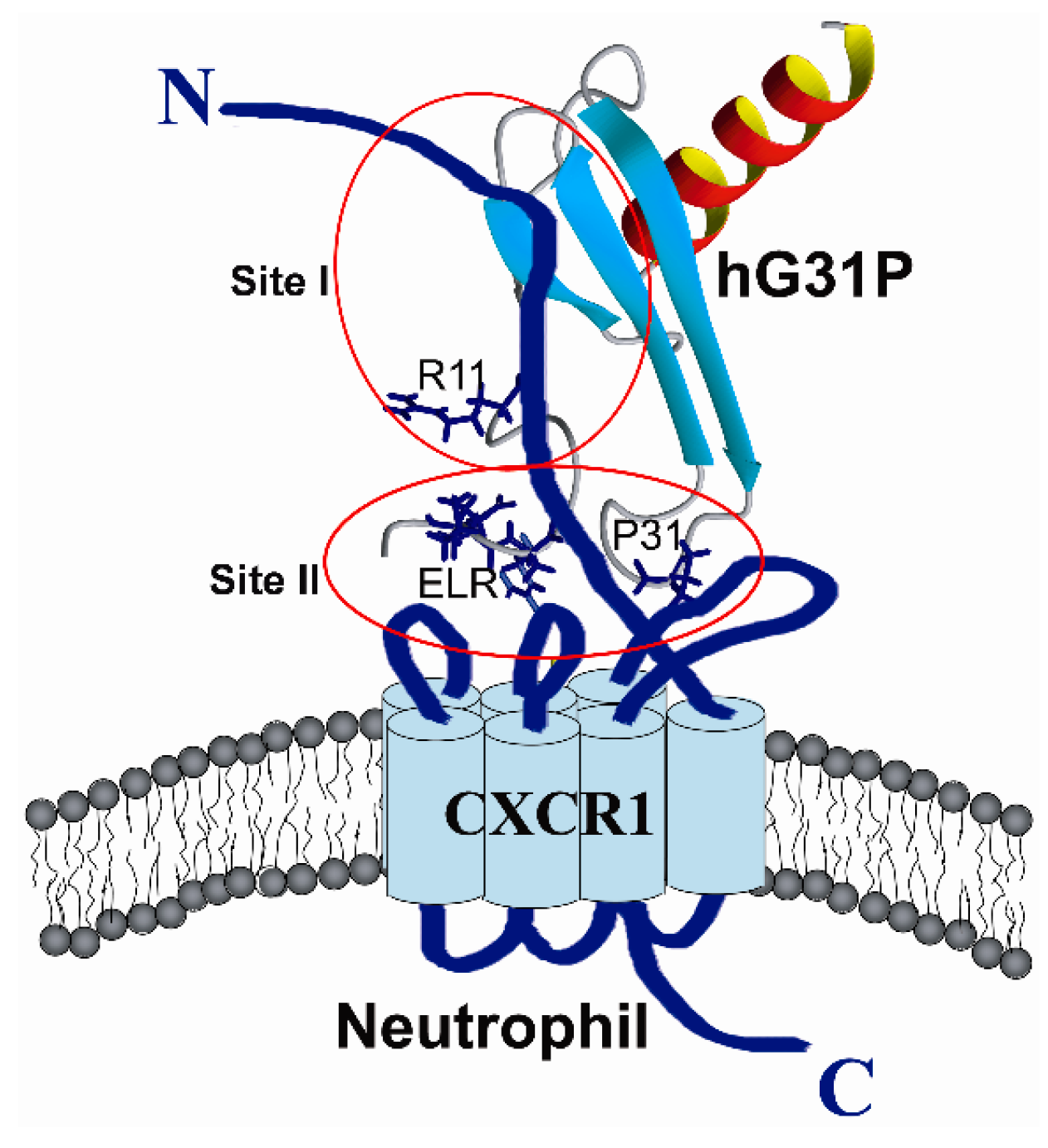
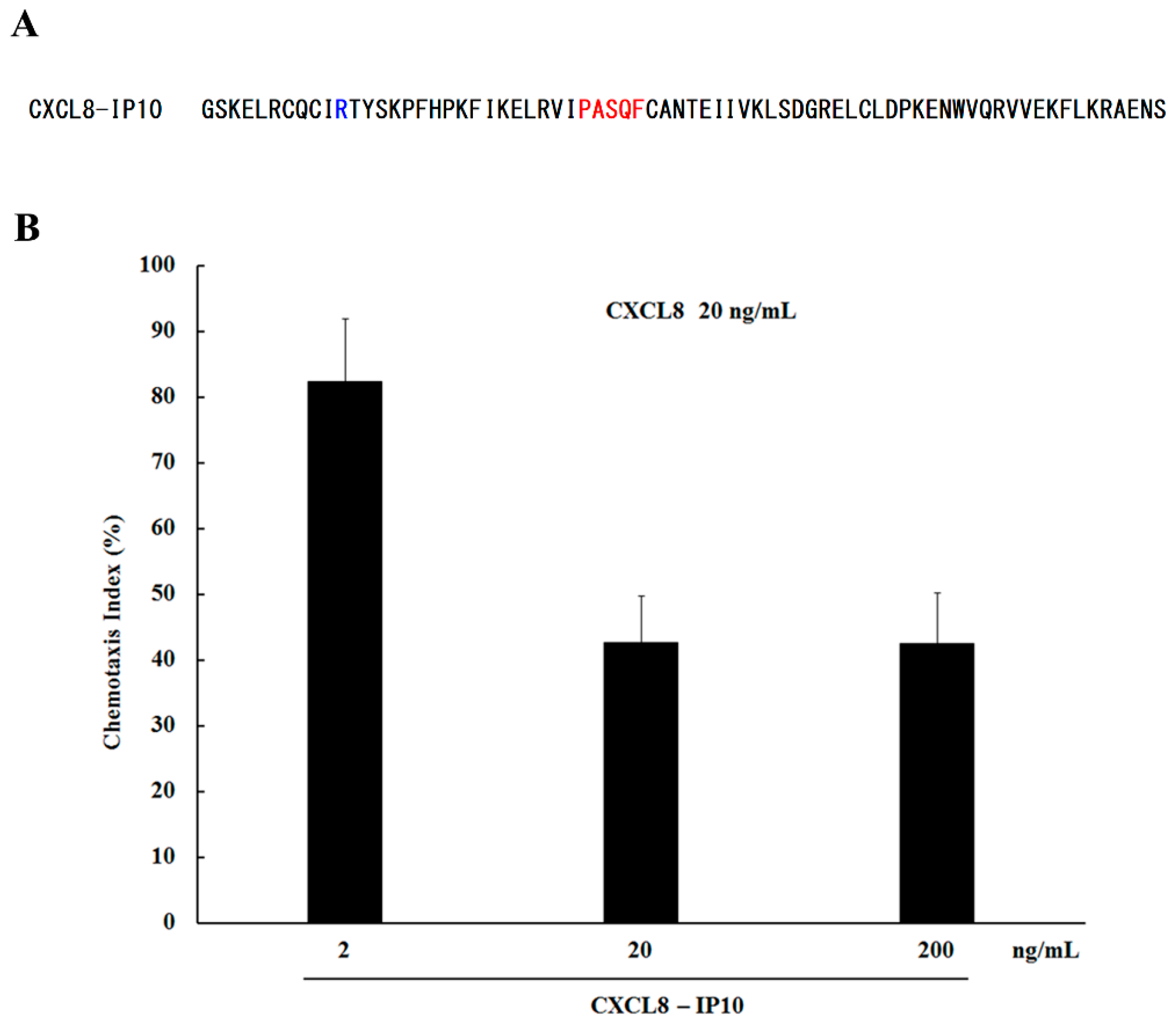
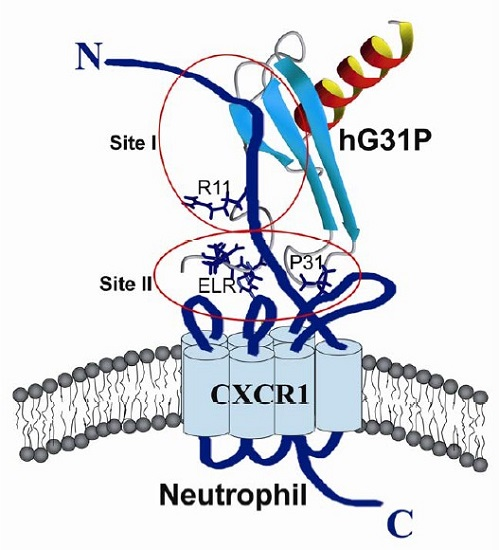
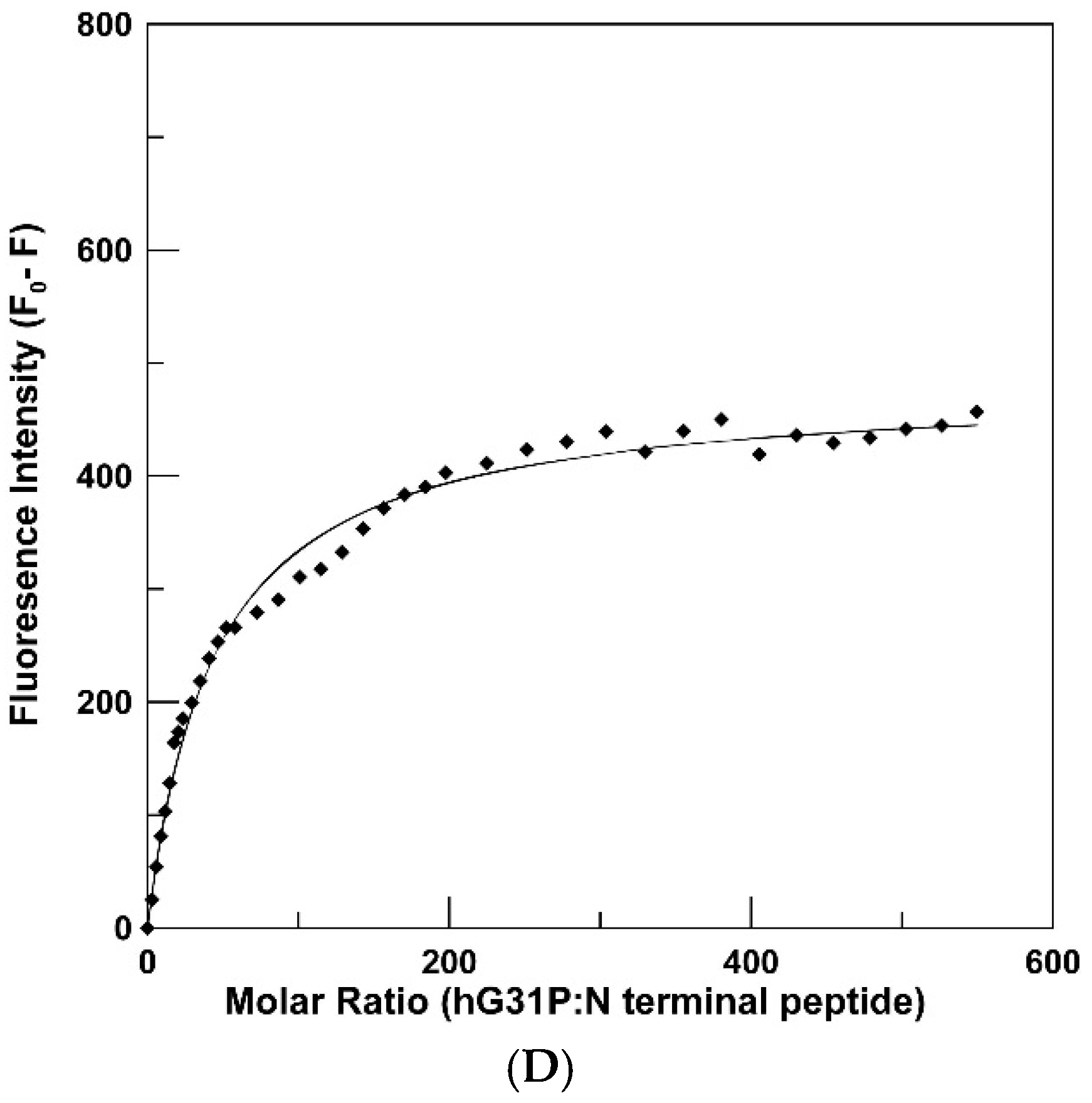
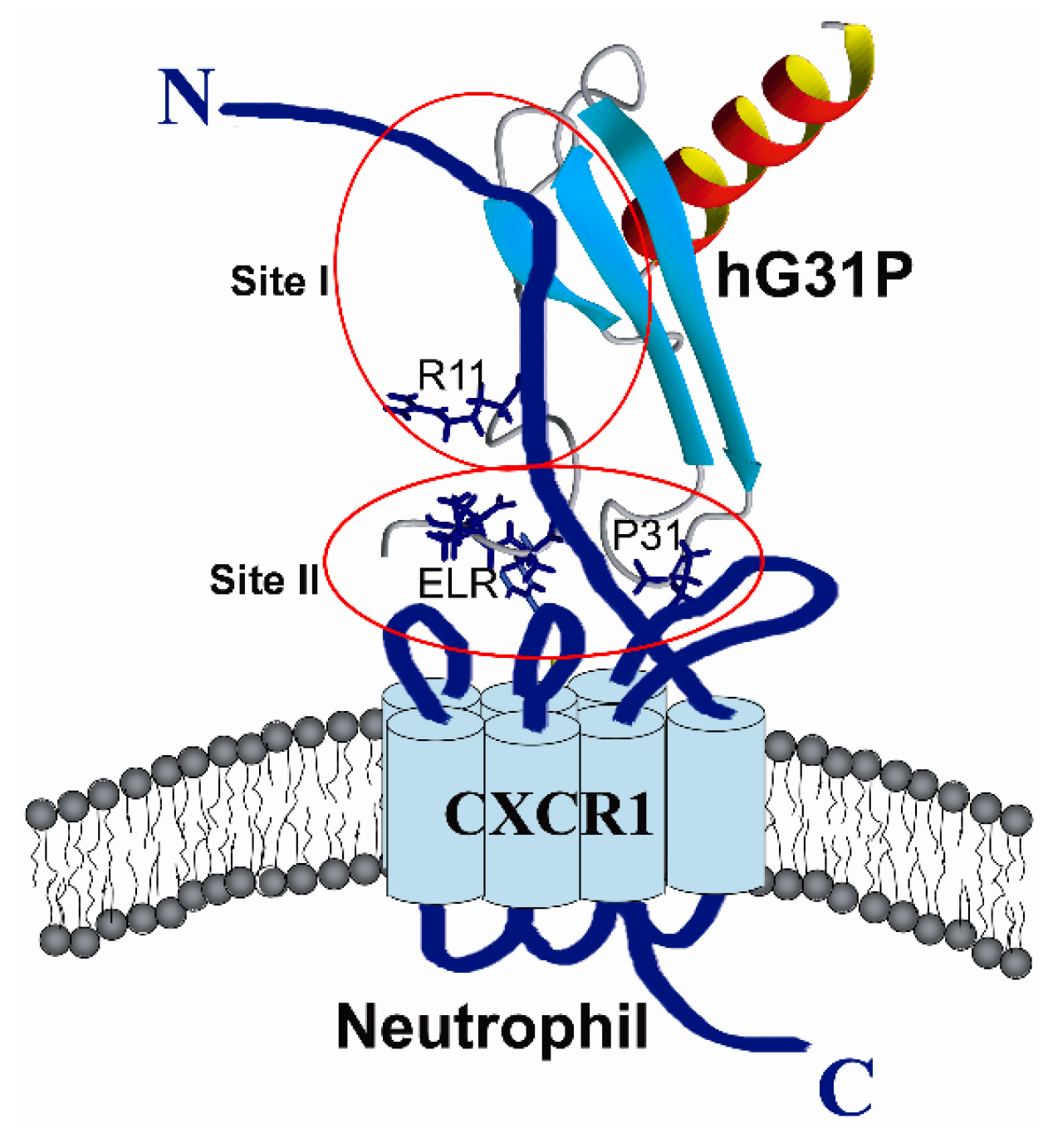
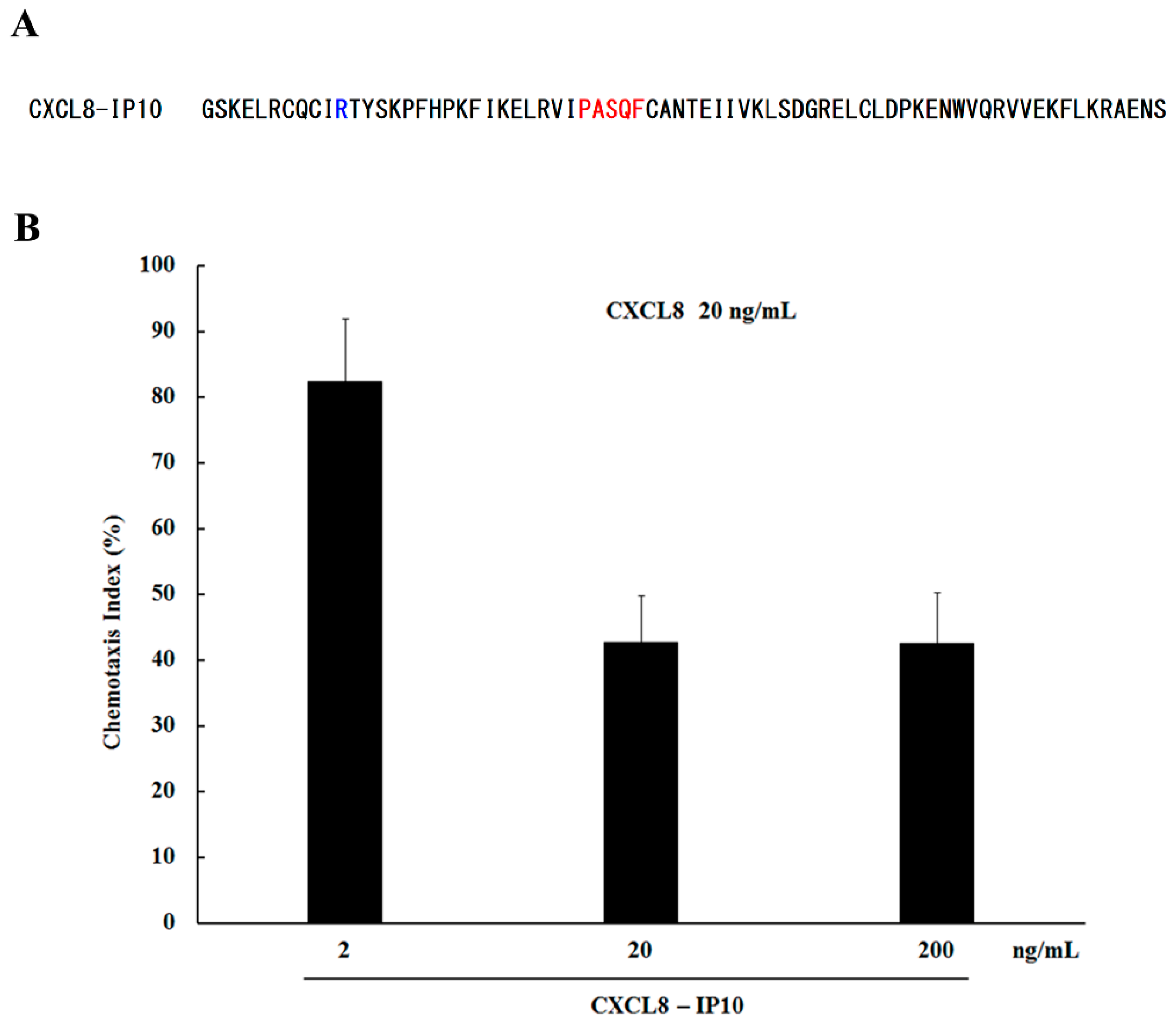
2.3. Peptide Binding
2.4. Biological Importance
3. Materials and Methods
3.1. Protein Preparation and Purification [24]
3.2. Neutrophil Chemotaxis Assay
3.3. NMR Spectroscopy
3.4. Structure Calculation
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Bizzarri, C.; Beccari, A.R.; Bertini, R.; Cavicchia, M.R.; Giorgini, S.; Allegretti, M. ELR+ CXC chemokines and their receptors CXCR1 and CXCR2 as new therapeutic targets. Pharmacol. Ther. 2006, 112, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Busch-Petersen, J. Small molecule antagonists of the CXCR2 and CXCR1 chemokine receptors as therapeutic agents for the treatment of inflammatory diseases. Curr. Top. Med. Chem. 2006, 6, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.M.; Maxwell, P.J.; Waugh, D.J.J. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals 2013, 6, 929–959. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.T.; Weber, I.T.; St Charles, R.; Xuan, J.C.; Appella, E.; Yamada, M.; Matsushima, K.; Edwards, B.F.; Clore, G.M.; Gronenborn, A.M. Crystal structure of interleukin 8: Symbiosis of NMR and crystallography. Proc. Natl. Acad. Sci. USA 1991, 88, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Schumacher, C.; Baggiolini, M.; Moser, B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J. Biol. Chem. 1991, 266, 23128–23134. [Google Scholar] [PubMed]
- Hebert, C.A.; Vitangcol, R.V.; Baker, J.B. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J. Biol. Chem. 1991, 266, 18989–18994. [Google Scholar] [PubMed]
- Hammond, M.E.; Shyamala, V.; Siani, M.A.; Gallegos, C.A.; Feucht, P.H.; Abbott, J.; Lapointe, G.R.; Moghadam, M.; Khoja, H.; Zakel, J.; et al. Receptor recognition and specificity of interleukin-8 is determined by residues that cluster near a surface-accessible hydrophobic pocket. J. Biol. Chem. 1996, 271, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Dewald, B.; Loetscher, M.; Moser, B.; Baggiolini, M. Structural requirements for interleukin-8 function identified by design of analogs and CXC chemokine hybrids. J. Biol. Chem. 1994, 269, 16075–16081. [Google Scholar] [PubMed]
- Skelton, N.J.; Quan, C.; Reilly, D.; Lowman, H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 1999, 7, 157–168. [Google Scholar] [CrossRef]
- Gordon, J.R.; Li, F.; Zhang, X.; Wang, W.B.; Zhao, X.; Nayyar, A. The combined CXCR1/CXCR2 antagonist CXCL8(3–74)K11R/G31P blocks neutrophil infiltration, pyrexia, and pulmonary vascular pathology in endotoxemic animals. J. Leukoc. Biol. 2005, 78, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gordon, J.R. IL-8(3-73) K11R is a high affinity agonist of the neutrophil CXCR1 and CXCR2. Biochem. Biophys. Res. Commun. 2001, 286, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, X.; Gordon, J.R. CXCL8(3-73)K11R/G31P antagonizes ligand binding to the neutrophil CXCR1 and CXCR2 receptors and cellular responses to CXCL8/IL-8. Biochem. Biophys. Res. Commun. 2002, 293, 939–944. [Google Scholar] [CrossRef]
- Zhao, X.; Town, J.R.; Li, F.; Zhang, X.; Cockcroft, D.; Gordon, J.R. ELR-CXC chemokine receptor antagonism targets inflammatory responses at multiple levels. J. Immunol. 2009, 182, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Town, J.R.; Yang, A.; Zhang, X.; Sawicki, G.; Gordon, J.R. A novel ELR-CXC chemokine antagonist reduces intestinal ischemia reperfusion-induced mortality and local and remote organ injury. J. Surg. Res. 2009, in press. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Wang, B.; Zhang, W.J.; Li, Q.; Luan, X.; Cheng, J.W.; Gordon, J.R.; Li, F.; Liu, H. CXCR1/2 antagonism with CXCL8/Interleukin-8 analogue CXCL8(3-72)K11R/G31P restricts lung cancer growth by inhibiting tumor cell proliferation and suppressing angiogenesis. Oncotarget 2015, 6, 21315–21327. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.R.; Borkakoti, N.; Bottomley, G.A.; Conway, E.A.; Gowan, I.; Fallowfield, A.G.; Handa, B.K.; Jones, P.S.; Keech, E.; Kirtland, S.J.; et al. Identification and characterisation of an inhibitor of interleukin-8: A receptor based approach. Bioorg. Med. Chem. Lett. 1996, 6, 1869–1874. [Google Scholar] [CrossRef]
- Attwood, M.R.; Conway, E.A.; Dunsdon, R.M.; Greening, J.R.; Handa, B.K.; Jones, P.S.; Jordan, S.C.; Keech, E.; Wilson, F.X. Peptide based inhibitors of interleukin-8: Structural simplification and enhanced potency. Bioorg. Med. Chem. Lett. 1997, 7, 429–432. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Rajagopalan, L.; Rajarathnam, K. Structural basis of chemokine receptor function—A model for binding affinity and ligand selectivity. Biosci. Rep. 2006, 26, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Dewald, B.; Geiser, T.; Moser, B.; Baggiolini, M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc. Natl. Acad. Sci. USA 1993, 90, 3574–3577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Town, J.R.; Li, F.; Li, W.; Zhang, X.; Gordon, J.R. Blockade of neutrophil responses in aspiration pneumonia via ELR-CXC chemokine antagonism does not predispose to airway bacterial outgrowth. Pulm. Pharmacol. Ther. 2010, 23, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Huang, K.C.; Yu, H.Y.; Gao, K.J.; Zhao, X.; Li, F.; Town, J.R.; Gordon, J.R.; Cheng, J.W. A new protocol for high-yield purification of recombinant human CXCL8(3-72)K11R/G31P expressed in Escherichia coli. Protein Expr. Purif. 2008, 61, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wuthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Biomol. NMR 1998, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ferentz, A.E.; Wagner, G. NMR spectroscopy: A multifaceted approach to macromolecular structure. Q. Rev. Biophys. 2000, 33, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Cornilescu, G.; Delaglio, F.; Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 1999, 13, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T. X-PLOR Version 3.1: A system for X-ray Crystallography and NMR; Yale University Press: New Haven, CT, USA, 1992. [Google Scholar]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds hG31Pare available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NOE Restraints | 2343 |
|---|---|
| Intraresidue (|i-j|=0) | 532 |
| Sequential (|i-j|=1) | 669 |
| Medium range (2≦|i-j|≦4) | 352 |
| Long range (|i-j|>4) | 465 |
| Hydrogen bond constraints | 120 |
| Dihedral angles a | 106 |
| Intermolecular | 99 |
| Final energy (kcal mol−1) | |
| E (total) | 209.8468 ± 13.8168 |
| E (bond) | 10.20659 ± 1.57751 |
| E (angle) | 100.5601 ± 5.99895 |
| E (improper) | 9.874589 ± 0.97132 |
| E (van der Waals) | 51.37347 ± 5.53424 |
| E (NOE) | 37.72119 ± 3.85691 |
| E (cdih) | 0.110884 ± 0.24450 |
| Structural Statistics (20 structures) | |
| NOE violations, number > 0.3 Å | 0 |
| Dihedral angel violations, number > 5° | 0 |
| RMSD for geometrical analysis | |
| Bond lengths (Å) | 0.002 ± 0.0002 |
| Bond angles (degree) | 0.383 ± 0.0116 |
| Impropers (degree) | 0.228 ± 0.0144 |
| Atomic RMSD for protein b | |
| All heavy atoms | 1.38 ± 0.08 |
| Backbone | 0.68 ± 0.09 |
| Ramachandran statistics b | |
| Most favoured region (%) | 88.8 |
| Additionally allowed (%) | 11.1 |
| Generously allowed (%) | 0.1 |
| Disallowed (%) | 0.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.-T.; Yu, H.-Y.; Gordon, J.R.; Li, F.; Cheng, J.-W. Effects of K11R and G31P Mutations on the Structure and Biological Activities of CXCL8: Solution Structure of Human CXCL8(3-72)K11R/G31P. Molecules 2017, 22, 1229. https://doi.org/10.3390/molecules22071229
Cheng H-T, Yu H-Y, Gordon JR, Li F, Cheng J-W. Effects of K11R and G31P Mutations on the Structure and Biological Activities of CXCL8: Solution Structure of Human CXCL8(3-72)K11R/G31P. Molecules. 2017; 22(7):1229. https://doi.org/10.3390/molecules22071229
Chicago/Turabian StyleCheng, Hsi-Tsung, Hui-Yuan Yu, John R. Gordon, Fang Li, and Jya-Wei Cheng. 2017. "Effects of K11R and G31P Mutations on the Structure and Biological Activities of CXCL8: Solution Structure of Human CXCL8(3-72)K11R/G31P" Molecules 22, no. 7: 1229. https://doi.org/10.3390/molecules22071229