Co-Transcriptional Folding and Regulation Mechanisms of Riboswitches

1
Hubei Key Laboratory of Economic Forest Germplasm Improvement and Resources Comprehensive Utilization, Hubei Collaborative Innovation Center for the Characteristic Resources Exploitation of Dabie Mountains, Huanggang Normal University, Huanggang 438000, Hubei, China
2
Department of Physics, Wuhan University, Wuhan 430072, Hubei, China
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(7), 1169; https://doi.org/10.3390/molecules22071169
Submission received: 18 June 2017
/
Revised: 7 July 2017
/
Accepted: 9 July 2017
/
Published: 13 July 2017
(This article belongs to the Section Bioorganic Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Riboswitches are genetic control elements within non-coding regions of mRNA. These self-regulatory elements have been found to sense a range of small metabolites, ions, and other physical signals to exert regulatory control of transcription, translation, and splicing. To date, more than a dozen riboswitch classes have been characterized that vary widely in size and secondary structure. Extensive experiments and theoretical studies have made great strides in understanding the general structures, genetic mechanisms, and regulatory activities of individual riboswitches. As the ligand-dependent co-transcriptional folding and unfolding dynamics of riboswitches are the key determinant of gene expression, it is important to investigate the thermodynamics and kinetics of riboswitches both in the presence and absence of metabolites under the transcription. This review will provide a brief summary of the studies about the regulation mechanisms of the pbuE, SMK, yitJ, and metF riboswitches based on the ligand-dependent co-transcriptional folding of the riboswitches.
1. Introduction
Riboswitches, as self-regulatory elements, regulate downstream gene expression through conformational changes driven by sensing specific metabolites [1,2,3,4,5,6], ions, and other physicalsignals [2,7]. Metabolite-specific riboswitches, the most widespread class that has been previously validated, are involved in many important biological processes, such as vitamin biosynthesis [8,9], nucleotide and amino acid metabolism [10,11], sulfur metabolism, and metal transport [1,12,13]. Most riboswitches are characterized by two domains: a most-conserved aptamer domain responsible for ligand binding by forming the aptamer structure, and an expression platform that converts folding changes in the aptamer domain into changes in gene expression [14,15,16]. The structure arrangement of riboswitches is able to modulate the expression of downstream coding sequences at the levels of transcription [17,18,19], translation [3,20], and RNA splicing [21,22]. However, there are several riboswitches, such as the SMK riboswitch, that only utilize a single domain for both ligand binding and gene regulation [3,23].
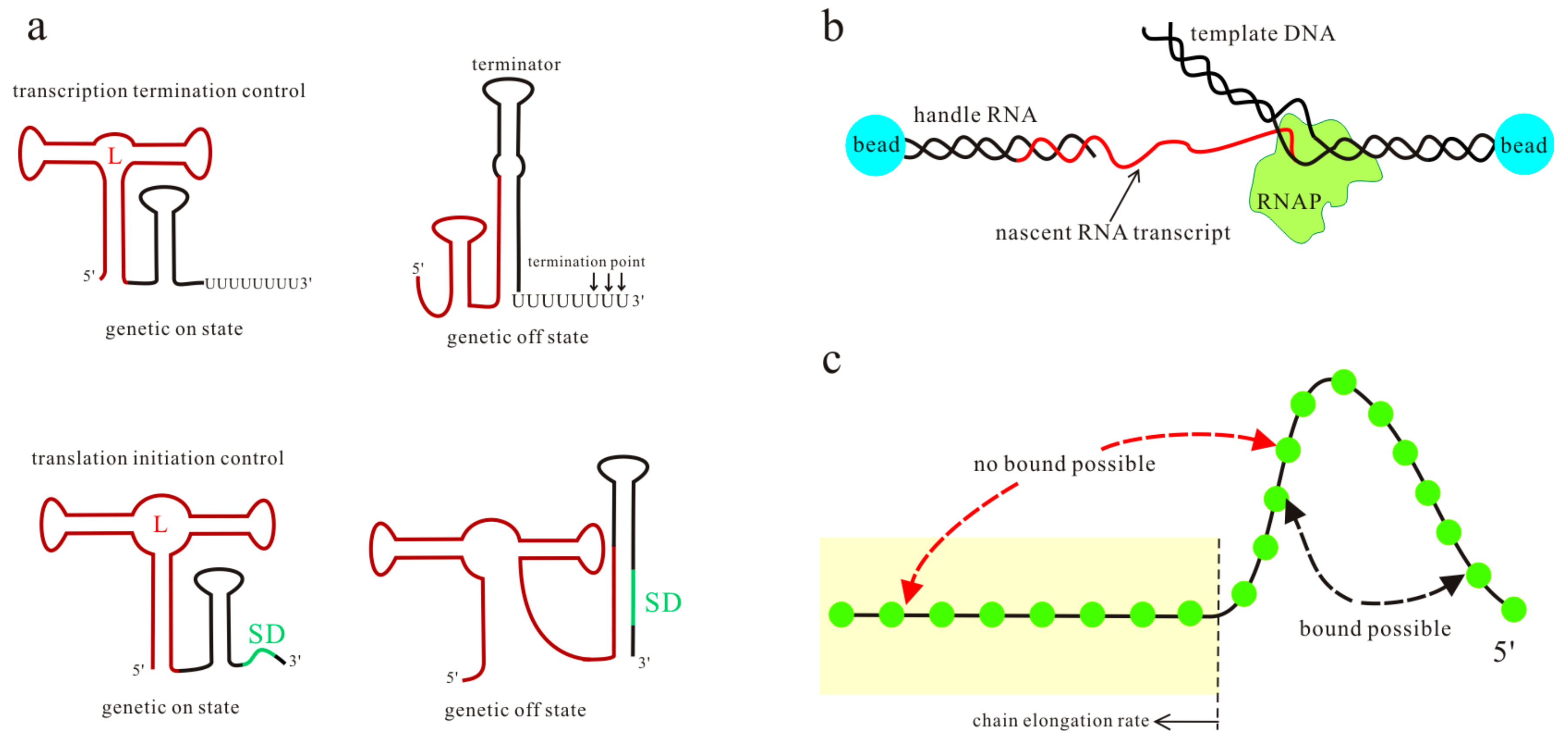
The commonly accepted molecular mechanism for riboswitch function proposes a signal-dependent RNA structural shift, usually between two distinct functional states, i.e., ligand bound state and unbound state (Figure 1a). One of the alternative states serves as the genetic off state, which may contain an intrinsic terminator hairpin (T hairpin) or a paired region covering the Shine-Dalgarno (SD) sequence [24,25,26,27]. The other state serves as the genetic on state that destroys these regulatory elements, and then the downstream genes are expressed. Translational control is critical for gene regulation in bacteria [28], while alternative pre-mRNA splicing is a central mode of gene regulation in eukaryotes [29]. For the riboswitches acting at the level of RNA splicing, the main difference in the two functional states is the structural flexibility near the relevant splice site [22,30]. Depending on ligand concentration and other intracellular factors, one of the two functional states is adopted by riboswitches to regulate downstream gene expression.
Previous studies have investigated many features of riboswitches to explore their regulation mechanisms, such as ligand recognition [8,31,32], folding landscape [3,33,34], the rates and energies for ligand binding and dissociation [19,24], and ligand specificity and its structural basis [35,36,37,38,39]. Experiments such as in-line probing [40,41,42,43], fluorescence spectroscopy [44,45,46], and single-round transcription assay [47,48], in conjunction with theoretical simulations [20,26,49], have established the basic principles that underpin riboswitch function in all three kingdoms of life. Most of these studies focus on the aptamer domain or the transcript products. However, in vivo nascent mRNAs fold as they are transcribed, and this sequential process affects the folding efficiency and the predominant structures. Thus, for some riboswitches, even though the high-resolution structures of the bound aptamer have been solved, the unbound functional structures formed during the transcription and the detailed regulation mechanisms are not well known [50,51,52,53]. As the ligand-dependent co-transcriptional folding and unfolding dynamics of riboswitches play an important role in exerting their functions [25,54,55], it is important to investigate the folding behaviors of riboswitches both in the presence and absence of metabolites in the transcription context. Recently, the optical-trapping assay (Figure 1b) has been developed to monitor co-transcriptional folding during the individual transcript of the pbuE adenine riboswitch [56]. Since the extension changes can be related to the number of nucleotides involved in the folding, by measuring the separation between beads, this assay furnishes a tool for probing structure formation and transition with force during the transcription. The ligand-dependent co-transcriptional folding of riboswitches has also been studied by using the kinetic Monte Carlo simulation to include RNA growth, by sequentially enlarging the subset of contacts that can be formed (Figure 1c) [54]. The effect of a bound ligand is mimicked by modifying the free energy stabilizing the multiloop region, but the method only considers the base pairs in the native structures. The analytical theory, which combines the master equation with the free energy landscape, has been proven to be a reliable tool to calculate co-transcriptional folding kinetics [57]. To explore the detailed regulation mechanisms of riboswitches, ligand binding kinetics have been incorporated into the theory [17]. By using this theory, the regulatory behaviors of several riboswitches under different transcription conditions have been investigated [17,58,59]. All of these studies produce a framework for describing the function of riboswitches at the system level. Here, we will provide a brief overview of the studies about the regulation mechanisms of the pbuE, SMK, yitJ, and metF riboswitches, based on their ligand-dependent co-transcriptional folding behaviors [17,58,59].
2. The Systematic Co-Transcriptional Folding Theory
From the early 1980s, key experiments showed that structure formation happens co-transcriptionally [60,61]. Many experiments have since substantiated this view [62,63,64]. For example, the co-transcriptional folding of the genomic ribozyme is assayed by monitoring the co-transcriptional self-cleavage of transcripts with variable lengths [65]. Likewise, a number of computational methods, such as RNAkinetics [66], Kinefold [67], and COFOLD [61], are developed to explicitly simulate co-transcriptional folding pathways of mRNAs as a series of structural changes over time. However, in order to investigate the riboswitch-mediated regulation mechanisms, ligand binding should be taken into account.
RNA’s secondary structure is stabilized mainly by the base-stacking interactions. As the rate for the formation of a base stack is usually larger than that of disrupting the stack, except the loop-closing stack under the folding condition, once the first few stacks in a helix are closed and stabilized, the zipping of the subsequent stacks in the helix would be fast [57]. It is proper to use the helices as building blocks for the study of the overall (slower) folding kinetics. The systematic helix-based computational theory, in which all structures are constructed according to helices as building blocks and a kinetic move is an addition or a deletion of a helix or an exchange between two helices, is successfully used to study the folding kinetics of the Hepatitis delta virus ribozyme and other mRNAs [57,68,69]. To simulate the co-transcriptional folding of mRNAs, this theory has been further developed to include RNA growth [70]. In this model, releasing one nucleotide by the transcription elongation complex to freely form structures is treated as one transcriptional step. If the transcription speed of an RNA sequence is ν nucleotides per second, the folding time window for each step will be seconds. From a time t when an N-nt chain is free to form structures to a time when the (N + 1)-th nucleotide can freely form structures, the N-nt chain samples the conformation space at step N. Within this step, the population distribution is relaxed from (] to [), where and are the population of state i at the beginning and end of step N, respectively, and is the number of conformations for the N-nt chain. This is defined as the N-th step. At each such transcriptional step, the population kinetics are calculated in the following manner: first, the conformation space is generated according to helices as building blocks, so a kinetic move is the addition or the deletion of a helix, or an exchange between two helices. Then, the transition rate from state i to state j in the conformation space is calculated according to the structure relationship between the two states [57], whose free energies are obtained based on the nearest-neighbor model [71,72]. Finally, the population kinetics within the folding time window () are calculated by solving the master equation , where the initial condition is determined by the final population distribution at the previous step [70]. is the rate matrix with elements and , and is the vector for the population distribution. For consecutive steps, the folding results of the current step turn into the initial condition of the next step. By applying this method from the first step to the end of the transcription, the co-transcriptional folding population kinetics of mRNAs can be calculated. To simulate transcription pausing at a specific site, a large number of effective time steps are assigned to the corresponding (paused) step. Co-transcriptional folding under different transcription rates can also be mimicked by changing .
Ligand binding kinetics are incorporated into the model to predict the ligand-dependent co-transcriptional folding behaviors of riboswitches [17]. As the ligand concentration is much larger than that of the mRNA in cells, the second-order ligand binding kinetics: mRNA+ligand → the ligand bound state, can be approximated as a linear relation. When the ligand is present, the ligand bound states are added to the conformation space. The ligand free state containing the aptamer structure can transit to the corresponding ligand bound state with the effective binding rate , and the reverse transition with the rate , where [L] is the ligand concentration, and and are the experimentally measured association rate and dissociation rate, respectively. The ligand binding can stabilize the corresponding bound state by a free energy of , where is the Boltzmann constant, and is the temperature. We have studied the co-transcriptional folding behaviors of the pbuE [17], SMK [58], yitJ, and metF riboswitches by using this method [59].
3. The Kinetic Regulation Regime of the pbuE and metF Riboswitches
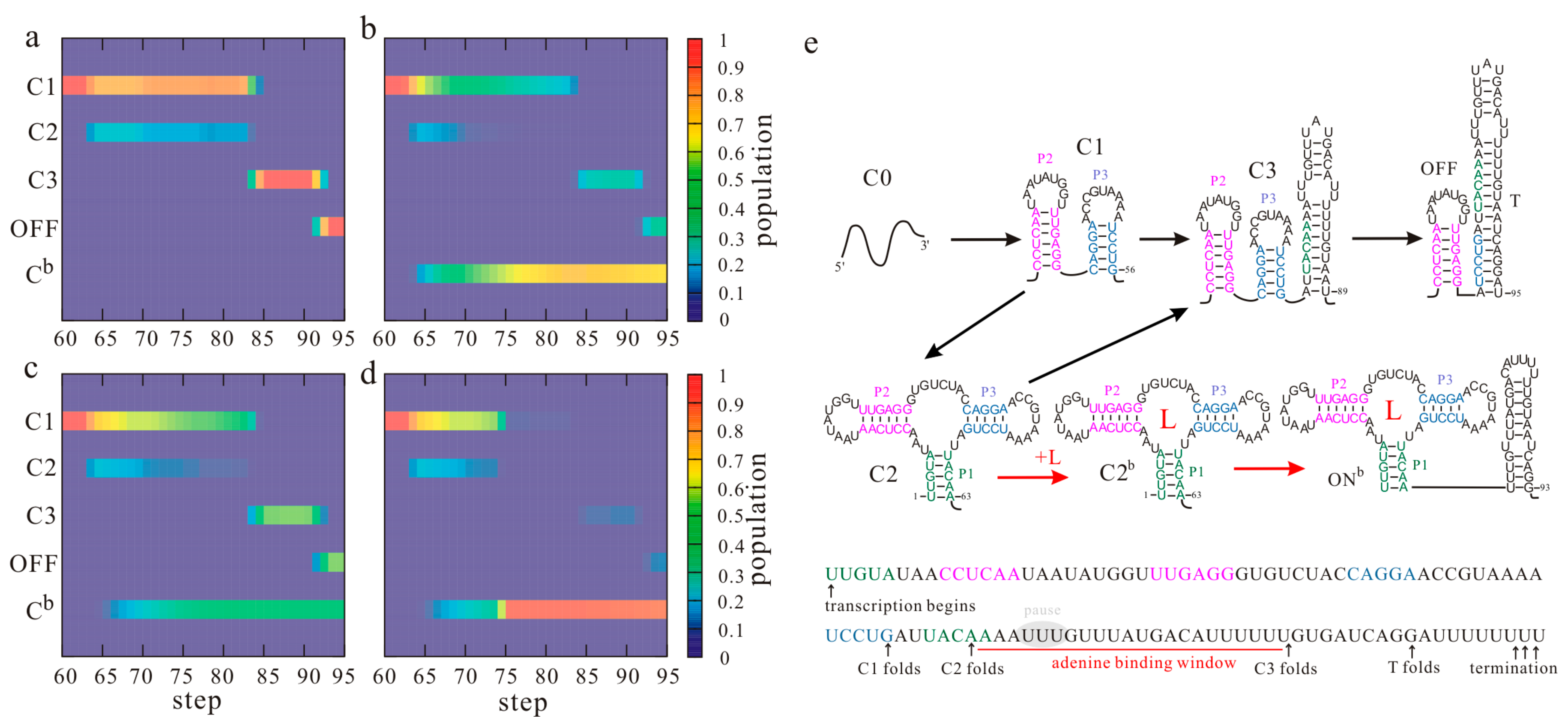
Among the riboswitches known to date, the pbuE adenine riboswitch is a rare type of “on” switch that activates gene expression upon ligand binding by controlling transcription termination [55]. Its aptamer structure is characterized by a three-way junction consisting of helices P1, P2, and P3, which are further stabilized by tertiary interactions in the folded state. The full-length pbuE riboswitch has been found to be fixed at a ligand-incompetent structure [24,55], and the aptamer structure is not formed during the refolding process [17], suggesting that the transcription context plays a key role in its function. The folding events of the Bacillus subtilis pbuE riboswitch during the transcription were monitored by the helix-based co-transcriptional folding method [17]. Figure 2 shows the population kinetics of the main structures (a–d) and folding pathways (e) under different transcription conditions. The folding of helices P2 and P3, which occurs in turns, is not affected by the transcription conditions. However, when helix P1 is nucleated, adenine can bind to the riboswitch. Thus, the population kinetics of the main states and folding pathways from step 60 are shown in Figure 2 for further discussion. As state C1, consisting of helices P2 and P3, is the most stable state before the expression platform is transcribed, almost all the riboswitches populate at this state at step 60 (Figure 2a). When helix P1 is nucleated from step 62, state C1 can transit to the aptamer structure C2 by forming helix P1 with the rate . As forming this short nonlocal helix needs to close the big multiloop, which leads to an increase in the free energy (, ), only about one fifth of the RNAs equilibrate into state C2. In the absence of adenine, the aptamer structure C2 will be gradually invaded by the T hairpin to form state C3 with elongation of the nascent RNA chain. At the end of the transcription, about 98% of the riboswitches fold through C3 to the OFF state with the T hairpin, which prompts transcription release at the terminator U tract (termination point) [14,73]. Given that factors such as RNA polymerase (RNAP) and nucleoside triphosphate (NTP) may affect the termination efficiency (TE) [74,75], the result agrees well with a TE of 89% given by the gel-based assay [56].
Conversely, when the ligand is present, adenine binding stabilizes the aptamer structure C2 to form the bound state C2b (the superscript “b” denotes the state with ligand bound, Figure 2e) by introducing tertiary interactions. With the growth of the nascent RNA chain, the bound aptamer will prevent the formation of the T hairpin. Instead, it will direct the folding of the ligand bound ON state (ONb) by forming a hairpin, which can introduce about −1.61 kcal/mol free energies. The results in Figure 2b suggest that with 100 µM adenine, about 67% of the riboswitches populate at the ONb state at the end of the transcription. Even if adenine binding would contribute a free energy as low as (4 ± 1) kcal/mol [76], this state is still much more unstable than the OFF state (, ). However, due to the slow dissociation rate of the bound aptamer, the RNAP is able to successfully pass the termination signal before the ONb state equilibrates into a low-energy OFF state, consistent with the riboswitches operating under a kinetic regulation regime. Hence, the formation of the two functional states (ONb and OFF) are both irreversible events for effective riboswitch-mediated gene regulation.
During the transcription process, the aptamer structure is formed before the OFF state. In the absence of the ligand, the aptamer structure would quickly transit to the OFF state. However, in the presence of the ligand, adenine can bind to the aptamer structure and then preclude the structure from forming an OFF state. The ligand binding must occur during the transcription process, and the action of the riboswitch is sensitive to co-transcriptional folding. During the transcription, the time window allowed for adenine binding is from step 62 when the aptamer structure can be formed to step 84 when state C3 becomes much more stable (). This binding time window provides the riboswitch with only one chance to switch genetic on or off. Excepting the termination region, there is a short series of uridine (U) residues within the template, which may induce transcription pausing at around step 67 [24,55], considering about 12 nt enclosed by the RNAP [77,78]. If the pause duration is s, the time allowed for adenine binding will be () s, where is the transcription rate. When the transcription rate increases from 20 nt/s to 50 nt/s, the population of the ONb state decreases from 67% (Figure 2b) to 43% (Figure 2c) with 100 µM adenine. Nevertheless, even under the same conditions, the population of the ONb state will increase to about 86% if the transcription pause occurs (Figure 2d). Obviously, a fast transcription rate yields a short binding time , thereby repressing the formation of the bound state, whereas the transcription pausing has the opposite effect. In other words, the ligand concentration, the transcription speed, and pausing are coupled to modulate the pbuE riboswitch to perform regulatory activities. Since the transcription rate and pausing are affected by intracellular factors such as NTP concentration and proteins [19,79,80], the expression of the pbuE riboswitch downstream gene is largely dependent on the intracellular environment.
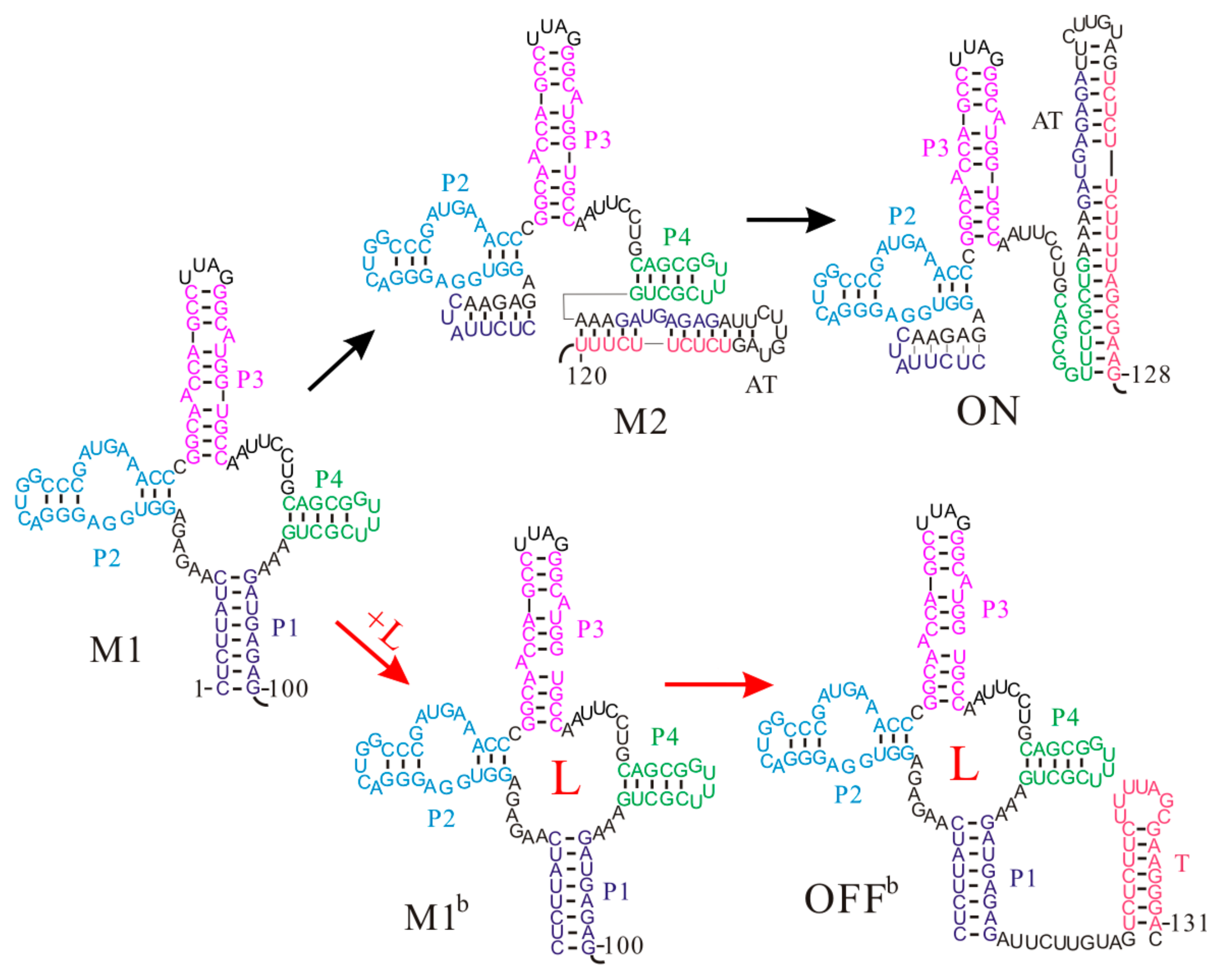
Even though the aptamer structure of the S-box riboswitches is a four-way junction structure, the Thermoanaerobacter tengcongensis metF S-box riboswitch shows similar co-transcriptional folding behaviors to that of the pbuE riboswitch [59]. Once the aptamer sequence is released during the transcription, the aptamer structure M1 consisting of helices P1, P2, P3, and P4 is formed without any trapped states (Figure 3). As the nascent RNA chain grows, the upper part of the anti-terminator (AT) hairpin nucleates and gradually invades into the aptamer structure M1. Soon, this structure will be replaced by state M2, in which helix P1 is disrupted. By the time the RNAP reaches the termination point, a low-energy ON state with the AT hairpin is fully formed, and at the same time helix P4 is broken as well (, ). In this case, the RNAP can run through to the end of the template and the gene is expressed. On the contrary, if the ligand S-adenosylmethionine (SAM) successfully binds to the aptamer structure before the expression platform is transcribed, the bound aptamer M1b will fold to an OFFb state by directly forming the T hairpin. Due to the long half-life of the SAM-RNA complex [47], the transcript has been terminated before the riboswitch folds into the thermodynamically favored ON state [59], suggesting a kinetic regulation regime. Since the formation of the ON state disrupts the aptamer structure and it is very stable, SAM binding only can occur before formation of this state. This demonstrates that the ligand binding window is limited during the transcription process, and the fate of downstream gene expression depends on the folding of the riboswitch within this time period. Thus, the regulatory activities of the metF riboswitch are also tied to the intracellular environment. In addition, although both states M2 and ON have been proposed as the genetic on state of the metF riboswitch previously [52,53], our results suggest that state M2 is just an intermediate structure and persists for several transcriptional steps, in agreement with the recent study [51].
4. The Association of Thermodynamic and Kinetic Regulation Regimes of the yitJ Riboswitch
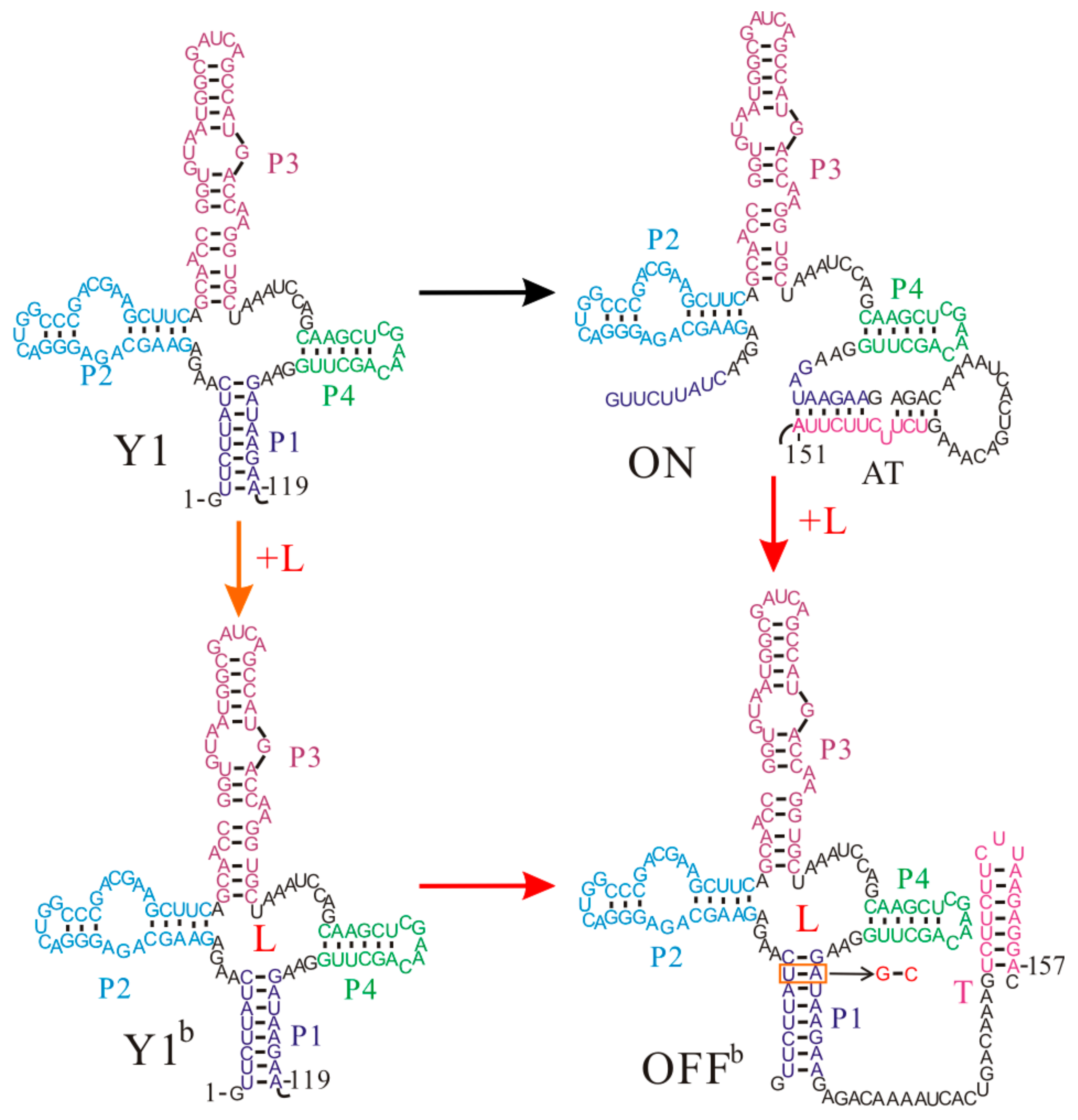
The Bacillus subtilis yitJ riboswitch is another SAM-specific riboswitch that has been intensively studied [13,81]. The two S-box (yitJ and metF) riboswitches have similar OFF state structures and share some features, such as the reliance of termination efficiency on SAM concentration, but their regulation mechanisms are different [59]. The aptamer structure Y1 is formed as soon as the relevant nucleotides are synthesized. When SAM is present at sufficient concentrations, SAM binding stabilizes the aptamer structure Y1, which induces the formation of the T hairpin (OFFb) as the nascent RNA grows (Figure 4) [59]. However, if the ligand cannot bind to the riboswitch before the expression platform is synthesized, the AT hairpin (ON) will be formed instead, which allows the RNAP to successfully pass the termination point. As the formation of the AT hairpin only disrupts helix P1 in the aptamer structure, an ON state can quickly equilibrate into an OFF state () through the tunneling pathway within one transcriptional step. This implies that their population distribution is thermodynamically controlled when the RNAP reaches the termination point. In addition, because the two states (, ) have similar free energies, a single base pair mutation by changing UA to GC in helix P1, can result in high termination efficiency even without the ligand [13], suggesting thermodynamic control characteristics.
Since the transition from an OFF state containing the aptamer structure to an ON state is very fast, even if the yitJ riboswitch predominantly folds into an ON state during the transcription, this fold is in equilibrium with a small population of the OFF state. Upon SAM binding, the equilibrium is shifted toward the thermodynamically favored OFFb state. The free energy of the ON state is about −1.17 kcal/mol lower than that of the OFF state, and only about 0.13 µM ligand is required to make an OFFb state slightly more stable than an ON state (, , ) [47,59]. However, the ligand binding is a kinetic process, and whether it reaches equilibrium depends on the binding time window and the effective binding rate (). For this riboswitch, the ligand binding could occur from the 117th step when the aptamer structure is formed to the termination point. At the end of the transcription, the ligand bound states can occupy about of the population, where is the equilibrium distribution of the ligand bound states. Even at 3 μM SAM, an OFFb state only can obtain about 11% of the population at 50 nt/s, far less than the equilibrium value [59], because of the slow effective binding rate. Namely, a much higher ligand concentration may be required to trigger riboswitch regulation relative to the dissociation constant. Additionally, the amount of the ligand required to trigger the switch is dependent on the transcription rate. Thus, the transcription process should also be determinant for riboswitch activation. All these results strongly suggest the yitJ riboswitch operates under a combination of thermodynamic and kinetic regulationmechanisms [59].
5. The Thermodynamic Regulation Regime of the SMK Riboswitch
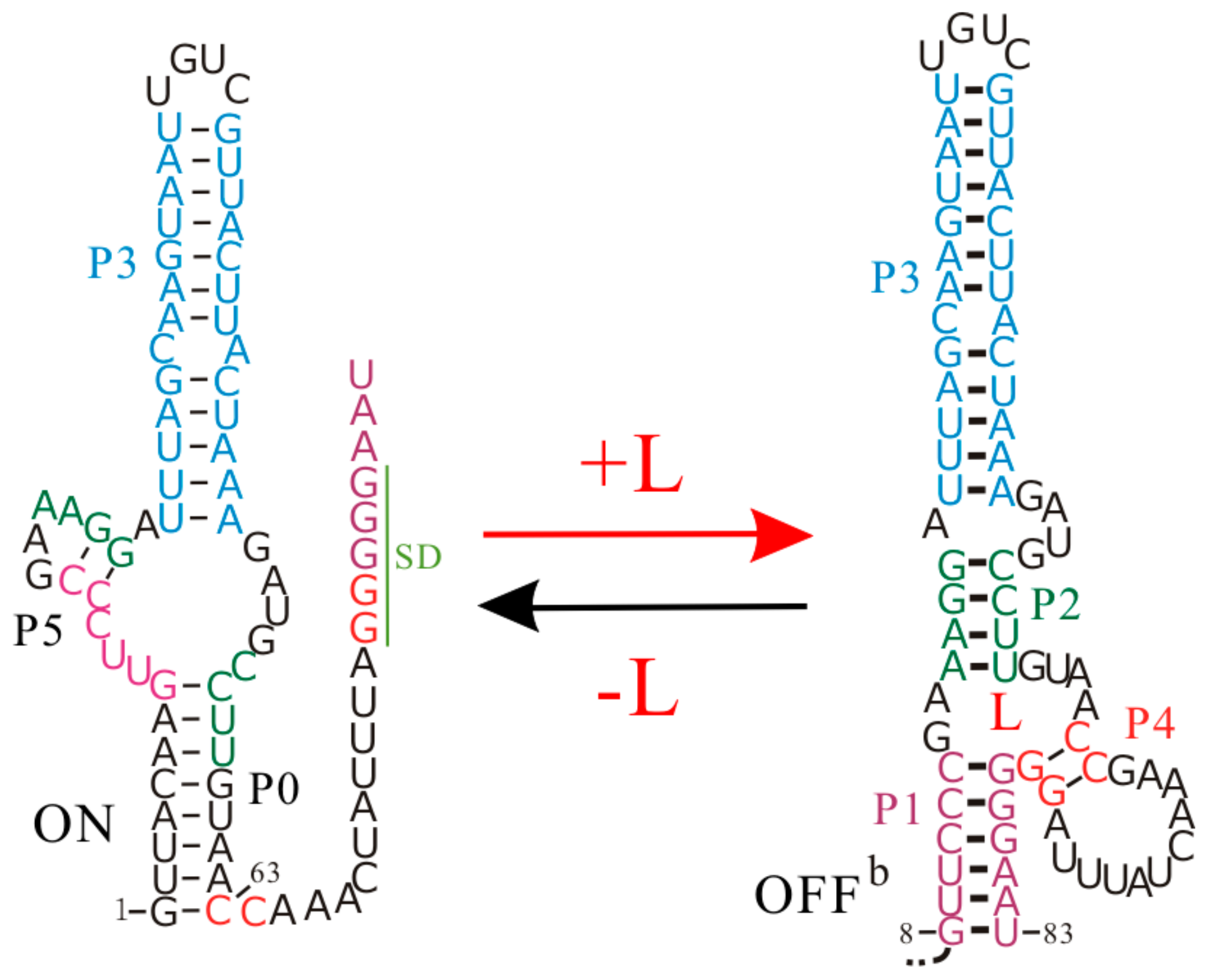
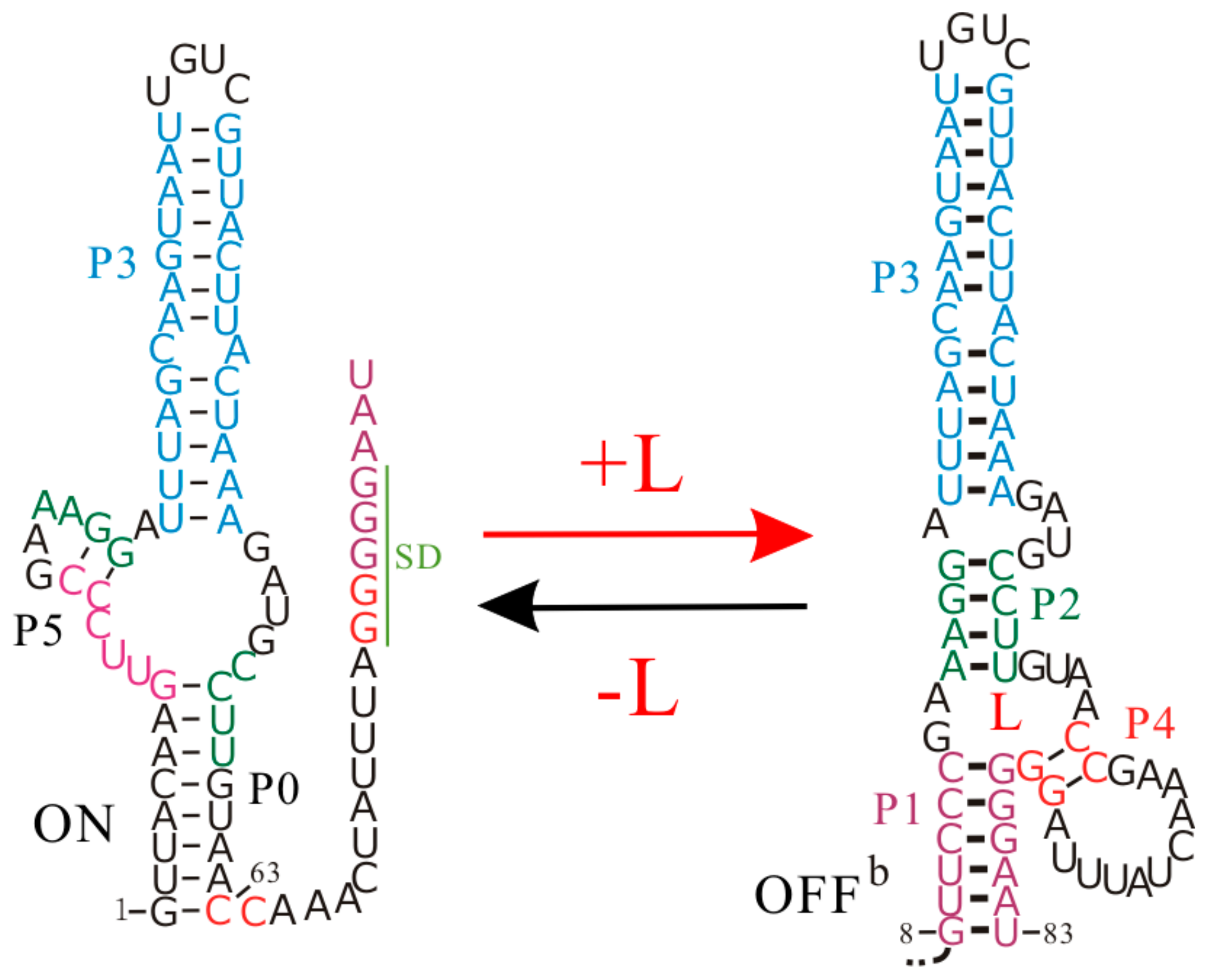
The Enterococcus faecalis SAM-III (SMK) riboswitch is one of five distinct classes of SAM-specific riboswitches that have been discovered [35]. This riboswitch senses the intracellular SAM or its derivative S-adenosylhomocysteine to regulate the translation of the metK gene [36,82], which encodes the synthetase of SAM, a cofactor in the methylation reactions of proteins, nucleic acids, and other biomolecules [82,83]. It is a typical riboswitch that operates at the translation level, and to be more specific, its ligand binding and regulatory domains are coincident [3,26]. The SD sequence directly takes part in binding SAM, and is sequestered by base pairing with the anti-SD sequence in the presence of the ligand (Figure 5).
The co-transcriptional folding and refolding behaviors of the SMK riboswitch as well as its shortened construct have been studied by the recently developed method [58]. In contrast to most riboswitches containing two separate domains, this riboswitch, with a single domain to perform gene regulation, displays many unique features in its regulation activities. During the free folding and co-transcriptional folding processes without SAM, the SMK riboswitch quickly folds into the low-energy ON state (), while the SAM binding pocket structure (OFF state) consisting of helices P1, P2, P3, and P4 is not formed. As a co-transcriptional fold follows a sequential process, an ON state with the unstructured SD sequence is fully formed (step 63) prior to an OFF state (step 83) during the transcription (Figure 5), and it is more stable than the OFF state (). Thus, even with SAM at a saturating level, the SMK riboswitch still first folds to an ON state, instead of the binding pocket structure OFF, which is formed near the end of the transcription. For this riboswitch, whether the ligand is present or not, the main co-transcriptional folding pathway is the same. Unlike the pbuE and two S-box riboswitches, sequential folding during the transcription process is not necessary for the SAM-III riboswitch to efficiently perform gene regulation.
Although an ON state is first formed, it will fold into the bound state OFFb when SAM is abundant, since the ligand binding can make the OFFb state more stable than the ON state. The transition from ON state to OFF state requires it to undergo conformational rearrangements, as it needs to break helices P0 and P5 and form helices P1, P2, and P4. Compared to the mRNA degradation rate () [26], the tunneling pathway from an ON state to an OFF state yields a faster transition rate (). If SAM binding to the full-length riboswitch is described as a three-state transition model: , the population of ON and OFFb states will be thermodynamically distributed [58]. Its switch efficiency is linked to the stability of the two function structures instead of the transcription context. Therefore, the SMK riboswitch can quickly switch back and forth to regulate the translation of its host gene in response to the ligand concentration, strongly supporting a pure thermodynamic regulation mechanism.
The thiamine pyrophosphate (TPP) riboswitch within the NMT1 gene from Neurospora crassa is a typical riboswitch that controls RNA splicing [84]. The TPP riboswitch aptamer forms a tuning fork architecture, in which the prongs are formed by two parallel stacking helices and arranged via a central three-way junction [85,86,87]. TPP binding stabilizes the aptamer structure, increasing the structural flexibility near the 5′ splice site. The effect of the change yields the long spliced mRNAs carrying the short upstream open reading frames that compete with the translation of the main open reading frame. In the absence of TPP, the riboswitch adopts an alternative structure that occludes the 5′ splice site, leading to a short spliced mRNA, and then inducing the expression of the NMT1 gene. This riboswitch also utilizes a single domain for both ligand binding and gene regulation, so we can speculate that the SMK and TPP riboswitches may share some common regulatory features.
6. Conclusions
Riboswitches, which adopt alternative RNA folds to regulate gene expression, are sensitive to co-transcriptional folding events. Co-transcriptional folding following a sequential progression, with associated structures forming within seconds (or less) of the times that their sequences clear the RNAP footprint, poses a serious challenge in the quest for detecting these structures in an experimental context. By incorporating the ligand binding kinetics into the helix-based co-transcriptional folding theory, the regulation mechanisms of the riboswitches can be attained through simulating the co-transcriptional folding behaviors of the riboswitches in the presence/absence of ligands under different conditions. The good agreement of the results with the experiments demonstrates that it is a reliable tool to study regulation mechanisms for riboswitches and the functions of other RNAs.
For the riboswitches under the same regulation mechanisms, their regulatory behaviors and functional states exhibit many common features. The kinetically controlled riboswitches, such as the pbuE and metF riboswitches, usually exert regulatory control of transcription. Their ON states are much more stable (unstable) than their OFF states, so the ligand binding does not offset the free energy difference even at saturating concentrations. In addition, because the formation of the most stable functional state, such as the OFF state of the pbuE riboswitch, largely disrupts the aptamer structure, the full-length riboswitches often resist the ligand binding. Hence, the ligand binding is linked to the transcription context and it must occur prior to the formation of this stable state during the transcription. That is to say, the time window allowed for the ligand binding is limited. This limited time window can be modulated by the transcription process. For example, proper transcription pausing and slow elongation rates could give the aptamer structure more time to bind to the ligand. For these riboswitches, even the bound functional state, such as the ONb state of the pbuE riboswitch, is not the most stable state, and it will remain folded before the RNAP has made the genetic decision because of the slow dissociation rate. Therefore, the riboswitches are always irreversible genetic switches, and their intracellular environments play a key role in their regulation; this has also been found in other systems [19].
Co-transcriptional folding is fundamental for kinetically controlled riboswitches to exert modulatory effects far from equilibrium. Conversely, for the riboswitches operating under a pure thermodynamic regime, such as the SMK and add adenine riboswitches, the transcription context is not required for riboswitch function, as the ligand can bind post-transcriptionally [25,58,88]. These riboswitches are likely to function at the translation level. As their unbound functional states allow a large portion of the aptamer structures to form, the responsiveness of the full-length riboswitches to the ligand can be retained. Contrary to kinetically controlled riboswitches, the ON and OFF states of these riboswitches have similar free energies and much faster transition rates compared to the mRNA degradation rate. Thus, the two functional states, such as the ON and OFFb states of the SMK riboswitch, are thermodynamically distributed. The riboswitches reversibly interconvert between the two functional states, with the outcome ultimately determined by thermodynamic changes due to ligand binding. For the yjtJ riboswitch acting at the transcription level, its ON and OFF states have similar free energies and fast transition rates. However, the amount of the ligand required to trigger the switch is not only dependent on the dissociation constant, but also on the transcription process, so it functions under an association of thermodynamic and kinetic regimes.
Acknowledgments
This work was partly supported by the National Natural Science Foundation of China under Grants No. 31600592 (to S.G.), 31270761 and 11574234 (to W.B.Z.). It was supported in part from Hubei Key Laboratory of Economic Forest Germplasm Improvement and Resources Comprehensive Utilization, Hubei Collaborative Innovation Center for the Characteristic Resources Exploitation of Dabie Mountains [2017BX08].
Author Contributions
S.G. and W.Z. wrote and designed the paper. Y.W. and Z.W. discussed the results and reviewed the manuscript. All authors have contributed in the improving of the manuscript, and read and approved its final version.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Serganov, A.; Nudler, E. A Decade of Riboswitches. Cell 2013, 152, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Reining, A.; Nozinovic, S.; Schlepckow, K.; Buhr, F.; Fürtig, B.; Schwalbe, H. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 2013, 499, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.C.; Thirumalai, D. Kinetics of allosteric transitions in S-adenosylmethionine riboswitch are accurately predicted from the folding landscape. J. Am. Chem. Soc. 2013, 135, 16641–16650. [Google Scholar] [CrossRef] [PubMed]
- Perdrizet, G.A., II; Artsimovitch, I.; Furman, R.; Sosnick, T.R.; Pan, T. Transcriptional pausing coordinates folding of the aptamer domain and the expression platform of a riboswitch. Proc. Natl. Acad. Sci. USA 2012, 109, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Boese, B.; Barrick, J.E.; Winkler, W.C.; Breaker, R.R. Riboswitches Control Fundamental Biochemical Pathways in Bacillus subtilis and Other Bacteria. Cell 2003, 113, 577–586. [Google Scholar] [CrossRef]
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 2003, 10, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Ramesh, A.; Zhou, Z.; Weinberg, Z.; Vallery, T.; Winkler, W.C.; Breaker, R.R. Bacterial Riboswitches Cooperatively Bind Ni2+ or Co2+ Ions and Control Expression of Heavy Metal Transporters. Mol. Cell 2015, 57, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Huang, L.; Patel, D.J. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature 2009, 458, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Mellin, J.R.; Koutero, M.; Dar, D.; Nahori, M.-A.; Sorek, R.; Cossart, P. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science 2014, 345, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Abbas, C.A.; Sibirny, A. Genetic control of biosynthesis and transport of riboflavin and flavin nucleotides and construction of robust biotechnological producers. Microbiol. Mol. Biol. Rev. 2011, 75, 321–360. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Breaker, R.R. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005, 59, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.M.; Hamelberg, D. Atomistic basis for the on-off signaling mechanism in SAM-II riboswitch. Nucleic Acids Res. 2010, 38, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ding, F.; Chowdhury, A.; Pradhan, V.; Tomsic, J.; Holmes, W.M.; Henkin, T.M.; Ke, A. SAM recognition and conformational switching mechanism in the Bacillus subtilis yitJ S box/SAM-I riboswitch. J. Mol. Biol. 2010, 404, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Breaker, R.R. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004, 11, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Breaker, R.R. Genetic control by metabolite-binding riboswitches. ChemBioChem 2003, 4, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Vitreschak, A.G.; Rodionov, D.A.; Mironov, A.A.; Gelfand, M.S. Riboswitches: The oldest mechanism for the regulation of gene expression? TRENDS Genet. 2004, 20, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Zhang, W. Kinetic regulation mechanism of pbuE riboswitch. J. Chem. Phys. 2015, 142, 15103. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.N.; Breaker, R.R. Purine sensing by riboswitches. Biol. Cell 2008, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wickiser, J.K.; Winkler, W.C.; Breaker, R.R.; Crothers, D.M. The Speed of RNA Transcription and Metabolite Binding Kinetics Operate an FMN Riboswitch. Mol. Cell 2005, 18, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Zhao, Y.; Chen, C.; Xiao, Y. Role of Ligand Binding in Structural Organization of Add A-riboswitch Aptamer: A Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2011, 29, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Ríos, J. The THI-box Riboswitch, or How RNA Binds Thiamin Pyrophosphate. Structure 2007, 15, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kubodera, T.; Watanabe, M.; Yoshiuchi, K.; Yamashita, N.; Nishimura, A.; Nakai, S.; Gomi, K.; Hanamoto, H. Thiamine-regulated gene expression of Aspergillus oryzae thiA requires splicing of the intron containing a riboswitch-like domain in the 5′-UTR. FEBS Lett. 2003, 555, 516–520. [Google Scholar] [CrossRef]
- Lu, C.; Smith, A.M.; Ding, F.; Chowdhury, A.; Henkin, T.M.; Ke, A. Variable sequences outside the SAM-binding core critically influence the conformational dynamics of the SAM-III/SMK box riboswitch. J. Mol. Biol. 2011, 409, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Lang, K.; Graber, D.; Micura, R. Ligand-Induced Folding of the Adenosine Deaminase A-Riboswitch and Implications on Riboswitch Translational Control. ChemBioChem 2007, 8, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Lemay, J.-F.; Desnoyers, G.; Blouin, S.; Heppell, B.; Bastet, L.; St-Pierre, P.; Massé, E.; Lafontaine, D.A. Comparative Study between Transcriptionally- and Translationally-Acting Adenine Riboswitches Reveals Key Differences in Riboswitch Regulatory Mechanisms. PLoS Genet. 2011, 7, e1001278. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Yoon, J.; Hyeon, C.; Thirumalai, D. Using simulations and kinetic network models to reveal the dynamics and functions of riboswitches. Methods Enzymol. 2015, 553, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.J.; Lund, P.E.; Blanco, M.R.; Walter, N.G. The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat. Commun. 2016, 7, 8976. [Google Scholar] [CrossRef] [PubMed]
- Nudler, E.; Mironov, A.S. The riboswitch control of bacterial metabolism. Trends Biochem. Sci. 2004, 29, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Barrick, J.E.; Breaker, R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007, 8, R239. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Serganov, A.; Patel, D.J. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol. Cell 2010, 40, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Patel, D.J. Molecular recognition and function of riboswitches. Curr. Opin. Struct. Biol. 2012, 22, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Anthony, P.C.; Perez, C.F.; García-García, C.; Block, S.M. Folding energy landscape of the thiamine pyrophosphate riboswitch aptamer. Proc. Natl. Acad. Sci. USA 2012, 109, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.; Hyeon, C.; Thirumalai, D. Sequence-dependent folding landscapes of adenine riboswitch aptamers. Phys. Chem. Chem. Phys. 2014, 16, 6376–6382. [Google Scholar] [CrossRef] [PubMed]
- Montange, R.K.; Mondragón, E.; van Tyne, D.; Garst, A.D.; Ceres, P.; Batey, R.T. Discrimination between Closely Related Cellular Metabolites by the SAM-I Riboswitch. J. Mol. Biol. 2010, 396, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Priyakumar, U.D. Atomistic details of the ligand discrimination mechanism of S MK/SAM-III riboswitch. J. Phys. Chem. B 2010, 114, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Doshi, U.; Kelley, J.M.; Hamelberg, D. Atomic-level insights into metabolite recognition and specificity of the SAM-II riboswitch. RNA 2012, 18, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Reiss, C.W.; Xiong, Y.; Strobel, S.A. Structural Basis for Ligand Binding to the Guanidine-I Riboswitch. Structure 2017, 25, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, J.; Lilley, D.M.J. The Structure of the Guanidine-II Riboswitch. Cell Chem. Biol. 2017, 24, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.E.; Breaker, R.R. Biochemical Validation of a Third Guanidine Riboswitch Class in Bacteria. Biochemistry 2017, 56, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hwang, X.Y.; Stav, S.; Breaker, R.R. The yjdF riboswitch candidate regulates gene expression by binding diverse azaaromatic compounds. RNA 2016, 22, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, V.K.; Huang, W.; Spedale, J.; Aboul-Ela, F. Basis for ligand discrimination between ON and OFF state riboswitch conformations: The case of the SAM-I riboswitch. RNA 2012, 18, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Liberman, J.A.; Wedekind, J.E. Riboswitch structure in the ligand-free state. Wiley Interdiscip. Rev. RNA 2012, 3, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Allnér, O.; Nilsson, L.; Villa, A. Loop-loop interaction in an adenine-sensing riboswitch: A molecular dynamics study. RNA 2013, 19, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Prychyna, O.; Leung, J.; Avdic, D.; O’Neill, M.A. Ligand-directed dynamics of adenine riboswitch conformers. J. Am. Chem. Soc. 2007, 129, 11308–11309. [Google Scholar] [CrossRef] [PubMed]
- Kulshina, N.; Edwards, T.E.; Ferré-D’Amaré, A.R. Thermodynamic analysis of ligand binding and ligand binding-induced tertiary structure formation by the thiamine pyrophosphate riboswitch. RNA 2010, 16, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Tomšič, J.; McDaniel, B.A.; Grundy, F.J.; Henkin, T.M. Natural variability in S-adenosylmethionine (SAM)-dependent riboswitches: S-box elements in bacillus subtilis exhibit differential sensitivity to SAM in vivo and in vitro. J. Bacteriol. 2008, 190, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Sosnick, T.R.; Pan, T. Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. Proc. Natl. Acad. Sci. USA 2007, 104, 17995–18000. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bulusu, G.; Mitra, A. MD simulations of ligand-bound and ligand-free aptamer: Molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA 2009, 15, 1673–1692. [Google Scholar] [CrossRef] [PubMed]
- Rentmeister, A.; Mayer, G.; Kuhn, N.; Famulok, M. Conformational changes in the expression domain of the Escherichia coli thiM riboswitch. Nucleic Acids Res. 2007, 35, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Kim, J.; Jha, S.; Aboul-ela, F. Conformational heterogeneity of the SAM-I riboswitch transcriptional ON state: A chaperone-like role for S-adenosyl methionine. J. Mol. Biol. 2012, 418, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Hennelly, S.P.; Sanbonmatsu, K.Y. Tertiary contacts control switching of the SAM-I riboswitch. Nucleic Acids Res. 2011, 39, 2416–2431. [Google Scholar] [CrossRef] [PubMed]
- Montange, R.K.; Batey, R.T. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature 2006, 441, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Lutz, B.; Faber, M.; Verma, A.; Klumpp, S.; Schug, A. Differences between cotranscriptional and free riboswitch folding. Nucleic Acids Res. 2014, 42, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Lemay, J.-F.; Penedo, J.C.; Tremblay, R.; Lilley, D.M.J.; Lafontaine, D.A. Folding of the Adenine Riboswitch. Chem. Biol. 2006, 13, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Frieda, K.L.; Block, S.M. Direct Observation of Cotranscriptional Folding in an Adenine Riboswitch. Science 2012, 338, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, W.-B.; Chen, S.-J. Predicting Secondary Structural Folding Kinetics for Nucleic Acids. Biophys. J. 2010, 98, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Wang, Z.; Wang, Y.; Zhang, W. Reversible-Switch Mechanism of the SAM-III Riboswitch. J. Phys. Chem. B 2016, 120, 12305–12311. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Zhang, W. The regulation mechanism of yitJ and metF riboswitches. J. Chem. Phys. 2015, 143, 45103. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.R.; Mills, D.R. Secondary structure formation during RNA synthesis. Nucleic Acids Res. 1981, 9, 5109–5124. [Google Scholar] [CrossRef] [PubMed]
- Proctor, J.R.; Meyer, I.M. CoFold: An RNA secondary structure prediction method that takes co-transcriptional folding into account. Nucleic Acids Res. 2013, 41, e102. [Google Scholar] [CrossRef] [PubMed]
- Heilman-Miller, S.L.; Woodson, S.A. Effect of transcription on folding of the Tetrahymena ribozyme. RNA 2003, 9, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Chadalavada, D.M.; Cerrone-Szakal, A.L.; Bevilacqua, P.C. Wild-type is the optimal sequence of the HDV ribozyme under cotranscriptional conditions. RNA 2007, 13, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Mahen, E.M.; Watson, P.Y.; Cottrell, J.W.; Fedor, M.J. mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol. 2010, 8, e1000307. [Google Scholar] [CrossRef] [PubMed]
- Diegelman-Parente, A.; Bevilacqua, P.C. A Mechanistic Framework for Co-transcriptional Folding of the HDV Genomic Ribozyme in the Presence of Downstream Sequence. J. Mol. Biol. 2002, 324, 1–16. [Google Scholar] [CrossRef]
- Danilova, L.V.; Pervouchine, D.D.; Favorov, A.V.; Mironov, A. a RNAKinetics: A web server that models secondary structure kinetics of an elongating RNA. J. Bioinform. Comput. Biol. 2006, 4, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Xayaphoummine, A.; Bucher, T.; Isambert, H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005, 33, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, W. Kinetic analysis of the effects of target structure on siRNA efficiency. J. Chem. Phys. 2012, 137, 225102. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, S.; Wang, Y.; Zhang, W. Kinetic partitioning mechanism of HDV ribozyme folding. J. Chem. Phys. 2014, 140, 25102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, W.; Chen, S.-J. Cotranscriptional folding kinetics of ribonucleic acid secondary structures. J. Chem. Phys. 2011, 135, 245101. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; SantaLucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of RNA Duplexes with Watson-Crick Base Pairs †. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.S.; von Hippel, P.H. Transcription termination at intrinsic terminators: The role of the RNA hairpin. Proc. Natl. Acad. Sci. USA 1995, 92, 8793–8797. [Google Scholar] [CrossRef] [PubMed]
- Artsimovitch, I.; Svetlov, V.; Anthony, L.; Burgess, R.R.; Landick, R. RNA Polymerases from Bacillus subtilis and Escherichia coli Differ in Recognition of Regulatory Signals In Vitro. J. Bacteriol. 2000, 182, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Mooney, R.A.; Artsimovitch, I.; Landick, R. Information processing by RNA polymerase: Recognition of regulatory signals during RNA chain elongation. J. Bacteriol. 1998, 180, 3265–3275. [Google Scholar] [PubMed]
- Greenleaf, W.J.; Frieda, K.L.; Foster, D.A.N.; Woodside, M.T.; Block, S.M. Direct Observation of Hierarchical Folding in Single Riboswitch Aptamers. Science 2008, 319, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Monforte, J.A.; Kahn, J.D.; Hearst, J.E. RNA folding during transcription by Escherichia coli RNA polymerase analyzed by RNA self-cleavage. Biochemistry 1990, 29, 7882–7890. [Google Scholar] [CrossRef] [PubMed]
- Komissarova, N.; Kashlev, M. Functional topography of nascent RNA in elongation intermediates of RNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95, 14699–14704. [Google Scholar] [CrossRef] [PubMed]
- Artsimovitch, I.; Landick, R. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl. Acad. Sci. USA 2000, 97, 7090–7095. [Google Scholar] [CrossRef] [PubMed]
- Tolić-Nørrelykke, S.F.; Engh, A.M.; Landick, R.; Gelles, J. Diversity in the rates of transcript elongation by single RNA polymerase molecules. J. Biol. Chem. 2004, 279, 3292–3299. [Google Scholar] [CrossRef] [PubMed]
- Grundy, F.J.; Henkin, T.M. The S box regulon: A new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria. Mol. Microbiol. 1998, 30, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.T. Recognition of S-adenosylmethionine by riboswitches. Wiley Interdiscip. Rev. RNA 2011, 2, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.T.; Grundy, F.J.; Henkin, T.M. S-adenosylmethionine directly inhibits binding of 30S ribosomal subunits to the SMK box translational riboswitch RNA. Proc. Natl. Acad. Sci. USA 2007, 104, 4876–4880. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Nahvi, A.; Roth, A.; Collins, J.A.; Breaker, R.R. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004, 428, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.T.; Moulin, M.; Webb, M.E.; Smith, A.G. Thiamine biosynthesis in algae is regulated by riboswitches. Proc. Natl. Acad. Sci. USA 2007, 104, 20770–20775. [Google Scholar] [CrossRef] [PubMed]
- Haller, A.; Altman, R.B.; Souliere, M.F.; Blanchard, S.C.; Micura, R. Folding and ligand recognition of the TPP riboswitch aptamer at single-molecule resolution. Proc. Natl. Acad. Sci. USA 2013, 110, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Cressina, E.; Chen, L.; Moulin, M.; Leeper, F.J.; Abell, C.; Smith, A.G. Identification of novel ligands for thiamine pyrophosphate (TPP) riboswitches. Biochem. Soc. Trans. 2011, 39, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Neupane, K.; Yu, H.; Foster, D.A.N.; Wang, F.; Woodside, M.T. Single-molecule force spectroscopy of the add adenine riboswitch relates folding to regulatory mechanism. Nucleic Acids Res. 2011, 39, 7677–7687. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The transcription and translation control mechanisms of riboswitches (a), where the aptamer sequence and Shine–Dalgarno (SD) sequence are colored brown and green respectively; L is the ligand. A schematic of the optical-trapping assay and the kinetic Monte Carlo simulation is shown in (b,c). In (c), nucleotides (green circles) out of the tube (yellow) are possible to form base pairs. The kinetic Monte Carlo method grows the RNA chain with a constant rate, allowing more and more base pairs to form or open in the available sequence.
Figure 1.
The transcription and translation control mechanisms of riboswitches (a), where the aptamer sequence and Shine–Dalgarno (SD) sequence are colored brown and green respectively; L is the ligand. A schematic of the optical-trapping assay and the kinetic Monte Carlo simulation is shown in (b,c). In (c), nucleotides (green circles) out of the tube (yellow) are possible to form base pairs. The kinetic Monte Carlo method grows the RNA chain with a constant rate, allowing more and more base pairs to form or open in the available sequence.
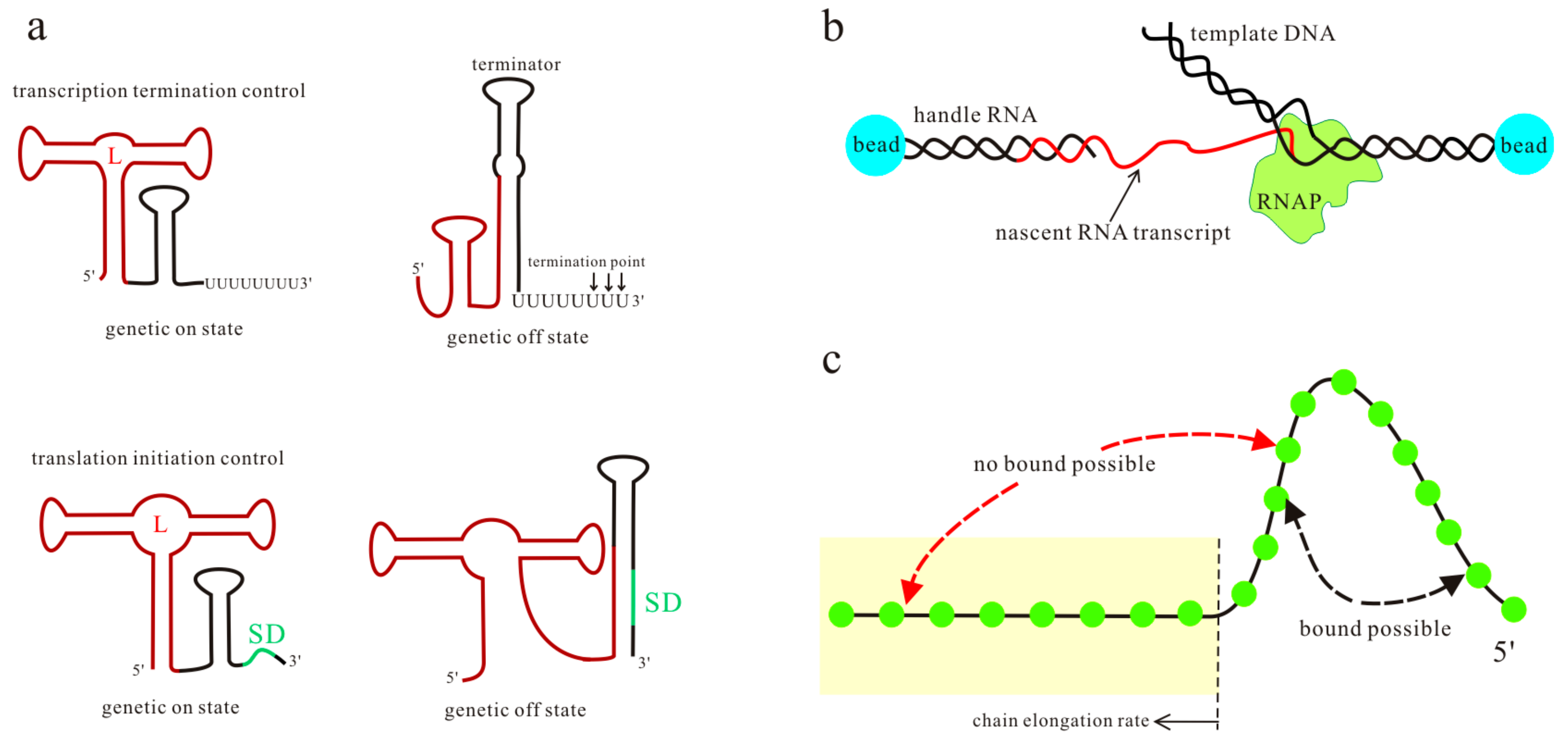
Figure 2.
The ligand-dependent co-transcriptional folding behaviors of the pbuE riboswitch. The population kinetics of main structures from step 60 under different transcription conditions are shown: 20 nt/s, 0 µM adenine (a); 20 nt/s, 100 µM adenine (b); 50 nt/s, 100 µM adenine (c); 50 nt/s, 100 µM adenine with pausing (60 s) at step 67 (d). The superscript “b” denotes the corresponding state with the ligand bound and Cb denotes the ensemble of all the bound states. Main co-transcriptional folding pathways and folding events are mapped in (e). T is the terminator hairpin; L is the ligand (adenine). Nucleotides within paired regions of the aptamer structure are colored differently.
Figure 2.
The ligand-dependent co-transcriptional folding behaviors of the pbuE riboswitch. The population kinetics of main structures from step 60 under different transcription conditions are shown: 20 nt/s, 0 µM adenine (a); 20 nt/s, 100 µM adenine (b); 50 nt/s, 100 µM adenine (c); 50 nt/s, 100 µM adenine with pausing (60 s) at step 67 (d). The superscript “b” denotes the corresponding state with the ligand bound and Cb denotes the ensemble of all the bound states. Main co-transcriptional folding pathways and folding events are mapped in (e). T is the terminator hairpin; L is the ligand (adenine). Nucleotides within paired regions of the aptamer structure are colored differently.
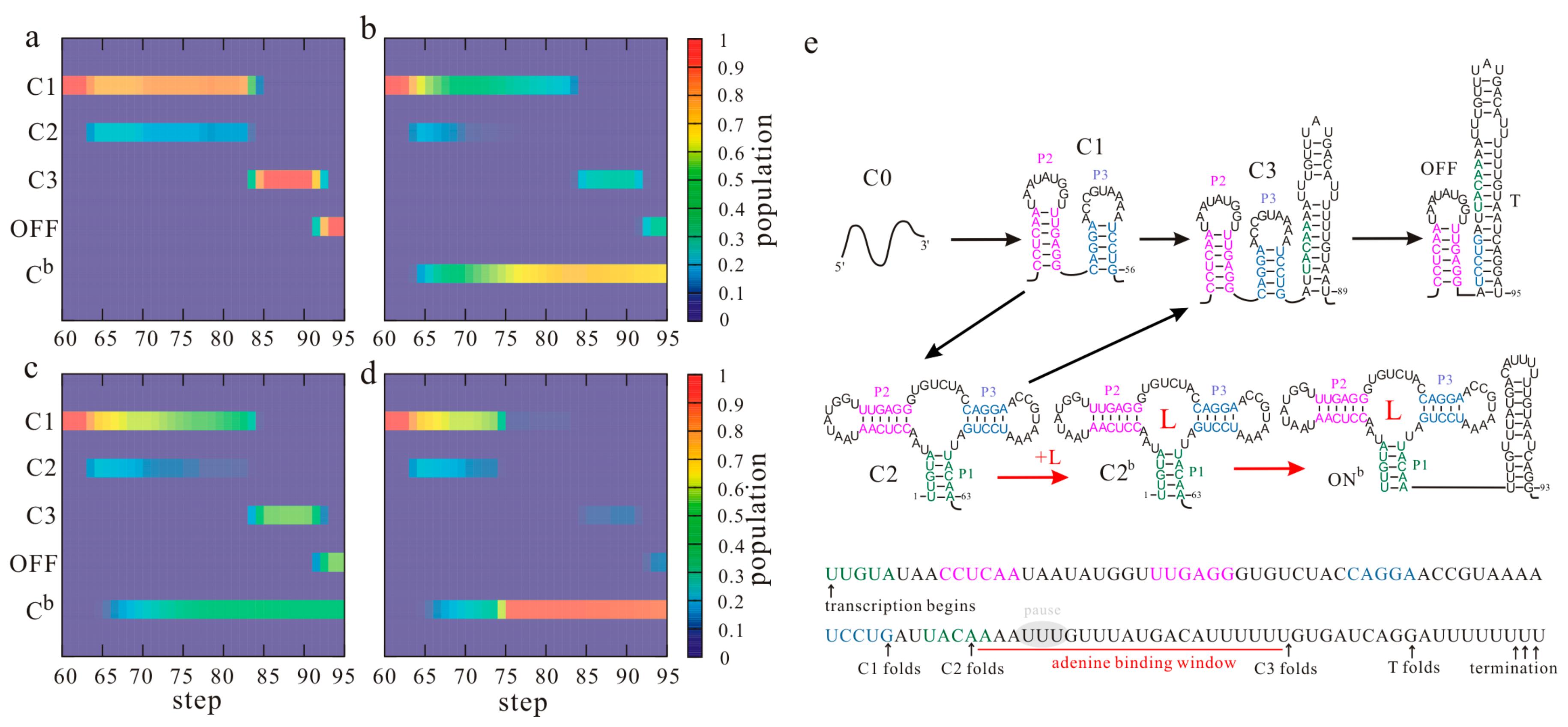
Figure 3.
The main co-transcriptional folding pathways of the metF S-box riboswitch. Helices P1, P2, P3, and P4 and the T hairpin are colored differently. AT is the anti-terminator hairpin; L is the ligand S-adenosylmethionine (SAM).
Figure 3.
The main co-transcriptional folding pathways of the metF S-box riboswitch. Helices P1, P2, P3, and P4 and the T hairpin are colored differently. AT is the anti-terminator hairpin; L is the ligand S-adenosylmethionine (SAM).

Figure 4.
The main co-transcriptional folding pathways of the yitJ S-box riboswitch. helices P1, P2, P3, P4 and the T hairpin are colored differently. L is the ligand SAM. The AU base pair depicted in the red box within OFFb state denotes the mutated base pair (GC).
Figure 4.
The main co-transcriptional folding pathways of the yitJ S-box riboswitch. helices P1, P2, P3, P4 and the T hairpin are colored differently. L is the ligand SAM. The AU base pair depicted in the red box within OFFb state denotes the mutated base pair (GC).
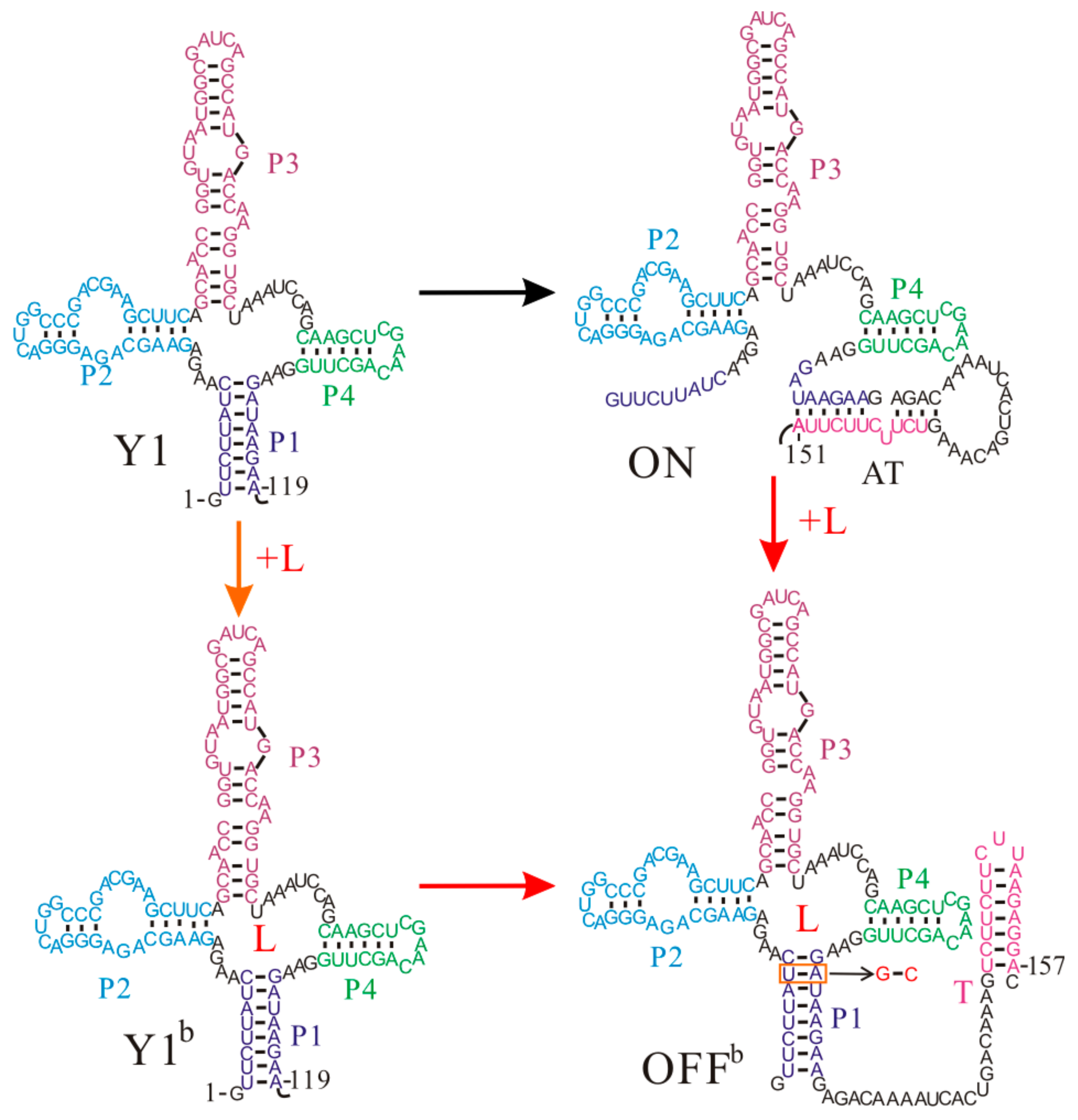
Figure 5.
The regulation mechanisms of the SMK riboswitch. L is the ligand SAM.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gong, S.; Wang, Y.; Wang, Z.; Zhang, W. Co-Transcriptional Folding and Regulation Mechanisms of Riboswitches. Molecules 2017, 22, 1169. https://doi.org/10.3390/molecules22071169
AMA Style
Gong S, Wang Y, Wang Z, Zhang W. Co-Transcriptional Folding and Regulation Mechanisms of Riboswitches. Molecules. 2017; 22(7):1169. https://doi.org/10.3390/molecules22071169
Chicago/Turabian StyleGong, Sha, Yanli Wang, Zhen Wang, and Wenbing Zhang. 2017. "Co-Transcriptional Folding and Regulation Mechanisms of Riboswitches" Molecules 22, no. 7: 1169. https://doi.org/10.3390/molecules22071169