
Dichlorotrifluoromethoxyacetic Acid: Preparation and Reactivity †

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
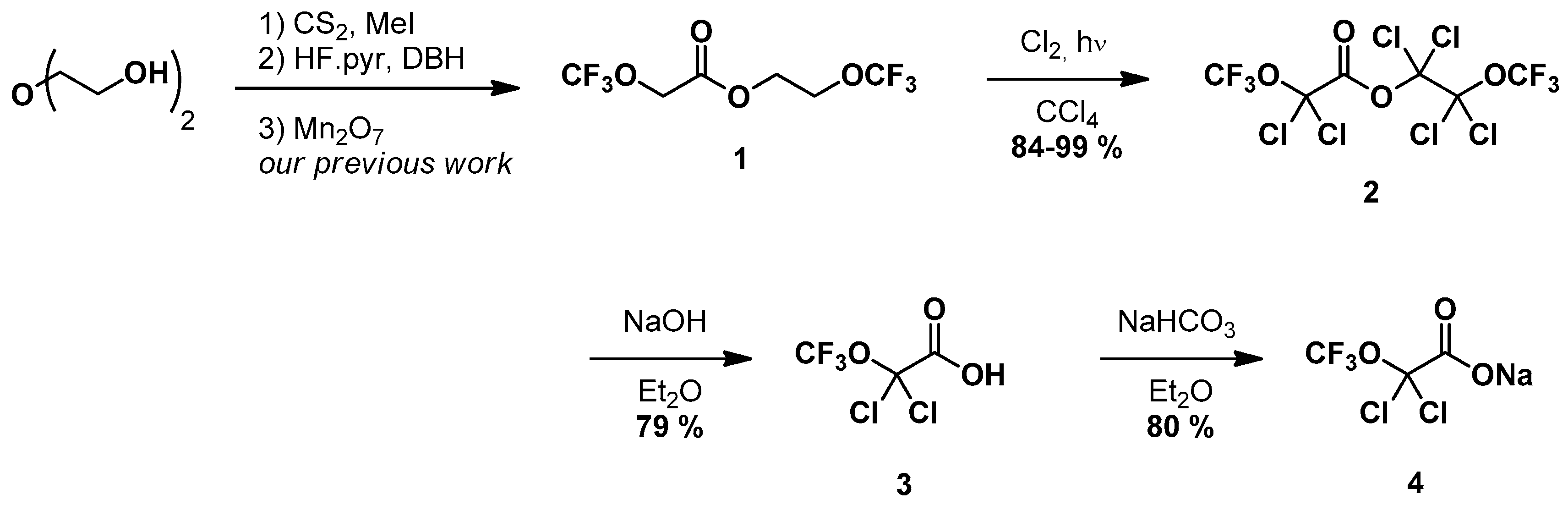
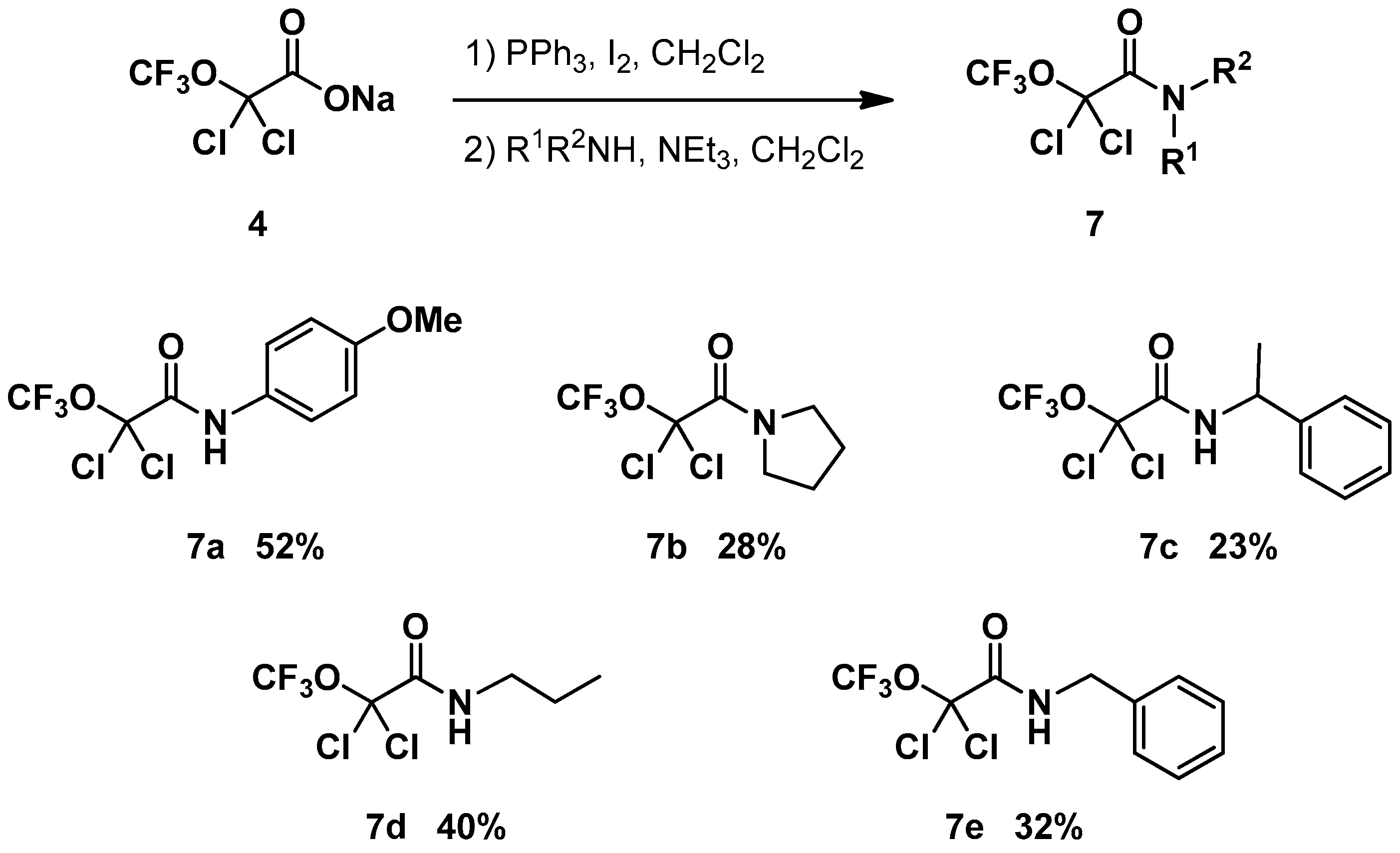
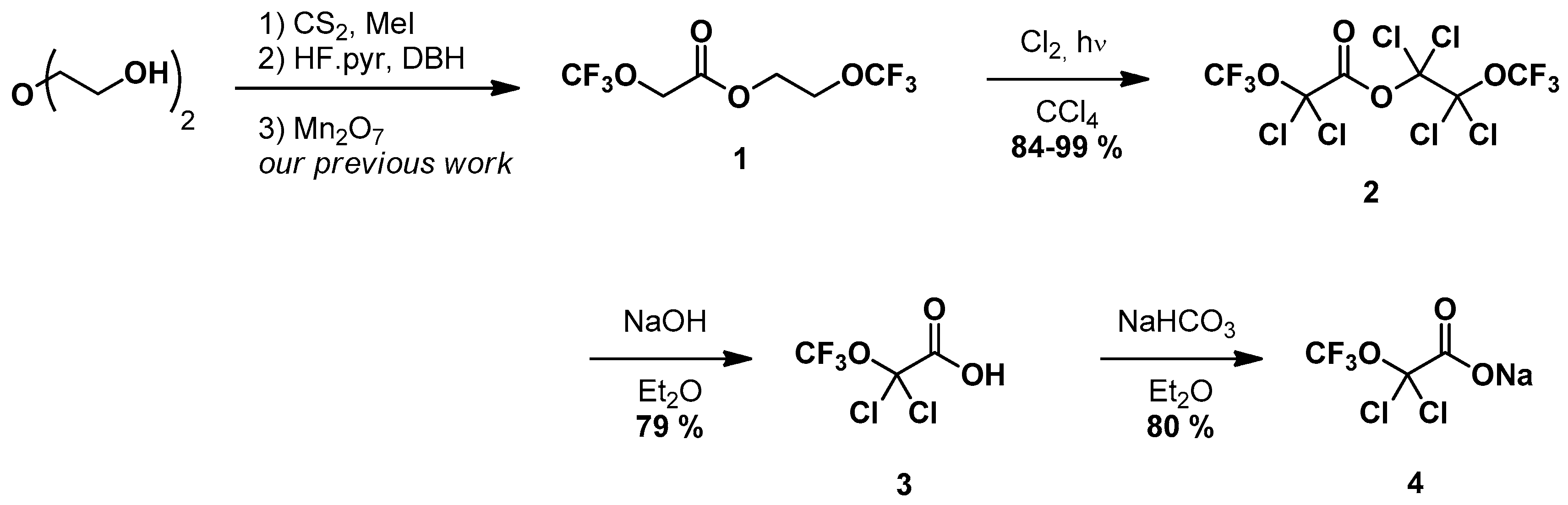
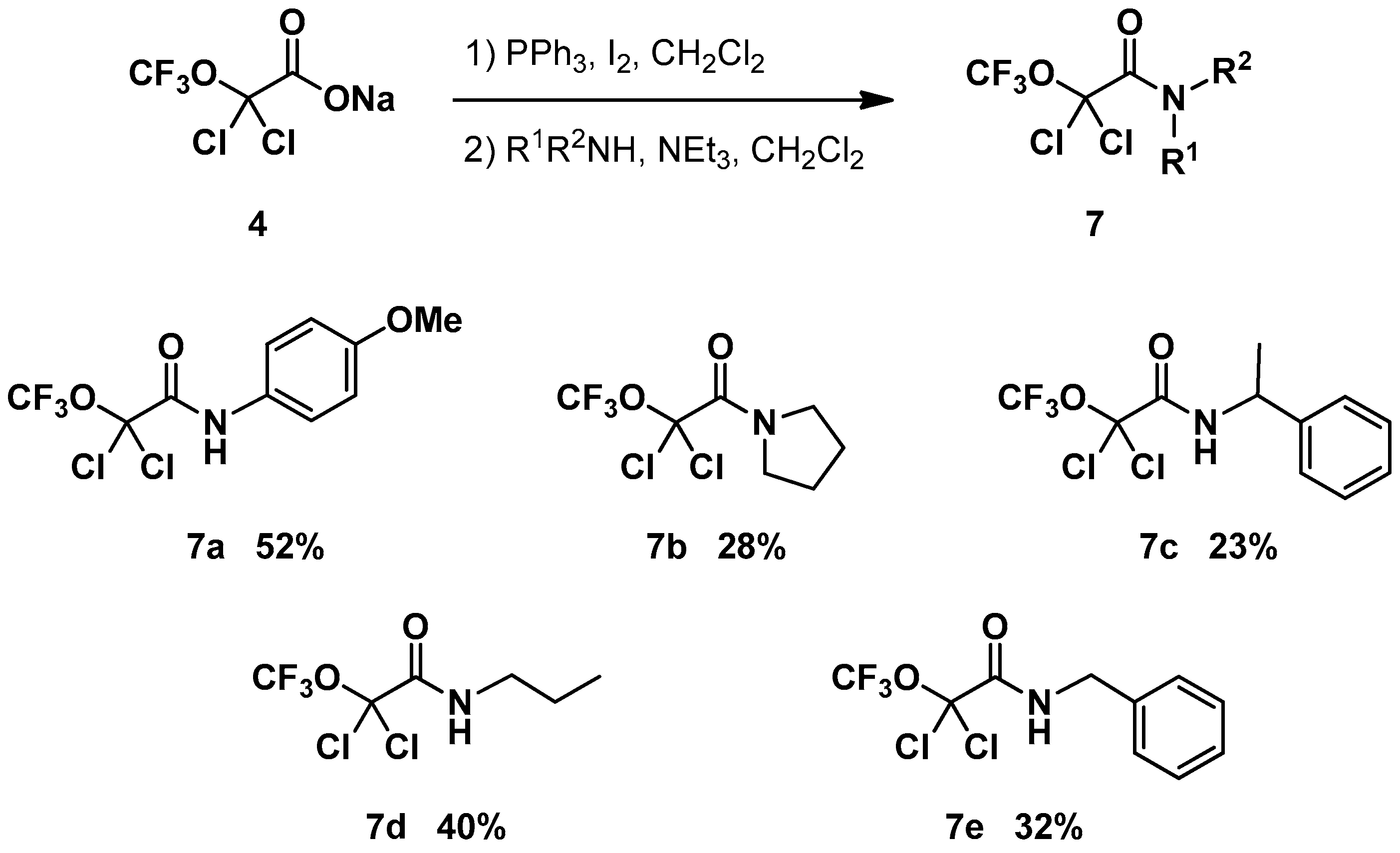
2. Results and Discussion
3. Materials and Methods
3.1. General Information
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yagupolskii, L.M. Synthesis of derivatives of phenyl trifluoromethyl ethers. Dokl. Acad. Nauk SSSR 1955, 105, 100–102. Chem. Abstr. 1956, 50, 11270b. [Google Scholar]
- Zeng, Z.; Gao, T.; Li, Y.; Wang, X.; Yang, X.; Wu, M. Synthesis and biological activity of arylsulfonamide derivatives containing 2-arylamino-4(3H)-quinazolinone. J. Pest. Sci. 2016, 41, 171–174. [Google Scholar] [CrossRef]
- Xu, D.; Guan, J.; Xu, X.; Gong, S.; Xu, H. A New Method for the Synthesis of Oxadiazine Insecticide Indoxacarb. J. Heterocycl. Chem. 2016, 63, 1469–1473. [Google Scholar] [CrossRef]
- Liu, A.; Wang, X.; Liu, X.; Li, J.; Chen, H.; Hu, L.; Yu, W.; He, L.; Liu, W.; Huang, M. Synthesis and Fungicidal Activity of Novel 2-Heteroatomthiazole-based Carboxanilides. J. Heterocycl. Chem. 2017, 54, 1625–1629. [Google Scholar] [CrossRef]
- Alverez, C.; Arkin, M.R.; Bulfer, S.L.; Colombo, R.; Kovaliov, M.; LaPorte, M.G.; Lim, C.; Liang, M.; Moore, W.J.; Neitz, R.J.; et al. Structure−Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS Med. Chem. Lett. 2015, 6, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, X.; Feng, C.; Liu, X.; Wang, H.; Feng, F.; Zhang, J. Proton-Coupled Organic Cation Antiporter Contributes to the Hepatic Uptake of Matrine. J. Pharm. Sci. 2016, 105, 1301–1306. [Google Scholar] [CrossRef]
- Yamamoto, S.; Ohta, H.; Abe, K.; Kambe, D.; Tsukiyama, N.; Kawakita, Y.; Moriya, M.; Yasuhara, A. Identification of 1-Methyl-N-(propan-2-yl)-N-({2-[4-(trifluoromethoxy)-phenyl]pyridin-4-yl}methyl)-1H-imidazole-4-carboxamide as a Potent and Orally Available Glycine Transporter 1 Inhibitor. Chem. Pharm. Bull. 2016, 64, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Satam, V.S.; Pedada, S.R.; Kamraj, P.; Antao, N.; Singh, A.; Hindupur, R.M.; Pati, H.N.; Thompson, A.M.; Launay, D.; Martin, D. Development of a Scalable Process for the Synthesis of DNDI-VL-2098: A Potential Preclinical Drug Candidate for the Treatment of Visceral Leishmaniasis. Org. Proc. Res. Dev. 2017, 21, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, Y.M.; Kim, R.Y.; Seo, M.J.; No, Z.; Nam, K.; Kim, S.; Kim, J. Synthesis and structure-activity studies of side chain analogues of the anti-tubercular agent, Q203. Eur. J. Med. Chem. 2017, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, P.E.; Sheppard, W. α-Fluorinated Ethers. II. Alkyl Fluoroalkyl Ethers. J. Org. Chem. 1964, 29, 11–15. [Google Scholar] [CrossRef]
- Haas, A. The Element Displacement Principle: A New Guide in P-Block Element Chemistry. Adv. Inorg. Chem. 1984, 28, 167–202. [Google Scholar]
- Olah, G.A.; Yamamoto, T.; Hashimoto, T.; Shih, J.G.; Trivedi, N.; Singh, B.P.; Piteau, M.; Olah, J.A. Aromatic substitution. 53. Electrophilic nitration, halogenation, acylation, and alkylation of α,α,α-trifluoromethoxybenzene. J. Am. Chem. Soc. 1987, 109, 3708–3713. [Google Scholar] [CrossRef]
- Kondratov, I.S.; Logvinenko, I.G.; Tolmachova, N.A.; Morev, R.N.; Kliachyna, M.A.; Clausen, F.; Daniliuc, C.G.; Haufe, G. Synthesis and physical chemical properties of 2-amino-4-(trifluoromethoxy)butanoic acid –CF3O-containing analogue of natural lipophilic amino acids. Org. Biomol. Chem. 2017, 15, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Lee, K.N.; Lee, J.W.; Zhen, C.; Ngai, M.-Y. Access to a New Class of Synthetic Building Blocks via Trifluoromethoxylation of Pyridines and Pyrimidines. Chem. Sci. 2016, 7, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Manteau, B.; Vors, J.-P.; Pazenok, S. Trifluoromethyl ethers-synthesis and properties of an unusual substituent. Beilstein J. Org. Chem. 2008, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Besset, T.; Jubault, P.; Pannecoucke, X.; Poisson, T. New entries toward the synthesis of OCF3-containing molecules. Org. Chem. Front. 2016, 3, 1004–1010. [Google Scholar] [CrossRef]
- Tlili, A.; Toulgouat, F.; Billard, T. Synthetic Approaches to Trifluoromethoxy-Substituted Compounds. Angew. Chem. Int. Ed. 2016, 55, 11726–11735. [Google Scholar] [CrossRef] [PubMed]
- Kolomeitsev, A.A.; Vorobyev, M.; Gillandt, A. Versatile application of trifluoromethyl triflate. Tetrahedron Lett. 2008, 49, 449–454. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.-P.; Pazenok, S.; Langlois, B. A New and Direct Trifluoromethoxylation of Aliphatic Substrates with 2,4-Dinitro(trifluoromethoxy)benzene. Adv. Synth. Catal. 2010, 352, 2831–2837. [Google Scholar] [CrossRef]
- Huang, C.; Liang, T.; Harada, S.; Lee, E.; Ritter, T. Silver-Mediated Trifluoromethoxylation of Aryl Stannanes and Arylboronic Acids. J. Am. Chem. Soc. 2011, 133, 13308–13310. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, T.M.; Davydova, Y.A.; Yagupolskii, Y.L. Efficient synthesis of 5′-fluoroalkoxythiazoles via α-bromo-α-fluoroalkoxyacetophenones Hantzsch type cyclization with thioureas or thioamides. J. Fluor. Chem. 2012, 136, 20–25. [Google Scholar] [CrossRef]
- Chen, C.; Chen, P.; Liu, G. Palladium-Catalyzed Intramolecular Aminotrifluoromethoxylation of Alkenes. J. Am. Chem. Soc. 2015, 137, 15648–15651. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Cong, F.; Guo, S.; Wang, L.; Tang, P. Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes with a new trifluoromethoxylation reagent. Nat. Chem. 2017, 9, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Fantasia, S.; Welch, J.M.; Togni, A. Reactivity of a Hypervalent Iodine Trifluoromethylating Reagent toward THF: Ring Opening and Formation of Trifluoromethyl Ethers. J. Org. Chem. 2010, 75, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-B.; Chen, C.; Chu, L.; Chen, Z.-H.; Xu, X.-H.; Qing, F.-L. Silver-Mediated Oxidative Trifluoromethylation of Phenols: Direct Synthesis of Aryl Trifluoromethyl Ethers. Angew. Chem. Int. Ed. 2015, 54, 11839–11842. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-B.; Xu, X.-H.; Qing, F.-L. Silver-Mediated Oxidative Trifluoromethylation of Alcohols to Alkyl Trifluoromethyl Ethers. Org. Lett. 2015, 17, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.; Han, S.; Liu, Z.; Wang, L.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Regioselective Synthesis of N-Heteroaromatic Trifluoromethoxy Compounds by Direct O−CF3 Bond Formation. Chem. Eur. J. 2016, 22, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ni, C.; He, Z.; Hu, J. O-Trifluoromethylation of Phenols Access to Aryl Trifluoromethyl Ethers by O-Carboxydifluoromethylation and Decarboxylative Fluorination. Org. Lett. 2016, 18, 3754–3757. [Google Scholar] [CrossRef] [PubMed]
- Blazejewski, J.-C.; Anselmi, E.; Wakselman, C. 2-Trifluoromethoxyethyl Triflate: A Versatile Trifluoromethoxyethyl Carrier. J. Org. Chem. 2001, 66, 1061–1063. [Google Scholar] [CrossRef]
- Blazejewski, J.-C.; Anselmi, E.; Wernicke, A.; Wakselman, C. Synthesis of 2-trifluoromethoxyethyl trifluoromethoxyacetate and derived 2-trifluoromethoxyacrylates. J. Fluorine Chem. 2002, 117, 161–166. [Google Scholar] [CrossRef]
- Magnier, E.; Diter, P.; Blazejewski, J.-C. The trifluoromethoxy group as a fluorine twin in the Diels-Alder reactions of halogenated quinones. Tetrahedron Lett. 2008, 49, 4575–4578. [Google Scholar]
- Zriba, R.; Magnier, E.; Blazejewski, J.-C. Preparation and reactivity of some new keto and styrene based trifluoromethoxylated synthons. Synlett 2009, 7, 1131–1135. [Google Scholar]
- Brahms, D.L.S.; Dailey, W.P. Fluorinated Carbenes. Chem. Rev. 1996, 96, 1585–1632. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.A.; Zhang, M.; Krogh-Jespersen, K. A New Synthesis of Difluorodiazirine and the Absolute Reactivity of Difluorocarbene. J. Am. Chem. Soc. 2009, 131, 2128–2130. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.A. “Carbon Dichloride”: Dihalocarbenes Sixty Years after Hine. J. Org. Chem. 2010, 75, 5773–5783. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.A.; Zhang, M. Activation Parameters for Additions of Ambiphilic Methoxychlorocarbene to Alkenes. Org. Lett. 2008, 10, 4045–4048. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.A.; Zhang, M.; Krogh-Jespersen, K. The Nucleophilic Reactivity of Fluoromethoxycarbene. Org. Lett. 2010, 12, 3476–3479. [Google Scholar] [CrossRef] [PubMed]
- Grygorenko, O.O.; Artamonov, O.S.; Komarov, I.V.; Mykhailiuk, P.K. Trifluoromethyl-substituted cyclopropanes. Tetrahedron 2011, 67, 803–823. [Google Scholar] [CrossRef]
- Only Claimed in the Following Patent: Scherer, O.; Millauer, H. Fluoro ethers. ZA 6804804, 18 December 1968. [Google Scholar]
- Bordeau, M.; Frébault, F.; Gobet, M.; Picard, J.-P. Ethyl Difluoro(trimethylsilyl)acetate and Difluoro(trimethylsilyl)acetamides-Precursors of 3,3-Difluoroazetidinones. Eur. J. Org. Chem. 2006, 4147–4154. [Google Scholar] [CrossRef]
Sample Availability: Not available |

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zriba, R.; Desmarchelier, A.; Cadoret, F.; Bouvet, S.; Barthelemy, A.-L.; Pégot, B.; Diter, P.; Dagousset, G.; Blazejewski, J.-C.; Anselmi, E.; et al. Dichlorotrifluoromethoxyacetic Acid: Preparation and Reactivity. Molecules 2017, 22, 966. https://doi.org/10.3390/molecules22060966
Zriba R, Desmarchelier A, Cadoret F, Bouvet S, Barthelemy A-L, Pégot B, Diter P, Dagousset G, Blazejewski J-C, Anselmi E, et al. Dichlorotrifluoromethoxyacetic Acid: Preparation and Reactivity. Molecules. 2017; 22(6):966. https://doi.org/10.3390/molecules22060966
Chicago/Turabian StyleZriba, Riadh, Alaric Desmarchelier, Frédéric Cadoret, Sébastien Bouvet, Anne-Laure Barthelemy, Bruce Pégot, Patrick Diter, Guillaume Dagousset, Jean-Claude Blazejewski, Elsa Anselmi, and et al. 2017. "Dichlorotrifluoromethoxyacetic Acid: Preparation and Reactivity" Molecules 22, no. 6: 966. https://doi.org/10.3390/molecules22060966