
Synthesis, Characterization and Protonation Behavior of Quinoxaline-Fused Porphycenes
by
 ,
,
Daiki Kuzuhara
*,† ,
,
Mika Sakaguchi
,
Wataru Furukawa
,
Takuya Okabe
,
Naoki Aratani
and
Hiroko Yamada
* Graduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama-cho, Ikoma 630-0192, Japan
*
Authors to whom correspondence should be addressed.
†
Current address: Faculty of Science and Engineering, Iwate University, 4-3-5 Ueda, Morioka 020-8551, Japan.
Molecules 2017, 22(6), 908; https://doi.org/10.3390/molecules22060908
Submission received: 13 April 2017
/
Revised: 15 May 2017
/
Accepted: 24 May 2017
/
Published: 31 May 2017
(This article belongs to the Special Issue Porphyrins and Phthalocyanines: Synthesis, Properties and Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:9,10-Quinoxaline-fused porphycenes 1a-H2 and 1b-H2 were synthesized by intramolecular McMurry coupling. As a result of the annulation of the quinoxaline moiety on the porphycene skeleton, 1a-H2 and 1b-H2 display absorption and fluorescence in the near infra-red (NIR) region. Additionally, the quinoxaline moieties of 1a-H2 and 1b-H2 act as electron-withdrawing groups, introducing lower reduction potentials than for pristine porphycene. The protonation occurred at the nitrogen atoms in the cavity of freebase porphycenes and at the quinoxaline moieties for their nickel complexes to give diprotonic species.
1. Introduction
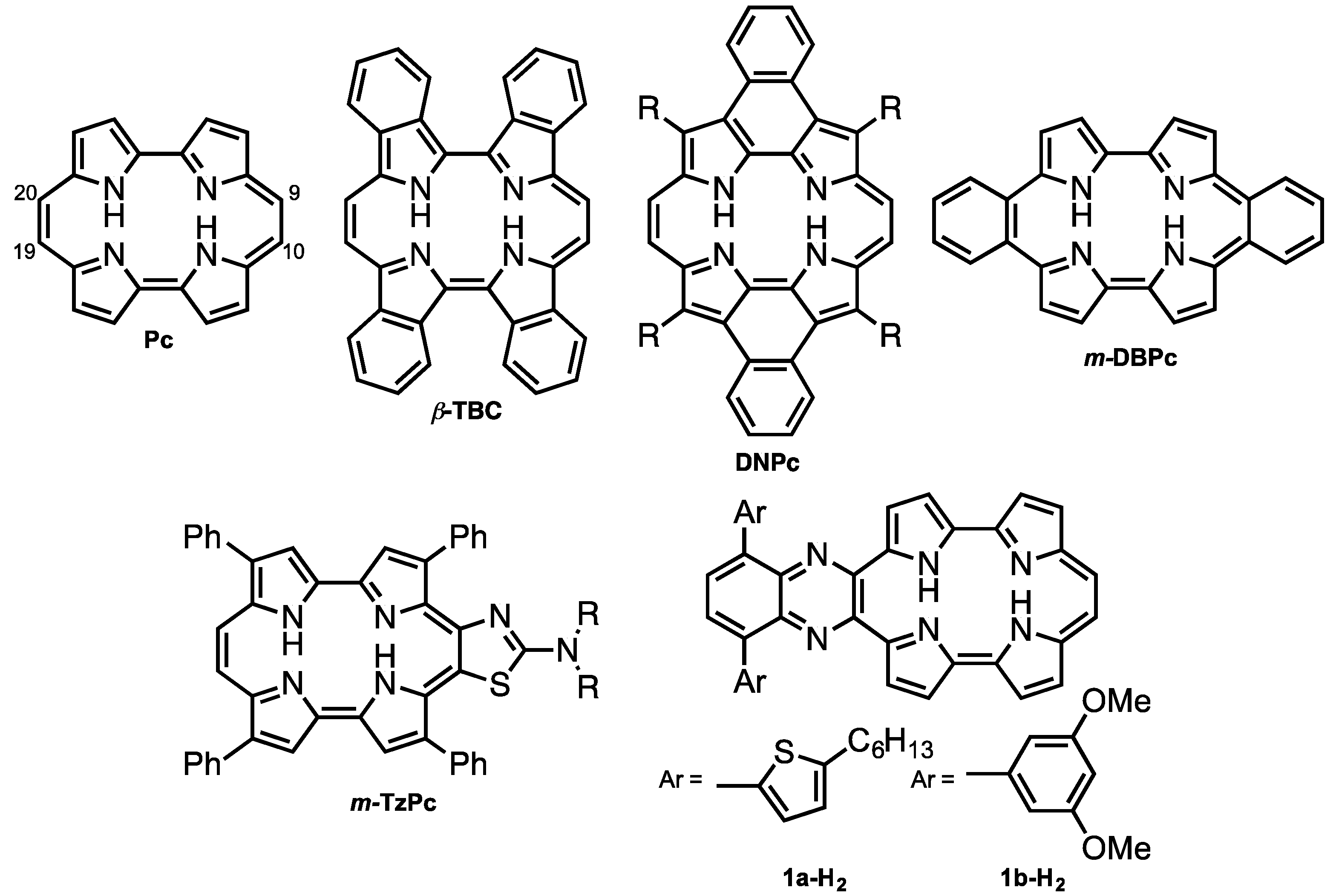
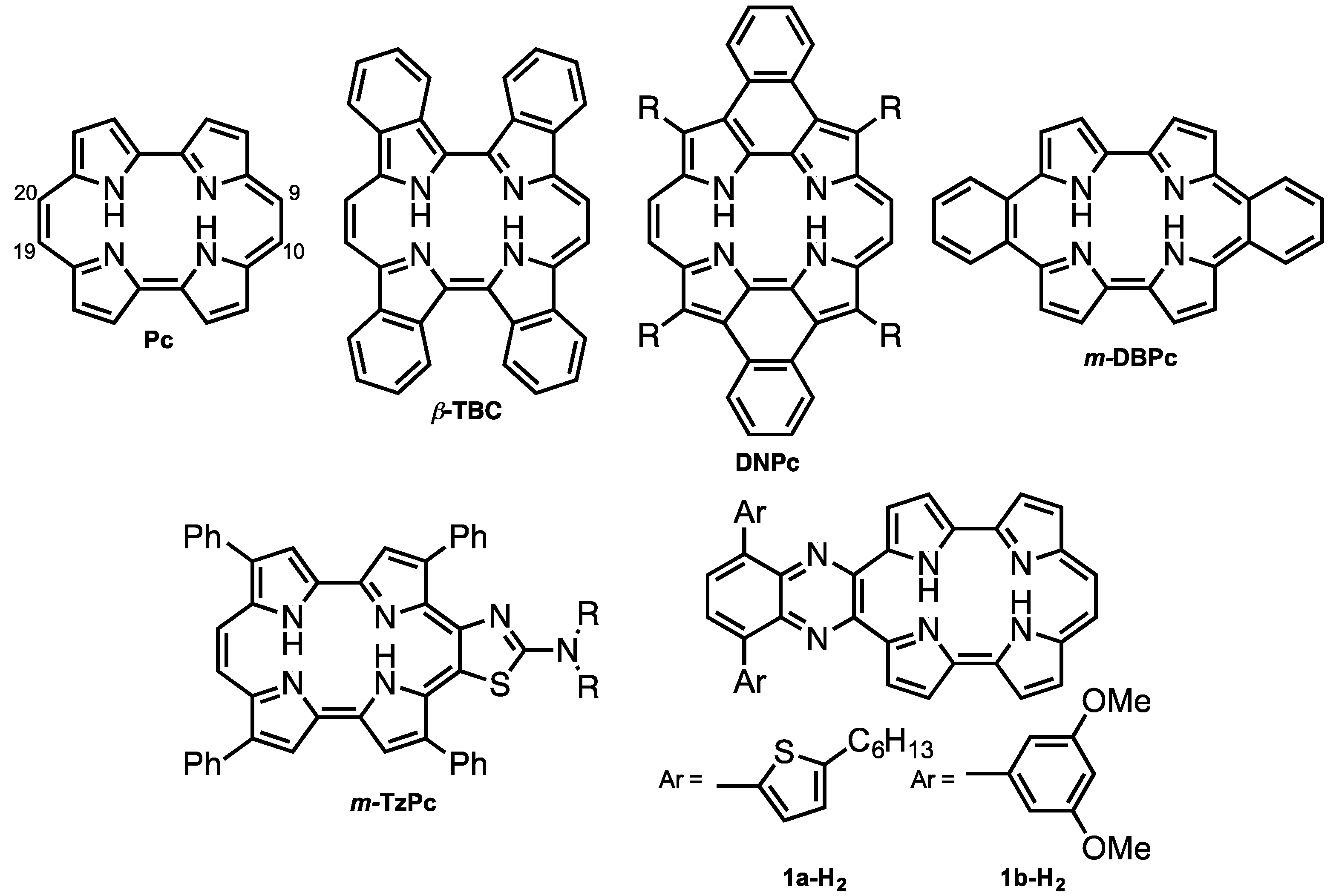
Porphycene (Pc < Figure 1) is the first reported constitutional isomer of porphyrin [1]. It has a rectangular-shaped structure with two bipyrrole units connected by two ethylene bridges. This structural change leads to a lower LUMO level and narrower HOMO-LUMO energy difference compared with those of porphyrin [2]. In addition, porphycene derivatives and their metal complexes are capable of generating singlet oxygen efficiently [3,4]. As a result of these electronic and optical properties, porphycene derivatives are applied as photosensitizers for photodynamic therapy [5,6] and non-linear optical materials [7,8,9,10,11]. However, there have been only a few reports on porphycenes having near infra-red (NIR) absorption, although NIR absorbing materials can be utilized in organic solar cells, biomarkers and photodynamic reagents. Benzoannulation has great potential to expand the π-conjugations and achieve NIR absorptions [12]. We have previously reported on tetrabenzo-porphycene (β-TBPc, Figure 1) [13] and tetranaphthoporphycene (β-TNPc) [14], which demonstrated red-shifted absorption from pristine Pc with an increasing number of benzene rings. Organic solar cells (OSC) based on β-TBPc as a p-type material and PC61BM as an n-type material exhibited 1.54% power-conversion efficiency [15,16,17]. Sessler and Panda independently reported dinaphthoporphycenes (DNPcs), in which absorptions reached around 800 nm [8,18]. Furthermore, the π-conjugation of porphycenes can be expanded by benzoannulation at meso-positions (9, 10, 19 and 20-positions, Figure 1). meso-Monobenzobenzoporphycene (m-MBPc) showed an absorption peak at 762 nm [19]. Recently, Hayashi reported meso-dibenzoporphycene (m-DBPc), displaying an extraordinarily red-shifted absorption peak at 1047 nm [20], indicating that benzoannulation at the meso-positions earns the largest perturbations to π-conjugation of porphycenes. In addition, Nonell et al. reported meso-thiazole-fused porphycenes (m-TzPc), which also showed NIR absorption and fluorescence [21]. In this context we have designed meso-quinoxaline-fused porphycene and its nickel complex to attain NIR absorption and fluorescence. Quinoxaline can be employed not only as an electron-withdrawing group to stabilize LUMO energy but also for the expansion of the π-conjugations. Moreover, since modification of the quinoxaline moiety could impact electronic properties and increase solubility, 5-hexylthienyl (a) and 3,5-dimethoxyphenyl (b) groups are introduced on the quinoxaline unit. Investigation of optical and electrochemical properties, X-ray diffraction analysis, and protonation characteristics are also described.
2. Results and Discussion
2.1. Synthesis and Characterization of Quinoxaline-Fused Porphycenes and Their Nickel Complexes
Scheme 1 shows the synthetic scheme of 1a-H2, 1b-H2 and their nickel complexes, 1a-Ni and 1b-Ni. The starting materials, 2a and 2b, were synthesized according to established procedures [22,23]. The bromination of 2a and 2b with N-bromosuccinimide (NBS) gave 3a and 3b in 98 and 96% yield, respectively, and then protection at the pyrrolic NH groups with tert-butoxycarbonyl (Boc) groups gave 4a and 4b in 96% and 89%, respectively. To synthesize the 2,2′-bipyrrole units, Suzuki-Miyaura coupling conditions were optimized. After screening coupling conditions, we found that 5a and 5b were obtained with Boc-protected pyrrole-2-boronic acid, Pd(OAc)2, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos) and K3PO4 in 1-butanol in 74% and 97%, respectively. The similar coupling conditions were reported by Buchwald [24]. After deprotection of the Boc groups of 5a and 5b under heating conditions in ethylene glycol, formylation by Vilsmeier-Haack reaction of 6a and 6b gave 7a and 7b, in 99% and 58%, respectively. Finally, an intramolecular McMurry coupling of 7a and 7b afforded the quinoxaline-fused porphycenes, 1a-H2 and 1b-H2, in 13% and 7%, respectively. The corresponding nickel complexes, 1a-Ni and 1b-Ni, were synthesized with Ni(OAc)2 in 1,2-dichlorobenzene in the presence of pyridine in 70% and 45%, respectively. These compounds were characterized by 1H-, 13C-NMR and mass spectroscopies and X-ray crystallography. The high-resolution mass spectroscopy detected parent ion peaks of 1a-H2 and 1b-H2 at m/z = 745.3147 (calcd. for C46H42N6S2: 745.3169 [M]+) and 685.4412 (calcd. for C40H35O4N5Na: 685.4426 [M + Na]+), respectively.
The single crystal X-ray structures of 2a and 2b are shown in Figure S1 in Supplementary Materials. The NH groups of pyrroles face nitrogen atoms in the quinoxaline moiety in the formation of the hydrogen bondings. The thiophenes and quinoxaline of 2a consist of relatively planar structures with twisted angles 12.35° and 10.38° facilitating nitrogen-sulfur interactions. On the other hand, 2b shows larger twisted angles (40.82° and 47.07°) between dimethoxybenzenes and quinoxaline.
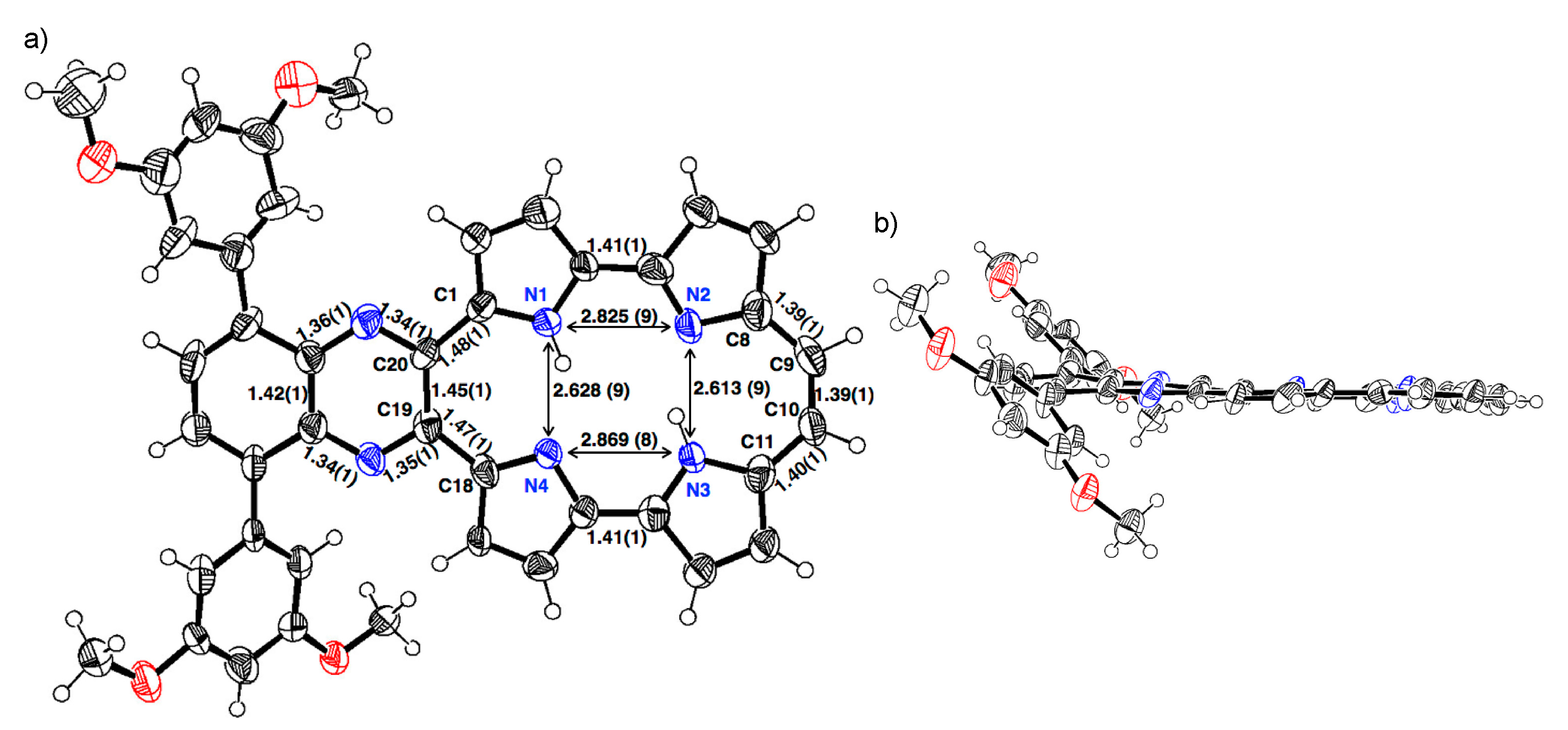
The single crystal of 1b-H2 was obtained from a mixture of THF/methanol/trifluoroacetic acid (TFA) (Figure 2). The porphycene core forms a highly planar structure in which a mean-plane deviation of 24 porphycene atoms is 0.046 Å. The porphycene and quinoxaline units are slightly tilted and the dihedral angle of each plane is 8.07°. The 3,5-dimethoxybenzene on quinoxaline moiety makes the angles of 45.10° and 45.38°, which are consistent with those of 2b. The distances of N1–N4 and N2–N3 are 2.628(9) and 2.613(9) Å, respectively, which values are comparable with that of Pc (2.63 Å) [1] and longer than those of m-MBPc (2.578 and 2.589 Å) and m-DBPc (2.54 Å) [20]. The distances of C8–C9 (1.39(1) Å) and C10–C11 (1.40(1) Å) are consistent with those of Pc (<1.40 Å) [1]. In contrast, the bond lengths of the opposite side, C1–C20 and C18–C19 exhibit slightly longer distances (1.48(1) and 1.47(1) Å).
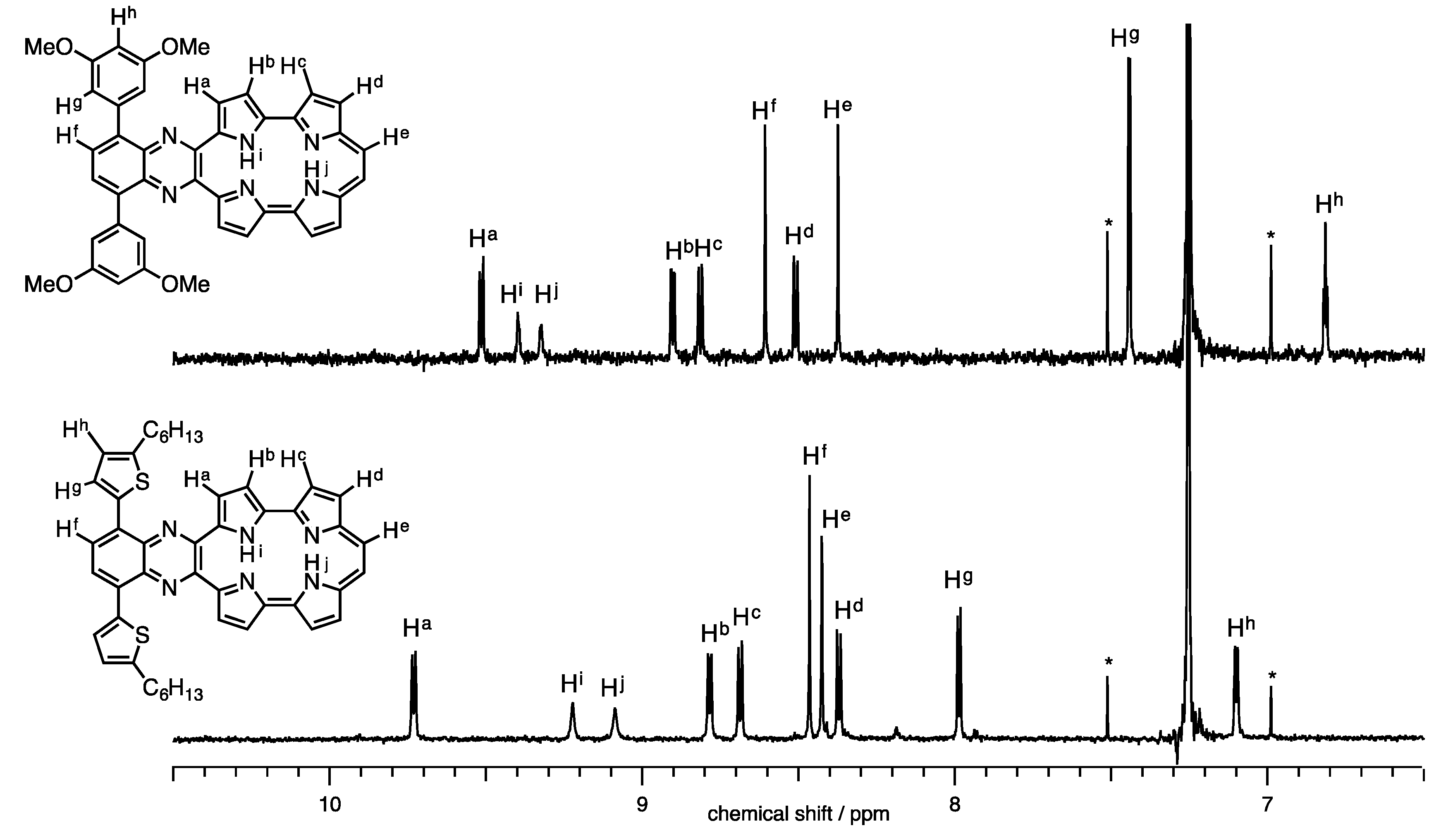
The 1H-NMR spectra of 1a-H2 and 1b-H2 in CDCl3 are shown in Figure 3. Full characterizations were carried out with H-H COSY and NOE techniques (Figures S2 and S3). The NH proton peaks were observed at 9.05 and 9.17 ppm for 1a-H2 and 9.32 and 9.40 ppm for 1b-H2. Generally, NH chemical shifts of porphycenes display in the range of 1 to 7 ppm depending on the substituents with change in cavity sizes and the effect of the hydrogen bond [20,25]. The NH chemical shifts of 1a-H2 and 1b-H2 remarkably downfield-shifted from the pristine Pc, while the distances of 1b-H2 (2.628(9) and 2.613(9) Å), which were obtained by X-ray diffraction, are comparable to Pc (2.63 Å). Notably, the chemical shift of the inner NH protons of porphyrinoids is also affected with macrocyclic diatropic ring currents. To interpret the aromaticity of 1a-H2 and 1b-H2, the nucleus-independent chemical shift (NICS(0)) values were calculated at the B3LYP/6-31G* level (Figure 4) [26]. The NICS(0) values of the midpoint made by the four nitrogen atoms are –8.71 ppm for 1a-H2, −8.57 ppm for 1b-H2 and −15.62 ppm for Pc. The result of the attachments of quinoxaline to porphycene induces the decrease in the aromaticities of 1a-H2 and 1b-H2, reflecting downfield shifts of the NH protons compared with the pristine Pc. In addition, the peaks of ethylenic protons of 1a-H2 and 1b-H2 were observed at 8.42 and 8.37 ppm, respectively. These peaks exhibit an upfield shift compared with the pristine Pc (δ = 9.67 ppm) [1]. This result also coincides with the NICS values.
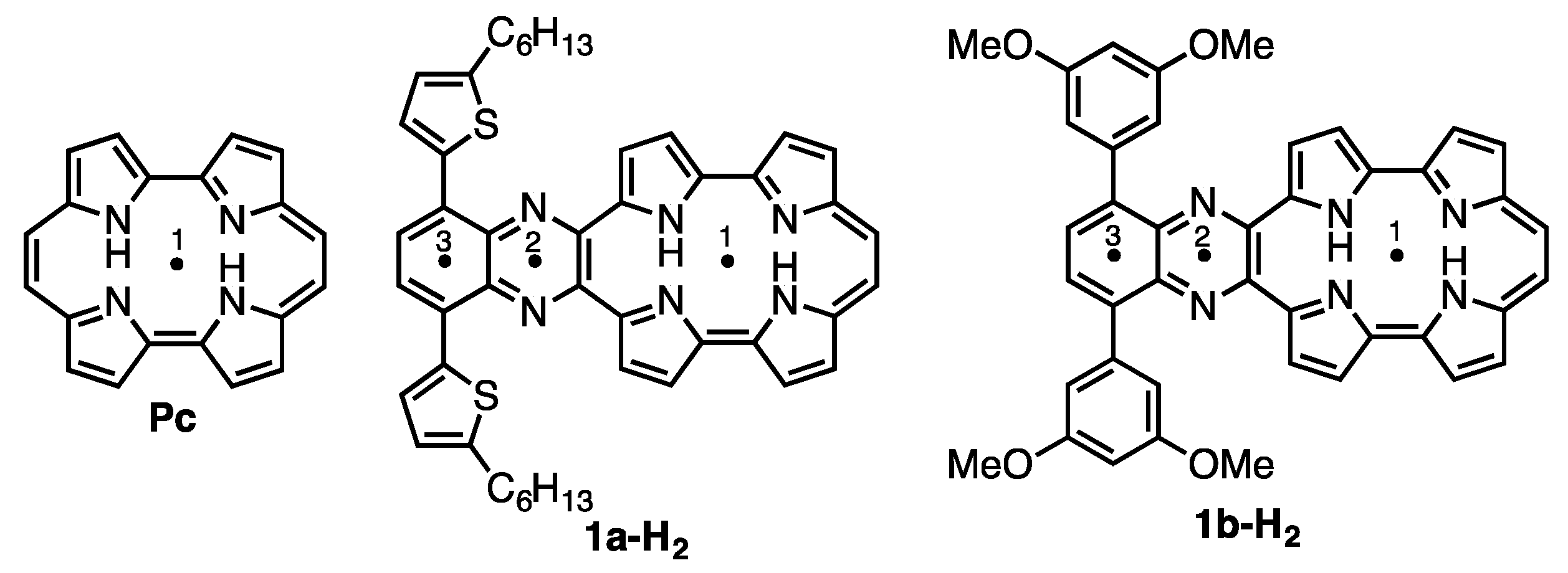
The absorption and fluorescence spectra of 1a-H2 and 1b-H2 were measured in CH2Cl2 (Figure 5). Typically, porphycene exhibits strong Soret bands around 370 nm and three Q-bands peak-tops at 550 to 660 nm. The 1a-H2 and 1b-H2 show significantly red-shifted absorption compared with Pc due to the expansion of the π-conjugation attached with quinoxaline moieties on the porphycene core. The Soret bands of 1a-H2 and 1b-H2 are observed at 407 and 397 nm, respectively, associated with four Q-bands peak-tops. The longest absorption peaks are detected at 814 nm for 1a-H2 and 781 nm for 1b-H2. Furthermore, the absorption of 1a-H2 shows red-shift compared with that of 1b-H2 since the coplanarity between thiophene and quinoxaline moiety is higher than that of the 3,5-dimethoxy-benzene of 1b-H2. This indicates that the optical property could be customized by the substitutions on the quinoxaline moiety. The compounds 1a-H2 and 1b-H2 show fluorescence at the NIR regions. The fluorescence peak tops of 1a-H2 and 1b-H2 are observed at 935 and 911 nm with quantum yields at 0.16% and 0.20%, as determined by the indocyanine green standards [27].
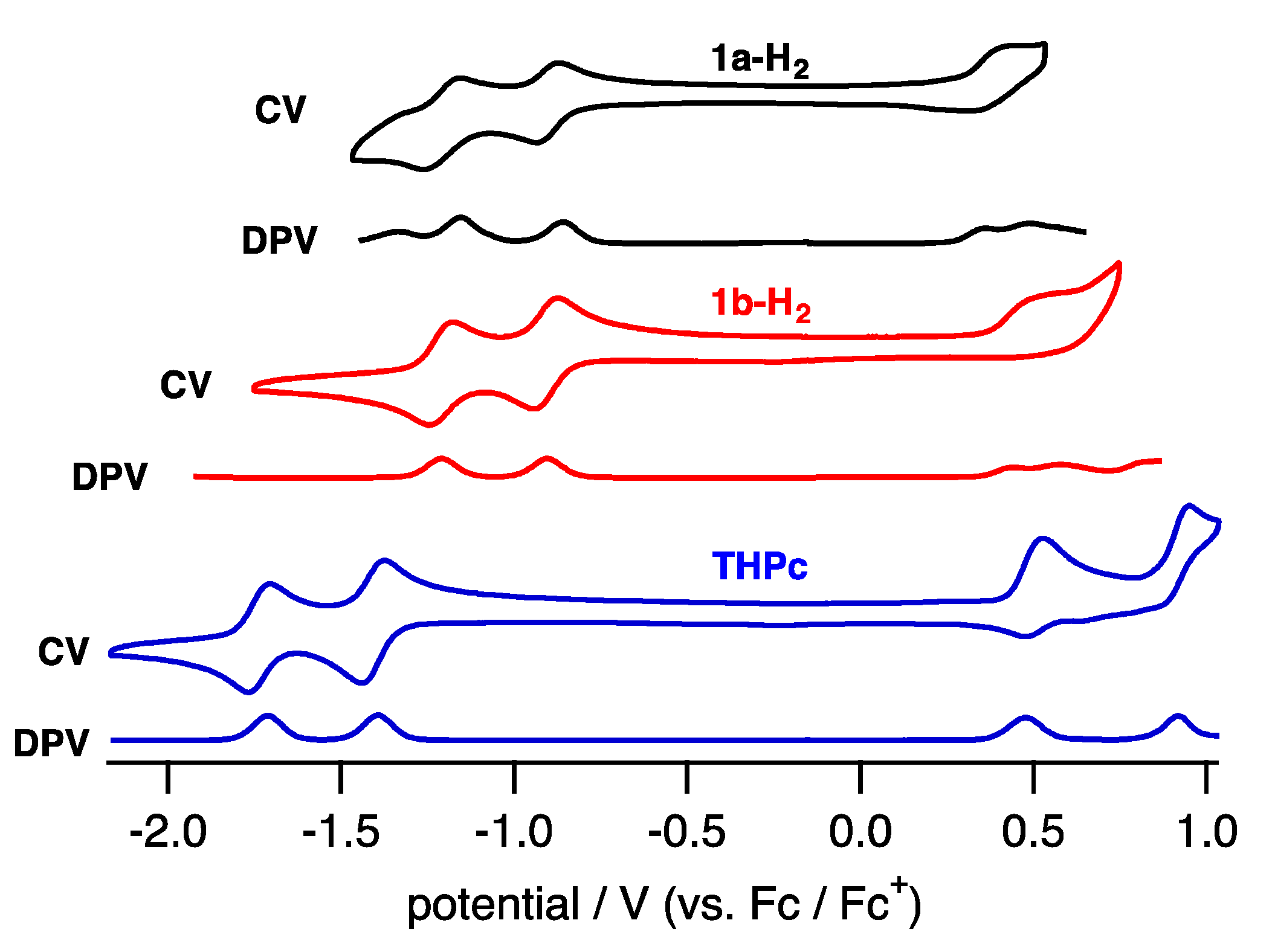
To elucidate the electronic properties, cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in CH2Cl2 containing tetrabutylammonium hexafluorophosphate (TBAPF6) as an electrolyte were performed (Figure 6). The compounds 1a-H2 and 1b-H2 exhibited two reversible reduction waves and one quasi-reversible oxidation wave.
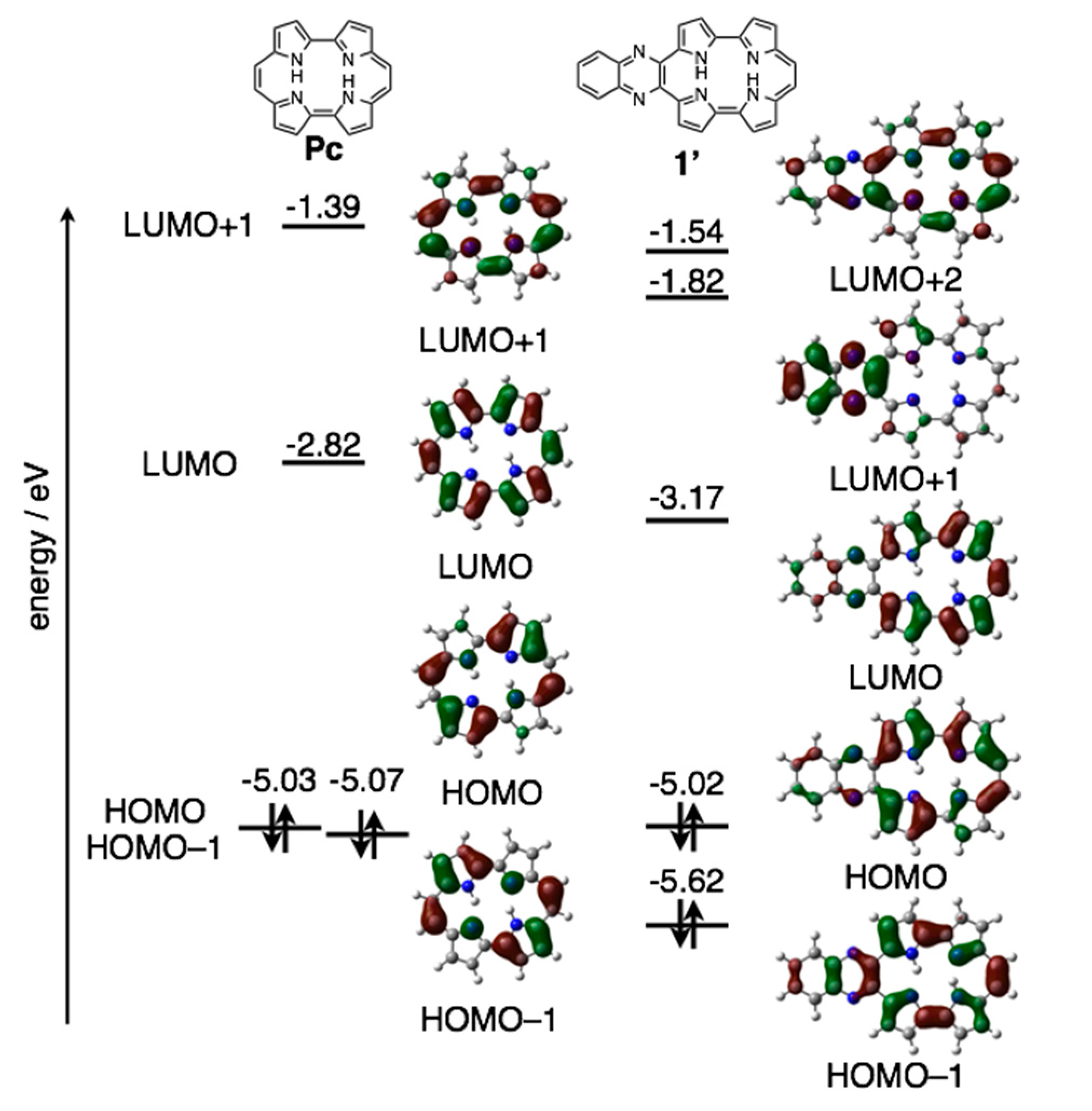
The reduction waves are observed at –0.85 and −1.16 V (vs. ferrocene/ferrocenium cation) for 1a-H2 and −0.93 and −1.23 V for 1b-H2, while oxidation waves are observed at 0.36 V for 1b-H2 and 0.44 V for 1b-H2. In particular, reduction potentials are positively shifted compared with 2,7,12,17-tetrahexylporphycene (−1.42 and −1.74 V) as a reference compound at the same conditions, affecting the attached quinoxaline as an electron-withdrawing group. To further understand the electronic features, DFT calculations were carried out at the B3LYP/6-31G* level using the Gaussian 09 program (Figure 7) [28]. Geometry optimization calculations were carried out using the structure of 1′ with the substituents on quinoxaline moiety removed for simplicity. The energy level of 1′ LUMO is largely stabilized from porphycene (ΔeV = −0.35 eV), while that of HOMO is comparable (ΔeV = −0.01 eV). These values are consistent with the CV results.
2.2. Protonation of Quinoxaline-Fused Porphycenes and Their Nickel Complexes
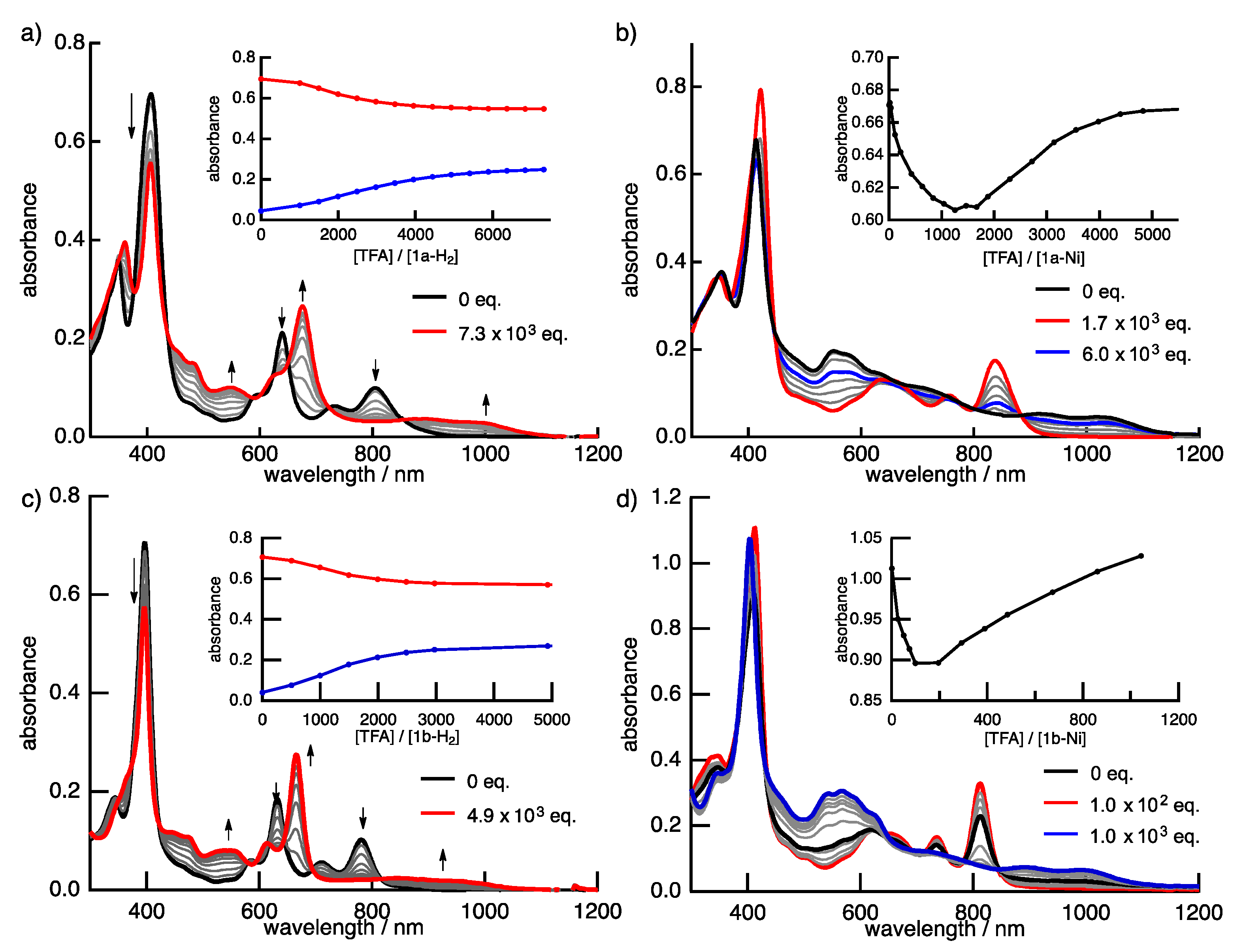
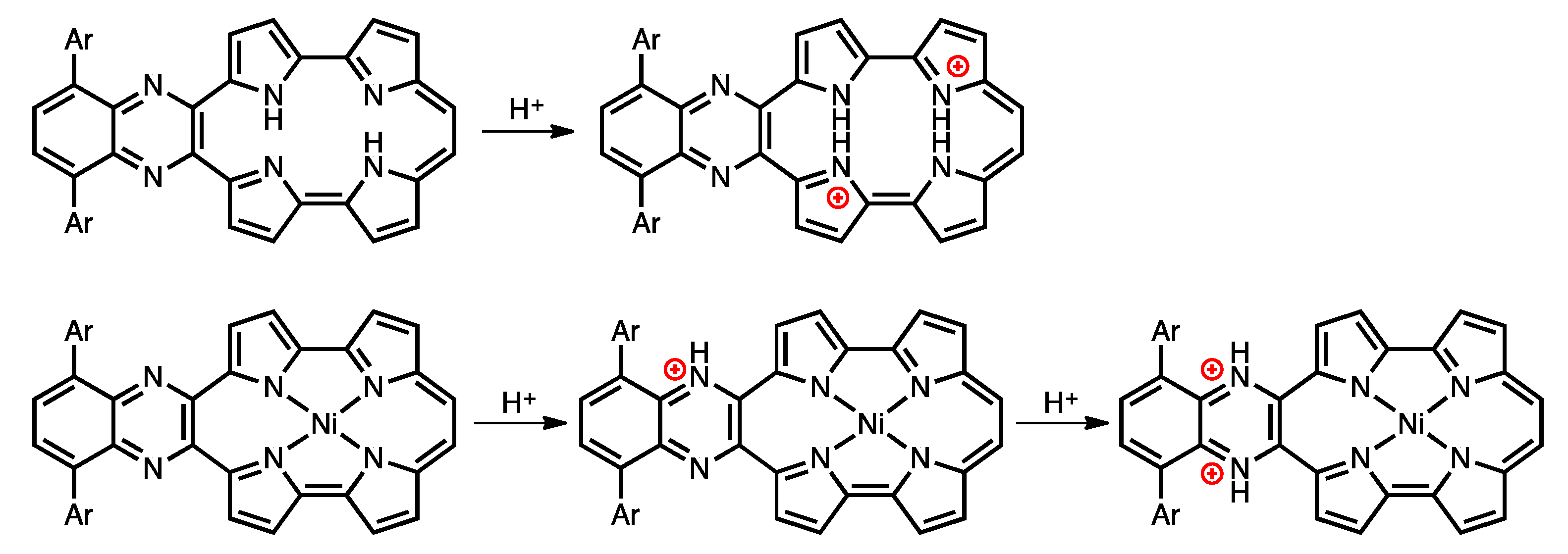
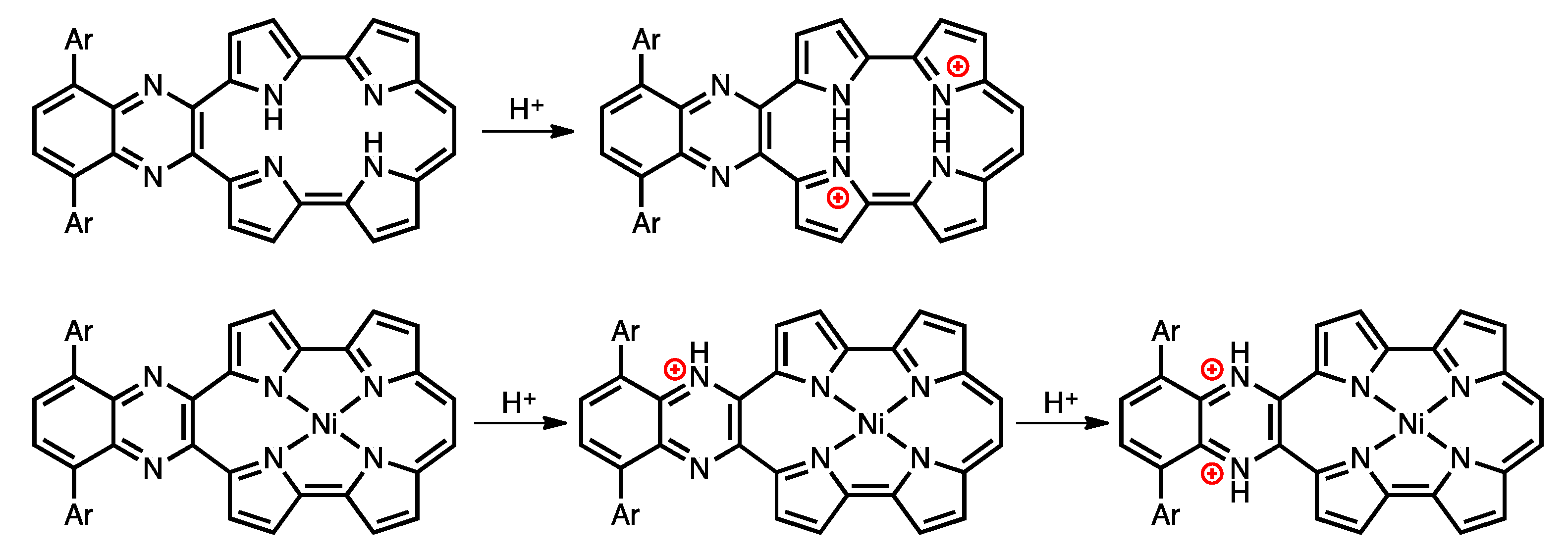
Protonation is an important phenomenon in porphyrin and related compounds because of its influence on optical and electronic properties [29]. Fukuzumi reported the protonation of distorted octaphenylphthalocyanine and its zinc complex [30]. Interestingly, they reported different phenomena: the freebase octaphenylphthalocyanine was protonated on nitrogen atoms on the inside of the cavity, while the zinc complex was protonated on the meso-nitrogen atoms, depending on the basicity. Furthermore, the protonation behavior of porphycene is known to be different from that of porphyrin. The protonation of porphyrin proceeds step-by-step, but the protonation of porphycene gives a porphycene dication immediately. This is because the monoprotonated porphycene is unstable due to the steric repulsion between the neighboring hydrogen atoms and the loss of the hydrogen bonding between NH and N [31,32]. Encouraged by these results, the protonation of 1a-H2, 1b-H2 and their nickel complexes was investigated, since the compounds have two different imine-type nitrogen atoms at the cavity and quinoxaline parts. Firstly, protonation was performed for the reference compounds, THPc and its nickel complex (THPc-Ni). THPc showed single absorption change according to the increase in the amount of TFA, while THPc-Ni exhibited no spectral changes after the addition of 2120 eq. of TFA because of the absence of protonation sites. (Figures S4 and S5) The changes in the 1a-H2 and 1a-Ni absorption spectra by the TFA titrations are shown in Figure 8. Upon addition of TFA to a solution of 1a-H2 in CH2Cl2, the Soret band at 397 nm decreases simultaneously with the observed Q-like bands at 656 nm and broadened peak over 1000 nm. The isosbestic points are observed at 437, 649, 721 and 853 nm. After the addition of 7300 equivalents of TFA, there are no further changes. In contrast, when TFA is added to a solution of 1a-Ni, a two–step spectral change is observed. The first spectral change is observed until the addition of 1700 eq. TFA. The Soret band peak at 422 nm decreases and shifts to 419 nm associated with the generation of the broad band around 1100 nm. Subsequently, the second spectral change occurs with the increase in the amount of TFA (1700 eq. to 6000 eq.). The peak at 419 nm is shifted to 412 nm, which slightly increases intensity. The compounds 1b-H2 and 1b-Ni exhibited similar results (Figure 8). These findings suggest that the protonation of the freebase 1a-H2 and 1b-H2 provide diprotonated porphycene species because these trends correspond to general freebase porphycenes [31,32], while the protonation of nickel complexes give diprotonated quinoxaline species as stepwise reactions (Scheme 2).
3. Materials and Methods
3.1. General Information
Commercially available reagents and solvents for syntheses were of reagent grade and used without further purification. TLC and gravity column chromatography were performed on Art. 5554 (Merck KGaA, Darmstadt, Germany) plates and silica gel 60N (Kanto Chemical, Chuo, Japan), respectively. For spectral measurements, spectral-grade dichloromethane was purchased from Nacalai Tesque. 1H- and 13C-NMR spectra were recorded with a JNM-ECX 400 spectrometer JEOL Akishima, Japan) at ambient temperature using tetramethylsilane (TMS) as an internal standard. Electrospray ionization (ESI) mass spectra were recorded on a JEOL JMS-MS T100LC spectrometer. Matrix assisted laser desorption/ionization (MALDI)-TOF-MS spectra were recorded on a JEOL SpiralTOF JMS-S3000 spectrometer (JEOL Akishima, Japan). FAB mass spectra were measured on a JEOL JMS-700 MStation spectrometer. UV-vis spectra were measured using a V-670 UV/VIS/NIR Spectro-photometer (JASCO, Hachioji, Japan). The fluorescence spectra were measured with a HORIBA (Kyoto, Japan) Jobin Yvon Spectrofluorometer Fluorolog-3 (Model: FL3-11-NIR).
3.2. X-ray Crystallography
X-ray crystallographic data for 2a and 2b were recorded at 90 K on an APEX II X-ray diffractometer (Bruker, Billerica, MA, USA) equipped with a large area CCD detector and graphite monochromated Mo-Kα radiation and the data for 1b-H2 and 1a-H2 were recorded at 103 K on a R-AXIS RAPID/S instrument (Rigaku, Akishima, Japan) using Mo-Kα radiation. The structure was solved by direct methods with SHELXS-97 [33] and refined by full-matrix least-squares techniques against F2 (SHELXL-97) [33]. The intensities were corrected for Lorentz and polarization effects. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. All calculations were performed using Crystal Structure 4.0 or Yadokari-XG 2011. Crystallographic data for 1b-H2: C42H32N6O4, Mw = 684.74, orthorhombic, space group Pna21 (#33), a = 28.452, b = 30.414, c = 3.827 Å, V = 3311.3 Å3, T = 103(2) K, Z = 4, reflections measured 39,848, 5127 unique. The final R1 was 0.0880 (I > 2σ(I)), and the final wR on F2 was 0.2299 (all data), GOF = 1.019. CCDC No. 1543213 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
3.3. Cyclic Voltammetry and Differential Pulse Voltammetry Measurements
CV and DPV measurements were examined in a solution of 0.1 M TBAPF6 in dry CH2Cl2 with a scan rate of 100 mV s−1 at room temperature in an argon-flowed cell. A glassy carbon electrode and platinum wire were used as a working and counter electrode, respectively. An Ag/AgNO3 electrode was used as a reference electrode, which was externally calibrated with the half-wave potential of ferrocene/ferrocenium redox couple.
3.4. DFT Calculations
Geometries were fully optimized using the density functional theory (DFT). The functional and basis set used in the DFT calculations were the Becke’s three-parameter hybrid functional combined with the Lee–Yang–Parr correlation functional (B3LYP) and the 6-31G(d) basis set, respectively. Equilibrium geometries were verified via frequency calculations, where no imaginary frequency was found. The excitation energies and oscillator strengths were computed with time-dependent density functional theory (TD-DFT) based on the B3LYP/6-31G(d) optimized geometries. All the calculations were carried out using the Gaussian 09 program suite [28].
3.5. Synthesis of 5,8-Bis(5-hexylthiophen-2-yl)-2,3-di(1H-pyrrol-2-yl)quinoxaline (2a)
5,8-Dibromo-2,3-di(1H-2-pyrrolyl)quinoxaline [34] (300 mg, 0.72 mmol), tributyl(5-hexylthiophen-2-yl)stannane (1.27 g, 2.8 mmol) and Pd(PPh3)4 (80 mg, 0.07 mmol) were dissolved in degassed toluene (15 mL) under an argon atmosphere. The resulting solution was refluxed for 20 h. After cooling to room temperature, the solvent was removed under a reduced pressure. The crude product was purified by silica gel column chromatography (CH2Cl2/hexanes = 1/1) to give 2a as an orange solid. Yield: 80% (340 mg, 0.57 mmol). 1H-NMR (DMSO-d6, 400 MHz): δ 10.94 (2H, brs, NH), 8.04 (2H, s), 7.77 (2H, d, J = 3.7 Hz), 7.08 (2H, m), 6.89 (2H, d, J = 3.7 Hz), 6.39 (2H, m), 6.19 (2H, m), 2.85 (4H, t, J = 7.8 Hz), 1.66 (4H, quin., J = 7.8 Hz), 1.38–1.26 (12H, m), 0.84 (6H, t, J = 7.8 Hz); 13C-NMR (CDCl3, 100 MHz): δ 148.78, 141.39, 136.15, 135.54, 130.02, 129.70, 125.64, 125.58, 123.91, 121.72, 113.44, 110.08, 32.80, 31.77, 30.31, 29.02, 22.73, 14.25; HRMS (ESI): m/z 615.2592 (calcd. for C36H40N4NaS2 [M + Na]+, 615.2593).
3.6. Synthesis of 2,3-Bis(5-bromo-1H-pyrrol-2-yl)-5,8-bis(5-hexylthiophen-2-yl)quinoxaline (3a)
NBS (180 mg, 1.0 mmol) was added to a solution of 2a (300 mg, 0.50 mmol) in CHCl3 (30 mL) under an argon atmosphere. The solution was stirred for 3 h at room temperature under dark condition. The reaction was quenched with water and extracted with CHCl3. The combined organic layer was washed with brine and dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was purified by silica gel column chromatography (CH2Cl2). Recrystallization from CH2Cl2/methanol gave 3a as a yellow solid. Yield: 98% (350 mg, 0.47 mmol). 1H-NMR (CDCl3, 400 MHz): δ = 10.04 (2H, brs, NH), 7.88 (2H, s), 7.46 (2H, d, J = 3.7 Hz), 7.18 (2H, dd, J = 4.1 and 2.7 Hz), 6.85 (2H, d, J = 3.7 Hz), 6.24 (2H, dd, J = 4.1 and 2.7 Hz), 2.93 (4H, t, J = 7.8 Hz), 1.84 (4H, quin., J = 7.8 Hz), 1.49–1.31 (12H, m), 0.90 (6H, t, J = 7.8 Hz); 13C-NMR (CDCl3, 100 MHz): δ 149.26, 139.69, 135.49, 135.20, 130.79, 129.72, 125.49, 125.39, 123.80, 114.47, 112.10, 103.63, 31.88, 31.75, 30.44, 29.19, 22.76, 14.24; HRMS (ESI): m/z 771.0802 (calcd. for C36H38Br2N4NaS2 [M + Na]+, 771.0807).
3.7. Synthesis of Di-tert-butyl 5,5′-(5,8-bis(5-hexylthiophen-2-yl)quinoxaline-2,3-diyl) bis(2-bromo-1H-pyrrole-1-carboxylate) (4a)
A solution of 3a (150 mg, 0.22 mmol), DMAP (2 mg, 0.02 mmol) and Boc2O (105 mg, 0.48 mmol) in CH2Cl2 (10 mL) was stirred for 5 h at room temperature under an argon atmosphere. After removal of solvent under a reduced pressure, the crude product was purified by silica gel column chromatography (CH2Cl2/hexanes = 1/1) to give 4a as an orange solid. Yield: 96% (184 mg, 0.19 mmol). 1H-NMR (CDCl3, 400 MHz): δ 7.97 (2H, s), 7.67 (2H, d, J = 3.7 Hz), 6.81 (2H, d, J = 3.7 Hz), 6.32 (2H, d, J = 3.7 Hz), 6.26 (2H, d, J = 3.7 Hz), 2.84 (4H, t, J = 7.3 Hz), 1.72 (4H, quin., J = 7.8 Hz), 1.42–1.30 (12H, m), 1.16 (18H, s), 0.89 (6H, t, J = 7.8 Hz); 13C-NMR (CDCl3, 100 MHz): δ 148.67, 147.75, 144.70, 136.90, 136.40, 133.25, 131.08, 127.48, 127.22, 124.47, 116.30, 115.42, 104.33, 85.36, 31.73, 31.65, 30.30, 29.01, 27.49, 22.73, 14.25; HRMS (FAB): m/z 948.1953 (calcd. for C46H54O4Br2N4S2 [M]+, 948.1948).
3.8. Synthesis of Tetra-tert-butyl 5,5″-(5,8-bis(5-hexylthiophen-2-yl)quinoxaline-2,3-diyl) bis(1H,1′H-[2,2′-bipyrrole]-1,1′-dicarboxylate) (5a)
A degassed solution of (1-(tert-butoxycarbonyl)-1H-pyrrol-2-yl)boronic acid [26] (19 mg, 0.09 mmol), 4a (30 mg, 0.031 mmol), Pd(OAc)2 (0.3 mg, 0.001 mmol), SPhos (0.8 mg, 0.002 mmol), K3PO4 (25.5 mg, 0.12 mmol) in water (0.8 mL) and in 1-butanol (2.0 mL) was heated at 80 °C for 20 h under an argon atmosphere. After cooling to room temperature, the reaction mixture was extracted with CHCl3. The combined organic layer was washed with brine and dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was purified by silica gel column chromatography (CH2Cl2/hexanes = 1/1) to give 5a as an orange solid. Yield: 74% (24 mg, 0.02 mmol). 1H-NMR (CDCl3, 400 MHz): δ 7.95 (2H, s), 7.75 (2H, d, J = 3.7 Hz), 7.43 (2H, m), 6.77 (2H, d, J = 3.2 Hz), 6.45 (2H, s), 6.20–6.25 (6H, m), 2.82 (4H, t, J = 7.8 Hz), 1.70 (4H, m), 1.42–1.29 (30H, m), 1.06 (18H, s), 0.89 (6H, t, J = 6.9 Hz); 13C-NMR (CDCl3, 100 MHz) (typical signals because some signals were overlapped]: δ 178.7, 144.6, 142.3, 137.8, 137.7 137.5, 132.3, 129.7, 128.2, 126.0, 121.7, 110.9, 45.7, 21.1; HRMS (ESI): m/z 1145.5220 (calcd. for C64H78O8N6NaS2 [M + Na]+, 1145.52212)
3.9. Synthesis of 2,3-Di(1H,1′H-[2,2′-bipyrrol]-5-yl)-5,8-bis(5-hexylthiophen-2-yl)quinoxaline (6a)
A solution of 5a (30 mg, 0.027 mmol) in ethylene glycol (10 mL) was degassed for 1 h under a reduced pressure and then purged with an argon gas and heated at 170 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with CHCl3. The combined organic layer was washed with water and brine, then dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was recrystallized from CH2Cl2/methanol to give 6a as a red solid. Yield: 92% (18 mg, 0.025 mmol). 1H-NMR (DMSO-d6, 400 MHz): δ 11.34 (2H, brs), 10.88 (2H, brs), 8.11 (2H, s), 7.81 (2H, d, J = 3.7 Hz), 6.95 (2H, d, J = 3.7 Hz), 6.87 (2H, brs), 6.77 (2H, m), 6.49 (4H, m), 6.13 (2H, dd, J = 5.7, 2.5 Hz), 2.89 (4H, t, J = 7.8 Hz), 1.71 (6H, t, J = 7.6 Hz), 1.34 (12H, m), 0.87 (6H, t, J = 7.1 Hz); 13C-NMR (CDCl3, 100 MHz): δ 148.86, 142.81, 135.67, 135.43, 130.67, 129.52, 128.40, 126.69, 125.70, 125.16, 124.77, 119.40, 114.67, 109.23, 105.61, 104.95, 31.95, 31.49, 30.11, 28.92, 22.64, 14.50; HRMS (ESI): m/z 745.3123 (calcd. for C44H46N6NaS2 [M + Na]+, 745.31252).
3.10. Synthesis of 5′,5‴-(5,8-Bis(5-hexylthiophen-2-yl)quinoxaline-2,3-diyl)-bis((1H,1′H-[2,2′-bipyrrole]-5-carbaldehyde)) (7a)
POCl3 (0.04 mL, 0.46 mmol) was slowly added to DMF (0.3 mL, 3.9 mmol) at 0 °C under an argon atmosphere to prepare the Vilsmeier’s reagent. A solution of 6a (17 mg, 0.02 mmol) in DMF (1.0 mL) was added to the Vilsmeier’s reagent at 0 °C. The mixture was stirred for 1 h at 0 °C. The concentrated aqueous acetyl acetate was added to the reaction mixture, followed by stirring for 4 h at room temperature. The reaction mixture was extracted with CHCl3. The reaction mixture was extracted with CHCl3 and the combined organic layer was washed with water and brine, and then dried over Na2SO4. The solvent was removed under a reduced pressure. The crude product was recrystallization from CH2Cl2/methanol to give 7a as a red solid. Yield: 99% yield (15 mg, 0.02 mmol). 1H-NMR (DMSO-d6, 400 MHz): δ 12.74 (2H, s), 11.52 (2H, s), 9.44 (2H, s), 8.18 (2H, s), 7.85 (2H, d, J = 3.8 Hz), 7.09 (2H, dd, J = 3.8, 2.2 Hz) 6.90 (2H, t, J = 3.0 Hz), 6.75 (2H, dd, J = 3.8, 2.2 Hz), 6.70 (2H, dd, J = 3.8, 2.2 Hz), 2.84 (4H, t, J = 7.7 Hz), 1.64 (6H, t, J = 7.1 Hz), 1.35–1.19 (16H, m), 0.83 (6H, t, J = 6.9 Hz); 13C-NMR could not be observed because of low solubility for common solvents.; HRMS (FAB): m/z 779.3196 (calcd. for C46H47N6O2S2 [M + H]+, 779.3141).
3.11. Synthesis of Porphycene 1a-H2
In a 100 mL three-necked round bottom flask, TiCl4 (0.4 mL, 3.7 mmol) was added dropwise to a THF (15 mL) solution containing Zn dust (0.48 g) and CuCl (28 mg, 0.29 mmol) at room temperature under an argon atmosphere, and then the mixture was refluxed for 2 h. Subsequently a solution of 7a (100 mg, 0.13 mmol) in THF (15 mL) was added dropwise to the boiling reaction mixture. The mixture was stirred at the same temperature for another 1 h. After cooling to 0 °C, an aqueous 10% K2CO3 solution (18 mL) was added dropwise. After filtration the precipitate was washed with CH2Cl2. The combined organic layer was then dried with Na2SO4 and the solution was removed under reduced pressure. The crude product was purified by alumina column chromatography (CH2Cl2) and silica gel column chromatography (CH2Cl2−hexanes = 4:1). Recrystallization from CHCl3/methanol provided 1a-H2 as a green solid. Yield: 13% (13 mg, 0.02 mmol). λabs [nm] (ε/M–1·cm–1): 349 (4.32 × 104), 407 (8.48 × 104), 597 (1.03 × 104), 640 (2.63 × 104), 733 (7.57 × 103), 805 (1.24 × 104); λfl [nm] (Φfl): 935 (0.16%); 1H-NMR (CDCl3, 400 MHz): δ 9.71 (2H, d, J = 4.0 Hz), 9.18 (1H, s), 9.03 (1H, s), 8.77 (2H, d, J = 4.4 Hz), 8.67 (2H, d, J = 4.0 Hz), 8.47 (2H, s), 8.41 (2H, s), 8.36 (2H, d, J = 4.4 Hz), 7.99 (2H, d, J = 3.3 Hz), 7.11 (2H, d, J = 3.3 Hz), 3.15 (4H, t, J = 7.5 Hz), 2.01 (4H, t, J = 7.7 Hz), 1.64 (4H, m), 1.50 (8H, m), 1.01 (6H, t, J = 7.0 Hz); 13C-NMR (CDCl3, 100 MHz) [typical signals because some signals were overlapped]: δ = 149.39, 148.09, 147.84, 145.42, 136.95, 135.45, 134.69, 133.46, 131.87, 131.15, 127.12, 126.68, 126.59, 126.11, 124.41, 123.57, 116.45, 31.93, 31.84, 30.57, 29.11, 22.76, 14.27; HRMS (FAB): m/z 745.3147 (calcd. for C46H44N6S2 [M]+, 745.3169).
3.12. Synthesis of Porphycene 1a-Ni
A mixture of 1a-H2 (3.0 mg, 4.0 μmol), Ni(OAc)2·4H2O (13 mg, 54 μmol) and pyridine (0.6 mL) in o-dichlorobenzene (2 mL) was heated at 180 °C for 5 h under an argon atmosphere. After cooling to room temperature, the solvent was removed under a reduced pressure. The crude product was purified by silica gel column chromatography (CH2Cl2/hexanes = 1/1) to give 1a-Ni as a green solid. Yield: 70% yield (1.5 mg, 1.9 μmol). λabs [nm] (ε/M–1 cm–1): 348 (3.17 × 104), 422 (6.90 × 104), 637 (1.13 × 104), 755 (8.29 × 103), 837 (1.52 × 104); 1H-NMR (CDCl3, 400 MHz): δ 9.93 (2H, d, J = 4.6 Hz), 8.52 (2H, d, J = 4.6 Hz), 8.42 (2H, s), 8.35 (2H, d, J = 5.0 Hz), 8.19 (4H, d, J = 3.7 Hz), 7.94 (2H, d, J = 3.7 Hz), 7.09 (2H, d, J = 3.7 Hz), 3.11 (4H, t, J = 7.3 Hz), 2.04–1.96 (4H, m), 1.50–1.46 (8H, m), 1.00 (6H, t, J = 6.9 Hz); 13C-NMR could not be assigned correctly because of low solubility. HRMS (MALDI-TOF): m/z 801.2339 (calcd. for C46H43N6NiS2 [M + H]+, 801.2331).
3.13. Synthesis of 2,3-Bis(5-bromo-1H-pyrrol-2-yl)-5,8-bis(3,5-dimethoxyphenyl)quinoxaline (3b)
NBS (307 mg, 1.72 mmol) was added to a solution of 2b (550 mg, 1.03 mmol) in CHCl3 (55 mL) at room temperature under an argon atmosphere. The solution was stirred for 5 h under the dark and then water was added. The separated organic layer was washed with brine and dried over Na2SO4. After removal of the solvent, the crude product was purified by silica gel column chromatography (CH2Cl2). Recrystallization from CH2Cl2/methanol gave 3b as a yellow solid. Yield: 86% (615 mg, 0.890 mmol). 1H-NMR (CDCl3, 400 MHz): δ = 9.46 (2H, s), 7.76 (2H, s), 7.10 (2H, dd, J = 3.8, 2.7 Hz), 6.94 (4H, J = 2.4 Hz), 6.61 (2H, t, J = 2.3 Hz), 6.20 (2H, dd, J = 3.8, 2.7 Hz), 3.90 (12H, s); 13C-NMR (CDCl3, 100 MHz): δ = 160.53, 140.24, 139.76, 138.46, 130.60, 129.04, 114.10, 112.19, 108.83, 103.34, 100.01, 55.73; HRMS (EI): m/z 688.0321 (calcd. for C32H26Br2N4O2 [M]+, 688.0321).
3.14. Synthesis of Di-tert-butyl 5,5′-(5,8-bis(3,5-dimethoxyphenyl)quinoxaline-2,3-diyl) bis(2-bromo-1H-pyrrole-1-carboxylate) (4b)
A mixture of 3b (735 mg, 1.1 mmol), DMAP (13 mg, 0.1 mmol) and Boc2O (1.4 mL, 6.0 mmol) in CH2Cl2 (80 mL) was stirred for 11 h at room temperature under argon atmosphere. After removal of the solvent, the crude product was purified by recrystallization from methanol to give 4b as a yellow solid. Yield: 89% yield (842 mg, 0.95 mmol). 1H-NMR (CDCl3, 400 MHz): δ 7.86 (2H, s), 6.98 (4H, d, J = 2.3 Hz), 6.55 (2H, d, J = 2.1 Hz), 6.27 (4H, q, J = 3.7 Hz), 3.81 (12H, s), 1.16 (18H, s); 13C-NMR (CDCl3, 100 MHz): δ = 160.22, 147.64, 144.53, 140.09, 139.00, 138.08, 133.57, 130.15, 116.22, 115.17, 108.98, 104.04, 100.51, 85.34, 55.37, 27.27; HRMS (ESI): m/z 911.1267 (calcd. for C42H43Br2N4NaO8 [M + Na]+, 911.1262).
3.15. Synthesis of Di-tert-butyl 5,5′-(5,8-bis(3,5-dimethoxyphenyl)quinoxaline-2,3-diyl)-bis(2-bromo-1H-pyrrole-1-carboxylate) (5b)
A degassed solution of 4b (500 mg, 0.561 mmol), (1-(tert-butoxycarbonyl)-1H-pyrrol-2-yl)boronic acid (621 mg, 2.94 mmol), Pd(OAc)2 (5.0 mg, 0.0223 mmol), SPhos (16.0 mg, 0.0390 mmol), K3PO4 (467 mg, 2.20 mmol) in 1-butanol (10 mL) and H2O (3.6 mL) was heated at 80 °C for 24 h under an argon atmosphere. After cooling to room temperature, the reaction mixture was diluted with CHCl3. The organic layer was washed with water and brine, and dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was purified by silica gel column chromatography (CH2Cl2). Recrystallization from methanol gave 5b as an orange solid. Yield: 97% (576 mg, 0.542 mmol). 1H-NMR (CDCl3, 400 MHz): δ 7.77 (2H, s), 7.39 (2H, m), 6.90 (4H, d, J = 2.3 Hz), 6.48 (2H, t, J = 2.3 Hz), 6.30 (2H, m), 6.21 (4H, m), 6.13 (2H, m), 3.79 (12H, s), 1.30 (18H, s), 1.00 (18H, s); HRMS (ESI): m/z 1063.4817 (calcd. for C60H67N6O12 [M + H]+, 1063.4811).
3.16. Synthesis of 2,3-Di(1H,1′H-[2,2′-bipyrrol]-5-yl)-5,8-bis(3,5-dimethoxyphenyl)quinoxaline (6b)
A suspended solution of 5b (560 mg, 0.58 mmol) in ethylene glycol (60 mL) was degassed for 1 h then purged with an argon atmosphere. The resulting mixture was heated at 180 °C for 2 h. After cooling to room temperature, the reaction mixture was extracted with CHCl3. The organic layer was washed with water and brine, and dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was recrystallized from CH2Cl2/methanol to give 6b as a red solid. Yield: 89% (310 mg, 0.47 mmol). 1H-NMR (DMSO-d6, 400 MHz): δ 9.74 (2H, s), 8.56 (2H, s), 7.73 (2H, s), 7.31 (2H, dd, J = 3.7, 2.3 Hz), 7.05 (2H, d, J = 2.3 Hz), 6.85 (2H, m), 6.65 (2H, t, J = 2.3 Hz), 6.40–6.42 (2H, m), 6.32–6.28 (4H, m), 3.92 (12H, s); 13C-NMR (DMSO-d6, 100 MHz): δ 160.02, 142.91, 140.02, 137.78, 136.99, 129.35, 128.83, 127.93, 124.58, 118.53, 113.01, 108.82, 108.70, 104.80, 103.96, 99.52, 79.17, 55.25, 22.07; HRMS (ESI): m/z 663.2720 (calcd. for C40H35N6O4 [M + H]+, 663.2714).
3.17. Synthesis of 5′,5‴-(5,8-Bis(5-hexylthiophen-2-yl)quinoxaline-2,3-diyl) bis((1H,1′H-[2,2′-bipyrrole]-5-carbaldehyde)) (7b)
POCl3 (0.2 mL, 2.1 mmol) was slowly added to DMF (1.4 mL, 17.9 mmol) at 0 °C under an argon atmosphere to prepare the Vilsmeier’s reagent. A solution of 6b (60 mg, 0.090 mmol) in DMF (4.0 mL) was carefully added to the Vilsmeier’s reagent at 0 °C. The resulting mixture was stirred for 1 h at 0 °C. The concentrated aqueous acetyl acetate was added and stirred for 4 h at room temperature. The reaction mixture was extracted with CHCl3 and the combined organic layer was washed with water and brine, then dried over Na2SO4. After removal of the solvent under a reduced pressure, the crude product was recrystallized from CH2Cl2/methanol to give 7b as a red sold. Yield: 57% (37 mg, 0.051 mmol). 1H-NMR (DMSO-d6, 400 MHz): δ 12.37 (2H, s), 11.41 (2H, s), 9.40 (2H, s), 7.92 (2H, s), 7.08–7.05 (6H, m), 6.86–6.84 (2H, m), 6.64–6.62 (4H, m), 6.38–6.36 (2H, m), 3.80 (12H, s); 13C-NMR could not be observed because of low solubility for common solvents.; HRMS (ESI): m/z 719.2618 (calcd. for C42H35N6O6 [M + H]+, 719.2613).
3.18. Synthesis of Porphycene 1b-H2
In a 100 mL three-necked round bottom flask, TiCl4 (0.2 mL, 2.2 mmol) was added dropwise to a THF (8 mL) solution containing Zn dust (0.30 g) and CuCl (17 mg, 0.29 mmol) at room temperature under argon atmosphere, then the mixture was refluxed for 2 h. Subsequently a solution of 7b (55 mg, 0.077 mmol) in THF (8 mL) was added dropwise to the boiling reaction mixture. The mixture was stirred at the same temperature for another 1 h. After cooling to 0 °C, an aqueous 10% K2CO3 solution (10 mL) was added dropwise. After filtration the precipitate was washed with CH2Cl2, the combined organic layer was then dried with Na2SO4, and the solution was removed under reduced pressure. The crude product was purified by alumina column chromatography (CH2Cl2) and silica gel column chromatography (CH2Cl2−hexanes = 1:4). Recrystallization from CHCl3/methanol provided 1b-H2 as a green solid. Yield: 17% (9 mg, 0.013 mmol). λabs [nm] (ε/M−1 cm–1): 344 (3.86 × 104), 397 (1.46 × 105), 585 (1.23 × 104), 632 (3.80 × 104), 710 (1.14 × 104), 781 (2.11 × 104); λfl [nm] (Φfl): 911, 980 (0.20%); 1H-NMR (CDCl3, 400 MHz): δ 9.52 (2H, d, J = 5.0 Hz), 8.91 (2H, d, J = 4.1 Hz), 8.82 (2H, d, J = 4.1 Hz), 8.62 (2H, s), 8.52 (2H, d, J = 4.6 Hz), 8.38 (2H, s), 7.45 (4H, d, J = 2.3 Hz), 6.82 (4H, t, J = 2.5 Hz), 4.04 (12H, s); 13C-NMR could not be assigned correctly because of low solubility.
3.19. Synthesis of Porphycene 1b-Ni
A mixture of 1b-H2 (1.0 mg, 1.5 μmol), Ni(OAc)2·4H2O (7.5 mg, 30 μmol) and pyridine (0.3 mL) in o-dichlorobenzene (1 mL) was heated at 180 °C for 6 h under an argon atmosphere. After cooling to room temperature, the solvent was removed under a reduced pressure. The crude product was purified by silica gel column chromatography (CH2Cl2/hexanes = 1/1) to give 1b-Ni as a green solid. Yield: 45% yield (0.5 mg, 0.674 μmol). λabs [nm] (ε/M−1 cm−1): 347 (1.07 × 104), 412 (2.88 × 104), 622 (4.86 × 103), 735 (4.29 × 103), 812 (8.53 × 103); 1H-NMR (400 MHz, CDCl3): δ 9.53 (2H, d, J = 5.0 Hz), 9.41 (2H, s), 9.33 (2H, s), 8.92 (2H, d, J = 3.2 Hz), 8.83 (2H, d, J = 4.1 Hz), 8.62 (2H, s), 8.52 (2H, d, J = 2.3 Hz), 8.38 (2H, s), 7.45 (4H, d, J = 2.3 Hz), 6.83 (2H, t, J = 2.5 Hz), 4.04 (12H, s): 13C-NMR could not be assigned correctly because of low solubility.
4. Conclusions
In summary, the quinoxaline-fused porphycenes 1a-H2 and 1b-H2 and their nickel complexes were successfully synthesized and characterized. These porphycenes displayed absorption and fluorescence at the NIR regions by the introduction of quinoxaline parts onto the porphycenes because of the effective expansion of the π-conjugation. The attached quinoxaline moiety also contributes to a decrease in the reduction potentials. We revealed that protonation by addition of TFA conducted for the dicationic porphycene from free base 1a-H2 and 1b-H2 and dicationic quinoxaline from nickel complexes associated with generation of the NIR absorptions. These results demonstrated that the synthesis of porphycene–based NIR absorbing dyes offers the practical applications for PDT, non-linear optical and OSC materials.
Supplementary Materials
The following are available online: Table S1 and S2, and Figure S1: Crystal data of 2a and 2b; Figure S2 and S3: H-H COSY and NOE spectra of 1a-H2; Figure S4 and S5: Changing of the absorption spectra of THPc and THPc-Ni upon addition of TFA.
Acknowledgments
This work was partly supported by JSPS KAKENHI Grant Numbers JP16H02286 and JP26105004 to H.Y., JP26288038 to N.A., and JP16K17950 to D.K. and Grants-in-Aid for JSPS Research Fellow (No. 16J05324 to T.O.), Izumi and Science and technology, supported by MEXT. The authors thank Yoshiko Nishikawa, Fumio Asanoma and Shouhei Katao for the measurement of mass spectra, NMR spectra and single-crystal structure analysis, respectively. The authors thank to Leigh McDowell in NAIST for the check of English.
Author Contributions
D.K. conceived and designed the experiments, and wrote the paper. M.S., W.F. and T.O. carried out syntheses and characterization parts. N.A. performed the X-ray diffraction analysis. N.A. and H.Y. supervised all experiments and manuscript drafting. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts interest.
References
- Vogel, E.; Köcher, M.; Schmickler, H.; Lex, J. Porphycene—A novel porphin isomer. Angew. Chem. Int. Ed. 1986, 25, 257–259. [Google Scholar] [CrossRef]
- Waluk, J. Structure, Spectroscopy, Photophysics, and Tautomerism of Free-Base Porphycenes and Other Porphyrin Isomers. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K., Guilard, R., Eds.; World Scientific: Singapore, 2010; Volume 7, pp. 359–435. [Google Scholar]
- Shimakoshi, H.; Baba, T.; Iseki, Y.; Aritome, I.; Endo, A.; Adachi, C.; Hisaeda, Y. Photophysical and photosensitizing properties of brominated porphycenes. Chem. Commun. 2008, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Panda, P.K. β-Octamethoxyporphycenes. Org. Lett. 2014, 16, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Braslavsky, S.; Muller, M.; Mártire, D.; Mártire, D.; Pörting, S.; Pörting, S.; Bertolotti, S.; Bertolotti, S.; Chakravorti, S.; Chakravorti, S.; et al. Photophysical properties of porphycene derivatives (18π porphyrinoids). J. Photochem. Photobiol. B 1997, 40, 191–198. [Google Scholar] [CrossRef]
- Stockert, J.C.; Cañete, M.; Juarranz, A.; Villanueva, A.; Horobin, R.W.; Borrell, J.I.; Teixidó, J.; Nonell, S. Porphycenes: Facts and prospects in photodynamic therapy of cancer. Curr. Med. Chem. 2007, 14, 997–1026. [Google Scholar] [CrossRef] [PubMed]
- Arnbjerg, J.; Jiménez-Banzo, A.; Paterson, M.J.; Nonell, S.; Borrell, J.I.; Christiansen, O.; Ogilby, P.R. Two-Photon Absorption in Tetraphenylporphycenes: Are Porphycenes Better Candidates than Porphyrins for Providing Optimal Optical Properties for Two-Photon Photodynamic Therapy? J. Am. Chem. Soc. 2007, 129, 5188–5199. [Google Scholar] [CrossRef] [PubMed]
- Sarma, T.; Panda, P.K.; Anusha, P.T.; Rao, S.V. Dinaphthoporphycenes: Synthesis and Nonlinear Optical Studies. Org. Lett. 2011, 13, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.V.; Shuvan Prashant, T. Two-photon and three-photon absorption in dinaphthoporphycenes. Chem. Phys. Lett. 2011, 514, 98–103. [Google Scholar]
- Kim, K.S.; Sung, Y.M.; Matsuo, T.; Hayashi, T.; Kim, D. Investigation of Aromaticity and Photophysical Properties in [18]/[20]π Porphycene Derivatives. Chem. Eur. J. 2011, 17, 7882–7889. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Lee, S.; Kim, D.; Panda, P.K. β-Octakis(methylthio)porphycenes: Synthesis, characterisation and third order nonlinear optical studies. Chem. Commun. 2015, 51, 7705–7708. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.; Yamada, H.; Okujima, T. Synthesis of Porphyrins Fused with Aromatic Rings. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2010; Volume 2, pp. 1–102. [Google Scholar]
- Kuzuhara, D.; Mack, J.; Yamada, H.; Okujima, T.; Ono, N.; Kobayashi, N. Synthesis, Structures, and Optical and Electrochemical Properties of Benzoporphycenes. Chem. Eur. J. 2009, 15, 10060–10069. [Google Scholar] [CrossRef] [PubMed]
- Kuzuhara, D.; Yamada, H.; Mori, S.; Okujima, T.; Uno, H. Synthesis, structures and properties of benzoporphycenes and naphthoporphycenes. J. Porphyr. Phthalocya. 2011, 15, 930–942. [Google Scholar] [CrossRef]
- Saeki, H.; Kurimoto, O.; Nakaoka, H.; Misaki, M.; Kuzuhara, D.; Yamada, H.; Ishida, K.; Ueda, Y. Effect of crystallinity in small molecular weight organic heterojunction solar cells. J. Mater. Chem. C 2014, 2, 5357–5364. [Google Scholar] [CrossRef]
- Saeki, H.; Misaki, M.; Kuzuhara, D.; Yamada, H.; Ueda, Y. Fabrication of Phase-Separated Benzoporphycene/[6,6]-Phenyl-C61-Butyric Acid Methyl Ester Films for Use in Organic Photovoltaic Cells. Jpn. J. Appl. Phys. 2013, 52, 111601. [Google Scholar] [CrossRef]
- Saeki, H.; Kurimoto, O.; Misaki, M.; Kuzuhara, D.; Yamada, H.; Ueda, Y. Thermal Conversion Behavior and Morphology Control of Benzoporphycene from a Novel Soluble Precursor. Appl. Phys. Express 2013, 6, 035601. [Google Scholar] [CrossRef]
- Roznyatovskiy, V.; Lynch, V.; Sessler, J.L. Dinaphthoporphycenes. Org. Lett. 2010, 12, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, D.; Sessler, J.L. Porphycenes: Synthesis and derivatives. Chem. Soc. Rev. 2008, 37, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Ogawa, A.; Fukuda, T.; Onoda, A.; Hasegawa, J.-Y.; Hayashi, T. meso-Dibenzoporphycene has a Large Bathochromic Shift and a Porphycene Framework with an Unusual cis Tautomeric Form. Angew. Chem. Int. Ed. 2015, 54, 6227–6230. [Google Scholar] [CrossRef] [PubMed]
- Planas, O.; Gallavardin, T.; Nonell, S. A novel fluoro-chromogenic click reaction for the labelling of proteins and nanoparticles with near-IR theranostic agents. Chem. Commun. 2015, 51, 5586–5589. [Google Scholar] [CrossRef] [PubMed]
- Aldakov, D.; Anzenbacher, P., Jr. Dipyrrolyl quinoxalines with extended chromophores are efficient fluorimetric sensors for pyrophosphate. Chem. Commun. 2003, 1394–1395. [Google Scholar] [CrossRef]
- Anzenbacher, P., Jr.; Jursikova, K.; Aldakov, D.; Marquezb, M.; Pohl, R. Materials chemistry approach to anion-sensor design. Tetrahedron 2004, 60, 11163–11168. [Google Scholar] [CrossRef]
- Billingsley, K.; Buchwald, S.L. Highly Efficient Monophosphine-Based Catalyst for the Palladium-Catalyzed Suzuki−Miyaura Reaction of Heteroaryl Halides and Heteroaryl Boronic Acids and Esters. J. Am. Chem. Soc. 2007, 129, 3358–3366. [Google Scholar] [CrossRef] [PubMed]
- Kuzuhara, D.; Yamada, H.; Yano, K.; Okujima, T.; Mori, S.; Uno, H. First Synthesis of Dodecasubstituted Porphycenes. Chem. Eur. J. 2011, 17, 3376–3383. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.E. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Peng, X.; Song, F.; Lu, E.; Wang, Y.; Zhou, W.; Fan, J.; Gao, Y. Heptamethine cyanine dyes with a large stokes shift and strong fluorescence: A paradigm for excited-state intramolecular charge transfer. J. Am. Chem. Soc. 2005, 127, 4170–4171. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stone, A.; Fleischer, E.B. The molecular and crystal structure of porphyrin diacids. J. Am. Chem. Soc. 1968, 90, 2735–2748. [Google Scholar] [CrossRef]
- Honda, T.; Kojima, T.; Kobayashi, N.; Fukuzumi, S. Crystal Structures and Electronic Properties of Saddle-Distorted and Protonated Phthalocyanines. Angew. Chem. Int. Ed. 2011, 50, 2725–2728. [Google Scholar] [CrossRef] [PubMed]
- Waluk, J.; Muller, M.; Swiderek, P.; Köcher, M.; Vogel, E.; Hohlneicher, G.; Michl, J. Electronic states of porphycenes. J. Am. Chem. Soc. 1991, 113, 5511–5527. [Google Scholar] [CrossRef]
- Sessler, J.L.; Brucker, E.A.; Lynch, V.; Choe, M.; Sorey, S.; Vogel, E. Solution Phase and Single Crystal Diffraction X-ray Analyses of Diprotonated Porphyrin Isomers—Etioporphyrin, Etioporphycene, and Etiocorrphycene Bishydroperchlorate Salts. Chem. Eur. J. 1996, 2, 1527–1532. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112. [Google Scholar] [CrossRef] [PubMed]
- Aldakov, D.; Palacios, M.A.; Anzenbacher, P. Benzothiadiazoles and Dipyrrolyl Quinoxalines with Extended Conjugated Chromophores-Fluorophores and Anion Sensors. Chem. Mater. 2005, 17, 5238–5241. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available. |
Figure 1.
Structures of porphycene (Pc) and π-expanded porphycenes.
Scheme 1.
Synthesis of 1a-H2, 1b-H2, 1a-Ni and 1b-Ni.
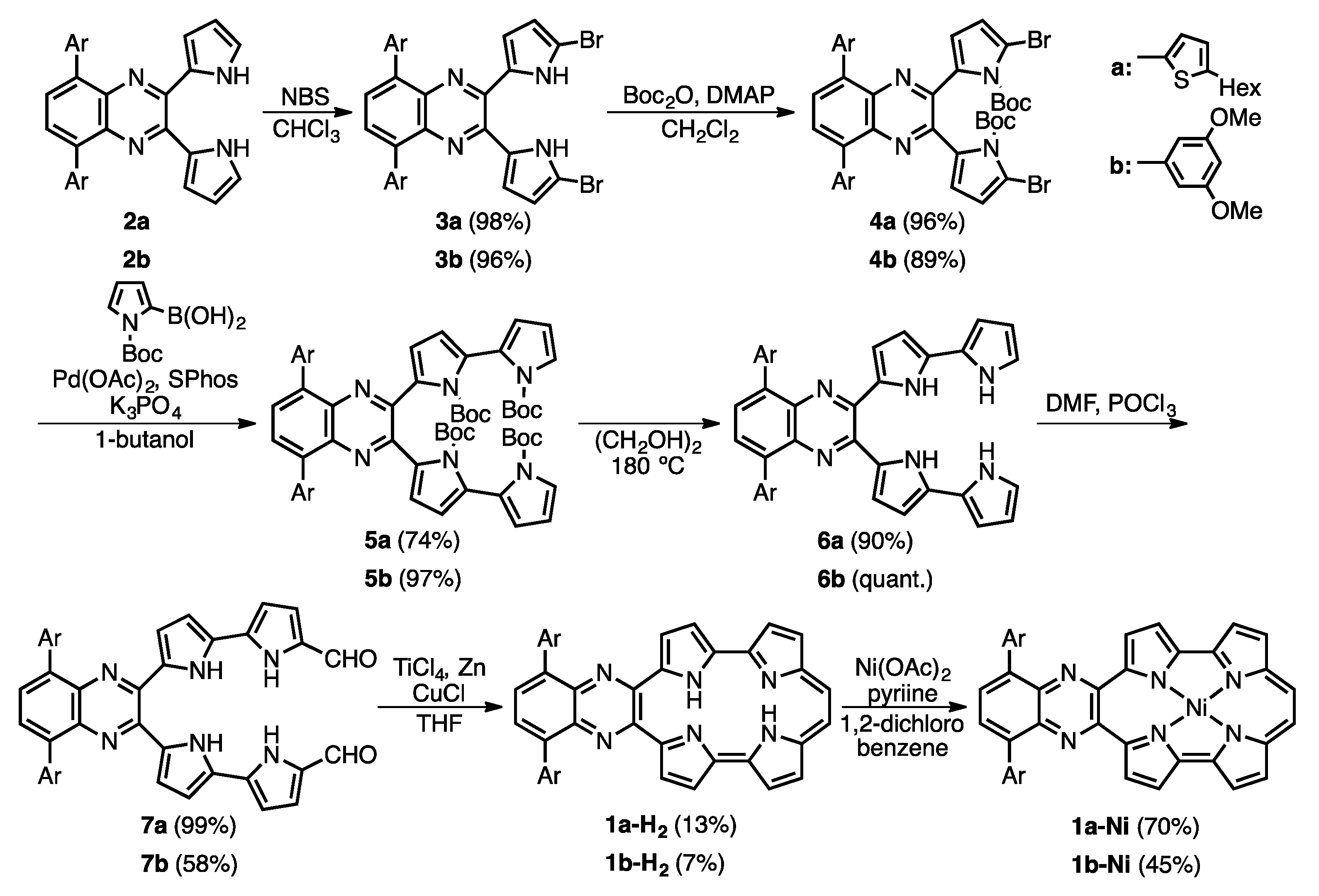
Figure 2.
Crystal structure of 1b-H2. (a) Top view and selected bond lengths; (b) side view. Thermal ellipsoids represent for 50% probability.
Figure 2.
Crystal structure of 1b-H2. (a) Top view and selected bond lengths; (b) side view. Thermal ellipsoids represent for 50% probability.
Figure 3.
1H-NMR spectra of 1a-H2 and 1b-H2 in CDCl3.

Figure 4.
NICS(0) values of Pc, 1a-H2 and 1b-H2.
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Pc | –15.62 | – | – |
| 1a-H2 | –8.51 | –12.56 | –8.57 |
| 1b-H2 | –8.57 | –12.67 | –9.03 |
Figure 5.
(a) Absorption and (b) fluorescence spectra of 1a-H2 (black) and 1b-H2 (red) in CH2Cl2. The fluorescence was obtained by excitation at 411 nm for 1a-H2 and 377 nm for 1b-H2.
Figure 5.
(a) Absorption and (b) fluorescence spectra of 1a-H2 (black) and 1b-H2 (red) in CH2Cl2. The fluorescence was obtained by excitation at 411 nm for 1a-H2 and 377 nm for 1b-H2.
Figure 6.
CV and DPV of 1a-H2 (black), 1b-H2 (red) and THPc (blue) in CH2Cl2.
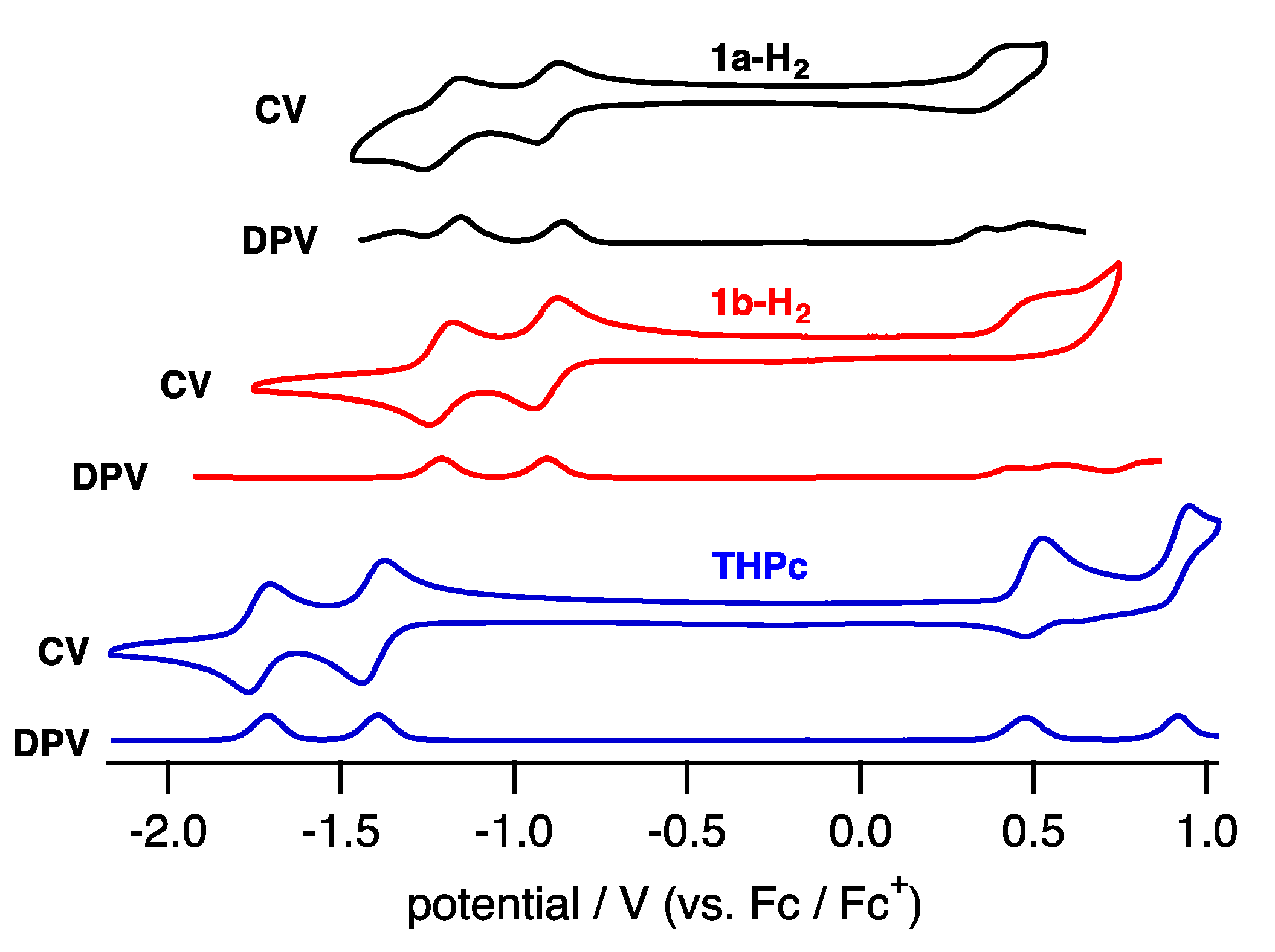
Figure 7.
Kohn-Sham molecular orbitals and energy diagrams of Pc and 1′.
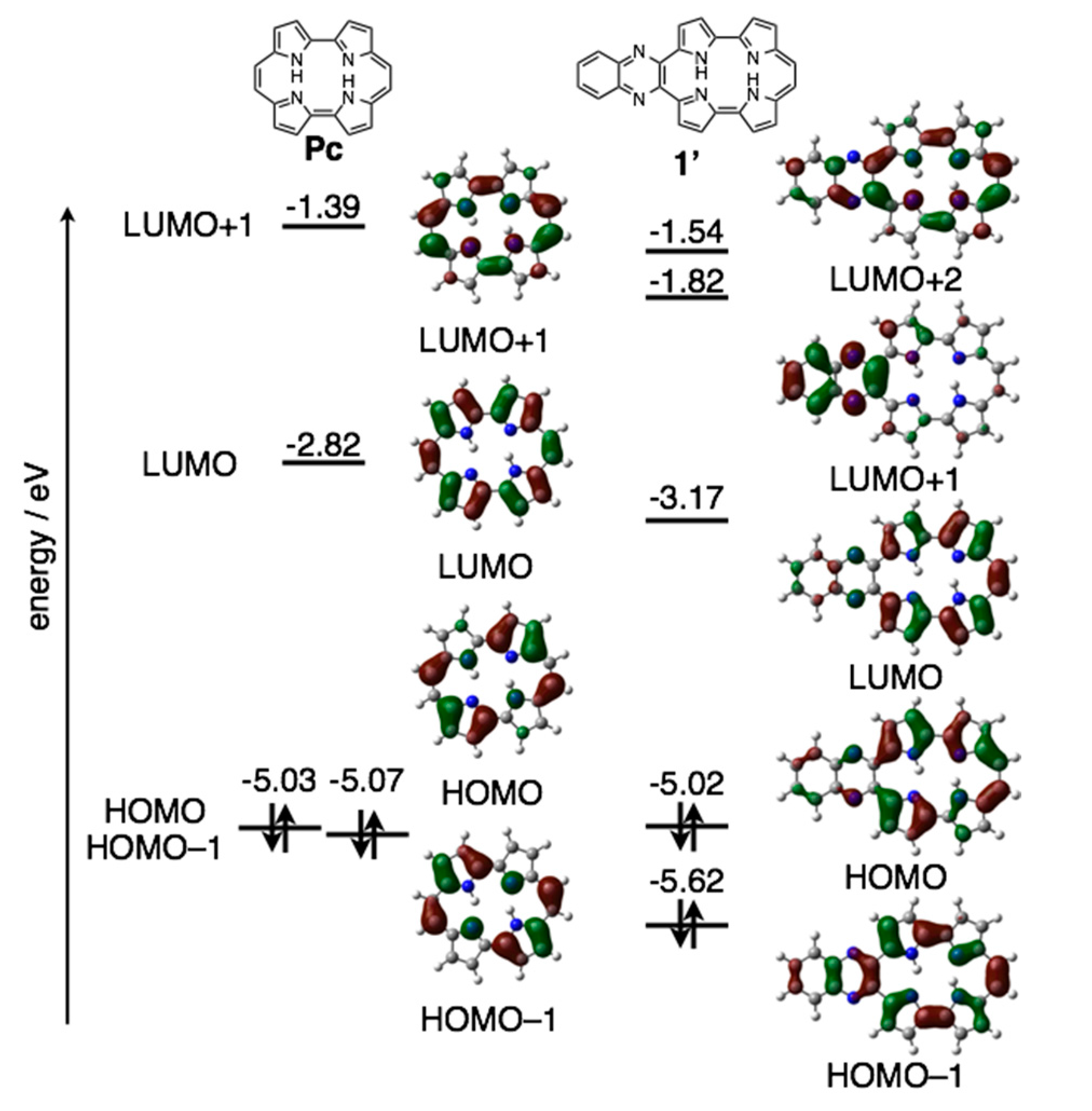
Figure 8.
The TFA titration of the absorption spectra of (a) 1a-H2, (b) 1a-Ni, (c) 1b-H2 and (d) 1b-Ni in CH2Cl2. Inset: absorbance changes at (a) 405 (red) and 667 nm (blue); (b) at 411 nm; (c) 397 (red) and 665 nm (blue), and (d) 407 nm.
Figure 8.
The TFA titration of the absorption spectra of (a) 1a-H2, (b) 1a-Ni, (c) 1b-H2 and (d) 1b-Ni in CH2Cl2. Inset: absorbance changes at (a) 405 (red) and 667 nm (blue); (b) at 411 nm; (c) 397 (red) and 665 nm (blue), and (d) 407 nm.

Scheme 2.
Protonation of free-base and nickel complex of porphycenes.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuzuhara, D.; Sakaguchi, M.; Furukawa, W.; Okabe, T.; Aratani, N.; Yamada, H. Synthesis, Characterization and Protonation Behavior of Quinoxaline-Fused Porphycenes. Molecules 2017, 22, 908. https://doi.org/10.3390/molecules22060908
AMA Style
Kuzuhara D, Sakaguchi M, Furukawa W, Okabe T, Aratani N, Yamada H. Synthesis, Characterization and Protonation Behavior of Quinoxaline-Fused Porphycenes. Molecules. 2017; 22(6):908. https://doi.org/10.3390/molecules22060908
Chicago/Turabian StyleKuzuhara, Daiki, Mika Sakaguchi, Wataru Furukawa, Takuya Okabe, Naoki Aratani, and Hiroko Yamada. 2017. "Synthesis, Characterization and Protonation Behavior of Quinoxaline-Fused Porphycenes" Molecules 22, no. 6: 908. https://doi.org/10.3390/molecules22060908