Synthesis and Evaluation of Phenylxanthine Derivatives as Potential Dual A2AR Antagonists/MAO-B Inhibitors for Parkinson’s Disease

 ,
,
Abstract
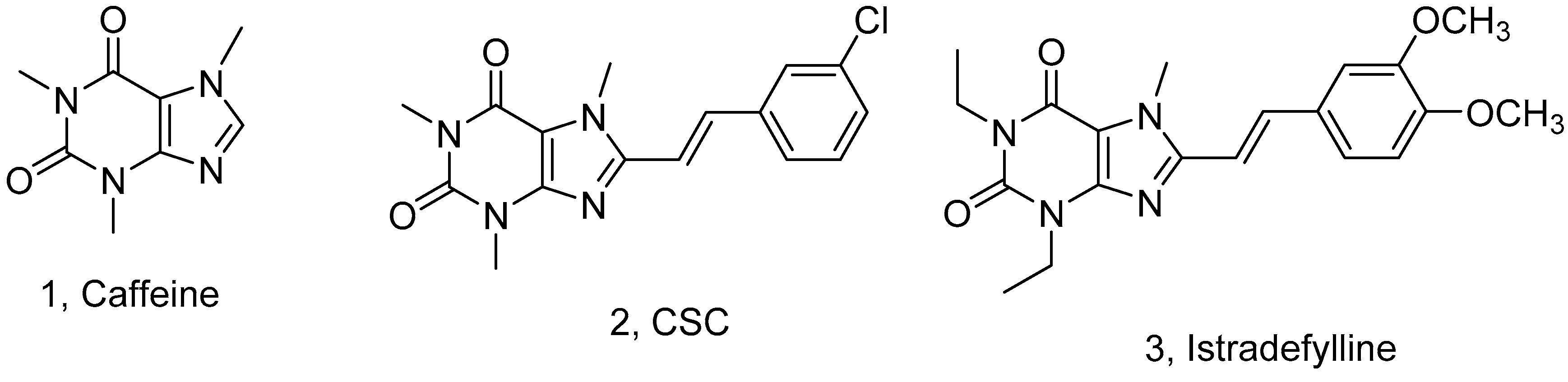
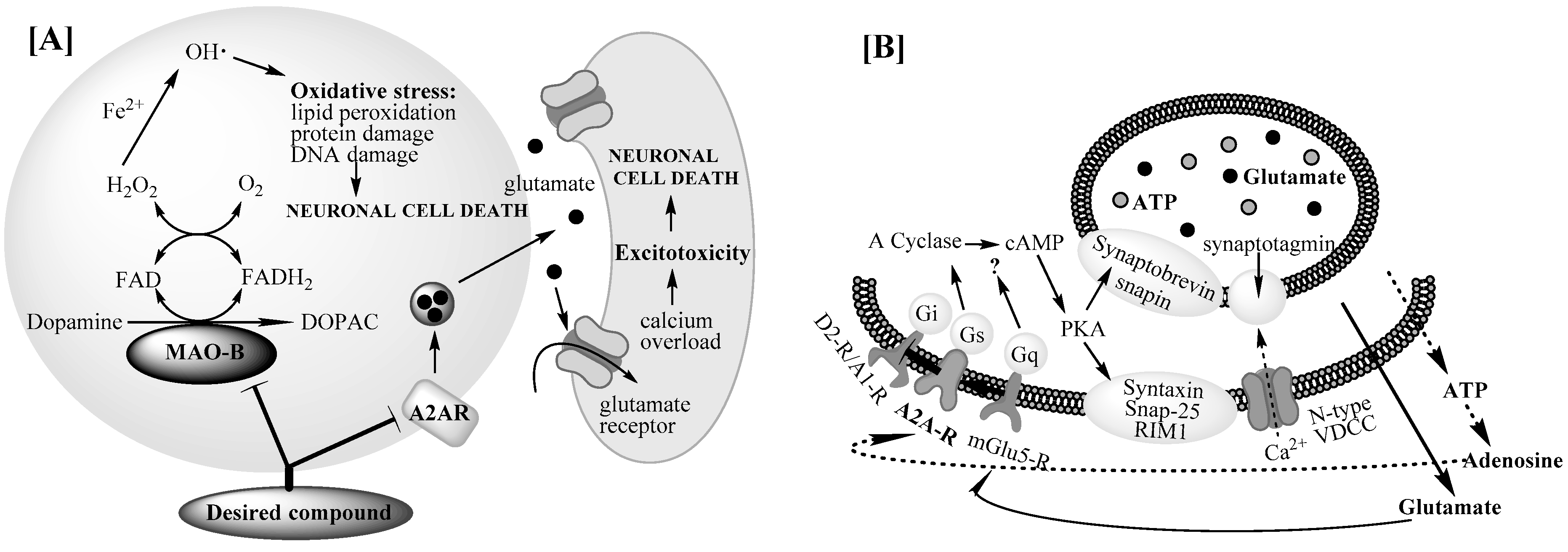
:1. Introduction
2. Results and Discussion
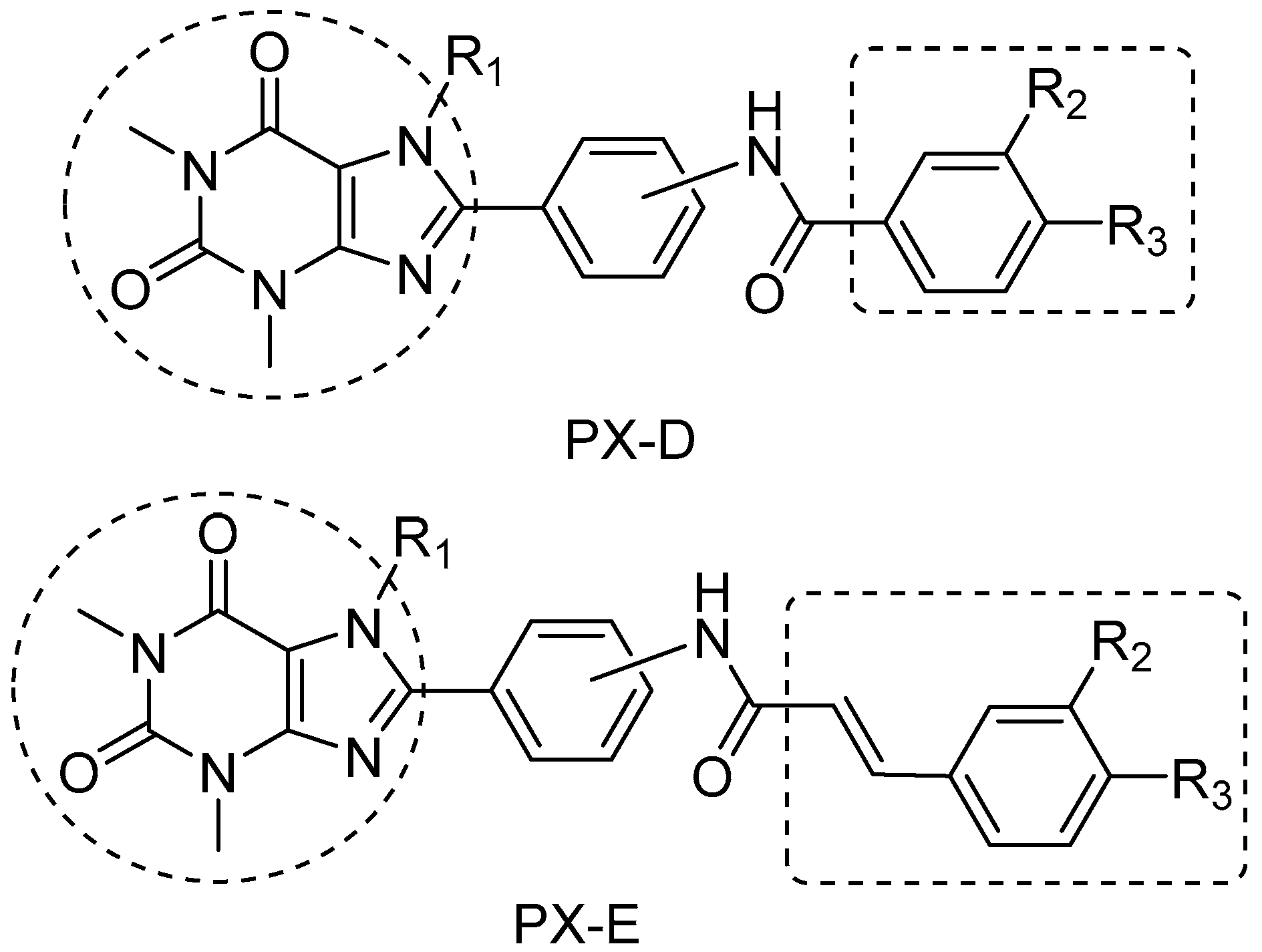
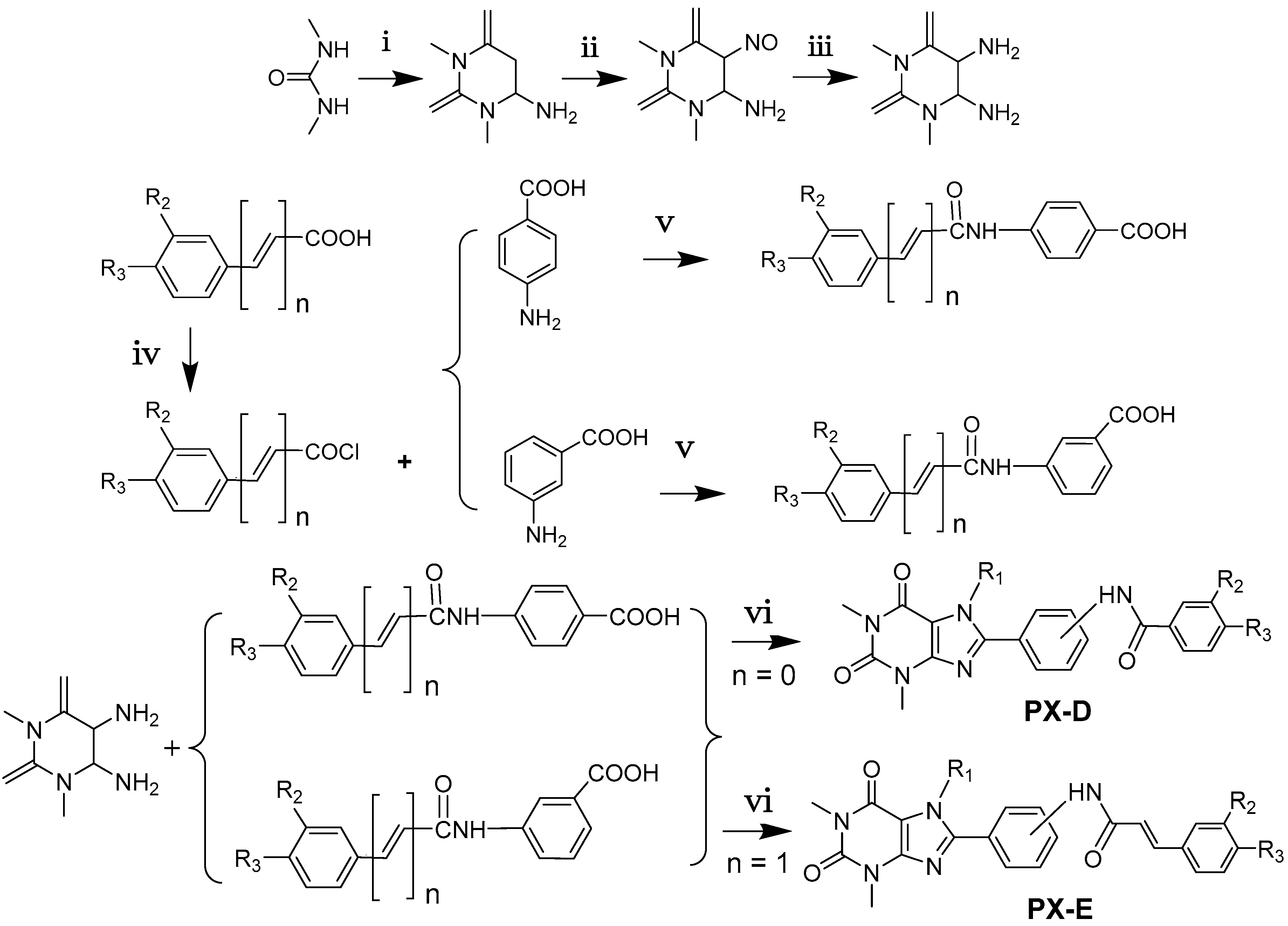
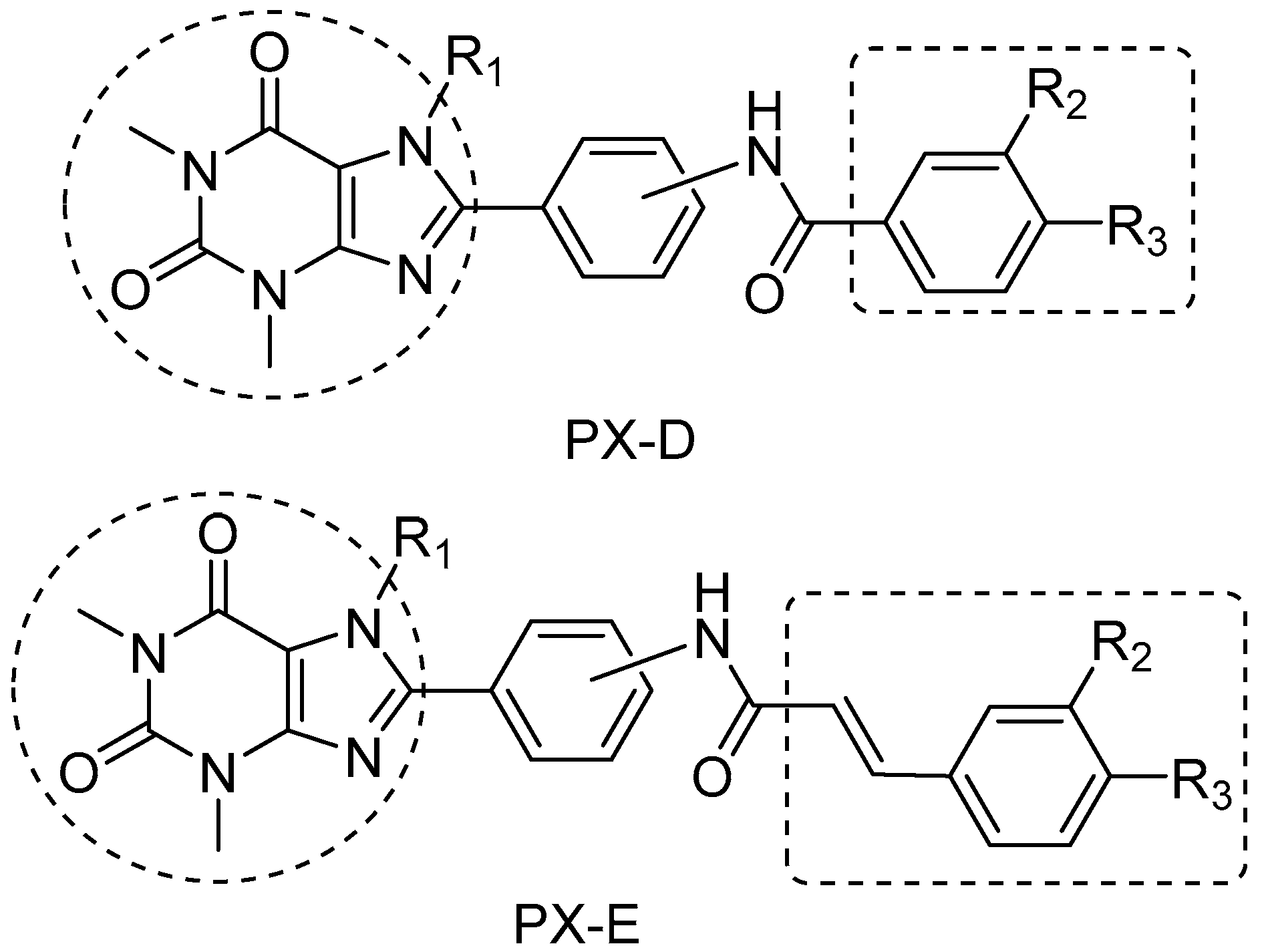
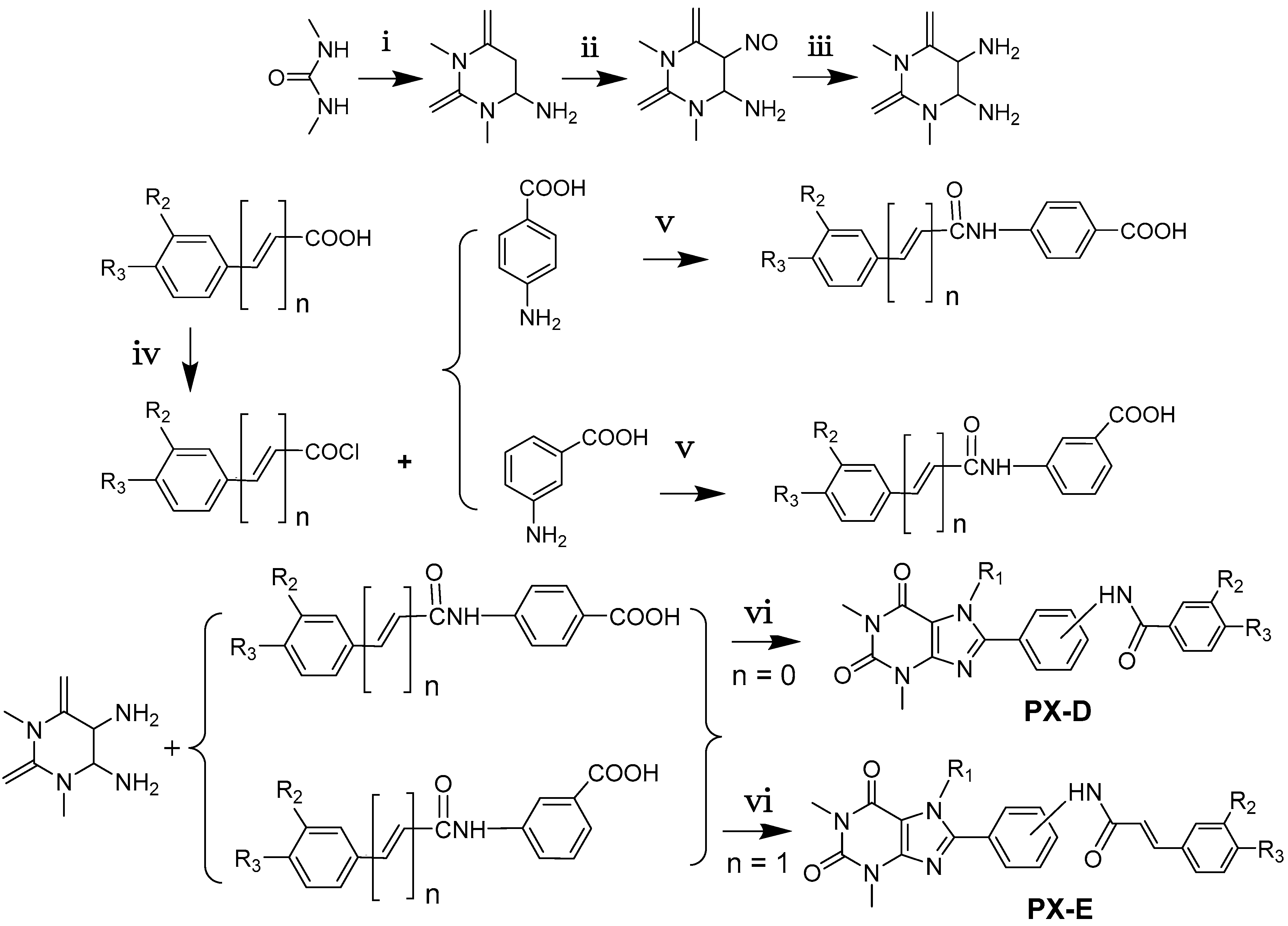
2.1. Chemistry
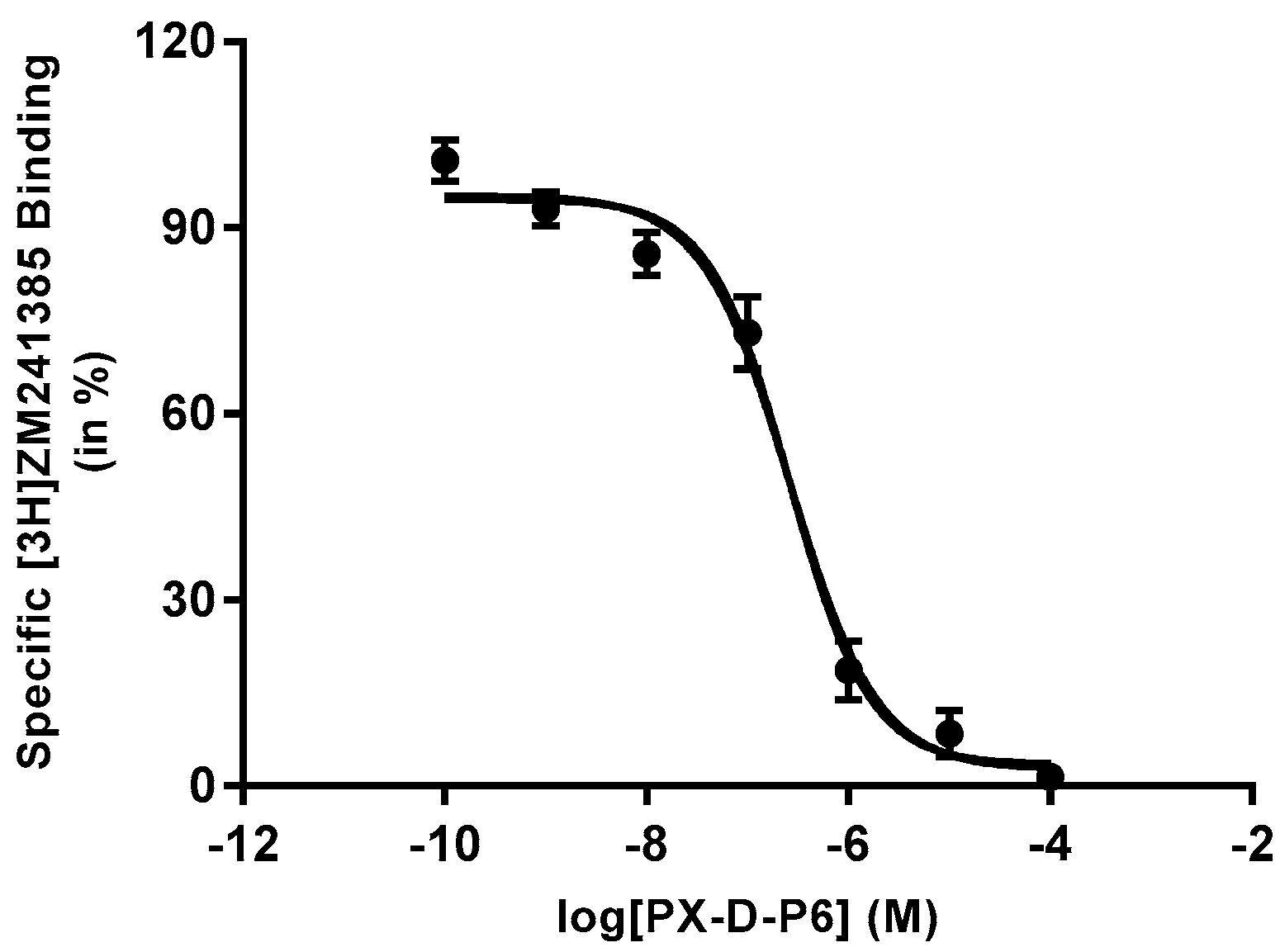
2.2. Structure−Activity Relationship between Test Compounds and A2AR
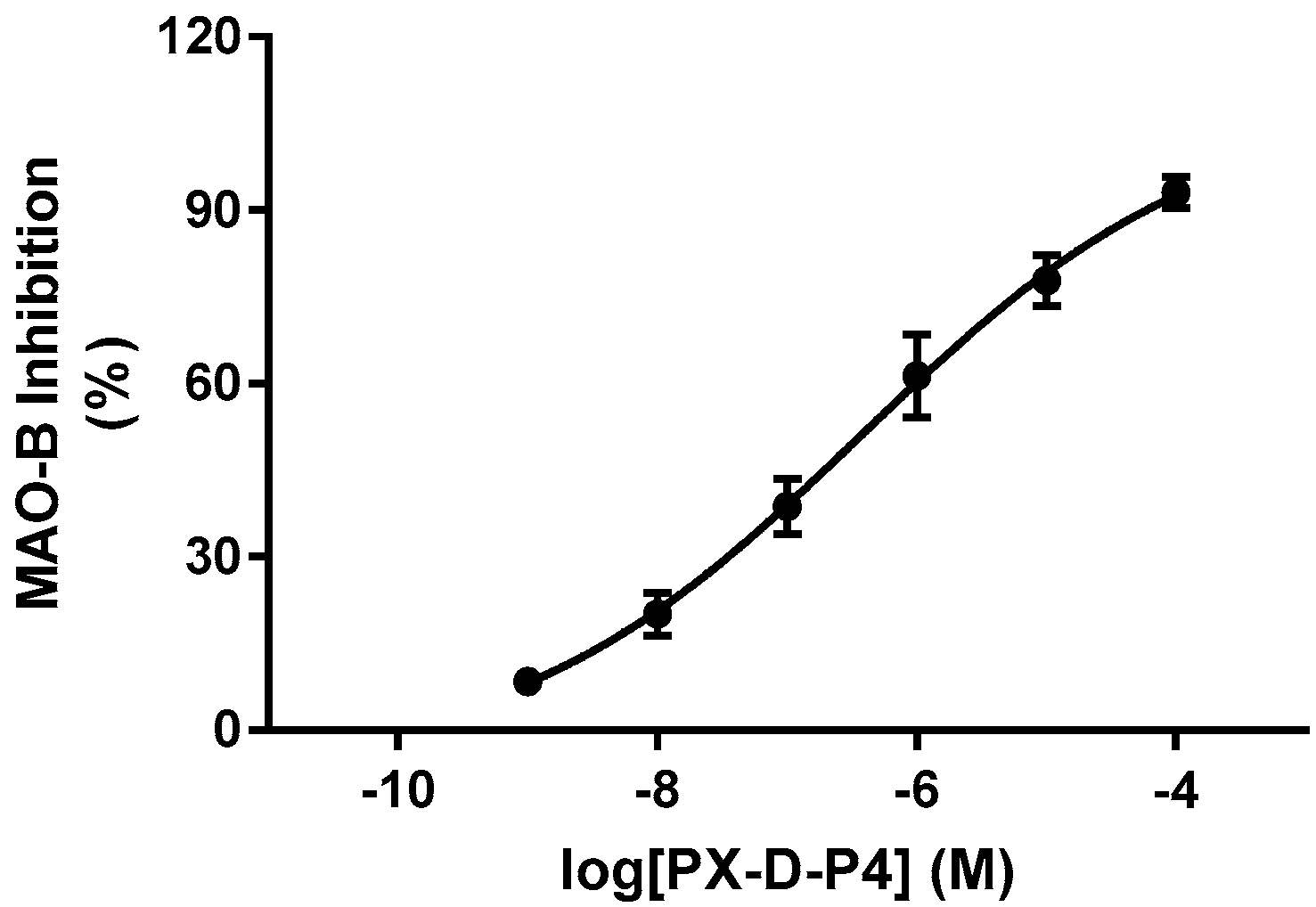
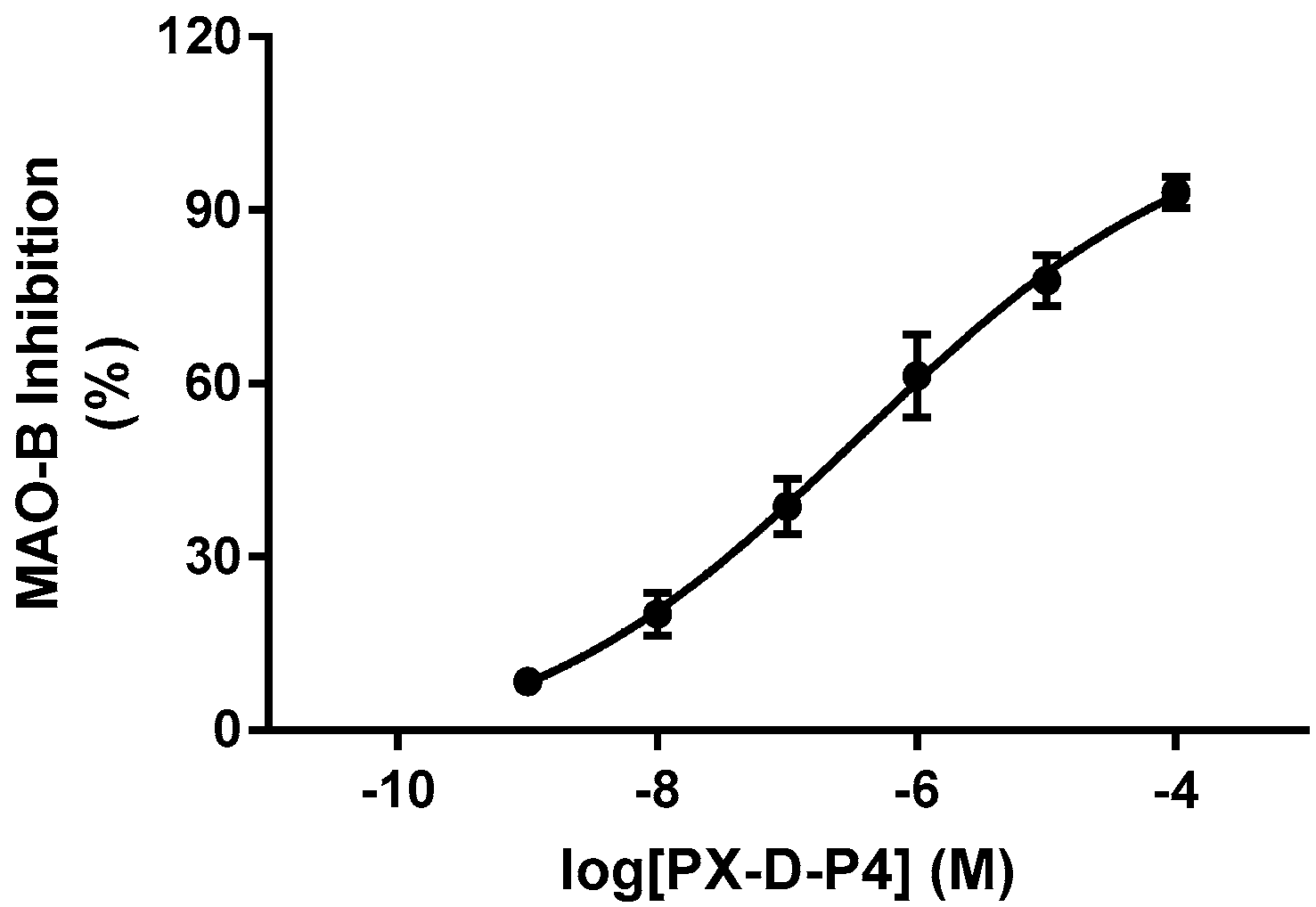
2.3. Structure−Activity Relationship between Compounds and MAO-B
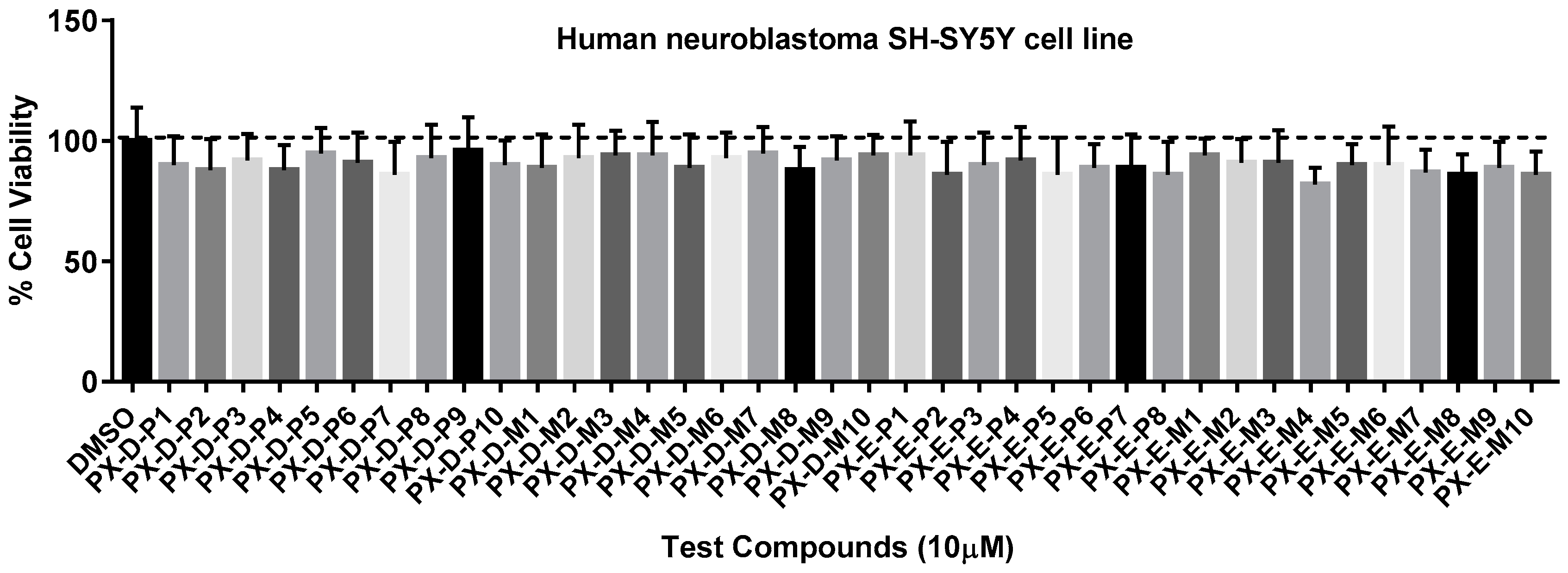
2.4. Cell Cytotoxicity
2.5. Pharmacokinetics and Brain Distribution of PX-D-P6 and PX-E-P8
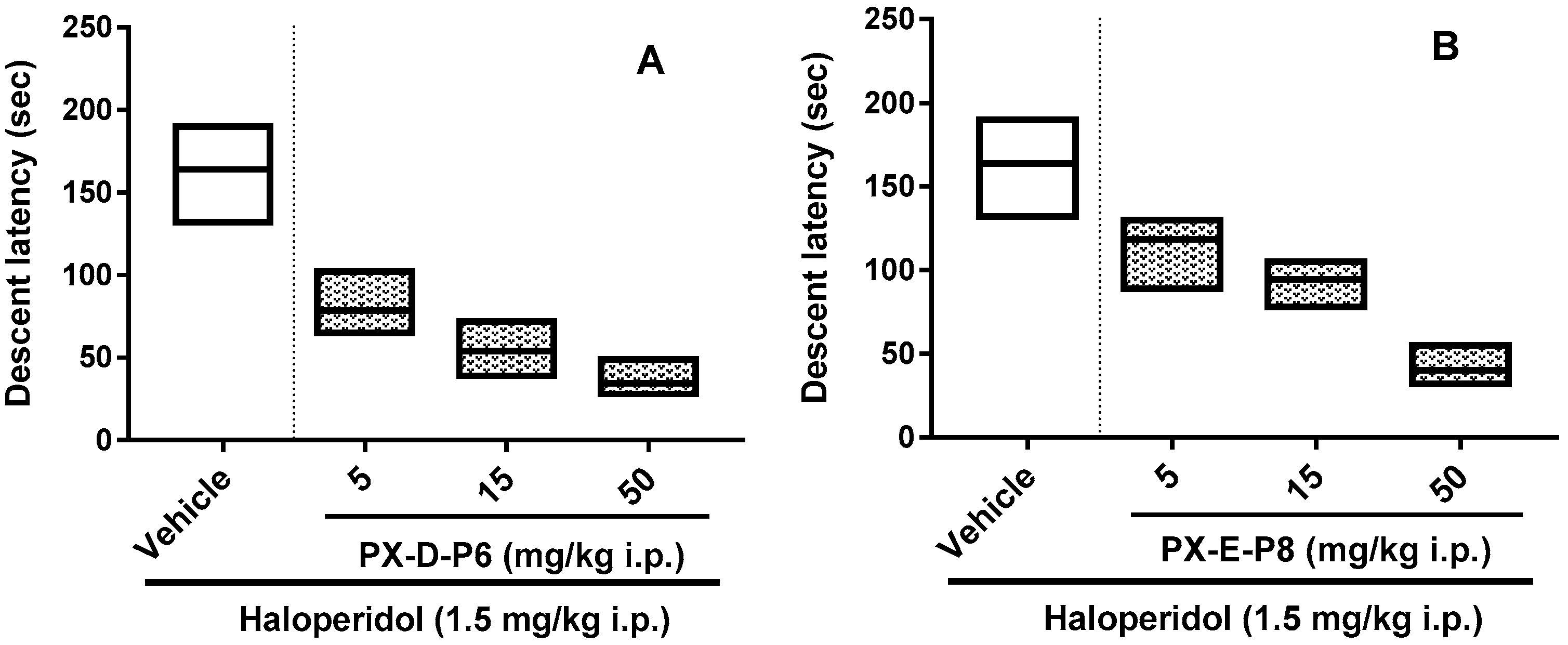
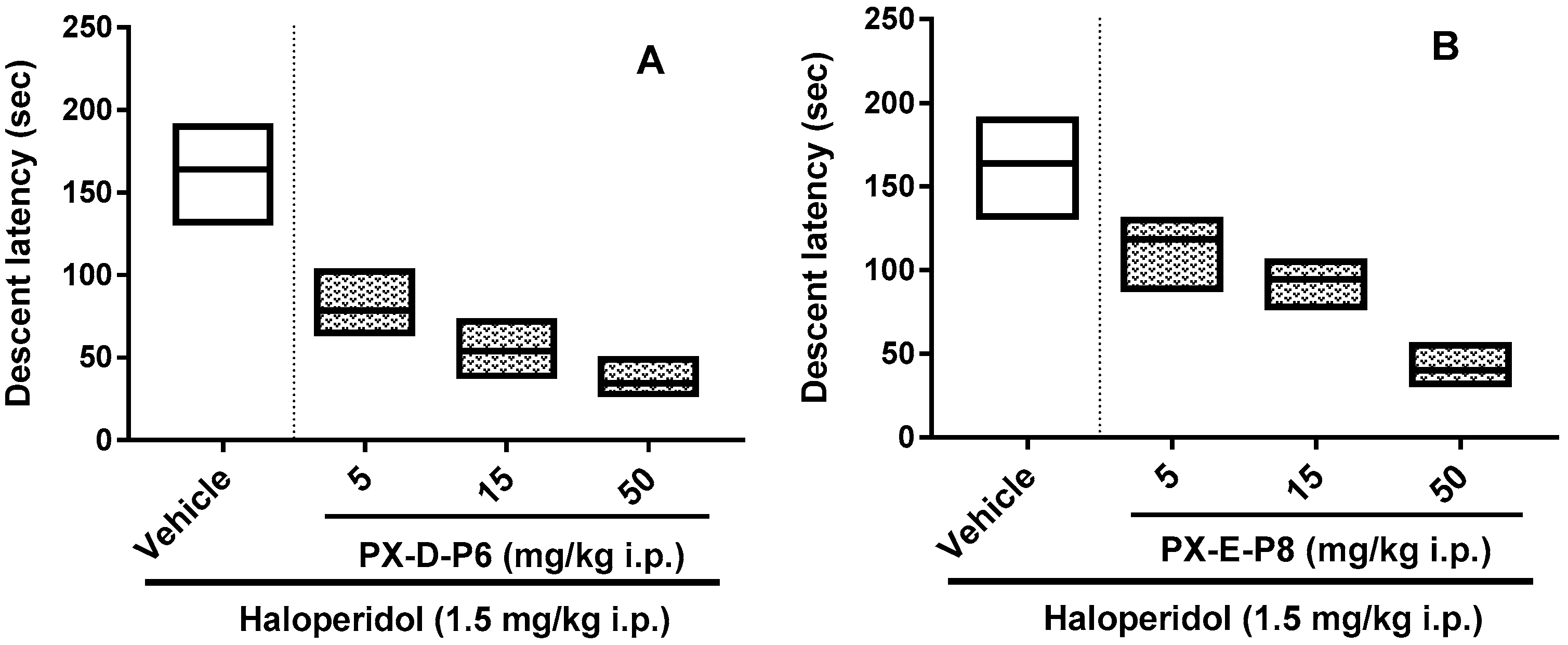
2.6. Haloperidol Induced Catalepsy Study
3. Materials and Methods
3.1. Materials
3.2. Synthesis of PX-D and PX-E Analogues
3.3. Affinity Toward A2AR
3.4. MAO-B Inhibition Studies
3.5. MTT Cytotoxicity Assay
3.6. Pharmacokinetic Experiments
3.7. Pharmacological Potential: Catalepsy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dwolatzky, T. Cognitive Impairment and dementia in Parkinson disease. J. Am. Med. Assoc. 2011, 305, 2231–2234. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef]
- Allain, H.; Bentuéferrer, D.; Akwa, Y. Disease-modifying drugs and Parkinson′s disease. Prog. Neurobiol. 2008, 84, 25–39. [Google Scholar] [CrossRef]
- Villalba, R.M.; Mathai, A.; Smith, Y. Morphological changes of glutamatergic synapses in animal models of Parkinson′s disease. Front Neuroanat. 2015, 9, 117. [Google Scholar] [CrossRef]
- Politis, M.; Niccolini, F. Serotonin in Parkinson′s disease. Behav. Brain. Res. 2014, 277, 136–145. [Google Scholar] [CrossRef]
- Shimada, J.; Suzuki, F. Medicinal Chemistry of Adenosine Receptors in the Brain and Periphery. In Adenosine Receptors and Parkinson′s Disease; Academic Press: New York, NY, USA, 2000; pp. 31–50. [Google Scholar]
- Ciruela, F.; Casado, V.; Rodrigues, R.J.; Lujan, R.; Burgueno, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; Cortes, A.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferré, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef]
- Morelli, M.; Carta, A.R.; Jenner, P. Adenosine A2A receptors and Parkinson′s disease. Handb. Exp. Pharmacol. 2009, 193, 589–615. [Google Scholar] [CrossRef]
- Freitas, M.E.; Fox, S.H. Nondopaminergic treatments for Parkinson′s disease, current and future prospects. Neurodegener. Dis. Manag. 2016, 1, 491–512. [Google Scholar] [CrossRef]
- A 12-week Randomized Study to Evaluate Oral Istradefylline in Subjects With Moderate to Severe Parkinson′s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01968031 (accessed on 13 June 2017).
- Song, B.; Xiao, T.; Qi, X.L.; Li, L.N.; Qin, K.Y.; Nian, S.Y.; Hu, G.X.; Yu, Y.F.; Liang, G.; Ye, F.Q. Design and synthesis of 8-substituted benzamido-phenylxanthine derivatives as MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1739–1742. [Google Scholar] [CrossRef]
- Hu, S.W.; Nian, S.Y.; Qin, K.Y.; Xiao, T.; Li, L.N.; Qi, X.L.; Ye, F.Q.; Liang, G.; Hu, G.X.; He, J.C.; et al. Design, synthesis and inhibitory activities of 8-(substituted styrol-formamido) phenyl-xanthine derivatives on monoamine oxidase B. Chem. Pharm. Bull. 2012, 60, 385–390. [Google Scholar] [CrossRef]
- Riederer, P.; Lachenmayer, L.; Laux, G. Clinical applications of MAO-inhibitors. Curr. Med. Chem. 2004, 11, 2033–2043. [Google Scholar] [CrossRef]
- Youdim, M.B.H; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Chen, J.F.; Steyn, S.; Staal, R.; Petzer, J.P.; Xu, K.; Van der Schyf, C.J.; Castagnoli, K.; Sonsalla, P.K.; Castagnoli, N.; Schwarzschild, M.A. 8-(3-Chlorostyryl)caffeine may attenuate MPTP neurotoxicity through dual actions of monoamine oxidase inhibition and A2A receptor antagonism. J. Biol. Chem. 2002, 277, 36040–36044. [Google Scholar] [CrossRef]
- Youdim, M.B.H. Why do we need multifunctional neuroprotective and neurorestorative drugs for Parkinson′s and Alzheimer's diseases as disease modifying agents. Rambam. Maimonides. Med. J. 2010, 1, e0011. [Google Scholar] [CrossRef]
- Rivara, S.; Piersanti, G.; Bartoccini, F.; Diamantini, G.; Pala, D.; Riccioni, T.; Stasi, M.A.; Cabri, W.; Borsini, F.; Mor, M.; et al. Synthesis of (E)-8-(3-chlorostyryl)caffeine analogues leading to 9-deazaxanthine derivatives as dual A2A antagonists/MAO-B inhibitors. J. Med. Chem. 2013, 56, 1247–1261. [Google Scholar] [CrossRef]
- Lazareno, S.; Birdsall, N.J. Estimation of antagonist Kb from inhibition curves in functional experiments, alternatives to the Cheng-Prusoff equation. Trends. Pharmacol. Sci. 1993, 14, 237–239. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson′s disease, a source of novel treatments and clues 468 to the cause of the disease. Brit. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- Cieślak, M.; Komoszyński, M.; Wojtczak, A. Adenosine A2A receptors in Parkinson′s disease treatment. Purinerg. Signal. 2008, 4, 305–312. [Google Scholar] [CrossRef]
- Barodia, S.K.; Mishra, C.B.; Prakash, A.; Senthil Kumar, J.B.; Kumari, N.; Luthra, P.M. 8-(furan-2-yl)-3-phenethylthiazolo [5,4-e] [1,2,4] triazolo [1,5-c] pyrimidine-2(3H)-thione as novel, selective and potent adenosine A2A receptor antagonist. Neurosci. Lett. 2013, 558, 203–207. [Google Scholar] [CrossRef]
- Kinoshita, H.; Hasegawa, T.; Katsumata, Y.; Kameyama, T.; Yamamoto, I.; Nabeshima, T. Effect of dizocilpine (MK-801) on the catalepsy induced by delta 9-tetrahydrocannabinol in mice. J. Neural. Transm. 1994, 95, 137–143. [Google Scholar] [CrossRef]
Sample Availability: Sample of the compound PX-D-P6 is available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | -HNCO | R1 | R2 | R3 | Ki hA2AR (μM) 1 | Ki hMAO-B (μM) 1 |
|---|---|---|---|---|---|---|
| Istradefylline | 0.05 ± 0.01 | >10 | ||||
| PX-D-P1 | para- | H | H | H | 2.31 ± 0.31 | 1.32 ± 0.11 |
| PX-D-P2 | para- | H | H | CH3 | 2.87 ± 0.21 | 2.06 ± 0.25 |
| PX-D-P3 | para- | H | H | OCH3 | 2.67 ± 0.14 | 2.94 ± 0.31 |
| PX-D-P4 | para- | H | Cl | H | 1.52 ± 0.16 | 0.25 ± 0.05 |
| PX-D-P5 | para- | H | OCH3 | OCH3 | 4.27 ± 0.51 | 1.92 ± 0.25 |
| PX-D-P6 | para- | CH3 | H | H | 0.33 ± 0.09 | 0.29 ± 0.03 |
| PX-D-P7 | para- | CH3 | H | CH3 | 0.39 ± 0.06 | 1.21 ± 0.15 |
| PX-D-P8 | para- | CH3 | H | OCH3 | 1.19 ± 0.12 | 0.47 ± 0.12 |
| PX-D-P9 | para- | CH3 | Cl | H | 0.27 ± 0.08 | 1.51 ± 0.19 |
| PX-D-P10 | para- | CH3 | OCH3 | OCH3 | 1.34 ± 0.16 | 2.97 ± 0.37 |
| PX-D-M1 | meta- | H | H | H | 2.91 ± 0.21 | 3.11 ± 0.42 |
| PX-D-M2 | meta- | H | H | CH3 | 4.51 ± 0.57 | 2.17 ± 0.29 |
| PX-D-M 3 | meta- | H | H | OCH3 | 3.77 ± 0.55 | 2.57 ± 0.37 |
| PX-D-M 4 | meta- | H | Cl | H | 3.61 ± 0.41 | 1.27 ± 0.18 |
| PX-D-M5 | meta- | H | OCH3 | OCH3 | 4.99 ± 0.75 | 2.36 ± 0.31 |
| PX-D-M6 | meta- | CH3 | H | H | 2.03 ± 0.25 | 2.26 ± 0.29 |
| PX-D-M7 | meta- | CH3 | H | CH3 | 2.78 ± 0.33 | 2.57 ± 0.31 |
| PX-D-M8 | meta- | CH3 | H | OCH3 | 3.05 ± 0.36 | 2.86 ± 0.21 |
| PX-D-M9 | meta- | CH3 | Cl | H | 2.44 ± 0.21 | 0.76 ± 0.11 |
| PX-D-M10 | meta- | CH3 | OCH3 | OCH3 | 3.59 ± 0.36 | 2.54 ± 0.31 |
| PX-E-P1 | para- | H | H | H | 4.97 ± 0.59 | 8.11 ± 1.03 |
| PX-E-P2 | para- | H | H | F | 3.41 ± 0.47 | 9.36 ± 1.21 |
| PX-E-P3 | para- | H | CF3 | H | 5.18 ± 0.67 | 4.22 ± 0.55 |
| PX-E-P4 | para- | H | Cl | H | 6.43 ± 0.98 | 1.75 ± 0.15 |
| PX-E-P5 | para- | CH3 | H | H | 1.46 ± 0.17 | >10 |
| PX-E-P6 | para- | CH3 | H | F | 0.79 ± 0.11 | 5.04 ± 0.53 |
| PX-E-P7 | para- | CH3 | CF3 | H | 1.98 ± 0.15 | >10 |
| PX-E-P8 | para- | CH3 | Cl | H | 0.85 ± 0.11 | 0.63 ± 0.11 |
| PX-E-M1 | meta- | H | H | H | 9.65 ± 0.98 | 3.42 ± 0.32 |
| PX-E-M2 | meta- | H | H | F | 7.23 ± 0.86 | 2.48 ± 0.23 |
| PX-E-M3 | meta- | H | CF3 | H | 7.44 ± 0.91 | 2.63 ± 0.31 |
| PX-E-M4 | meta- | H | Cl | H | 9.58 ± 1.13 | >10 |
| PX-E-M5 | meta- | H | OCH3 | OCH3 | >10 | 2.11 ± 0.19 |
| PX-E-M6 | meta- | CH3 | H | H | 5.25 ± 0.65 | 3.72 ± 0.52 |
| PX-E-M7 | meta- | CH3 | H | F | 7.74 ± 0.92 | 2.58 ± 0.37 |
| PX-E-M8 | meta- | CH3 | CF3 | H | 7.1 ± 0.84 | 4.76 ± 0.68 |
| PX-E-M9 | meta- | CH3 | Cl | H | 9.18 ± 1.23 | 2.93 ± 0.41 |
| PX-E-M10 | meta- | CH3 | OCH3 | OCH3 | 6.24 ± 0.77 | 2.55 ± 0.24 |
| Parameters | PX-D-P6 | PX-E-P8 |
|---|---|---|
| Tmax (h) | 0.51 ± 0.15 | 0.45 ± 0.21 |
| Cmax (ng/mL) | 2112.6 ± 508.4 | 1642.1 ± 435.9 |
| t1/2 (h) | 4.51 ± 0.61 | 3.27 ± 0.39 |
| AUC0→t (ng/mL·h) | 30526.2 ± 1206.7 | 15564.2 ± 1537.4 |
| CL (L/h/kg) | 5.47± 1.23 | 10.28 ± 2.19 |
| Compound | Plasma (ng/mL) | Brain (ng/g) | B/P Ratio |
|---|---|---|---|
| PX-D-P6 | 1126 ± 207 | 1289 ± 132 | 1.14 |
| PX-E-P8 | 915 ± 231 | 887 ± 113 | 0.97 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Han, C.; Xu, Y.; Wu, K.; Chen, S.; Hu, M.; Wang, L.; Ye, Y.; Ye, F. Synthesis and Evaluation of Phenylxanthine Derivatives as Potential Dual A2AR Antagonists/MAO-B Inhibitors for Parkinson’s Disease. Molecules 2017, 22, 1010. https://doi.org/10.3390/molecules22061010
Wang X, Han C, Xu Y, Wu K, Chen S, Hu M, Wang L, Ye Y, Ye F. Synthesis and Evaluation of Phenylxanthine Derivatives as Potential Dual A2AR Antagonists/MAO-B Inhibitors for Parkinson’s Disease. Molecules. 2017; 22(6):1010. https://doi.org/10.3390/molecules22061010
Chicago/Turabian StyleWang, Xuebao, Chao Han, Yong Xu, Kaiqi Wu, Shuangya Chen, Mangsha Hu, Luyao Wang, Yun Ye, and Faqing Ye. 2017. "Synthesis and Evaluation of Phenylxanthine Derivatives as Potential Dual A2AR Antagonists/MAO-B Inhibitors for Parkinson’s Disease" Molecules 22, no. 6: 1010. https://doi.org/10.3390/molecules22061010