
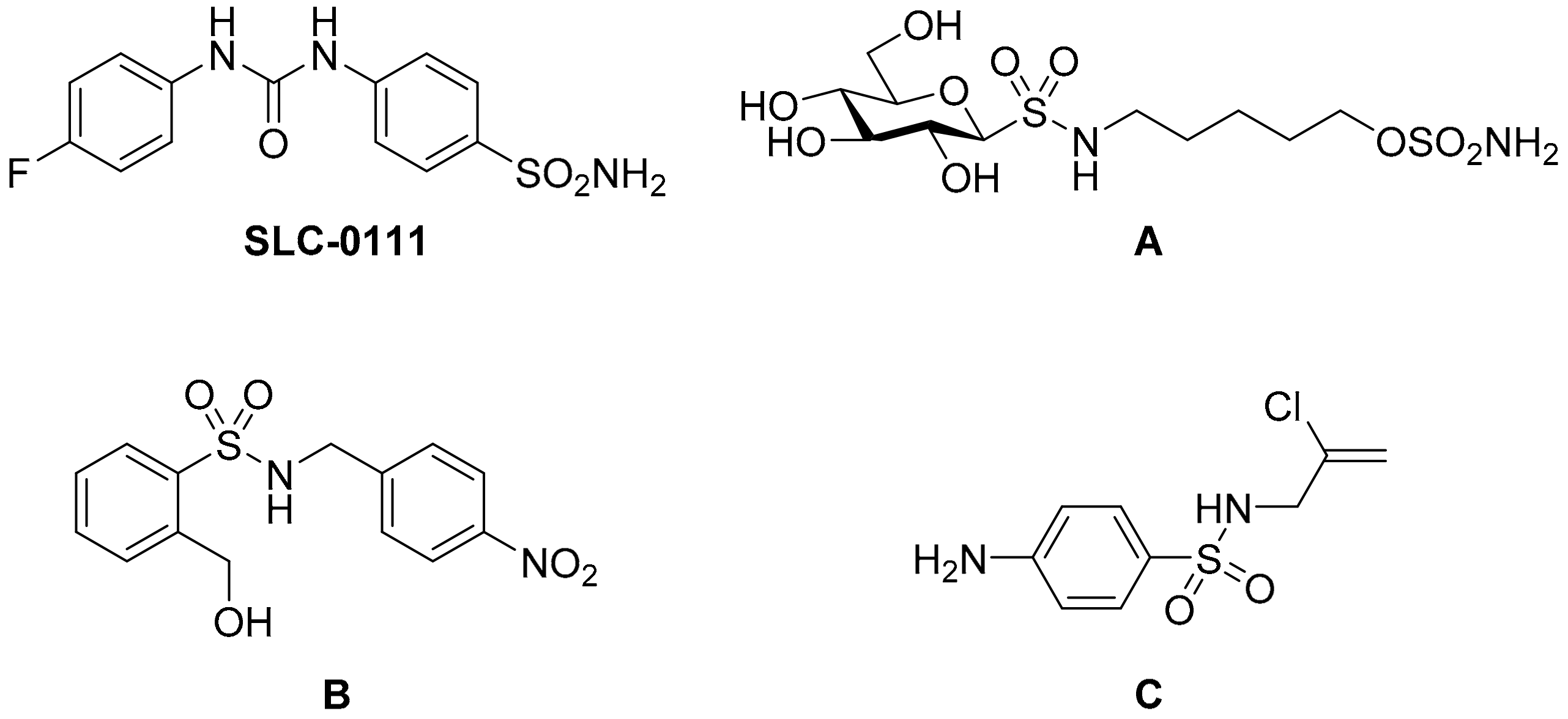
Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides

 and
and
Abstract
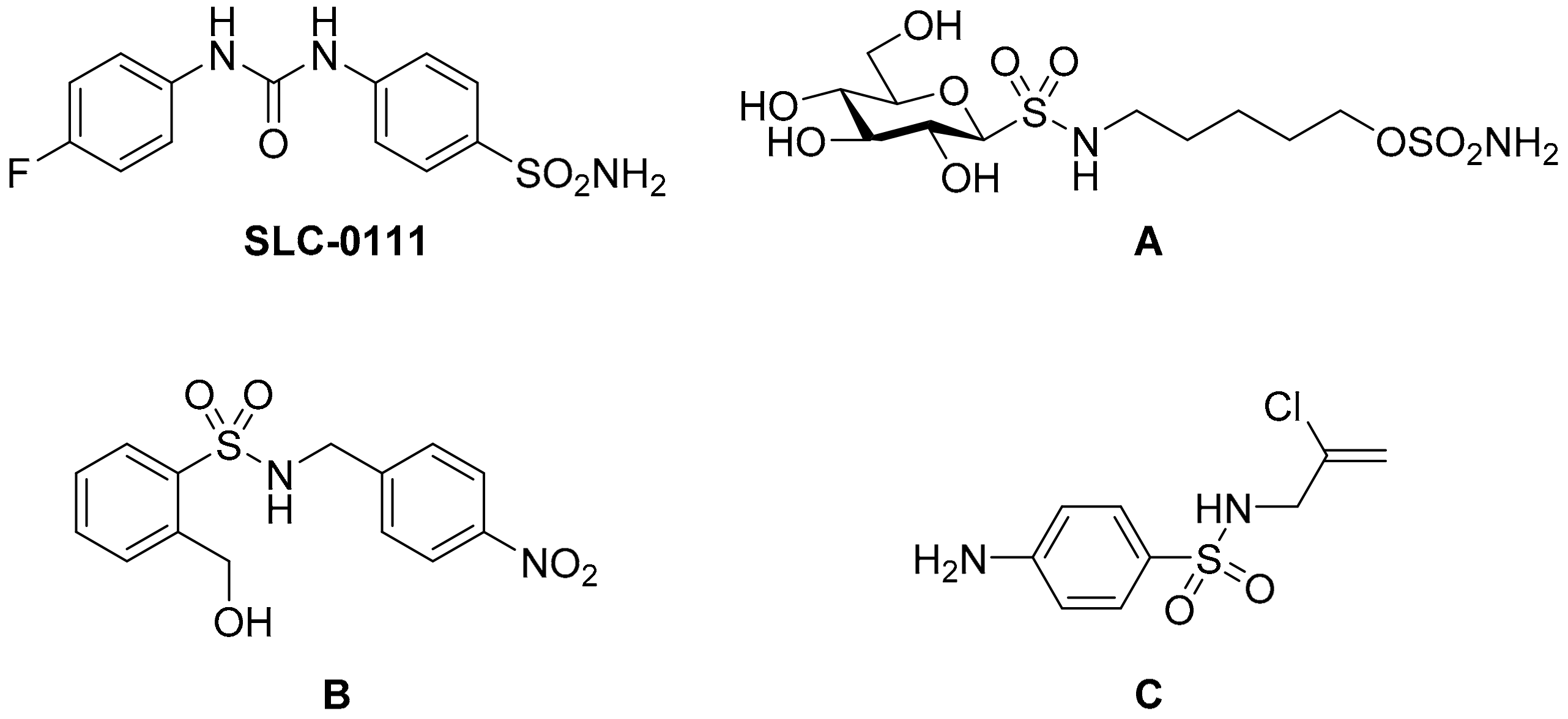
:1. Introduction
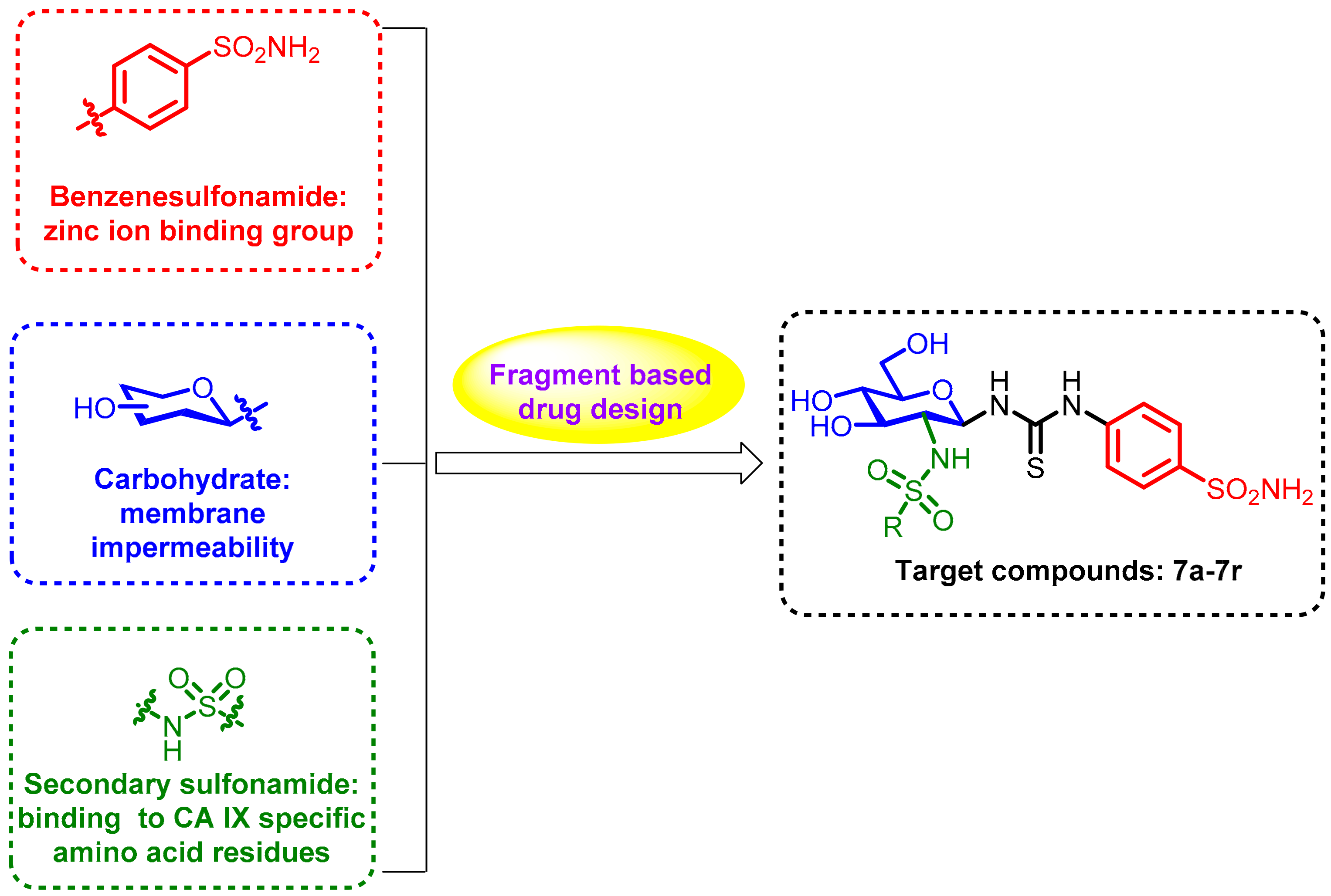
2. Results and Discussion
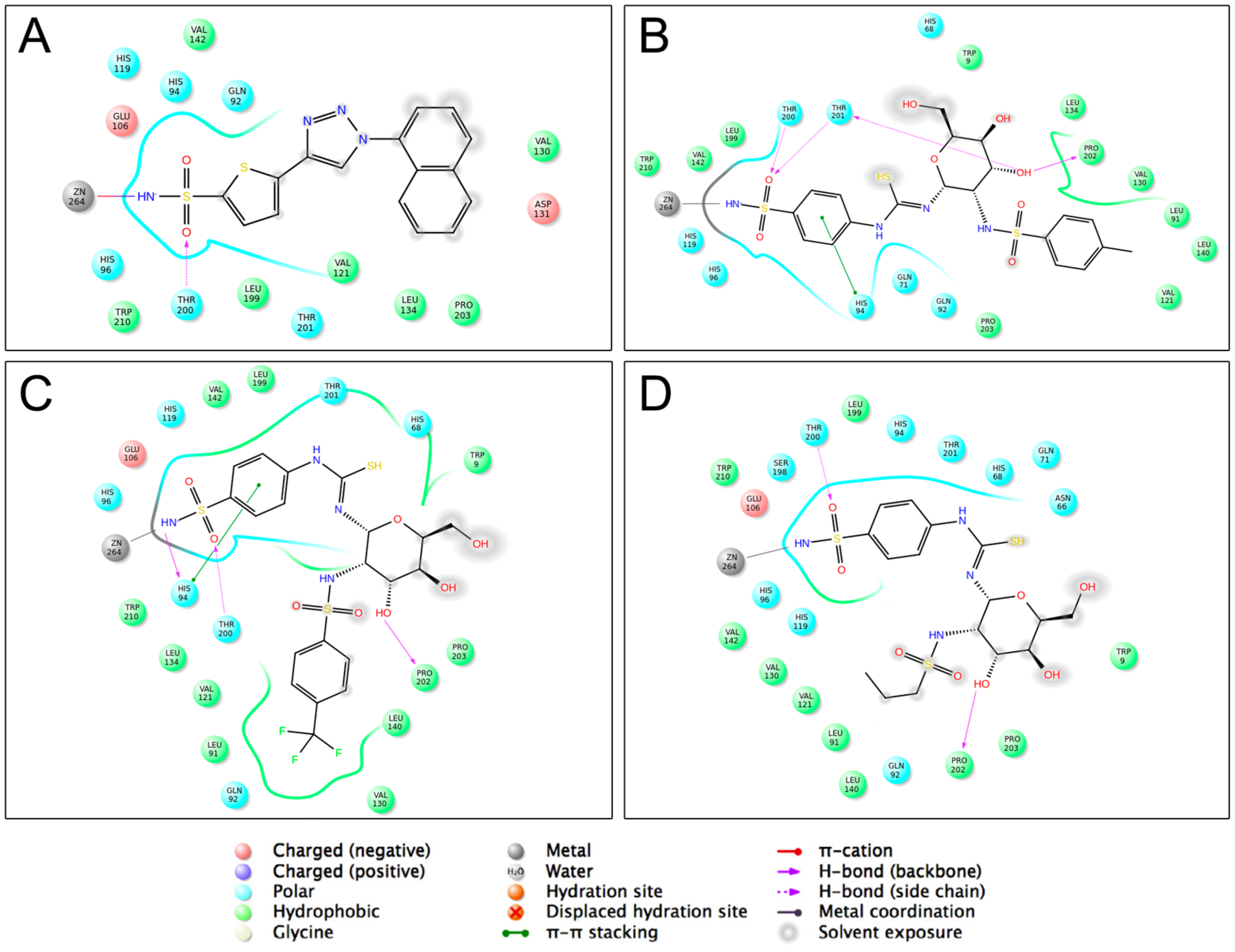
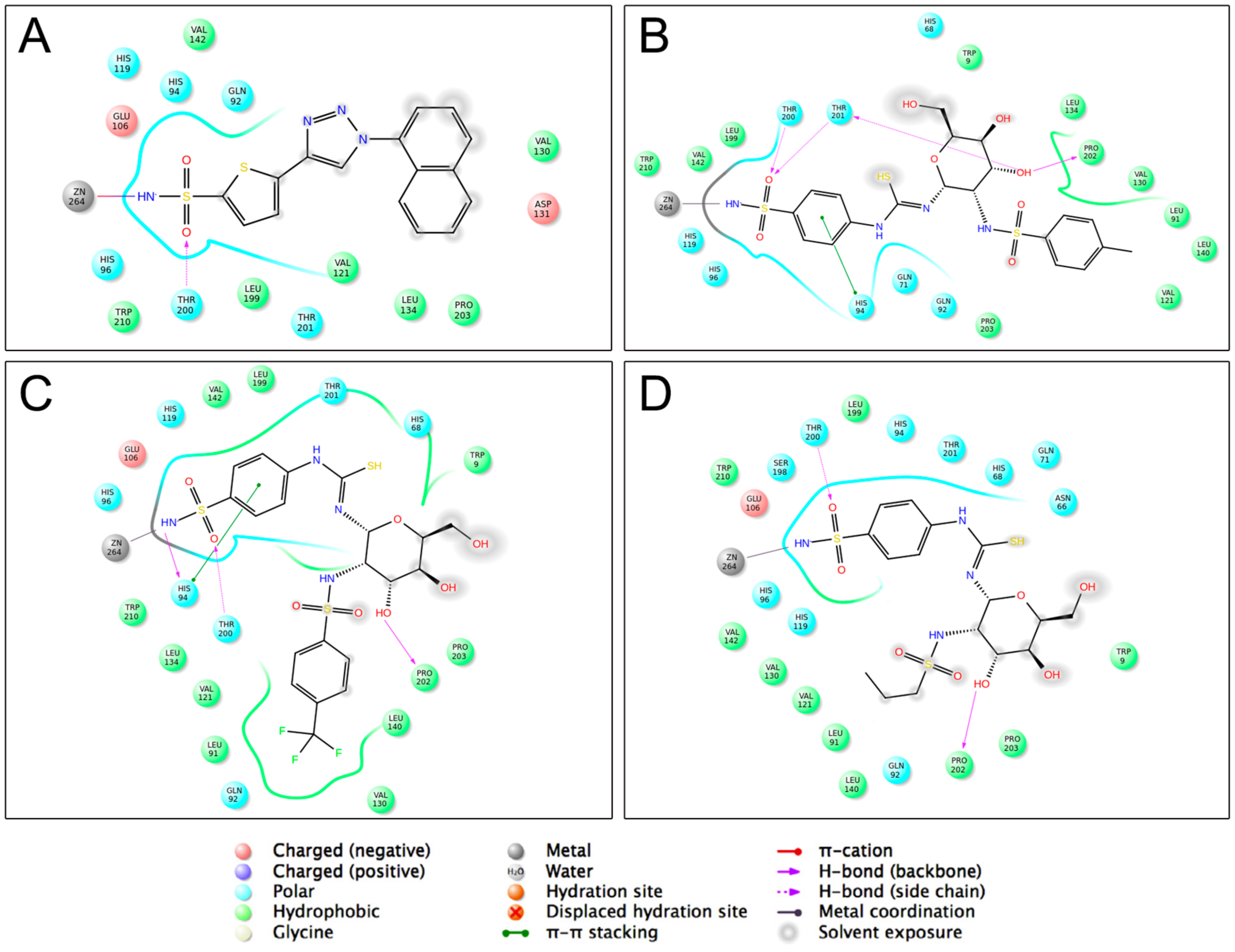
2.1. Molecular Docking Studies
2.2. Predictions by cLog P and TPSA
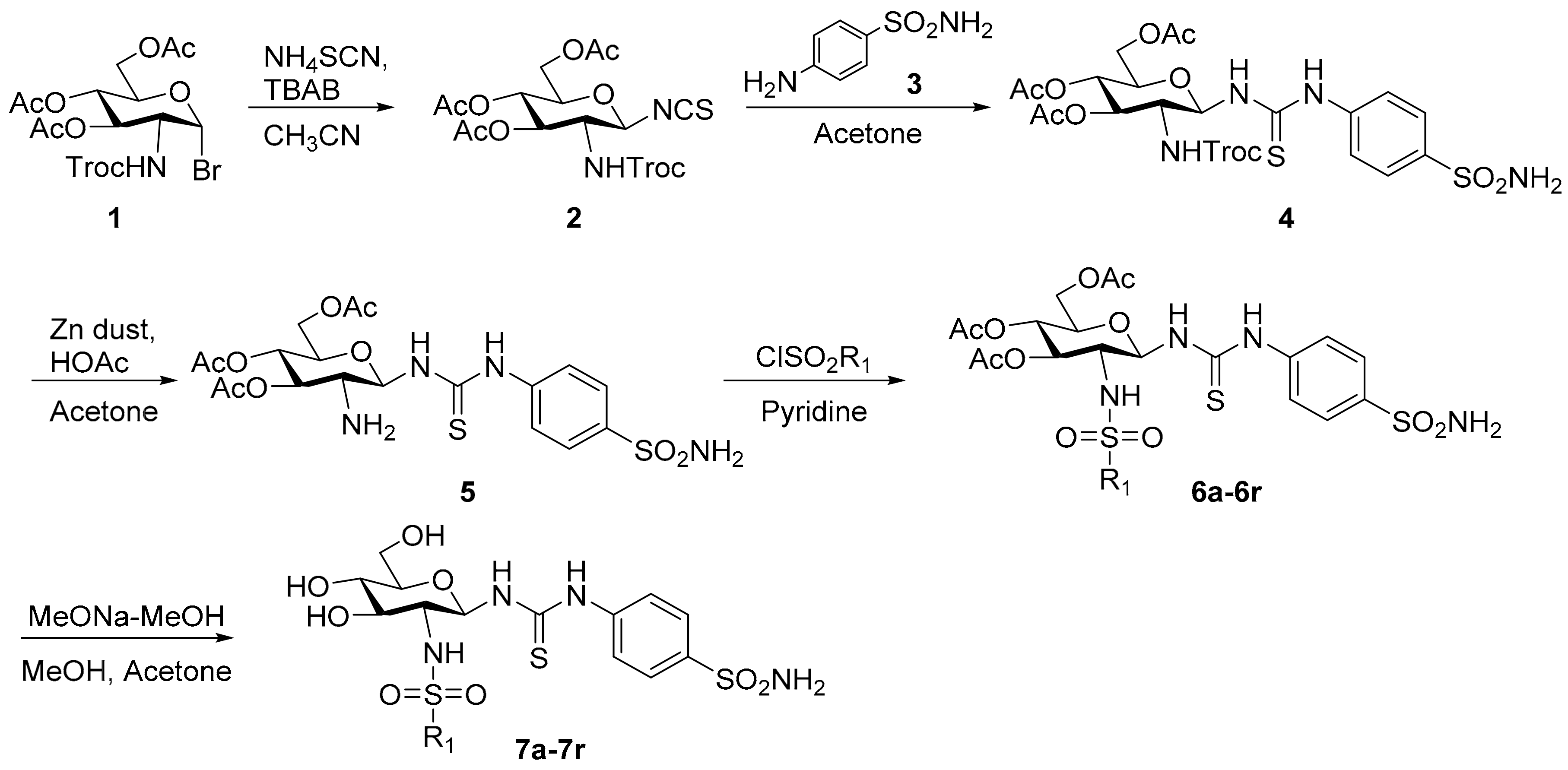
2.3. Chemistry
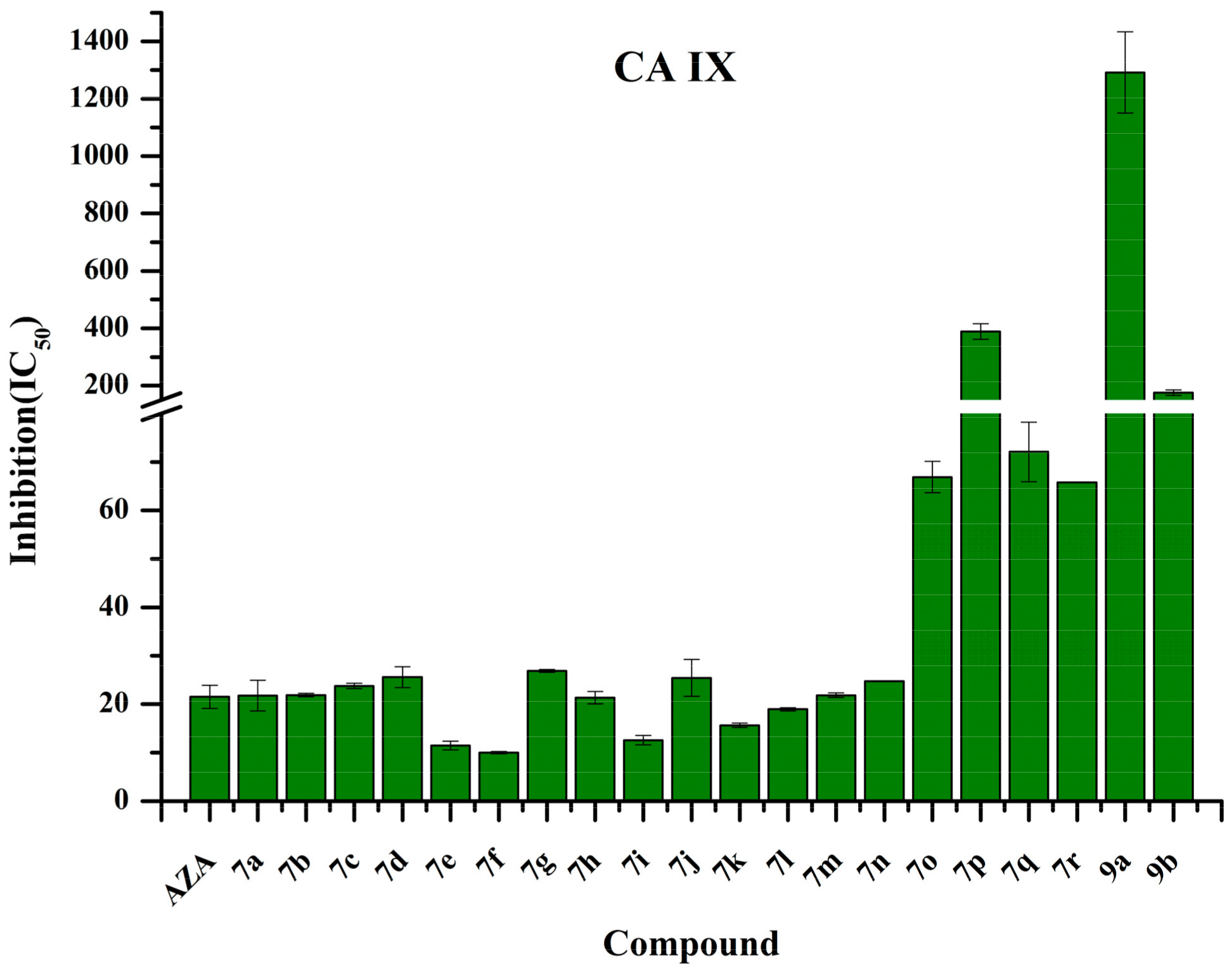
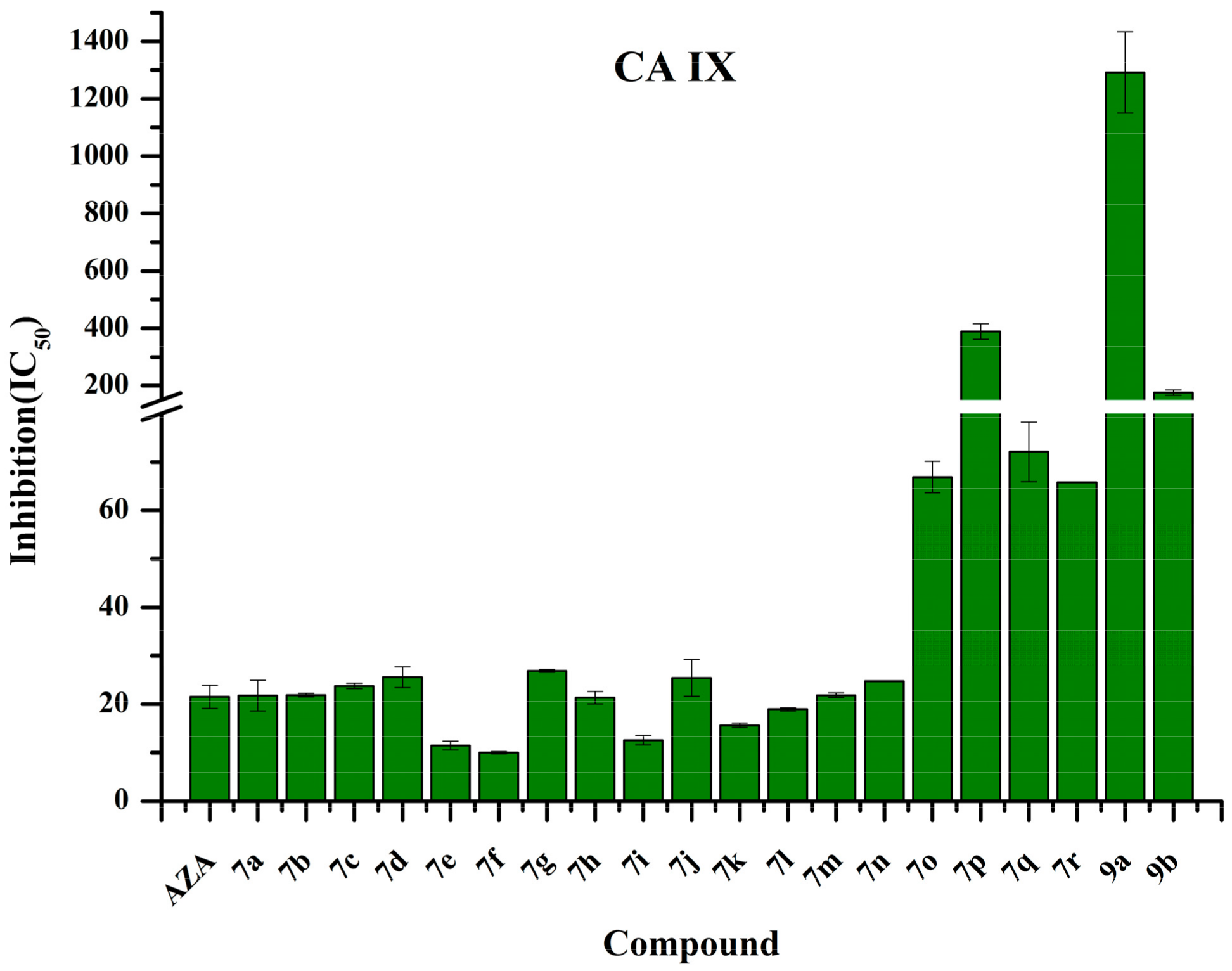
2.4. Human Carbonic Anhydrase IX Inhibition Studies
3. Materials and Methods
3.1. Molecular Docking and Predictions by cLog P and TPSA
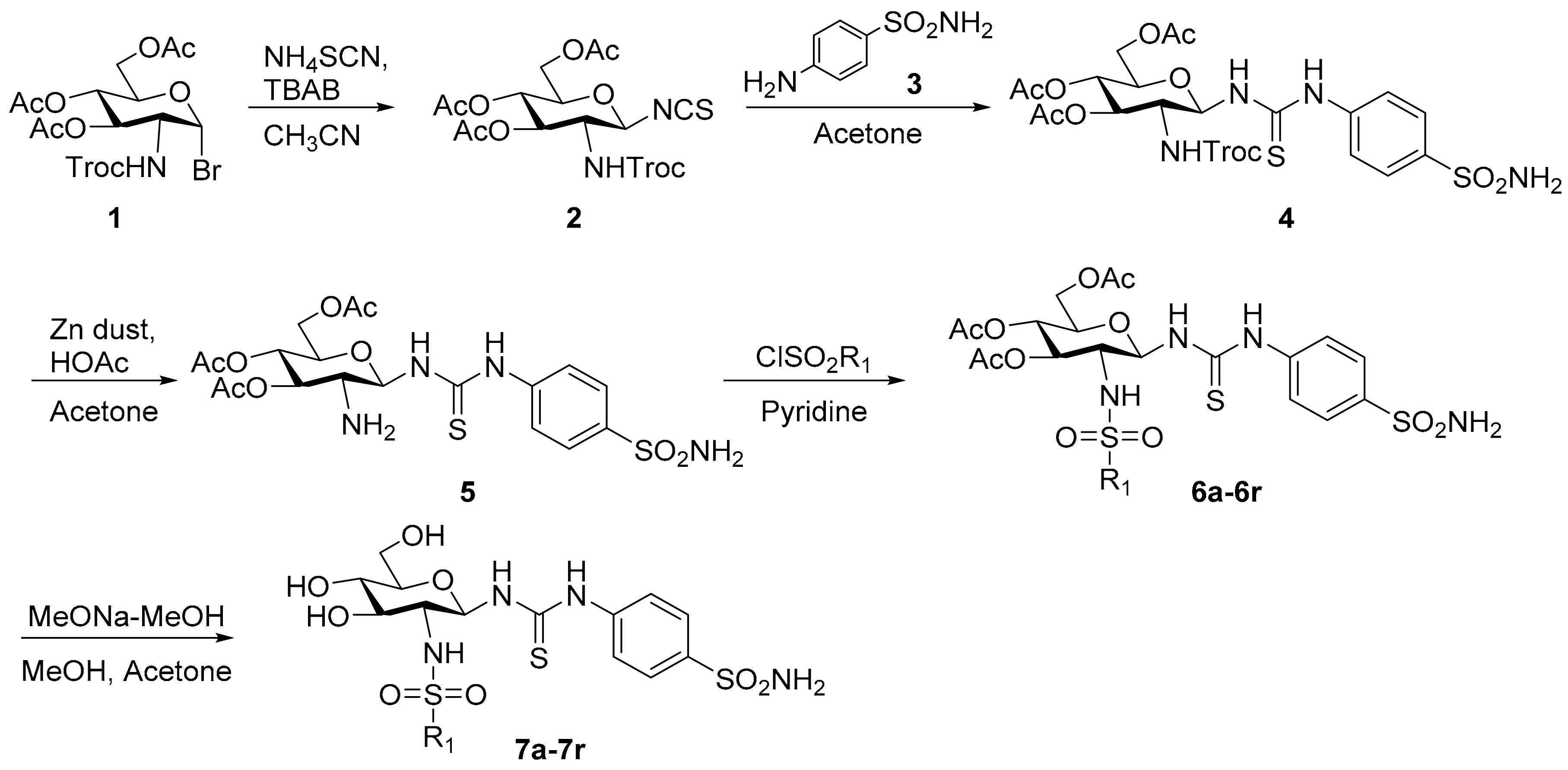
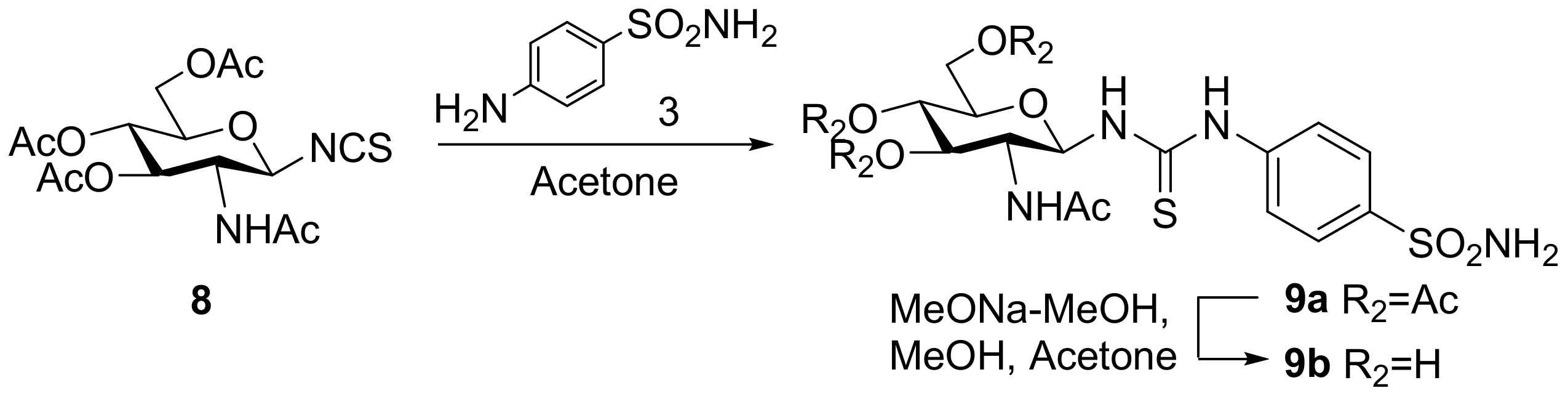
3.2. Chemistry
3.2.1. 2-(2,2,2-Trichloroethoxycarbonylamino)-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl isothiocyanate (2)
3.2.2. N-[4-(Aminosulfonyl)phenyl]-N′-[2′-(2′,2′,2′-trichloroethoxycarbonylamino)-3′,4′,6′-tri-O-acetyl-2′-deoxy-β-d-glucopyranosyl]thiourea (4)
3.2.3. General Procedure for the Synthesis of Compounds 7a–7r
3.2.4. N-[4-(Aminosulfonyl)phenyl]-N′-(2′-acetylamino-3′,4′,6′-tri-O-acetyl-2′-deoxy-β-d-glucopyranosyl)thiourea (9a)
3.2.5. N-[4-(Aminosulfonyl)phenyl]-N′-(2′-acetylamino-2′-deoxy-β-d-glucopyranosyl)thiourea (9b)
3.3. Carbonic Anhydrase Inhibition Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Rami, M.; Scozzafava, A.; Montero, J.L.; Supuran, C.T. Carbonic anhydrase IX: A new druggable target for the design of antitumor agents. Med. Res. Rev. 2008, 28, 445–463. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Supuran, C.T.; Krasavin, M. Multicomponent chemistry in the synthesis of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Karioti, A.; Carta, F.; Supuran, C.T. Phenols and polyphenols as carbonic anhydrase inhibitors. Molecules 2016, 21, 1649. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Pinard, M.A.; McKenna, R. Targeting carbonic anhydrase IX activity and expression. Molecules 2015, 20, 2323–2348. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Zhang, G.J.; Wang, X.M.; Li, X.F. Is carbonic anhydrase IX a validated target for molecular imaging of cancer and hypoxia? Future Oncol. 2015, 11, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Brahimi-Horn, M.C.; Pouysségur, J. Membrane-bound carbonic anhydrases are key pH regulators controlling tumor growth and cell migration. Adv. Enzyme Regul. 2010, 50, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; Mahon, B.P.; McKenna, R.; Carta, F.; Supuran, C.T. Kinetic and X-ray crystallographic investigations on carbonic anhydrase isoforms I, II, IX and XII of a thioureido analog of SLC-0111. Bioorg. Med. Chem. 2016, 24, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.; McKenna, R. Carbonic anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Thiry, A.; Supuran, C.T.; Masereel, B.; Dogne, J.M. Recent developments of carbonic anhydrase inhibitors as potential anticancer drugs. J. Med. Chem. 2008, 51, 3051–3056. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Poulsen, S.A.; Supuran, C.T. Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med. Res. Rev. 2009, 29, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Carroux, C.J.; Rankin, G.M.; Moeker, J.; Bornaghi, L.F.; Katneni, K.; Morizzi, J.; Charman, S.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. A prodrug approach toward cancer-related carbonic anhydrase inhibition. J. Med. Chem. 2013, 56, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Ombouma, J.; Vullo, D.; Supuran, C.T.; Winum, J.Y. Ferrier sulfamidoglycosylation of glycals catalyzed by nitrosonium tetrafluoroborate: Towards new carbonic anhydrase glycoinhibitors. Bioorg. Med. Chem. 2014, 22, 6353–6359. [Google Scholar] [CrossRef] [PubMed]
- Riafrecha, L.E.; Rodríguez, O.M.; Vullo, D.; Supuran, C.T.; Colinas, P.A. Attachment of carbohydrates to methoxyaryl moieties leads to highly selective inhibitors of the cancer associated carbonic anhydrase isoforms IX and XII. Bioorg. Med. Chem. 2014, 22, 5308–5314. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Colinas, P.A.; Supuran, C.T. Glycosidic carbonic anhydrase IX inhibitors: A sweet approach against cancer. Bioorg. Med. Chem. 2013, 21, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Moeker, J.; Mahon, B.P.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; McKenna, R.; Poulsen, S.A. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J. Med. Chem. 2014, 57, 8635–8645. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Carta, F.; Supuran, C.T. Secondary and tertiary sulfonamides: A patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Coviello, V.; Marchi, B.; Sartini, S.; Quattrini, L.; Marini, A.M.; Simorini, F.; Taliani, S.; Salerno, S.; Orlandi, P.; Fioravanti, A.; et al. 1,2-Benzisothiazole derivatives bearing 4-, 5-, or 6-alkyl/arylcarboxamide moieties inhibit carbonic anhydrase isoform IX (CAIX) and cell proliferation under hypoxic conditions. J. Med. Chem. 2016, 59, 6547–6552. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Secci, D.; De Monte, C.; Mollica, A.; Ceruso, M.; Akdemir, A.; Sobolev, A.P.; Codispoti, R.; De Cosmi, F.; Guglielmi, P.; et al. A novel library of saccharin and acesulfame derivatives as potent and selective inhibitors of carbonic anhydrase IX and XII isoforms. Bioorg. Med. Chem. 2016, 24, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzio, M.; Guglielmi, P.; Carradori, S.; Secci, D.; Florio, R.; Mollica, A.; Ceruso, M.; Akdemir, A.; Sobolev, A.P.; Supuran, C.T. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related caibonic anhydrase IX and XII isoforms. J. Enzyme Inhib. Med. Chem. 2017, 33, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Compain, G.; Martin-Mingot, A.; Maresca, A.; Thibaudeau, S.; Supuran, C.T. Superacid synthesis of halogen containing N-substituted-4-aminobenzene sulfonamides: New selective tumor-associated carbonic anhydrase inhibitors. Bioorg. Med. Chem. 2013, 21, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Academic Press: London, UK, 2008; pp. 43–47. [Google Scholar]
- Hron, R.J.; Jursic, B.C.; Neumann, D.M. Synthesis of N-aryl and N-arylcarbamoylamino derivatives of 1,3-diazinane-5-carboxamide and their activity against glioblastoma LN-229 cell line. Bioorg. Med. Chem. 2016, 24, 6183–6193. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.A.; Pratt, M.R.; Hruby, V.J.; Polt, R. Solid-phase synthesis of O-linked glycopeptide analogues of enkephalin. J. Org. Chem. 2001, 66, 2327–2342. [Google Scholar] [CrossRef] [PubMed]
- Moeker, J.; Teruya, K.; Rossit, S.; Wilkinson, B.L.; Lopez, M.; Bornaghi, L.F.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. Design and synthesis of thiourea compounds that inhibit transmembrane anchored carbonic anhydrases. Bioorg. Med. Chem. 2012, 20, 2392–2404. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.F.; Cui, S.S.; Luan, W.J.; Wang, S.; Hou, Z.; Liu, Y.X.; Liu, Y.; Cheng, M.S. Natural product-based design, synthesis and biological evaluation of Albiziabioside A derivatives that selectively induce HCT116 cell death. Eur. J. Med. Chem. 2016, 113, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Wang, Y.S.; Liu, X.; Cheng, M.S. Syntheses and structure-activity relationship studies of N-substituted-β-d-glucosaminides as selective cytotoxic agents. Bioorg. Med. Chem. Lett. 2012, 22, 7110–7113. [Google Scholar] [CrossRef] [PubMed]
- Liav, A.; Angala, S.K.; Brennan, P.J. N-Glycosyl-N′-[p-(isoamyloxy)phenyl]-thiourea derivatives: Potential anti-TB therapeutic agents. Synth. Commun. 2008, 38, 1176–1183. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio User Manual; Accelrys Inc.: San Diego, CA, USA, 2008.
- Verpoorte, J.A.; Meiita, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 7a–7r and 9a–9b are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | Docking Scores (Kcal/mol) | cLog P | TPSA | hCA IX (IC50, nM) a,b | |
|---|---|---|---|---|---|---|---|
| Autodock | X-Score | ||||||
| AZA c | −6.34 | −6.81 | −0.9809 | 113.77 | 21.52 ± 2.36 | ||
| 7a | H |  | −7.46 | −8.56 | 0.1321 | 205.55 | 21.75 ± 3.18 |
| 7b | H |  | −7.67 | −9.03 | 0.1321 | 205.55 | 21.90 ± 0.37 |
| 7c | H |  | −7.38 | −8.39 | −0.0893 | 214.48 | 23.78 ± 0.51 |
| 7d | H |  | −7.10 | −8.33 | 0.0785 | 205.55 | 25.57 ± 2.15 |
| 7e | H |  | −7.05 | −8.08 | 0.3273 | 205.55 | 11.47 ± 0.93 |
| 7f | H |  | −7.10 | −8.66 | 1.0453 | 205.55 | 10.01 ± 0.23 |
| 7g | H |  | −7.17 | −8.75 | 0.6485 | 205.55 | 26.85 ± 0.28 |
| 7h | H |  | −7.53 | −8.81 | 0.7985 | 205.55 | 21.34 ± 1.29 |
| 7i | H |  | −7.52 | −8.72 | 0.7985 | 205.55 | 12.56 ± 0.96 |
| 7j | H |  | −8.63 | −8.51 | −3.6589 | 248.37 | 25.42 ± 3.80 |
| 7k | H |  | −8.51 | −8.23 | −3.6589 | 248.37 | 15.63 ± 0.44 |
| 7l | H |  | −8.17 | −7.52 | −0.2319 | 248.37 | 18.95 ± 0.33 |
| 7m | H |  | −7.74 | −7.46 | 0.8071 | 228.49 | 21.86 ± 0.49 |
| 7n | H |  | −8.15 | −8.18 | −1.0982 | 205.55 | 24.78 ± 0.05 |
| 7o | H |  | −6.69 | −8.14 | −0.5692 | 205.55 | 66.88 ± 3.26 |
| 7p | H |  | −6.58 | −7.94 | −1.1367 | 205.55 | 388.80 ± 27.70 |
| 7q | H |  | −7.39 | −8.18 | −0.4524 | 208.90 | 72.09 ± 6.15 |
| 7r | H |  | −7.43 | −8.33 | −3.6589 | 205.55 | 65.80 ± 0.08 |
| 9a |  |  | −7.61 | −8.05 | −0.6134 | 188.25 | 1292.00 ± 141.35 |
| 9b | H |  | −6.16 | −8.68 | −1.6898 | 204.50 | 175.80 ± 9.45 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.-R.; Fan, Z.-F.; Qi, S.-J.; Wang, Y.-S.; Wang, J.; Liu, Y.; Cheng, M.-S. Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides. Molecules 2017, 22, 785. https://doi.org/10.3390/molecules22050785
Li F-R, Fan Z-F, Qi S-J, Wang Y-S, Wang J, Liu Y, Cheng M-S. Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides. Molecules. 2017; 22(5):785. https://doi.org/10.3390/molecules22050785
Chicago/Turabian StyleLi, Feng-Ran, Zhan-Fang Fan, Su-Jiao Qi, Yan-Shi Wang, Jian Wang, Yang Liu, and Mao-Sheng Cheng. 2017. "Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides" Molecules 22, no. 5: 785. https://doi.org/10.3390/molecules22050785