
Multi-Anti-Parasitic Activity of Arylidene Ketones and Thiazolidene Hydrazines against Trypanosoma cruzi and Leishmania spp.

 ,
,  , ,
, ,  ,
,
Abstract
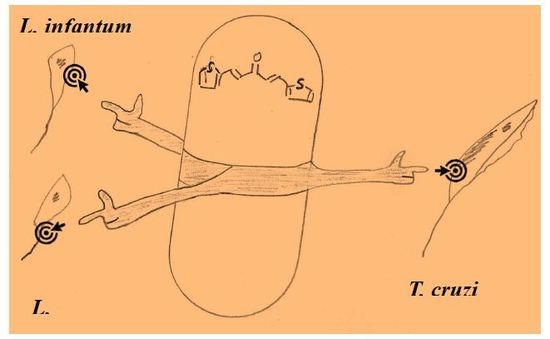
:
1. Introduction
2. Results and Discussion
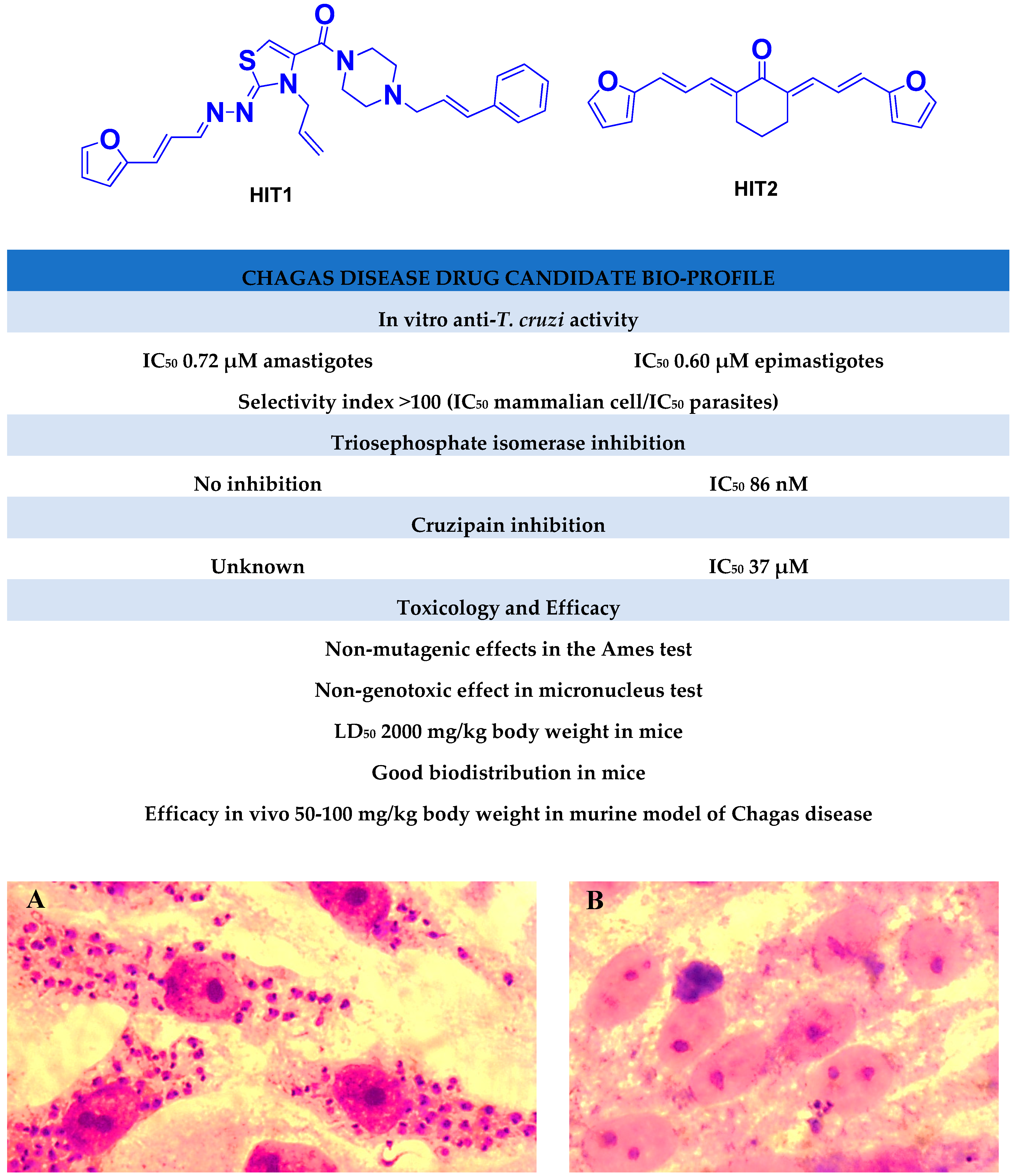
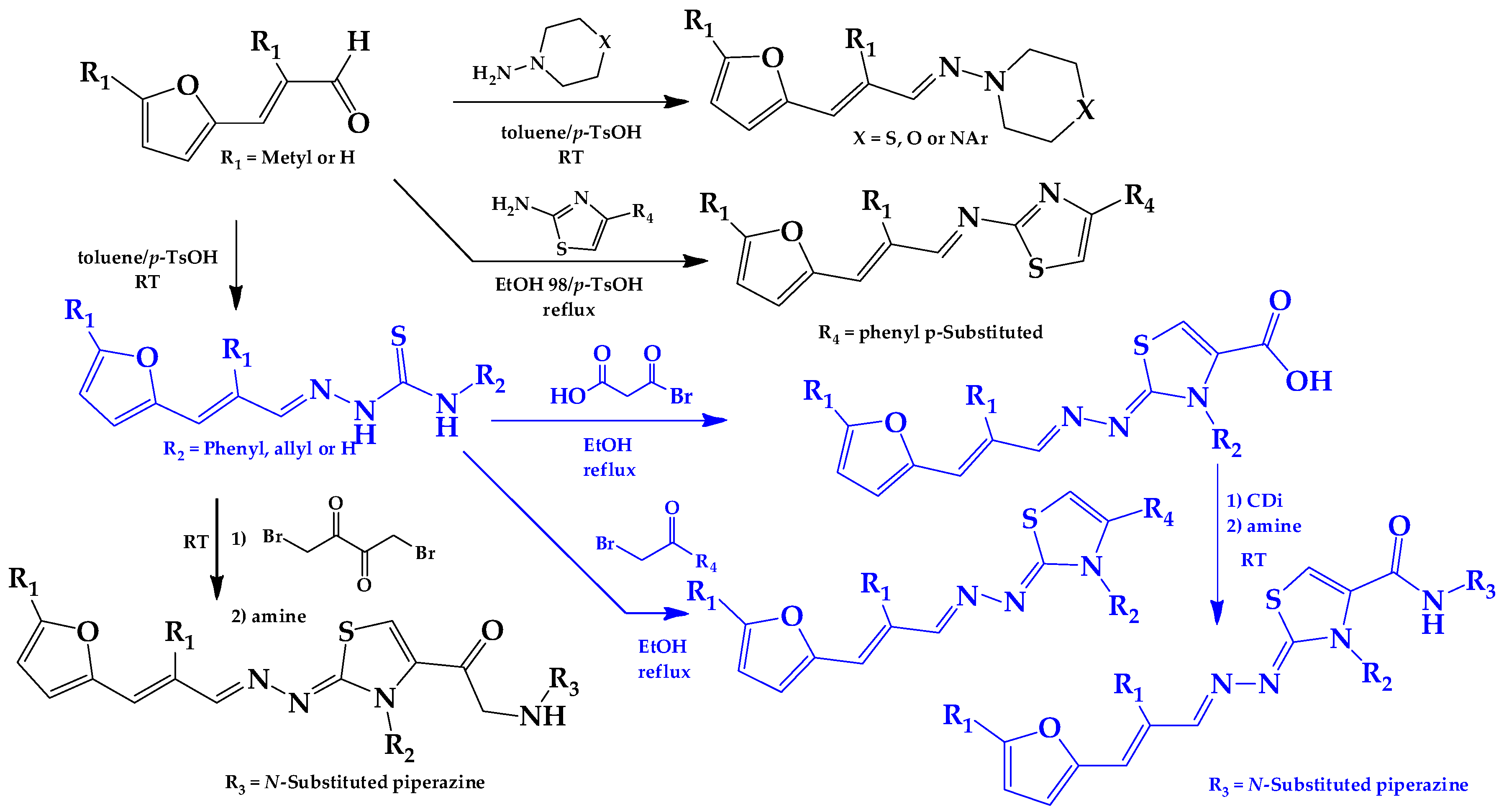
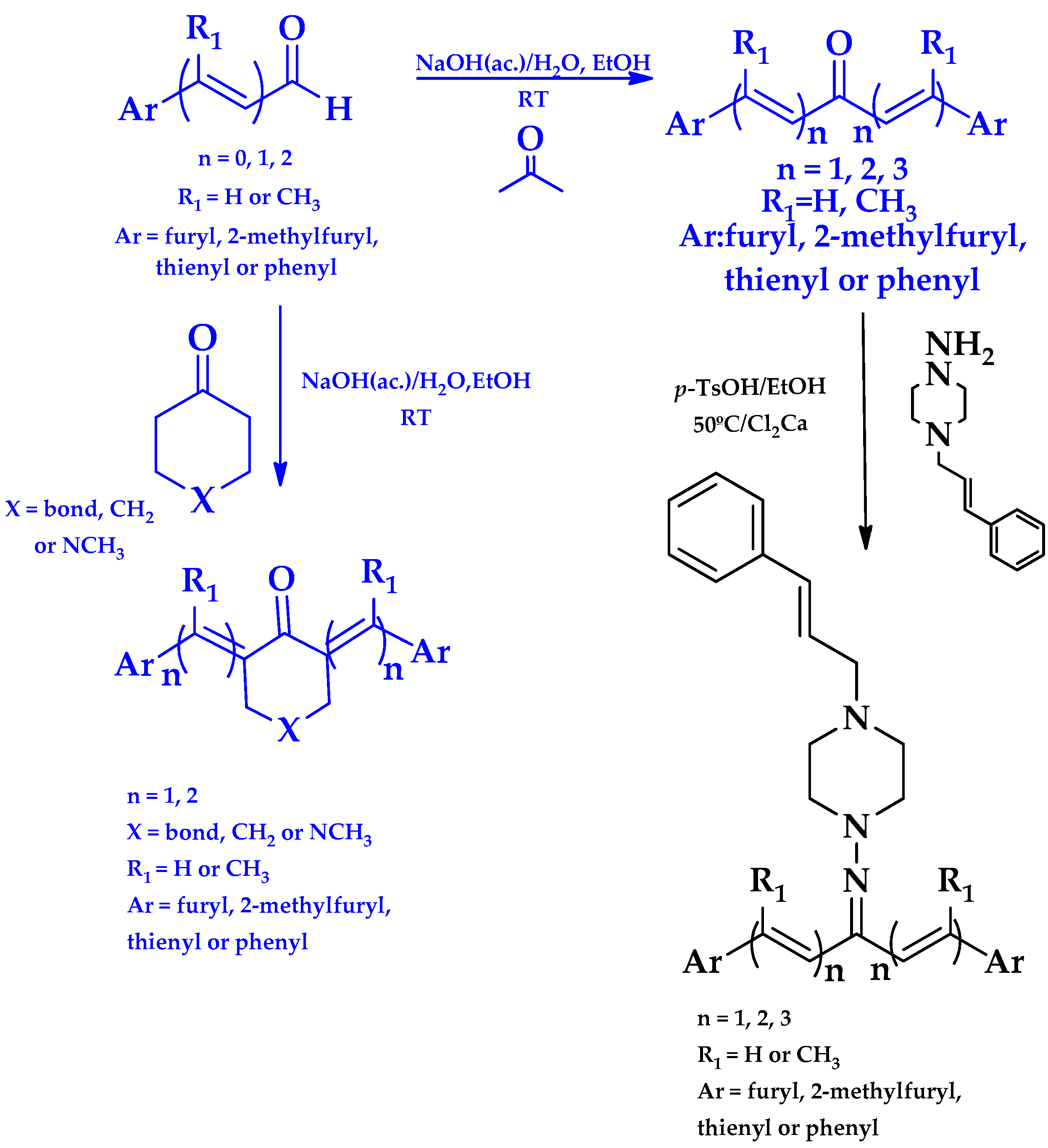
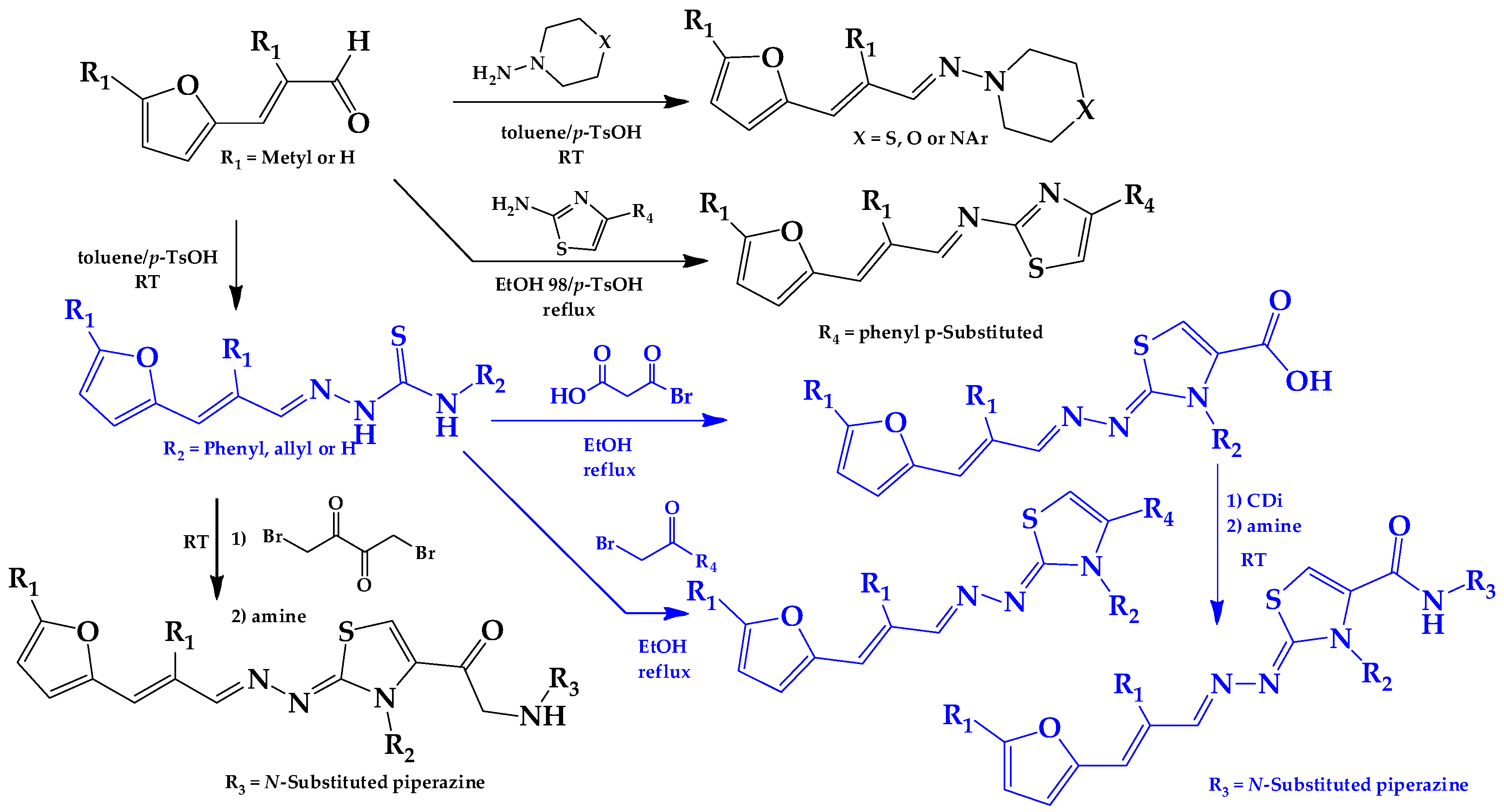
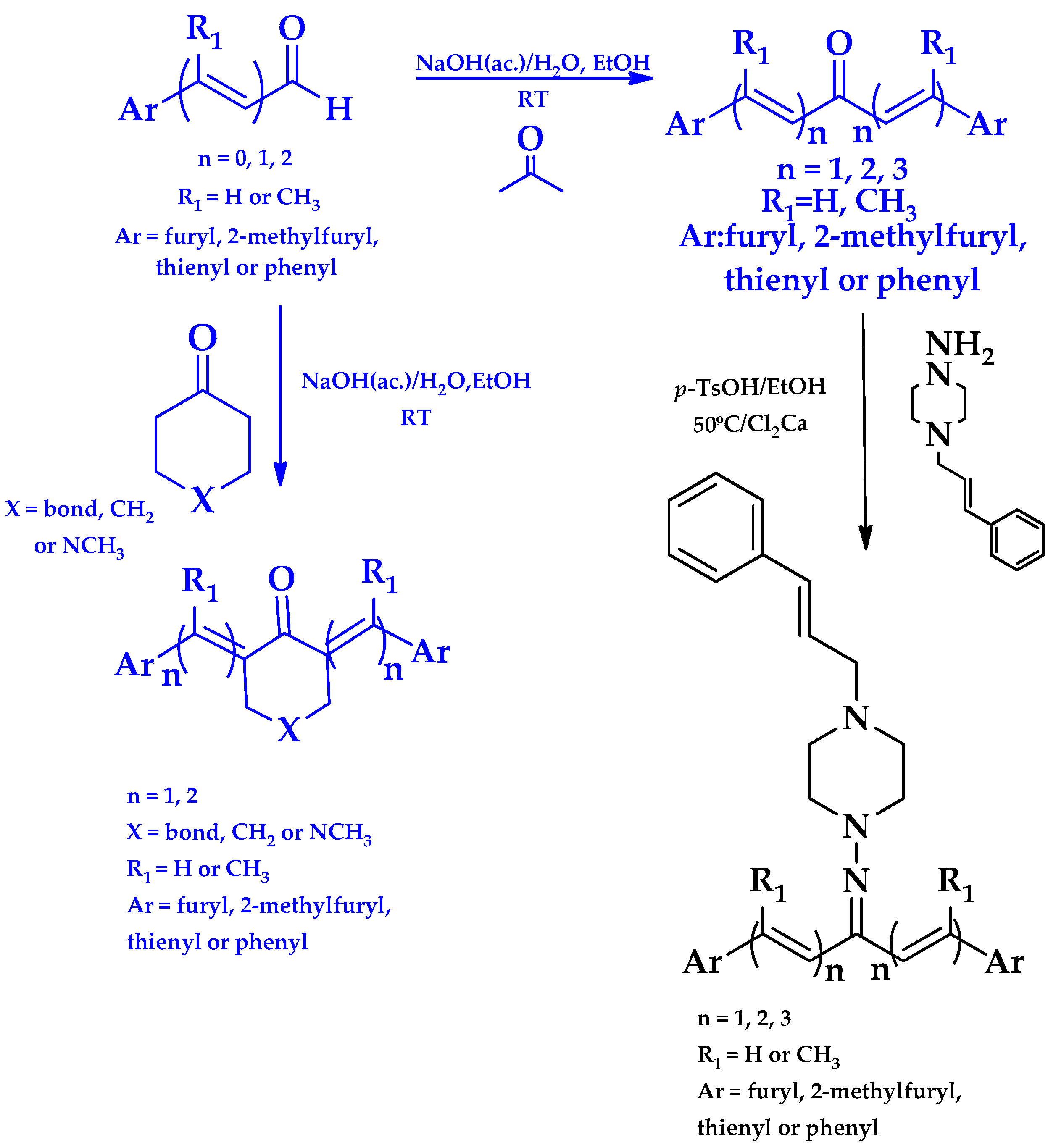
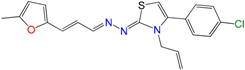
2.1. Synthesis of HIT1 and HIT2 Derivatives
In Vitro Biological Studies
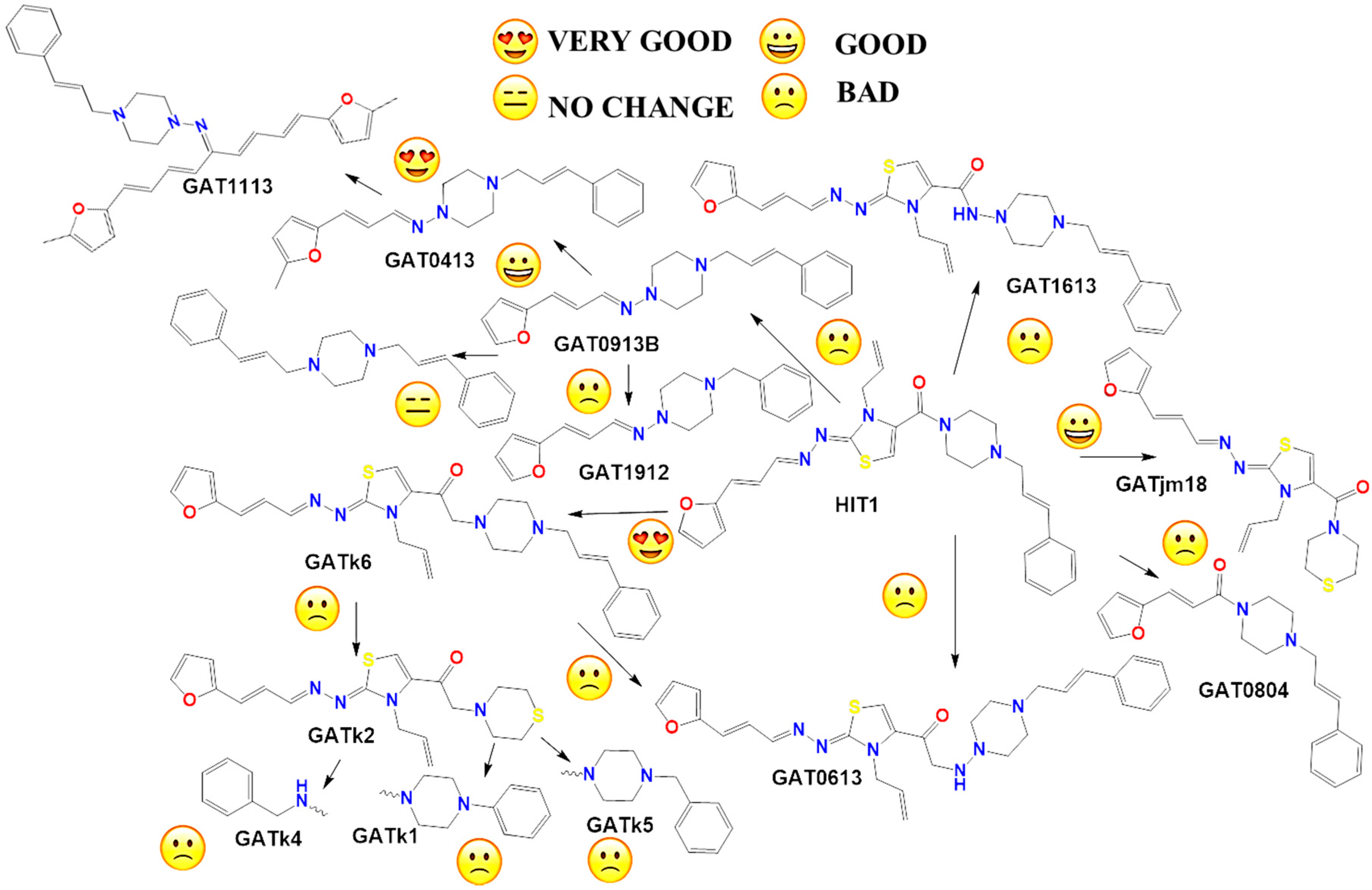
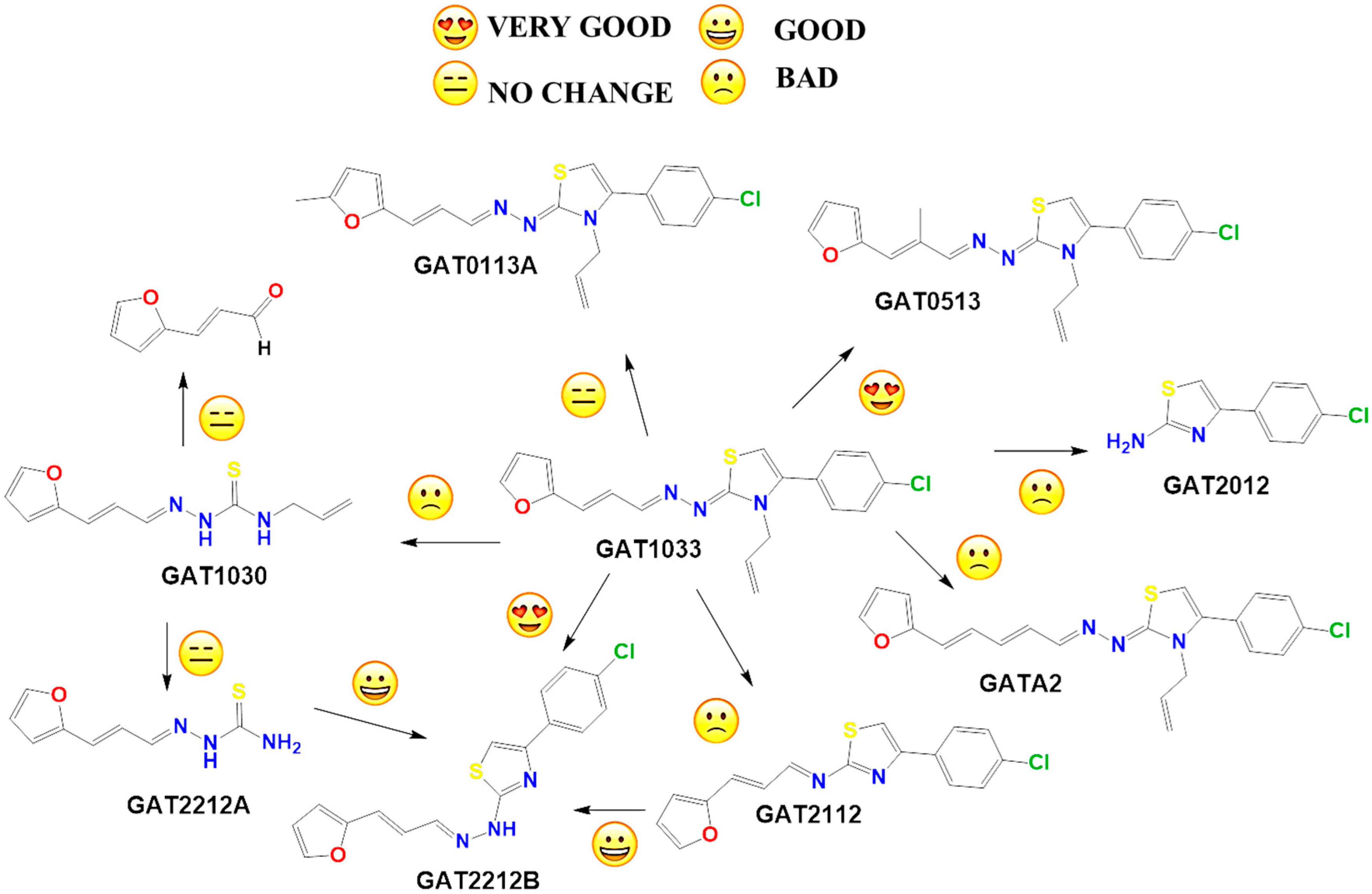
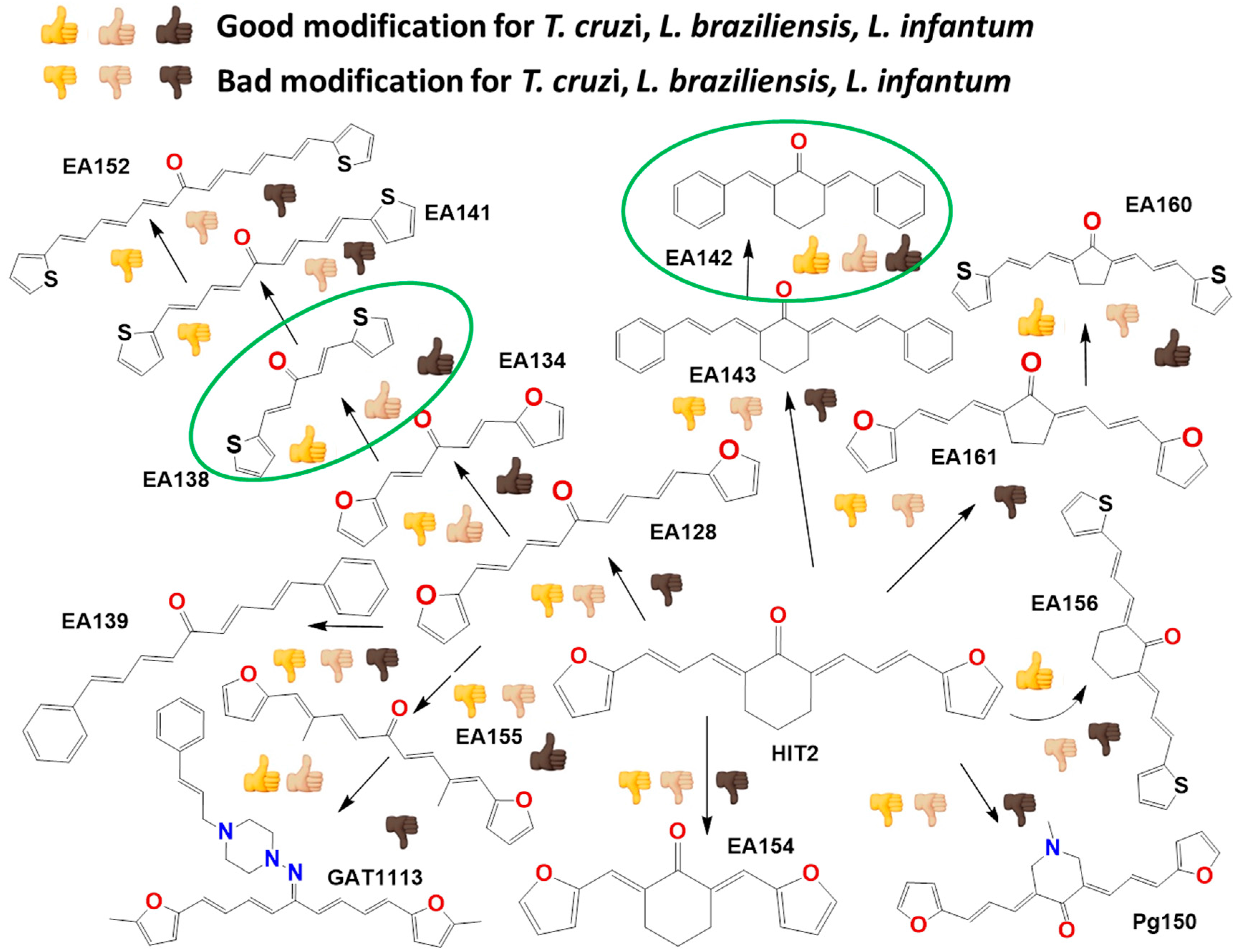
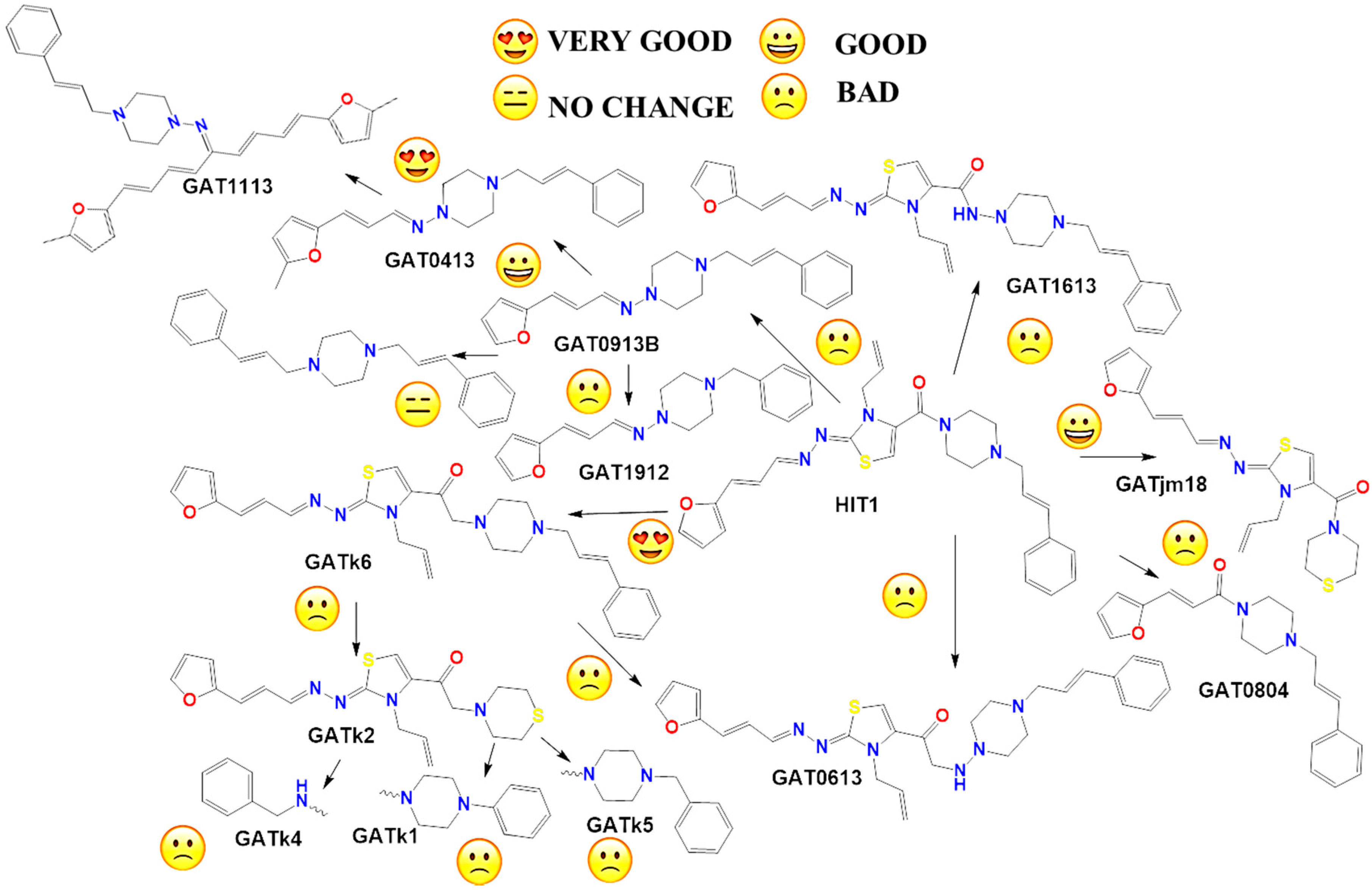
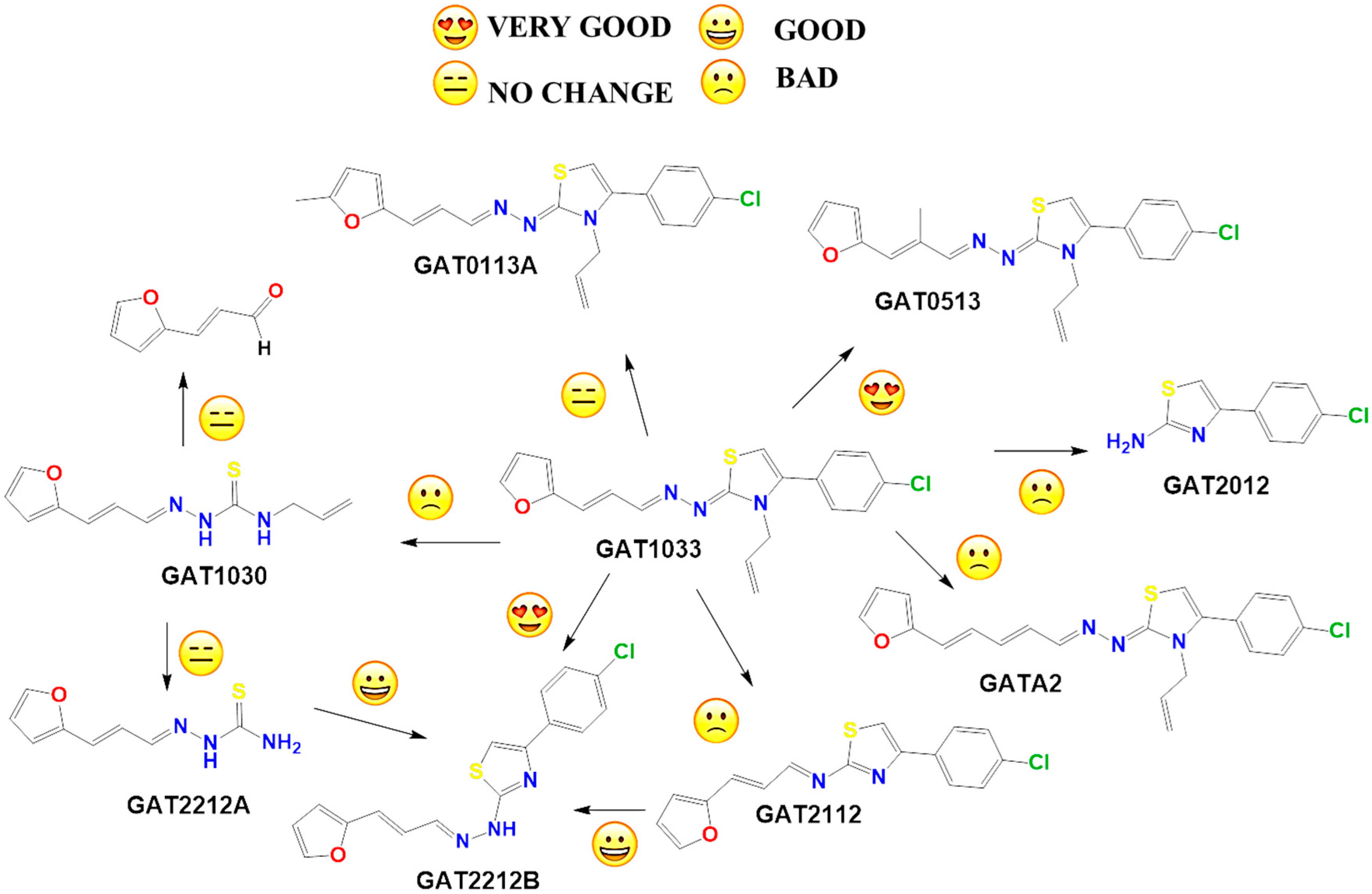
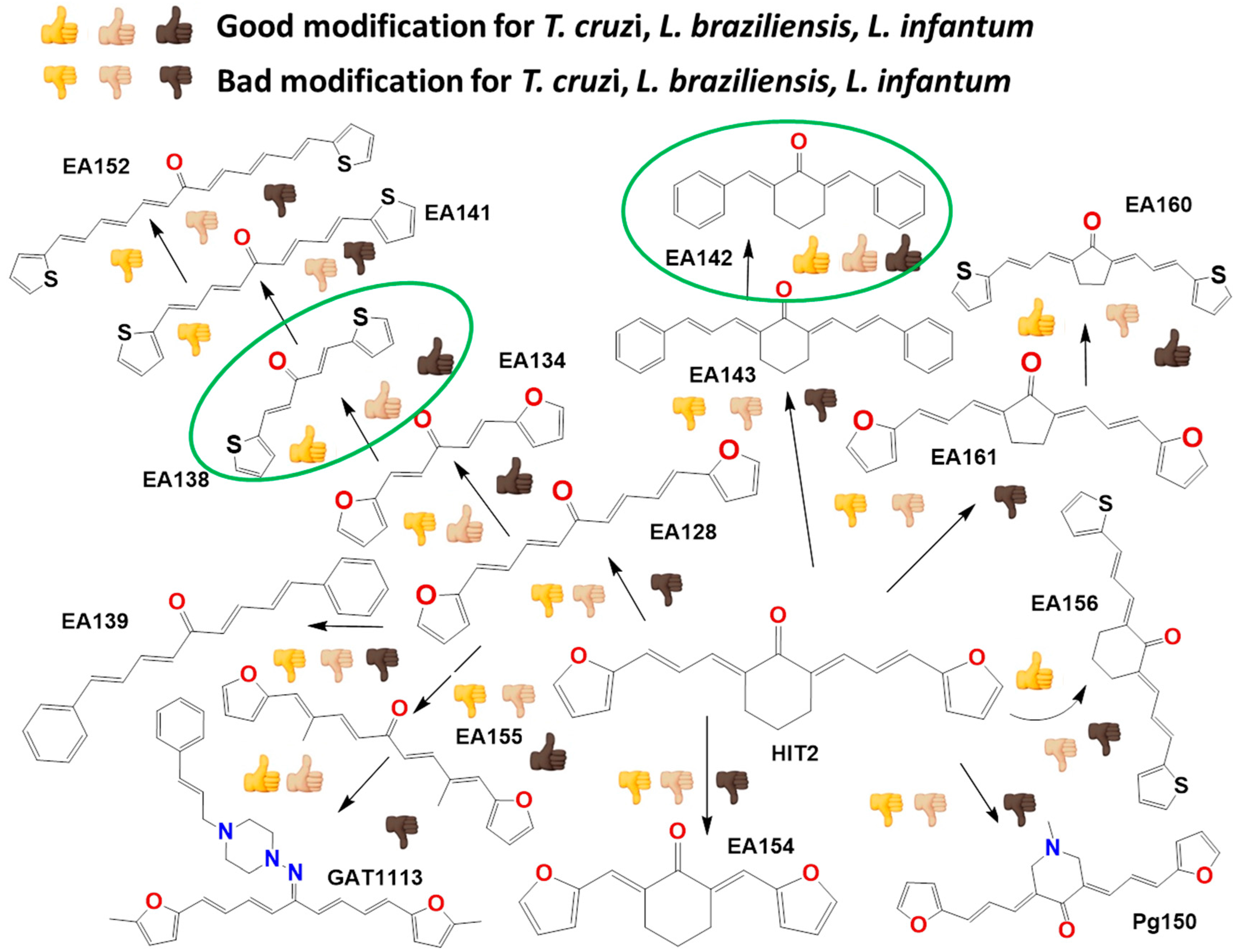
2.2. In Vitro Molecular Stripping for T. cruzi
2.3. Exploring the Mechanism of Action
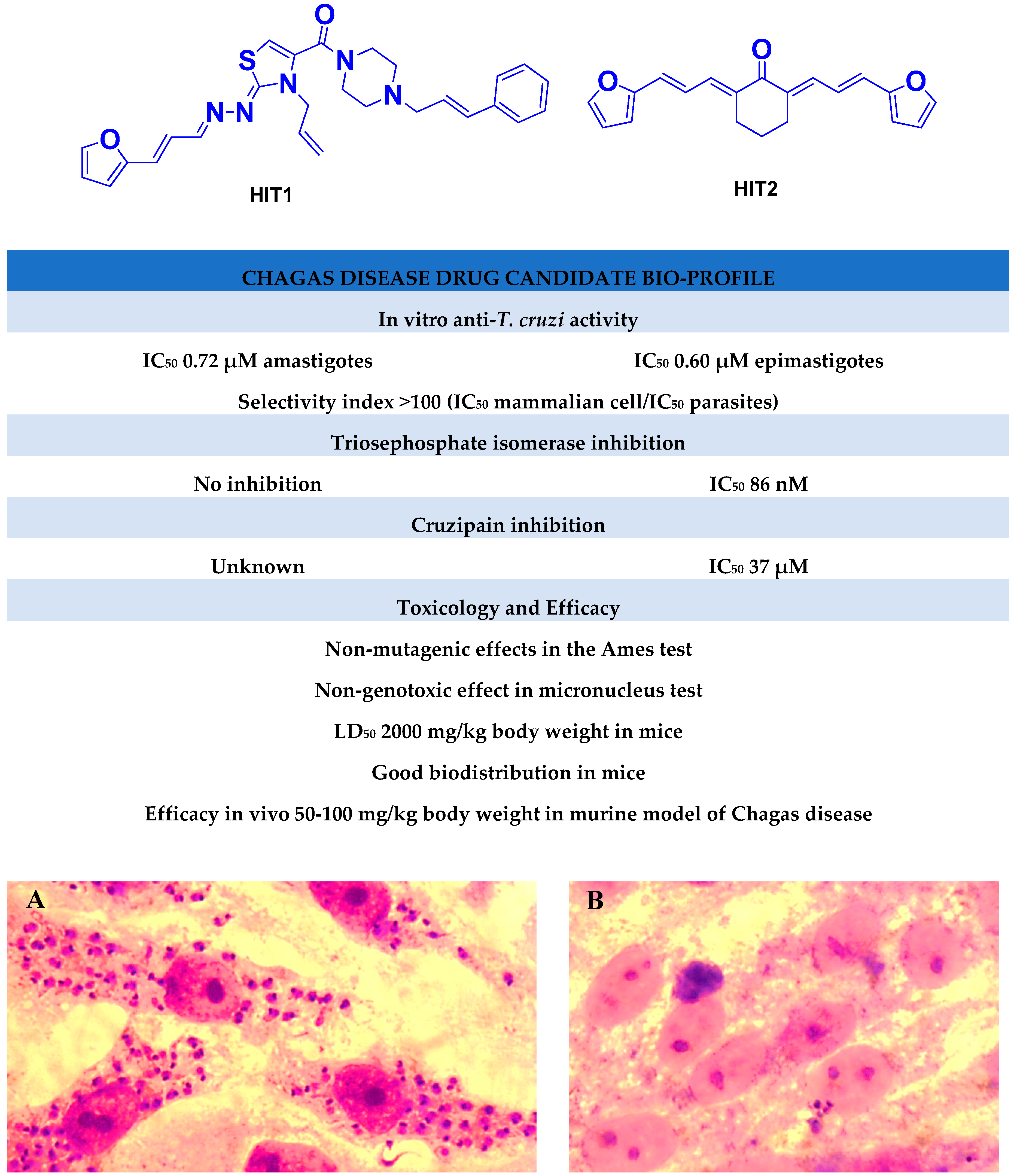
2.3.1. Inhibition of Triosephosphate Isomerase and Cruzipain
2.3.2. Selective Inhibitors
2.4. Toxicology In Vivo
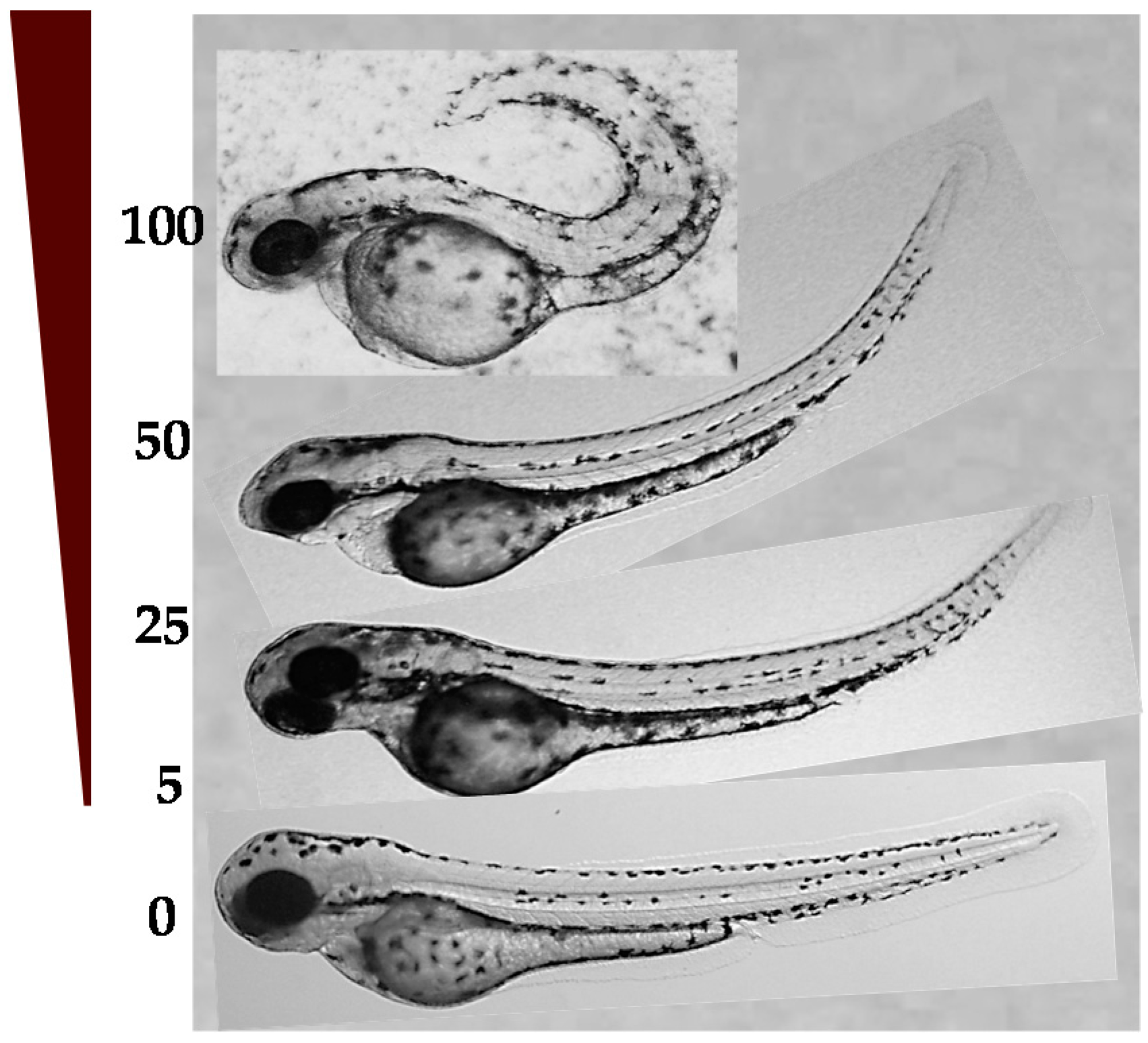
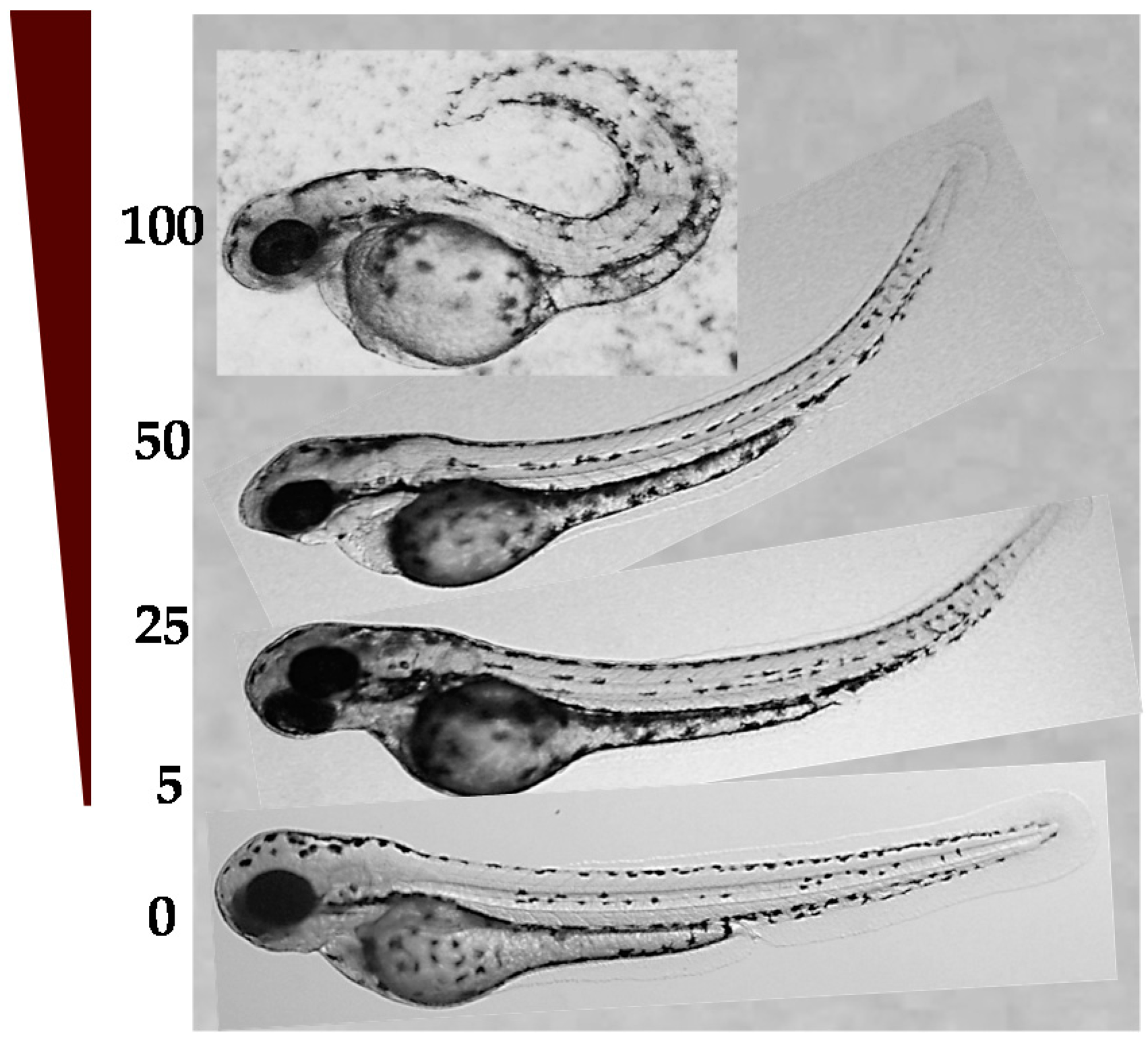
2.4.1. LD50 in Zebrafish Embryos and Developmental Toxicity
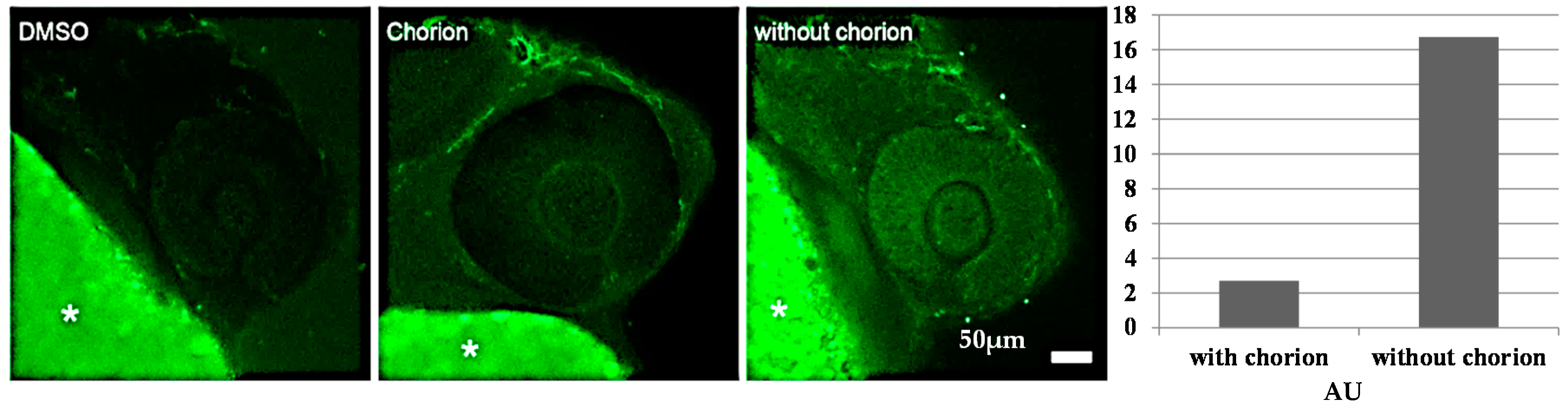
2.4.2. Biodistribution of the Compound in a Zebrafish Model
3. Experimental Section
3.1. General
3.1.1. General Synthetic-Procedure for Thiazolylidene Hydrazines GAT0113A, GAT0513, GAT2015, GATA2 and GAT2212b
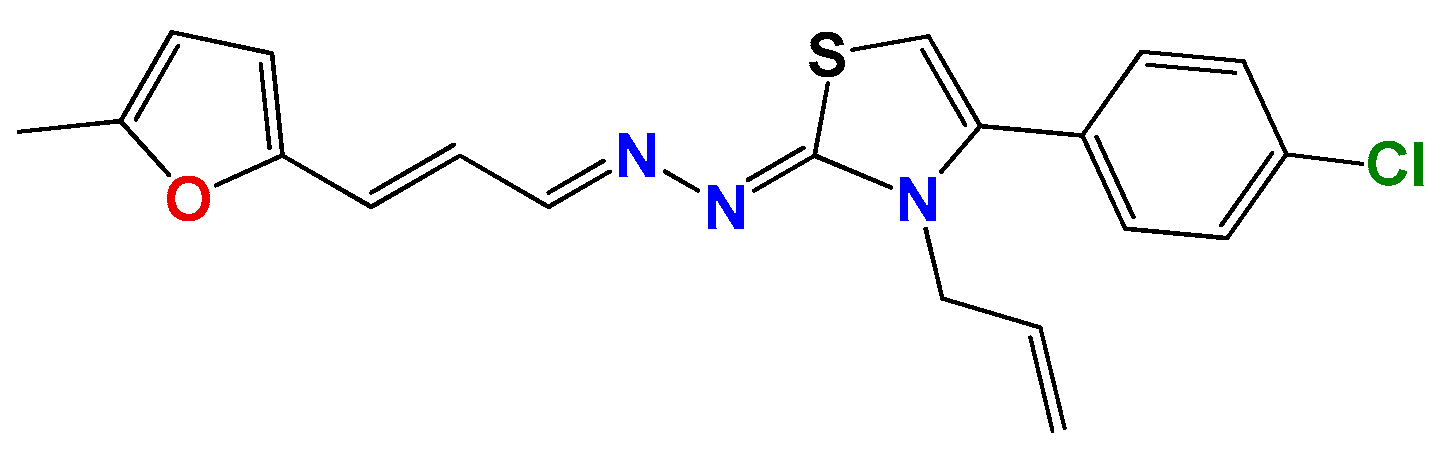
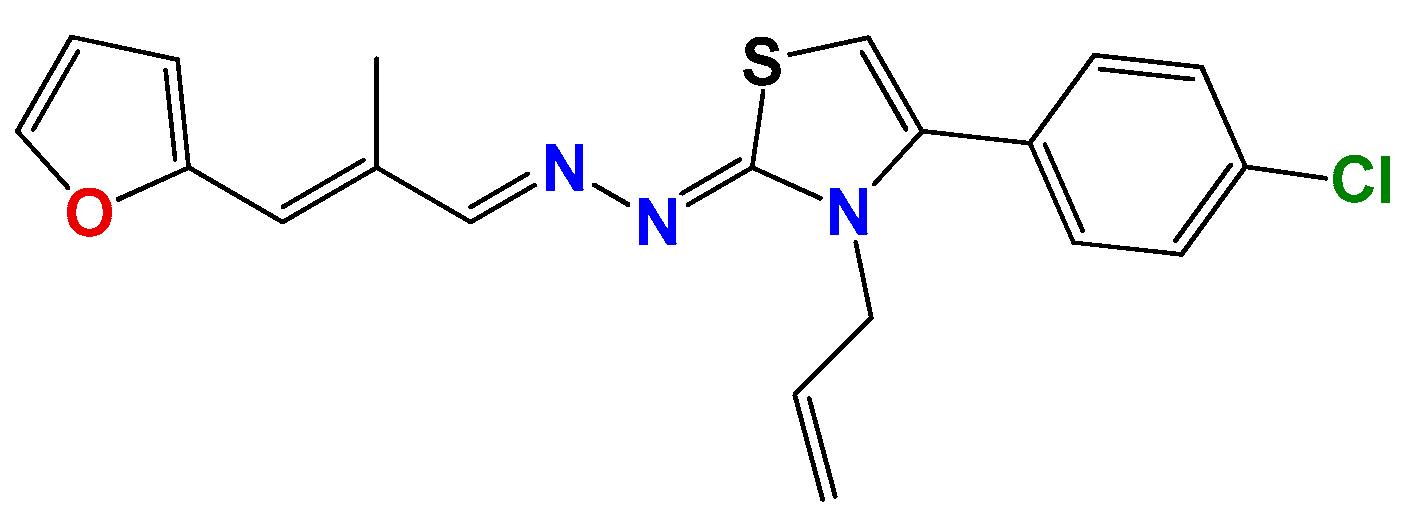
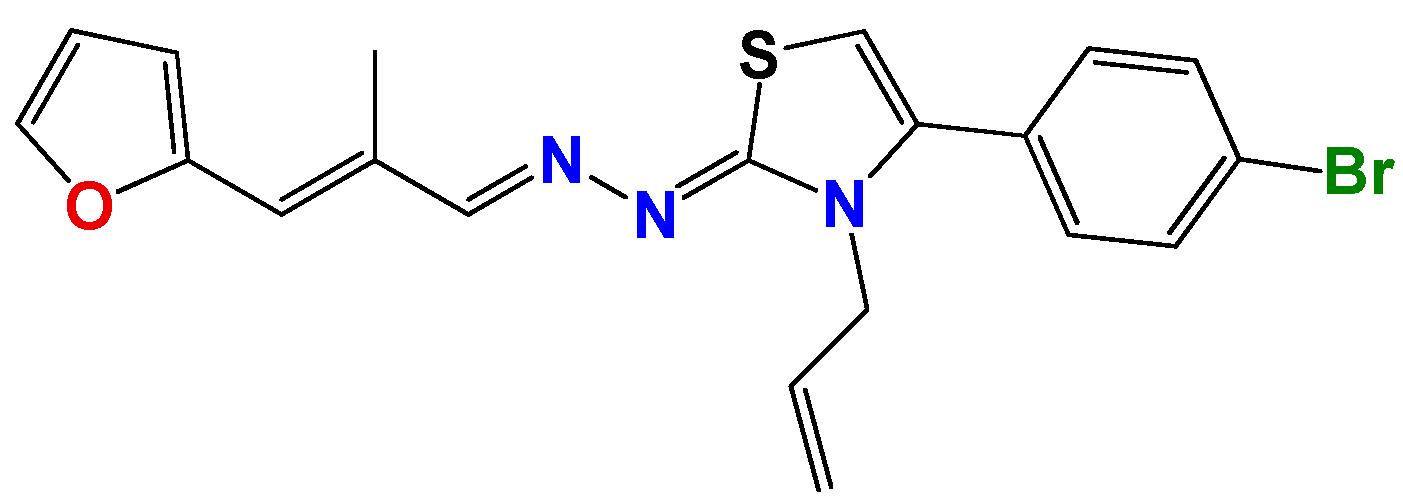
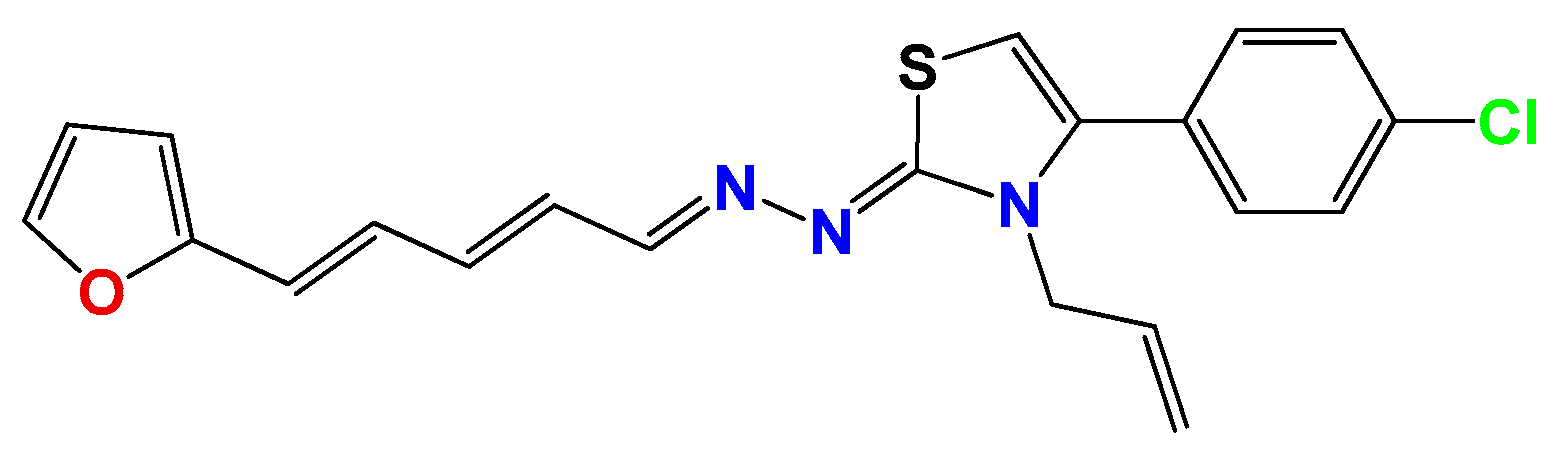
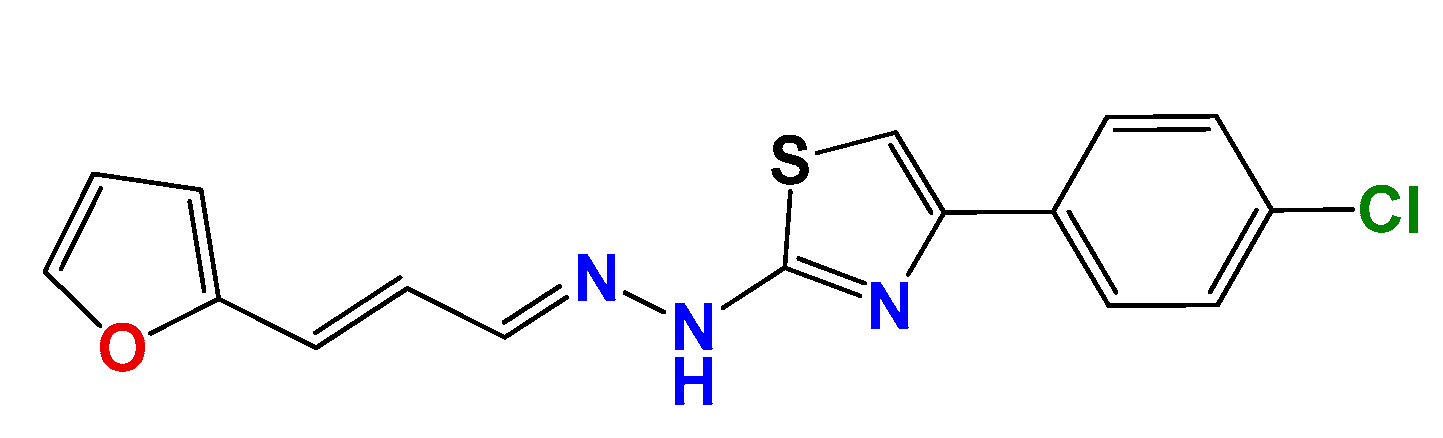
3.1.2. Synthetic Procedure for 4(4-Chlorophenyl)-thiazol-2(3H)-ylidene-2-amine (GAT2012)

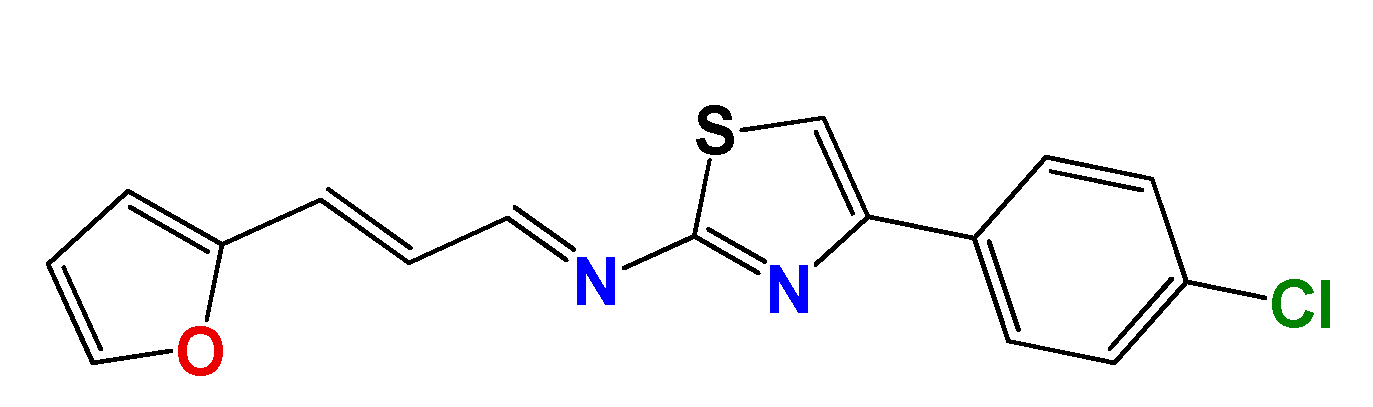
3.1.3. Synthetic Procedure of (E)-4-(4-Chlorophenyl)-N-((E)-3-(furan-2-yl)allylidene)thiazol-2-amine (GAT2112)
3.1.4. General Synthetic Procedure for Piperazine-Hydrazine Derivatives
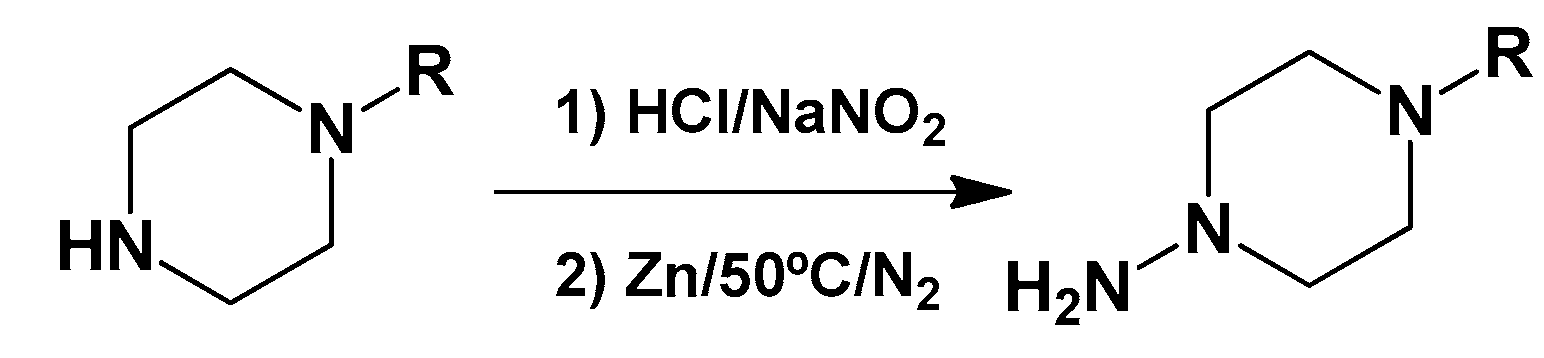
3.1.5. Synthetic Procedure for Thiazolylidene Hydrazides GATk1, GATk2, GATk4, GATk5, GATk6 and GAT0613
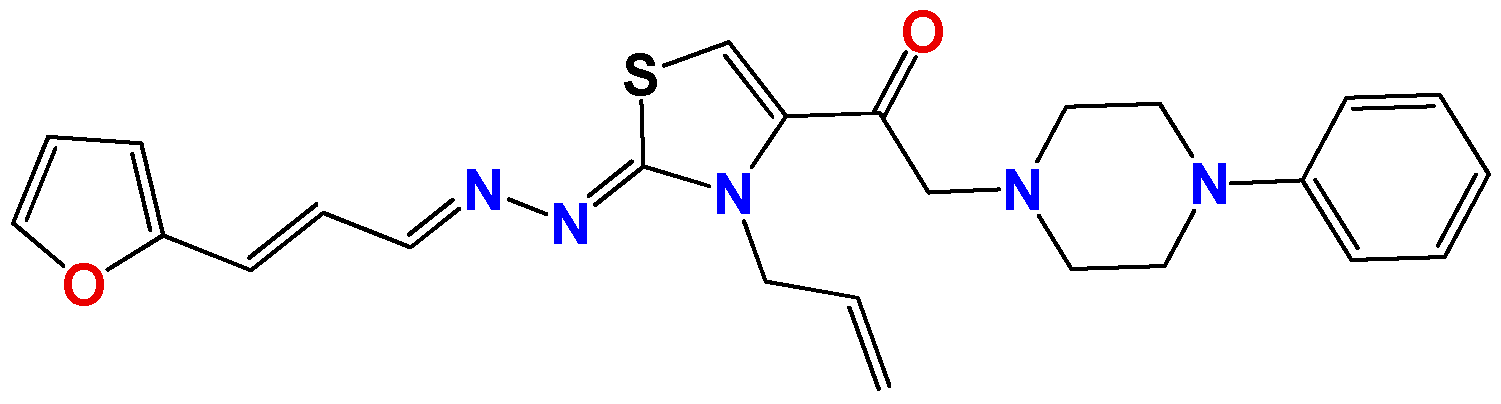
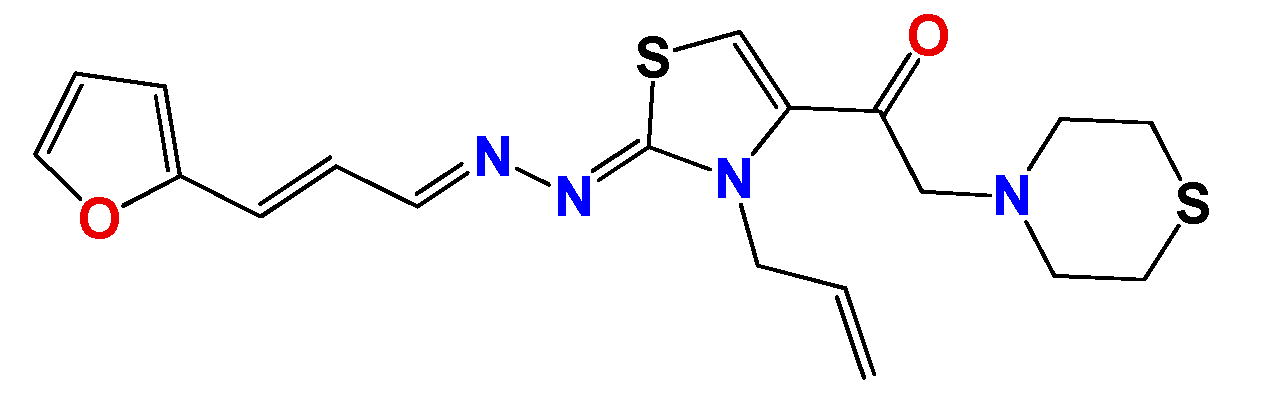
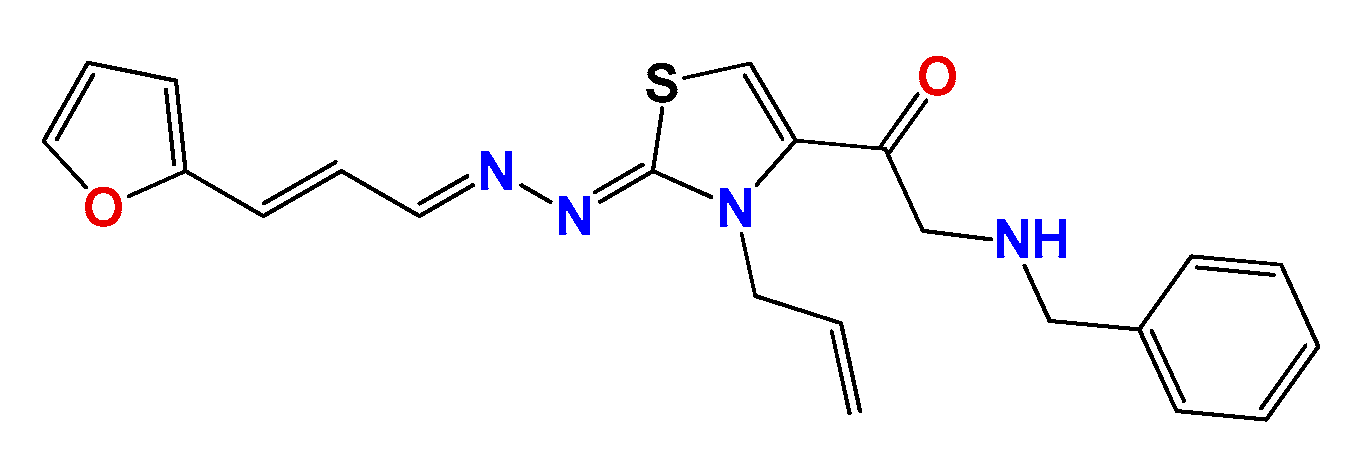
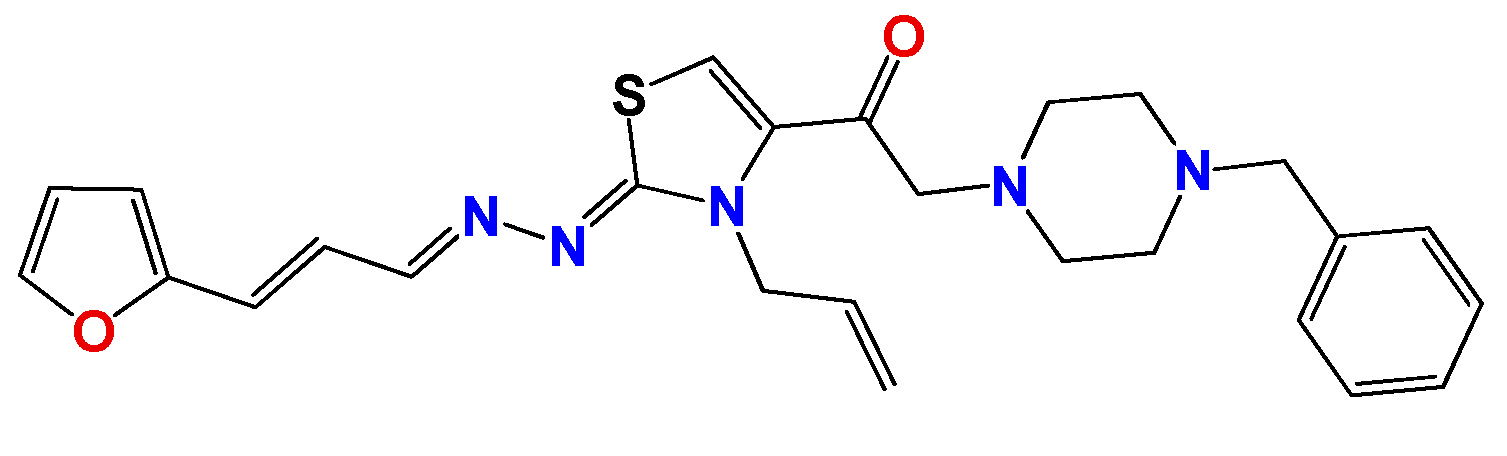

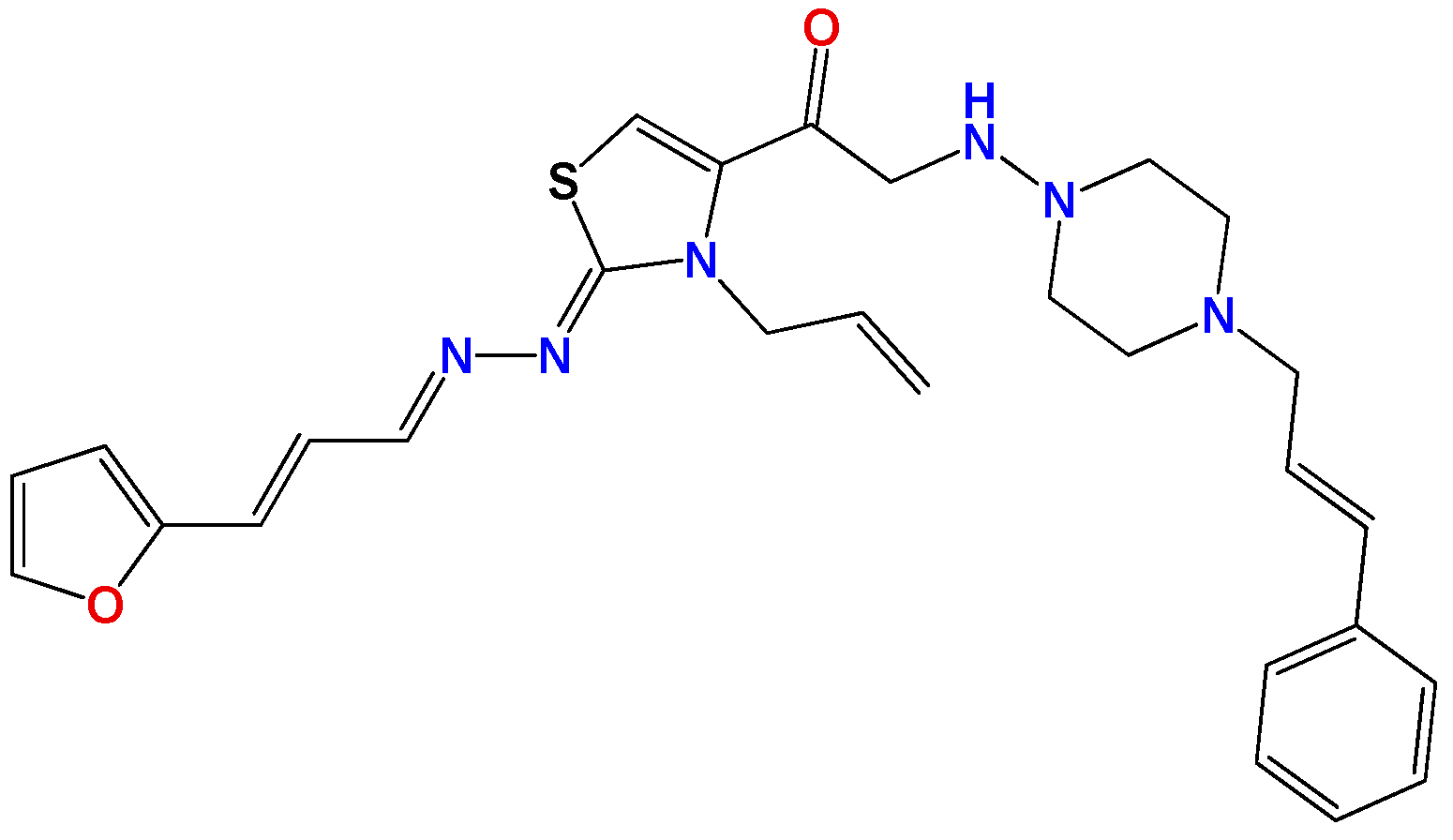
3.1.6. General Synthetic Procedure for Compounds GAT1113, GAT0413, GAT0913b and GAT1912
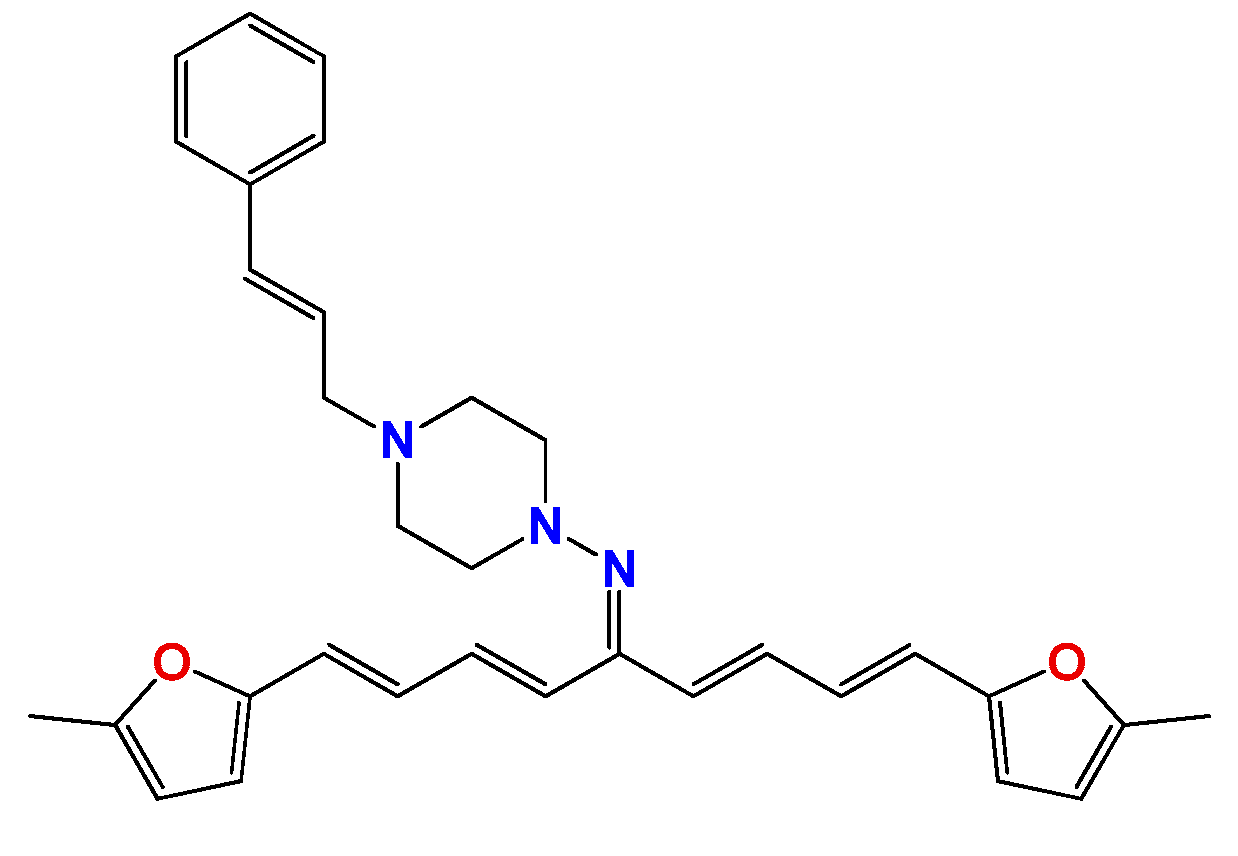
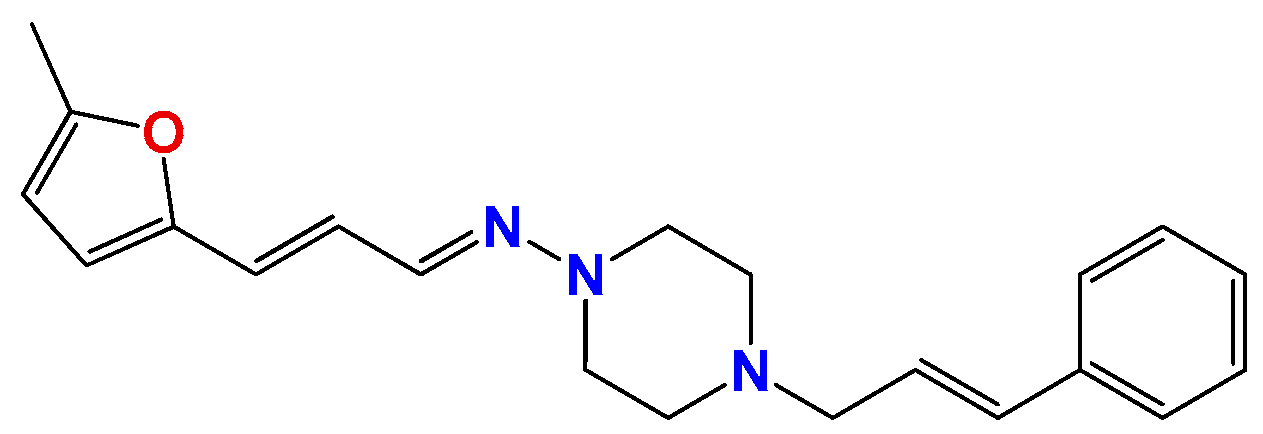

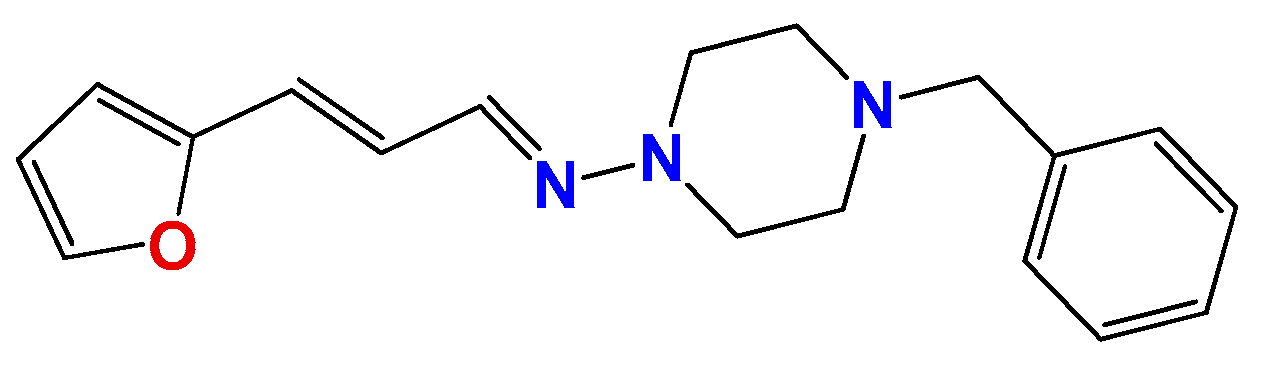
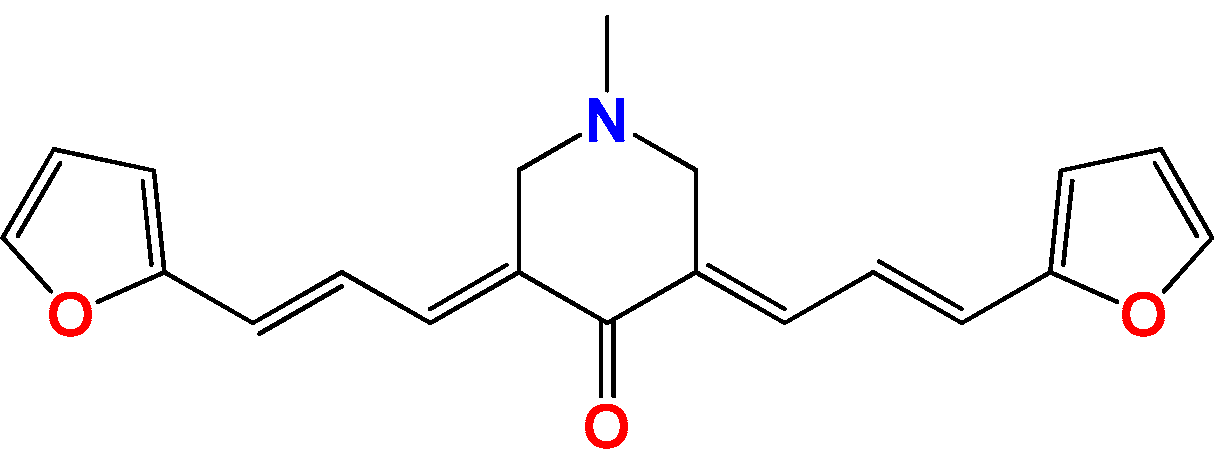
3.1.7. Synthetic Procedure for Compound (3E,5E)-3,5-Bis((E)-3-(furan-2-yl)allylidene)-1-methylpiperidin-4-one (Pg150) [12]
3.2. Anti-Parasitic Test In Vitro [15,37]
3.3. Nonspecific In Vitro Cytotoxicity of Mammalian Cells [15,38]
3.4. Inhibition of Triosephosphate Isomerase [12]
3.5. Inhibition of T. cruzi Cruzipain [12]
3.6. Nucleophile Challenge [41]
3.7. Adapting the Zebrafish Embryo Toxicity Test (FET) to Lipophilic Drugs
3.7.1. Embryo Toxicity Test [42,43]
3.7.2. Determination of Drug Bioconcentration [33]
3.7.3. Fluorescence Measurements Using Laser Scanning Confocal Microscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Esch, K.J.; Petersen, C.A. Transmission and epidemiology of zoonotic protozoal diseases of companion animals. Clin. Microbiol. Rev. 2013, 26, 58–85. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chagas Disease (American Trypanosomiasis); WHO: Geneva, Switzerland, 2017; Available online: http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed on 4 May 2017).
- Salerno, R.; Salvatella, R.; Issa, J.; Anzola, M.C. A regional fight against Chagas disease: lessons learned from a successful collaborative partnership. Rev. Panam. De Salud Públ. 2015, 37, 38–43. [Google Scholar]
- WHO. Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2016; Volume 21, pp. 421–428. [Google Scholar]
- Solano-Gallego, L.; Koutinas, A; Miró, G.; Cardoso, L.; Pennisi, M.G.; Ferrer, L.; Bourdeau, P.; Oliva, G.; Baneth, G. Directions for the diagnosis, clinical staging, treatment and prevention of canine leishmaniosis. Vet. Parasitol. 2009, 165, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Oddone Costanzo, R. Leishmaniosis visceral: A 101 años del primer caso diagnosticado en las Américas Visceral leishmaniosis: 101 years from the first case diagnosed in the Americas. Mem. Inst. Investig. Cienc. Salud 2012, 10, 100–104. [Google Scholar]
- Satragno, D.; Faral-Tello, P.; Canneva, B.; Verger, L.; Lozano, A.; Vitale, E.; Greif, G.; Soto, C.; Robello, C.; Basmadjián, Y. Autochthonous Outbreak and Expansion of Canine Visceral Leishmaniasis, Uruguay. Emerg. Infect. Dis. 2017, 23, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, J.C.; González-Pacanowska, D.; Croft, S.L.; Olesen, O.F.; Späth, G.F. Collaborative actions in anti-trypanosomatid chemotherapy with partners from disease endemic areas. Trends Parasitol. 2010, 26, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug Resistance in Leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L. Neglected tropical diseases: Multi-target-directed ligands in the search for novel lead candidates against Trypanosoma and Leishmania. J. Med. Chem. 2009, 52, 7339–7359. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; DeBilbao, N.V.; Aguirre-López, B.; Cabrera, N.; DíazMazariegos, S.; et al. Potent and Selective Inhibitors of Trypanosoma cruzi Triosephosphate Isomerase with Concomitant Inhibition of Cruzipain: Inhibition of Parasite Growth through Multitarget Activity. Chem. Med. Chem. 2016, 11, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, G.; Varela, J.; Márquez, P.; Gabay, M.; Arias Rivas, C.E.; Cuchilla, K.; Echeverría, G.A.; Piro, O.E.; Chorilli, M.; Leal, S.M.; et al. Optimization of antitrypanosomatid agents: Identification of nonmutagenic drug candidates with in vivo activity. J. Med. Chem. 2014, 57, 3984–3999. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, G.; Martínez, J.; Varela, J.; Birriel, E.; Cruces, E.; Gabay, M.; Leal, S.M.; Escobar, P.; Aguirre-López, B.; Gómez-puyou, M.T.; et al. Development of bis-thiazoles as inhibitors of triosephosphate isomerase from Trypanosoma cruzi. Identification of new non-mutagenic agents that are active in vivo. Eur. J. Med. Chem. 2015, 100, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, G.; Varela, J.; Cruces, E.; Fernández, M.; Gabay, M.; Leal, S.M.; Escobar, P.; Sanabria, L.; Serna, E.; Torres, S.T.; et al. Identification of a new amide-containing thiazole as a drug candidate for treatment of chagas’ disease. Antimicrob. Agents Chemother. 2015, 59, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lazarin-bidóia, D.; Desoti, C.; Martins, C.; Ribeiro, M.; Din, U.; Chem, B.M. Dibenzylideneacetones Are Potent Trypanocidal Compounds That Affect the Trypanosoma cruzi Redox System. Antimicrob. Agents Chemother. 2016, 60, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Viira, B.; Gendron, T.; Lanfranchi, D.A.; Cojean, S.; Horvath, D.; Marcou, G.; Varnek, A.; Maes, L.; Maran, U.; Loiseau, P.M.; et al. In silico mining for antimalarial structure-activity knowledge and discovery of novel antimalarial curcuminoids. Molecules 2016, 21, 853. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.; Lanfranchi, D.A.; Davioud-Charvet, E. Redox-active agents in reactions involving the trypanothione/trypanothione reductase-based system to fight kinetoplastidal parasites. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 405–428. [Google Scholar]
- Rodríguez, G.; Nargoli, J.; López, A.; Moyna, G.; Álvarez, G.; Fernández, M.; Osorio-Martínez, C.A.; González, M.; Cerecetto, H. Synthesis and in vivo proof of concept of a BODIPY-based fluorescent probe as a tracer for biodistribution studies of a new anti-Chagas agent. RSC Adv. 2017, 7, 7983–7989. [Google Scholar] [CrossRef]
- Cosentino, R.O.; Agüero, F. A simple strain typing assay for Trypanosoma cruzi: Discrimination of major evolutionary lineages from a single amplification product. PLoS Negl. Trop. Dis. 2012, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Ali, S.A.; Ashfaq, K.; Siddiqui, A.A.; Agha, A.; Yasinzai, M.M. Latent activity of curcumin against Leishmaniasis in vitro. Biol. Pharm. Bull. 2002, 25, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Braga, S.F.P.; Alves, É.V.P.; Ferreira, R.S.; Fradico, J.R.B.; Lage, P.S.; Duarte, M.C.; Ribeiro, T.G.; Júnior, P.A.S.; Romanha, A.J.; Tonini, M.L.; et al. Synthesis and evaluation of the antiparasitic activity of bis-(arylmethylidene) cycloalkanones. Eur. J. Med. Chem. 2014, 71, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.; Ma, P.; Elena, C.; Rivas, A.; Cuchilla, K.; Echeverr, G.A; Piro, O.E.; Chorilli, M.; Leal, S.M.; Escobar, P.; et al. Optimization of Antitrypanosomatid Agents: Identification of Nonmutagenic Drug Candidates with in Vivo Activity. J. Med. Chem. 2014, 57, 3984–3999. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Benitez, D.; Chavez, S.; Da Cunha, J.; Hernández, P.; Tinoco, L.W.; Campillo, N.E.; Páez, J.a.; Cerecetto, H.; González, M. Development of second generation amidinohydrazones, thio- and semicarbazones as Trypanosoma cruzi-inhibitors bearing benzofuroxan and benzimidazole 1,3-dioxide core scaffolds. Medchemcomm 2010, 1, 216–228. [Google Scholar] [CrossRef]
- Bellera, C.L.; Balcazar, D.E.; Vanrell, M.C.; Casassa, A.F.; Palestro, P.H.; Gavernet, L.; Labriola, C.A.; Gálvez, J.; Bruno-Blanch, L.E.; Romano, P.S.; et al. Computer-guided drug repurposing: Identification of trypanocidal activity of clofazimine, benidipine and saquinavir. Eur. J. Med. Chem. 2015, 93, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglini, M.L.; Bellera, C.L.; Fraccaroli, L.; Larocca, L.; Carrillo, C.; Talevi, A.; Alba Soto, C.D. Novel cruzipain inhibitors for the chemotherapy of chronic Chagas disease. Int. J. Antimicrob. Agents 2016, 48, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Mallari, J.P.; Hansell, E.J.; Mackey, Z.; Doyle, P.; Zhou, Y.M.; Gut, J.; Rosenthal, P.J.; McKerrow, J.H.; Guy, R.K. Discovery of potent thiosemicarbazone inhibitors of rhodesain and cruzain. Bioorg. Med. Chem. Lett. 2005, 15, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Buchanan-Kilbey, G.; Djumpah, J.; Papadopoulou, M.V; Bloomer, W.; Hu, L.; Wilkinson, S.R.; Ashworth, R. Evaluating the developmental toxicity of trypanocidal nitroaromatic compounds on zebrafish. Acta Trop. 2013, 128, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-T.; Tanguay, R.L. The role of chorion on toxicity of silver nanoparticles in the embryonic zebrafish assay. Environ. Health Toxicol. 2014, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.H.; Zhu, L; Gong, Z.Y. Bioaccumulation and toxicity test of diethylstilbestrol to zebrafish (Danio rerio) embryo. Huan Jing Ke Xue 2009, 30, 522–526. [Google Scholar] [PubMed]
- Redelstein, R.; Zielke, H.; Spira, D.; Feiler, U.; Erdinger, L.; Zimmer, H.; Wiseman, S.; Hecker, M.; Giesy, J.P.; Seiler, T.B.; et al. Bioaccumulation and molecular effects of sediment-bound metals in zebrafish embryos. Environ. Sci. Pollut. Res. 2015, 22, 16290–16304. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, J.; Ferling, H.; Mallow, U.; Wintermayr, H.; Negele, R.D. Toxic effects of the non-steroidal anti-inflammatory drug diclofenac. Part I: Histopathological alterations and bioaccumulation in rainbow trout. Aquat. Toxicol. 2004, 68, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug Metabolism and Pharmacokinetics, the Blood-Brain Barrier, and Central Nervous System Drug Discovery. J. Am. Soc. Exp. Neurother. 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Nanjwade, B.K.; Patel, D.J.; Udhani, R.a; Manvi, F.V. Functions of lipids for enhancement of oral bioavailability of poorly water-soluble drugs. Sci. Pharm. 2011, 79, 705–727. [Google Scholar] [CrossRef] [PubMed]
- García, F.P.; Araceli, O.; Ramírez, B.; Scoot, W.; Gaytán, J.C. Acumulación, toxicidad y teratogénesis provocada por presencia de arsénico en aguas en el pez cebra (Danio rerio). Rev. Toxicol. 2007, 24, 18–22. [Google Scholar]
- Ferreira, M.E.; Rojas de Arias, A.; Yaluff, G.; de Bilbao, N.V.; Nakayama, H.; Torres, S.; Schinini, A.; Guy, I.; Heinzen, H.; Fournet, A. AntiLeishmanial activity of furoquinolines and coumarins from Helietta apiculata. Phytomedicine 2010, 17, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Roldos, V.; Nakayama, H.; Rolón, M.; Montero-Torres, A.; Trucco, F.; Torres, S.; Vega, C.; Marrero-Ponce, Y.; Heguaburu, V.; Yaluff, G.; et al. Activity of a hydroxybibenzyl bryophyte constituent against Leishmania spp. and Trypanosoma cruzi: In silico, in vitro and in vivo activity studies. Eur. J. Med. Chem. 2008, 43, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Pérez-Montfort, R.; López-Calahorra, F.; Costas, M.; Rodríguez-Romero, A.; Tuena de Gómez-Puyou, M.; Gómez Puyou, A. Structural differences in triosephosphate isomerase from different species and discovery of a multitrypanosomatid inhibitor. Biochemistry 2006, 45, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Boiani, M.; Cerecetto, H.; González, M. 2-Benzyl-2-methyl-2H-benzimidazole 1,3-dioxide derivatives: Spectroscopic and theoretical study. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2007, 67, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Liang, Y.-R.; Xu, F.-L.; Wu, Q.; Lin, X.-F. Stereoselective synthesis of spiro[5.5]undecane derivatives via biocatalytic [5 + 1] double Michael additions. J. Mol. Catal. B Enzym. 2013, 97, 18–22. [Google Scholar] [CrossRef]
- Henn, K.; Braunbeck, T. Dechorionation as a tool to improve the fish embryo toxicity test (FET) with the zebrafish (Danio rerio). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2011, 153, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mandrell, D.; Truong, L.; Jephson, C.; Sarker, M.R.; Moore, A.; Lang, C.; Simonich, M.T.; Tanguay, R.L. Automated zebrafish chorion removal and single embryo placement: optimizing throughput of zebrafish developmental toxicity screens. J. Lab. Autom. 2012, 17, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Zolessi, F.R. Functional Diversification of the Four MARCKS Family Members in Zebrafish Neural Development. J. Exp. Zool. Part B Mol. Dev. Evol. 2017, 328, 119–138. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the studied compounds are available from the authors. |

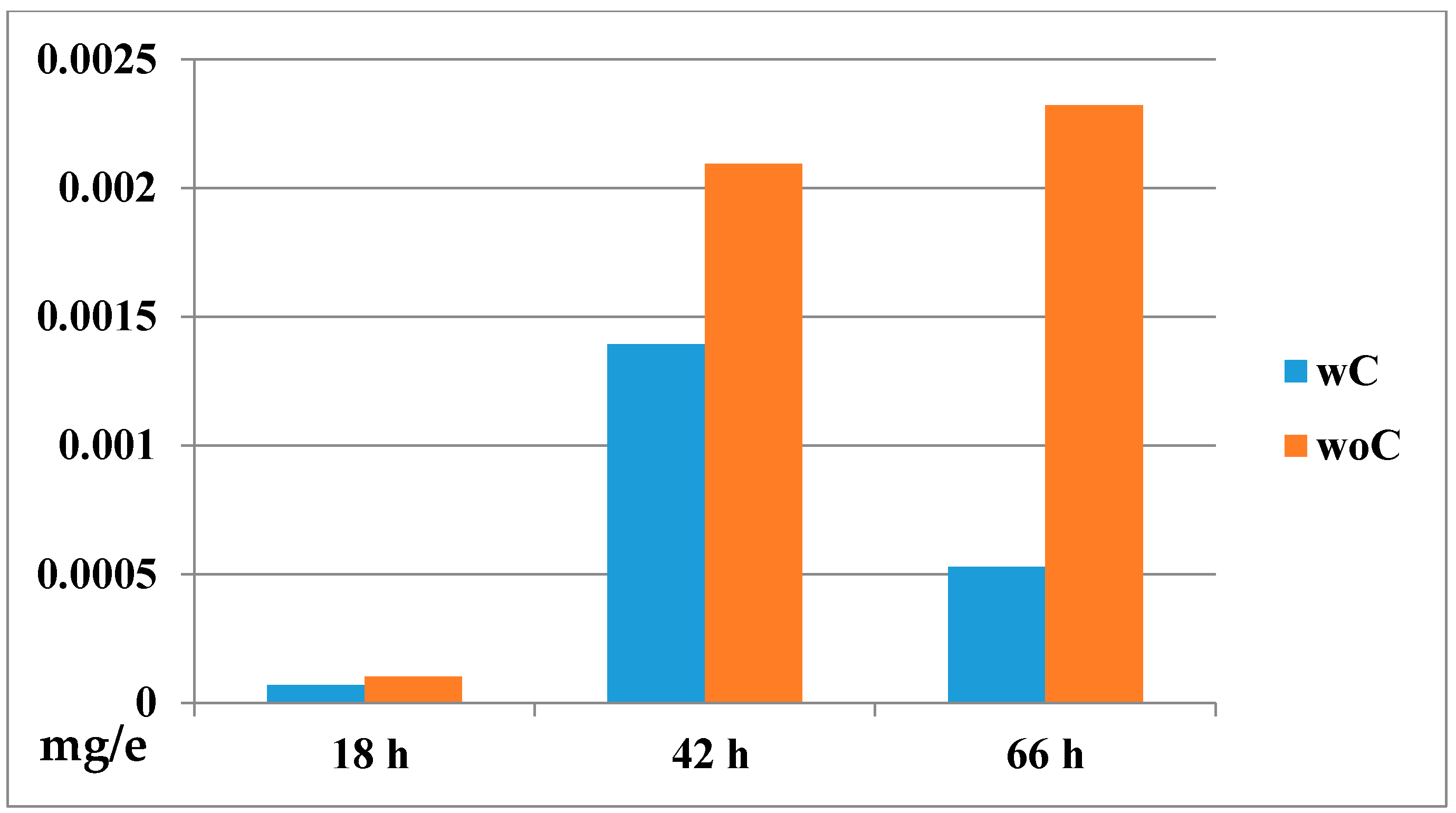

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
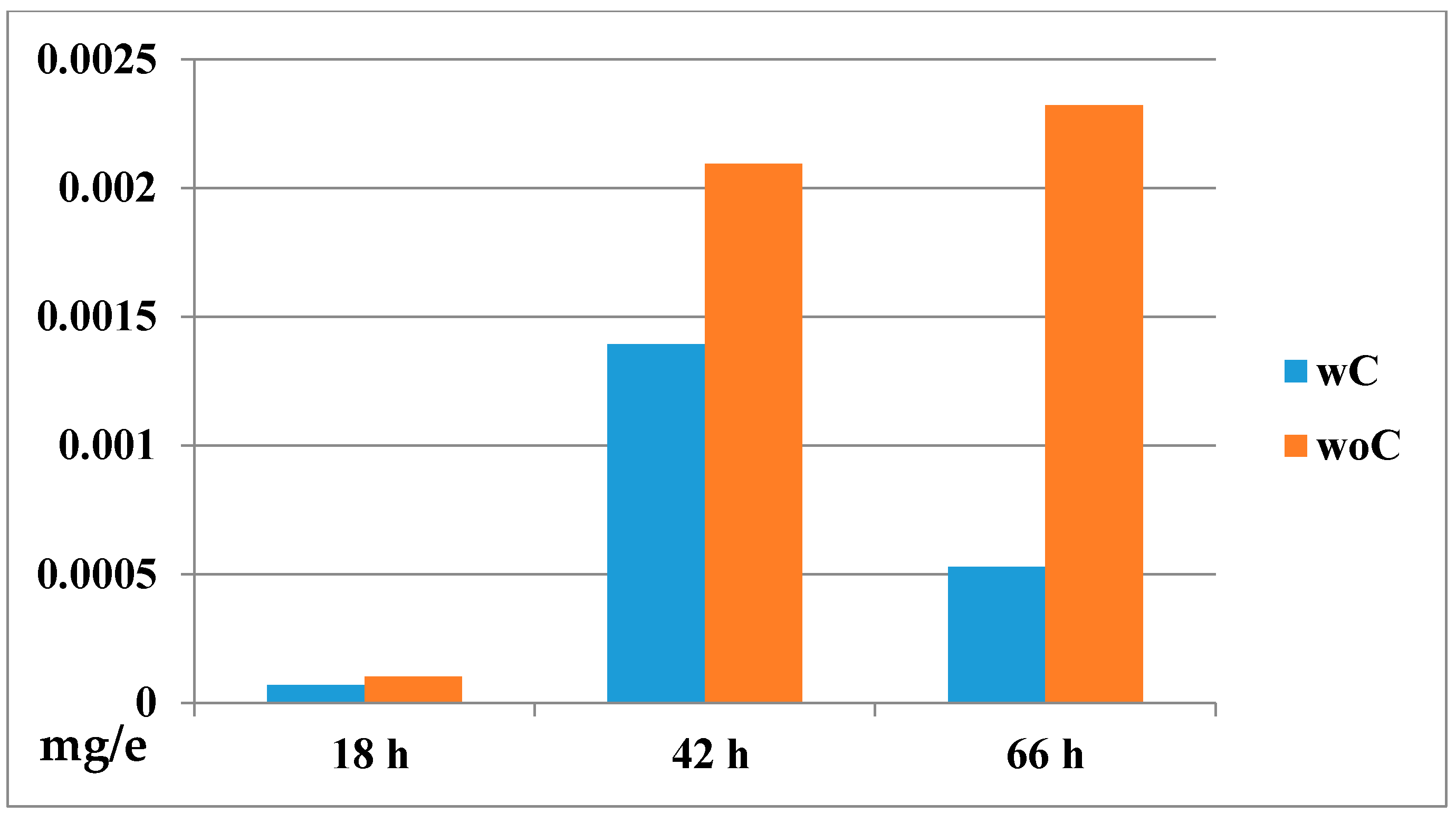{kind=link}
| A | ||||
| Structure | Compound | IC50 ± SD a (µM) Epimastigotes T. cruzi | IC50 ± SD (µM) Promastigotes L. braziliensis | IC50 ± SD (µM) Promastigotes L. infantum |
 | GAT1033 b | 1.6 ± 0.5 c | 7 ± 1 | 2.0 ± 0.2 |
 | GAT0113A | 3.0 ± 0.5 | 10 ± 1 | 8 ± 2 |
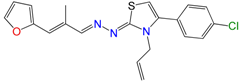 | GAT0513 | 0.09 ± 0.02 | 33 ± 11 | 58 ± 12 |
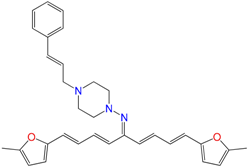
 | HIT1 | 3.1 ± 0.2 d | 12 ± 5 | 4 ± 1 |
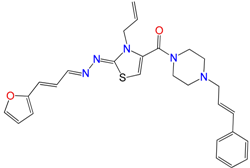
 | GAT1113 | 1.6 ± 0.3 | 16 ± 4 | 14 ± 2 |
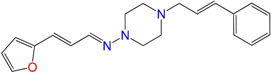 | GAT0913b | 25 ± 8 | 18 ± 5 | 21 ± 2 |
| Glucantime | - | 18 ± 2 | 26 ± 9 | |
| Miltefosine | 8 ± 1 | - | 0.9 ± 0.2 | |
| Benznidazole | 7 ± 1 | - | - | |
| B | ||||
 | Curcumin | 5.6 ± 1 e | - | 5.9 ± 0.3 b |
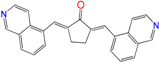 | Comp13 f | 31 ± 2 f | 0.9 ± 0.2 c | - |
 | EA142 g | 5.1 ± 0.3 h | 4.2 ± 0.7 | 9.6 ± 0.9 |
 | EA134 | 24 ± 2 h | 10 ± 6 | 6 ± 2 |
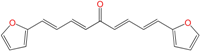 | EA128 | 5.0 ± 0.7 h | 36 ± 9 | 31 ± 9 |
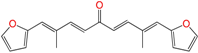 | EA155 | 8.2 ± 2.0 h | 16 ± 2 | 6 ± 1 |
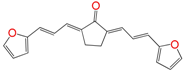 | EA161 | 5.4 ± 1.6 h | 18 ± 4 | 16 ± 4 |
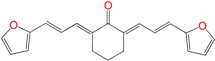 | HIT2 | 0.6 ± 0.2 h | 7 ± 1 | 13 ± 7 |
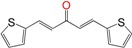 | EA138 | 5.0 ± 0.8 h | 8 ± 2 | 4.0 ± 0.5 |
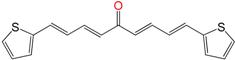 | EA141 | 12.6 ± 1.4 h | 36 ± 3 | 19 ± 5 |
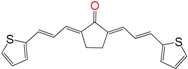 | EA160 | 0.04 ± 0.01 h | >100 | 11 ± 3 |
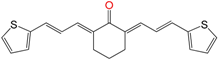 | EA156 | 0.6 ± 0.2 h | >100 | 16 ± 3 |
| Glucantime | - | 18 ± 2 | 26 ± 9 | |
| Miltefosine | 8 ± 3 | 0.9 ± 0.2 | - | |
| Benznidazole | 7 ± 1 | - | - | |
| Compound | IC50 ± SD (µM) Fibroblast NCTC929 | IC50 ± SD (µM) Murine Macrophages | SI e NCTC/ L. braziliensis | SI NCTC/ L. infantum | SI J774.1/ T. cruzi |
|---|---|---|---|---|---|
| GAT1033 | 405 ± 10 | 60 ± 6 a | 58 | 203 | 37 a |
| GAT0113A | 319 ± 16 | 66 ± 7 | 32 | 40 | 22 |
| GAT0513 | 1443 ± 30 | 55 ± 5 | 44 | 25 | 611 |
| HIT1 | 346 ± 9 | 30 ± 5 b | 29 | 87 | 10 |
| GAT1113 | 165 ± 5 | 45 ± 5 | 10 | 12 | 28 |
| GAT0913b | 160 ± 7 | 25 ± 3 | 9 | 8 | 1 |
| Curcumin | - | 10 ± 2 d | 2 | - | 2 |
| Comp13 | - | 21± 5 d | 23 | - | 1 |
| EA134 | 114 ± 2 | 115 ± 6 c | 11 | 19 | 5 c |
| EA128 | 494 ± 25 | 60 ± 3 c | 14 | 16 | 12 c |
| EA155 | 543 ± 15 | 33 ± 8 c | 34 | 91 | 4 c |
| EA161 | 756 ± 17 | 19 ± 2 c | 42 | 47 | 4 c |
| HIT2 | 160 ± 9 | 10 ± 2 c | 23 | 12 | 17 c |
| EA138 | 158 ± 5 | 38 ±7 c | 20 | 40 | 8 c |
| EA141 | 1704 ± 40 | 10 ± 2 c | 47 | 90 | 1 c |
| EA160 | 4909 ± 36 | 15 ± 1 c | nc | 446 | 375 c |
| EA156 | 1985 ± 20 | 20 ± 1 c | nc | 124 | 33 c |
| Glucantime | - | 15 ± 1 | 1 | 0.5 | - |
| Miltefosine | - | 50 ± 7 | - | 56 | 6 |
| Benznidazole | - | 400 ± 4 | - | - | 57 |
| Compound | Percentage of Inhibition a,b/IC50 ± SD (µM) TcTIM | Percentage of Inhibition a,b/IC50 ± SD (µM) LmTIM c | Percentage of Inhibition a,b/IC50 ± SD (µM) Cruzipain |
|---|---|---|---|
| GAT1033 | 0/- | 0/- | 0/- |
| HIT1 | 0/- | 0/- | 100/4.3 ± 0.4 |
| EA128 | 100/3.0 ± 0.7 d | 0/- | 48/- d |
| EA155 | 100/3.3 ± 0.5 d | 100/˂25 e | 0/- d |
| HIT2 | 100/0.086 ± 0.007 d | 100/˂25 e | 80/37.0 ± 1.1 d |
| EA138 | 0/- d | 100/˂25 e | 50/- d |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez, G.; Perdomo, C.; Coronel, C.; Aguilera, E.; Varela, J.; Aparicio, G.; Zolessi, F.R.; Cabrera, N.; Vega, C.; Rolón, M.; et al. Multi-Anti-Parasitic Activity of Arylidene Ketones and Thiazolidene Hydrazines against Trypanosoma cruzi and Leishmania spp. Molecules 2017, 22, 709. https://doi.org/10.3390/molecules22050709
Álvarez G, Perdomo C, Coronel C, Aguilera E, Varela J, Aparicio G, Zolessi FR, Cabrera N, Vega C, Rolón M, et al. Multi-Anti-Parasitic Activity of Arylidene Ketones and Thiazolidene Hydrazines against Trypanosoma cruzi and Leishmania spp. Molecules. 2017; 22(5):709. https://doi.org/10.3390/molecules22050709
Chicago/Turabian StyleÁlvarez, Guzmán, Cintya Perdomo, Cathia Coronel, Elena Aguilera, Javier Varela, Gonzalo Aparicio, Flavio R. Zolessi, Nallely Cabrera, Celeste Vega, Miriam Rolón, and et al. 2017. "Multi-Anti-Parasitic Activity of Arylidene Ketones and Thiazolidene Hydrazines against Trypanosoma cruzi and Leishmania spp." Molecules 22, no. 5: 709. https://doi.org/10.3390/molecules22050709