3.2. Preparation of Alkynes and Compound Characterization
Non-1-yn-5-ol (
1b). To dry THF (254 mL) was added
n-BuLi (2.5 M in
n-hexane, 9.74 mL, 24.4 mmol) at −78 °C. 4-pentyn-1-al [
8] (1.00 g, 12.2 mmol) was added slowly at −78 °C. Then the mixture was stirred for 3 h while its temperature reached 23 °C. Methanol (50 mL) was added to quench excess
n-BuLi. The mixture was concentrated. The residue was dissolved in 10% aqueous HCl and the resulting mixture was extracted with CH
2Cl
2. The combined organic layers were dried over Na
2SO
4, concentrated, and purified by silica gel column chromatography. Elution with cyclohexane/acetone (50/1) gave a pale yellow oil (0.38 g, 22%);
1H-NMR (300 MHz, CDCl
3) δ 3.75 (br, 1H, C
HOH), 2.34 (t,
J = 6.0 Hz, 2H), 1.98 (s, 1H, ≡C
H), 1.72–1.60 (m, 2H), 1.67 (br, 1H, O
H), 1.46–1.33 (m, 6H), 0.91 (t,
J = 6.3 Hz, 3H, C
H3).
13C-NMR (75 MHz, CDCl
3) δ 84.5 (
C≡CH), 71.0, 68.9, 37.3, 35.9, 28.0, 22.9, 15.2, 14.3. HRMS (ESI)
m/
z calcd. for C
9H
17O
+ [M + H]
+: 141.1274; found: 141.1269.
Dec-1-en-7-yn-4-ol (
1g). To a solution of
tert-butyldimethyl(oct-1-en-7-yn-4-yloxy)silane [
8] (3.00 g, 12.6 mmol) in dry THF (15 mL) cooled at −78 °C was added dropwise lithium diisopropylamide (LDA, 10.064 mL, 2.5 M in THF, 25.16 mmol) via syringe. The resulting mixture was stirred at −78 °C for 1 h followed by addition of iodoethane (5.056 mol, 62.9 mmol). The reaction solution allowed to warm to room temperature. After being stirred for 26 h, the mixture was passed through a silica-gel pad (eluted with CH
2Cl
2), and concentrated in vacuum. To the residue was added tetrabutylammonium fluoride (1 M in THF, 39.4 mL, 39.4 mmol) at room temperature. The reaction mixture was stirred for 10 h, and then was quenched with saturated aqueous NH
4Cl. The aqueous layer was extracted with CH
2Cl
2. The organic phase was then dried over Na
2SO
4, concentrated, and purified by silica gel column chromatography. Elution with cyclohexane/CH
2Cl
2 (30/1) gave a pale yellow oil (0.98 g, 51%);
1H-NMR (500 MHz, CDCl
3) δ 5.82 (m, 1H, C
H=
CH
2), 5.14–5.11 (m, 2H, CH=
CH2), 3.80 (m, 1H, C
H–OH), 2.32–2.26 (m, 3H), 2.21–2.11 (m, 3H), 2.00 (br, 1H, O
H), 1.66 (m, 1H), 1.60 (m, 1H), 1.10 (t,
J = 7.5 Hz, 3H, C
H3).
13C-NMR (125 MHz, CDCl
3) δ 134.6 (
CH=CH
2), 118.0 (CH=
CH
2), 82.4, 78.8, 70.0 (
CH–OH), 41.8, 35.6, 15.3, 14.2, 12.3. HRMS (ESI)
m/
z calcd. for C
10H
17O
+ [M + H]
+: 153.1274; found: 153.1269.
8-(Trimethylsilyl)oct-1-en-7-yn-4-ol (
1h). To a solution of 5-(trimethylsilyl)pent-4-ynal [
15] (9.17 g, 59.4 mmol) in CH
2Cl
2 (400 mL) cooled at −78 °C was added dropwise allyboronic acid pinacol ester (12.26 mL, 65.4 mmol). The resulting mixture was stirred at −78 °C for 1 h, and then at 0 °C for 3 h. The reaction was quenched by addition of water, extracted with CH
2Cl
2, and washed with brine. The organic layer was dried with Na
2SO
4 and concentrated. To the residue was added CH
2Cl
2 (100 mL) and triethanolamine (15 mL, 112 mmol) at room temperature. The mixture was stirred for 3 h, then passed through a silica gel pad (eluted with CH
2Cl
2), concentrated, and purified by silica gel column chromatography. Elution with cyclohexane/EtOAc (100/1) gave a yellow oil (11.13 g, 95%);
1H-NMR (400 MHz, CDCl
3) δ 5.82 (m, 1H, C
H=
CH
2), 5.17–5.12 (m, 2H, CH=
CH2), 3.79 (m, 1H, C
H–OH), 2.38 (t,
J = 7.2 Hz, 2H), 2.30 (m, 1H), 2.20 (m, 1H), 1.91 (br, 1H, O
H), 1.74–1.61 (m, 2H), 0.14 (s, 9H, C
H3 × 3).
13C-NMR (100 MHz, CDCl
3) δ 134.5 (
CH=CH
2), 118.2 (CH=
CH
2), 106.9 (≡
C–Si), 85.3 (
C≡C–Si), 70.0 (
CH–OH), 41.8, 35.2, 16.5, 0.08 (Si
CH
3 × 3). HRMS (ESI)
m/
z calcd. for C
11H
21OSi
+ [M + H]
+: 197.1356; found: 197.1357.
4-Chlorooct-1-en-7-yne (1i). To a solution of oct-1-en-7-yn-4-ol (1a, 300 mg, 2.4 mmol) in CH2Cl2 (18 mL) at 0 °C, pyridine (0.384 mL, 4.8 mmol) was then added, followed by triphosgene (356 mg, 1.2 mmol) in one portion. The solution was stirred for 5 min and then warmed to gentle reflux. After 6 h, the reaction mixture was poured into a separatory funnel containing 1 M aqueous HCl (20 mL), and the biphasic mixture was shaken vigorously. Upon separation of layers, the aqueous layer was re-extracted with CH2Cl2. The organic phase was dried over Na2SO4, concentrated, and purified by silica gel column chromatography. Elution with cyclohexane gave a pale yellow oil (213.6 mg, 62%); 1H-NMR (300 MHz, CDCl3) δ5.85 (m, 1H, CH=CH2), 5.18–5.13 (m, 2H, CH=CH2), 4.10 (m, 1H, CH–Cl), 2.56 (m, 2H), 2.41 (m, 2H), 2.05-1.82 (m, 2H), 1.98 (s, 1H, ≡CH). 13C-NMR (75 MHz, CDCl3) δ 133.7 (CH=CH2), 118.2 (CH=CH2), 82.8 (C≡CH), 69.1 (C≡CH), 60.8 (CH–Cl), 42.5, 36.2, 15.8. HRMS (ESI) m/z calcd. for C8H1235Cl+ [M + H]+: 143.0622; found: 143.0618.
4-Fluorodec-1-en-7-yne (1j). Dec-1-en-7-yn-4-ol (1g, 300 mg, 1.96 mmol) was added to a previously cooled (−45 °C) solution of diethylaminosulfurtrifluoride (594 mg, 3.69 mmol) in dry CH2Cl2 (2.84 mL) with vigorous stirring over a 10-min period. The solution was allowed to come to room temperature overnight after which time it was transferred into a separatory funnel containing water and CH2Cl2. The organic phase was then dried over Na2SO4, concentrated, and purified by silica gel chromatography. Elution with cyclohexane/CH2Cl2 (50/1) gave a yellow oil (15 mg, 5.0%); 1H-NMR (300 MHz, CDCl3) δ 5.82 (m, 1H, CH=CH2), 5.16–5.10 (m, 2H, CH=CH2), 4.66 (brd, J = 51.3 Hz, 1H, CH-F), 2.43–2.29 (m, 4H), 2.15 (q, J = 6.9 Hz, 2H, CH2CH3), 1.85–1.69 (m, 2H), 1.11 (t, J = 7.2 Hz, 3H, CH3). 13C-NMR (75 MHz, CDCl3) δ 133.0 (d, J = 6.0 Hz, CH=CH2), 117.9 (CH=CH2), 92.0 (d, J = 168.9 Hz, CH–F), 82.3, 78.1, 39.3 (d, J = 21.2 Hz), 34.0 (d, J = 20.9 Hz), 14.7 (d, J = 5.2 Hz), 14.2, 12.4. LRMS (ESI) m/z calcd. for C10H16F+ [M + H]+: 155.1; found: 155.1.
3.3. General Precedure for Preparation of Co2(CO)6–Alkyne Complexes and Compound Characterization
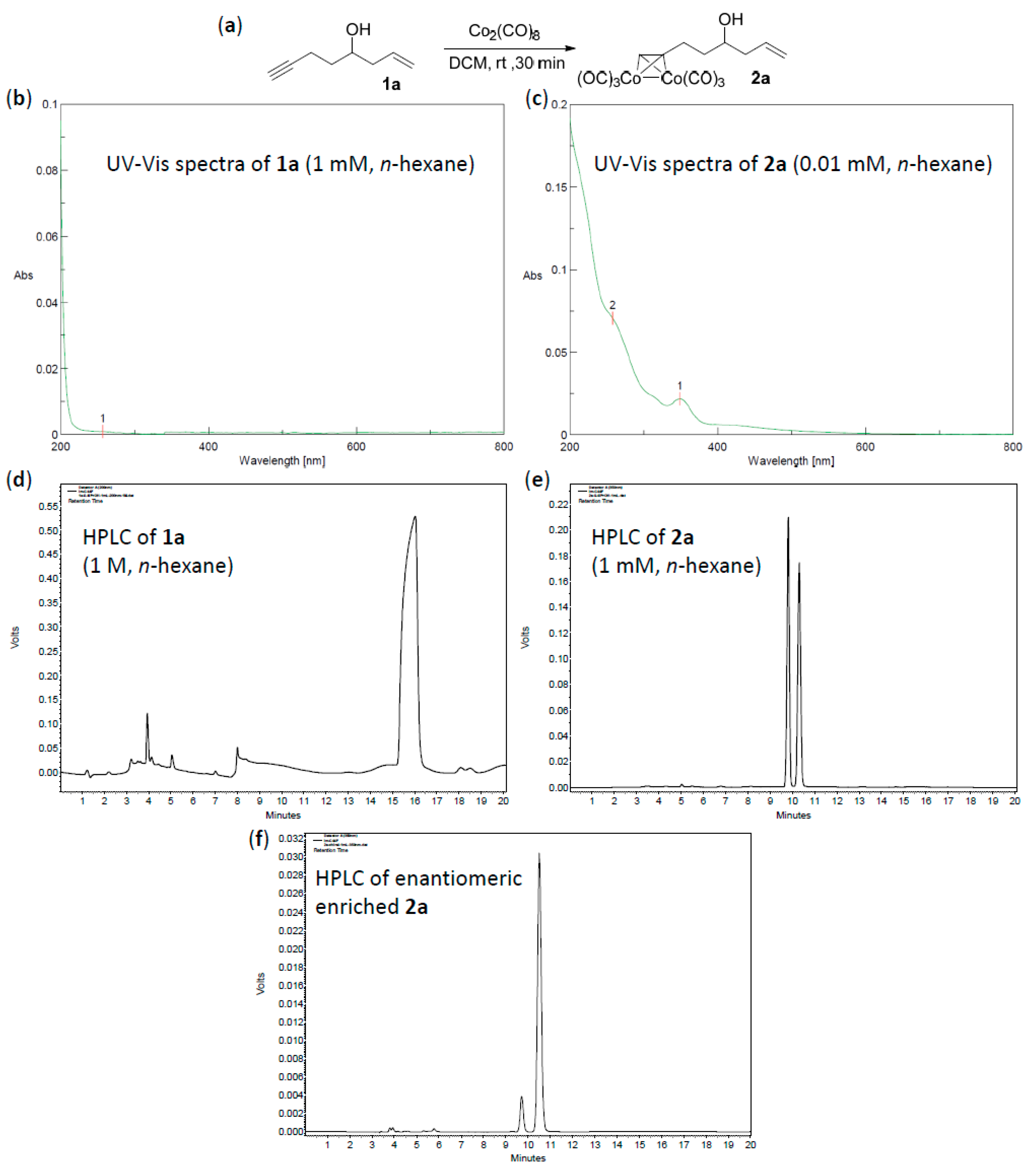
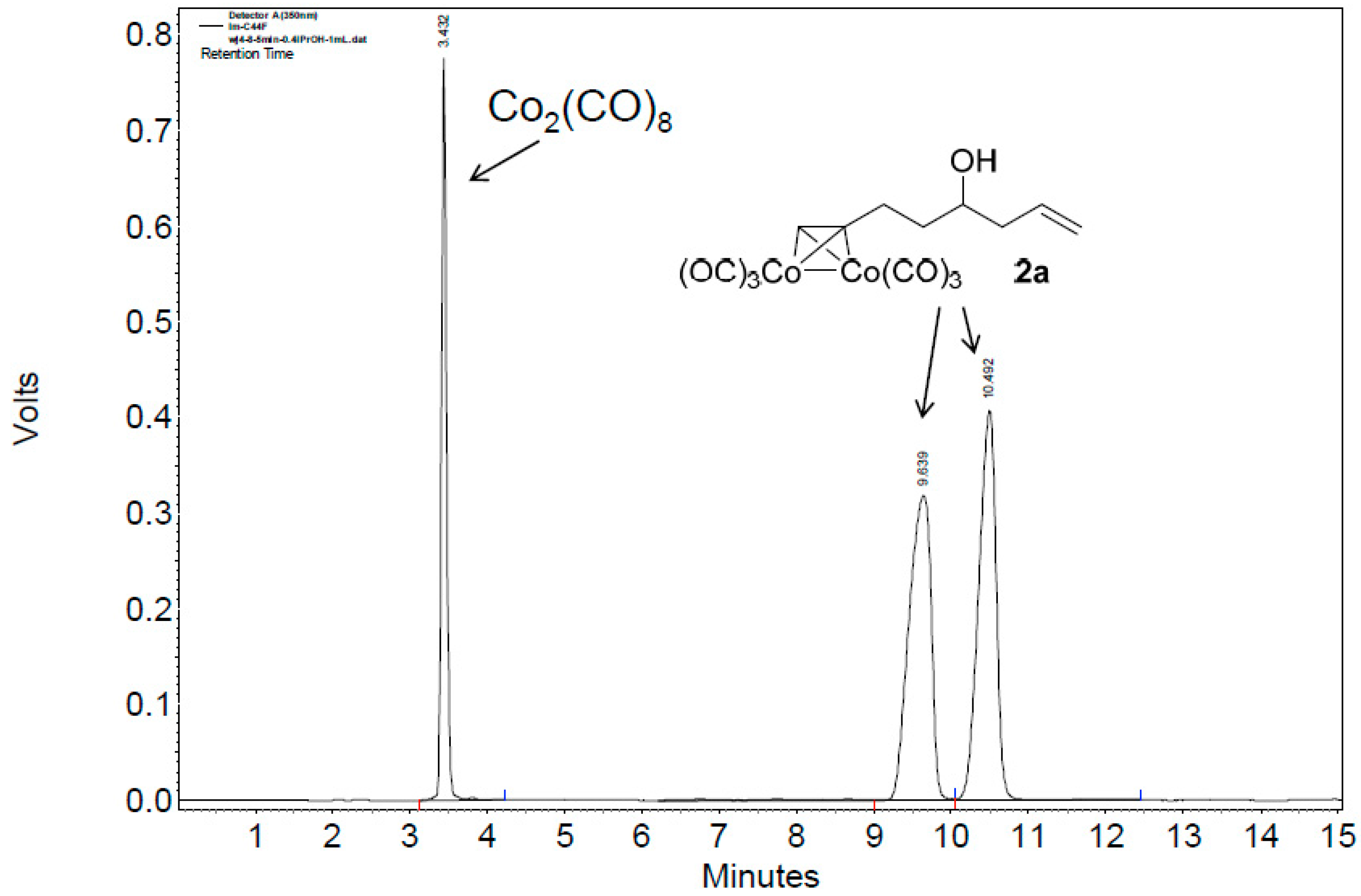
A mixture of alkyne 1 (2 mmol, 1.0 equiv.), Co2(CO)8 (2.2 mmol, 1.1 equiv.) in CH2Cl2 (0.5 mL) was stirred at room temperature under air atmosphere (balloon) until thin-layer chromatography (TLC) monitoring showed all alkyne consumed (about 1 h). The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to provide Co2(CO)6–alkyne complex 2.
Hexacarbonyl[µ-[(7,8-η:7,8-η)oct-1-en-7-yn-4-ol]]-dicobalt-(Co–Co) (2a): Dark red oil, yield 57%; 1H-NMR (500 MHz, CDCl3) δ 6.02 (s, 1H, ≡CH), 5.83 (m, 1H, CH=CH2), 5.19–5.15 (m, 2H, CH=CH2), 3.79 (m, 1H, CH-OH), 3.09 (m, 1H), 2.92 (m, 1H), 2.36 (m, 1H), 2.22 (m, 1H), 1.80 (m, 2H), 1.66 (d, J = 4.0 Hz, 1H, OH). 13C-NMR (125 MHz, CDCl3) δ 199.9 (br, CO × 6), 134.2 (C=CH), 118.8 (CH=CH2), 97.1 (C≡CH), 73.1 (C≡CH), 69.9 (CHOH), 42.1, 38.9, 30.4. HRMS (EI) m/z calcd. for C13H162Co2O6+ [M+ − CO]: 381.9292; found: 381.9300.
Hexacarbonyl[µ-[(1,2-η:1,2-η)non-1-yn-5-ol]]-dicobalt-(Co–Co) (2b): Dark red oil, yield 58%; 1H-NMR (300 MHz, CDCl3) δ 6.01 (s, 1H, ≡CH), 3.92 (m, 1H, CHOH), 3.07 (ddd, J = 15.6, 9.6, 6.3 Hz, 1H), 2.89 (ddd, J = 15.6, 9.9, 6.3 Hz, 1H), 1.76 (m, 2H), 1.49–1.43 (m, 4H), 1.38-1.33 (m, 3H), 0.92 (t, J = 6.6 Hz, 3H, CH3). 13C-NMR (75 MHz, CDCl3) δ 200.0 (br, CO × 6), 97.3 (C≡CH), 73.0 (C≡CH), 71.3 (CHOH), 39.5, 37.2, 30.3, 27.7, 22.7, 14.0 (CH3). HRMS (EI) m/z calcd. for C14H16Co2O6+ [M+ − CO]: 397.9605; found: 397.9615.
Hexacarbonyl[µ-[(4,5-η:4,5-η)pent-4-yn-2-ol]]-dicobalt-(Co–Co) (2c): Dark red oil, yield 67%; 1H-NMR (300 MHz, CDCl3) δ 6.10 (s, 1H, ≡CH), 3.95 (m, 1H, CHOH), 3.01 (d, J = 4.8 Hz, 2H, CH2), 1.57 (d, J = 6.3 Hz, 1H, OH), 1.35 (d, J = 5.4 Hz, 3H, CH3). 13C-NMR (75 MHz, CDCl3) δ 199.8 (br, CO × 6), 92.0 (C≡CH), 73.9 (C≡CH), 68.7 (CHOH), 43.7 (CH2), 23.7 (CH3). HRMS (EI) m/z calcd. for C10H8Co2O6+ [M+ − CO]: 341.8979; found: 341.8987.
Hexacarbonyl[µ-[(5,6-η:5,6-η)hex-5-yn-3-ol]]-dicobalt-(Co–Co) (2d): Dark red oil, yield 78%; 1H-NMR (300 MHz, CDCl3) δ 6.09 (s, 1H, ≡CH), 3.66 (m, 1H, CHOH), 3.00 (m, 2H, ≡CCH2), 1.76 (d, J = 3.3 Hz, 1H, OH), 1.63 (m, 2H, CH2CH3), 1.01 (t, J = 7.2 Hz, 3H, CH3). 13C-NMR (75 MHz, CDCl3) δ 199.8 (br, CO × 6), 92.5 (C≡CH), 74.1 (C≡CH), 73.9 (CHOH), 41.5 (≡CCH2), 30.2 (CH2CH3), 9.7 (CH3). HRMS (EI) m/z calcd. for C11H10Co2O6+ [M+ − CO]: 355.9136; found: 355.9142.
Hexacarbonyl[µ-[(1,2-η:1,2-η)pent-1-yn-3-ol]]-dicobalt-(Co–Co) (2e): Dark red oil, yield 84%; 1H-NMR (600 MHz, CDCl3) δ 6.06 (d, J = 0.6 Hz, 1H, ≡CH), 4.63 (ddd, J = 7.8, 5.4, 5.4 Hz, 1H, CHOH), 1.84 (d, J = 5.4 Hz, 1H, OH), 1.76 (m, 1H of CH2), 1.70 (m, 1H of CH2), 1.09 (t, J = 7.2 Hz, 3H, CH3). 13C-NMR (150 MHz, CDCl3) δ 199.6 (br, CO × 6), 99.8 (C≡CH), 73.9, 71.6, 32.9 (CH2), 10.6 (CH3). HRMS (EI) m/z calcd. for C10H8Co2O6+ [M+]: 369.8929; found: 369.8933.
Hexacarbonyl[µ-[(1,2-η:1,2-η)-3-methylpent-1-yn-3-ol]]-dicobalt-(Co–Co) (2f): Dark red oil, yield 78%; 1H-NMR (300 MHz, CDCl3) δ 6.06 (s, 1H, ≡CH), 1.78 (m, 2H, CH2), 1.73 (s, 1H, OH), 1.49 (s, 3H, CH3), 1.03 (t, J = 7.2 Hz, 3H, CH2CH3). 13C-NMR (75 MHz, CDCl3) δ 199.6 (br, CO × 6), 105.2 (C≡CH), 74.9, 72.1, 37.8 (CH2), 29.8 (CH3), 8.7 (CH2CH3). HRMS (EI) m/z calcd. for C12H10Co2O7+ [M+]: 383.9085; found: 383.9090.
Hexacarbonyl[µ-[(7,8-η:7,8-η)dec-1-en-7-yn-4-ol]]-dicobalt-(Co–Co) (2g): Dark red oil, yield 69%; 1H-NMR (300 MHz, CDCl3) δ 5.82 (m, 1H, CH=CH2), 5.20-5.15 (m, 2H, CH=CH2), 3.79 (brd, J = 3.9 Hz, 1H, CH-OH), 3.08 (m, 1H), 2.91 (m, 1H), 2.86 (q, J = 7.2 Hz, 2H, CH2CH3), 2.36 (m, 1H), 2.22 (m, 1H), 1.82 (m, 2H), 1.69 (d, J = 3.9 Hz, 1H, OH), 1.29 (t, J = 7.2 Hz, 3H, CH3). 13C-NMR (75 MHz, CDCl3) δ 200.3 (br, CO × 6), 134.2 (CH=CH2), 118.8 (CH=CH2), 101.8, 99.0, 70.0 (CH-OH), 42.0, 38.3, 30.1, 27.0, 15.6 (CH3). HRMS (EI) m/z calcd. for C15H16Co2O6+ [M+ − CO]: 409.9605; found: 409.9607.
Hexacarbonyl[µ-[(7,8-η:7,8-η)-8-(trimethylsilyl)oct-1-en-7-yn-4-ol]]-dicobalt-(Co–Co) (2h): Dark red oil, yield 49%; 1H-NMR (300 MHz, CDCl3) δ 5.82 (m, 1H, CH=CH2), 5.21-5.16 (m, 2H, CH=CH2), 3.80 (m, 1H, CH-OH), 3.20 (m, 1H), 2.96 (m, 1H), 2.40 (m, 1H), 2.23 (m, 1H), 1.82 (m, 2H), 1.68 (s, 1H, OH), 0.30 (s, 9H, CH3 × 3). 13C-NMR (75 MHz, CDCl3) δ 200.6 (br, CO × 6), 134.1 (CH=CH2), 118.8 (CH=CH2), 112.3 (≡C-Si), 79.1 (C≡C-Si), 70.0 (CH-OH), 42.0, 39.2, 31.4, 0.65 (SiCH3 × 3). HRMS (EI) m/z calcd. for C16H20Co2O6Si+ [M+ − CO]: 453.9688; found: 453.9693.
Hexacarbonyl[µ-[(7,8-η:7,8-η)-4-chlorooct-1-en-7-yne]]-dicobalt-(Co–Co) (2i): Dark red oil, yield 74%; 1H-NMR (300 MHz, CDCl3) δ 6.03 (s, 1H, ≡CH), 5.85 (m, 1H, CH=CH2), 5.18–5.13 (m, 2H, CH=CH2), 4.05 (m, 1H, CH-Cl), 3.11 (m, 1H), 2.97 (m, 1H), 2.57 (m, 2H), 2.05 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 199.9 (br, CO × 6), 133.6 (CH=CH2), 118.4 (CH=CH2), 95.7 (C≡CH), 73.1 (C≡CH), 61.2 (CH-Cl), 42.7, 39.5, 30.8. HRMS (EI) m/z calcd. for C13H11Co235ClO5+ [M+ − CO]: 399.8954; found: 399.8960.
Hexacarbonyl[µ-[(7,8-η:7,8-η)-4-fluorodec-1-en-7-yne]]-dicobalt-(Co–Co) (2j): Dark red oil, yield 90%; 1H-NMR (600 MHz, CDCl3) δ 5.84 (ddt, J = 24.6, 17.4, 7.2 Hz, 1H, CH=CH2), 5.17–5.13 (m, 2H, CH=CH2), 4.66 (brd, J = 48.6 Hz, 1H, CH-F), 3.06 (m, 1H), 2.91 (m, 1H), 2.86 (q, J = 7.2 Hz, 2H, CH2CH3), 2.46 (m, 2H), 1.93 (m, 2H), 1.29 (t, J = 7.2 Hz, 3H, CH3). 13C-NMR (150 MHz, CDCl3) δ 200.1 (br, CO × 6), 132.6 (d, J = 6.3 Hz, CH=CH2), 118.3 (CH=CH2), 101.8 (C≡CEt), 98.2 (C≡CEt), 92.5 (d, J = 170.1 Hz, CH-F), 39.4 (d, J = 21.3 Hz, CH2CH=CH2), 36.2 (d, J = 20.7 Hz, CH2CH2C≡C), 29.3 (d, J = 4.1 Hz, C≡CCH2CH2), 27.0 (CH2CH3), 15.6 (CH3). HRMS (EI) m/z calcd. for C15H15Co2FO5+ [M+ − CO]: 411.9562; found: 411.9570.
Hexacarbonyl[µ-[2-((3,4-η:3,4-η)but-3-yn-1-yloxy)tetrahydro-2H-pyran]]-dicobalt-(Co–Co) (2k): Dark red oil, yield 65%; 1H-NMR (500 MHz, CDCl3) δ 6.03 (s, 1H, ≡CH), 4.63 (t, J = 4.0 Hz, 1H, O–CH–O), 3.99 (m, 1H), 3.88 (m, 1H), 3.60 (m, 1H), 3.53 (m, 1H), 3.15 (m, 2H), 1.84 (m, 1H), 1.72 (m, 1H), 1.62–1.51 (m, 4H). 13C-NMR (125 MHz, CDCl3) δ 199.9 (br, CO × 6), 98.9 (O–CH–O), 92.9 (C≡CH), 73.7 (C≡CH), 67.3, 62.4, 34.1, 30.6, 25.4, 19.5. HRMS (EI) m/z calcd. for C14H14Co2O7+ [M+ − CO]: 411.9398; found: 411.9400.





{kind=link}
{kind=link}





























