
Solventless Synthesis of Poly(pyrazolyl)phenyl-methane Ligands and Thermal Transformation of Tris(3,5-dimethylpyrazol-1-yl)phenylmethane

 ,
,
Abstract
:
1. Introduction
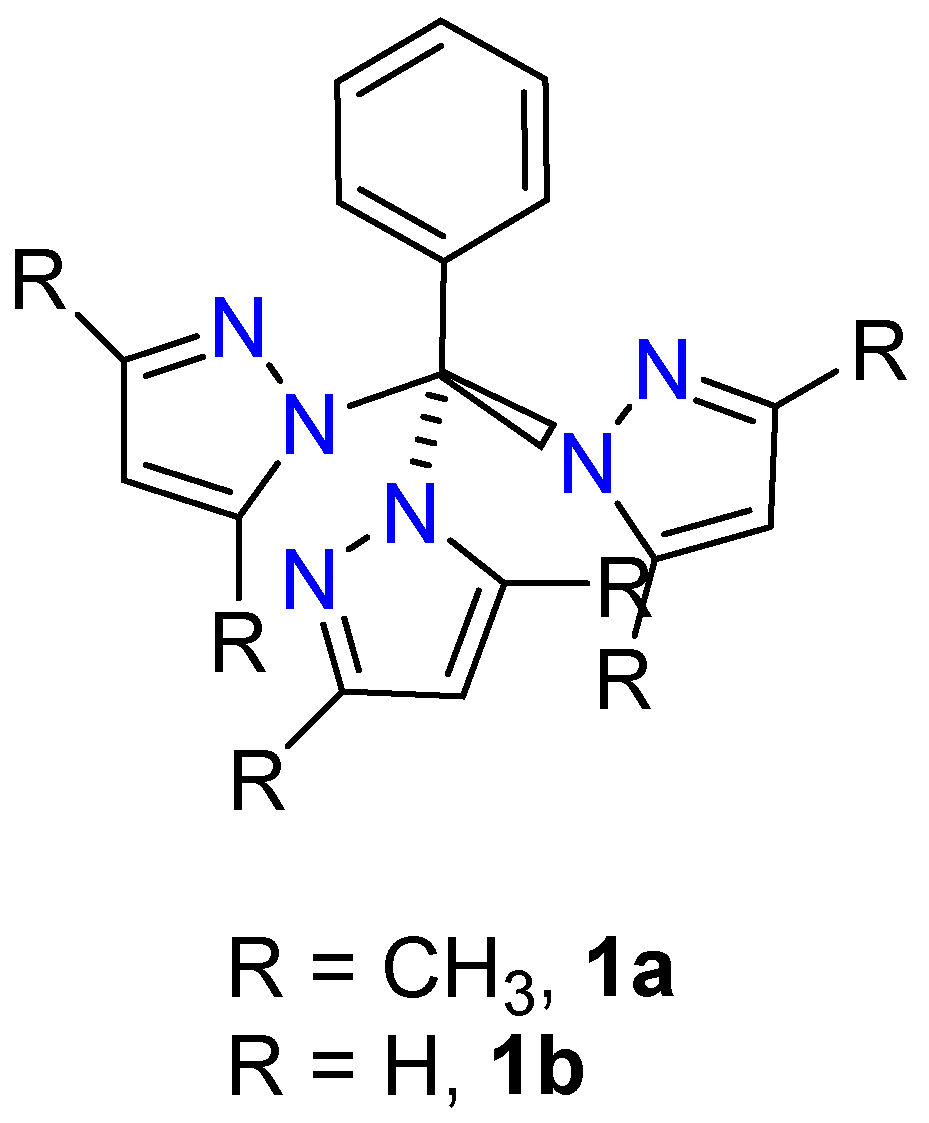
2. Results and Discussion
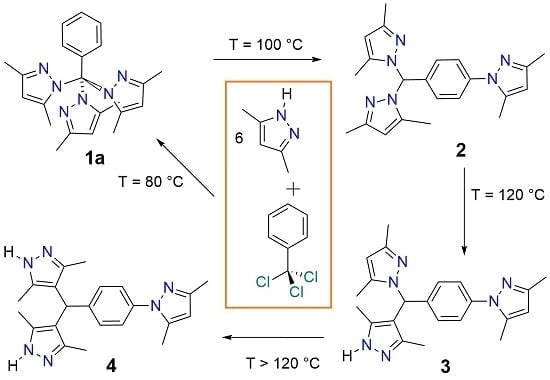
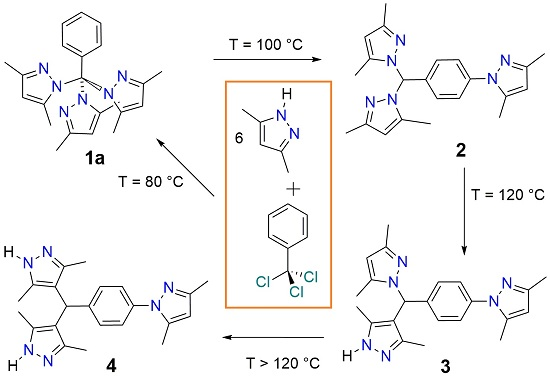
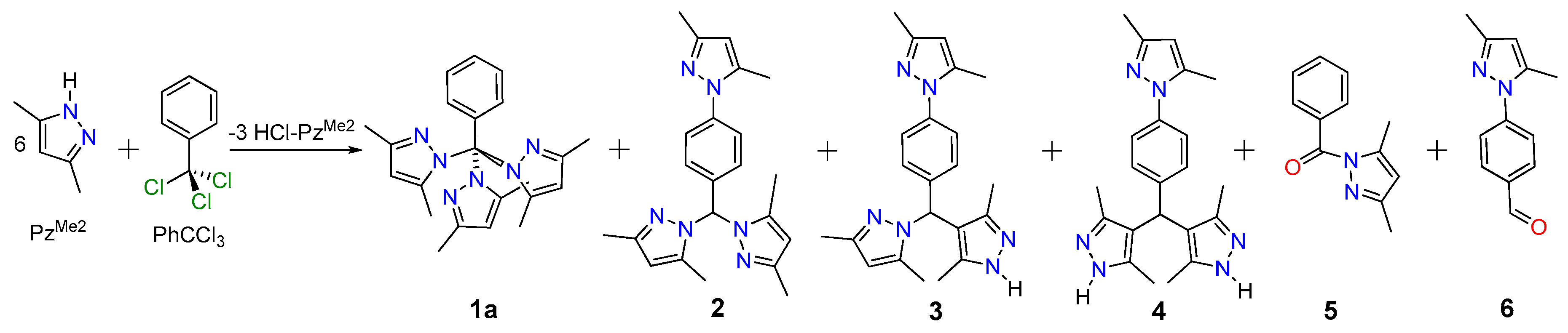
2.1. Screening Reactions with PzMe2
2.2. Effect of Temperature and Time on the Composition of the Reaction Mixtures
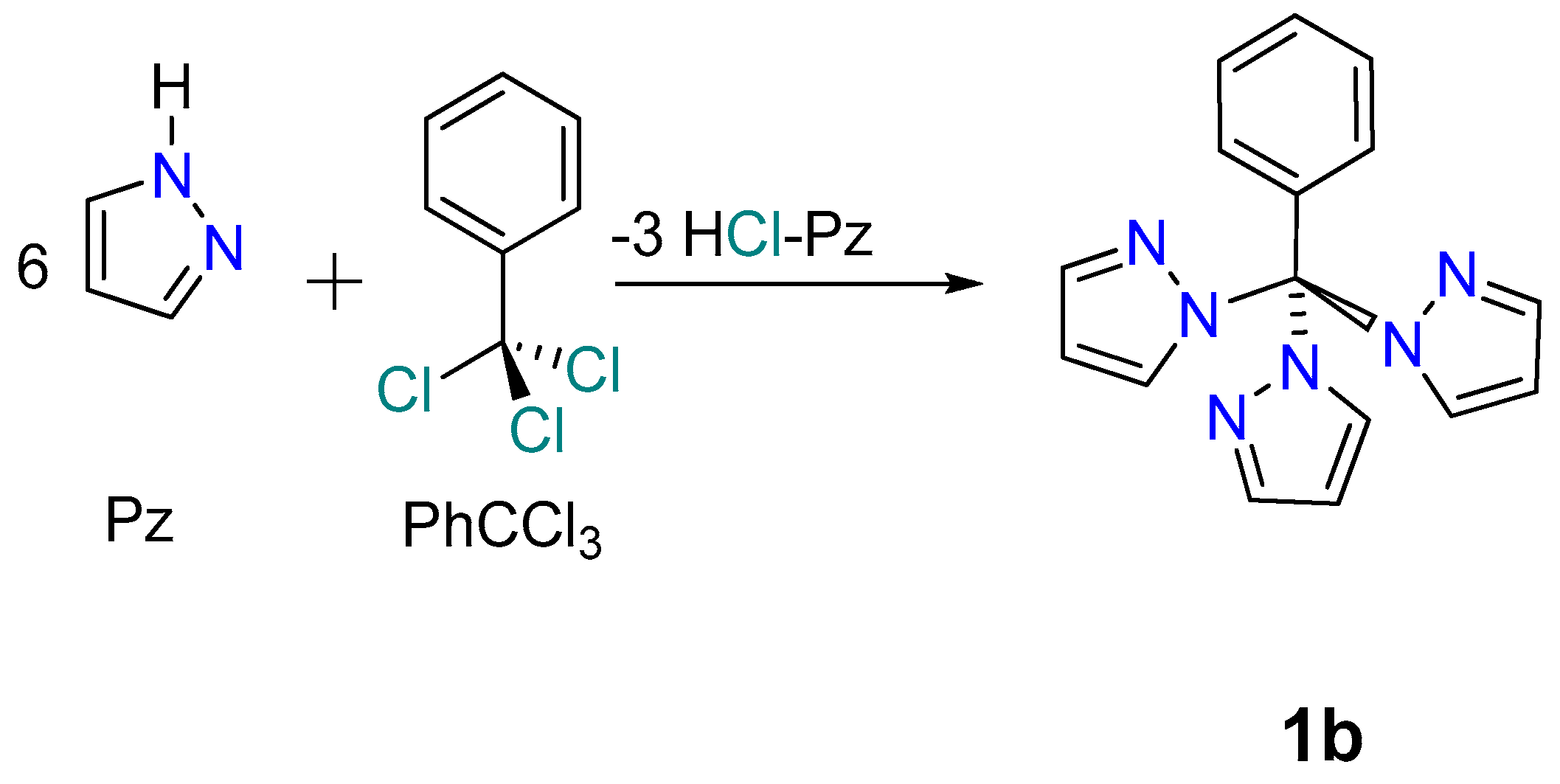
2.3. Reaction between PhCCl3 and Pz
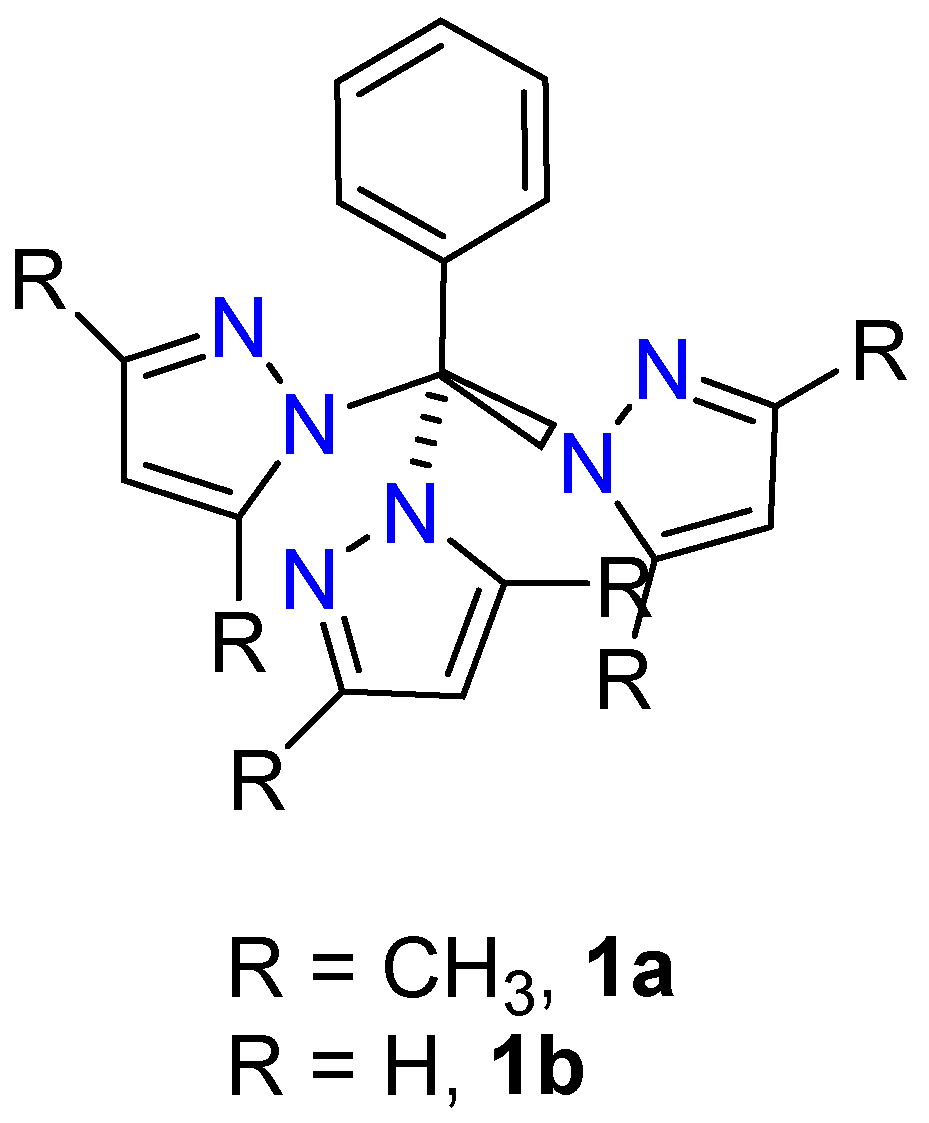
2.4. Stucture by NMR and IR
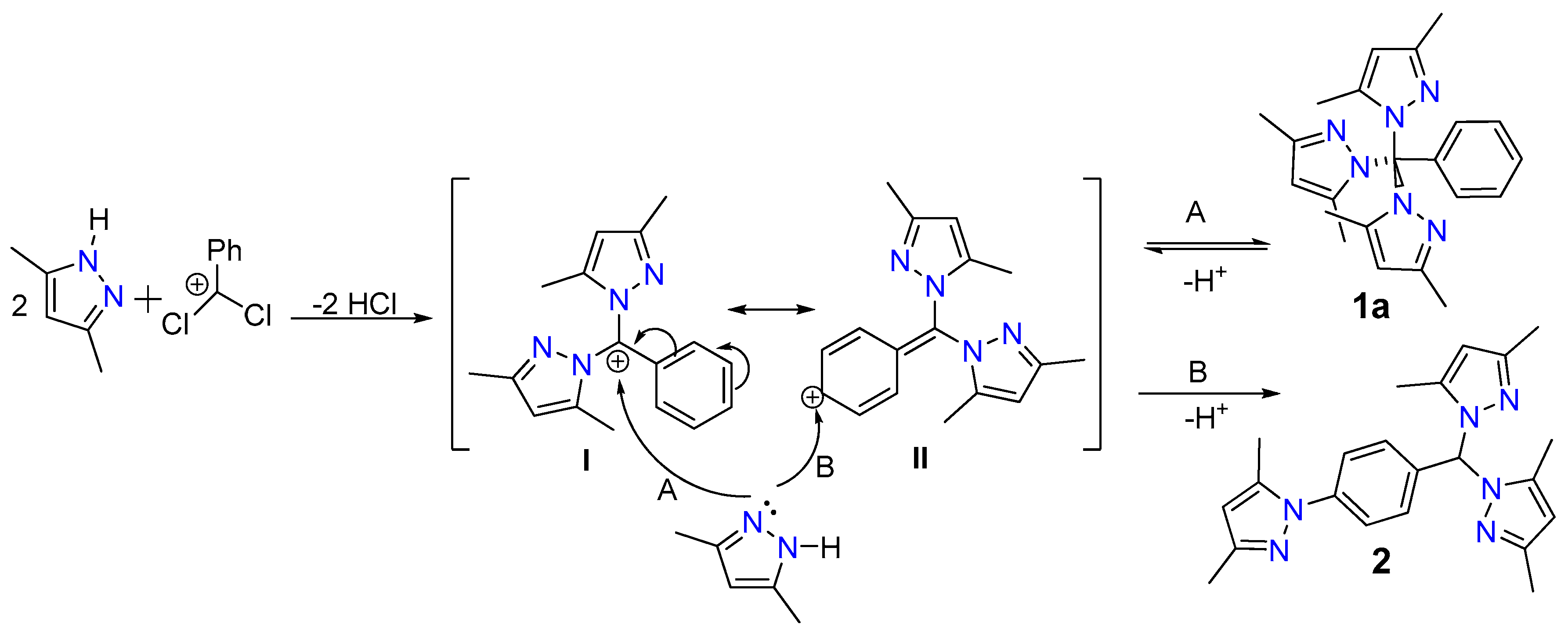
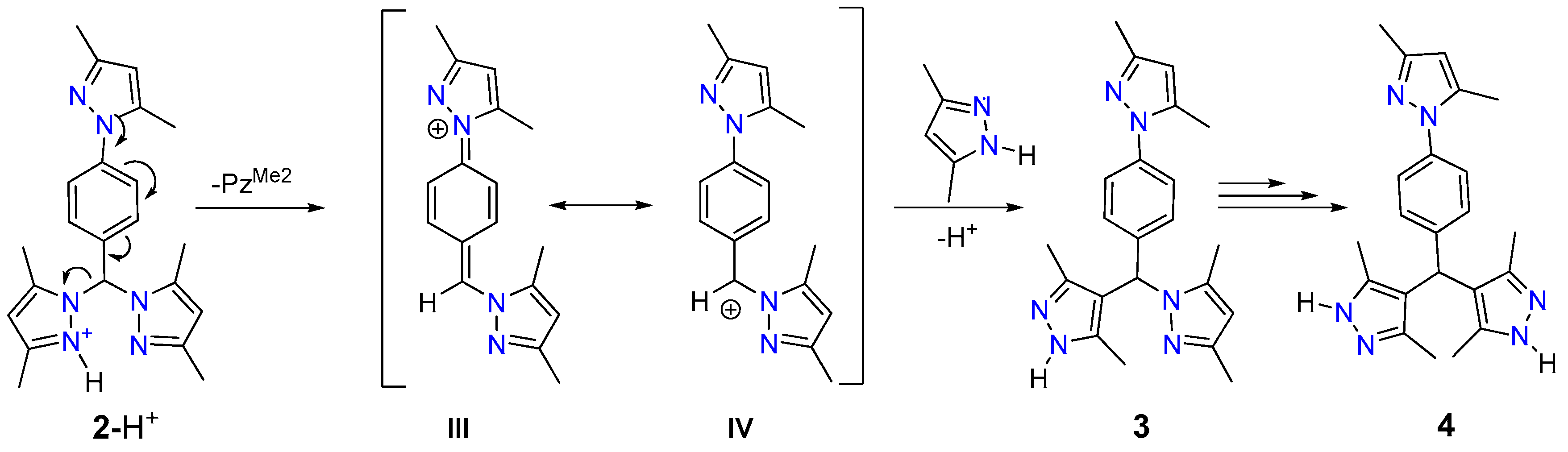
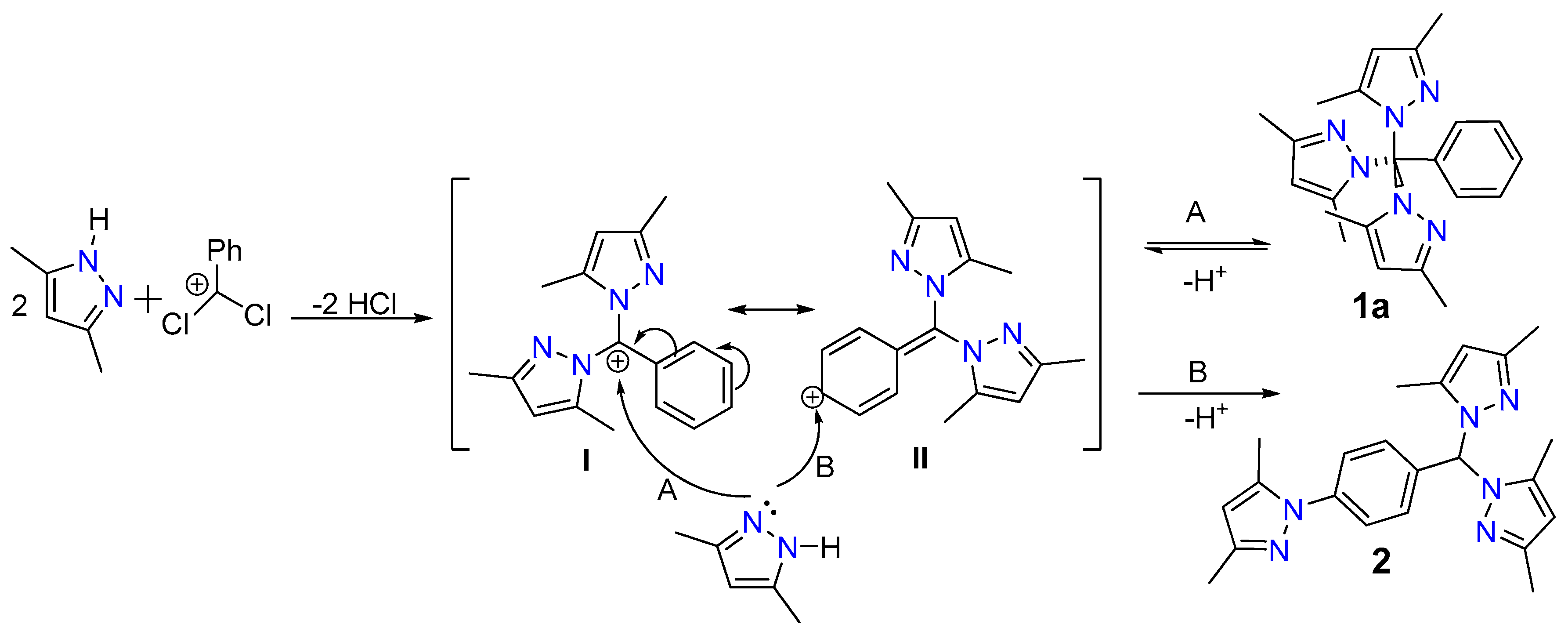
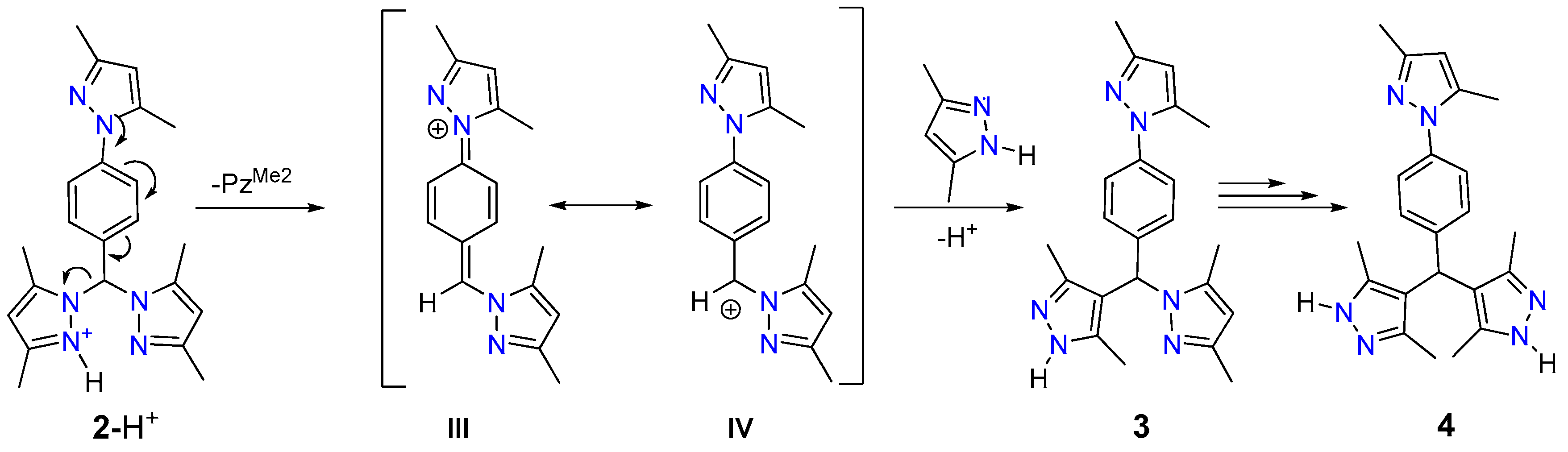
2.5. Proposed Reaction Mechanism
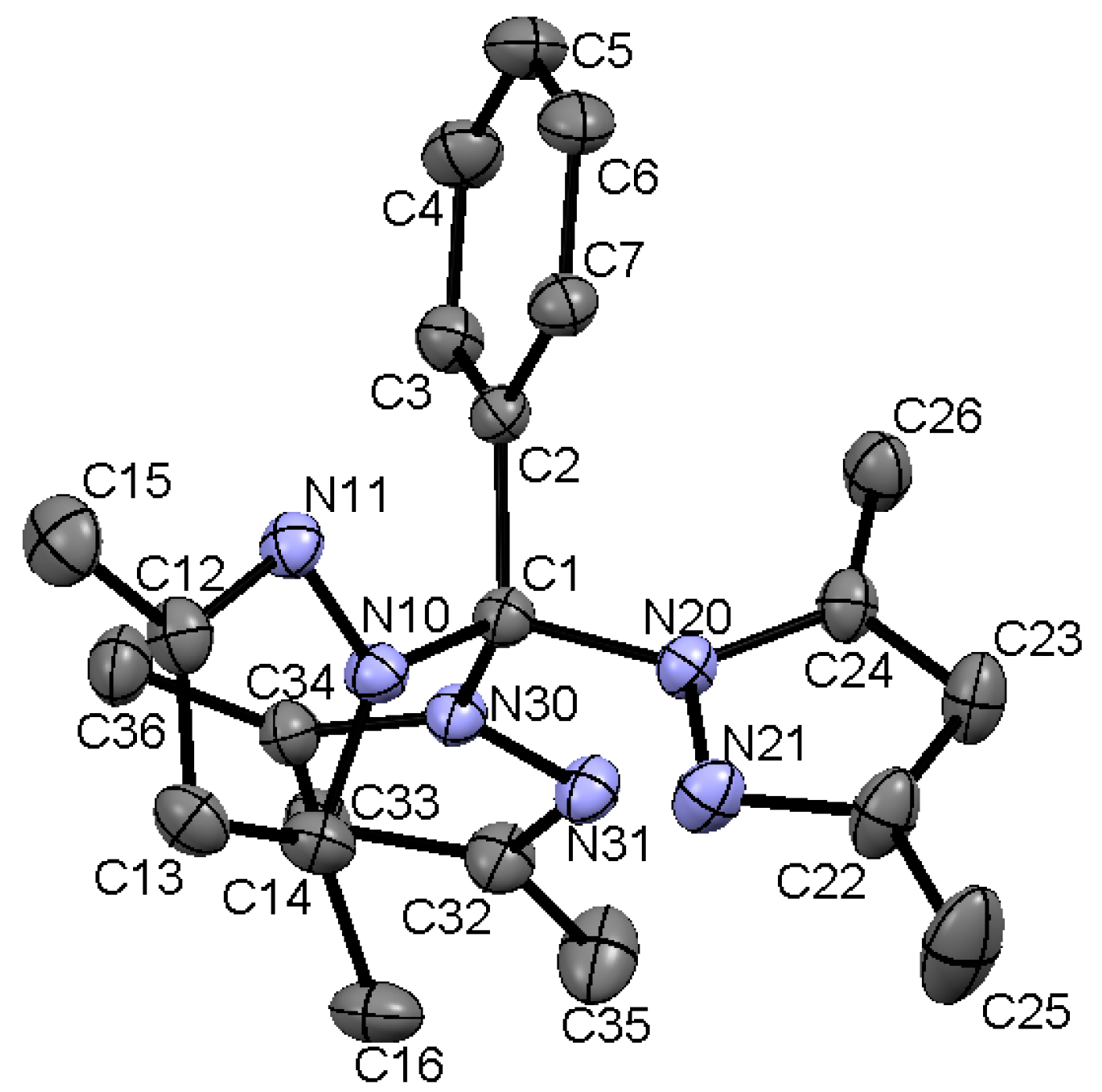
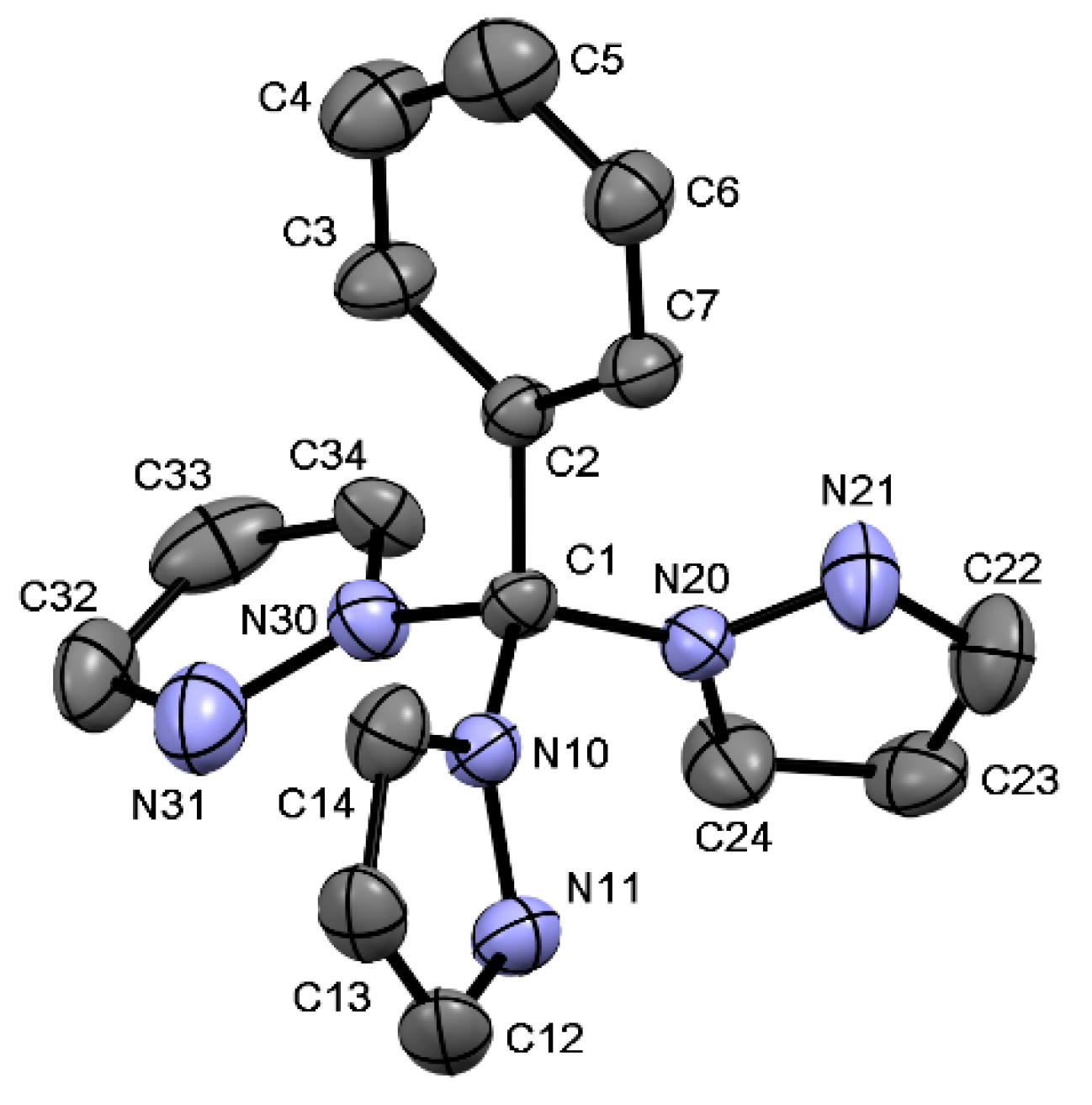
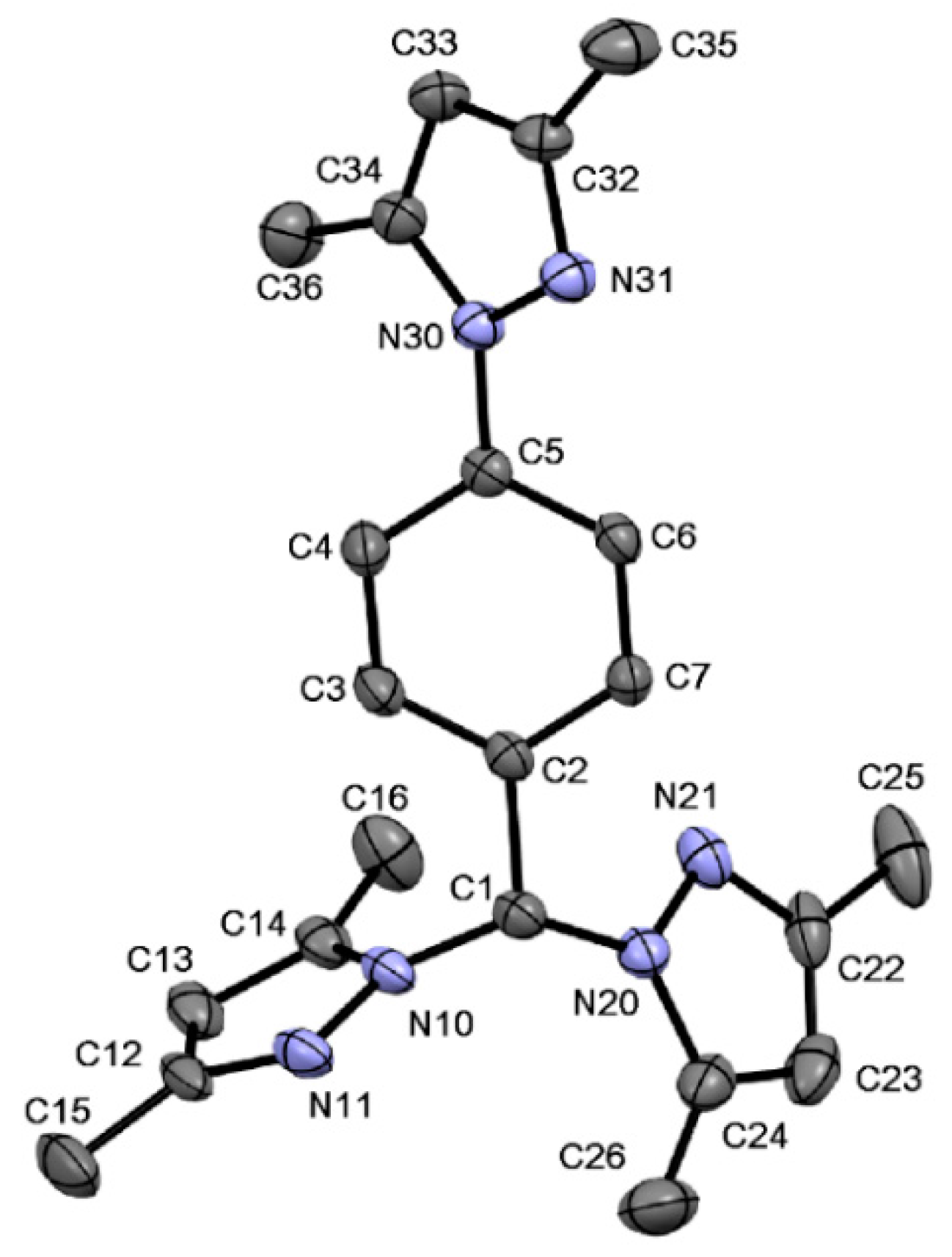
2.6. Single Crystal Structures of 1a–b and 2
3. Materials and Methods
3.1. Chemicals
3.2. Instrumental Methods
3.3. X-ray Diffraction Methods
3.4. Synthetic Procedures
3.4.1. Open Flask Procedure
3.4.2. Sealed Ampoule Procedure
3.4.3. Vacuum Sealed Ampoule Procedure
3.5. Synthesis of Compounds 1–6



3.6. Temperature Effects Experiments
3.7. Time Effect Experiments
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scorpionates, T.S. The Coordination Chemistry of Polypyrazolylborate Ligands; Imperial College Press: London, UK, 1999. [Google Scholar]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Muñoz-Hernández, M.A.; Montiel-Palma, V. Polypyrazolates of the heavier group 13 and 14 elements: A review. Inorg. Chim. Acta 2009, 362, 4328–4339. [Google Scholar] [CrossRef]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Semeniuc, R.F.; Reger, D.L. Metal Complexes of Multitopic, Third Generation Poly(pyrazolyl)methane Ligands: Multiple Coordination Arrangements. Eur. J. Inorg. Chem. 2016, 2253–2271. [Google Scholar] [CrossRef]
- Bigmore, H.R.; Sally, C.; Mountford, L.P.; Tredget, C.S. Coordination, organometallic and related chemistry of tris(pyrazolyl)methane ligands. Dalton Trans. 2005, 4, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Fernández-Baeza, J.; Antiñolo, A.; Tejeda, J.; Lara-Sánchez, A. Heteroscorpionate ligands based on bis(pyrazol-1-yl)methane: Design and coordination chemistry. Dalton Trans. 2004, 10, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Fernández-Baeza, J.; Lara-Sanchez, A.; Tejeda, J.; Sanchez-Barba, L.F. Microreview: Recent Advances in the Design and Coordination Chemistry of Heteroscorpionate Ligands Bearing Stereogenic Centres. Eur. J. Inorg. Chem. 2008, 5309–5326. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Sánchez-Barba, L.F. Metal complexes with heteroscorpionate ligands based on the bis(pyrazol-1-yl)methane moiety: Catalytic Chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Padilla-Martínez, I.I.; Poveda, M.L.; Carmona, E.; Monge, M.A.; Ruiz-Valero, C. Synthesis and Reactivity of [Ir(C2H4)2TpmMe2]PF6 (TpmMe2 = Tris(3,5-dimethylpyrazolyl)methane): Comparison with the Analogous TpMe2 Derivatives (TpMe2 = Hydrotris(3,5-dimethylpyrazolyl)borate). Organometallics 2002, 21, 93–104. [Google Scholar] [CrossRef]
- Hernández-Juárez, M.; Salazar, V.; Padilla-Martínez, I.I.; García-Báez, E.V. Carbonyl-carbonyl and carbonyl-π interactions as directing motifs in the supramolecular structure of carbonyl(3-oxopenta-1,4-diene-1,5-diyl)[tris(3,5-dimethyl-1H-pyrazol-1-yl-κN2)methane]iridium(III) trifluoromethanesulfonate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2012, 68, m367–m369. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Juárez, M.; Salazar, V.; García-Báez, E.V.; Padilla-Martínez, I.I.; Höpfl, H.; Rosales-Hoz, M.J. Synthesis and Crystal Structure of Iridium-1,4-benzoquinone Complexes of Tris(3,5-dimethylpyrazolyl)methane Ligand: Decarbonylation, Protonation and Substitution Reactions. Organometallics 2012, 31, 5439–5451. [Google Scholar] [CrossRef]
- Cushion, M.G.; Meyer, J.; Heath, A.; Schwarz, A.D.; Fernandez, I.; Breher, F.; Mountford, P. Syntheses and Structural Diversity of Group 2 and Group 12 Tris(pyrazolyl)methane and Zwitterionic Tris(pyrazolyl)methanide Compounds. Organometallics 2010, 29, 1174–1190. [Google Scholar] [CrossRef]
- Manzano, B.R.; Jalón, F.A.; Carrión, M.C.; Durá, G. Bis(pyrazol-1-yl)(pyridin-x-yl)methane Ligands-Mono- or Ditopic Ligands in Complexes and Supramolecular Frameworks. Eur. J. Inorg. Chem. 2016, 2272–2295. [Google Scholar] [CrossRef]
- Wang, L.; Chambron, J.-C. Synthesis of a Macrobicycle Incorporating the Tris(pyrazolyl)methane Ligand. Org. Lett. 2004, 6, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Bassanetti, I.; Comotti, A.; Sozzani, P.; Bracco, S.; Calestani, G.; Mezzadri, F.; Marchio, L. Porous Molecular Crystals by Macrocyclic Coordination Supramolecules. J. Am. Chem. Soc. 2014, 136, 14883–14895. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-Soluble C-Scorpionate Complexes–Catalytic and Biological Applications. Eur. J. Inorg. Chem. 2016, 2236–2252. [Google Scholar] [CrossRef]
- García, R.; Paulo, A.; Santos, I. Rhenium and technetium complexes with anionic or neutral scorpionates: An overview of their relevance in biomedical applications. Inorg. Chim. Acta 2009, 362, 4315–4327. [Google Scholar] [CrossRef]
- Zhang, J.; Li, A.; Hor, T.S.A. Ligand effect on ethylene trimerisation with [NNN]-heteroscorpionate pyrazolyl Cr(III) catalysts. Dalton Trans. 2009, 42, 9327–9333. [Google Scholar] [CrossRef] [PubMed]
- Julia, S.; del Mazo, J.M.; Avila, L.; Elguero, J. Improved synthesis of polyazolylmethanes under solid-liquid phase-transfer catalysis. Org. Prep. Proced. Int. 1984, 16, 299–307. [Google Scholar] [CrossRef]
- Reger, D.L.; Grattan, T.C.; Brown, K.J.; Little, C.A.; Lamba, J.J.S.; Rheingold, A.L.; Sommer, R.D. Syntheses of tris(pyrazolyl)methane ligands and {[tris(pyrazolyl)methane]Mn(CO)3}SO3CF3 complexes: Comparison of ligand donor properties. J. Organomet. Chem. 2000, 607, 120–128. [Google Scholar] [CrossRef]
- Fujisawa, K.; Ono, T.; Aoki, H.; Ishikawa, Y.; Miyashita, Y.; Okamoto, K.-I.; Nakazawa, H.; Higashimura, H. Copper(II) complexes with a novel tris(3,5-diisopropyl-1-pyrazolyl)methane ligand, [Cu(X2){HC(3,5-iPr2pz)3}] (X = Cl and NO3). Inorg. Chem. Commun. 2004, 7, 330–332. [Google Scholar] [CrossRef]
- Reger, D.L.; Grattan, T.C. Synthesis of Modified Tris(pyrazolyl)methane Ligands: Backbone Functionalization. Synthesis 2003, 350–356. [Google Scholar] [CrossRef]
- Goodman, M.S.; Bateman, M.A. Synthesis of tris(pyrazolyl)methanes of unprecedented complexity and functionality. Tetrahedron Lett. 2001, 42, 5–7. [Google Scholar] [CrossRef]
- Liddle, B.J.; Gardinier, J.R. A Practical Synthesis of Tris(pyrazolyl)methylaryls. J. Org. Chem. 2007, 72, 9794–9797. [Google Scholar] [CrossRef] [PubMed]
- Krieck, S.; Koch, A.; Hinze, K.; Müller, C.; Lange, J.; Görls, H.; Westerhausen, M. s-Block Metal Complexes with Bis- and Tris(pyrazolyl)methane and -methanide Ligands. Eur. J. Inorg. Chem. 2016, 15–16, 2332–2348. [Google Scholar] [CrossRef]
- Zhang, X.-M. Hydro(solvo)thermal in situ ligand syntheses. Coord. Chem. Rev. 2005, 249, 1201–1219. [Google Scholar] [CrossRef]
- Lupacchini, M.; Mascitti, A.; Giachi, G.; Tonucci, L.; d’Alessandro, N.; Martinez, J.; Colacino, E. Sonochemistry in non-conventional, green solvents or solvent-free reactions. Tetrahedron 2017, 73, 609–653. [Google Scholar] [CrossRef]
- Do, J.-L.; Friščić, T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J. Microwave Assisted Reactions in Organic Chemistry: A Review of Recent Advances. Int. J. Chem. 2012, 4, 29–43. [Google Scholar] [CrossRef]
- Escobedo, R.; Miranda, R.; Martínez, J. Infrared Irradiation: Toward Green Chemistry, a Review. Int. J. Mol. Sci. 2016, 17, 453. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, E.R.; Mann, K.L.V.; Reeves, Z.R.; Behrendt, A.; Jeffery, J.C.; Maher, J.P.; McCleverty, J.A.; Ward, M.D. Copper(II) complexes of new potentially hexadentate N3S3- or N6-donor podand ligands based on the tris(pyrazolyl)borate or tris(pyrazolyl)methane core. New J. Chem. 1999, 23, 417–423. [Google Scholar] [CrossRef]
- García-Sánchez, A.; Padilla-Martínez, I.I.; Martínez-Martínez, F.J.; Höpfl, H.; García-Báez, E.V. 1,1,2,2-Tetrachloro-1,2-diphenylethane. Acta Crystallogr. Sect. E Struct. Rep. 2005, 61, o678–o680. [Google Scholar] [CrossRef]
- Seidel, F.; Their, W.; Uber, A.; Dittmer, J. Über die Bildung des Triacetyl-essigesters (II. Mitteil.). Eur. J. Inorg. Chem. 1935, 68, 1913–1924. [Google Scholar] [CrossRef]
- Artico, M.; Silvestri, R.; Stefancich, G.; Avigliano, L.; di Giulio, A.; Maccarrone, M.; Agostinelli, E.; Mondovi, B.; Morpurgo, L. Aromatic hydrazides as specific inhibitors of bovine serum amine oxidase. Eur. J. Med. Chem. 1992, 27, 219–228. [Google Scholar] [CrossRef]
- Liao, P.-Q.; He, C.-T.; Zhou, D.-D.; Zhang, J.-P.; Chen, X.-M. Porous Metal Azole Frameworks. In The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications; Kaskel, S., Ed.; Wiley VCH: Weinheim, Germany, 2016; Volume 1, pp. 309–338. [Google Scholar]
- Cook, L.J.K.; Kearsey, R.; Lamb, J.V.; Pace, E.J.; Gould, J.A. Efficient and chromatography-free methodology for the modular synthesis of oligo-(1H-pyrazol-4-yl)-arenes with controllable size, shape and steric bulk. Tetrahedron Lett. 2016, 57, 895–898. [Google Scholar] [CrossRef]
- Sanjeev, R.; Jagannadham, V. Substituent effects on the spontaneous cleavage of benzyl-gem-dichlorides in aqueous solution. Indian J. Chem. Sect. B Org. Chem. Incl. Med. Chem. 2002, 41B, 2145–2149. [Google Scholar]
- Merga, G.; Aravindakumar, C.T.; Rao, B.S.M.; Mohan, H.; Mittal, J.P. Pulse radiolysis study of the reactions of SO4− with some substituted benzenes in aqueous solution. J. Chem. Soc. Faraday Trans. 1994, 90, 597–604. [Google Scholar] [CrossRef]
- Jude, H.; Rein, F.N.; Chen, W.; Scott, B.L.; Dattelbaum, D.M.; Rocha, R.C. Pyrazole and Pyrazolyl Complexes of cis-Bis(2,2′-bipyridine)chlororuthenium(II): Synthesis, Structural and Electronic Characterization and Acid-Base Chemistry. Eur. J. Inorg. Chem. 2009, 5, 683–690. [Google Scholar] [CrossRef]
- AL-Attas, A.S.; Habeeb, M.M.; Al-Raimi, D.S. Synthesis and spectroscopic studies of charge transfer complexes between chloranilic acid and some heterocyclic amines in etanol. J. Mol. Struct. 2009, 928, 158–170. [Google Scholar] [CrossRef]
- Ehlert, M.K.; Storr, A.; Thompson, R.C. Metal pyrazolate polymers. Part 3. Synthesis and study of Cu(I) and Cu(II) complexes of 4-Xdmpz (where X = H, Cl, Br, I, and CH3 for Cu(I) and X = H, Cl, Br, and CH3 for Cu(II); dmpz = 3,5-dimethylpyrazolate). Can. J. Chem. 1992, 70, 1121–1128. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Typical Interatomic Distances: Organic Compounds. International Tables for Crystallography; Wilson, A.J.C., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992; Volume C, pp. 685–706. [Google Scholar]
- Harding, D.J.; Harding, P.; Plant, S.E. Tris(5-methyl-3-phenyl-1H-pyrazol-1-yl)methane. Acta Crystallogr. Sect. E Struct. Rep. 2008, 64, o896. [Google Scholar] [CrossRef] [PubMed]
- Ochando, L.E.; Rius, J.; Louer, D.; Claramunt, R.M.; Lopez, C.; Elguero, J.; Amigo, J.M. Phase Transitions in Tris(3,5-dimethylpyrazol-1-yl)methane. The Structure of the High-Temperature Phase from X-ray Powder Diffraction. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 939–944. [Google Scholar] [CrossRef]
- Declercq, J.-P.; Van Meerssche, M. Tris(3,5-dimethyl-1-pyrazolyl)methane, C16H22N6. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1984, 40, 1098–1101. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Yang, X.; Holliday, B. J. 1,1′,1″-{[4-(3,4-Ethylenedioxythiophen-2-yl)phenyl]methanetriyl}tris(1H-pyrazole). Acta Cryst. Sect. 2011, 67, o3021. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Yang, X.; Holliday, B. J. 4-[Tris(1H-pyrazol-1-yl)methyl]phenol. Acta Crystallogr. Sect. E Struct. Rep. 2011, 67, o3045. [Google Scholar] [CrossRef] [PubMed]
- Mclauchlan, C.C.; Smith, B.L.; Pippins, R.S.; Nelson, B.M. 2,2,2-Tris(pyrazol-1-yl)ethanol. Acta Crystallogr. Sect. E Struct. Rep. 2011, 67, o1133–o1134. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, T.; Pust, P.; Betz, R.; Klüfersand, P.; Mayer, P. Trispyrazol-1-ylmethane. Acta Crystallogr. Sect. E Struct. Rep. 2009, 65, o108. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a, 1b, 2–4 are available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Vessel | Ratio 1 | Temperature (°C)/Time (h) | Products Isolated Yields (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Step 1 | Step 2 | 1a | 2 | 3 | 4 | 5 | 6 | |||
| 1 | Open flask | 6:1 | 100/10 | none | --- | 11 | --- | --- | 6 | 10 |
| 2 | SA 2 | 6:1 | 120/24 | none | --- | --- | --- | 8 | Trace | Trace |
| 3 | SA | 6:1 | 80/48 | none | --- | --- | --- | --- | 7 | Trace |
| 4 | SA | 6:1 | 80/24 | 120/24 | --- | 53 | 27 | --- | 6 | Trace |
| 5 | SA | 8:1 | 80/24 | 120/24 | --- | 48 | 25 | --- | 8 | Trace |
| 6 | SA | 4:1 | 80/24 | 120/24 | --- | 27 | 7 | --- | 9 | Trace |
| 7 | VSA 3 | 6:1 | 80/24 | none | 15 | --- | --- | --- | --- | --- |
| 8 | VSA | 6:1 | 80/48 | none | 30 | --- | --- | --- | --- | --- |
| 9 | VSA | 6:1 | 80/48 | 120/24 | --- | 70 | 2 | --- | --- | Trace |
| 10 | VSA | 6:1 | 160/72 | --- | --- | --- | 14 | --- | --- | |
| Comp. | 80 °C to T2 (°C) | ||
|---|---|---|---|
| 100 | 120 | 160 | |
| 1a | 26 | 18 | 0 |
| 2 | 11 | 55 | 0 |
| 3 | 0 | 12 | 0 |
| 4 | 0 | 0 | 11 |
| Comp. | Heating Time (Days) | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| 1a | 13 | 8 | 0 |
| 2 | 7 | 0 | 0 |
| 3 | 5 | 3 | 0 |
| 4 | 23 | 35 | 40 |
 | |||
|---|---|---|---|
| Comp. | Substituents | Torsion Angle (°) R3CNN | PzR1R2 Conformation 1 |
| 1a | R1 = R2 = CH3, R3 = C6H5 | −8.97(17), 101.46(15), 141.34(13) | sp, ac, ac |
| 7 [44] | R1 = CH3, R2 = C6H5, R3 = H | −18.3, 133.4, 148.6 | sp, ac, ac |
| 8 2 [45,46] | R1 = R2 = CH3, R3 = H | (±)25(5), (±)116(5), (±)152(5) | sp, ac, ap |
| 1b | R1 = R2 = H, R3 = C6H5 | −87.2(4), −104.2(4), −166.6(4) | sc, ac, ap |
| 9 [25] | R1 = R2 = H, R3 = o-C6H4-NH2 | 29.89(13), 87.54(11), 179.85(9) | sp, sc, ap |
| 10 [25] | R1 = R2 = H, R3 = p-C6H4-NH2 | −25(11), −83(14), 173(7) | sp, sc, ap |
| 11 [47] | R1 = R2 = H, R3 = p-C6H4-C6O2S | 42.6(7), 73.6(8), 166.4(2) | sc, sc, ap |
| 12 [48] | R1 = R2 = H, R3 = p-C6H4OH | −45.02(14), −68.86(14), 173.04(14) | sc, sc, ap |
| 13 [49] | R1 = R2 = H, R3 = CH2OH | −34.49(13), −62.50(12), 174.56(9) | sc, sc, ap |
| 14 [24] | R1 = H, R2 = CH3, R3 = H | 24.48, 27.97, 170.87 | sp, sp, ap |
| 15 [50] | R1 = R2 = R3 = H | 13.6, 15.7, 176.0 | sp, sp, ap |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Venegas, E.; García-Báez, E.V.; Martínez-Martínez, F.J.; Cruz, A.; Padilla-Martínez, I.I. Solventless Synthesis of Poly(pyrazolyl)phenyl-methane Ligands and Thermal Transformation of Tris(3,5-dimethylpyrazol-1-yl)phenylmethane. Molecules 2017, 22, 441. https://doi.org/10.3390/molecules22030441
Rodríguez-Venegas E, García-Báez EV, Martínez-Martínez FJ, Cruz A, Padilla-Martínez II. Solventless Synthesis of Poly(pyrazolyl)phenyl-methane Ligands and Thermal Transformation of Tris(3,5-dimethylpyrazol-1-yl)phenylmethane. Molecules. 2017; 22(3):441. https://doi.org/10.3390/molecules22030441
Chicago/Turabian StyleRodríguez-Venegas, Edith, Efrén V. García-Báez, Francisco J. Martínez-Martínez, Alejandro Cruz, and Itzia I. Padilla-Martínez. 2017. "Solventless Synthesis of Poly(pyrazolyl)phenyl-methane Ligands and Thermal Transformation of Tris(3,5-dimethylpyrazol-1-yl)phenylmethane" Molecules 22, no. 3: 441. https://doi.org/10.3390/molecules22030441