
Synthesis of Reusable Silica Nanosphere-Supported Pt(IV) Complex for Formation of Disulfide Bonds in Peptides

Abstract
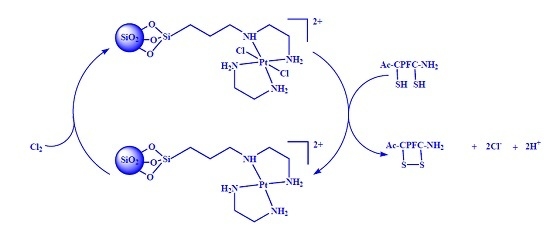
:
1. Introduction
2. Results and Discussion
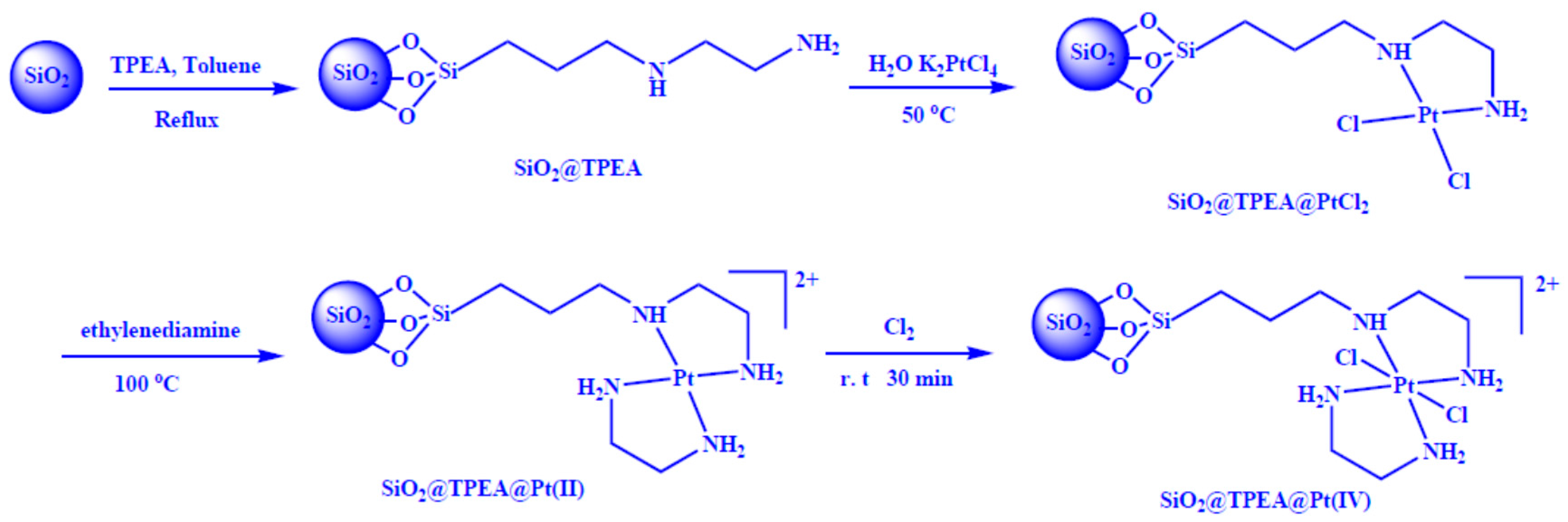
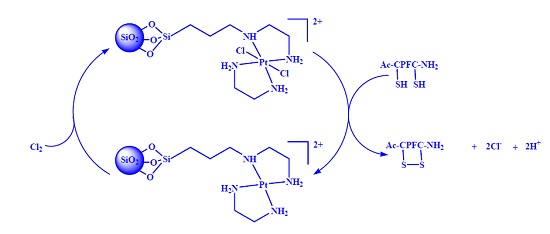
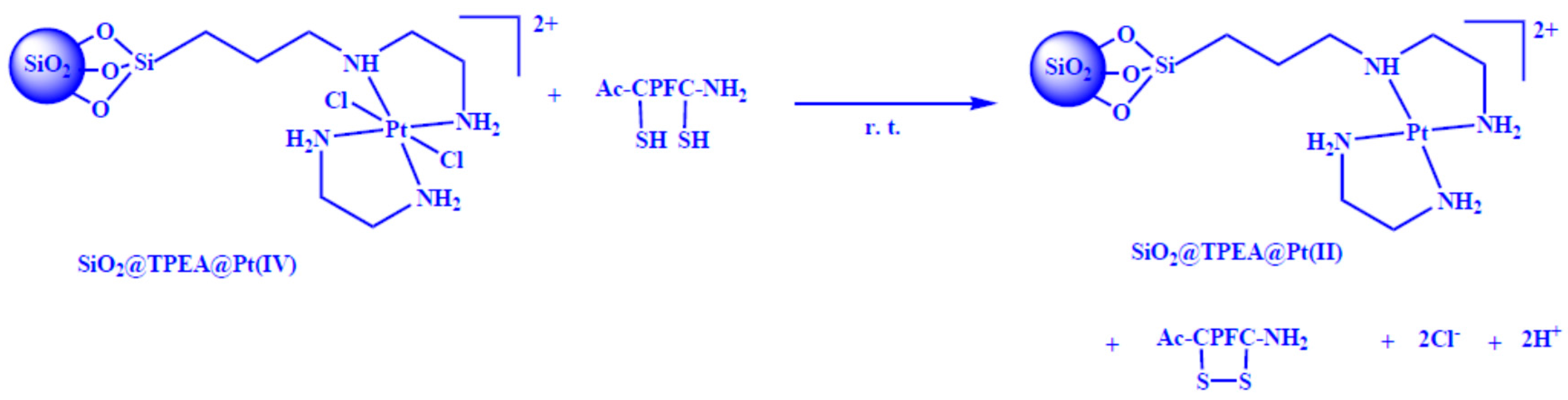
2.1. Synthesis and Characterization of SiO2@TPEA@Pt(IV)
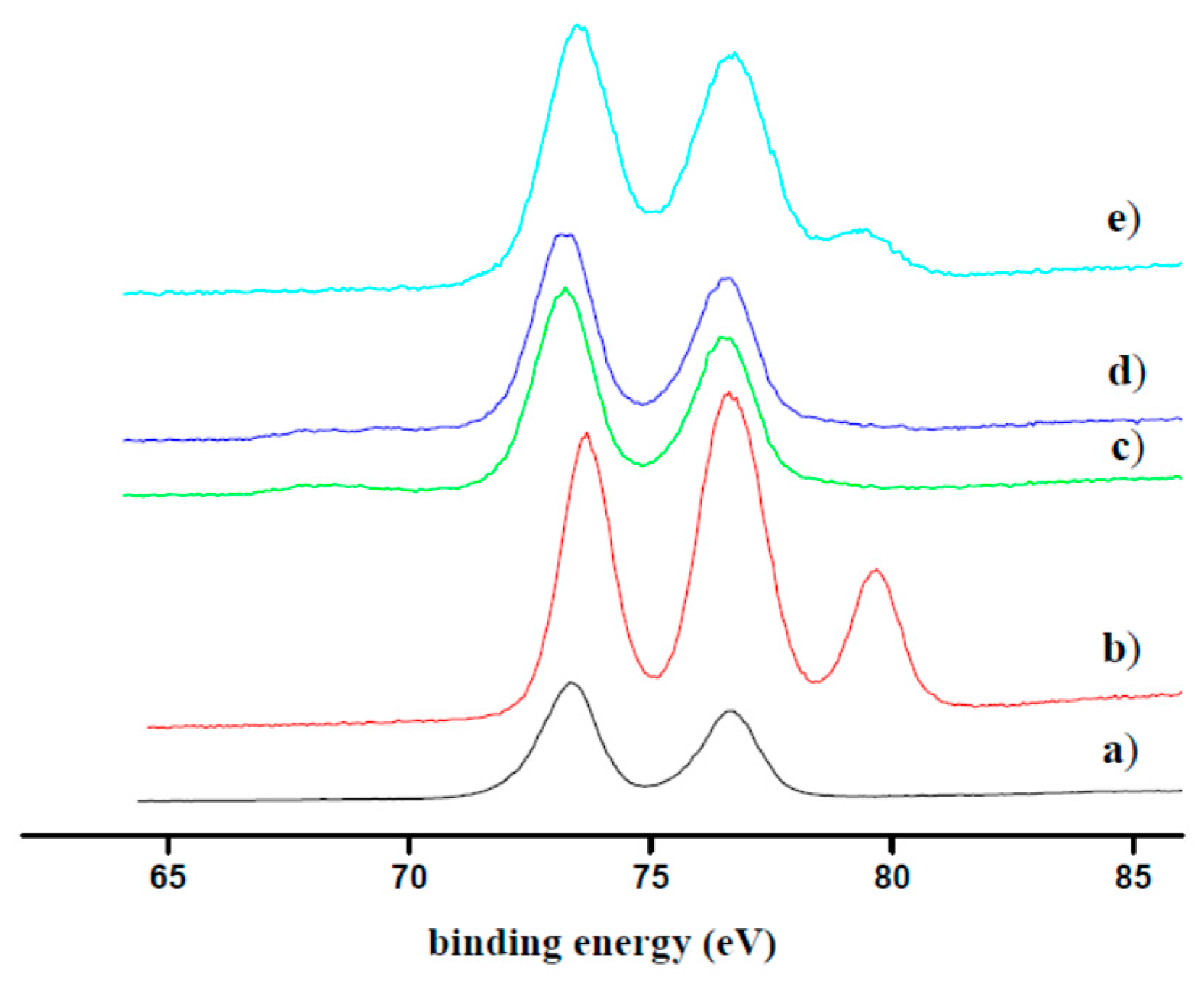
2.2. Pt(IV) Loading Determination of SiO2@TPEA@Pt(IV)
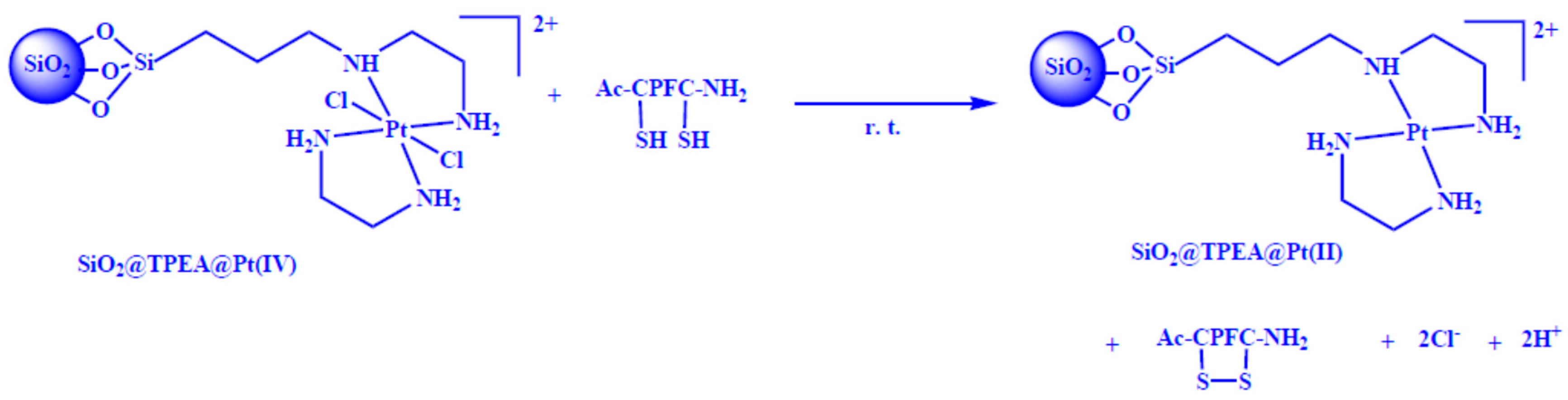
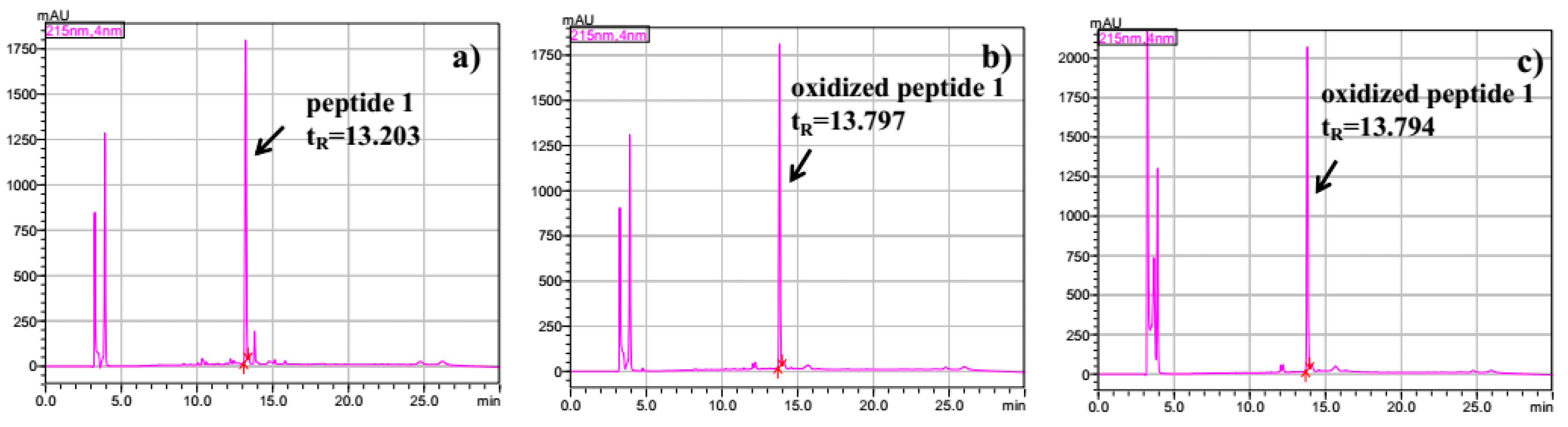
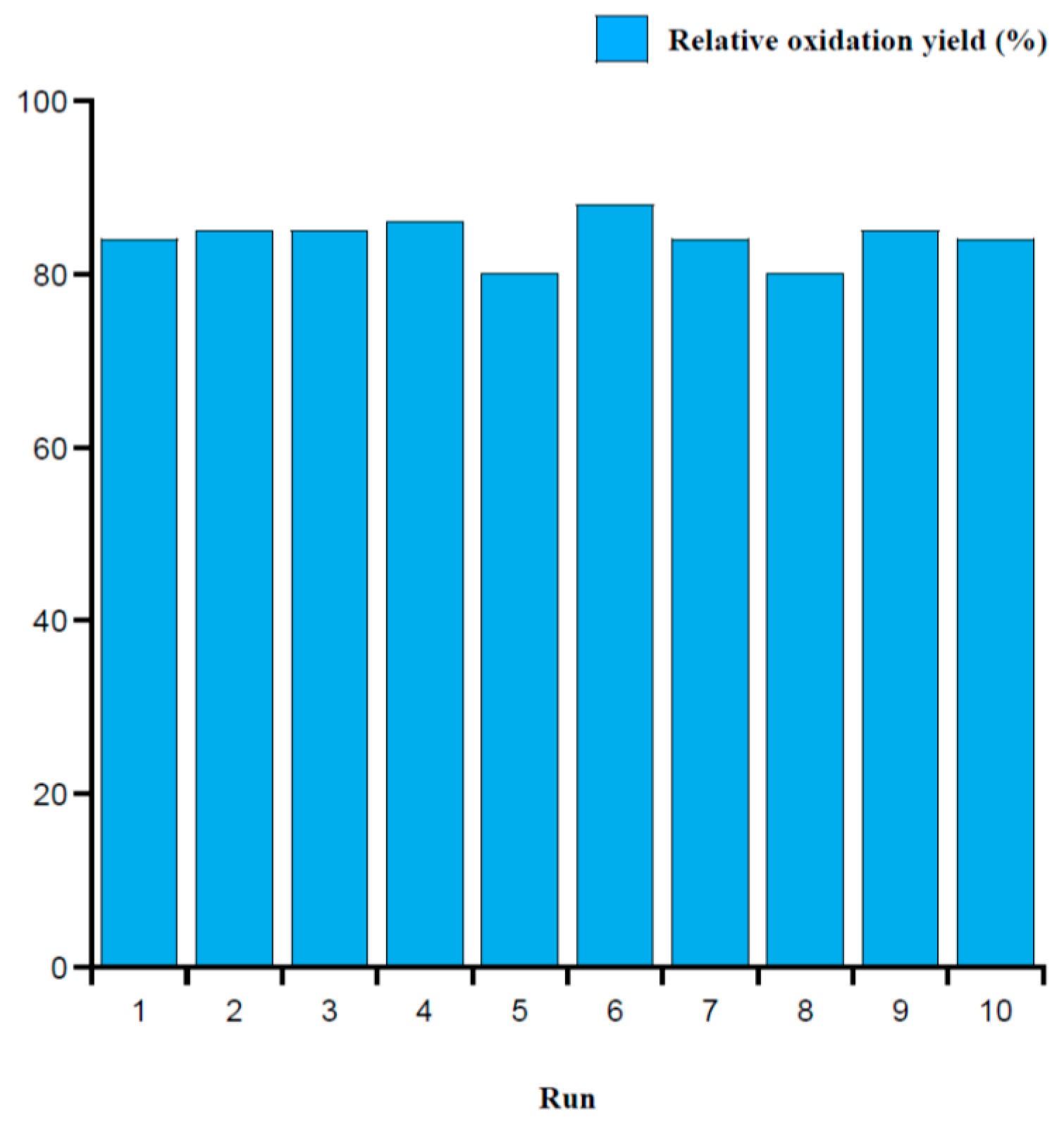
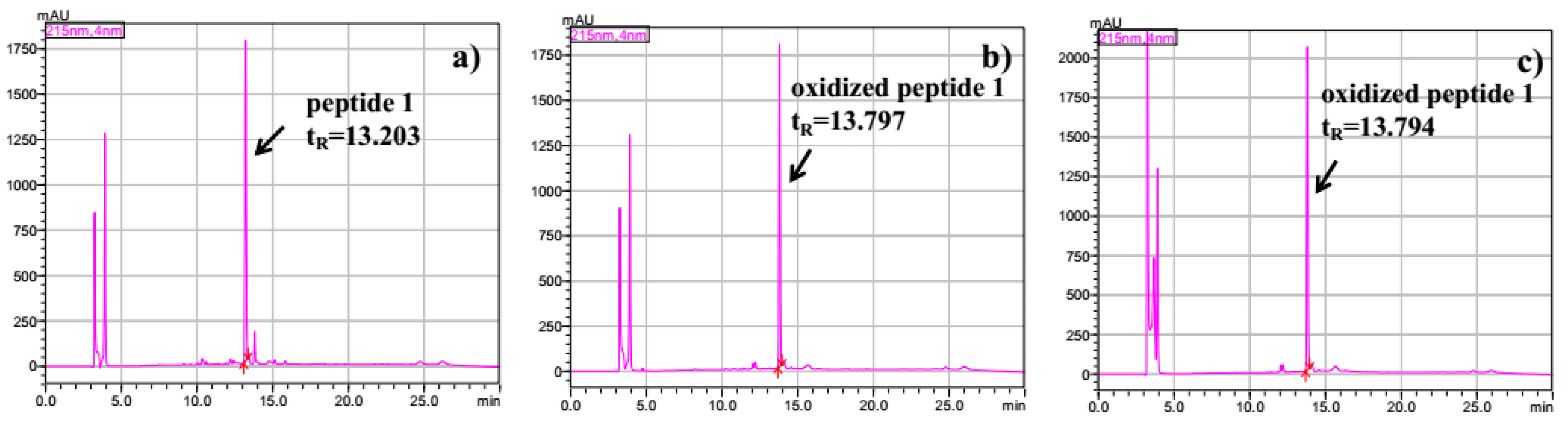
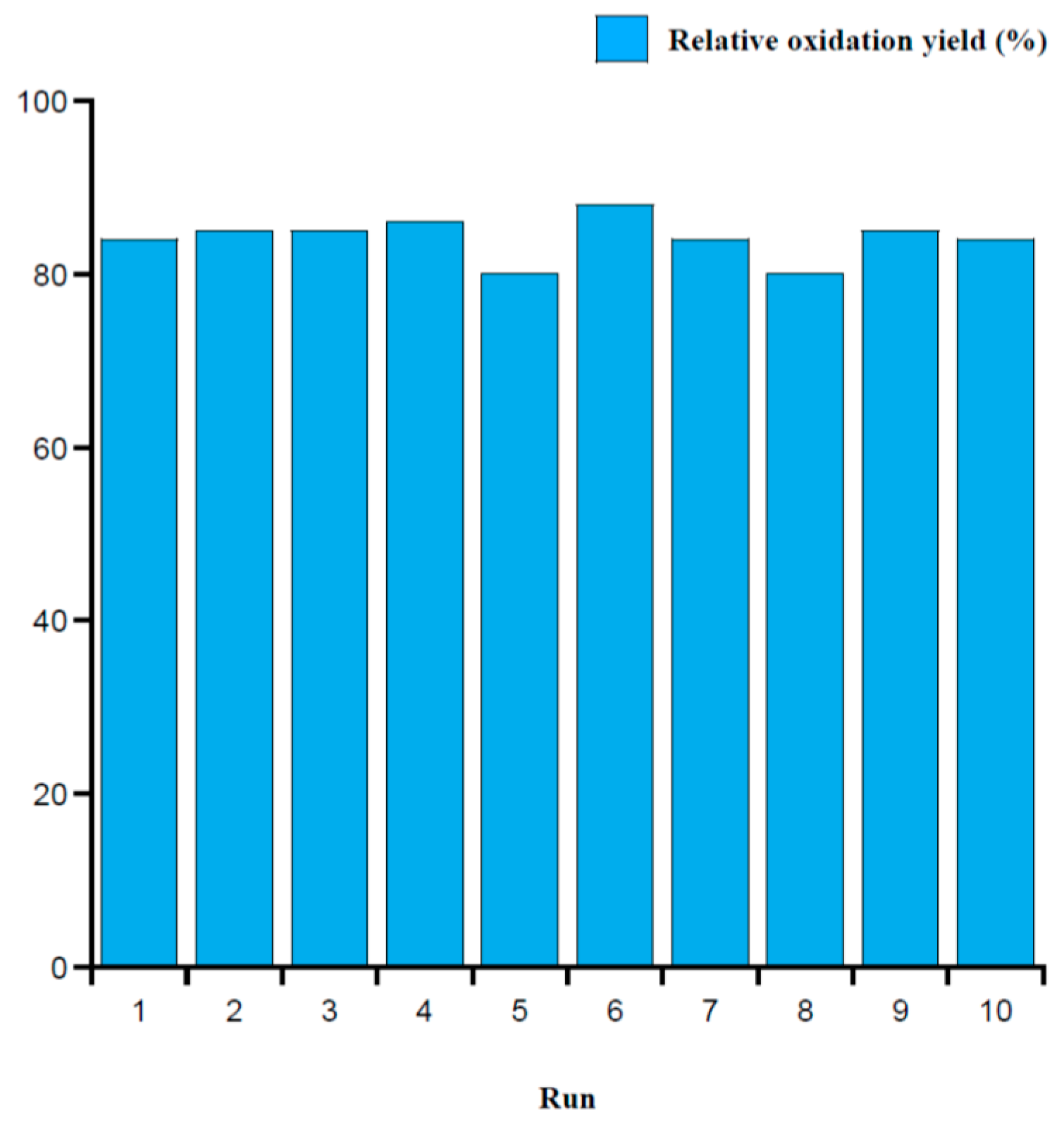
2.3. Intramolecular Disulfide Formation in Peptides by SiO2@TPEA@Pt(IV)
3. Experimental Procedures
3.1. Materials
3.2. Instrumentation
3.3. Preparation of TPEA-Modified Silica Nanospheres (SiO2@TPEA)
3.4. Synthesis of SiO2@TPEA@Pt(IV)
3.5. Determination of Pt(IV) Loading in the Synthesized SiO2@TPEA@Pt(IV)
3.5.1. Stoichiometry and Product Analysis
3.5.2. Determination of Pt(IV) Loading in SiO2@TPEA@Pt(IV)
3.6. Disulfide Bond Formation in Peptides
3.6.1. Synthesis and Purification of Peptides
3.6.2. General Method of Disulfide Formation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gongora-Benitez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef] [PubMed]
- Northfield, S.E.; Wang, C.K.; Schroeder, C.I.; Durek, T.; Kan, M.W.; Swedberg, J.E.; Craik, D.J. Disulfide-rich macrocyclic peptides as templates in drug design. Eur. J. Med. Chem. 2014, 77, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gopalakrishnan, R.; Schaer, T.; Marger, F.; Hovius, R.; Bertrand, D.; Pojer, F.; Heinis, C. Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nat. Chem. 2014, 6, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Zompra, A.A.; Galanis, A.S.; Werbitzky, O.; Albericio, F. Manufacturing peptides as active pharmaceutical ingredients. Future Med. Chem. 2009, 1, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Mandal, B.; Basu, B. Recent advances in S-S bond formation. RSC Adv. 2014, 4, 13854–13881. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv. 2005, 23, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, E.V.; Sidorova, M.V.; Evstigneeva, R.P. Some peculiarities of synthesis of cysteine-containing peptides. Russ. Chem. Rev. 1998, 67, 545–562. [Google Scholar] [CrossRef]
- Annis, I.; Hargittai, B.; Barany, G. Disulfide Bond Formation in Peptides. Methods Enzymol. 1997, 289, 198–221. [Google Scholar] [PubMed]
- Shi, T.S.; Rabenstein, D.L. trans-Dichlorotetracyanoplatinate(IV) as a Reagent for the Rapid and Quantitative Formation of Intramolecular Disulfdie Bonds in Peptides. J. Org. Chem. 1999, 64, 4590–4595. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.S.; Rabenstein, D.L. Discovery of a Highly Selective and Efficient Reagent for Formation of Intramolecular Disulfide Bonds in Peptides. J. Am. Chem. Soc. 2000, 122, 6809–6815. [Google Scholar] [CrossRef]
- Shi, T.S.; Rabenstein, D.L. Formation of multiple intramolecular disulfide bonds in peptides using the reagent trans-[Pt(ethylenediamine)2Cl2]2+. Tetrahedron Lett. 2001, 42, 7203–7206. [Google Scholar] [CrossRef]
- Shi, T.S.; Rabenstein, D.L. Convenient Synthesis of Human Calcitonin and Its Methionine Sulfoxide Derivative. Bioorg. Med. Chem. Lett. 2002, 12, 2237–2240. [Google Scholar] [CrossRef]
- Nguyen, K.; Iskandar, M.; Rabenstein, D. L. Kinetics and Equilibria of Cis/Trans Isomerization of Secondary Amide Peptide Bonds in Linear and Cyclic Peptides. J. Phys. Chem. B 2010, 114, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.M.; Albericio, F. N-chlorosuccinimide, an efficient peptide disulfide bond-forming reagent in aqueous solution. RSC Adv. 2013, 3, 14277–14280. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. N-chlorosuccinimide, an efficient for on-resin disulfide formation in solid-phase peptide synthesis. Org. Lett. 2013, 15, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Dedachi, K.; Iwaoka, M. Rapid and quantitative disulfide bond formation for a polypeptide chain using a cyclic selenoxide reagent in an aqueous medium. Chem. Eur. J. 2011, 17, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, M.; Tomoda, S. trans-3,4-Dihydroxy-1-selenolane Oxide: A New Reagent for Rapid and Quantitative Formation of Disulfide Bonds in Polypeptides. Chem. Lett. 2000, 29, 1400–1401. [Google Scholar] [CrossRef]
- Annis, I.; Chen, L.; Barany, G. Novel Solid-Phase Reagents for Facile Formation of Intramolecular Disulfide Bridges in Peptides under Mild Conditions. J. Am. Chem. Soc. 1998, 120, 7226–7238. [Google Scholar] [CrossRef]
- Verdie, P.; Ronga, L.; Cristau, M.; Amblard, M.; Cantel, S.; Enjalbal, C.; Puget, K.; Martinez, J.; Subra, G. Oxyfold: A Simple and Efficient Solid-Supported Reagent for Disulfide Bond Formation. Chem. Asian J. 2011, 6, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Ronga, L.; Verdie, P.; Sanchez, P.; Enjabal, C.; Maurras, A.; Jullian, M.; Puget, K.; Martinez, J.; Subra, G. Supported oligomethionine sulfoxide and Ellman’s reagent for cysteine bridges formation. Amino Acids 2013, 44, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.M.; Albericio, F. Immobilized N-Chlorosuccinimide as a Friendly Peptide Forming Reagent. ACS Comb. Sci. 2014, 16, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, M.; Mezzetti, A. Site isolated complexes of late transition metals grafted on silica: Challenges and chances for synthesis and catalysis. Catal. Sci. Technol. 2014, 4, 2724–2740. [Google Scholar] [CrossRef]
- Conley, M.P.; Coperet, C.; Thieuleux, C. Mesostructured Hybrid Organic-Silica Materials: Ideal Supports for Well-Defined Heterogeneous Organometallic Catalysts. ACS Catal. 2014, 4, 1458–1469. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S.; Dutta, S.; Zboril, R.; Gawande, M.B. Silica-nanosphere-based organic-inorganic hybrid nanomaterials: Synthesis, functionalization and applications in catalysis. Green Chem. 2015, 17, 3207–3230. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S. Silica nanosphere-supported palladium(II) furfural complex as a highly efficient and recyclable catalyst for oxidative amination of aldehydes. Dalton Trans. 2014, 43, 1292–1304. [Google Scholar] [CrossRef] [PubMed]
- Atar, A.B.; Kim, J.S.; Lim, K.T.; Jeong, Y.T. Bridging homogeneous and heterogeneous catalysis with CAN·SiO2as a solid catalyst for four component reactions for the synthesis of tetrasubstituted pyrroles. New J. Chem. 2015, 39, 396–402. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, A.P. A covalently anchored 2,4,6-triallyloxy-1,3,5-triazine Pd(II) complex over a modified surface of SBA-15: Catalytic application in hydrogenation reaction. RSC Adv. 2014, 4, 58467–58475. [Google Scholar] [CrossRef]
- Stoeber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Ma, Q.; Li, Y.; Su, X. Silica-nanobead-based sensors for analytical and bioanalytical applications. Trends Anal. Chem. 2015, 74, 130–145. [Google Scholar] [CrossRef]
- Zheng, F.; Hu, B. Preparation of a high pH-resistant AAPTS-silica coating and its application to capillary microextraction (CME) of Cu, Zn, Ni, Hg and Cd from biological samples followed by on-line ICP-MS detection. Anal. Chim. Acta 2007, 605, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, Y.S.; Ghatak, J.; Bhatta, U.M.; Khushalani, D. One-step method for the self-assembly of metal nanoparticles onto facetted hollow silica tubes. J. Mater. Chem. 2006, 16, 3619–3623. [Google Scholar] [CrossRef]
- He, Y.; Huang, Y.; Jin, Y.; Liu, X.; Liu, G.; Zhao, R. Well-Defined Nanostructured Surface-Imprinted Polymers for Highly Selective Magnetic Separation of Fluoroquinolones in Human Urine. ACS Appl. Mater. Interfaces 2014, 6, 9634–9642. [Google Scholar] [CrossRef] [PubMed]
- Robillard, M.S.; Valentijn, A.; Rob, P.M.; Meeuwenoord, N.J.; van der Marel, G.A.; van Boom, J.H.; Reedijk, J. The First Solid-Phase Synthesis of a Peptide-Tethered Platinum(II) Complex. Angew. Chem. Int. Ed. 2000, 39, 3096–3099. [Google Scholar] [CrossRef]
- Robillard, M.S.; Bacac, M.; van den Elst, H.; Flamigni, A.; van der Marel, G.A.; van Boom, J.H.; Reedijk, J. Automated Parallel Solid-Phase Synthesis and Anticancer Screening of a Library of Peptide-Tethered Platinum(II) Complexes. J. Comb. Chem. 2003, 5, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Basolo, F.; Bailar, J.C., Jr.; Tarr, B.R. The Stereochemistry of Complex Inorganic Compounds. X. The Stereoisomers of Dichlorobis-(ethylenediamine)-platinum(IV) Chloride. J. Am. Chem. Soc. 1950, 72, 2433–2438. [Google Scholar] [CrossRef]
- Watt, G.W.; Thompson, J.S., Jr. Deprotonation of 2-(2-aminoethylamino)ethanol complexes of platinum(II, IV) and palladium(II) with liquid ammonia and potassium amide. J. Inorg. Nucl. Chem. 1974, 36, 1075–1078. [Google Scholar] [CrossRef]
- Liang, B.; Huo, S.; Ren, Y.; Sun, S.; Cao, Z.; Shen, S. A. platinum(IV)-based metallointercalator: Synthesis, cytotoxicity, and redox reactions with thiol-containing compounds. Transit. Met. Chem. 2015, 40, 31–37. [Google Scholar] [CrossRef]
- Wilson, J.J.; Lippard, S.J. Synthetic Methods for the Preparation of Platinum Anticancer Complexes. Chem. Rev. 2014, 114, 4470–4495. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for Design and Development of Platinum(IV) Anticancer Complexes. J. Med. Chem. 2007, 50, 3403. [Google Scholar] [CrossRef] [PubMed]
- Kolbeck, C.K.; Taccardi, N.; Paape, N.; Schulz, P.S.; Wasserscheid, P.; Steinruck, H.P.; Maier, F. Redox chemistry, solubility, and surface distribution of Pt(II) and Pt(IV) complexes dissolved in ionic liquids. J. Mol. Liq. 2014, 192, 103–113. [Google Scholar] [CrossRef]
- Burroughs, P.; Hamnett, A.; McGilp, J.F.; Orchard, A.F. Radiation damage in some platinum(IV) complexes produced during soft X-ray photoelectron spectroscopic studies. J. Chem. Soc. Faraday Trans. 2 1975, 71, 177–187. [Google Scholar] [CrossRef]
- Ling, X.; Shen, Y.; Sun, R.; Zhang, M.; Li, C.; Mao, J.; Xing, J.; Sun, C.; Tu, J. Tumor-targeting delivery of hyaluronic acid-platinum(IV) nanoconjugate to reduce toxicity and improve survival. Polym. Chem. 2015, 6, 1541–1552. [Google Scholar] [CrossRef]
- Lengke, M.F.; Fleet, M.E.; Southam, G. Synthesis of Platinum Nanoparticles by Reaction of Filamentous Cyanobacteria with Platinum(IV)-Chloride Complex. Langmuir 2006, 22, 7318–7323. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, X.; Zhao, X.; Liu, M.; Shen, F.; Ren, Y.; Liu, Y.; Huo, S.; Shen, S. Kinetics and mechanism of oxidation of 3,6-dioxa-1,8-octanedithiol and DL-dithiothreitol by a platinum(IV) complex. Transit. Met. Chem. 2016, 41, 45–55. [Google Scholar] [CrossRef]
- Coantic, S.; Subra, G.; Martinez, J. Microwave-assisted Solid Phase Peptide Synthesis on High Loaded Resins. Int. J. Pept. Res. Ther. 2008, 14, 143–147. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compound (SiO2@TPEA@Pt(IV)) is available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Relative Oxidation Yield |
|---|---|
| 1 Ac-CPFC-NH2 | 84% |
| 2 CGYCHKLHQMK-NH2 | 68% |
| 3 CYFQNCPRG-NH2 (reduced arginine vasopressin) | 68% |
| 4 CRGDKGPDC-NH2 (reduced iRGD peptide) | 50% |
| 5 CYINQCPLG-NH2 (reduced oxytocin) | 59% |
| 6 AGCKNFFWKTFTSC (reduced somatostatin) | 50% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Zhao, X.; Zhang, Y.; Han, A.; Huo, S.; Shen, S. Synthesis of Reusable Silica Nanosphere-Supported Pt(IV) Complex for Formation of Disulfide Bonds in Peptides. Molecules 2017, 22, 338. https://doi.org/10.3390/molecules22020338
Hou X, Zhao X, Zhang Y, Han A, Huo S, Shen S. Synthesis of Reusable Silica Nanosphere-Supported Pt(IV) Complex for Formation of Disulfide Bonds in Peptides. Molecules. 2017; 22(2):338. https://doi.org/10.3390/molecules22020338
Chicago/Turabian StyleHou, Xiaonan, Xiaowei Zhao, Yamei Zhang, Aiying Han, Shuying Huo, and Shigang Shen. 2017. "Synthesis of Reusable Silica Nanosphere-Supported Pt(IV) Complex for Formation of Disulfide Bonds in Peptides" Molecules 22, no. 2: 338. https://doi.org/10.3390/molecules22020338