De Novo RNA Sequencing and Expression Analysis of Aconitum carmichaelii to Analyze Key Genes Involved in the Biosynthesis of Diterpene Alkaloids

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Sample Preparation and High-Throughput Transcriptome Sequencing
2.2. De Novo Transcriptome Assembly
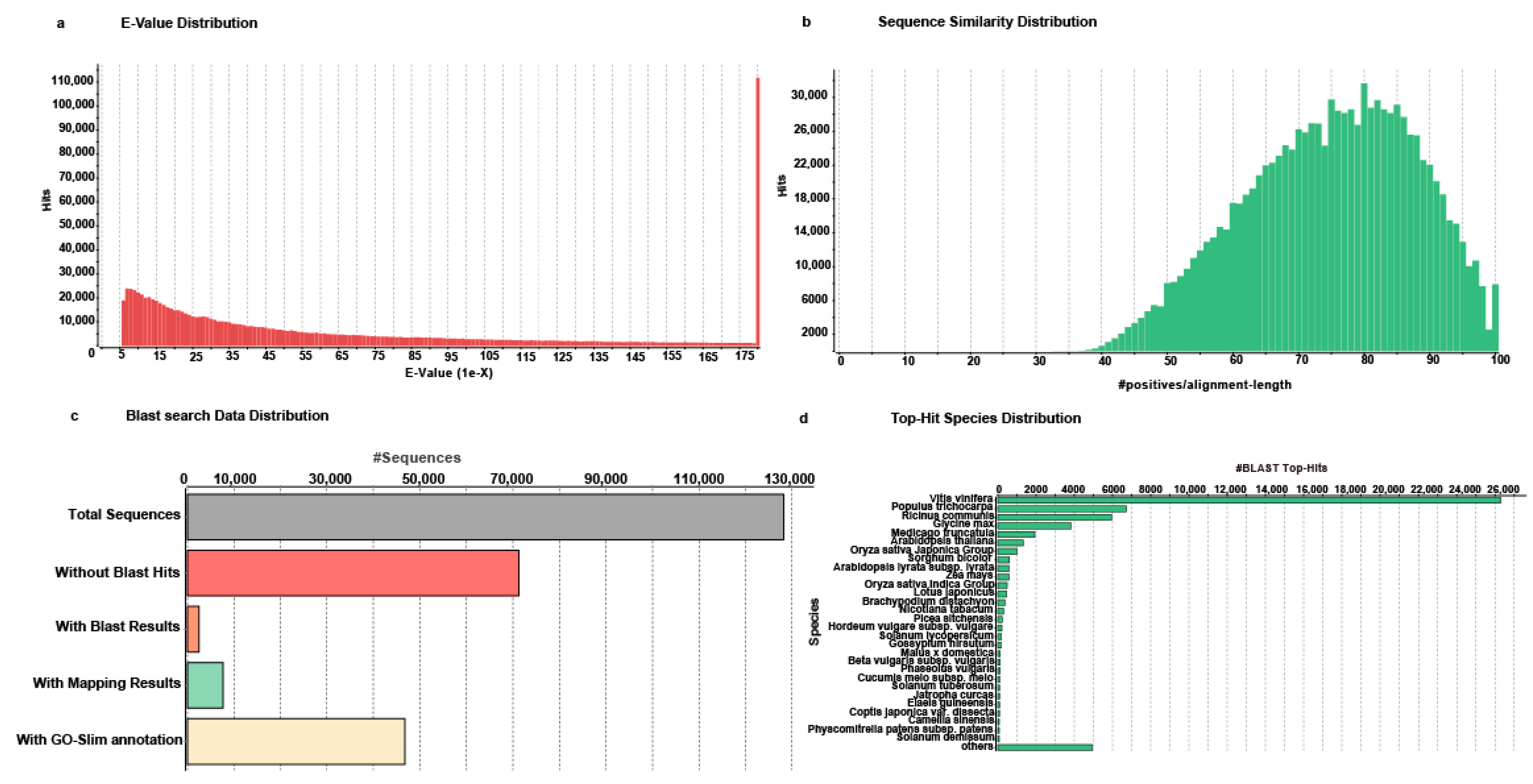
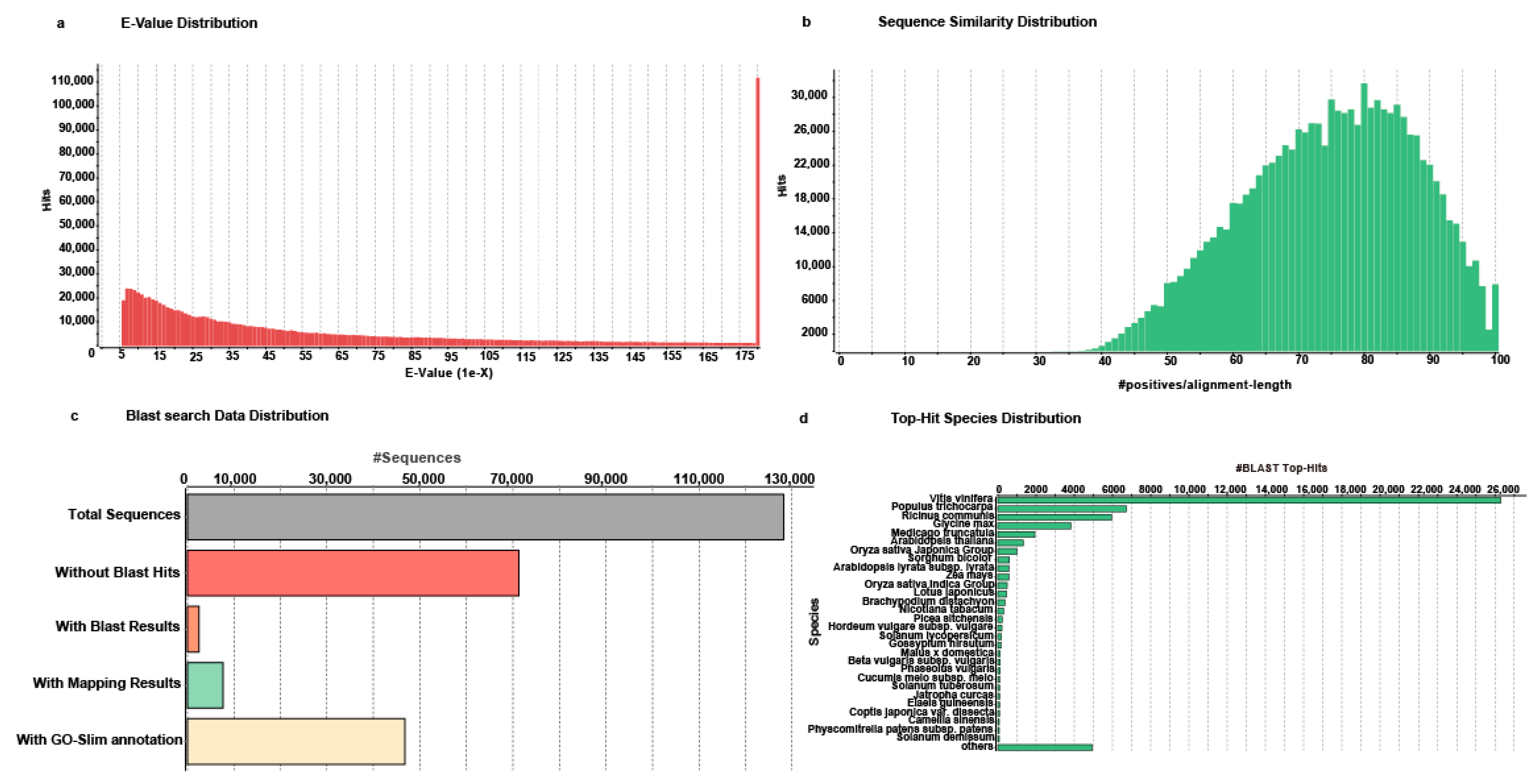
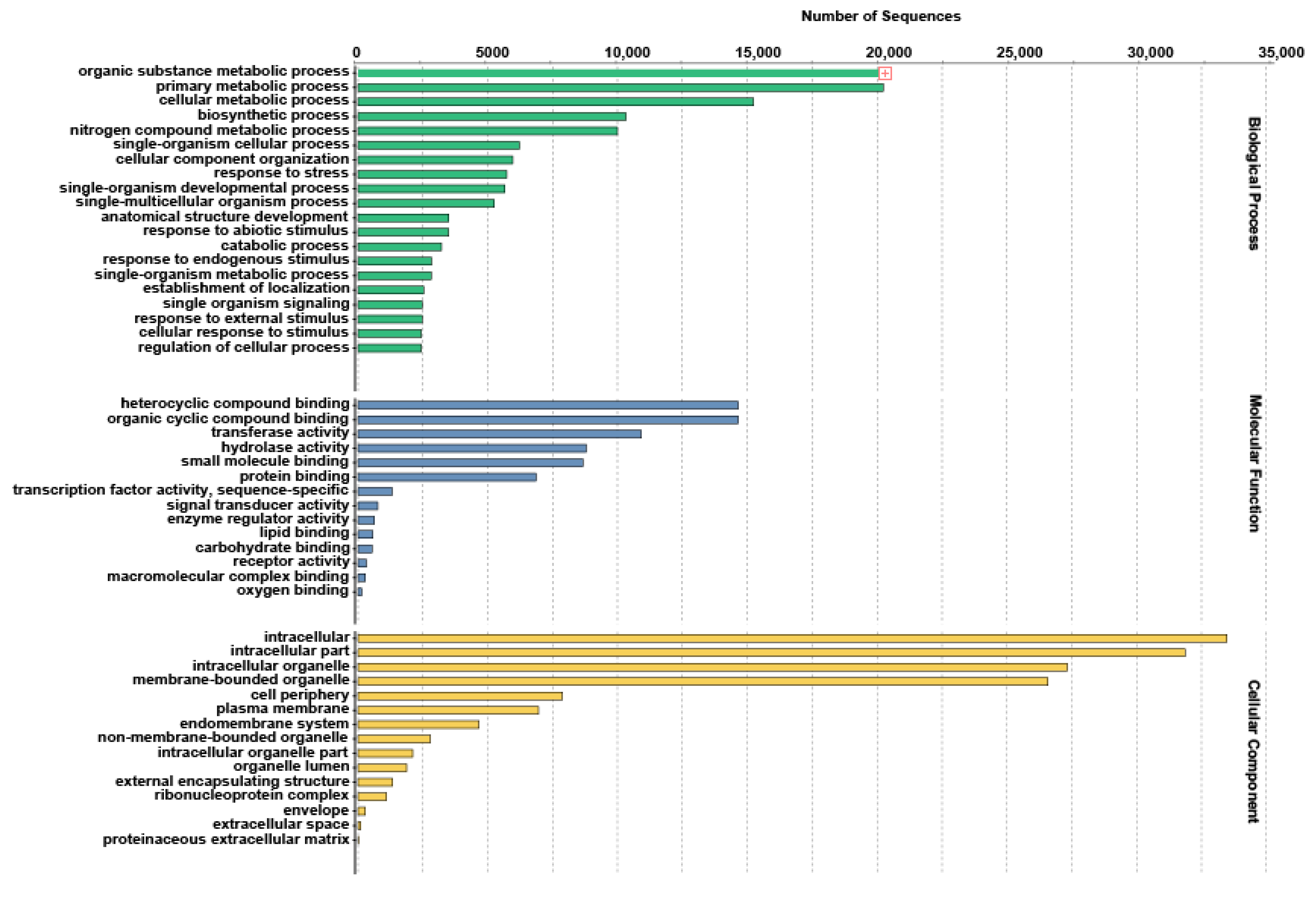
2.3. Functional Annotation of Unigenes
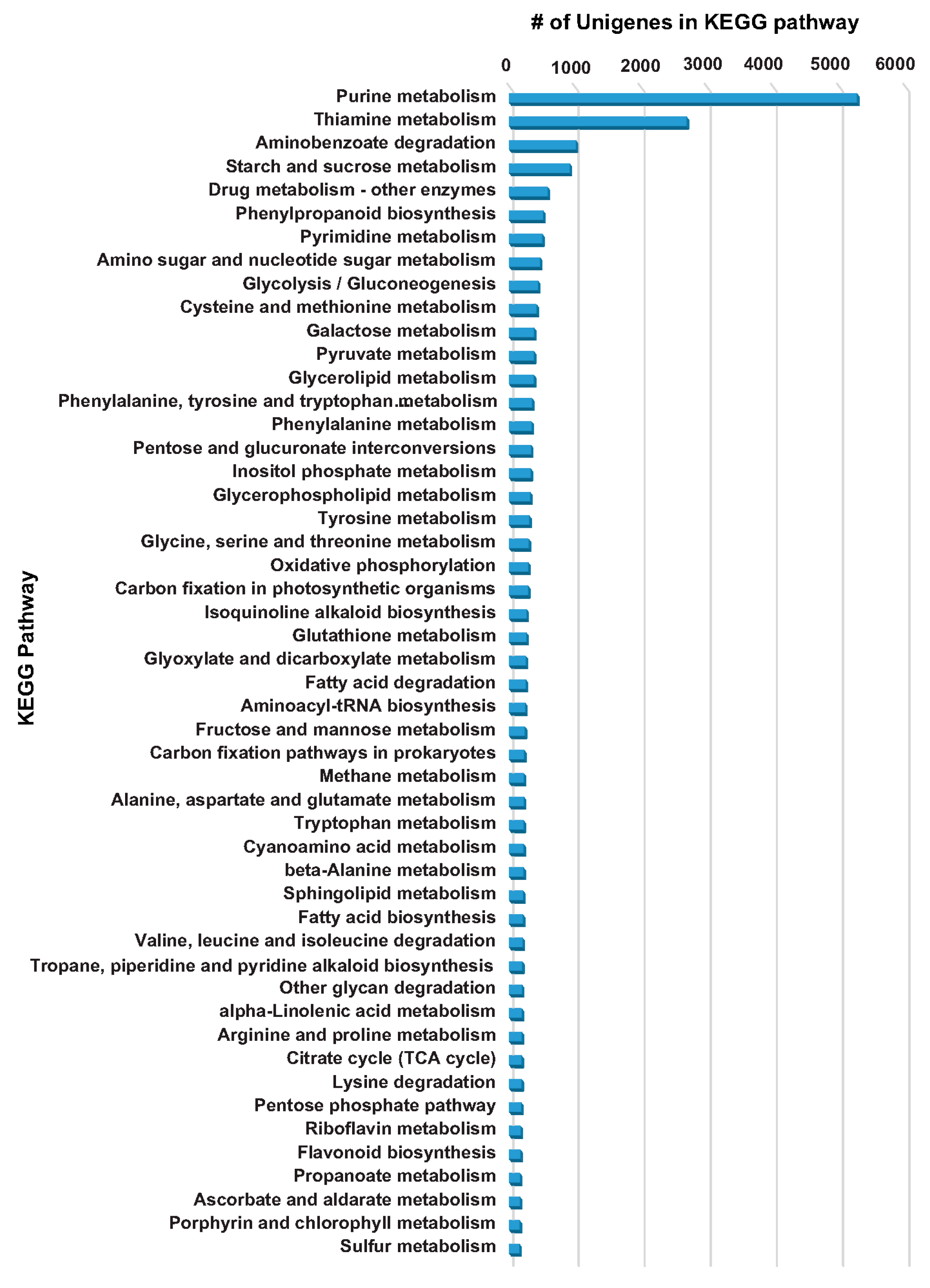
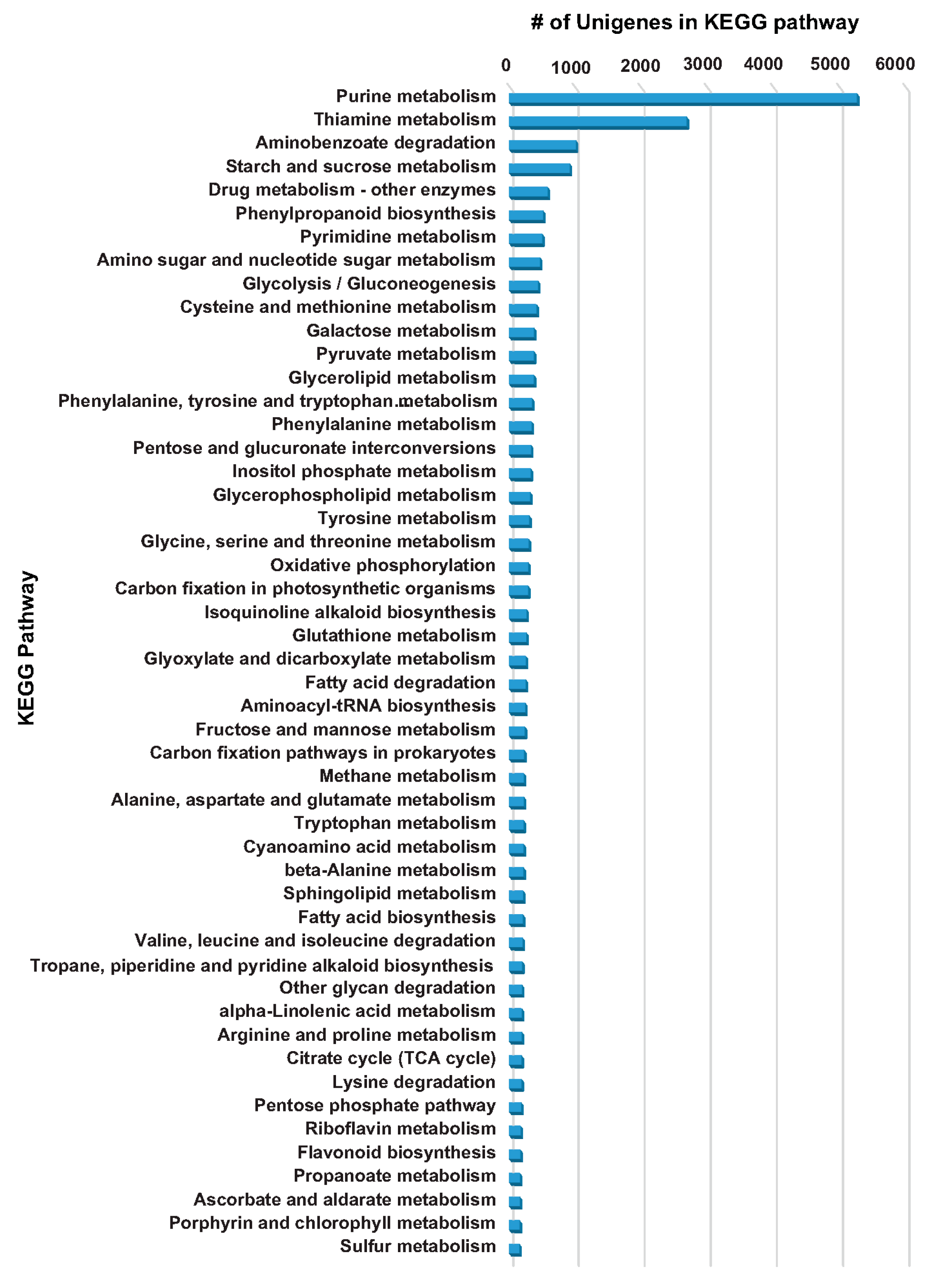
2.4. KEGG Database-Based Functional Characterization of A. carmichaelii Transcriptome Assembly
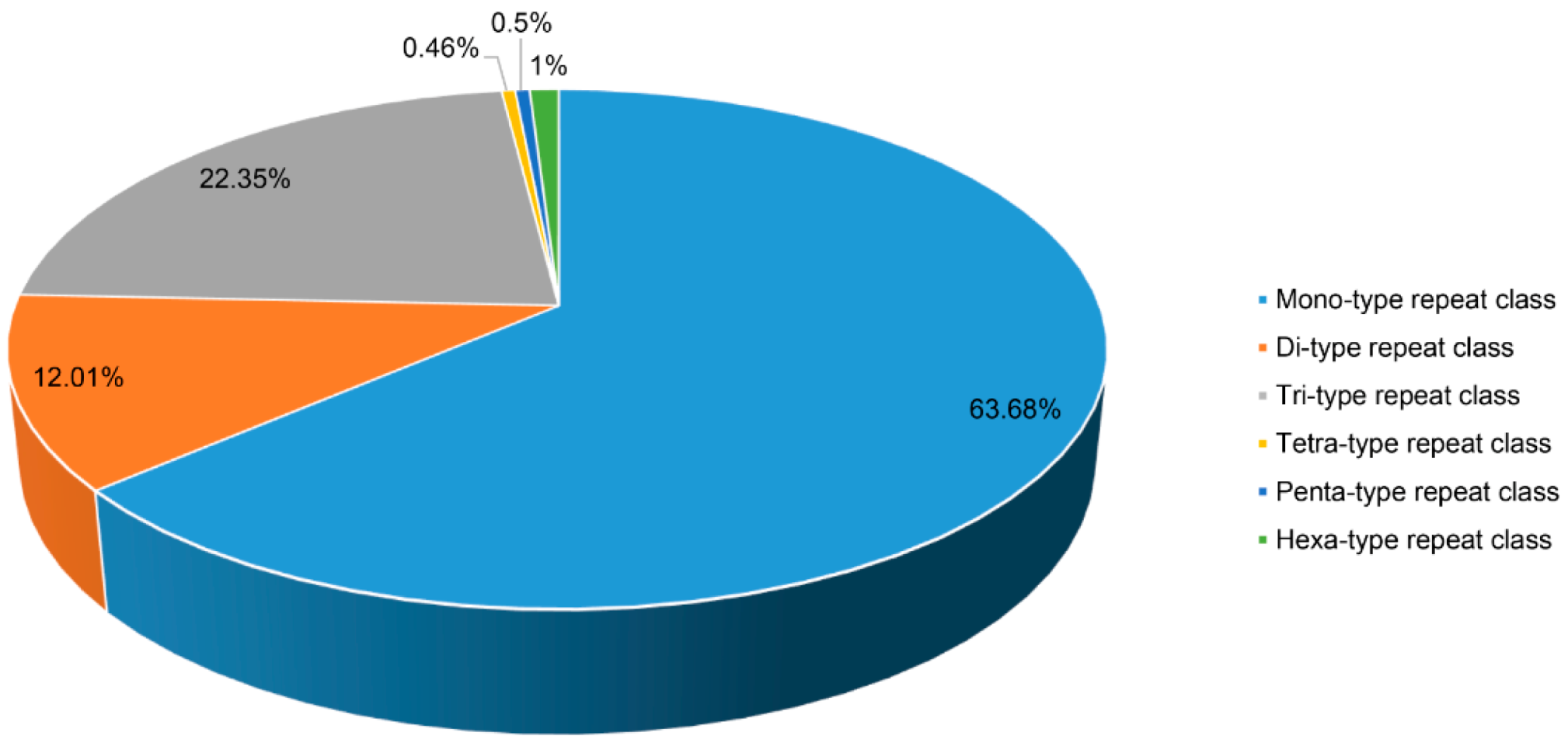
2.5. Identification of Simple Sequence Repeats (SSRs)
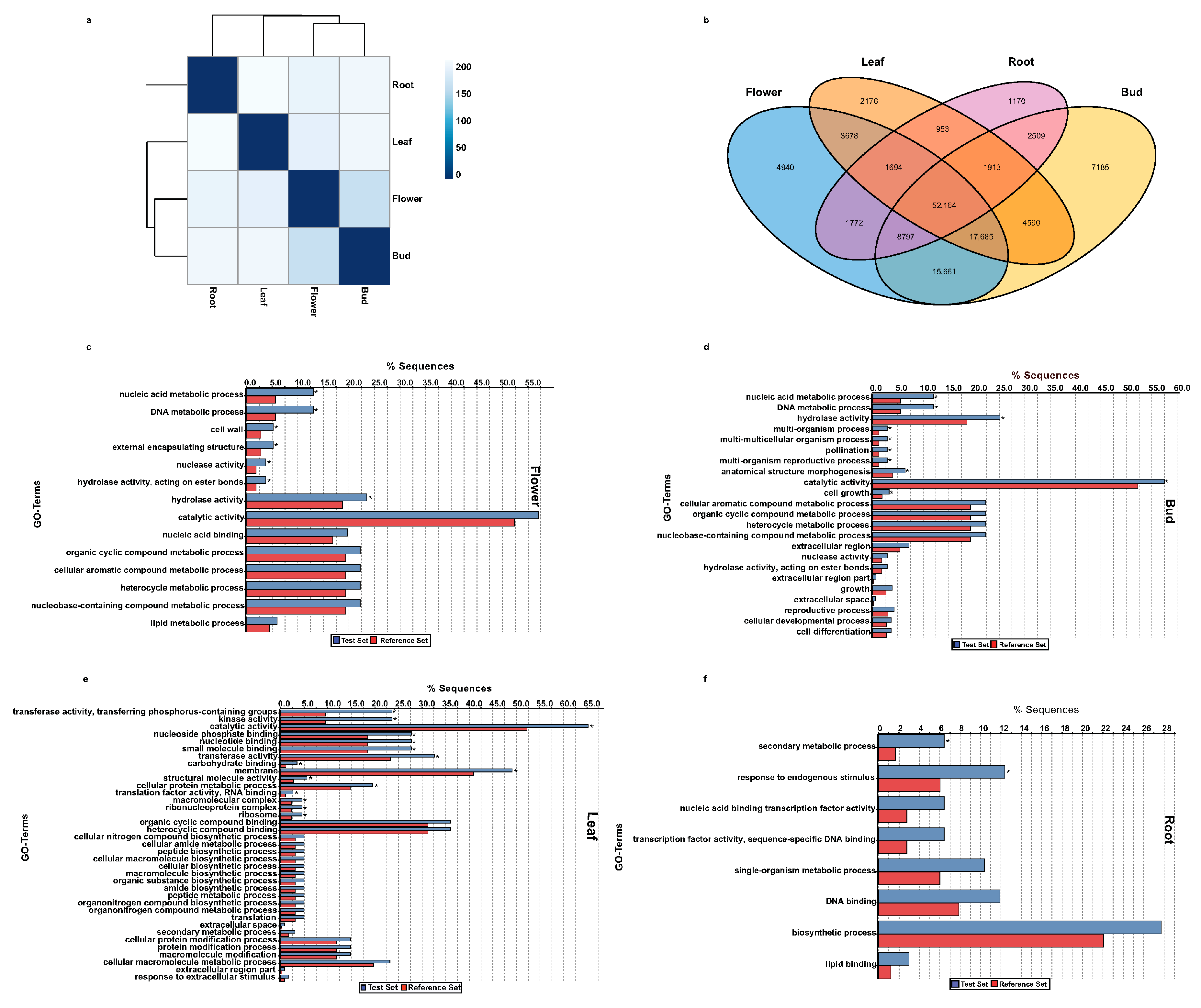
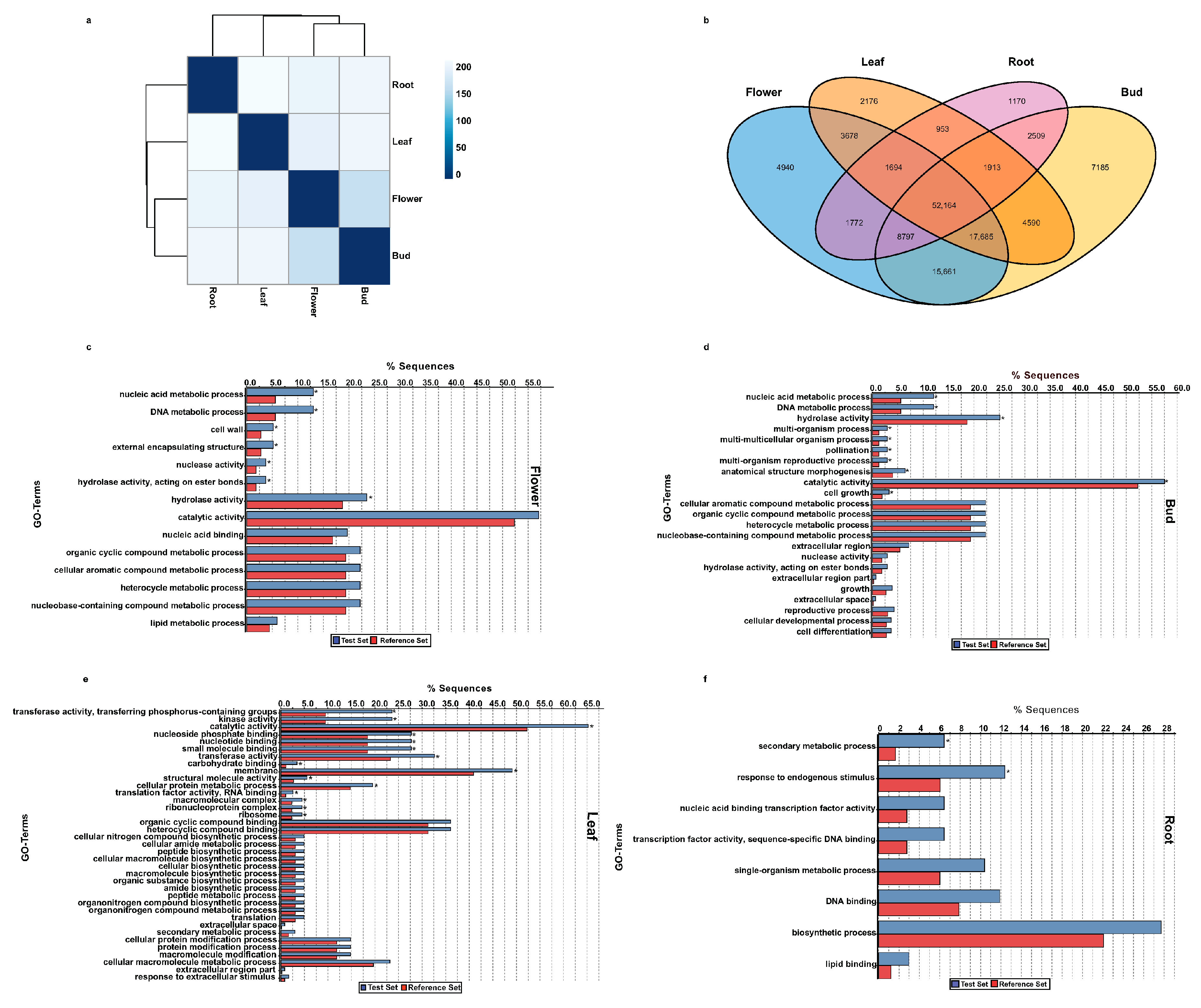
2.6. Functional Classification of Tissue-Specific Unigenes of A. carmichaelii
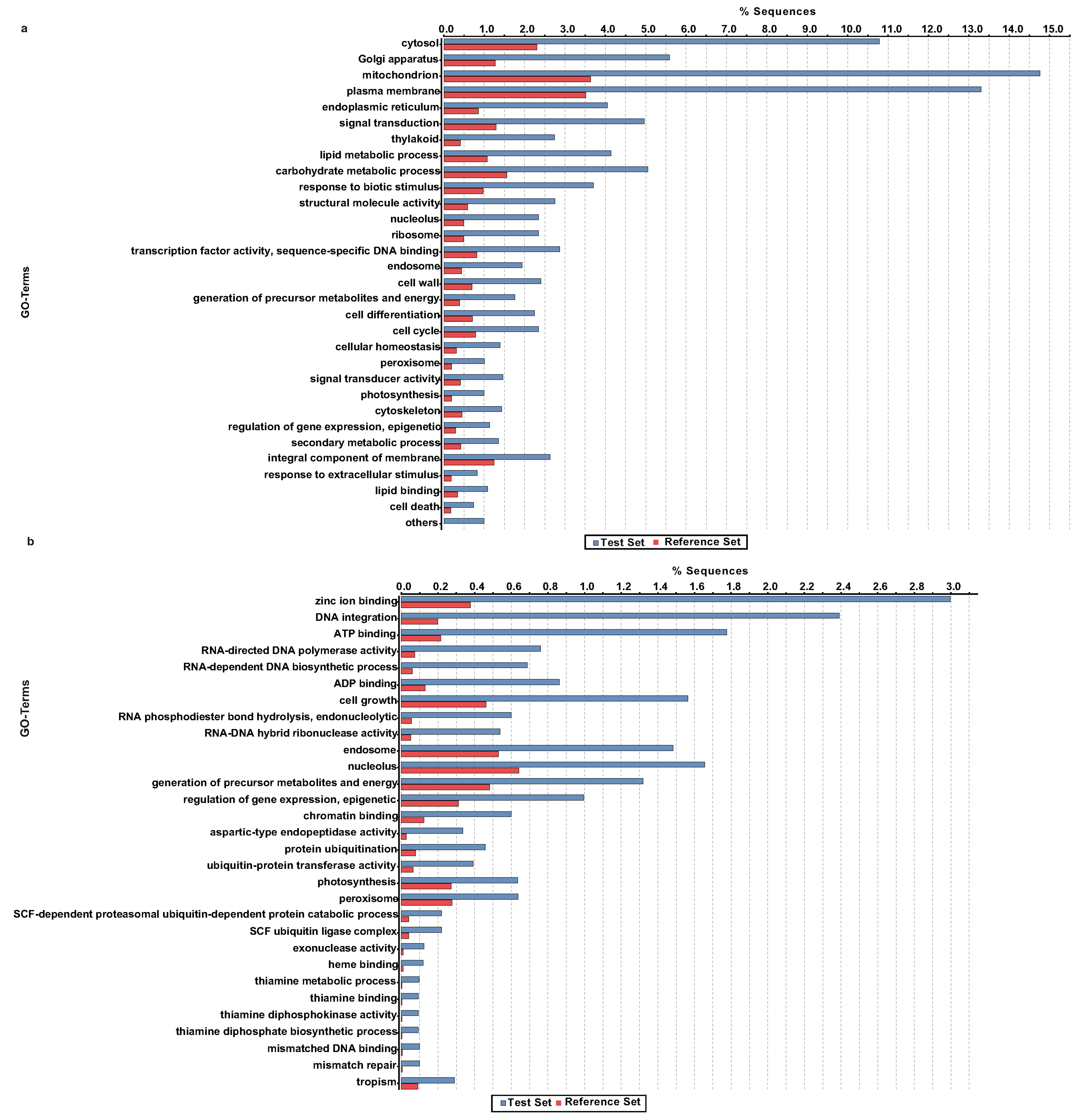
2.7. Identification of Orthologous Unigenes and Transcriptome Divergence between A. carmichaelii and A. heterophyllum
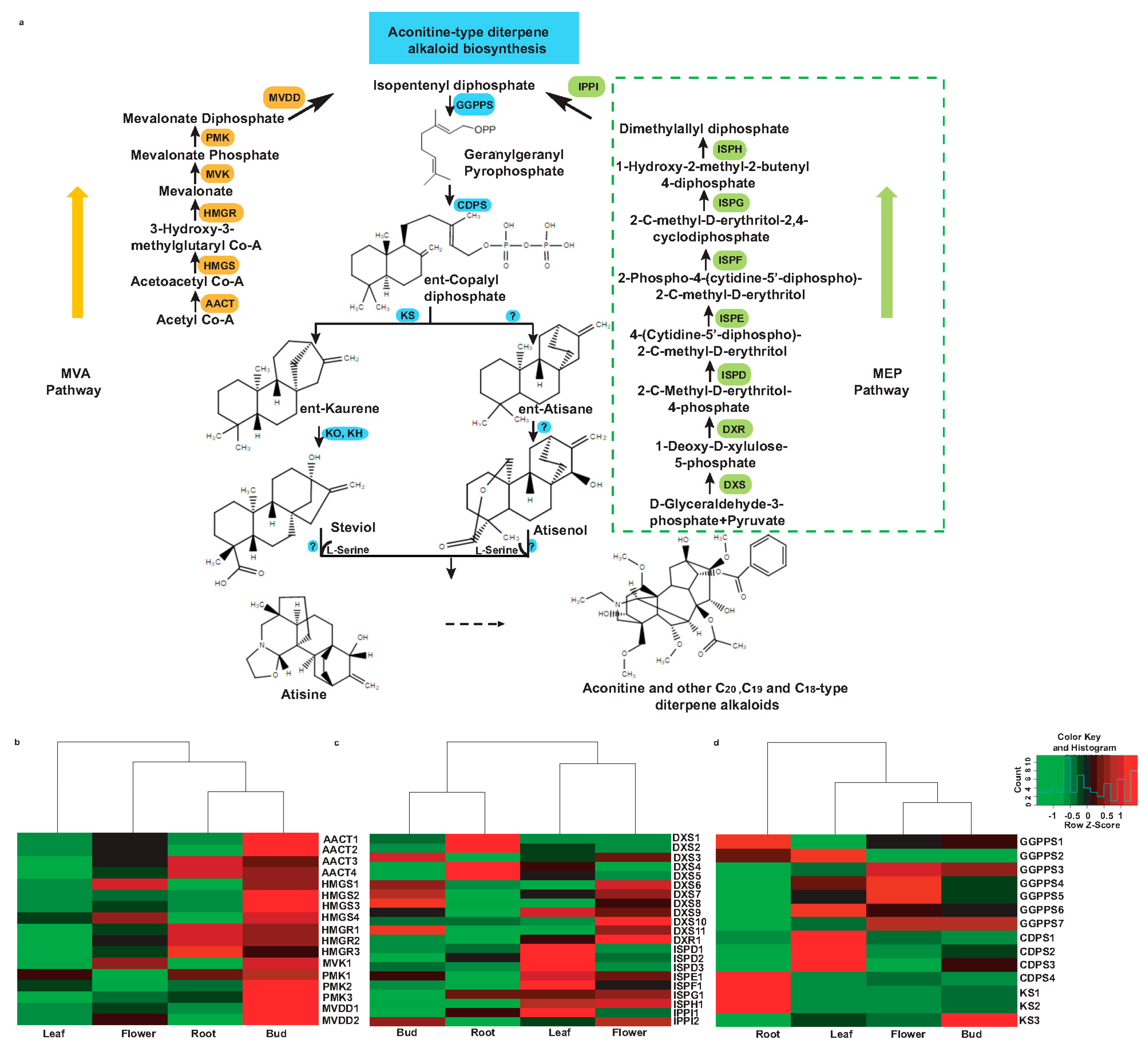
2.8. Identification of Putative Candidate Unigenes Associated with the Biosynthesis of Aconitine and Its Derivatives
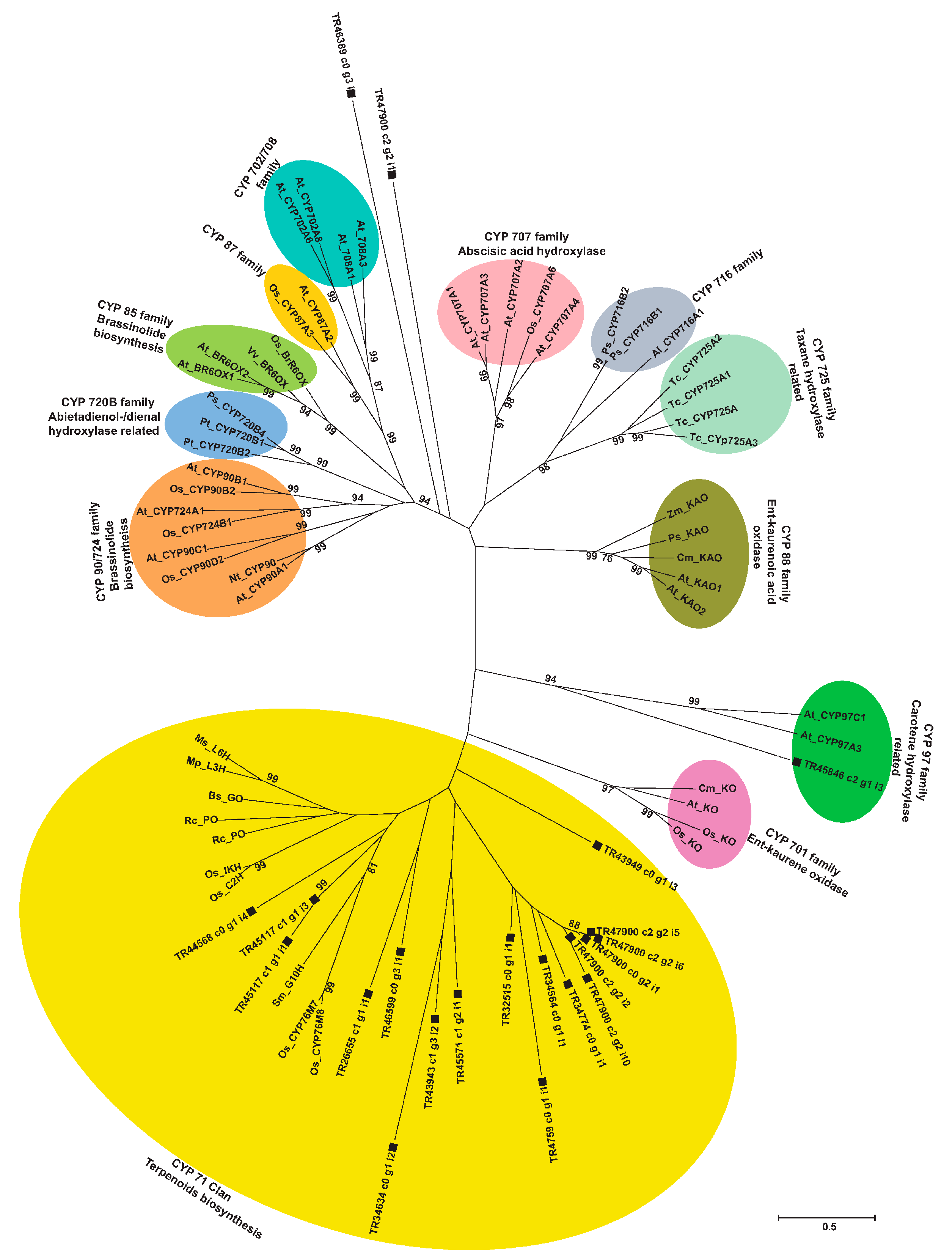
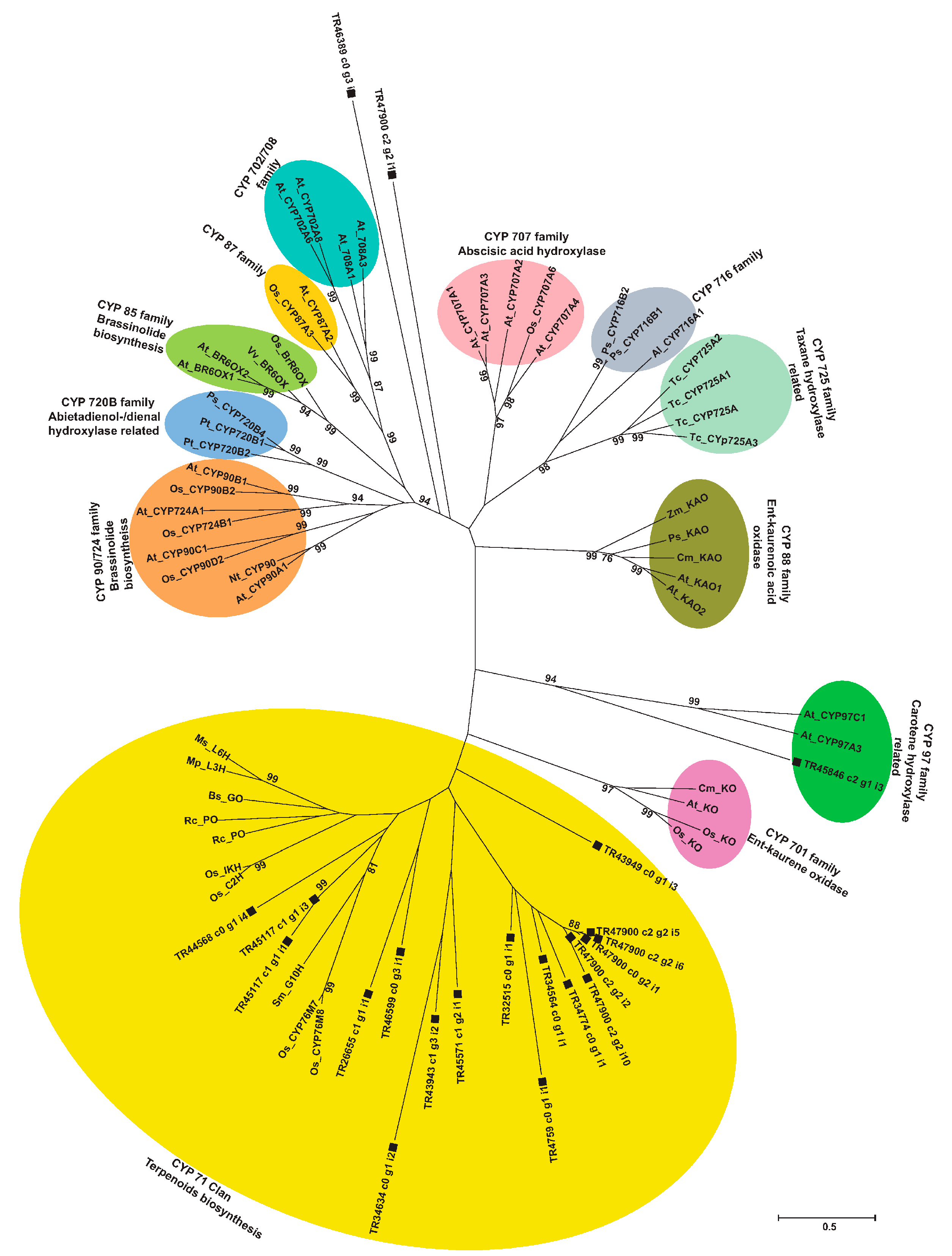
2.9. Phylogenetic Analysis of Unigenes Annotated as Cytochrome P450 with Highest Expression in Root
3. Materials and Methods
3.1. Plant Materials
3.2. RNA Isolation and cDNA Library Preparation
3.3. Illumina Sequencing
3.4. RNA-Seq Raw Reads Pre-Processing, De Novo Transcriptome Assembly, and Functional Classification of Unigenes
3.5. Identification of Simple Sequence Repeats (SSRs)
3.6. GO Enrichment Analysis
3.7. KEGG Pathway Enrichment Analysis
3.8. Comparative Transcriptome Analysis of A. carmichaelii and A. heterophyllum
3.9. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nyirimigabo, E.; Xu, Y.; Li, Y.; Wang, Y.; Agyemang, K.; Zhang, Y. A review on phytochemistry, pharmacology and toxicology studies of aconitum. J. Pharm. Pharmacol. 2015, 67, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Singhuber, J.; Zhu, M.; Prinz, S.; Kopp, B. Aconitum in traditional chinese medicine: A valuable drug or an unpredictable risk? J. Ethnopharmacol. 2009, 126, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yand, Y.-S.; Shu, X.-Y.; Huang, J.; Hou, D.-B. Aconitum carmichaelii debeaux, cultivated as a medicinal plant in Western China. Genet. Resour. Crop Evol. 2016, 63, 919–924. [Google Scholar] [CrossRef]
- Sun, H.; Ni, B.; Zhang, A.; Wang, M.; Dong, H.; Wang, X. Metabolomics study on fuzi and its processed products using ultra-performance liquid-chromatography/electrospray-ionization synapt high-definition mass spectrometry coupled with pattern recognition analysis. Analyst 2012, 137, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, R.; Motoya, W.; Atsumi, T.; Mouri, C.; Kakiuchi, N.; Mikage, M. The relationship between growth of the aerial part and alkaloid content variation in cultivated Aconitum carmichaeli Debeaux. J. Nat. Med. 2011, 65, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Tang, L.; Zhou, X.; Wang, T.; Kou, Z.; Wang, Z. A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii debeaux. J. Ethnopharmacol. 2015, 160, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Sugawara, T.; Fujishita, K.; Shinozaki, Y.; Matsukawa, T.; Suzuki, T.; Koizumi, S. The astrocyte-targeted therapy by bushi for the neuropathic pain in mice. PLoS ONE 2011, 6, e23510. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Goto, K.; Ishige, A.; Komatsu, Y.; Kamei, J. Antinociceptive effect of Gosha-jinki-gan, a Kampo medicine, in streptozotocin-induced diabetic mice. Jpn. J. Pharmacol. 1999, 79, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Murai, T.; Ueda, M.; Katsuura, M.; Oishi, M.; Miwa, Y.; Okamoto, Y.; Uejima, E.; Taguchi, T.; Noguchi, S.; et al. Clinical features of paclitaxel-induced peripheral neuropathy and role of Gosya-jinki-gan. Gan Kagaku Ryoho Cancer Chemother. 2009, 36, 89–92. [Google Scholar]
- Vranova, E.; Coman, D.; Gruissem, W. Structure and dynamics of the isoprenoid pathway network. Mol. Plant 2012, 5, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Zi, J.; Mafu, S.; Peters, R.J. To gibberellins and beyond! Surveying the evolution of (di)terpenoid metabolism. Ann. Rev. Plant Biol. 2014, 65, 259–286. [Google Scholar] [CrossRef] [PubMed]
- Cherney, E.C.; Baran, P.S. Terpenoid-alkaloids: Their biosynthetic twist of fate and total synthesis. Isr. J. Chem. 2011, 51, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Devkota, K.P.; Sewald, N. Terpenoid alkaloids derived by amination reaction. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 923–951. [Google Scholar]
- Csupor, D.; Wenzig, E.M.; Zupko, I.; Wolkart, K.; Hohmann, J.; Bauer, R. Qualitative and quantitative analysis of aconitine-type and lipo-alkaloids of Aconitum carmichaelii roots. J. Chromatogr. A 2009, 1216, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, B.; Zhang, Q.; Fang, J.; Ma, K.; Li, Y.; Chen, H.; Dong, F.; Gao, Y.; Li, F.; et al. Metabonomic study on the toxicity of hei-shun-pian, the processed lateral root of Aconitum carmichaelii debx (ranunculaceae). J. Ethnopharmacol. 2008, 116, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, M.; Zhang, A.; Ni, B.; Dong, H.; Wang, X. UPLC-Q-TOF-HDMS analysis of constituents in the root of two kinds of Aconitum using a metabolomics approach. Phytochem. Anal. PCA 2013, 24, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Huang, Z.; Tang, X.; Yi, J.; Chen, S.; Yang, A.; Yang, J. Dynamic variation patterns of Aconitum alkaloids in daughter root of Aconitum carmichaelii (fuzi) in the decoction process based on the content changes of nine Aconitum alkaloids by HPLC- MS- MS. Iran. J. Pharm. Res. IJPR 2016, 15, 263–273. [Google Scholar] [PubMed]
- Pal, T.; Malhotra, N.; Chanumolu, S.K.; Chauhan, R.S. Next-generation sequencing (NGS) transcriptomes reveal association of multiple genes and pathways contributing to secondary metabolites accumulation in tuberous roots of Aconitum heterophyllum wall. Planta 2015, 242, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, Y.; Liang, Z.; Ho, A.; Wong, L.; Yong, P.; Chen, H.; Zhao, Z. Distribution of toxic alkaloids in tissues from three herbal medicine Aconitum species using laser micro-dissection, UHPLC-QTOF MS and LC-MS/MS techniques. Phytochemistry 2014, 107, 155–174. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Claros, M.G.; Bautista, R.; Guerrero-Fernandez, D.; Benzerki, H.; Seoane, P.; Fernandez-Pozo, N. Why assembling plant genome sequences is so challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Honaas, L.A.; Wafula, E.K.; Wickett, N.J.; Der, J.P.; Zhang, Y.; Edger, P.P.; Altman, N.S.; Pires, J.C.; Leebens-Mack, J.H.; dePamphilis, C.W. Selecting superior de novo transcriptome assemblies: Lessons learned by leveraging the best plant genome. PLoS ONE 2016, 11, e0146062. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. Soapdenovo-trans: De novo transcriptome assembly with short RNA-seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, Y.; Raimondo, D.; Redfern, O.C.; Watson, J.; Frishman, D.; Linial, M.; Orengo, C.; Thornton, J.; Tramontano, A. Protein function annotation by homology-based inference. Genome Biol. 2009, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.L.; Beuning, P.J.; Ondrechen, M.J. Biochemical functional predictions for protein structures of unknown or uncertain function. Comput. Struct. Biotechnol. J. 2015, 13, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2go: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The kegg databases at genomenet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Gianfranceschi, L.; Seglias, N.; Tarchini, R.; Komjanc, M.; Gessler, C. Simple sequence repeats for the genetic analysis of apple. Theor. Appl. Genet. 1998, 96, 1069–1076. [Google Scholar] [CrossRef]
- Senthilvel, S.; Jayashree, B.; Mahalakshmi, V.; Kumar, P.S.; Nakka, S.; Nepolean, T.; Hash, C. Development and mapping of simple sequence repeat markers for pearl millet from data mining of expressed sequence tags. BMC Plant Biol. 2008, 8, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Thiel, T.; Stein, N.; Langridge, P.; Graner, A. In silico analysis on frequency and distribution of microsatellites in ests of some cereal species. Cell. Mol. Biol. Lett. 2002, 7, 537–546. [Google Scholar] [PubMed]
- Lambert, H.C.; Gisel, E.G.; Groher, M.E.; Wood-Dauphinee, S. Mcgill ingestive skills assessment (MISA): Development and first field test of an evaluation of functional ingestive skills of elderly persons. Dysphagia 2003, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Brodelius, P.E. Transport and accumulation of secondary metabolites. In Progress in Plant Cellular and Molecular Biology: Proceedings of the Viith International Congress on Plant Tissue and Cell Culture, Amsterdam, The Netherlands, 24–29 June 1990; Nijkamp, H.J.J., Van Der Plas, L.H.W., Van Aartrijk, J., Eds.; Springer: Dordrecht, The Netherlands, 1990; pp. 567–576. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Yamazaki, M.; Takahashi, H.; Nakamura, M.; Kojoma, M.; Suzuki, H.; Saito, K. RNA-seq transcriptome analysis of panax japonicus, and its comparison with other panax species to identify potential genes involved in the saponins biosynthesis. Front. Plant Sci. 2016, 7, 481. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. Orthofinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, C.; Mizutani, E.; Okada, K.; Nakagawa, H.; Fukushima, S.; Tanaka, A.; Maeda, S.; Kamakura, T.; Yamane, H.; Takatsuji, H.; et al. Diterpenoid phytoalexin factor, a bHLH transcription factor, plays a central role in the biosynthesis of diterpenoid phytoalexins in rice. Plant J. Cell Mol. Biol. 2015, 84, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Okada, K.; Miyamoto, K.; Koga, J.; Shibuya, N.; Nojiri, H.; Yamane, H. OsTGAP1, a bZIP transcription factor, coordinately regulates the inductive production of diterpenoid phytoalexins in rice. J. Biol. Chem. 2009, 284, 26510–26518. [Google Scholar] [CrossRef] [PubMed]
- Richman, A.S.; Gijzen, M.; Starratt, A.N.; Yang, Z.; Brandle, J.E. Diterpene synthesis in stevia rebaudiana: Recruitment and up-regulation of key enzymes from the gibberellin biosynthetic pathway. Plant J. Cell Mol. Biol. 1999, 19, 411–421. [Google Scholar] [CrossRef]
- Tholl, D.; Chen, F.; Petri, J.; Gershenzon, J.; Pichersky, E. Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from arabidopsis flowers. Plant J. Cell Mol. Biol. 2005, 42, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N.; Negre, F.; Nagegowda, D.A.; Orlova, I. Plant volatiles: Recent advances and future perspectives. Crit. Rev. Plant Sci. 2006, 25, 417–440. [Google Scholar] [CrossRef]
- Tholl, D.; Lee, S. Terpene specialized metabolism in arabidopsis thaliana. In Arabidopsis Book; American Society of Plant Biologists: Rockville, MD, USA, 2011; Volume 9, p. e0143. [Google Scholar]
- Misra, R.C.; Garg, A.; Roy, S.; Chanotiya, C.S.; Vasudev, P.G.; Ghosh, S. Involvement of an ent-copalyl diphosphate synthase in tissue-specific accumulation of specialized diterpenes in Andrographis paniculata. Plant Sci. 2015, 240, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Agrawal, L.; Misra, R.C.; Sharma, S.; Ghosh, S. Andrographis paniculata transcriptome provides molecular insights into tissue-specific accumulation of medicinal diterpenes. BMC Genom. 2015, 16, 659. [Google Scholar] [CrossRef] [PubMed]
- Toyomasu, T.; Usui, M.; Sugawara, C.; Kanno, Y.; Sakai, A.; Takahashi, H.; Nakazono, M.; Kuroda, M.; Miyamoto, K.; Morimoto, Y.; et al. Transcripts of two ent-copalyl diphosphate synthase genes differentially localize in rice plants according to their distinct biological roles. J. Exp. Bot. 2015, 66, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Malhotra, N.; Pal, T.; Chauhan, R.S. Molecular dissection of pathway components unravel atisine biosynthesis in a non-toxic Aconitum species, A. heterophyllum wall. 3 Biotech 2016, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, K.; Rasmussen, A.V.; Morant, M.; Nielsen, A.H.; Bjarnholt, N.; Zagrobelny, M.; Bak, S.; Moller, B.L. Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr. Opin. Plant Biol. 2005, 8, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Winkel, B.S. Metabolic channeling in plants. Ann. Rev. Plant Biol. 2004, 55, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Pateraki, I.; Heskes, A.M.; Hamberger, B. Cytochromes p450 for terpene functionalisation and metabolic engineering. Adv. Biochem. Eng. Biotechnol. 2015, 148, 107–139. [Google Scholar] [PubMed]
- Hamberger, B.; Bak, S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20120426. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Werck-Reichhart, D. A p450-centric view of plant evolution. Plant J. Cell Mol. Biol. 2011, 66, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hillwig, M.L.; Okada, K.; Yamazaki, K.; Wu, Y.; Swaminathan, S.; Yamane, H.; Peters, R.J. Characterization of CYP76M5–8 indicates metabolic plasticity within a plant biosynthetic gene cluster. J. Biol. Chem. 2012, 287, 6159–6168. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. Theor. Angew. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Transcripts |
|---|---|
| Number of transcripts | 128,183 |
| Total assembled bases | 78,623,549 |
| Average length (bps) | 613.37 |
| Median length (bps) | 383 |
| Maximum length (bps) | 13,485 |
| Minimum length (bps) | 224 |
| N50 (bps) | 830 |
| GC content | 42.35 |
| Results of SSR Searches | |
|---|---|
| Total number of sequences examined | 128,183 |
| Total number of identified SSRs | 16,068 |
| Number of SSR containing sequences | 14,168 |
| Number of sequences containing more than one SSR | 1675 |
| Number of SSRs present in the compound formation | 686 |
| Total number of orthogroups: 20,247 | |||
| Mean orthogroup size: 2.8 | |||
| Median orthogroup size: 2.0 | |||
| Description | Overall | A. heterophyllum | A. carmichaelii |
| Number of genes | 209,790 | 81,607 | 128,183 |
| Number of genes in orthogroups | 56,915 | 26,282 | 30,633 |
| Number of unassigned genes | 152,875 | 55,325 | 97,550 |
| Percentage of genes in orthogroups | 27.1 | 32.2 | 23.9 |
| Percentage of unassigned genes | 72.9 | 67.8 | 76.1 |
| Number of orthogroups containing species | 20,232 | 20,239 | 20,240 |
| Number of species-specific orthogroups | 15 | 8 | 7 |
| Number of genes in species-specific orthogroups | 87 | 39 | 48 |
| Enzymes | Enzyme Name | Unigene | Unigene Length | Similarity |
|---|---|---|---|---|
| Mevalonate Pathway (MVA Pathway) | ||||
| AACT1 | Acetyl-CoA acetyltransferase | TR41842|c0_g3_i5 | 950 | 96.21212 |
| AACT2 | TR41842|c0_g3_i3 | 957 | 93.61702 | |
| AACT3 | TR44607|c2_g6_i4 | 962 | 94.29658 | |
| AACT4 | TR44607|c2_g6_i2 | 1057 | 94.27481 | |
| HMGS1 | Hydroxymethylglutaryl-CoA synthase | TR19454|c0_g1_i1 | 571 | 85.81081 |
| HMGS2 | TR40295|c3_g2_i3 | 909 | 87.21805 | |
| HMGS3 | TR40295|c3_g2_i1 | 1761 | 91.45299 | |
| HMGS4 | TR40318|c0_g1_i1 | 1992 | 84.52656 | |
| HMGR1 | Hydroxymethylglutaryl-CoA reductase | TR44439|c4_g1_i5 | 784 | 72.42647 |
| HMGR2 | TR44439|c4_g1_i7 | 2118 | 89.20354 | |
| HMGR3 | TR44439|c4_g1_i1 | 2179 | 88.64028 | |
| MVK1 | Mevalonate kinase | TR45020|c0_g1_i1 | 1335 | 86.23377 |
| PMK1 | Phosphomevalonate kinase | TR45726|c0_g1_i3 | 575 | 83.50515 |
| PMK2 | TR45726|c0_g1_i1 | 641 | 83.50515 | |
| PMK3 | TR45726|c0_g1_i12 | 2237 | 82.47012 | |
| MVDD1 | Mevalonate diphosphate decarboxylase | TR44355|c0_g1_i1 | 1781 | 91.23223 |
| MVDD2 | TR44355|c0_g1_i3 | 1797 | 91.46919 | |
| Methylerythritol Pathway (MEP Pathway) | ||||
| DXS1 | 1-deoxy-d-xylulose-5-phosphate synthase | TR28848|c0_g1_i1 | 505 | 95.83333 |
| DXS2 | TR32192|c0_g1_i2 | 532 | 86.36364 | |
| DXS3 | TR47591|c0_g1_i2 | 554 | 77.77778 | |
| DXS4 | TR15579|c0_g1_i1 | 563 | 97.3262 | |
| DXS5 | TR32608|c0_g1_i1 | 578 | 96.33508 | |
| DXS6 | TR47591|c0_g1_i5 | 648 | 77.77778 | |
| DXS7 | TR29244|c0_g2_i2 | 668 | 78.18182 | |
| DXS8 | TR47591|c0_g1_i3 | 866 | 77.77778 | |
| DXS9 | TR47591|c0_g2_i1 | 2330 | 95.54235 | |
| DXS10 | TR43248|c0_g1_i2 | 2462 | 94.00279 | |
| DXS11 | TR42479|c0_g1_i1 | 2585 | 84.34903 | |
| DXR1 | 1-deoxy-d-xylulose-5-phosphate reductoisomerase | TR38806|c0_g1_i1 | 2065 | 92.11087 |
| ISPD1 | 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | TR34080|c0_g1_i3 | 1173 | 95.54455 |
| ISPD2 | TR34080|c0_g1_i1 | 1229 | 95.04505 | |
| ISPD3 | TR34080|c0_g1_i6 | 1355 | 95.47511 | |
| ISPE1 | 4-(cytidine-5′-diphospho)-2-C-methyl-d-erythritol kinase | TR43665|c0_g1_i2 | 1549 | 84.79381 |
| ISPF1 | 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase | TR34396|c0_g1_i1 | 945 | 90.37433 |
| ISPG | (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase | TR42806|c0_g1_i1 | 2806 | 94.22043 |
| ISPH1 | (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase | TR42285|c1_g1_i2 | 1718 | 91.16379 |
| IPPI1 | Isopentenyl diphosphate isomerase | TR37870|c0_g1_i1 | 1326 | 87.33333 |
| IPPI2 | TR44603|c0_g1_i3 | 2692 | 88.98848 | |
| Aconitine-Type Diterpene Alkaloid Biosynthetic Pathway | ||||
| GGPPS1 | Geranylgeranyl pyrophosphate synthase | TR47410|c1_g2_i2 | 1384 | 83.58663 |
| GGPPS2 | TR35223|c0_g1_i1 | 1172 | 88.60759 | |
| GGPPS3 | TR44055|c0_g1_i3 | 765 | 92.24806 | |
| GGPPS4 | TR44055|c0_g1_i1 | 1496 | 89.59732 | |
| GGPPS5 | TR46755|c1_g1_i1 | 1752 | 88.33819 | |
| GGPPS6 | TR42099|c0_g1_i1 | 1532 | 81.73077 | |
| GGPPS7 | TR46755|c0_g1_i3 | 592 | 75.71429 | |
| CDPS1 | ent-copalyl diphosphate synthase | TR48303|c1_g1_i4 | 748 | 71.42857 |
| CDPS2 | TR48303|c0_g1_i1 | 752 | 80.32787 | |
| CDPS3 | TR48303|c1_g1_i3 | 1512 | 79.66903 | |
| CDPS4 | TR36243|c0_g1_i1 | 2342 | 73.64238 | |
| KS1 | Kaurene synthase | TR44693|c0_g3_i1 | 1120 | 72.12121 |
| KS2 | TR44693|c0_g1_i2 | 1539 | 75.80645 | |
| KS3 | TR48214|c4_g1_i1 | 2144 | 77.95031 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, M.; Rai, A.; Kawano, N.; Yoshimatsu, K.; Takahashi, H.; Suzuki, H.; Kawahara, N.; Saito, K.; Yamazaki, M. De Novo RNA Sequencing and Expression Analysis of Aconitum carmichaelii to Analyze Key Genes Involved in the Biosynthesis of Diterpene Alkaloids. Molecules 2017, 22, 2155. https://doi.org/10.3390/molecules22122155
Rai M, Rai A, Kawano N, Yoshimatsu K, Takahashi H, Suzuki H, Kawahara N, Saito K, Yamazaki M. De Novo RNA Sequencing and Expression Analysis of Aconitum carmichaelii to Analyze Key Genes Involved in the Biosynthesis of Diterpene Alkaloids. Molecules. 2017; 22(12):2155. https://doi.org/10.3390/molecules22122155
Chicago/Turabian StyleRai, Megha, Amit Rai, Noriaki Kawano, Kayo Yoshimatsu, Hiroki Takahashi, Hideyuki Suzuki, Nobuo Kawahara, Kazuki Saito, and Mami Yamazaki. 2017. "De Novo RNA Sequencing and Expression Analysis of Aconitum carmichaelii to Analyze Key Genes Involved in the Biosynthesis of Diterpene Alkaloids" Molecules 22, no. 12: 2155. https://doi.org/10.3390/molecules22122155