Theoretical Study of ClOO + NO Reaction: Mechanism and Kinetics


1
Key Laboratory of Integrated Exploitation of Bayan Obo Multi-Metal Resources, Inner Mongolia University of Science and Technology, Baotou 014010, China
2
Institute of Theoretical Chemistry, State Key Laboratory of Theoretical and Computational Chemistry, Jilin University, Changchun 130023, China
3
College of Physics, Jilin University, Changchun 130012, China
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(12), 2121; https://doi.org/10.3390/molecules22122121
Submission received: 30 October 2017
/
Revised: 16 November 2017
/
Accepted: 20 November 2017
/
Published: 1 December 2017
(This article belongs to the Special Issue Radical Chemistry)
Abstract
:Theoretical investigations are performed on mechanism and kinetics of the reaction of halogen peroxy radical ClOO with NO radical. The electronic structure information for both of the singlet and triplet potential energy surfaces (PESs) is obtained at the MP2/6-311 + G(2df) level of theory, and the single-point energies are refined by the CCSD(T)/6-311 + G(2df) level. The rate constants for various product channels of the reaction in the pressure range of 1-7600 Torr are predicted. The main results are as follows: On the singlet surface, the addition-elimination mechanism is the most important. First, the N atom of the NO radical can attack the O atom of the ClOO radical to form an energy-riched intermediate IM1 ClOONOtp (21.3 kcal/mol) barrierlessly, then IM1 could isomerizes to IM2 ClOONOcp (22.1 kcal/mol) via a low energy barrier. Both IM1 and IM2 can dissociate to the primary product P1 ClNO + 1O2 and the secondary product P2 ClO + NO2. On the triplet surface, the direct Cl-abstraction reaction is the most feasible pathway. The Cl-abstraction can take place via a van der Waals complex, 3IM1 ONClOO (4.1 kcal/mol), then it fragments readily to give P1’ ClNO + 3O2 with a small barrier. The kinetic calculations show that at low temperatures, the singlet bimolecular product P1 is the primary product, while at high temperatures, the triplet product P1’ becomes the primary one; only at high pressures and low temperatures, the unimolecular products IM1 and IM2 can be found with quite small yields. At experimentally measured temperature 213 K, ClNO is the primary product in the whole pressure range, which is consistent with the previous experiment. The present study may be useful for further experimental studies for the title reaction.
1. Introduction
Halogen oxide has attracted wide experimental and theoretical attentions for its important role in many chemical processes, such as stratospheric ozone depletion, water disinfection, pulp bleaching, food preservation, etc. [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. In atmosphere, halogen oxide mainly exists in free radicals, such as XO, OXO and XOO. Asymmetrical XOO is the most stable structure of halogen oxide XO2 [22]. The free radical (ClOO) contains a very weak chlorine-oxygen bond (Cl-O) with a bond energy of smaller than 19.5 KJ∙mol−1. ClOO is often described as a helium atom “accompanied with” an oxygen molecule [23].
In the stratosphere, there’s a reaction that will destroy the ozone:
Cl + O3→ClO + O2
This reaction is accompanied with:
Cl + O2 + M→ClOO + M
And
ClO + ClO→ClOO + C
In these reactions, ClOO is an important intermediate and is vital to circulation of ozone depletion [23]. Therefore, elimination reactions of ClOO by other chemical elements like NO, NO2 and hydrocarbon in the atmosphere will influence the circulation of such chlorine-induced ozone depletion significantly.
On the other hand, NO, one of the major oxynitrides with wide distributions in atmosphere, not only is closely related with atmospheric pollutions (e.g., acid rain and photochemical smog), but also will destroy the ozone sphere [24]. Experimental and theoretical research on the generation and elimination reaction mechanism of NO has been one of important topic in atmosphere and combustion chemistry [25,26,27,28,29,30,31,32,33,34,35]. As a result, the ClOO + NO reaction has important potential significance to eliminate atmospheric pollutants and reduce ozone depletion.
For ClOO + NO reaction, there are three feasible reaction channels in thermodynamics:
ClOO + NO→ClO + NO2
ClOO + NO→ClNO + O2
ClOO + NO + M→ClONO2 + M
However, there has only been one experimental research project on kinetic of ClOO + NO reaction as far as we know. In 2006, Enami et al. [36] measured ClOO + NO reaction when temperature ranges from 205–243 K and pressure ranges from 50–150 Torr by using spectrographic technique under cavity environment. They discovered no obvious temperature dependence and concluded that the reaction rate remains a constant at 213 K, k (ClOO + NO) = (4.5 ± 0.9) × 10−11 cm3 mol−1 s−1. They speculated that the major product of ClOO + NO reaction is ClNO whose branching ratio is about 0.8. But the observed product is NO2 whose branching ratio is 0.18 ± 0.02 at 213 K and 0.15 ± 0.02 at 223 K. Their experimental research failed to elaborate the mechanism of this complicated ClOO + NO reaction with multiple quantum well and multiple channels, pressure and temperature dependence of its product within a wider measuring range, and product distribution. According to our knowledge, Yang et al. [37] carried out a theoretical study on this reaction at CCSD (T)/6-311 + G(2d)//B3LYP/6-311 + G(2d) level of theory in 2012. Although they have got relative detailed singlet potential energy surface, they didn’t considered triplet potential energy surface, and they didn’t calculate the kinetics of the ClOO + NO reaction. Kinetics information within other temperature and pressure range of the reaction is still not clear. In addition, recently, we have investigated the mechanism for the reaction of radical FOO with NO [38]. The calculated results show that starting from the energy-riched intermediate FOONO, FNO is the exclusive product at room temperature 298 K and at 1 Torr. Similarly, we speculate that ClNO is the primary product of the ClOO + NO reaction at experimentally measured temperature 213 K. Considering the potential significance of ClOO + NO reaction in atmospheric chemistry, it is necessary to make comprehensive theoretical studies on its mechanism and kinetic. In this paper, potential energy surface of ClOO + NO reaction was explored based on quantum chemistry calculation. A kinetic calculation was conducted by using Rice-Ramsperger-Kassel-Marcus (RRKM) unimolecular reaction rate theory of microcanonical ensemble [39], getting the reaction rate constants and branching ratios of many product channels under different temperatures and pressures.
2. Calculation Methods
The geometries of all of the reactants, products, intermediates, and transition states involved in the ClOO + NO reaction were optimized using the second order level of closed shell MΦller-Pleset MP2 [40] perturbation theory in conjunction with the 6-311 + G(2df) basis set. Frequency calculations were performed at the same level to check whether the obtained species is an equilibrium species (with all real frequencies) or a transition state (with one and only one imaginary frequency). To confirm that the transition states connect designated intermediates, we also performed intrinsic reaction coordinate (IRC) [41,42,43,44] calculations at the MP2/6-311 + G(2df) level. To obtain more reliable energetic data, single-point energy calculations were performed at the CCSD (T)/6-311 + G(2df) level using the MP2/6-311 + G(2df) optimized geometries of all the species. Unless noted, the CCSD(T) energies with inclusion of MP2 zero-point vibrational energies (ZPE) are used throughout. All calculations were carried out using the Gaussian 03 program packages [45].
According to the variational transition-state and RRKM [39] theories, the kinetic calculations for this multi-channel and multi-well reaction were carried out via the MultiWell 2011 [46,47] program on the basis of the PES obtained above in order to identify the likely mechanism and the branching ratios of various product channels.
3. Results and Discussion
3.1. Potential Energy Surface and Reaction Mechanism
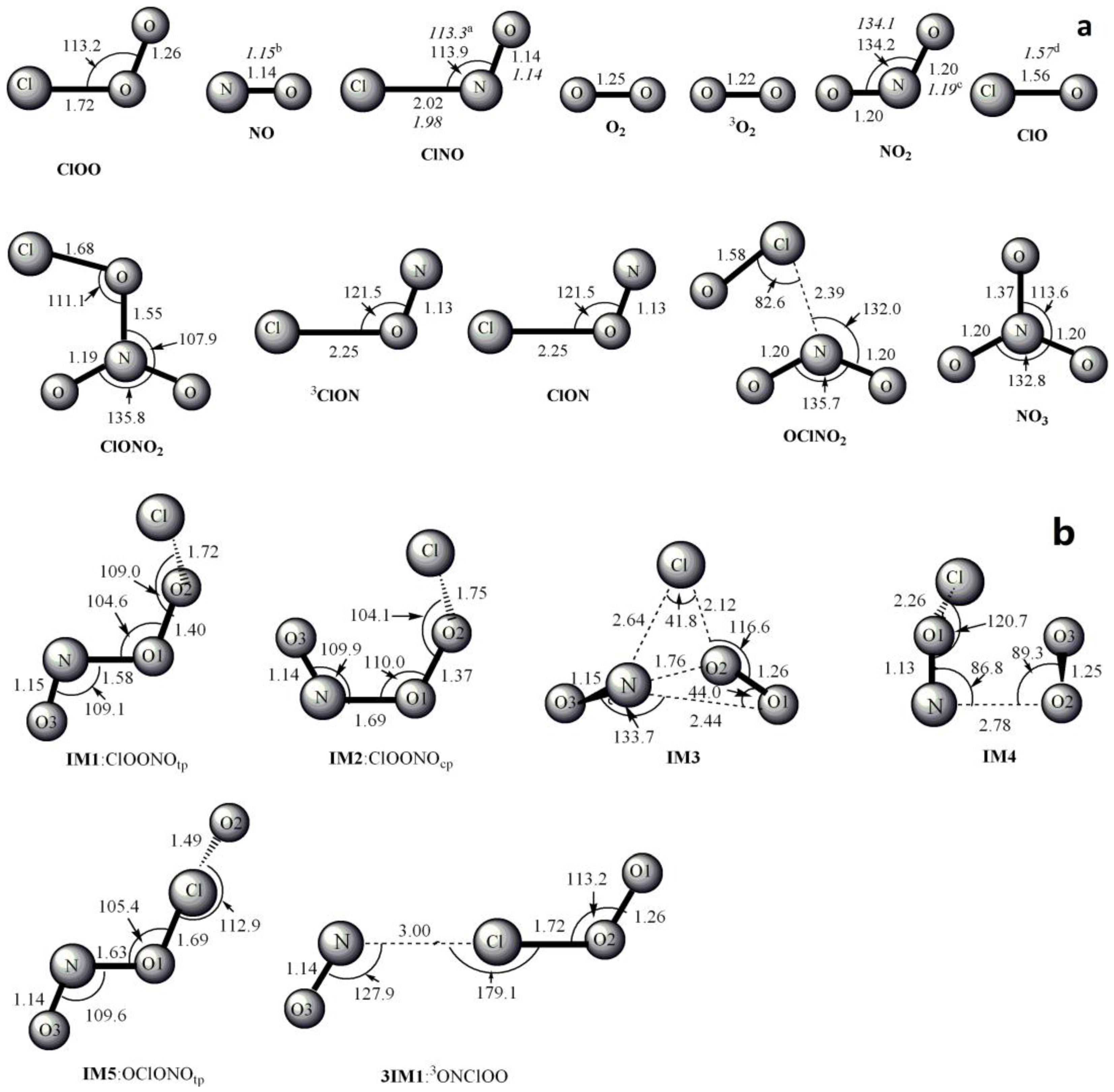
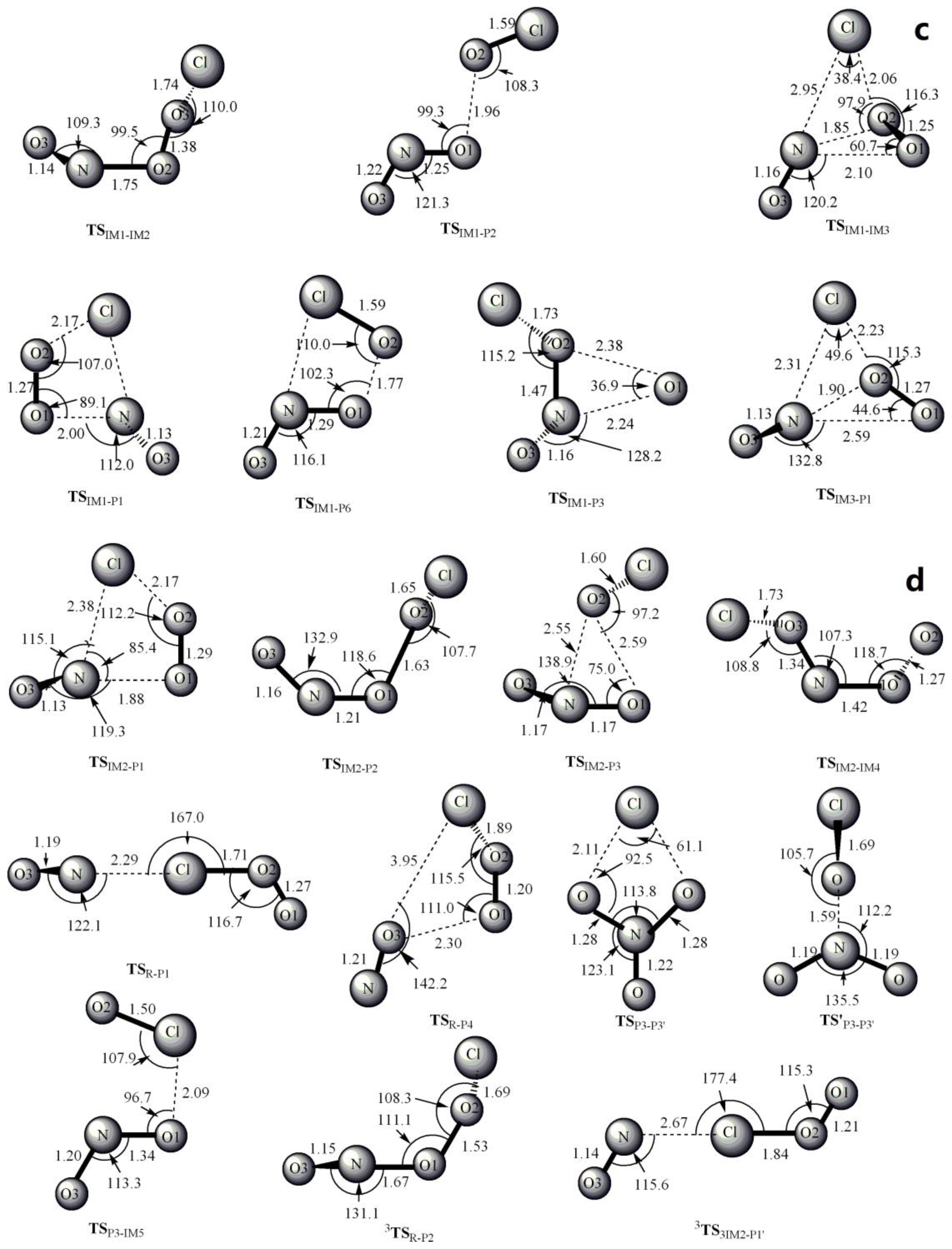
The optimized geometries of the reactants, products, intermediates and transition states for ClOO + NO reaction are shown in Figure 1, respectively, along with the available experimental data from the literature. It is found that when comparison is available, the agreement between theoretical and experimental results is good, with the largest discrepancy within a factor of 0.9%. The acronyms “cp” and “tp” are used to denote the cis-perp and trans-perp forms of the isomers. The schematic profile of the PESs is depicted in Figure 2. The total energy of the reactant R (ClOO + NO) is set to be zero for reference.
3.1.1. Addition Reaction Channels
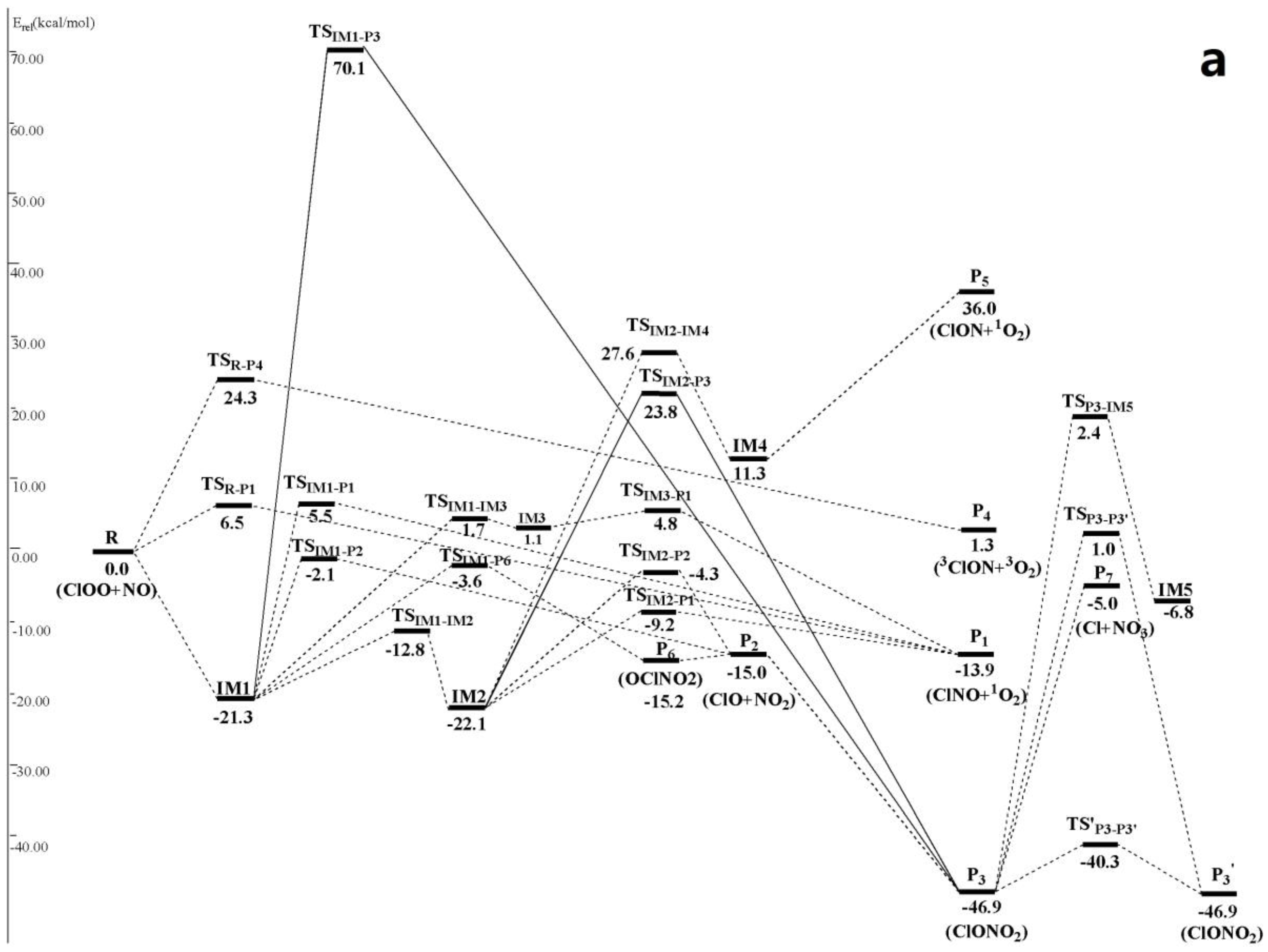
It can be seen from Figure 2 that on singlet potential energy surface, the ClOO + NO reaction could be added onto O atom of ClOO• through the N-atom of NO• to form entrance intermediate with rich energies IM1 ClOONOtp (−21.3 kcal/mol). Such addition reaction will release abundant heats, making IM1 gain high chemical activity and easy to be further isomerized and dissociated. This is a typical free radical-free radical reaction mechanism. We should note that IM1 can be converted into its cis-perp conformer IM2 ClOONOcp (−22.1 kcal/mol) only by overcoming a small energy barrier 8.5 kcal/mol and passing through a transition state TSIM1-IM2 of –NO group rotation.
Five available dissociation and isomerization reaction channels were found from IM1 ClOONOtp (Figure 2): (1) generation channel of P1 ClNO + 1O2 (−13.9 kcal/mol) through a four-membered ring transition state TSIM1-P1 (5.5 kcal/mol) of 1, 3 Cl-atom transfer; (2) generation channel of P2ClO + NO2 (−15.0 kcal/mol) through the transition state TSIM1-P2 (−2.1 kcal/mol) of direct O–O bond breakage; (3) generation channel of P6 OClNO2 (−15.2 kcal/mol) through a four-membered ring transition state TSIM1-P6 (−3.6 kcal/mol) of synergetic O–O bond breakage and N–Cl bond formation. P6 could make secondary dissociation reaction and produce P2 through direct N–Cl bond pyrolysis; (4) generation channel of IM3 (1.1 kcal/mol) through the transition state TSIM1-IM3 (1.7 kcal/mol) of synergetic N–O and O–Cl bond breakages. IM3 is a complex with loose structure, which passes through the transition state TSIM3-P1 and produces P1 after overcoming a 3.7 kcal/mol energy barrier. (5) Based on structure optimization on MP2 theoretical level combined with 6-311G(d) basis set and single-point energy correction on CCSD(T)/6-311 + G(2df) level, another reaction channel could be found. Based on O–Cl transfer, IM1 passes through a three-membered ring transition state TSIM1-P3 (70.1 kcal/mol) and produces P3 ClONO2 (−46.9 kcal/mol). P2 and P3 could convert mutually. P3 could be further converted to produce IM5 OlONOtp (−6.8 kcal/mol) (through TSP3-IM5 (2.4 kcal/mol)), P7 Cl• + NO3 (−5.0 kcal/mol) (through generation of loose energy barrier-free variational transition state) and P3′ (through TSP3-P3′ (1.0 kcal/mol) or TS′P3-P3′ (−40.3 kcal/mol)). Obviously, since energies of TSIM1-P1, TSIM1-IM3 and TSIM1-P3 of rate-limiting step in channel (1), (4) and (5) are all positive and far higher than those of TSIM1-P2and TSIM1-P6 in channel (2) and (3), channel (1), (4) and (5) couldn’t compete with channel (2) and (3). Therefore, they are not involved in the following kinetic calculation.
Four available dissociation and isomerization reaction channels were determined from IM2 ClOONOcp (Figure 2). Firstly, IM2 could make 1,3 Cl-atom transfer through a four-membered ring transition state TSIM2-P1 (−9.2 kcal/mol) and produce P1. Secondly, IM2 also could make direct O–O bond breakage through the transition state TSIM2-P2 (−4.3 kcal/mol) to produce P2. Moreover, IM2 can be converted into isomer IM4 ClONOO (11.3 kcal/mol) through a transition state TSIM2-IM4 (27.6 kcal/mol) of 1,4 Cl-transfer. IM4 can be decomposed directly into P5 ClON + 1O2 (36.0 kcal/mol) without energy barrier. Additionally, we also discovered a reaction channel of IM2 on CCSD(T)/6-311 + G(2df)//B3LYP/6-311 + G(2df) theoretical level, which produces P3 through 1,2-OCl transfer after passing through a three-membered ring transition state TSIM2-P3 (23.8 kcal/mol). It is easy to know that channel (3) and (4) have to overcome big energy barriers, which are impractical in energy. As a result, they are neglected in the following kinetic calculation.
3.1.2. Abstraction Reaction Channels
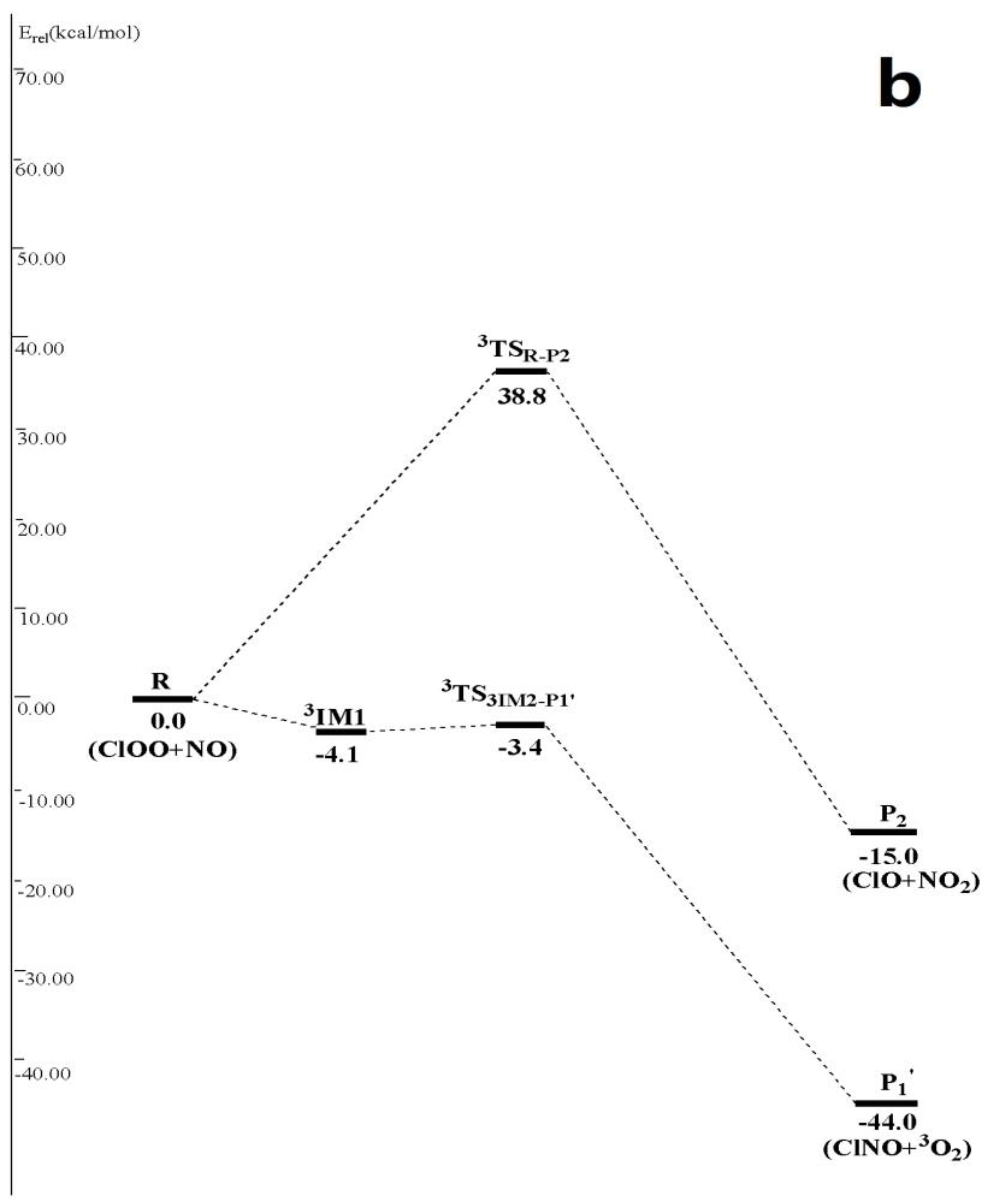
In Figure 2, ClOO + NO reaction has another two abstraction reaction channels on singlet and triplet potential energy surfaces. On the singlet potential energy surface, both N atom and O atom in NO• can abstract Cl atom in ClOO• directly to generate P1 and P4 3ClON + 3O2 (1.3 kcal/mol), respectively. Their transition states are TSR-P1 (6.5 kcal/mol), forming TSR-P4 (24.3 kcal/mol). On the triplet potential energy surface, N atom in NO• can either abstract O atom in ClOO• directly and produces P2 through a high-energy transition state 3TSR-P2 (38.8 kcal/mol), or abstract Cl atoms and produces P1′ ClNO + 3O2 (−44.0 kcal/mol) after passing through a triplet van der Waals complex 3IM1 3ONClOO (−4.1 kcal/mol) and then the transition state 3TS3IM2-P1′ (−3.4 kcal/mol). Since previous three abstraction reaction channels contain very high reaction energy barrier, the last abstraction reaction channel that produces P1′ is the only one feasible channel, which is considered in the following kinetic calculation.
To sum up, available channels for ClOO + NO reaction are:
R (ClOO + NO)→IM1* ClOONOtp→IM1
→P2ClO + NO2
→P6OClNO2→P2ClO + NO2
→IM2 ClOONOcp→P1ClNO + 1O2
→P2ClO + NO2
R (ClOO + NO)→3IM1* 3ONClOO→P1′ClNO + 3O2
On singlet potential energy surface, generation of IM1-2, P6, P1 and P2 as well as direction Cl- abstraction product P1′ through a triplet potential energy surface is the most feasible in view of energy (Figure 2). However, since the rate-limiting steps in their generation channels only have slight energy differences, it is difficult to determine possible reaction channels and feasible products under different temperature and pressure ranges only from the perspective of energy. Hence, the following text uses RRKM calculation to calculate rate constants and branching ratio of these competitive channels.
3.2. Kinetic Calculations
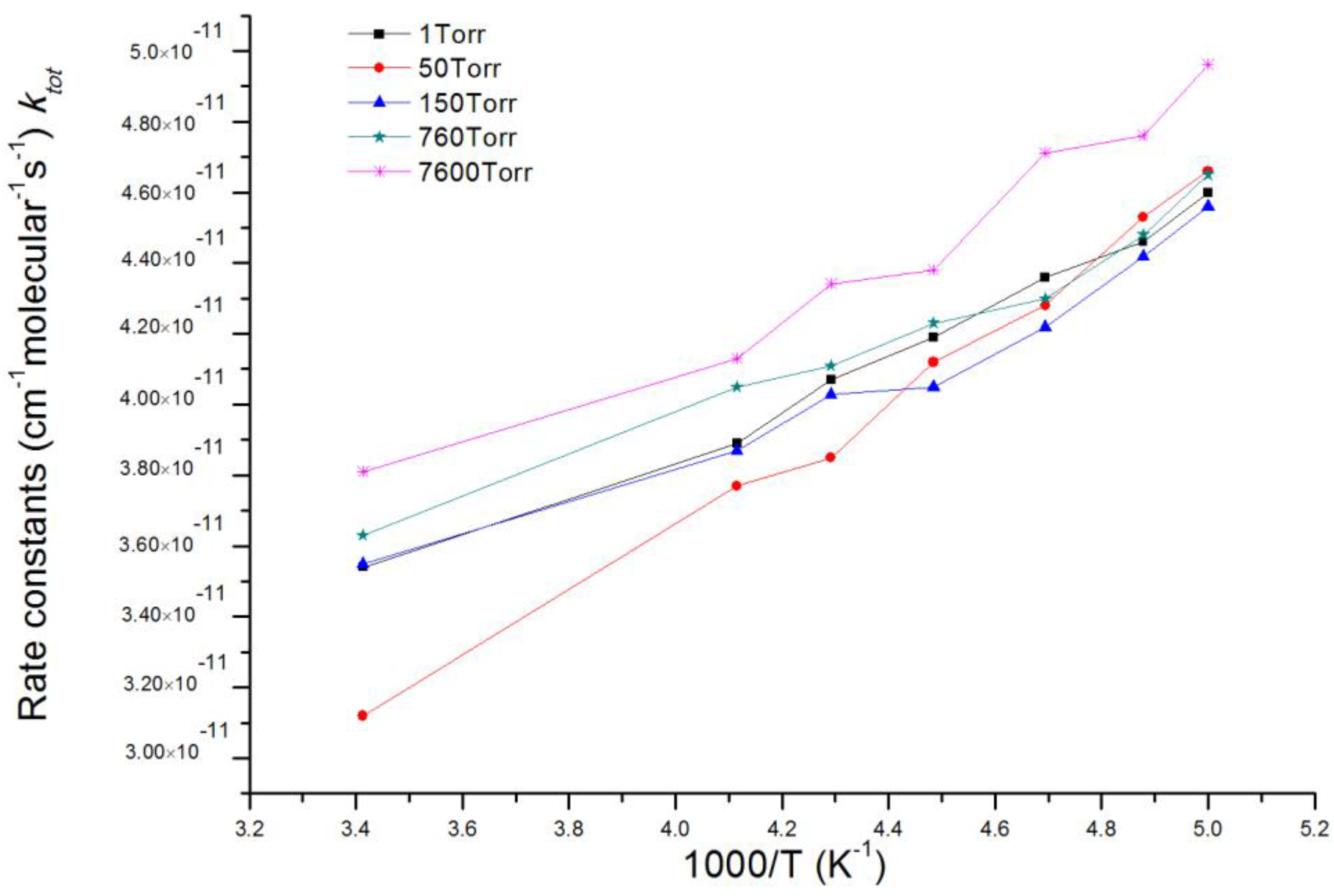
Based on acquired potential energy surfaces of ClOO + NO reaction, rate constants of the overall reaction and multiple reaction channels as well as branching ratio of various products under the temperature range 200–293 K and pressure range 1~7600 Torr were calculated with MultiWell 2011 program [46,47]. Energies and molecular parameters (reaction energy barrier, rotational inertia and vibration frequency) of reactants, products, intermediates and transition states which were calculated from ab initio were used in kinetic calculation. c bottleneck of energy barrier-free reaction channels, for example, entrance channel of IM1 ClOONOtp with chemical activity, was identified using variational transition state theory (VTST) [52,53]. Therefore, we carried out restricted optimization calculation under fixed length of N-O bond in IM1 ClOONOtp and multiple reference states CASSCF(8,6)/aug-cc-pvdz. Single-point total energy usage along the reaction coordinates was corrected by using CASPT2(8,6)/aug-cc-pvdz. CASPT2//CASSCF computation was accomplished by using MOLPRO 2006 program [54,55]. Other necessary information about potential energy surfaces in the kinetic calculation were acquired on CCSD(T)/6-311 + G(2df)//MP2/6-311 + G(2df) theoretical levels. The total reaction rate constant (ktot) is the sum of rate constants of corresponding reaction channels. Table 1 shows that the RRKM theoretical values agree well with experimental values of Enami et al. [36]. within the studying temperature range. The total reaction rate constant (ktot) at various pressures of 1, 50, 150, 760 and 7600 Torr in a temperature range of 200–293 K is presented in Figure 3. It is seen that under different pressures, ktot decreases slightly with the increase of temperature.
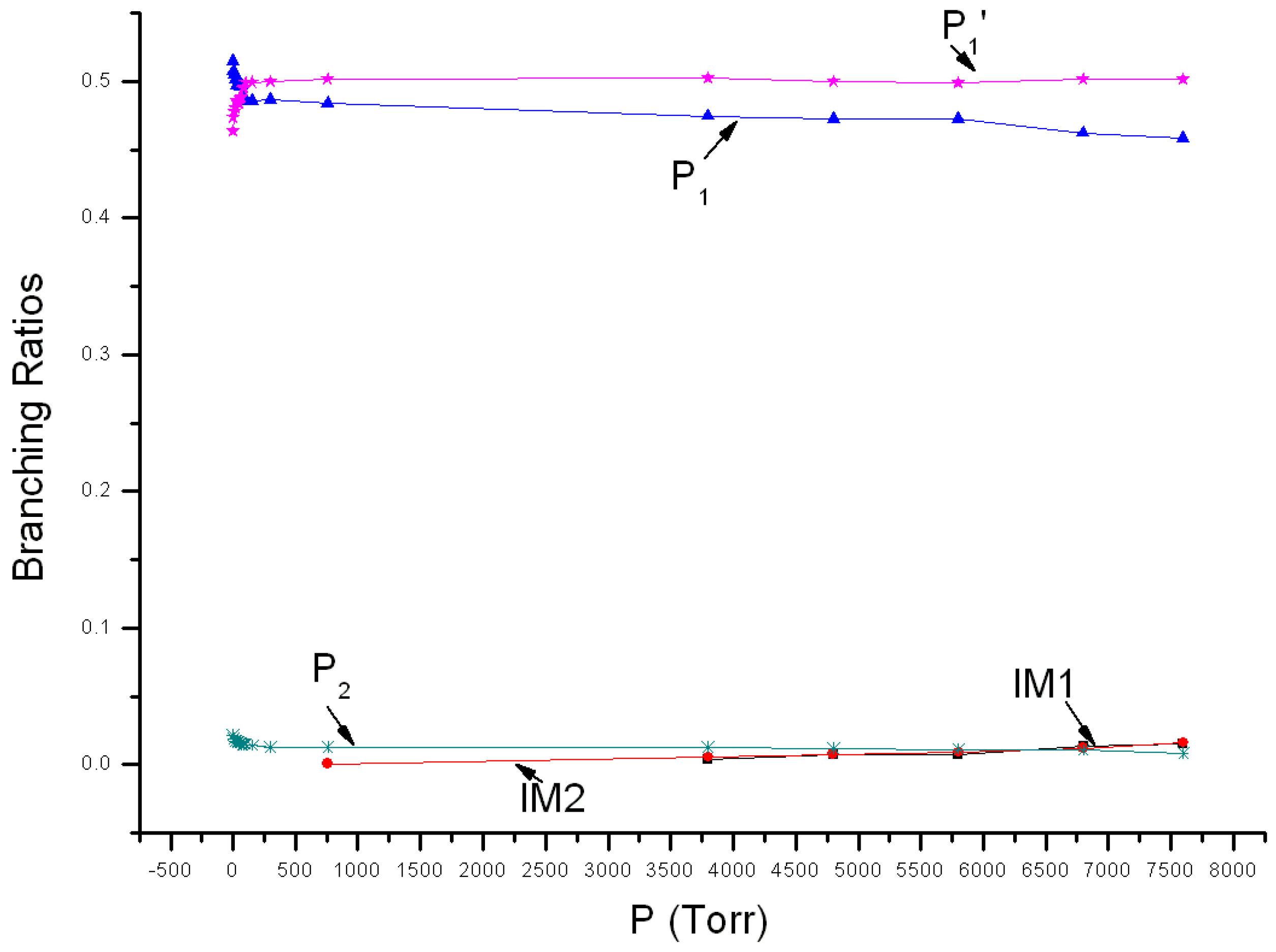
Variations of reaction branching ratios of the overall reaction channel and reaction channels against pressure (1~7600 Torr) under 213 K are shown in Figure 4. At 213 K, P1 is the major product when pressure <77 Torr, showing increasing yield from 0.52 at 1 Torr to 0.49 at 77 Torr. P1′ is the secondary major product and its yield increases continuously from 0.46 at 1 Torr to 0.49 at 77 Torr. When pressure >77 Torr, P1 become the secondary major product, having 0.46 yield at 7600 Torr, while P1′ become the major product, having 0.50 yield at 7600 Torr. The yield of P2 reduces gradually with the increase of pressure. It reduces from 0.02 at 1 Torr to 0.01 at 7600 Torr. Under 213 K, the stabilization effect of intermediate could be neglected, and yields of IM1 and IM2 are smaller than 0.02. Theoretical values were compared with previous experimental data. The experiment speculated that the major product is ClNO, which echoes with the theoretical result. However, the theoretical yield of ClNO from 1~7600 Torr is about 0.98–0.96, higher than the experimental estimation (0.80). The theoretical branching ratio of secondary product NO2 is lower than the observed 0.18 ± 0.02. Moreover, the kinetic calculation also revealed that yields of ClOONOtp and ClOONOcp under 7600 Torr are very small and their branching ratios are about 0.02. No associated experimental data is available yet.
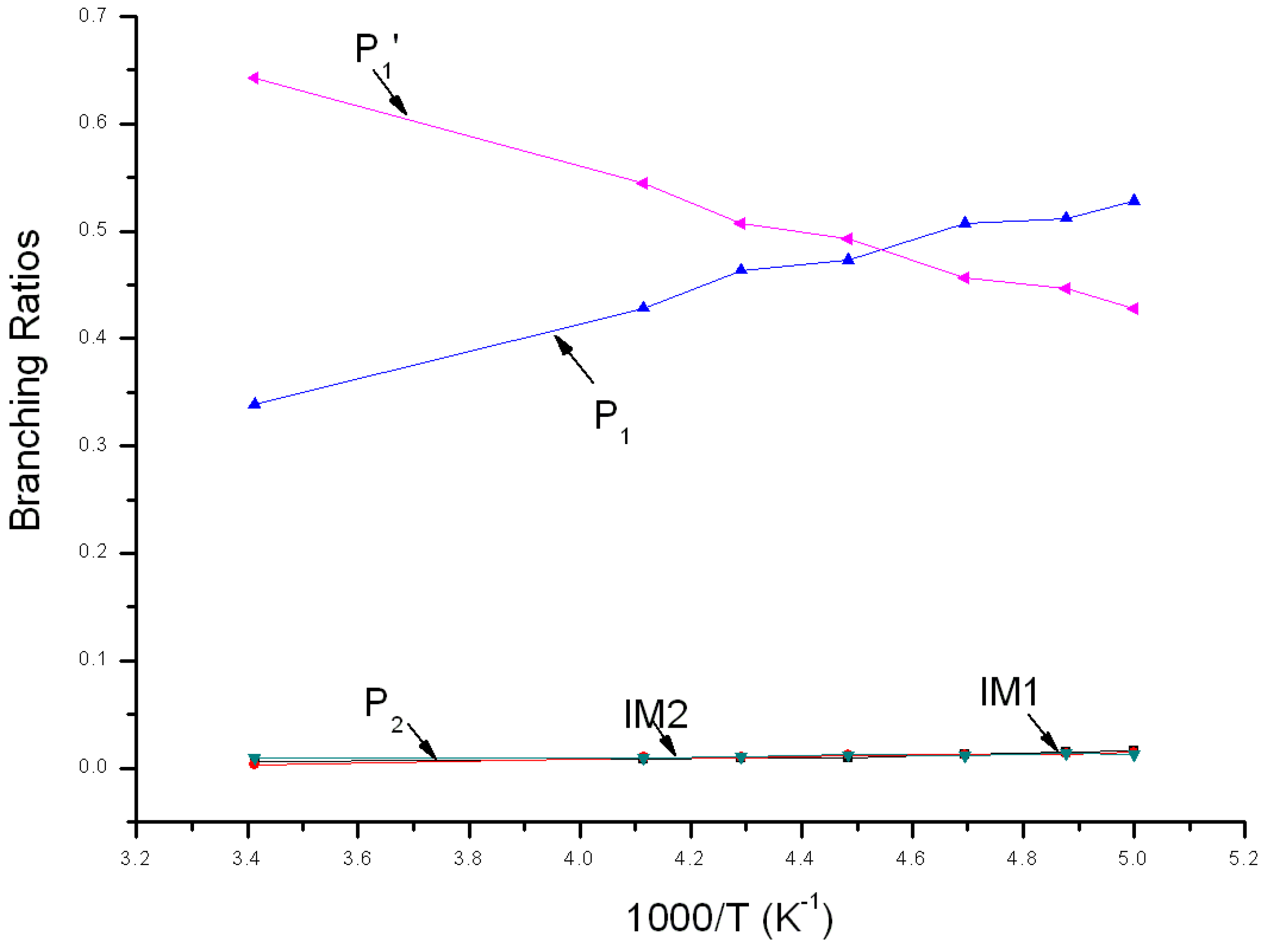
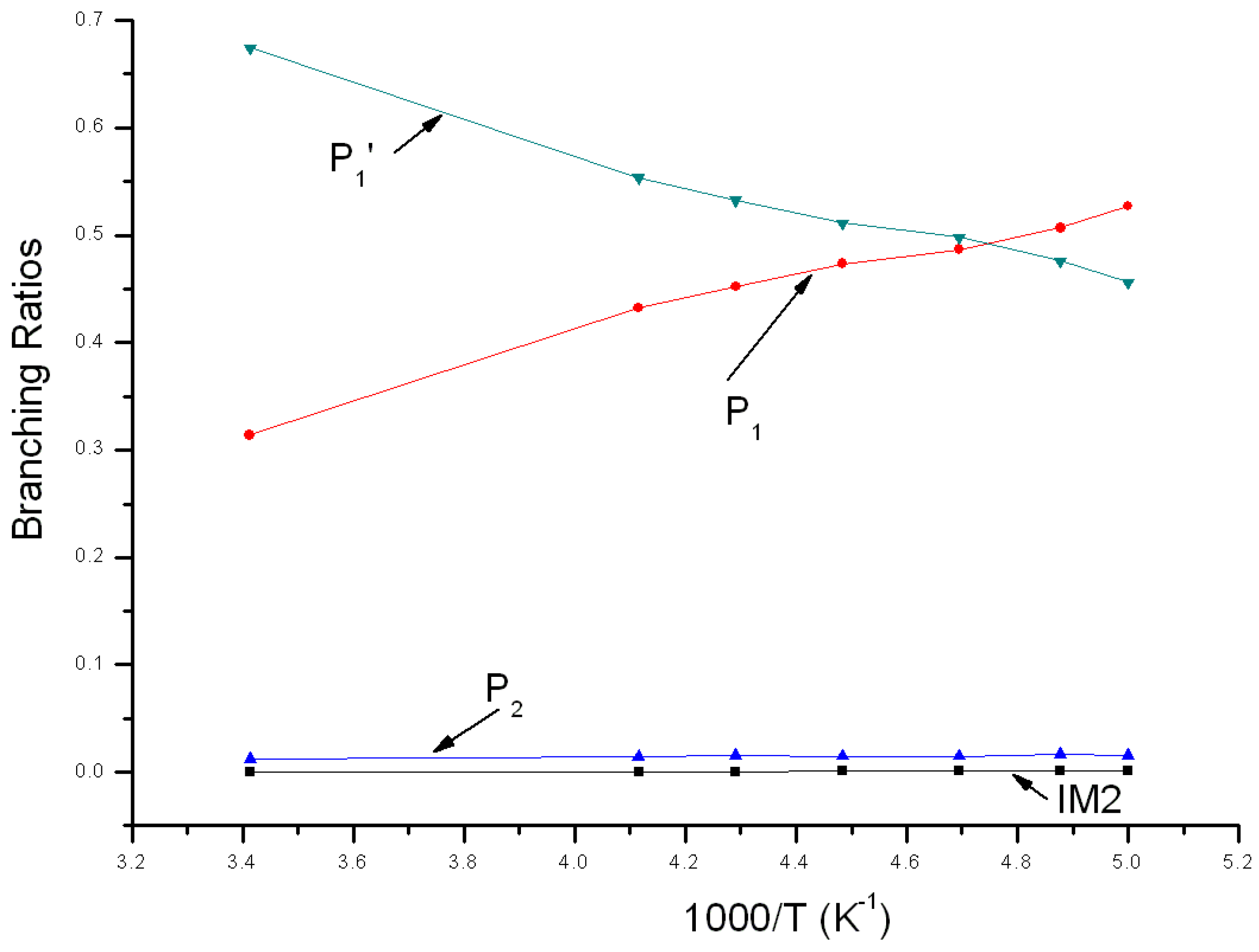
Reaction branching ratios of reaction channels at 1 Torr when temperature changes between 200~293 K are shown in Figure 5. Obviously, the impact stabilization effect of intermediate at 1Torr can be neglected completely. IM1 and IM2 transformed into P1 and P2 completely. In Figure 5, branching ratio of P1 is inversely proportional to temperature, reducing gradually from 0.52 at 200 K to 0.29 at 293 K. Branching ratio of P2 is also inversely proportional to temperature, reducing gradually from 0.02 at 200 K to about 0.01 at 293 K. On the contrary, branching ratio of P1′ is proportional to temperature, increasing continuously from 0.46 at 200 K to 0.70 at 293 K. When temperature <212 K, P1 is the major product and the branching ratio order of products is: P1 > P1′ > P2. When temperature >212 K, P1′ is the major product and the branching ratio order of products is: P1′ > P1 > P2.
Variations of the reaction branching ratio of reaction channels at 760 Torr when temperature varies between 200~293 K are shown in Figure 6. When temperature >210 K, P1′ is the major product, with an increasing yield from 0.46 at 200 K to the peak (about 0.67) at 293 K. When temperature <210 K, P1 is the major product and its yield reduces gradually from 0.53 at 200 K to 0.31 at 293 K. The branching ratio of bimolecular product P2 declines gradually from 0.02 at 200 K to 0.01 at 293 K. The branching ratio of unimolecular product IM2 remains smaller than 0.001 within this temperature variation range, which could be neglected.
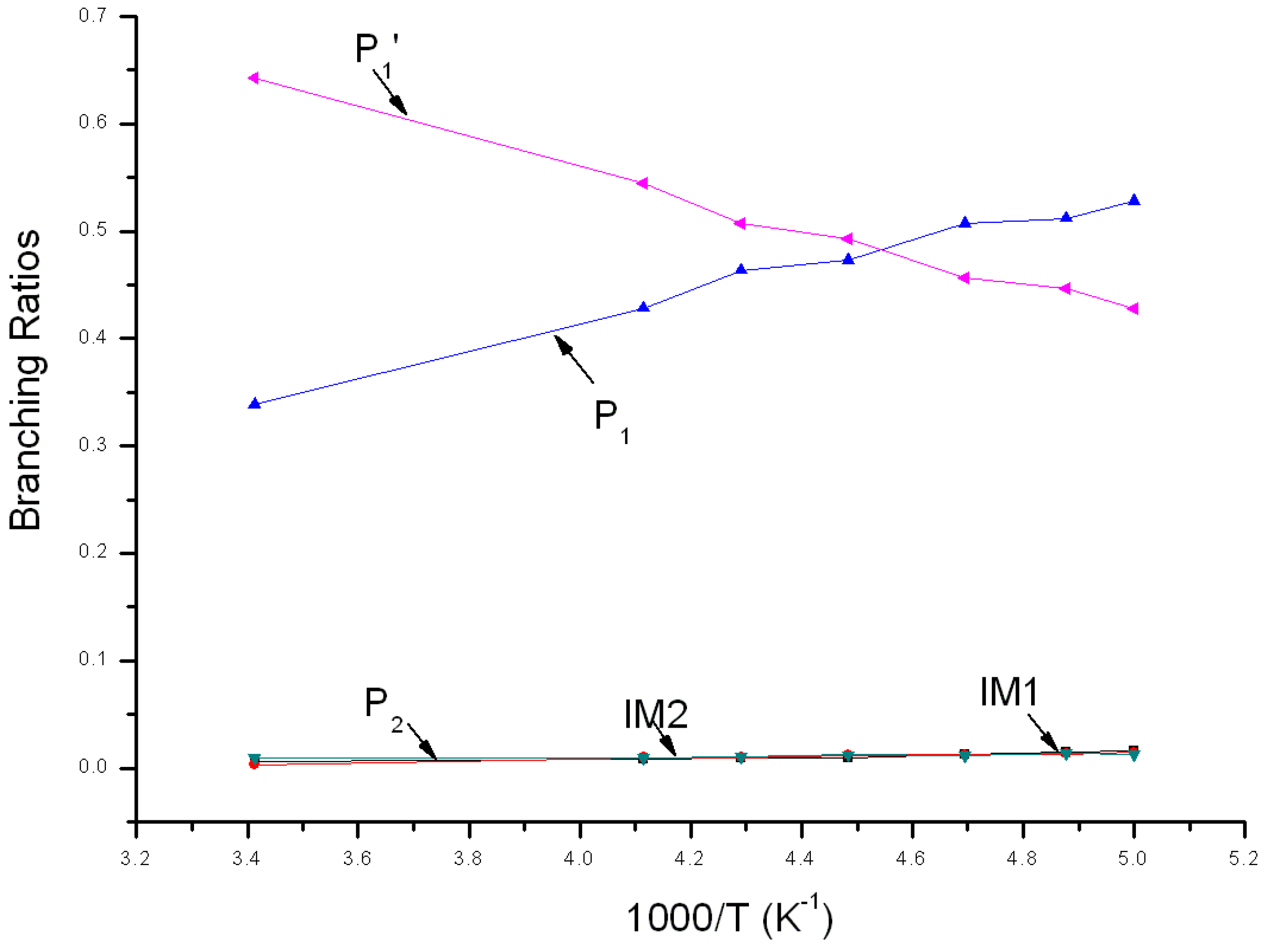
Variations of the reaction branching ratio of reaction channels at 7600 Torr when temperature varies between 200~293 K are shown in Figure 7. When temperature >220 K, P1′ is the major product and its yield increases continuously from 0.43 at 200 K to the peak (about 0.64) at 293 K. As temperature increases from 200 K to 293 K, branching ratios of both P1 and P2 decrease, from 0.53 to 0.34 and from 0.02 to 0.01, respectively. IM1 has some yield and its branching ratio decreases from 0.02 to 0.01. The yield of IM2 ≤ 0.01 and can be neglected within this temperature range. When temperature <220 K, the branching ratio order of products is: P1 > P1′ > IM1 > P2 ≈ IM2. Branching ratios of products at 200 K are: 0.53 > 0.43 > 0.02 > 0.01 ≈ 0.01.
4. Conclusions
On singlet potential energy surface, addition-dissociation is main mechanism of ClOO + NO reaction. Firstly, N atom in NO can attack Cl atom in ClOO to generate entrance intermediate IM1ClOONOtp with rich energy, influenced by no energy barrier. IM1 can transform into its cis isomer IM2 ClOONOcp easily. Viewed from IM1 and IM2, P1ClNO + 1O2 is the major product and P2 ClO + NO2 is the secondary product. On triplet potential energy surface, the channel that forms loose Van Der Waals complex 3IM1 3ONClOO and then overcome a small energy barrier to make Cl-abstraction reaction which produces P1′ClNO + 3O2 is the most important. The further kinetic calculation demonstrates that the total reaction rate constant (ktot) seems independent from pressure, but has a small negative temperature dependence. Within the studying pressure range, singlet biomolecular product P1 is the major product under low temperature, and triplet product fragment P1′ is the major product under high temperature. Monomolecular products IM1 and IM2 only have very small yields under low temperature and high pressure. The stabilization effect of intermediate at 213 K could be neglected. The yields of IM1 and IM2 remain smaller than 0.02. Within the studying pressure range, ClNO is the major product, which agrees with the experimental expectation.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (No. 11364027, 21363013, 11564031), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region, China (No. NJYT-17-B10) and the Scientific Apparatus Development of Special Item of Inner Mongolia University of Science and Technology, China (No. 201 5 KYYQ06).
Author Contributions
Nan-Nan Wu designed the research, performed the experimental work and wrote the manuscript. Nan-Nan Wu and Shun-Li Ou-Yang provided direction and contributed to the revisions of the manuscript. Liang Li read and approved the final manuscript.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- Finalyson-Pitts, B.J.; Pitts, J.N., Jr. Atmospheric Chemistry; Wiley Interscience: New York, NY, USA, 1986; Part 9. [Google Scholar]
- Rowland, F.S. Stratospheric Ozone Depletion. Phys. Chem. 1994, 32, 769–790. [Google Scholar]
- Sander, S.P.; Friedl, R.R.; Yung, Y.L. Quantum yield of chlorine-atom formation in the photodissociation of chlorine peroxide (ClOOCl) at 308 nm. Science 2013, 589, 52–55. [Google Scholar]
- Katz, A.; Narkis, N. Removal of chlorine dioxide disinfection by-products by ferrous salts. Water Res. 2001, 35, 101–108. [Google Scholar] [CrossRef]
- Hart, P.W.; Hsieh, J.S. Competing Parallel Reaction of Chlorine and Chlorine Dioxide during Kraft Pulp Bleaching. Chem. Eng. Commun. 1993, 126, 27–41. [Google Scholar] [CrossRef]
- Tsai, L.S.; Huxsoll, C.C.; Robertson, G.J. Prevention of Potato Spoilage during Storage by Chlorine Dioxide. Food Sci. 2001, 66, 472–477. [Google Scholar] [CrossRef]
- Ritter, S.K. Publication Ethics: Rights and Wrongs. Chem. Eng. News 2001, 79, 24–26. [Google Scholar] [CrossRef]
- Arkell, A.; Schwager, I. Matrix infrared study of the chlorine superoxide radical. J. Am. Chem. Soc. 1967, 89, 5999–6006. [Google Scholar] [CrossRef]
- Abramowitz, S.; Chase, M.W. Thermodynamic properties of gas phase species of importance to ozone depletion. Pure Appl. Chem. 1991, 63, 1449–1454. [Google Scholar] [CrossRef]
- Baer, S.; Hippler, H.; Rahn, R.; Siefke, M.; Seitzinger, N.; Troe, J. Thermodynamic and kinetic properties of the reaction Cl + O2 + M = ClOO + M in the range 160–300 K and 1–1000 bar. J. Chem. Phys. 1991, 95, 6463–6470. [Google Scholar] [CrossRef]
- Nicovich, J.M.; Krufter, K.D.; Shockolford, C.J.; Wine, P.H. Thermochemistry and kinetics of the Cl + O2 association reaction. Chem. Phys. Lett. 1991, 179, 367–373. [Google Scholar] [CrossRef]
- Mueller, H.S.P.; Willner, H. Vibrational and electronic spectra of chlorine dioxide, OClO, and chlorine superoxide ClOO, isolated in cryogenic matrixes. J. Phys. Chem. 1993, 97, 10589–10598. [Google Scholar] [CrossRef]
- Chase, M.W. NIST-JANAF Thermochemical Tables for the Bromine Oxides. J. Phys. Chem. Ref. Data 1996, 25, 1069–1111. [Google Scholar] [CrossRef]
- Wang, X.-B.; Wang, L.-S. The electronic structure and electron affinities of higher chlorine oxide radicals ClOx (x = 2–4) from photoelectron spectroscopy of ClO−x anions. J. Chem. Phys. 2000, 113, 10928–10933. [Google Scholar] [CrossRef]
- Distelrath, V.; Boesl, U. Mass selective gas phase study of ClO, OClO, ClOO and ClAr by anion-ZEKE-photoelectron spectroscopy. Faraday Discuss. 2000, 115, 161–174. [Google Scholar] [CrossRef]
- Kopitzky, R.; Grothe, H.; Willner, H. Chlorine Oxide Radicals ClOx (x = 1–4) Studied by Matrix Isolation Spectroscopy. Chem. Eur. J. 2002, 8, 5601–5621. [Google Scholar] [CrossRef]
- Elder, T. Reactions of Lignin Model Compounds with Chlorine Dioxide. Mol. Orbital Calc. Holzforsch. 1998, 52, 371–384. [Google Scholar] [CrossRef]
- Aloisio, S.; Francisco, J.S. Radical-Water Complexes in Earth’s Atmosphere. Acc. Chem. Res. 2000, 33, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Werner, H.J. Multireference configuration interaction calculations of the low-lying electronic states of ClO2. J. Chem. Phys. 1992, 96, 8948–8961. [Google Scholar] [CrossRef]
- Alcami, M.; Cooper, I.L. Ab initio calculations on bromine oxide and dioxides and their corresponding anions. J. Chem. Phys. 1998, 108, 9414–9424. [Google Scholar] [CrossRef]
- Alcami, M.; Mó, O.; Yáñez, M.; Cooper, I.L. The performance of density-functional theory in challenging cases: Halogen oxides. J. Chem. Phys. 2000, 112, 6131–6140. [Google Scholar] [CrossRef]
- Filatov, M.; Cremer, D. Bonding in the ClOO(2A’’) and BrOO(2A’’) radical: Nonrelativistic single-reference versus relativistic multi-reference descriptions in density functional theory. Phys. Chem. Chem. Phys. 2003, 5, 2320–2326. [Google Scholar] [CrossRef]
- Jafri, J.A.; Iii, B.H.L.; Bauschlicher, C.W., Jr.; Phillips, D.H. The lower electronic states of ClOO: A computational investigation. J. Chem. Phys. 1985, 83, 1693–1701. [Google Scholar] [CrossRef]
- Wood, S.C. Select the Right NOx Control Technology—Consider the Degree of Emission Reduction Needed, the Type of Fuel, Combustion Device Design, and Operational Factors. Chem. Eng. Prog. 1994, 90, 32–38. [Google Scholar]
- Brunet, S.M.K.; Guo, J.Z.; Carrington, T.; Filseth, S.V.; Sadowski, C.M. State-to-state and total rotational energy transfer rate constants for CN(B2Σ+, v = 0, N) + H2, CN(X2Σ+, v = 2, N) + H2, D2, and CN(X2Σ+, v = 3, N) + NO. J. Chem. Phys. 2002, 116, 3617. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Li, Z.-S.; Liu, J.-Y.; Sun, C.-C. Theoretical Study on Reaction Mechanism of the Cyanogen Radical with Nitrogen Dioxide. J. Phys. Chem. A 2005, 109, 10307–10313. [Google Scholar] [CrossRef] [PubMed]
- Baren, R.E.; Hershberger, J.F. Kinetics of the NCN Radical. J. Phys. Chem. A 2002, 106, 11093–11097. [Google Scholar] [CrossRef]
- Huang, C.-L.; Tseng, S.-Y.; Wang, T.-Y.; Wang, N.-S.; Xu, Z.-F.; Lin, M.-C. Reaction mechanism and kinetics of the NCN + NO reaction: Comparison of theory and experiment. J. Chem. Phys. 2005, 122. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-T.; Ho, J.-J. Theoretical Investigation of the Mechanisms of Reaction of NCN with NO and NS. J. Phys. Chem. A 2005, 109, 2564. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-G.; Li, Q.-S.; Zhang, S.-W.; Sun, Y.-B.; Sun, C.-C. Quantum mechanical studies on the potential energy surface of the reactions 3NCN/3CNN + NO, 1NNO + CN and 2N3 + CO. J. Mol. Struct. THEOCHEM 2005, 722, 139–146. [Google Scholar] [CrossRef]
- Jin, L.; Ding, Y.-H.; Wang, J.; Sun, C.-C. Reaction mechanism of the CCN radical with nitric oxide. J. Comput. Chem. 2006, 27, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Baren, R.E.; Hershberger, J.F. Kinetics of the NCS Radical. J. Phys. Chem. A 1999, 103, 11340–11344. [Google Scholar] [CrossRef]
- Wei, Z.-G.; Huang, X.-R.; Sun, Y.-B.; Sun, C.-C. On the Theoretical Study of the Potential Energy Surface for the C2H + NO2 Reaction. Chem. J. Chin. Univ. 2003, 24, 2224–2226. [Google Scholar]
- Chen, H.-L.; Wu, C.-W.; Ho, J.-J. Theoretical Investigation of the Mechanisms of Reactions of H2CN and H2SiN with NO. J. Phys. Chem. A 2006, 110, 8893–8900. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-L.; Ho, J.-J. Computational investigation of the reaction of NO with imine, silanimine, and its substituted derivatives. J. Mol. Struct. THEOCHEM 2006, 772, 93–102. [Google Scholar] [CrossRef]
- Enami, S.; Hoshino, Y.; Ito, Y.; Hashimoto, S.; Kawasaki, M. Kinetic Study of the ClOO + NO Reaction Using Cavity Ring-Down Spectroscopy. J. Phys. Chem. A 2006, 110, 3546–3551. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, M. Theoretical study of the mechanism for the ClOO + NO reaction on the singlet potential energy surface. Struct. Chem. 2012, 23, 107–114. [Google Scholar] [CrossRef]
- Ou-Yang, S.-L.; Wu, N.-N.; Liu, J.-Y.; Han, X.-G. Theoretical mechanism study on the reaction of FOO radical with NO. J. Chem. 2016, 2016. [Google Scholar] [CrossRef]
- Holbrook, K.A.; Pilling, M.J.; Robertson, S.H. Unimolecular Reactions; Wiley: Chichester, UK, 1972; Volume 15, pp. 357–362. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.v.R.; Pople, J. AB INITIO Molecular Orbital Theory; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1986; Volume 9, pp. 399–406. [Google Scholar]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 471–476. [Google Scholar] [CrossRef]
- Doubleday, C., Jr.; Page, M.; Mclver, J.W., Jr. Singlet biradicals as intermediates. Canonical variational transition-state theory results for trimethylene. J. Chem. Phys. 2002, 92, 4367–4371. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Frisch, M.J.; Truck, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. GAUSSIAN 03, Revision A.1; Guassian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Barker, J.R. Multiple-Well, multiple-path unimolecular reaction systems. I. MultiWell computer program suite. Int. J. Chem. Kinet. 2001, 33, 232–245. [Google Scholar] [CrossRef]
- Barker, J.R.; Ortiz, N.F.; Preses, J.M.; Lohr, L.L.; Maranzana, A.; Stimac, P.J. MultiWell-2011 Software; University of Michigan: Ann Arbor, MI, USA, 2007; Available online: http://aoss.engin.umich.edu/multiwell/ (accessed on 1 January 2011).
- Hellwege, K.H.; Hellwege, A.M. (Eds.) Landolt-Bornstein: Group II: Atomic and Molecular Physics Volume 7: Structure Data of Free Polyatomic Molecule; Springer: Berlin/Heidelberg, Germany, 1976. [Google Scholar]
- Huber, K.P.; Herzberg, G. Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules; Van Nostrand Reinhold Co.: New York, NY, USA, 1979. [Google Scholar]
- Herzberg, G. Electronic Spectra and Electronic Structure of Polyatomic Molecules; Van Nostrand: New York, NY, USA, 1966; pp. 273–283. [Google Scholar]
- Irikura, K.K. Experimental Vibrational Zero-Point Energies: Diatomic Molecules. J. Phys. Chem. Ref. Data 2007, 36, 389–397. [Google Scholar] [CrossRef]
- Klippenstein, S.J. An Efficient Procedure for Evaluating the Number of Available States within a Variably Defined Reaction Coordinate Framework. J. Phys. Chem. 1994, 98, 11459–11464. [Google Scholar] [CrossRef]
- Klippenstein, S.J. A bond length reaction coordinate for unimolecular reactions. II. Microcanonical and canonical implementations with application to the dissociation of NCNO. J. Chem. Phys. 1991, 94, 6469–6482. [Google Scholar] [CrossRef]
- Francisco, J.S. An ab initio investigation of the significance of the HOOF intermediate in coupling reactions involving FOOx and HOx species. J. Chem. Phys. 1993, 98, 2198–2207. [Google Scholar] [CrossRef]
- Francisco, J.S.; Su, Y. An ab initio study of the reaction between FO radicals and H2O. Chem. Phys. Lett. 1993, 215, 58–62. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
MP2/6-311 + G(2df) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of ClOO + NO reaction. The values in parentheses are the pertinent experimental data from the literature [48,49,50,51] and (a–d) represent refs [48,49,50,51], respectively. Bond lengths are in Å and bond angles are in degree.
Figure 1.
MP2/6-311 + G(2df) optimized geometries for the reactants, products, intermediates (IM) and the corresponding transition states (TS) of ClOO + NO reaction. The values in parentheses are the pertinent experimental data from the literature [48,49,50,51] and (a–d) represent refs [48,49,50,51], respectively. Bond lengths are in Å and bond angles are in degree.
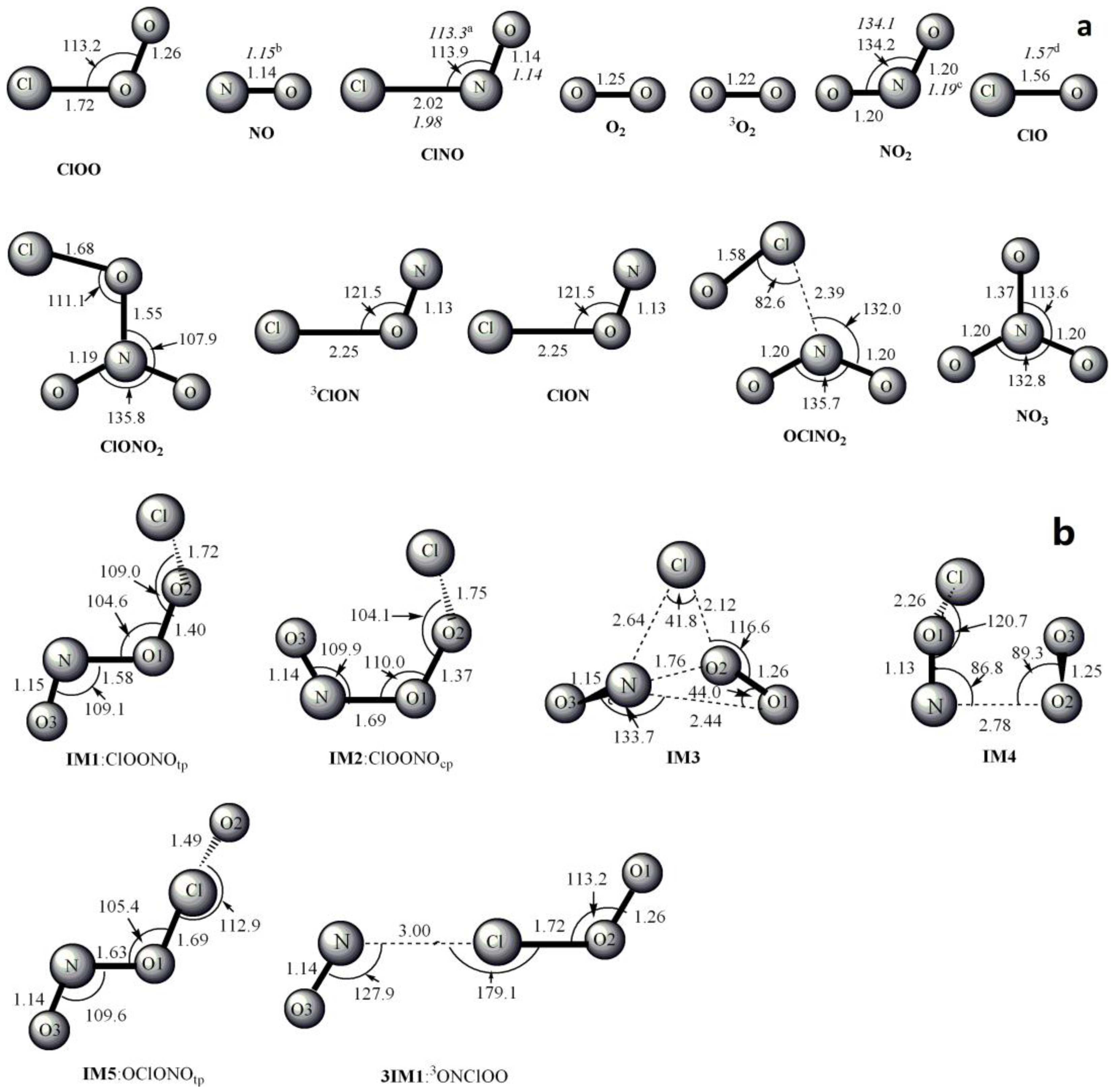
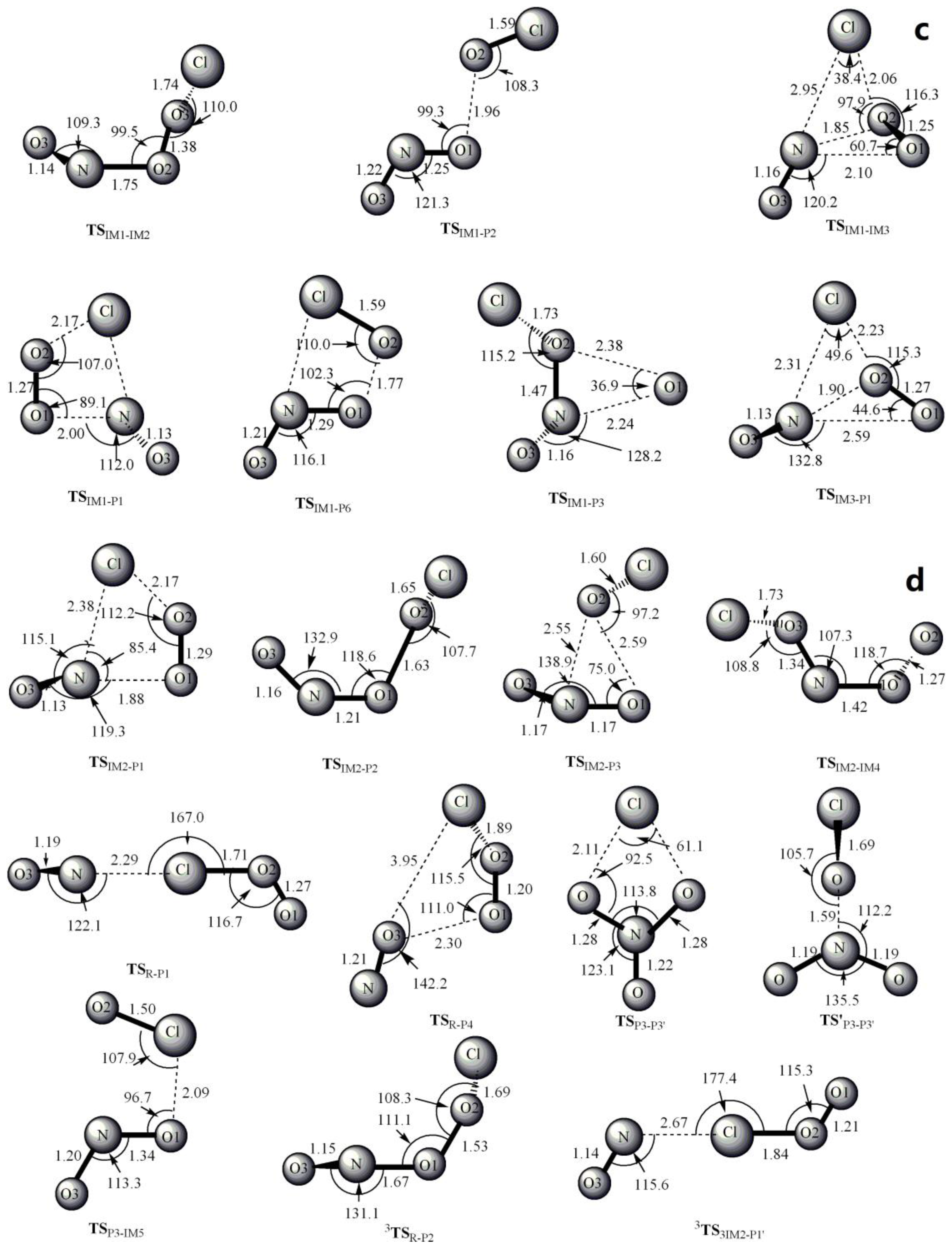
Figure 2.
(a) Schematic singlet potential energy surfaces of each product channels for ClOO + NO reaction at the CCSD(T)/6-311 + G(2df)//MP2/6-311 + G(2df) + ZPE level. TSIM1-P3 is obtained at the CCSD (T)/6-311 + G(2df)//MP2/6-311G(d) + ZPE level, and TSIM2-P3 is obtained at the CCSD (T)/6-311 + G(2df)//B3LYP/6-311 + G(2df) + ZPE level; (b) Schematic triplet potential energy surfaces of each product channels for ClOO + NO reaction at the CCSD(T)/6-311 + G(2df)//MP2/6-311 + G(2df) + ZPE level.
Figure 2.
(a) Schematic singlet potential energy surfaces of each product channels for ClOO + NO reaction at the CCSD(T)/6-311 + G(2df)//MP2/6-311 + G(2df) + ZPE level. TSIM1-P3 is obtained at the CCSD (T)/6-311 + G(2df)//MP2/6-311G(d) + ZPE level, and TSIM2-P3 is obtained at the CCSD (T)/6-311 + G(2df)//B3LYP/6-311 + G(2df) + ZPE level; (b) Schematic triplet potential energy surfaces of each product channels for ClOO + NO reaction at the CCSD(T)/6-311 + G(2df)//MP2/6-311 + G(2df) + ZPE level.
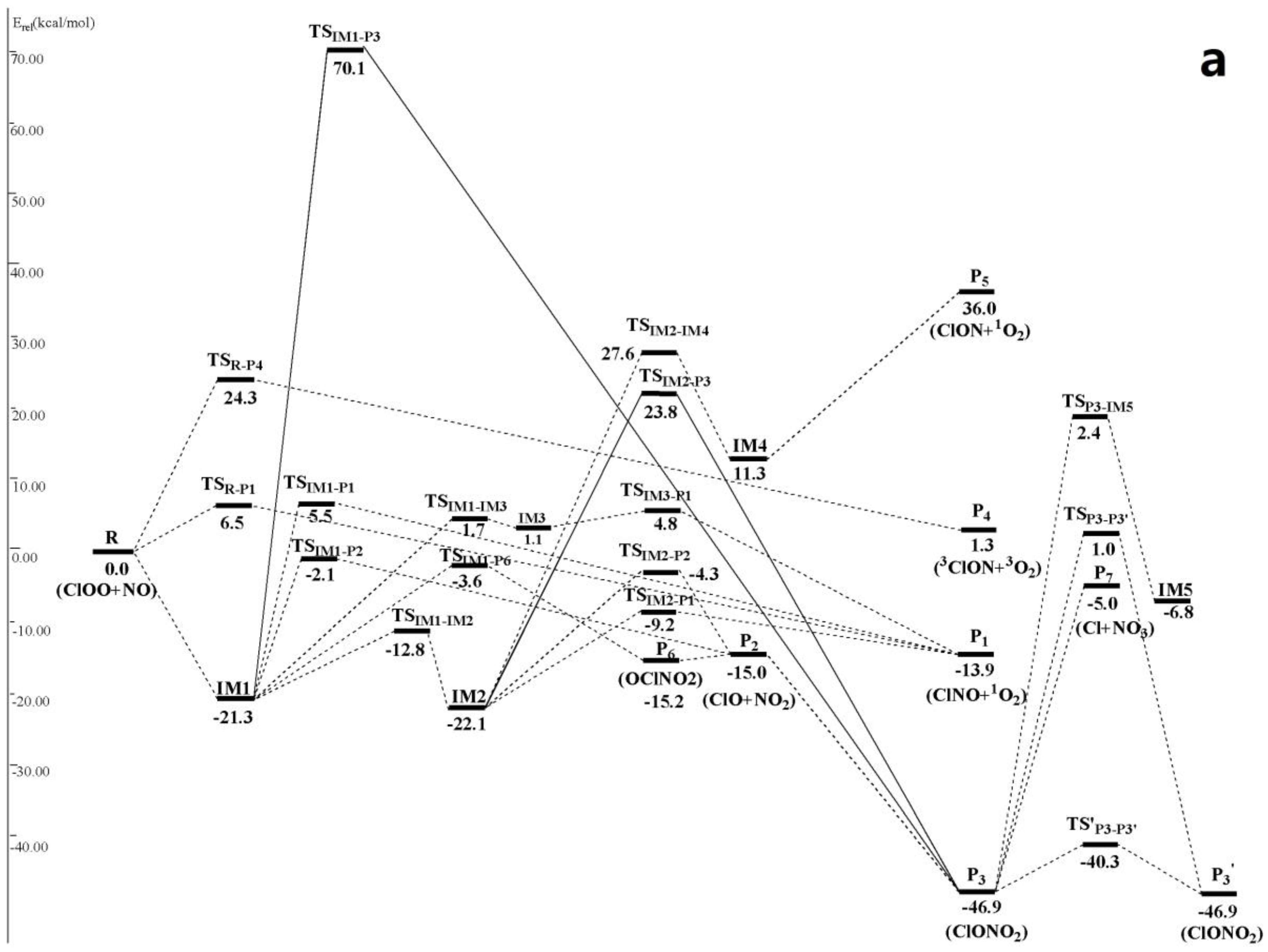
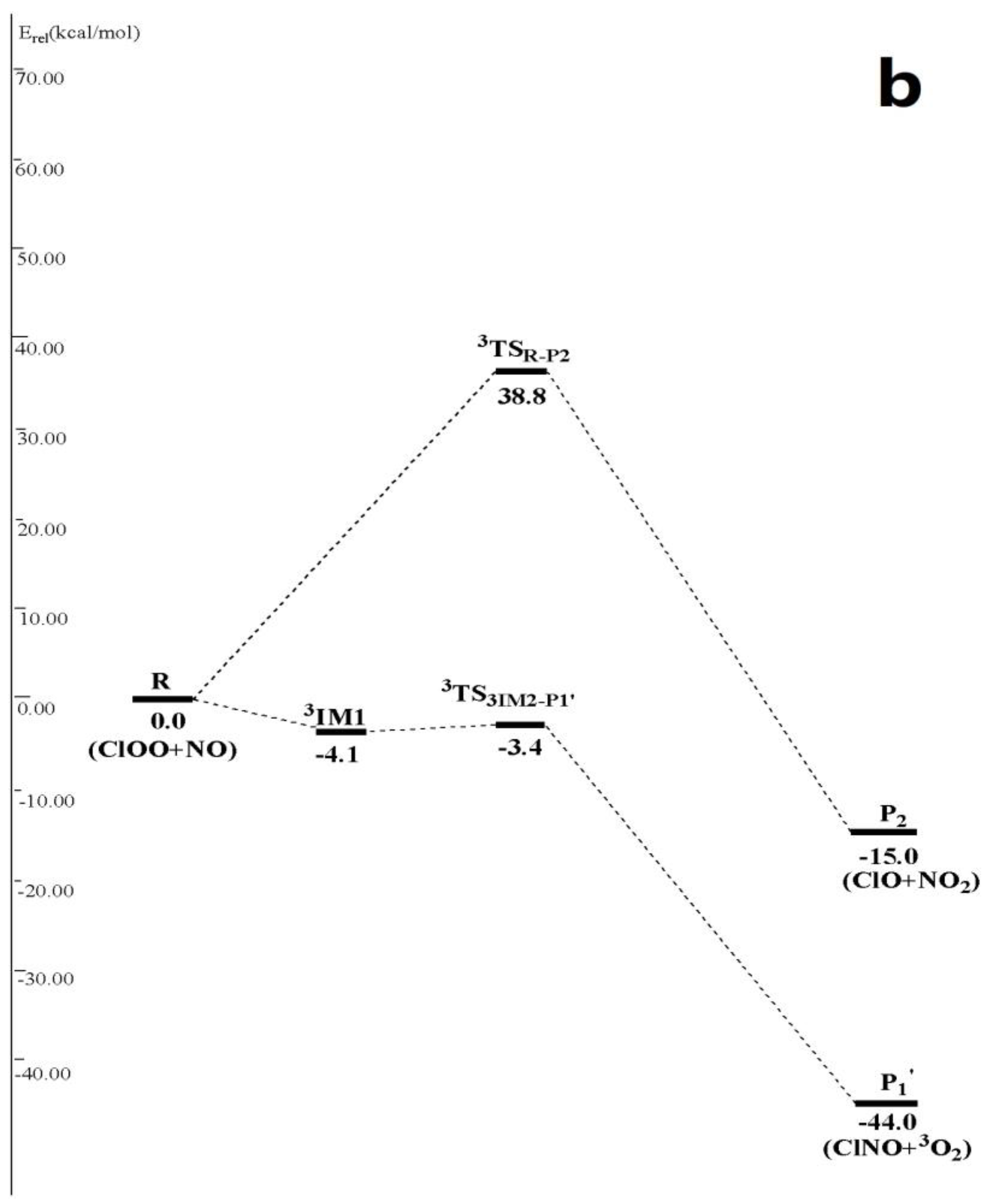
Figure 3.
The total reaction rate constant (ktot) at various pressures of 1, 50, 150, 760 and 7600 Torr in a temperature range of 200~293 K.
Figure 3.
The total reaction rate constant (ktot) at various pressures of 1, 50, 150, 760 and 7600 Torr in a temperature range of 200~293 K.
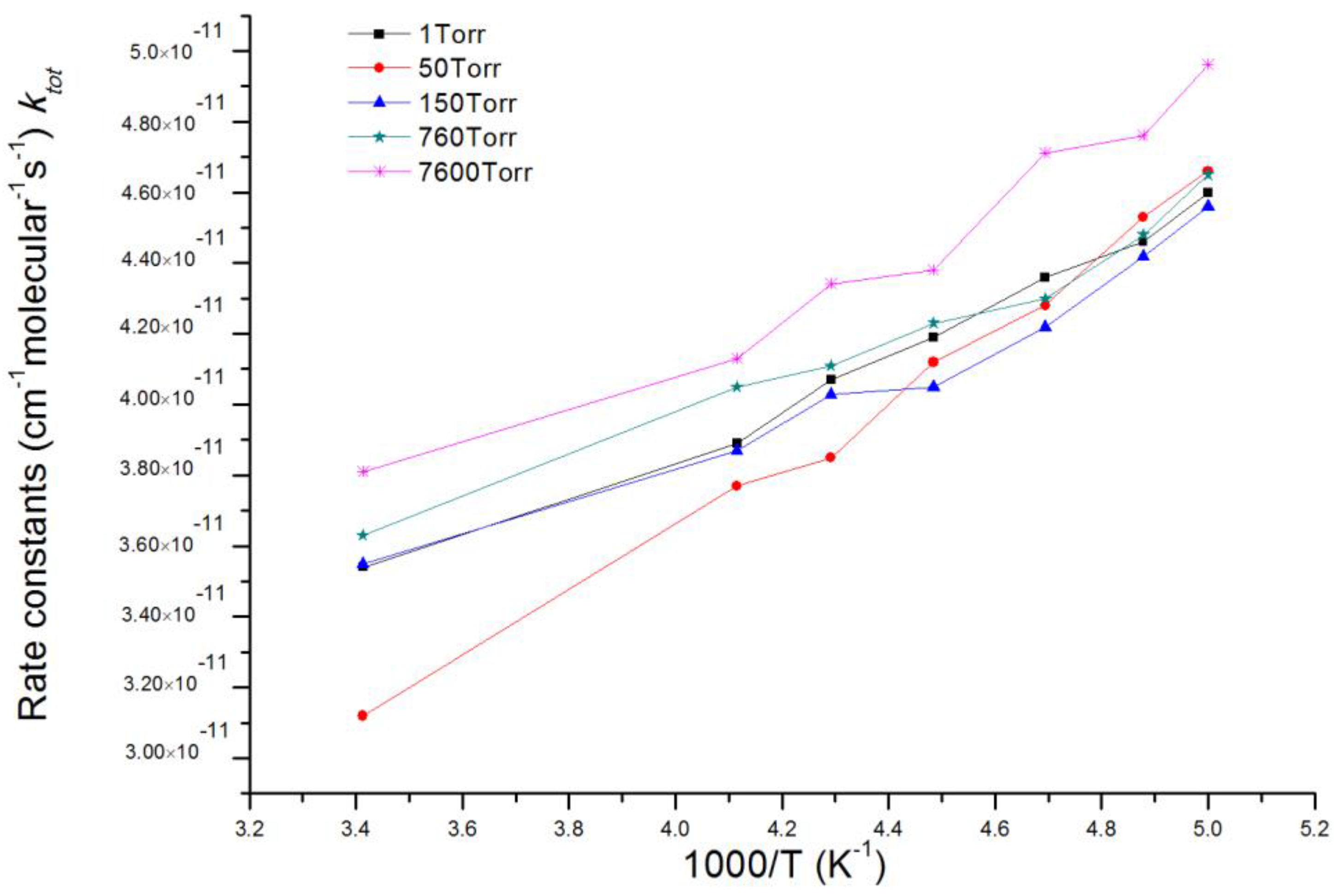
Figure 4.
Branching ratios of various reaction channels at 213 K in a pressure range from 1 Torr to 7600 Torr.
Figure 4.
Branching ratios of various reaction channels at 213 K in a pressure range from 1 Torr to 7600 Torr.
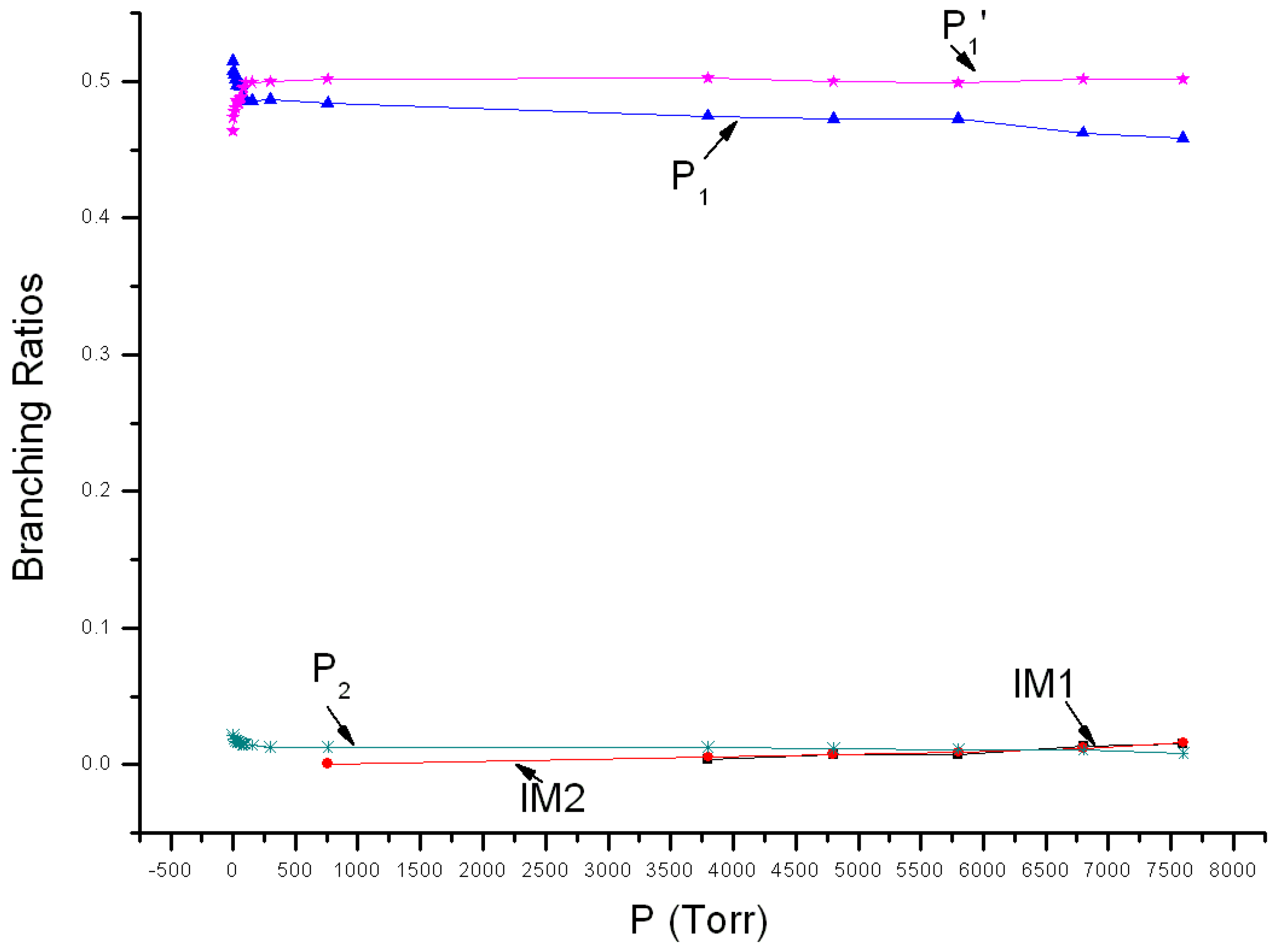
Figure 5.
Branching ratios of various reaction channels at 1 Torr in a temperature range from 200 K to 293 K.
Figure 5.
Branching ratios of various reaction channels at 1 Torr in a temperature range from 200 K to 293 K.
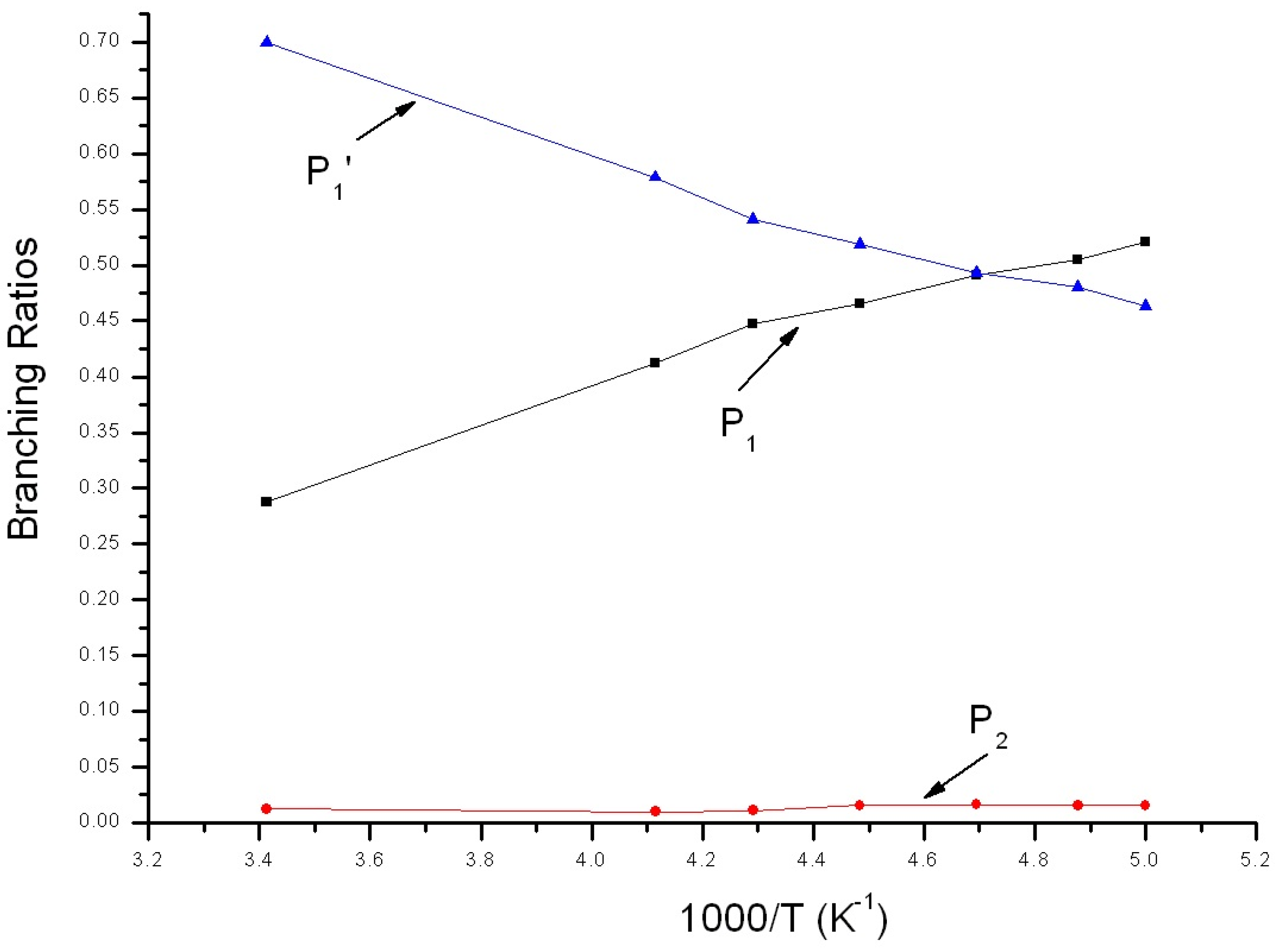
Figure 6.
Branching ratios of various reaction channels at 760 Torr in a temperature range from 200 K to 293 K.
Figure 6.
Branching ratios of various reaction channels at 760 Torr in a temperature range from 200 K to 293 K.
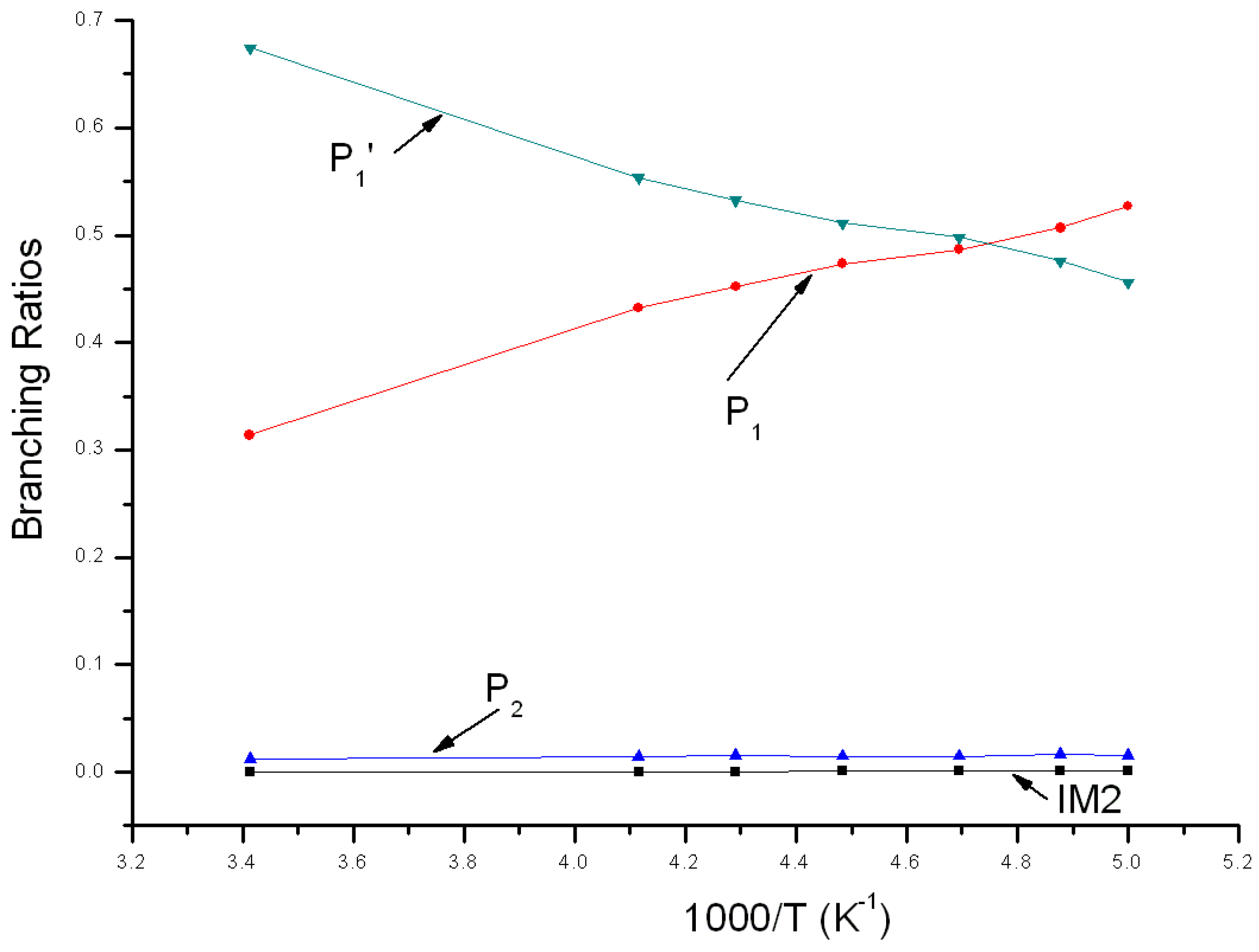
Figure 7.
Branching ratios of various reaction channels at 7600 Torr in a temperature range from 200 K to 293 K.
Figure 7.
Branching ratios of various reaction channels at 7600 Torr in a temperature range from 200 K to 293 K.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
At 150 Torr, experimental data and the theoretical calculation results of the total reaction rate constant (ktot).
Table 1.
At 150 Torr, experimental data and the theoretical calculation results of the total reaction rate constant (ktot).
| T(K)/ktot (×10−11 cm3 mol−1 s−1) | 205 | 213 | 223 | 233 | 243 |
|---|---|---|---|---|---|
| Experimental data a | 4.3 ± 0.8 | 4.5 ± 0.9 | 4.2 ± 0.8 | 4.9 ± 1.0 | 5.5 ± 1.2 |
| Theoretical calculation results (RRKM) | 4.4 | 4.2 | 4.1 | 4.0 | 3.9 |
a represent refs. [36].
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, N.-N.; Ou-Yang, S.-L.; Li, L. Theoretical Study of ClOO + NO Reaction: Mechanism and Kinetics. Molecules 2017, 22, 2121. https://doi.org/10.3390/molecules22122121
AMA Style
Wu N-N, Ou-Yang S-L, Li L. Theoretical Study of ClOO + NO Reaction: Mechanism and Kinetics. Molecules. 2017; 22(12):2121. https://doi.org/10.3390/molecules22122121
Chicago/Turabian StyleWu, Nan-Nan, Shun-Li Ou-Yang, and Liang Li. 2017. "Theoretical Study of ClOO + NO Reaction: Mechanism and Kinetics" Molecules 22, no. 12: 2121. https://doi.org/10.3390/molecules22122121