A Novel Strategy for Quantitative Analysis of Major Ginsenosides in Panacis Japonici Rhizoma with a Standardized Reference Fraction

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Preparation of TPJS
2.2. Sample Preparation
2.3. Optimization of the HPLC Conditions
2.4. Method Validaton
2.5. Quantification and Method Assessment
3. Experimental
3.1. General
3.2. Preparation of Total P. japonicus Saponin
3.3. Preparation of Standard Solutions
3.4. Preparation of Sample Solutions
3.5. HPLC Conditions
3.6. Calibration Curves, and Limits of Detection and Quantification
3.7. Precision, Repeatability, Stability, and Accuracy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- National Pharmacopeia Commission. Pharmacopoeia of the People's Republic of China, Part I; China Medical Science Press: Beijing, China, 2015; pp. 138–139. [Google Scholar]
- Li, S.P.; Qiao, C.F.; Chen, Y.W.; Zhao, J.; Cui, X.M.; Zhang, Q.W.; Liu, X.M.; Hu, D.J. A novel strategy with standardized reference extract qualification and single compound quantitative evaluation for quality control of Panax notoginseng used as a functional food. J. Chromatogr. A 2013, 1313, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.B.; Li, P.; Yang, R.L.; Zhang, Q.W.; Wang, Y.T. Separation and purification of 5 saponins from Panax notoginseng by Preparative High-performance Liquid Chromatography. J. Liquid Chromatogr. Relat. Technol. 2013, 36, 406–417. [Google Scholar]
- Wan, J.B.; Zhang, Q.W.; Hong, S.J.; Guan, J.; Ye, W.C.; Li, S.P.; Lee, M.Y.S.; Wang, Y.T. 5,6-Didehydroginsenosides from the Roots of Panax notoginseng. Molecules 2010, 15, 8169–8176. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Z.; Hu, Y.; Wu, W.Y.; Ye, M.; Guo, D.A. Saponins in the genus Panax L. (Araliaceae): A systematic review of their chemical diversity. Phytochemistry 2014, 106, 7–24. [Google Scholar] [PubMed]
- Yang, W.Z.; Ye, M.; Qiao, X.; Liu, C.F.; Miao, W.J.; Bo, T.; Tao, H.Y.; Guo, D.A. A strategy for efficient discovery of new natural compounds by integrating orthogonal column chromatography and liquid chromatography/mass spectrometry analysis: Its application in Panax ginseng, Panax quinquefolium and Panax notoginseng to characterize 437 potential new ginsenosides. Anal. Chim. Acta 2012, 739, 56–66. [Google Scholar] [PubMed]
- Yuan, C.; Xu, F.X.; Huang, X.J.; Li, S.P.; Zhang, Q.W. A novel 12, 23-epoxy dammarane saponin from Panax notoginseng. Chin. J. Nat. Med. 2015, 13, 303–306. [Google Scholar] [PubMed]
- Mancuso, C.; Santangelo, R. Panax ginseng and Panax quinquefolius: From pharmacology to toxicology. Food Chem. Toxicol. 2017, 107, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.R.; Hong, S.J.; Lee, S.M.Y.; Cong, W.H.; Wan, J.B.; Zhang, Z.R.; Zhang, Q.W.; Zhang, Y.; Wang, Y.T.; Lin, Z.X. Pro-angiogenic activity of notoginsenoside R1 in human umbilical vein endothelial cells in vitro and in a chemical-induced blood vessel loss model of zebrafish in vivo. Chin. J. Integr. Med. 2016, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Q.S.; Meng, F.C.; Tang, Z.H.; Chen, X.P.; Lin, L.G.; Chen, P.; Qiang, W.A.; Wang, Y.T.; Zhang, Q.W.; et al. Chikusetsusaponin IVa methyl ester induces G1 cell cycle arrest, triggers apoptosis and inhibits migration and invasion in ovarian cancer cells. Phytomedicine 2016, 23, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.M.; Wan, J.B.; Zhang, Q.W.; Li, S.P. Simultaneous determination of nucleobases, nucleosides and saponins in Panax notoginseng using multiple columns high performance liquid chromatography. J. Pharm. Biomed. Anal. 2008, 48, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.B.; Zhang, Q.W.; Hong, S.J.; Li, P.; Li, S.P.; Wang, Y.T. Chemical investigation of saponins in different parts of Panax notoginseng by pressurized liquid extraction and liquid chromatography-electrospray ionization-tandem mass spectrometry. Molecules 2012, 17, 5836–5853. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.B.; Zhang, Q.W.; Ye, W.C.; Wang, Y.T. Quantification and separation of protopanaxatriol and protopanaxadiol type saponins from Panax notoginseng with macroporous resins. Sep. Purif. Technol. 2008, 60, 198–205. [Google Scholar] [CrossRef]
- Wang, Y.; Choi, H.K.; Brinckmann, J.A.; Jiang, X.; Huang, L. Chemical analysis of Panax quinquefolius (North American ginseng): A review. J. Chromatogr. A 2015, 1426, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.S.; Wang, C.M.; Lu, J.J.; Lin, L.G.; Chen, P.; Zhang, Q.W. Simultaneous determination of six saponins in Panacis Japonici Rhizoma using quantitative analysis of multi-components with single-marker method. Curr. Pharm. Anal. 2017, 13, 289–295. [Google Scholar] [CrossRef]
- Wang, C.Q.; Jia, X.H.; Chen, J.; Xiao, X.Y.; Wang, X.; Cai, S.Q. Systematic study on QAMS method for quality control of Panax notoginseng. Zhongguo Zhongyao Zazhi 2012, 37, 3438–3445. [Google Scholar] [PubMed]
- Zhao, J.; Ma, S.C.; Li, S.P. Advanced strategies for quality control of Chinese medicines. J. Pharm. Biomed. Anal. 2018, 147, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Wu, X.; Li, L.; Sun, X.; Xiang, D. Optimization of separation and purification of total saponin from Panacis Japonici Rhizoma by macroporous resins. Zhongchengyao 2011, 33, 1163–1168. [Google Scholar]
- Wang, X.; Wang, L.; Liu, S.; Yang, L. Purification of total saponins of Panax japonicus. Yingyong Huagong 2012, 41, 781–783. [Google Scholar]
- Li, H.; Lee, J.H.; Ha, J.M. Effective purification of ginsenosides from cultured wild ginseng roots, red ginseng, and white ginseng with macroporous resins. J. Microbiol. Biotechnol. 2008, 18, 1789–1791. [Google Scholar] [PubMed]
- Mi, J.; Zhang, M.; Ren, G.; Zhang, H.; Wang, Y.; Hu, P. Enriched separation of protopanaxatriol ginsenosides, malonyl ginsenosides and protopanaxadiol ginsenosides from Panax ginseng using macroporous resins. J. Food Eng. 2012, 113, 577–588. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, J.; Gu, S.; Lin, L.; Chen, Y.; Ma, M.; Chen, B.; Liu, J. Orthogonal strategy development using reversed macroporous resin coupled with hydrophilic interaction liquid chromatography for the separation of ginsenosides from ginseng root extract. J. Sep. Sci. 2017, 40, 4128–4134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J.; Wang, Z.M.; Ma, X.Y.; Feng, W.H.; Zhang, Q.W. A Quantitative Method for simultaneous determination of four anthraquinones with one marker in Rhei Radix et Rhizoma. Chin. Herb. Med. 2012, 4, 157–163. [Google Scholar]
- Zhu, C.; Li, X.; Zhang, B.; Lin, Z. Quantitative analysis of multi-components by single marker—A rational method for the internal quality of Chinese herbal medicine. Integr. Med. Res. 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
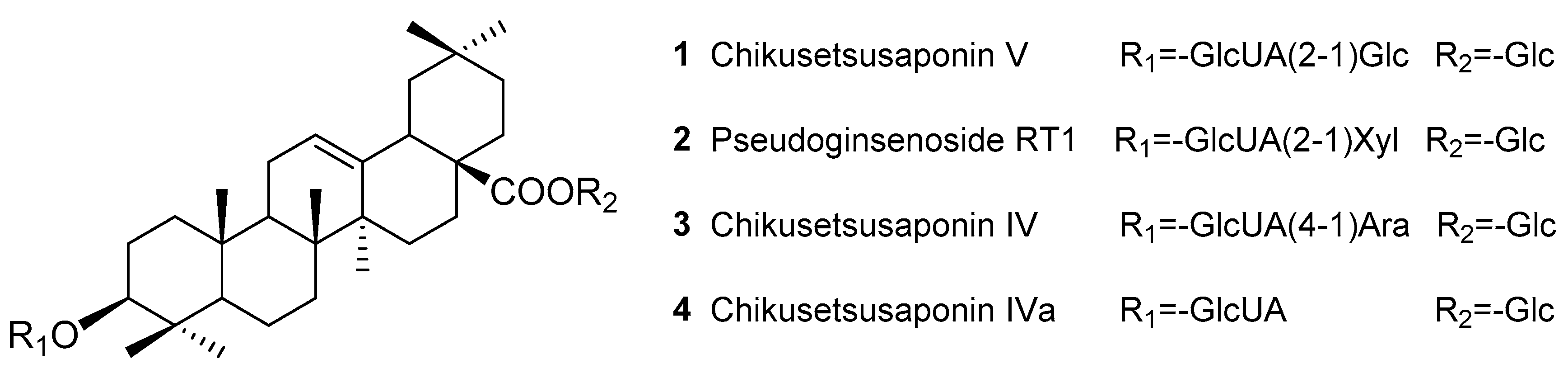
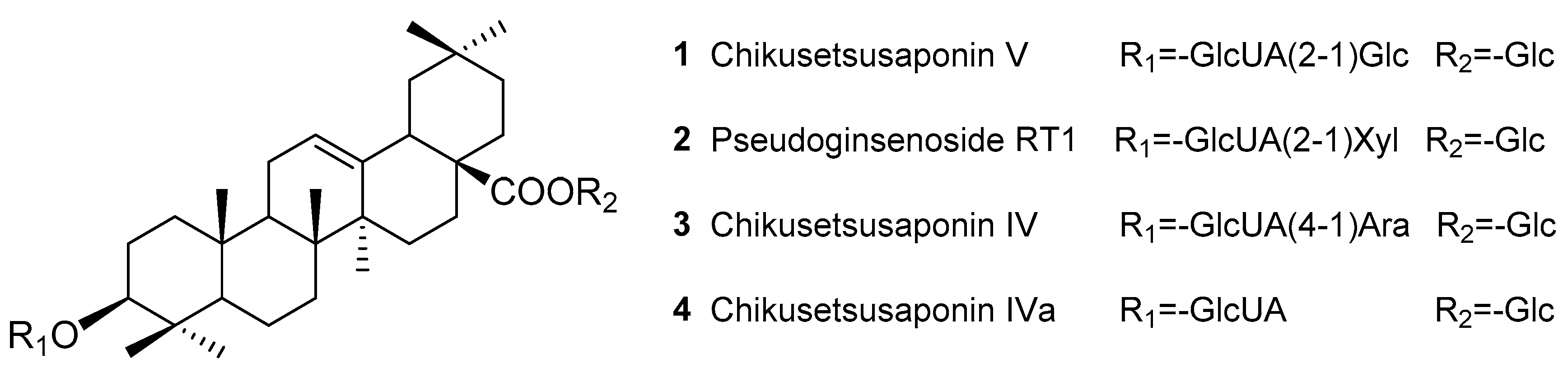
Sample Availability: Samples of the compounds Chikusetsusaponins IV, IVa, V, pseudoginsenoside RT1 and TPJS are available from the authors. |


{kind=link}
{kind=link}
| Method | Analytes | Regression Equation | R² | Linear Range (mg/mL) | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|---|---|
| M1 | CS-V | y = 5026.2x + 15.32 | 0.9998 | 0.004–1.025 | 0.7 | 2.9 |
| PG-RT1 | y = 5303.1x + 30.28 | 0.9998 | 0.004–1.010 | 0.5 | 1.9 | |
| CS-IV | y = 5157.5x + 30.64 | 0.9996 | 0.004–1.030 | 1.0 | 3.9 | |
| CS-IVa | y = 5140x + 14.70 | 0.9998 | 0.004–1.045 | 0.8 | 3.3 | |
| M2 | CS-V | y = 5193.6x + 42.02 | 0.9993 | 0.036–1.166 | 1.5 | 6.0 |
| PG-RT1 | y = 5233.9x + 34.39 | 0.9992 | 0.011–0.676 | 0.5 | 1.9 | |
| CS-IV | y = 5301.8x + 11.44 | 0.9999 | 0.024–0.793 | 1.0 | 3.9 | |
| CS-IVa | y = 5020x + 47.29 | 0.9993 | 0.020–1.286 | 0.7 | 2.8 |
| Method | Analytes | Recovery (%, RSD, n = 6) | Precision (%, RSD, n = 6) Intra-Day Inter-Day | Repeatability (%, RSD, n = 6) | Stability (%, RSD, n = 6) | |
|---|---|---|---|---|---|---|
| M1 | CS-V | 100.70, 1.11 | 1.69 | 0.95 | 0.94 | 0.58 |
| PG-RT1 | 98.15, 1.36 | 1.41 | 1.06 | 0.28 | 0.49 | |
| CS-IV | 102.26, 1.12 | 1.94 | 1.43 | 1.04 | 0.48 | |
| CS-IVa | 100.29, 2.35 | 2.53 | 1.06 | 1.47 | 0.54 | |
| M2 | CS-V | 98.66, 0.48 | 0.23 | 0.12 | 0.18 | 0.58 |
| PG-RT1 | 100.85, 0.69 | 0.72 | 1.33 | 0.57 | 0.49 | |
| CS-IV | 96.65, 0.41 | 0.40 | 0.55 | 0.34 | 0.48 | |
| CS-IVa | 103.33, 0.81 | 0.30 | 0.71 | 0.30 | 0.54 | |
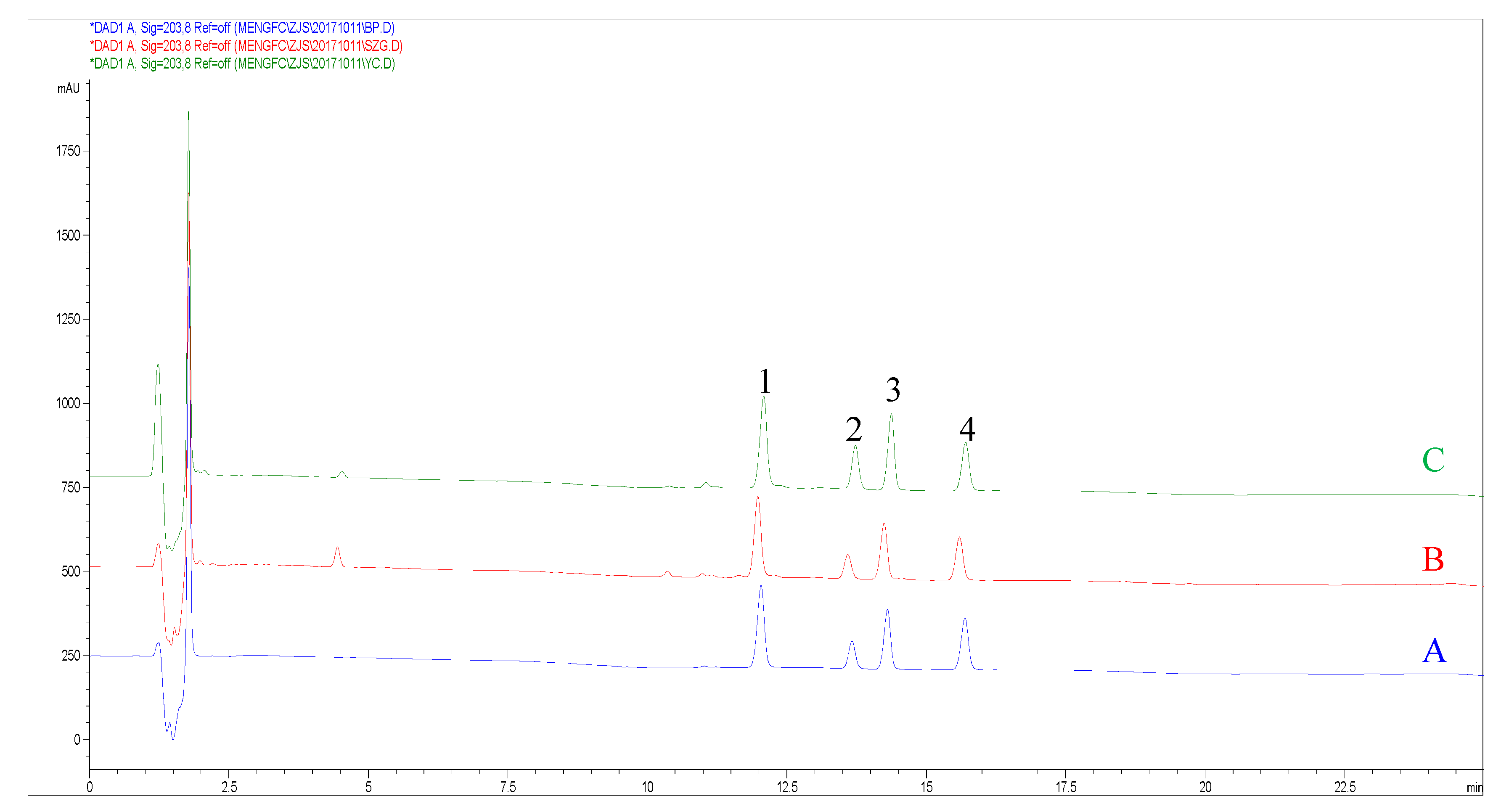
| No. | CS-V | PG-RT1 | CS-IV | CS-IVa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1 | M2 | PD % | M1 | M2 | PD % | M1 | M2 | PD % | M1 | M2 | PD % | |
| 1 | 6.40 | 6.35 | 0.78 | 2.88 | 2.91 | 1.03 | 4.85 | 4.91 | 1.23 | 3.34 | 3.39 | 1.49 |
| 2 | 5.27 | 5.21 | 1.15 | 3.12 | 3.17 | 1.59 | 4.08 | 4.13 | 1.22 | 1.48 | 1.49 | 0.67 |
| 3 | 8.92 | 8.91 | 0.11 | 2.45 | 2.48 | 1.21 | 5.22 | 5.30 | 1.52 | 4.57 | 4.65 | 1.74 |
| 4 | 11.33 | 11.35 | 0.18 | 2.21 | 2.24 | 1.34 | 1.87 | 1.87 | 0.00 | 7.52 | 7.68 | 2.11 |
| 5 | 8.74 | 8.72 | 0.22 | 3.87 | 3.93 | 1.54 | 4.03 | 4.07 | 0.99 | 7.07 | 7.22 | 2.10 |
| 6 | 5.81 | 5.76 | 0.86 | 0.94 | 0.95 | 1.05 | 2.56 | 2.57 | 0.39 | 3.14 | 3.18 | 1.27 |
| 7 | 4.19 | 4.12 | 1.70 | 2.17 | 2.19 | 0.91 | 4.42 | 4.48 | 1.35 | 2.47 | 2.49 | 0.81 |
| 8 | 8.41 | 8.40 | 0.12 | 2.66 | 2.70 | 0.0149 | 4.47 | 4.52 | 1.11 | 5.43 | 5.53 | 1.82 |
| 9 | 8.59 | 8.57 | 0.23 | 0.27 | 0.27 | 0.00 | 7.52 | 7.65 | 1.01 | 1.50 | 1.50 | 0.00 |
| 10 | 11.28 | 11.31 | 0.27 | 2.37 | 2.40 | 1.26 | 6.04 | 6.13 | 1.48 | 4.55 | 4.63 | 1.74 |
| 11 | 8.77 | 8.76 | 0.11 | 0.63 | 0.62 | 1.60 | 5.09 | 5.16 | 1.37 | 1.75 | 1.75 | 0.00 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, F.-C.; Wu, Q.-S.; Wang, R.; Li, S.-P.; Lin, L.-G.; Chen, P.; Zhang, Q.-W. A Novel Strategy for Quantitative Analysis of Major Ginsenosides in Panacis Japonici Rhizoma with a Standardized Reference Fraction. Molecules 2017, 22, 2067. https://doi.org/10.3390/molecules22122067
Meng F-C, Wu Q-S, Wang R, Li S-P, Lin L-G, Chen P, Zhang Q-W. A Novel Strategy for Quantitative Analysis of Major Ginsenosides in Panacis Japonici Rhizoma with a Standardized Reference Fraction. Molecules. 2017; 22(12):2067. https://doi.org/10.3390/molecules22122067
Chicago/Turabian StyleMeng, Fan-Cheng, Qiu-Shuang Wu, Ruibing Wang, Shao-Ping Li, Li-Gen Lin, Ping Chen, and Qing-Wen Zhang. 2017. "A Novel Strategy for Quantitative Analysis of Major Ginsenosides in Panacis Japonici Rhizoma with a Standardized Reference Fraction" Molecules 22, no. 12: 2067. https://doi.org/10.3390/molecules22122067