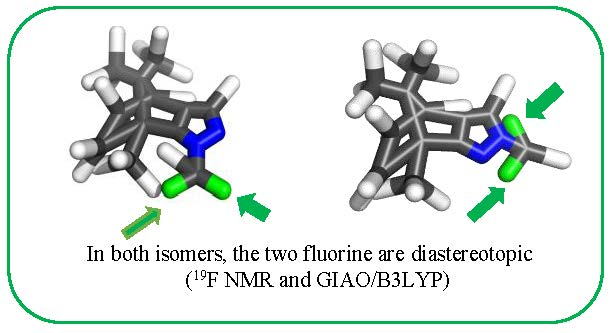
19F‐NMR Diastereotopic Signals in Two N-CHF2 Derivatives of (4S,7R)-7,8,8-Trimethyl-4,5,6,7-tetrahydro-4,7-methano-2H-indazole †

 , ,
, ,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
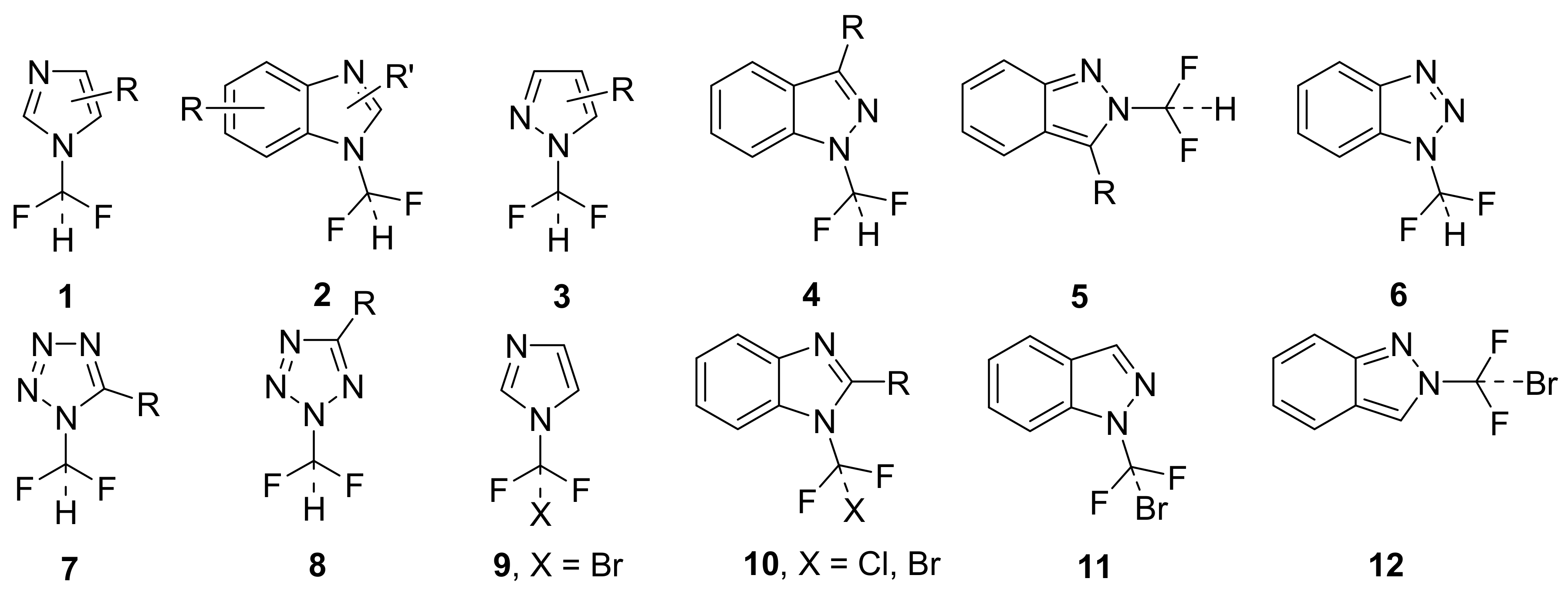
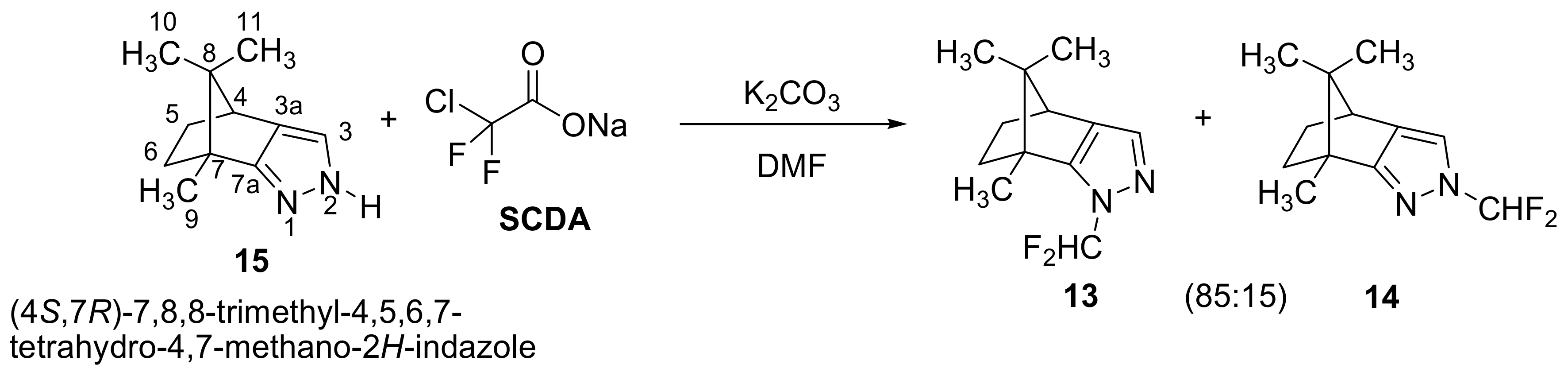
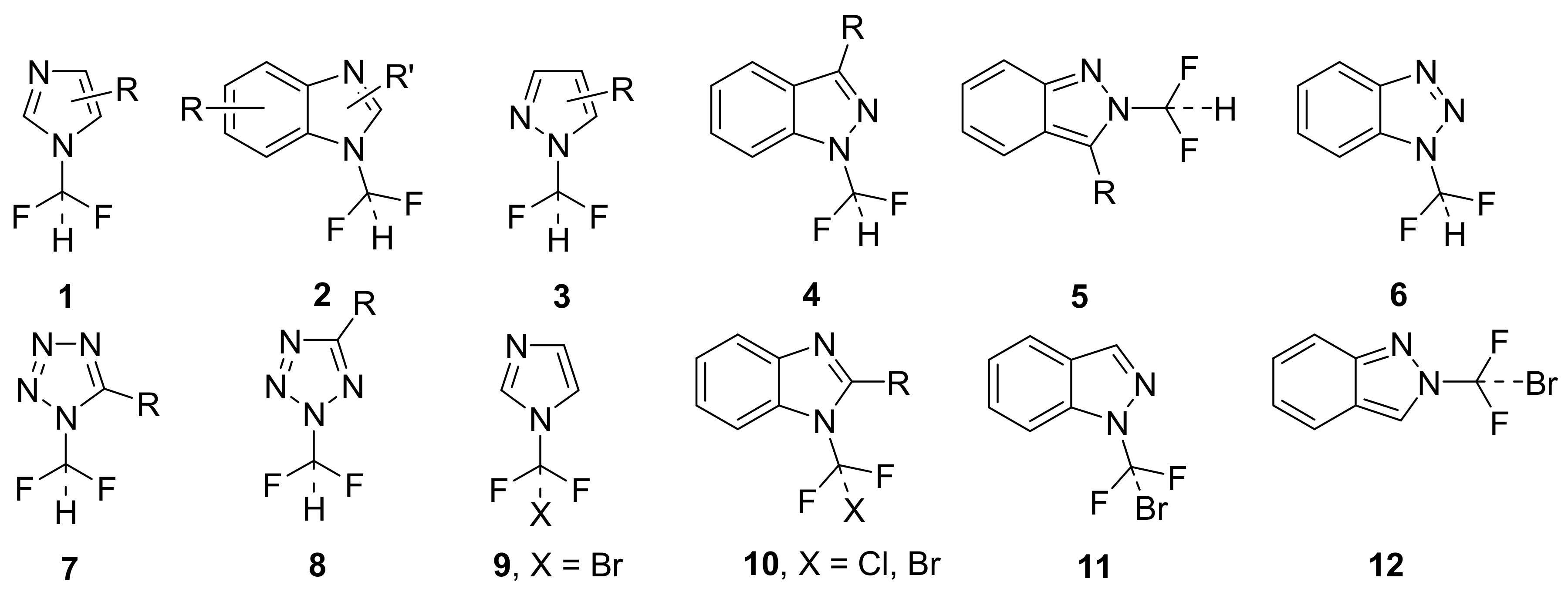
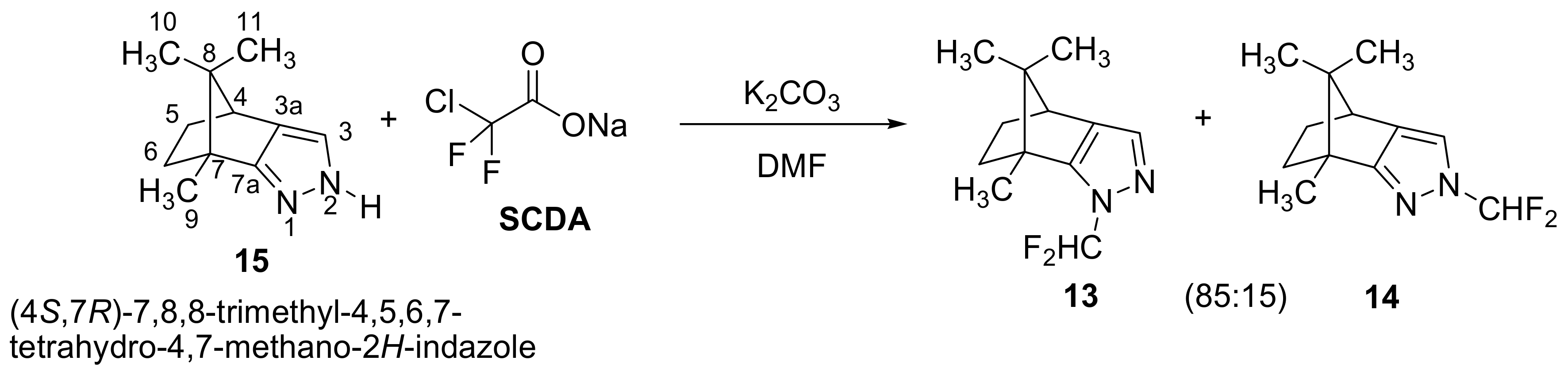
2.1. Chemistry
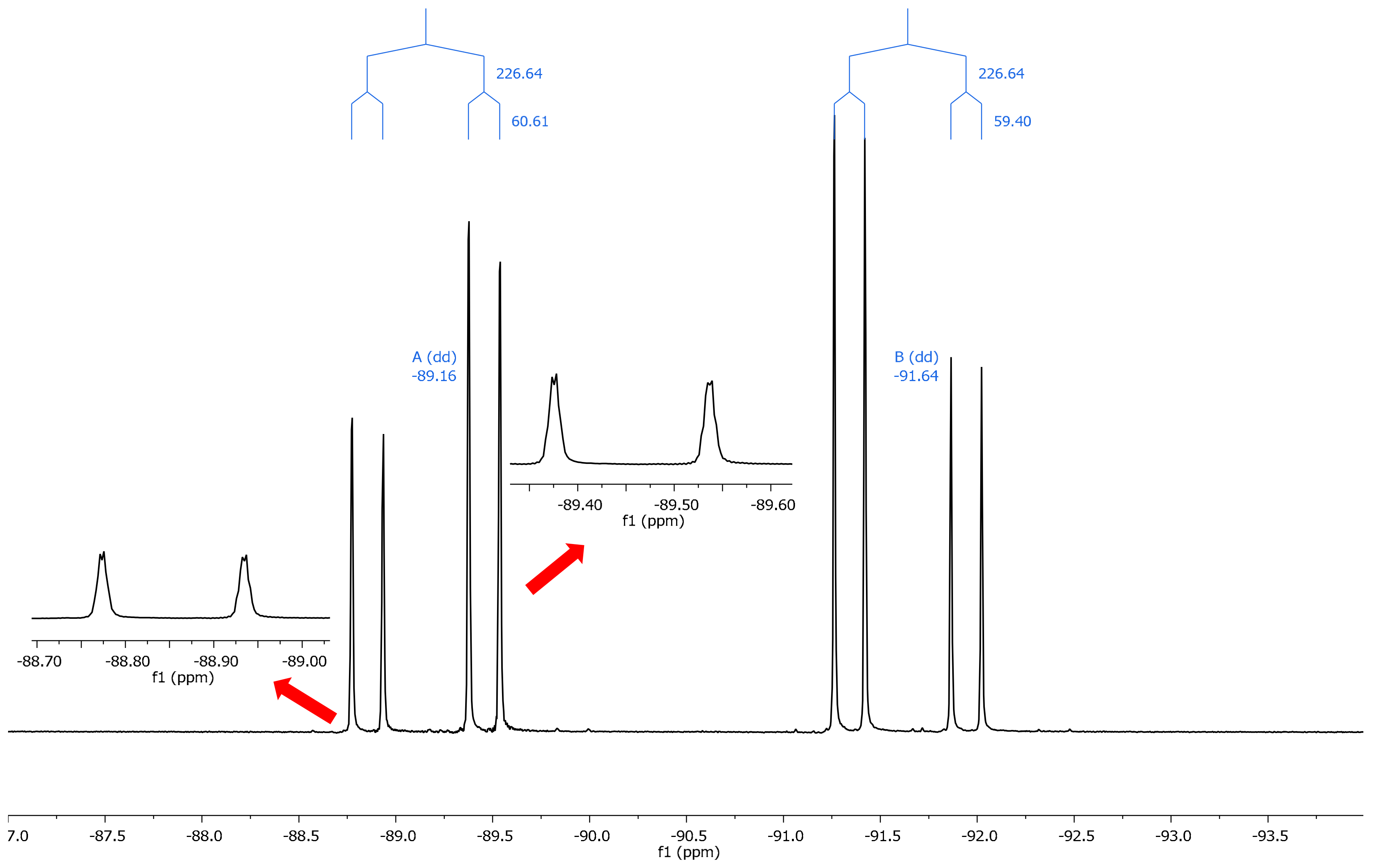
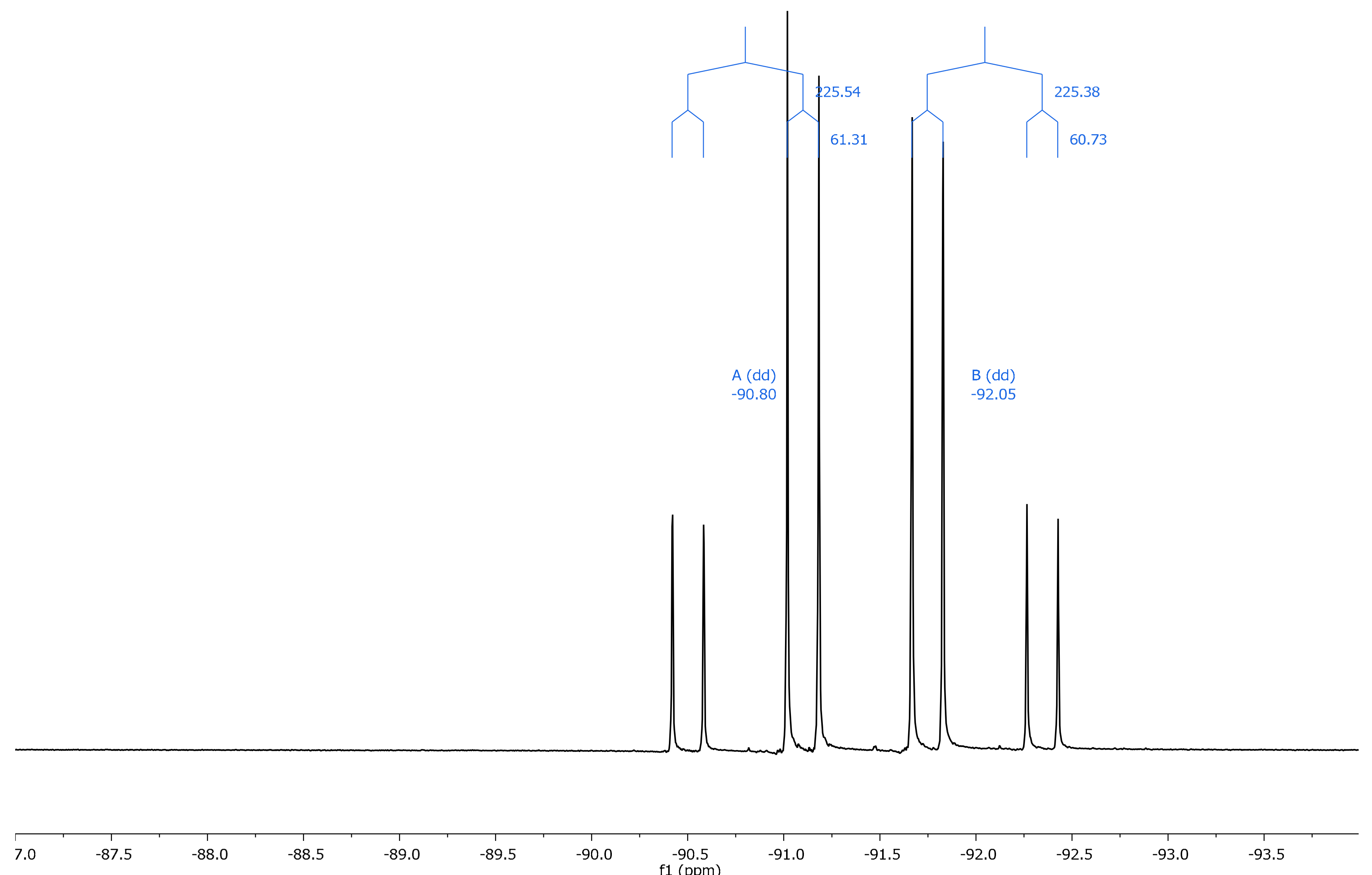
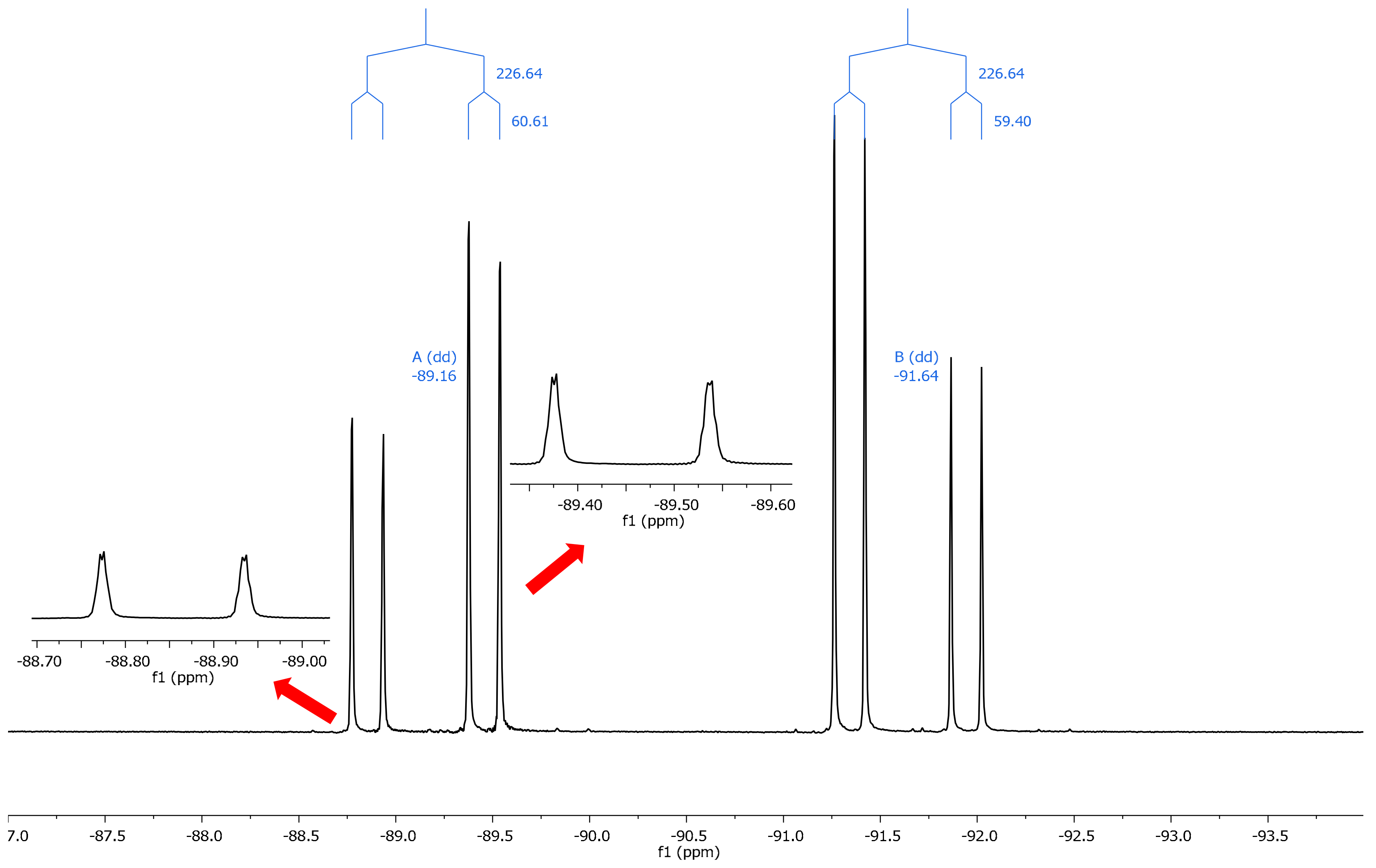
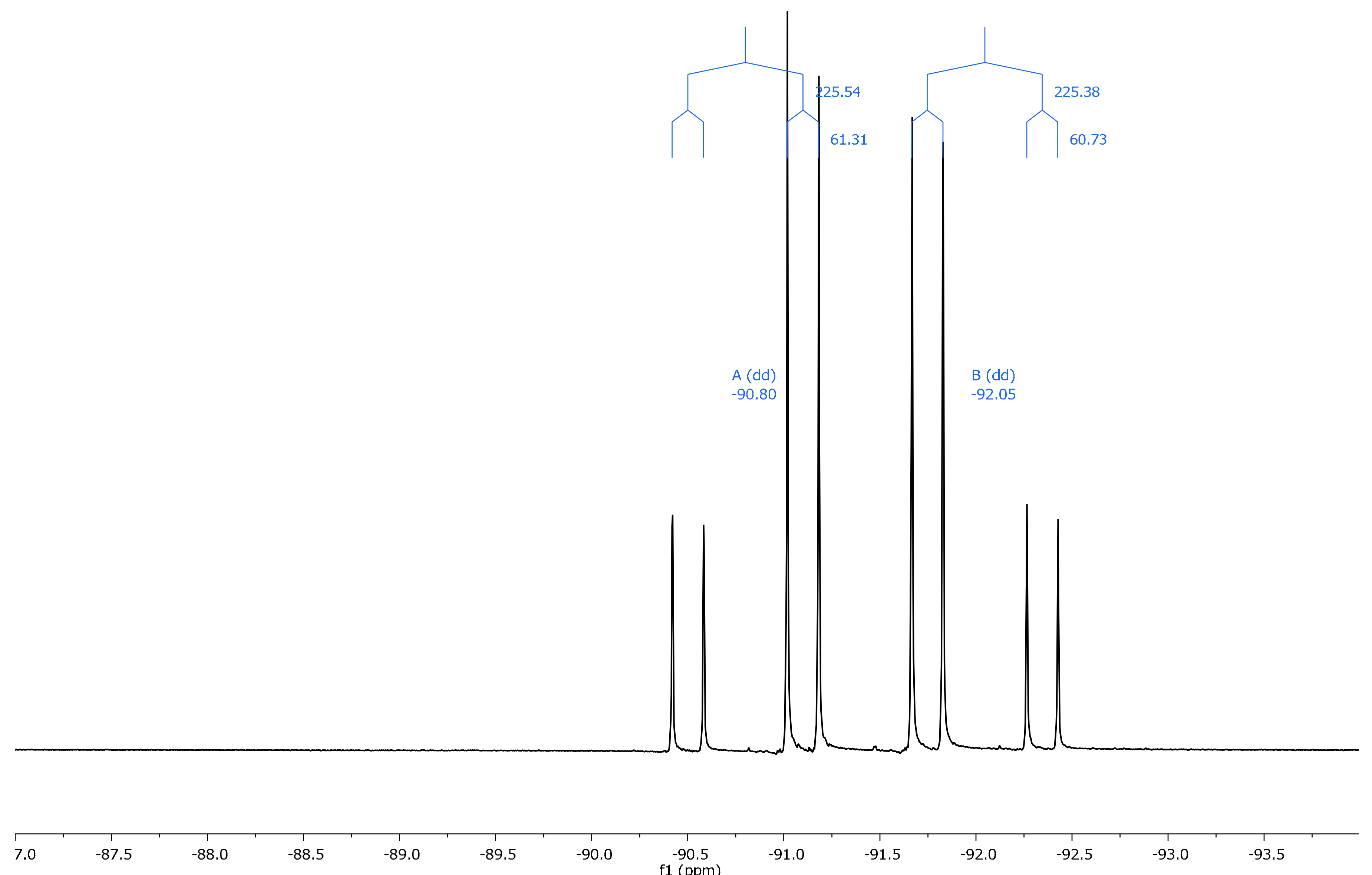
2.2. NMR Spectroscopy
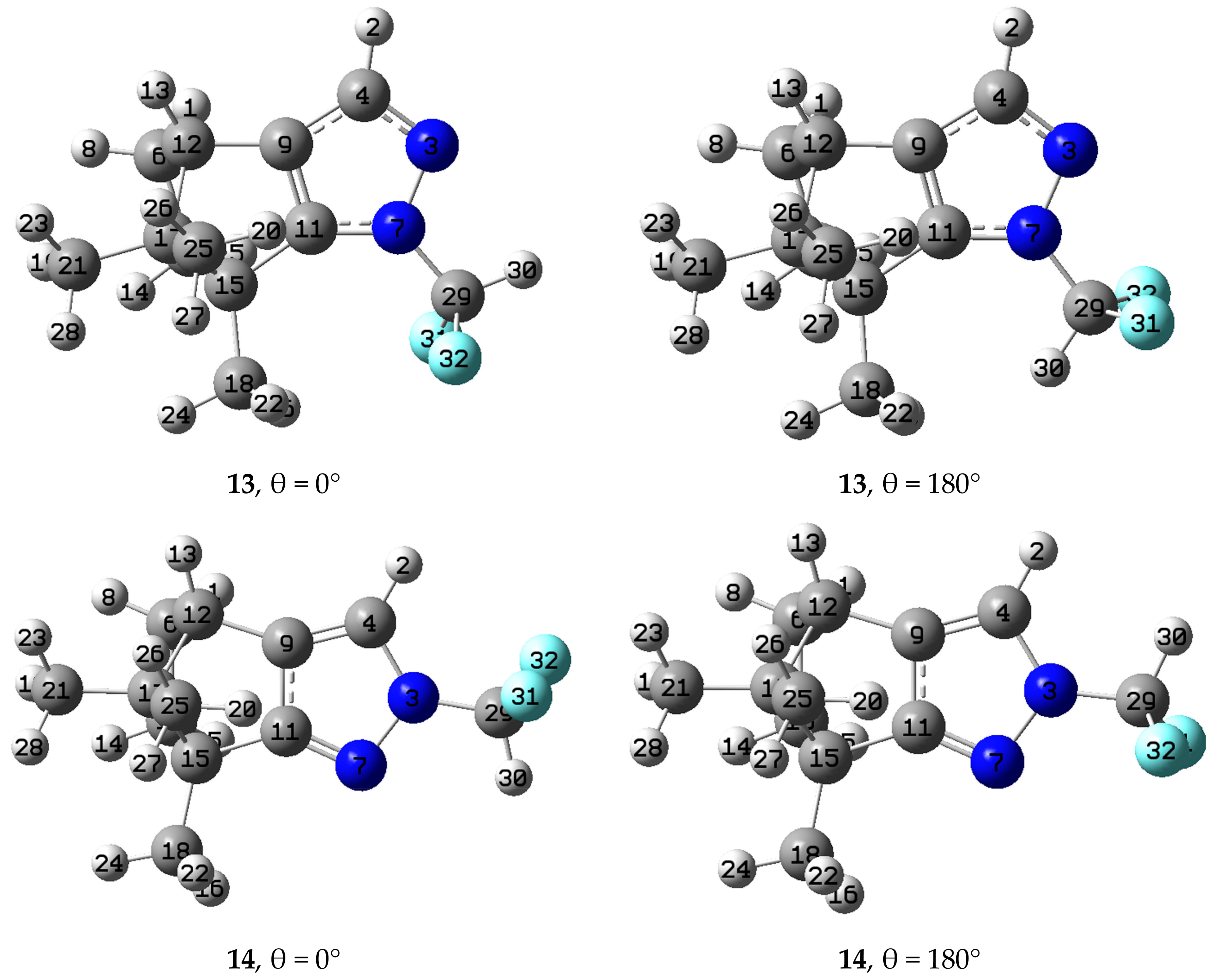
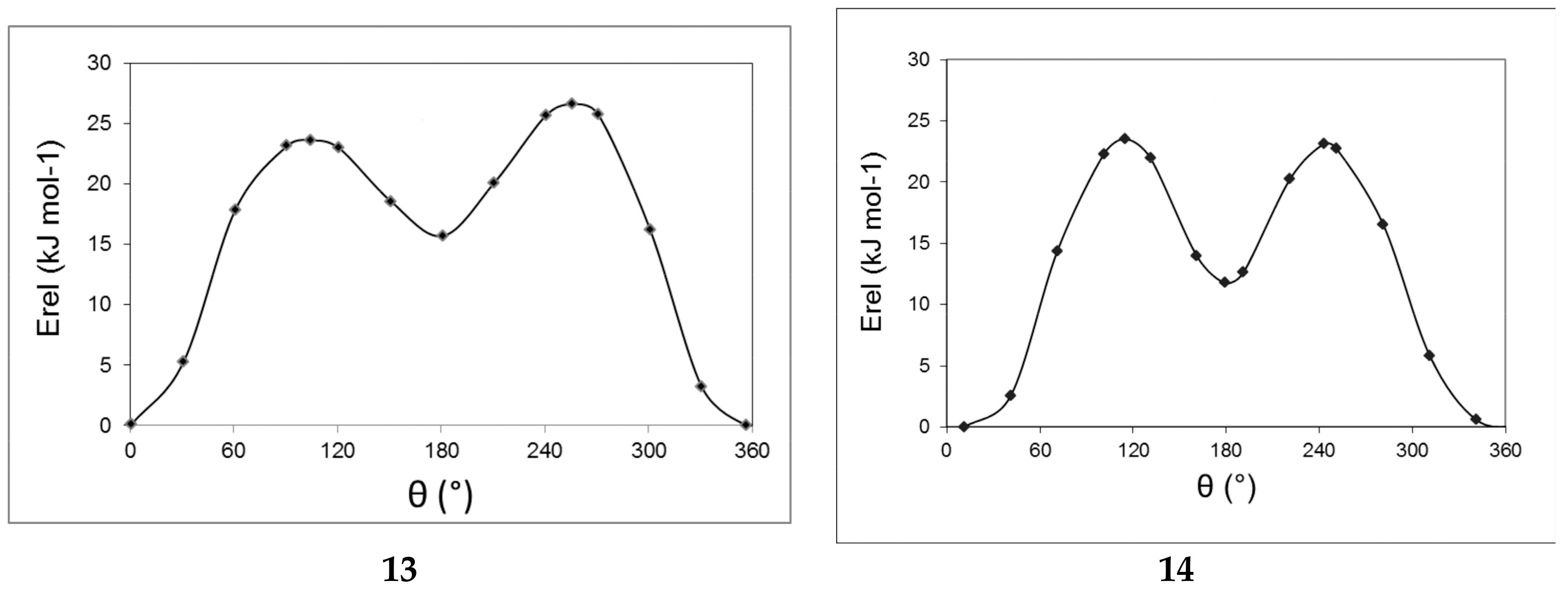
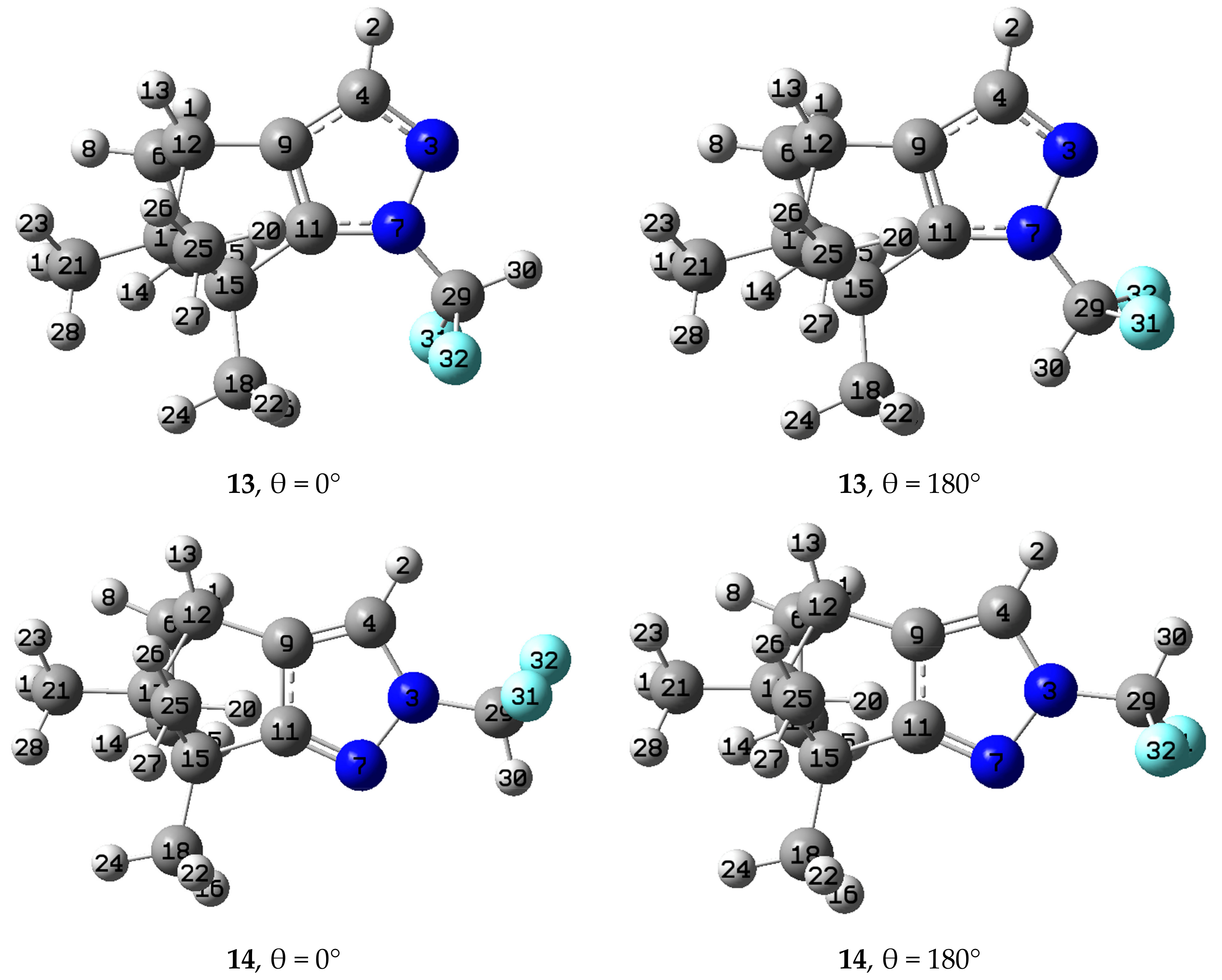
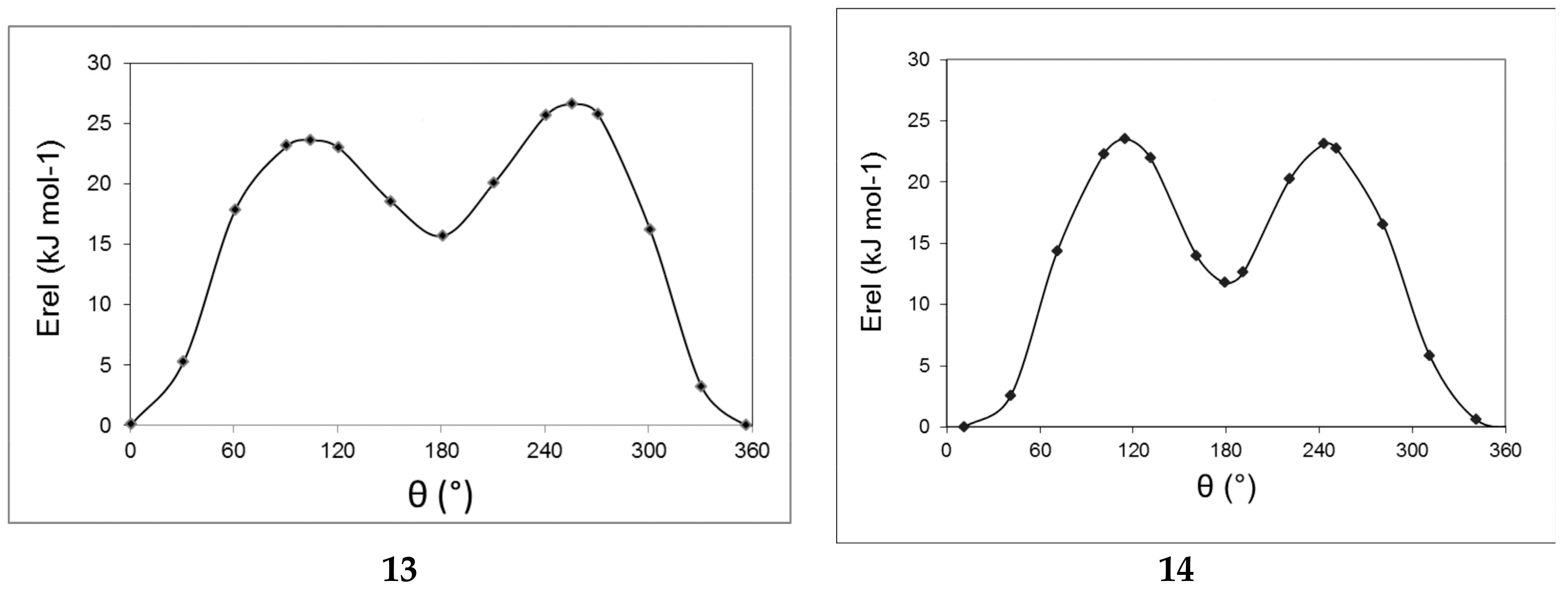
2.3. Computational Results
3. Experimental Section
3.1. Chemistry
3.2. NMR
3.3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Devriese, G.; Ottinger, R.; Zimmermann, D.; Reisse, J.; Mislow, K. On the sign of the anisochrony of diastereotopic groups. Bull. Soc. Chim. Belg. 1976, 85, 167–178. [Google Scholar] [CrossRef]
- Nasipuri, D. Stereochemistry of Organic Compounds. Principles and Applications, 2nd ed.; New Age International: New Dehli, India, 1994. [Google Scholar]
- Eliel, E.L.; Wilen, S.H. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Alkorta, I.; Elguero, J. Essential versus accidental isochrony of diastereotopic nuclei in NMR spectroscopy. Struct. Chem. 2016, 27, 671–679. [Google Scholar] [CrossRef]
- Elguero, J.; Marzin, C.; Tizané, D. Nuclear Magnetic Resonance of Asymmetrical Nitrogen in a Pentagonal Ring—1,2-Dimethylpyrazolidine. Tetrahedron Lett. 1969, 10, 513–514. [Google Scholar] [CrossRef]
- Elguero, J.; Marzin, C.; Tizané, D. Magnetic Non-equivalence and Nitrogen Inversion in a Series of Dinitrogen Pentagonal Herocycles, 2- and 3-Pyrazolines, Pyrazolidines and Pyrazolidones. Org. Magn. Reson. 1969, 1, 249–275. [Google Scholar] [CrossRef]
- Zvilichovsky, G. The Effect of Solvent on Chemical-shift Non-equivalence of Diastereotopic Geminal Nuclei in (pro)1-Chiral N,N-Disubstituted 5-Oxo-4-phenyl-2.5-dihydroisoxazol-2-ium-3-olates. J. Chem. Soc. Perkin Trans. 1988, 2, 2015–2019. [Google Scholar] [CrossRef]
- Molina, P.; Alajarín, M.; López-Leonardo, C.; Hernandez Cano, F.; Llamas-Saiz, A.L.; Concepcion Foces-Foces, C.; Rosa Maria Claramunt, R.M.; Elguero, J. 2,4-Bisimino-1,3-diazetidines: Iminophosphoranes, Carbodiimides and Related Betaines. J. Chem. Soc. Perkin Trans. 1992, 1, 199–210. [Google Scholar] [CrossRef]
- Fowler, K.G.; Littlefield, S.L.; Baird, M.C. Synthesis, Structures, and Properties of the Phosphonium-1-indenylide (PHIN) Ligands 1-C9H6PPh3, 1-C9H6PMePh2, and 1-C9H6PMe2Ph and of the Corresponding Ruthenium(II) Complexes [Ru(η5-C5H5)(η5-PHIN)]PF6. Organometallics 2011, 30, 6098–6107. [Google Scholar] [CrossRef]
- Powell, G.L.; Jacobus, J. The Nonequivalence of the Phosphorus Atoms in Cardiolipin. Biochemistry 1974, 13, 4024–4026. [Google Scholar] [CrossRef] [PubMed]
- Pastor, S.D.; Shum, S.P.; Rodebaugh, R.K.; Debellis, A.D.; Clarke, F.H. Sterically Congested Phosphite Ligands: Synthesis, Crystallographic Characterization, and Observation of Unprecedented Eight-Bond 31P, 31P Coupling in the 31P-NMR Spectra. Helv. Chim. Acta 1993, 76, 900–914. [Google Scholar] [CrossRef]
- King, S.A.; DiMichele, L. Multinuclear NMR Observation of a Ru(II)-BINAP Catalyst and Possible Intermediates in the Reduction of Ketoesters. In Catalysis of Organic Reactions; Scaros, M.G., Prunier, M.L., Eds.; Marcel Dekker: New York, NY, USA, 1995. [Google Scholar]
- Brunel, J.M.; Faure, B. A New 31P NMR Method for the Enantiomeric Excess Determination of Diols and Secondary Diamines with C2 Symmetry. Tetrahedron Asymmetry 1995, 6, 2353–2356. [Google Scholar] [CrossRef]
- Reich, H.J.; Kulicke, K.J. Dynamics of Solvent Exchange in Organolithium Reagents. Lithium as a Center of Chirality. J. Am. Chem. Soc. 1996, 118, 273–274. [Google Scholar] [CrossRef]
- Yamada, I.; Ohkouchi, M.; Yamaguchi, M.; Yamagishi, T. Asymmetric Hydrogenation of Acrylic Acid Derivatives by Novel Chiral Rhodium-Phosphinediamine Complex Catalysts by Selective Ligation Between Two Amino Units of the Ligand and Electrostatic Interaction. J. Chem. Soc. Perkin Trans. 1997, 1, 1869–1873. [Google Scholar] [CrossRef]
- Bilge, S.; Demiriz, S.; Okumus, A.; Kilic, Z.; Tercan, B.; Kökelek, T.; Buyukgungör, O. Phosphorus-Nitrogen Compounds. Part 13. Syntheses, Crystal Structures, Spectroscopic, Stereogenic, and Anisochronic Properties of Novel Spiro-Ansa-Spiro-, and Spiro-Crypta Phosphazene Derivatives. Inorg. Chem. 2006, 45, 8755–8767. [Google Scholar] [CrossRef] [PubMed]
- Pregosin, P.S. 31P- and 13C-NMR Studies on Metal Complexes of Phosphorus-donors: Recognizing Surprises. Coord. Chem. Rev. 2008, 252, 2156–2170. [Google Scholar] [CrossRef]
- Kruck, M.; Munoz, M.P.; Bishop, H.L.; Frost, C.G.; Chapman, C.J.; Kociok-Köhn, G.; Butts, C.P.; Lloyd-Jones, G.C. BINOL-3,3’-Triflone N,N-Dimethyl Phosphoramidites Through-Space 19F, 31P Spin-Spin Coupling with a Remarkable Dependency on Temperature and Solvent Internal Pressure. Chem. Eur. J. 2008, 14, 7808–7812. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Dardonville, C.; Elguero, J. Observation of Diastereotopic Signals in 15N-NMR Spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 3997–4000. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.C.; Spenser, I.D. 2H-NMR Spectroscopy as a Probe of the Stereochemistry of Enzymic Reactions at Prochiral Centres. Tetrahedron 1983, 39, 3549–3568. [Google Scholar] [CrossRef]
- Allen, B.D.; Cintrat, J.C.; Faucher, N.; Berthault, P.; Rousseau, B.; O'Leary, D.J. An Isosparteine Derivative for Stereochemical Assignment of Stereogenic (Chiral) Methyl Groups Using Tritium NMR: Theory and Experiment. J. Am. Chem. Soc. 2005, 127, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Salomone, A.; Perna, F.M.; Falcicchio, A.; Nilsson Lill, S.O.; Moliterni, A.; Michel, R.; Florio, S.; Stalke, D.; Capriati, V. Direct Observation of a Lithiated Oxirane: A Synergistic Study Using Spectroscopic, Crystallographic, and Theoretical Methods on the Structure and Stereodynamics of Lithiated ortho-Trifluoromethyl Styrene Oxide. Chem. Sci. 2014, 5, 528–538. [Google Scholar] [CrossRef]
- Powers, T.A.; Evans, S.A., Jr. Lanthanide Induced 17O NMR Shifts of Diastereotopic Oxygen Atoms in 1-Thiodecalin 1,1-Dioxide and Related Compounds. Tetrahedron Lett. 1990, 31, 5835–5838. [Google Scholar] [CrossRef]
- Drysdale, J.J.; Phillips, W.D. Restricted Rotation in Substituted Ethanes as Evidenced by Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1957, 79, 319–322. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, P.; Firestone, L.; Zhang, T.T. 19F Nuclear Magnetic Resonance Investigation of Stereoselective Binding of Isoflurane to Bovine Serum Albumin. Biophys. J. 1996, 70, 532–538. [Google Scholar] [CrossRef]
- Vaughan, M.D.; Cleve, P.; Robinson, V.; Duewel, H.S.; Honek, J.F. Difluoromethionine as a Novel 19F-NMR Structural Probe for Internal Amino Acid Packing in Proteins. J. Am. Chem. Soc. 1999, 121, 8475–8478. [Google Scholar]
- Qiu, X.L.; Qing, F.L. Synthesis of cis-4-Trifluoromethyl- and cis-4-Difluoromethyl-l-pyroglutamic Acids. J. Org. Chem. 2003, 68, 3614–3617. [Google Scholar] [CrossRef] [PubMed]
- Marchione, A.A.; Buck, R.C. Complete Multinuclear Magnetic Resonance Characterization of a Set of Polyfluorinated Acids and Alcohols. Magn. Reson. Chem. 2009, 47, 194–198. [Google Scholar] [CrossRef] [PubMed]
- El Dine, A.N.; Khalaf, A.; Grée, D.; Tasseau, O.; Fares, F.; Jaber, N.; Lesot, P.; Hachem, A.; Grée, R. Synthesis of Enones, Pyrazolines and Pyrrolines with gem-Difluoroalkyl Side Chains. Beilstein J. Org. Chem. 2013, 9, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Pike, S.J.; De Poli, M.; Zawodny, W.; Raftery, J.; Webb, S.J.; Clayden, J. Diastereotopic Fluorine Substituents as 19F NMR Probes of Screw-sense Preference in Helical Foldamers. Org. Biomol. Chem. 2013, 11, 3168–3176. [Google Scholar] [CrossRef] [PubMed]
- Mykhailiuk, P.K. In Situ Generation of Difluoromethyl Diazomethane for [3 + 2] Cycloadditions with Alkynes. Angew. Chem. Int. Ed. 2015, 54, 6558–6561. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Tian, Z.; Yang, D.; Li, X.; Li, H.; Jia, C.; Che, C.; Wang, M.; Qin, Z. Synthesis, Antifungal Activity and Structure-Activity Relationships of Novel 3-(Difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic Acid Amides. Molecules 2015, 20, 8395–8408. [Google Scholar] [CrossRef] [PubMed]
- Han, W.Y.; Zhao, J.; Wang, J.S.; Xiang, G.Y.; Zhang, D.L.; Bai, M.; Cui, B.D.; Wan, N.W.; Chen, Y.Z. Diastereoselective [3 + 2] cycloaddition of 3-ylideneoxindoles with in situ generated CF2HCHN2: Syntheses of CF2H-containing spirooxindoles. Org. Biomol. Chem. 2017, 15, 5571–5578. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Weizhou, H.; Jinbo, H. Difluoromethylation of O-, S-, N-, C-Nucleophiles Using Difluoromethyltri(n-butyl)ammonium Chloride as a New Difluorocarbene Source. Chin. J. Chem. 2011, 29, 2717–2721. [Google Scholar] [CrossRef]
- Thomoson, C.S.; Wang, L.; Dolbier, W.R. Use of Fluoroform as a Source of Difluorocarbene in the Synthesis of N-CF2H Heterocycles and Difluoromethoxypyridines. J. Fluor. Chem. 2014, 168, 34–39. [Google Scholar] [CrossRef]
- Prakash, G.K.; Krishnamoorthy, S.; Ganesh, S.K.; Kulkarni, A.; Haiges, R.; Olah, G.A. N-Difluoromethylation of Imidazoles and Benzimidazoles Using the Ruppert_Prakash Reagent under Neutral Conditions. Org. Lett. 2014, 16, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Makino, K.; Yamamoto, S.; Sakata, G. Synthesis of Fluoromethyl, Difluoromethyl and Trifluoromethyl Analogues of Pyrazosulfuron-ethyl as Herbicides. J. Heterocycl. Chem. 1990, 27, 807–810. [Google Scholar] [CrossRef]
- Petko, K.I.; Sokolenko, T.M.; Yagulpolskii, L.M. Chemical Properties of Derivatives of N-Difluoromethyl- and N-2H-Tetrafluoroethylpyrazoles. Chem. Heterocycl. Comp. 2006, 42, 1177–1184. [Google Scholar] [CrossRef]
- Pelc, M.; Huang, W.; Trujillo, J.; Baldus, J.; Turner, S.; Kleine, P.; Yang, S.; Thorarensen, A. An Efficient and Regioselective Difluoromethylation of 3-Iodoindazole with Chlorodifluoromethane. Synlett 2010, 219–222. [Google Scholar] [CrossRef]
- Yagupolskii, L.M.; Fedyuk, D.V.; Petko, K.I.; Troitskaya, V.I.; Rudyk, V.I.; Rudyuk, V.V. N-Trihalomethyl Derivatives of Benzimidazole, Benzotriazole and Indazole. J. Fluor. Chem. 2000, 106, 181–187. [Google Scholar] [CrossRef]
- Llamas-Saiz, A.; Foces-Foces, C.; Sobrados, I.; Elguero, J.; Meutermans, W. (4S,7R)-7,8,8-Trimethyl-4,5,6,7-Tetrahydro-4,7-methano-1H(2H)-indazole (Campho[2,3-c]Pyrazole): Comparison Between the X-Ray Structure and Carbon-13 NMR Data in the Solid State. Acta Crystallogr. Sect. C 1993, 49, 724–729. [Google Scholar] [CrossRef]
- Yap, G.P.A.; Claramunt, R.M.; López, C.; García, M.A.; Pérez-Nedina, C.; Alkorta, I.; Elguero, J. The Structures of Chiral and Racemate Campho[2,3-c]pyrazole: A Combined Crystallographic, Solid-State NMR and Computational Study. J. Mol. Struct. 2010, 965, 74–81. [Google Scholar] [CrossRef]
- Quesada-Moreno, M.M.; Avilés-Moreno, J.R.; López-González, J.J.; Claramunt, R.M.; López, C.; Alkorta, I.; Elguero, J. Chiral Self-Assembly of Enantiomerically Pure (4S,7R)-Campho[2,3-c]pyrazole in the Solid State: A Vibrational Circular Dichroism (VCD) and Computational Study. Tetrahedron Asymmetry 2014, 25, 507–515. [Google Scholar] [CrossRef]
- Webber, A.L.; Emsley, L.; Claramunt, R.M.; Brown, S.P. NMR Crystallography of Campho[2,3-c]pyrazole (Z’=6): Combining High-Resolution 1H-13C Solid-State MAS NMR Spectroscopy and GIPAW Chemical-Shift Calculations. J. Phys. Chem. A 2010, 114, 10435–10442. [Google Scholar] [CrossRef] [PubMed]
- Birchall, J.M.; Cross, G.W.; Haszeldine, R.N. Difluorocarbene. Proc. Chem. Soc. 1960, 81, 37–96. [Google Scholar]
- Mehta, V.P.; Greaney, M.F. S-, N-, and Se-Difluoromethylation Using Sodium Chlorodifluoroacetate. Org. Lett. 2013, 15, 5036–5039. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hua, M.; Huang, Y.; Zhang, Q.; Zhang, X.; Wu, J. Difluoromethylation of 2-Hydroxychalcones Using Sodium 2-Chloro-2,2-difluoroacetate as Difluoromethylating Agent. Chem. Res. Chin. Univ. 2015, 31, 362–366. [Google Scholar] [CrossRef]
- Cabildo, P.; Claramunt, R.M.; Cornago, P.; Lavandera, J.L.; Sanz, D.; Jagerovic, N.; Jimeno, M.L.; Elguero, J.; Gilles, I.; Aubagnac, J.L. Synthesis, structure (NMR and mass spectrometry) and conformational analysis of heterocyclic analogues of dibenzo[a,e]cycloocta-l,5-diene: 5,6,12,13-tetrahydrobispyrazolo[1,2-a:1’,2’-e] [1,2,5,6] tetraazocinediium dihalides. J. Chem. Soc. Perkin Trans. 1996, 2, 701–711. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S.; Kalinowski, H.O. NMR Spectroscopy of the Non-Elements; Wiley: New York, NY, USA, 1997; p. 635. [Google Scholar]
- Martínez, A.; Jimeno, M.L.; Elguero, J.; Fruchier, A. An Experimental Approach to the Mills-Nixon Effect: Tautomerism of 3(5),4-Polymethylenepyrazoles. New J. Chem. 1994, 18, 269–277. [Google Scholar]
- Alkorta, I.; Elguero, J. Tautomerism and the Mills-Nixon Effect. Struct. Chem. 1997, 8, 189–195. [Google Scholar] [CrossRef]
- Brand, D.J.; Steenkamp, J.A.; Brandt, E.V.; Takeuchi, Y. Conformational studies of (−)-epicatechin-Mosher ester. Tetrahedron Lett. 2007, 48, 2769–2773. [Google Scholar] [CrossRef]
- Brand, D.J.; Steenkamp, J.A.; Omata, K.; Kabuto, K.; Fujiwara, T.; Takeuchi, Y. The Origin of an Unusually Large 19F Chemical Shift Difference Between the Diastereomeric α-Cyano-α-Fluoro-p-Tolylacetic Acid (CFTA) Esters of 3’,4’,5,7-Tetra-O-Methylepicatechin. Chirality 2008, 20, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Corminboeuf, C.; Mo, Y. Direct Evaluation of the Hyperconjugative Interactions in 1,1,1-Trihaloethane (CH3CX3, X = F, Cl, and Br). J. Phys. Chem. A 2014, 118, 5743–5747. [Google Scholar] [CrossRef] [PubMed]
- Baranac-Stojanovic, M. Theoretical Analysis of the Rotational Barrier in Ethane: Cause and Consequences. Struct. Chem. 2015, 26, 989–996. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar]
- Ditchfield, R. Self-consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO (Linear Combination of Atomic Orbitals) Method for NMR Chemical Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- London, F. Quantum Theory of Interatomic Currents in Aromatic Compounds. J. Phys. Radium. 1937, 8, 397–409. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 84, 2027–2094. [Google Scholar] [CrossRef]
- Mennucci, B.; Cammi, R. (Eds.) Continuum Solvation Models in Chemical Physics. From Theory to Applications; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Silva, A.M.S.; Sousa, R.M.S.; Jimeno, M.L.; Blanco, F.; Alkorta, I.; Elguero, J. Experimental measurements and theoretical calculations of the chemical shifts and coupling constants of three azines (benzalazine, acetophenoneazine and cinnamaldazine). Magn. Reson. Chem. 2008, 46, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.; Alkorta, I.; Elguero, J. Statistical analysis of 13C- and 15N-NMR chemical shifts from GIAO/B3LYP/6-311++G** calculated absolute shieldings. Magn. Reson. Chem. 2007, 45, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Fresno, N.; Pérez-Fernández, R.; Jimeno, M.L.; Alkorta, I.; Sánchez-Sanz, G.; Elguero, J.; Del Bene, J.E. Multinuclear NMR Characterization of Cyanuric Fluoride (2,4,6-Trifluoro-1,3,5-triazine). J. Heterocycl. Chem. 2012, 49, 1257–1259. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor–Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
Sample Availability: Samples of the compounds 13, 14, 15, are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | θ (°) | 19F (31) | 19F (32) | Δδ (31–32) | 19F (a) | 19F (b) | Δδ (a–b) a |
|---|---|---|---|---|---|---|---|
| Calculated Values | Experimental Values | ||||||
| 13 | –3.6 | –93.18 | –89.73 | −3.45 | –91.64 | –89.16 | −2.48 |
| 13 | –179.3 | –90.07 | –98.52 | +8.45 | |||
| 14 | 11.1 | –92.83 | –88.66 | –4.17 | –92.05 | –90.80 | –1.25 |
| 14 | 179.3 | –85.73 | –97.40 | +11.67 | |||
| Comp. | θ (°) | 19F (31) | 19F (32) | Δδ (31–32) | 19F (a) | 19F (b) | Δδ (a–b) a |
|---|---|---|---|---|---|---|---|
| Calculated Values | Experimental Values | ||||||
| 13 | −4.0 | −94.07 | −90.23 | −3.84 | −91.64 | −89.16 | −2.48 |
| 13 | −179.5 | −91.97 | −99.34 | +7.37 | |||
| 14 | 11.2 | −94.21 | −90.68 | −3.53 | −92.05 | −90.80 | −1.25 |
| 14 | 179.4 | −86.35 | −97.99 | +11.64 | |||
| Comp. | 13 exp. CDCl3 | 13 calc. Gas | 13 calc. CHCl3 | 14 exp. CDCl3 | 14 calc. Gas | 14 calc. CHCl3 |
|---|---|---|---|---|---|---|
| C3 (C4) | 134.3 | 133.2 | 134.4 | 117.9 | 117.3 | 118.2 |
| C3a (C9) | 132.1 | 133.0 | 133.9 | 130.2 | 132.3 | 133.6 |
| C7a (C11) | 153.6 | 153.7 | 155.2 | 169.1 | 167.5 | 169.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pérez, D.; López, C.; Claramunt, R.M.; Alkorta, I.; Elguero, J. 19F‐NMR Diastereotopic Signals in Two N-CHF2 Derivatives of (4S,7R)-7,8,8-Trimethyl-4,5,6,7-tetrahydro-4,7-methano-2H-indazole. Molecules 2017, 22, 2003. https://doi.org/10.3390/molecules22112003
García-Pérez D, López C, Claramunt RM, Alkorta I, Elguero J. 19F‐NMR Diastereotopic Signals in Two N-CHF2 Derivatives of (4S,7R)-7,8,8-Trimethyl-4,5,6,7-tetrahydro-4,7-methano-2H-indazole. Molecules. 2017; 22(11):2003. https://doi.org/10.3390/molecules22112003
Chicago/Turabian StyleGarcía-Pérez, Diana, Concepción López, Rosa M. Claramunt, Ibon Alkorta, and José Elguero. 2017. "19F‐NMR Diastereotopic Signals in Two N-CHF2 Derivatives of (4S,7R)-7,8,8-Trimethyl-4,5,6,7-tetrahydro-4,7-methano-2H-indazole" Molecules 22, no. 11: 2003. https://doi.org/10.3390/molecules22112003