Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides

1
Key Laboratory of Chinese Materia Medica, Ministry of Education of Heilongjiang University of Chinese Medicine, Harbin 150040, China
2
Heilongjiang Institute for Food and Drug Control, Harbin 150001, China
3
College of Pharmacy, Harbin University of Commerce, Harbin 150001, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2017, 22(11), 1995; https://doi.org/10.3390/molecules22111995
Submission received: 28 October 2017
/
Accepted: 15 November 2017
/
Published: 22 November 2017
(This article belongs to the Section Natural Products Chemistry)
Abstract
:Four new compounds, aneglycoside A–C (1–3) and timosaponin U (4), were isolated from the rhizomes of Anemarrhena asphodeloides. Their structures were determined through extensive spectroscopic analysis, chemical characteristics, and high-resolution mass spectrometry (HRMS). All the isolations were evaluated for cytotoxicity against HepG2, Hela, and SGC7901 human cancer lines. Compounds 1, 2, and 4 showed weak antiproliferative activities on HepG2, Hela, and SGC7901 cells.

1. Introduction
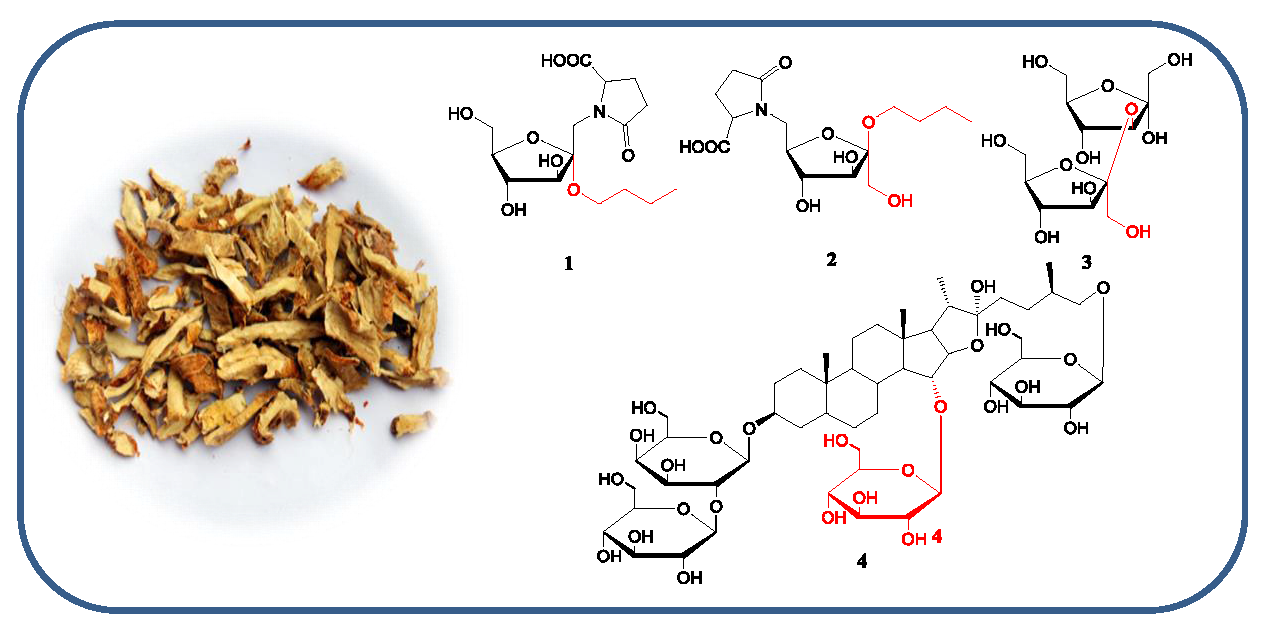
Anemarrhena asphodeloides Bunge (Asparagaceae) is a perennial herb, widely distributed in China, particularly in the Hebei and Anhui provinces. The dried rhizomes of A. asphodeloidesis is a commonly used traditional Chinese medicine known as “Zhimu”, used for its heat-clearing, fire-purging, Yin-nourishing, and dryness-moistening effects [1]. Steroidal saponins, flavonoids, and alkaloids are the major components of A. asphodeloidesis [2], resulting in various biological functions, including anti-tumor, anti-oxidant, anti-inflammation, anti-hypertension, and anti-hyperglycemic properties [3,4]. During further investigation of the bioactive constituents, four new compounds were found from the n-butanol layer of A. asphodeloidesis aqueous extract, including two new pyroglutamic acid-fructosides, aneglycoside A (1) and aneglycoside B (2), one new disaccharide, aneglycoside C (3), and one new steroidal saponin (4) (Figure 1). In this paper, we reported the isolations and structures of the new compounds 1–4, as well as their cytotoxicity activities.
2. Experimental Method
2.1. General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 instrument (JASCO, Tokyo, Japan). The infrared (IR) spectra were recorded on a Shimadzu FTIR-8400S (Shimadzu, Tokyo, Japan). High-resolution electrospray ionization mass spectrometry (HRESIMS) was conducted using a Waters Xevo-TOF-MS™ instrument (Waters, Bedford, MA, USA). The ultraviolet (UV) spectra were recorded on a UV-2450 spectrometer (Shimadzu, Kyoto, Japan). Semi-preparative and preparative high-performance liquid chromatography (HPLC) was performed on an Agilent 1100 liquid chromatography (Agilent Corporation, Waldbronn, Germany) with a Zorbax SB-C18 (9.4 mm × 25 cm) column (Agilent Corporation, Palo Alto, CA, USA). The hydrogen (1H), carbon (13C), and two-dimensional (2D) (1H-1H correlation spectroscopy (COSY), heteronuclear multiple bond correlation (HMBC), heteronuclear multiple quantum coherence (HSQC)) nuclear magnetic resonance (NMR) spectra were recorded on a JNM-ECA600 spectrometer (JEOL, Tokyo, Japan) using a standard pulse sequence. Column chromatography was performed using silica gel (100–120 mesh and 200–300 mesh, Qingdao Marine Chemical Co., Qingdao, China). The thin-layer chromatography used GF254 (MN), and spots were detected by spraying the plates with 10% H2SO4-EtOH reagent followed by heating at 105 °C for 5 min.
2.2. Plant Material
The rhizomes of A. asphodeloides were collected from the Anhui Province in China in April 2015, and identified by Ruifeng Fan of Heilongjiang University of Chinese Medicine. A voucher specimen (S 2015100803) was deposited at the Heilongjiang University of Chinese Medicine.
2.3. Extration and Isolation
Air-dried A. asphodeloides rhizomes (20 kg) were extracted under reflux with three times the amount of water for 2 h, then the residue was filtered, and extracted with seven times the amount of purified water for 1 h, then this process was repeated. The filtrates were combined and evaporated to a suitable concentration. Then, 95% ethanol was added four or five times to adjust the concentration of ethanol to 80%, allowed to stand for 1 day, filtered in vacuo, and centrifuged. The filtrate was concentrated in a vacuum to eliminate ethanol, and then purified with macroporous resin. The 95% ethanol fraction was evaporated in vacuo followed by suspension in water. The aqueous layer was further partitioned with ethyl acetate and n-butanol. The n-butanol-soluble was evaporated under reduced pressure to result in a residue of 751 g, which was chromatographed on a silica gel column of MeOH-CH2Cl2 (9:1, 5:1, 1:1 v/v) to create five fractions. Fraction 1 was fractioned into nine subfractions (Fractions 1.1–1.9) using a silica gel column eluted with CH2Cl2-MeOH (6:1). Fraction 1.7 was further separated by preparative HPLC MeOHåH2O (30:70) to yield compound 1 and 2. Fraction 5 was subjected to octadecylsilyl (ODS) chromatography (H2O/MeOH, 1:0 to 0:1) to create six subfractions. Fraction 5.1 was further separated by preparative HPLC MeOH-H2O (5:95) to yield compound 3. Fraction 4 was subjected to ODS chromatography (H2O/MeOH, 1:0 to 0:1) to create sevben subfrations. Fraction 7.5 was further separated by preparative HPLC MeOH-H2O (50:50) to yield compound 4.
2.3.1. Aneglycoside A—Compound 1
White amorphous powder. −23.7 (c = 2.1, MeOH); 1H- and 13C-NMR (MeOH, 400, 100 MHz) data are shown in Table 1, Figures S1 and S2; high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) m/z 348.1661 [M + H]+ (calcd. for C15H26NO8, 348.1658).
2.3.2. Aneglycoside B—Compound 2
White amorphous powder. −26.4 (c = 1.9, MeOH); 1H- and 13C-NMR (MeOH, 400, 100 MHz) data; see Table 1, Figures S3 and S4; HR-ESI-MS m/z 348.1643 [M + H]+ (calcd. for C15H26NO8, 348.1658).
2.3.3. Aneglycoside C—Compound 3
White amorphous powder. +19.8 (c = 1.7, D2O); 1H- and 13C-NMR (MeOH, 400, 100 MHz) data; see Table 2, Figures S5 and S6; HR-ESI-MS m/z 343.1203 [M + H]+ (calcd. for C12H23O11, 343.1240).
2.3.4. Timosaponin U—Compound 4
White amorphous powder. −43.2 (c = 1.4, MeOH); 1H- and 13C-NMR (MeOH, 400, 100 MHz) data, shown in Table 3, Figures S7 and S8; HR-ESI-MS m/z 1099.5528 [M + H]+ (calcd. for C51H87O25, 1099.5536).
2.4. Acid Hydrolysis, GC Analysis, and Opitcal Rotation Test
Compounds 1–4 (2.0 mg) were refluxed with HCl (2 mol/L, 5 mL) for 4 h at 90 °C. Then, the reagent was neutralized with sodium hydroxide (NaHCO3), and extracted with 5 mL methanol (MeOH) four times. The remaining aqueous layer was concentrated each time and then freeze-dried to provide a residue. The residue was dissolved in 1 mL pyridine and 0.7 mL silylation-derived agent, and added to the solution for shaking for 5 min, then placed at room temperature for 4 h, and 1.5 mL distilled water was added. After centrifugation, the supernatant was detected by gas chromatography (GC) [5,6]. The glycosyl configurations of compounds 1–4 were determined by the same retention time (tR) of standard d-fructose (tR = 6.3 min) for compounds 1–3, and d-galactose and d-glucose for compound 4 (tR = 32.5 min and tR = 16.8 min). Meanwhile, the MeOH extract layer was concentrated by rotary evaporation and then the optical rotation (OR) test was conducted. The configurations of pyroglutamic acid in compounds 1 and 2 were determined by the same [α] of standard l-pyroglutamic acid ( −27.5 (c = 10, NaOH)) [7].
2.5. Cytotoxicity Assays
The isolations were evaluated for their antiproliferative activities using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) method in vitro on HepG2, Hela, and SGC7901 cells obtained from the Shanghai Institute of Biochemistry and Cell Biology. They were cultured in Roswell Park Memorial Institute (RPMI) 1640 (10% Fetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin) in 5% CO2 at 37 °C. Then, the cells were cultured in 96-well plates for 24 h with 100 μL complete medium, and the compounds were added with varying concentrations of 5.0, 10.0, 25.0, 50.0 and 100 μg/mL. MTT (20 μL) with 5 mg/mL phosphate buffer saline was added for another 4 h in the 96-well plates, then dissolved in dimethyl sulfoxide (DMSO) and assayed at 490 nm by the VICTOR-X3 ELISA instrument (PerkinElmer, Massachusetts, USA) [6]. The cytotoxicity of the compounds against HepG2, Hela, and SGC7901 was calculated and expressed as an IC50 value. Doxorubicin was used as the positive control (Table 4).
3. Results
3.1. Structure Determination
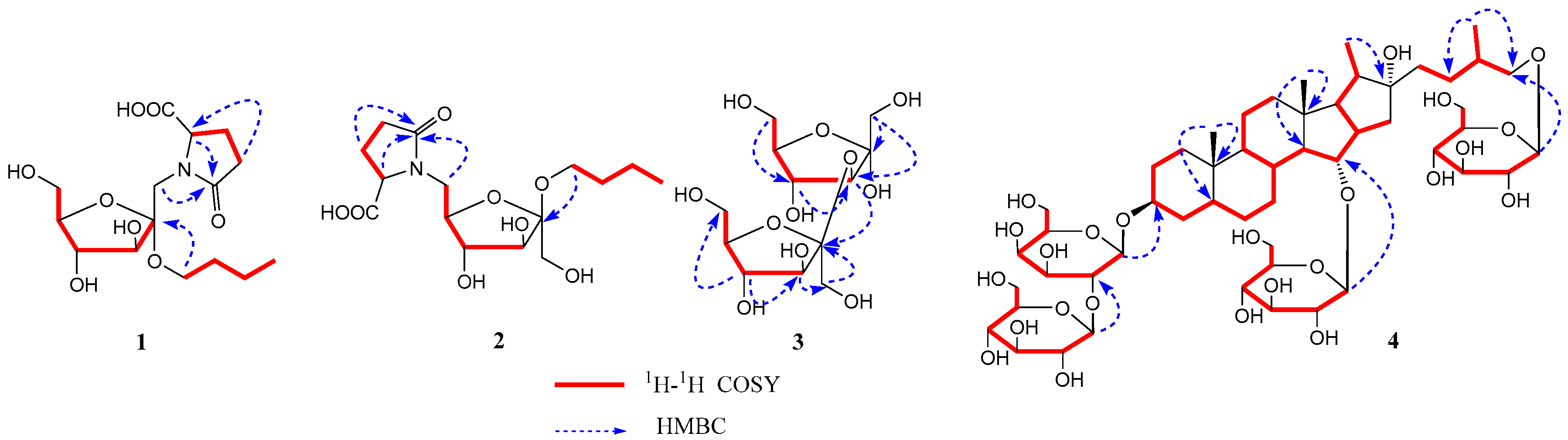
Compound 1 was isolated as a white powder. Its molecular formula C15H25NO8 was determined from data of the positive-ion HRESIMS (m/z: 348.1661 [M + H]+, (calcd. 348.1658). The 1H-NMR spectrum of compound 1 (Table 1) showed the signals of δH 4.32 (1H, dd, J = 8.0, 4.0 Hz, –CH), 2.49 (1H, m, –CH2-a), 2.33 (2H, m, –CH2), and 2.23 (1H, m, –CH2-b), assigned to l-pyroglutamic acid protons. Additionally, a group of the signals of d-fructose was observed at δH 4.35 (1H, d, J = 11.6 Hz, –OCH2-1a), 4.24 (1H, d, J = 11.6 Hz, –OCH2-1b), 3.96 (1H, d, J = 8.0 Hz, H-3), 3.91 (1H, d, J = 8.0 Hz, H-4), 3.73 (1H, o, H-6a), 3.71 (1H, o, H-5), and 3.56 (1H, o, H-6b). A group of n-butoxyl moiety proton signals were found at δH 3.74 (1H, m, –OCH2-a), 3.52 (1H, m, –OCH2-b), 1.53 (2H, m, –CH2), 1.37 (2H, m, –CH2), and 0.92 (3H, t, J = 7.2 Hz, –CH3). The 13C-NMR spectrum also exhibited 15 carbon signals, in three groups of carbon signals (δC 102.9, 83.5, 80.2, 76.8, 64.7, and 64.0; δC 62.3, 33.2, 20.3, and 14.3; δC 181.2, 173.4, 57.1, 30.2 and 25.8), attributed to d-fructose, n-butoxyl moiety, and l-pyroglutamic acid, respectively. Six carbon signals (δC 102.9, 83.5, 80.2, 76.8, 64.7 and 64.0) were similar to those of the sugar moiety of α-d-fructofuranoside [4]. Furthermore, glutamic acid that appeared in the form of l-pyroglutamic acid was corroborated by the HMBC of the correlation of δH 4.32 (1H, dd, J = 8.0, 4.0 Hz, pyr-H-5) with carbon signals at δC 181.2 (C-6′) and 173.4 (C-2′). The sugar linkage sites were determined by the HMBC correlation (Figure 2) between δH 4.35 (1H, d, J = 11.6 Hz, Fruc-H-1-a), 4.24 (1H, d, J = 11.6 Hz, Fruc-H-1-b), and δC 173.4 (pyr-C-2′), suggesting that pyroglutamic acid was linked by N to form a glycoside with C-1 of the fructosyl moiety. In addition, the correlation between δH 3.74 (1H, m, n-butoxyl-OCH2-a) and δC 102.9 (Fruc-C-2) indicated that the n-butoxyl group was linked to the fructosyl moiety at C-2. Additionally, the absolute configurations of fructofuranose and pyroglutamic acid were determined as d- and l-configurations by the acid hydrolysis, GC analysis, and OR test. From the above data and prior research [7,8], the structure of compound 1 was identified as 1-deoxy-1-[l-pyroglutamic acid]-2-n-butoxy-α-d-furanofructoside, named aneglycoside A.
Compound 2 was a white powder. Its molecular formula C15H25NO8 was deduced from the data of the positive-ion HRESIMS at m/z 348.1643 [M + H]+ (calcd. 348.1658). The 1H- and 13C-NMR data (Table 1) indicated that compound 2 was an n-butyl-(pyroglutamic acid) six-carbon sacchroside. By comparing the 1H-NMR and 13C-NMR spectra data of compounds 1 and 2, the configuration of the fructosyl moiety was found to represent the difference. The 13C-NMR data of the sugar moiety of compound 2 were similar to those of β-d-fructofuranoside [4]. Comparing the 13C-NMR spectrum data of compound 2 with that of fructose, fruc-C-6 was approximately up-field shifted by 3.0 ppm. The HMBC correlation (Figure 2), between δH 4.28 (2H, o, Fruc-H-6) and δC 173.4 (pyr-C-2′), suggested that pyroglutamic acid was linked by N to form a glycoside with C-6 of the fructosyl moiety. Furthermore, the correlation between δH 3.61 (1H, m, n-butoxyl-OCH2-a) and δC 105.6 (Fruc-C-2) indicated that the n-butoxyl group was linked to the fructosyl group at C-2. Like compound 1, the absolute configuration of fructofuranose and pyroglutamic acid in compound 2 was determined as d- and l-configuration. Therefore, the structure of compound 2 was established as 6-deoxy-6-[l-pyroglutamic acid]-2-n-butoxy-α-d-furanofructoside, and named aneglycoside B.
Compound 3 was a white powder, and its molecular formula was C12H22O11 according to HRESIMS at m/z 343.1203 [M + H]+ (calcd. 343.1240). The 13C-NMR (Table 2) spectrum of compound 3 showed the two characteristic signals of anomeric carbon at δC 98.8 and δC 102.5, which were deduced to be a disaccharide. Combined with the Distortionless Enhancement by Polarization Transfer (DEPT), the 13C-NMR spectrum of compound 3 showed 12 signals, in two groups of fructofuranosyl at δC 98.8, 81.8, 77.7, 83.5, 61.2, and 61.7, and δC 62.5, 102.5, 76.9, 74.5, 81.2 and 62.6. The α-configuration of fructofuranosyl was determined by the chemical shift of C-3 (δC 81.8) and C-5 (δC 83.5), and the β-configuration of fructofuranosyl was determined by the chemical shift of C-5′ (δC 81.2) based on the literature [9]. The connectivity of the two fructofuranosyls was mainly established on the HMBC correlation of H-3 (δH 3.94, d, J = 2.4 Hz) with C-2′ (δC 102.5) (Figure 2), which suggested the attachment position of β-fructofuranosyl at C-2 of α-fructofuranosyl. The absolute configuration of fructofuranosyl groups of compound 3 was determined as D and D by GC analysis after derivatization. Thus, the structure of compound 3 was determined to be α-d-fructofuranosyl-(3→2)-β-d-fructofuranose, and named aneglycoside C.
Compound 4 was isolated as a white amorphous powder, with the molecular formula of C51H86O25 determined by HRESIMS at m/z 1099.5528 [M + H]+ (calcd. for 1099.5536). The 1H-NMR spectrum of compound 4 (Table 3) showed four methyl proton signals of a typical steroidal skeleton at δH 1.03 (3H, d, J = 6.7 Hz, H3-27), δH 1.32 (3H, d, J = 6.8 Hz, H3-21), δH 0.89, and 0.95 (3H each, both s, H3-19, 18). In the 13C-NMR spectrum of compound 4 (Table 3), four methyl groups (δC 18.2, 24.3, 16.6, and 17.6) and quaternary carbon (δC 110.5) suggested that compound 4 was a furostanol saponin. The 13C-NMR data of compound 4 were similar to those of timosaponin E1 [10], obtained previously from the rhizomes of Anemarrhena asphodeloides. The major difference between them was that an additional signal of glucose in compound 4 was missing in timosaponin E1. The glucose linkage was established by the existence of long-range HMBC correlations between δH 4.80 (H-1′′′) and δC 79.2 (C-15), suggesting the attachment position of the glucose at C-15. Assignments of all groups of compound 4 were achieved through 1H-1H COSY, HSQC, and HMBC (Figure 2). The absolute configuration of the glycosyls group of compound 4 was determined by GC analysis after derivatization. The nuclear overhauser effect (NOESY) correlation of Me-19/H-1β, H-5; H-1α/H-3, Me-18/H-15, indicated the α-orientation of H-3 and β-orientation of H-15. According to the literature [11], the absolute configuration of C-25 was determined by the gap between two hydrogen protons (∆ab) in C-26. The absolute configuration of C-25 in compound 4 was R, based on the ∆ab = 0.45 gap (∆ab ≤ 0.48 was R, ∆ab ≥ 0.59 was S). The absolute configuration of glucopyranosyl groups of compound 4 was determined as D by GC analysis. Synthesizing the above results, the structure of compound 4 was determined to be (25R)-15-O-β-d-glucopyranosyl-26-O-β-d-glucopyranosyl-22- hydroxy-5β-furost-3β,15α,26-diol-3-O-β-d-glucopyranosyl(1→2)-β-d-galactopyranoside, and named timosaponin U.
3.2. Cytotoxic Activity
The compounds 1–4 were evaluated against three human tumor cell lines (HepG2, Hela, and SGC7901) for their cytotoxic activities using the MTT method. The research results showed that aneglycoside A and B (compounds 1 and 2) showed weak cytotoxicity against HepG2 and Hela cells with an IC50 value of 38.4, 29.7, and 41.7, 34.2 μM, respectively. Timosaponin U (compound 4) exhibited weak cytotoxicity against HepG2, Hela, and SGC7901 cells with IC50 values of 61.8, 39.7, and 44.5 μM, respectively. However, compound 3 displayed no cytotoxicity activity against these three human tumor cells, and compounds 1 and 2 did not show significant cytotoxicity activity on SGC7901 (Table 4).
4. Conclusions
A. asphodeloidesis possesses many kinds of bioactivities, such as anti-oxidant, anti-tumor, anti-inflammation, and blood sugar reduction capabilities, and has been applied to the treatment of febrile diseases with high fever. This study obtained four new glycosides compounds from A. asphodeloidesis, including pyroglutamic acid fructosides and steroidal saponins, and their cytotoxicity activities were evaluated. These results indicated these glycoside compounds could be the pharmacodynamic material basis for A. asphodeloidesis. Further study on the chemical constituents of A. asphodeloidesis could contribute to discovering active ingredients and leading compounds, and provide an experimental and scientific basis for drug design and drug discovery.
Supplementary Materials
Supplementary Materials are available online.
Acknowledgments
This project was supported by the Major State Basic Research Development Program (973 Program) of China (2013CB531801) and the National Natural Science Foundation of China (No. 81274103, 81773904).
Author Contributions
B.-Y.Y. and H.-X.K. designed the experiments; X.-Y.B. performed the experiments; Y.L. and G.Y.L wrote the paper. X.Y. modified the paper; all authors read and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peng, Y.; Zhang, Y.J.; Ma, Z.Q.; Pan, W.S.; Sun, Y.Q.; Song, S.J. Two new saponins from Anemarrhena asphodeloides Bge. Chin. Chem. Lett. 2007, 18, 171–174. [Google Scholar] [CrossRef]
- Saito, S.; Nagase, S.; Ichinose, K. New steroidal saponins from the Rhizomes of Anemarrhena asphodeloides Bunge (Liliaceae). Chem. Pharm. Bull. 1994, 42, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Xu, S.X.; Huang, S.; Wang, Z.X. A Study on the chemical constituents of Anemarrhena asphodeloides Bge. J. Shenyang Pharm. Univ. 1996, 13, 34–39. [Google Scholar]
- Zhang, C.; Xu, X.Z.; Li, C. Fructosides from Cynomorium songaricum. Phytochemistry 1996, 41, 975–976. [Google Scholar] [CrossRef]
- Wu, S.B.; Meyer, R.S.; Whitaker, B.D.; Litt, A.; Kennelly, E.J. Antioxidant glucosylated caffeoylquinic acid derivatives in the invasive tropica soda apple, Solanum viarum. J. Nat. Prod. 2012, 75, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Guo, R.; Li, T.; Liu, Y.; Wang, C.F.; Shu, Z.P.; Wang, Z.B.; Zhang, J.; Xia, Y.G.; Jiang, H.; et al. Five withanolides from the leaves of Datura metel L. and their inhibitory effects on nitric oxide production. Molecules 2014, 19, 4548–4559. [Google Scholar] [CrossRef] [PubMed]
- Greenaway, W.; Whatley, F.R. Enzymic synthesis of l-glutamic acid-15N and 4-aminobutyric acid-15N and the preparation of l-pyroglutamic acio-15N. J. Label. Compd. Radiopharm. 1975, 11, 395–400. [Google Scholar] [CrossRef]
- Hao, X.Y.; Tan, N.H.; Zhou, J. Chemical Constituents of Gastrodia elata Bl. Yunnan Plant Res. 2000, 22, 81–84. [Google Scholar]
- Zhang, Z.; Wang, D.; Zhao, Y.; Gao, H.; Hu, Y.H.; Hu, J.F. Fructose-derived carbohydrates from Alisma orientalis. Nat. Prod. Res. 2009, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Zhang, J.; Liu, Y.; Kuang, H.X. Steroidal saponins from the rhizomes of Anemarrhena asphodeloides. Molecules 2016, 21, 1075. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, A.; Mimaki, Y. Steroidal saponins from the whole plants of Agave utahensis and their cytotoxic activity. Phytochemistry 2009, 70, 807–815. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–4 are available from the authors. |
Figure 1.
Structures of compounds 1–4 from Anemarrhena asphodeloides.

Figure 2.
Key heteronuclear multiple bond correlation (HMBC) and 1H-1H correlation spectroscopy (COSY) correlations of compounds 1–4.
Figure 2.
Key heteronuclear multiple bond correlation (HMBC) and 1H-1H correlation spectroscopy (COSY) correlations of compounds 1–4.

{kind=link}
{kind=link}
{kind=link}
Table 1.
1H-nuclear magnetic resonance (NMR) and 13C-NMR data for compounds 1 and 2 (CD3OD).
| No. | Compound 1 | Compound 2 | ||
|---|---|---|---|---|
| δC | δH Mult (J, Hz) | δC | δH Mult (J, Hz) | |
| Sugar | ||||
| 1 | 64.0 | 4.35 (d, 11.6) | 61.7 | 3.65 (d, 11.8) |
| 4.24 (d, 11.6) | 3.52 (d, 11.8) | |||
| 2 | 102.9 | 105.6 | ||
| 3 | 80.2 | 3.96 (d, 8.0) | 78.1 | 4.12 (d, 8.0) |
| 4 | 76.8 | 3.91 (d, 8.0) | 77.4 | 3.99 (d, 8.0) |
| 5 | 83.5 | 3.71 overlap | 80.0 | 3.92 overlap |
| 6 | 64.7 | 3.73 overlap | 67.5 | 4.28 overlap |
| 3.56 (m) | ||||
| Pyroglutamic Acid | ||||
| 2′ | 173.4 | 173.8 | ||
| 3′ | 25.8 | 2.49 (m) | 25.9 | 2.49 (m) |
| 2.23 (m) | 2.21 (m) | |||
| 4′ | 30.2 | 2.33 (m) | 30.2 | 2.34 (m) |
| 5′ | 57.1 | 4.32 (dd, 8.0, 4.0) | 57.0 | 4.30 (m) |
| 6′ | 181.2 | 181.1 | ||
| n-butyl | ||||
| 1″ | 62.3 | 3.74 (m) | 62.3 | 3.61 (m) |
| 3.52 (m) | 3.45 (m) | |||
| 2″ | 33.2 | 1.53 (m) | 33.4 | 1.52 (m) |
| 3″ | 20.3 | 1.37 (m) | 20.3 | 1.37 (m) |
| 4″ | 14.3 | 0.92 (3H, t, 7.2) | 14.4 | 0.92 (3H, t, 7.2) |
Table 2.
The 1H- and 13C-NMR data of compound 3 (D2O).
| No. | δC | δH Mult (J, Hz) | No. | δC | δH Mult (J, Hz) |
|---|---|---|---|---|---|
| α-Fruf | β-Fruf | ||||
| 1 | 98.8 | 3.60 (d, 12.3) | 1′ | 62.5 | 3.52 (d, 12.3) |
| 4.13 (d, 12.3) | 4.06 (d, 12.3) | ||||
| 2 | 81.8 | 2′ | 102.5 | ||
| 3 | 77.7 | 3.94 (d, 2.4) | 3′ | 76.9 | 3.67 (d, 7.8) |
| 4 | 83.5 | 3.86 (dd, 2.4, 5.7) | 4′ | 74.5 | 4.02 (d, 7.8) |
| 5 | 61.2 | 3.92 (m) | 5′ | 81.2 | 3.83 (m) |
| 6 | 61.7 | 3.68 (d, 12.3) | 6′ | 62.6 | 3.58 (d, 12.4) |
| 3.77 (d, 12.3) | 3.77 (d, 12.4) |
Table 3.
1H- and 13C-NMR data for compound 4 (C5D5N).
| No. | δC | δH Mult (J, Hz) | No. | δC | δH Mult (J, Hz) |
|---|---|---|---|---|---|
| 1 | 31.2 | 1.86 (m) | 3-gal | ||
| 1.48 (m) | 1′ | 102.4 | 4.89 (d, 7.6) | ||
| 2 | 27.2 | 1.84 (m) | 2′ | 81.6 | 4.65 (m) |
| 1.29 (m) | 3′ | 76.9 | 4.03 (m) | ||
| 3 | 75.5 | 4.32 (m) | 4′ | 69.9 | 4.54 (m) |
| 4 | 31.0 | 1.48 (m) | 5′ | 76.6 | 4.07 (m) |
| 1.85 (m) | 6′ | 62.2 | 2.06 (m) | ||
| 5 | 37.0 | 2.15 (m) | 4.42 (m) | ||
| 6 | 27.1 | 1.92 (m) | glc | ||
| 1.52 (m) | 1″ | 106.0 | 5.27 (d, 7.6) | ||
| 7 | 27.0 | 1.87 (m) | 2″ | 75.3 | 4.37 (m) |
| 1.28 (m) | 3″ | 78.1 | 4.18 (m) | ||
| 8 | 36.5 | 1.85 (m) | 4″ | 71.8 | 4.27 (m) |
| 9 | 40.9 | 1.21 (m) | 5″ | 78.4 | 3.83 (m) |
| 10 | 35.5 | 6″ | 62.8 | 4.52 (m) | |
| 11 | 21.3 | 1.26 (m) | 4.32 (m) | ||
| 1.37 (m) | 15-glc | ||||
| 12 | 41.5 | 1.21 (m) | 1′′′ | 105.1 | 4.80 (d, 7.8) |
| 1.69 (m) | 2′′′ | 75.3 | 4.27 (m) | ||
| 13 | 41.4 | 3′′′ | 78.5 | 3.82 (m) | |
| 14 | 61.0 | 1.62 (m) | 4′′′ | 71.8 | 4.18 (m) |
| 15 | 79.2 | 4.38 (m) | 5′′′ | 78.6 | 3.95 (m) |
| 16 | 91.4 | 5.06 (dd, 3.6, 8.7) | 6′′′ | 62.8 | 4.52 (m) |
| 17 | 61.5 | 2.25 (m) | 4.32 (m) | ||
| 18 | 18.2 | 0.95 (3H, s) | 26-glc | ||
| 19 | 24.3 | 0.89 (3H, s) | 1′′′′ | 105.2 | 4.83 (d, 7.8) |
| 20 | 40.5 | 1.42 (m) | 2′′′′ | 75.3 | 4.27 (m) |
| 21 | 16.6 | 1.32 (3H, d, 6.8) | 3′′′′ | 78.7 | 4.19 (m) |
| 22 | 110.5 | 4′′′′ | 71.8 | 4.23 (m) | |
| 23 | 37.2 | 1.98 (m) | 5′′′′ | 78.6 | 3.95 (m) |
| 2.12 (m) | 6′′′′ | 62.9 | 4.53 (m) | ||
| 24 | 28.5 | 1.72 (m) | 4.36 (m) | ||
| 2.09 (m) | |||||
| 25 | 34.5 | 1.92 (m) | |||
| 26 | 75.1 | 3.55 (m) | |||
| 4.00 (m) | |||||
| 27 | 17.6 | 1.03 (3H, d, 6.7) |
Table 4.
Cytotoxicity of compounds 1–4.
| Compound | IC50 (μM) | Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| HepG2 | Hela | SGC7901 | HepG2 | Hela | SGC7901 | ||
| 1 | 38.4 ± 2.4 | 29.7 ± 1.9 | >100 | 4 | 61.8 ± 4.1 | 39.7 ± 3.7 | 44.5 ± 2.0 |
| 2 | 41.8 ± 3.5 | 34.2 ± 3.6 | >100 | doxorubicin | 8.4 ± 2.2 | 9.0 ± 1.4 | 6.7 ± 1.8 |
| 3 | >100 | >100 | >100 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, B.-Y.; Bi, X.-Y.; Liu, Y.; Li, G.-Y.; Yin, X.; Kuang, H.-X. Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides. Molecules 2017, 22, 1995. https://doi.org/10.3390/molecules22111995
AMA Style
Yang B-Y, Bi X-Y, Liu Y, Li G-Y, Yin X, Kuang H-X. Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides. Molecules. 2017; 22(11):1995. https://doi.org/10.3390/molecules22111995
Chicago/Turabian StyleYang, Bing-You, Xue-Yan Bi, Yan Liu, Guo-Yu Li, Xin Yin, and Hai-Xue Kuang. 2017. "Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides" Molecules 22, no. 11: 1995. https://doi.org/10.3390/molecules22111995