Furanones and Anthranilic Acid Derivatives from the Endophytic Fungus Dendrothyrium variisporum


Abstract
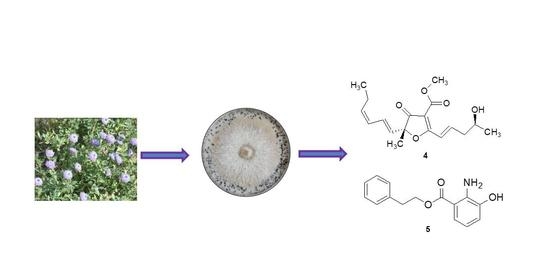
:
1. Introduction
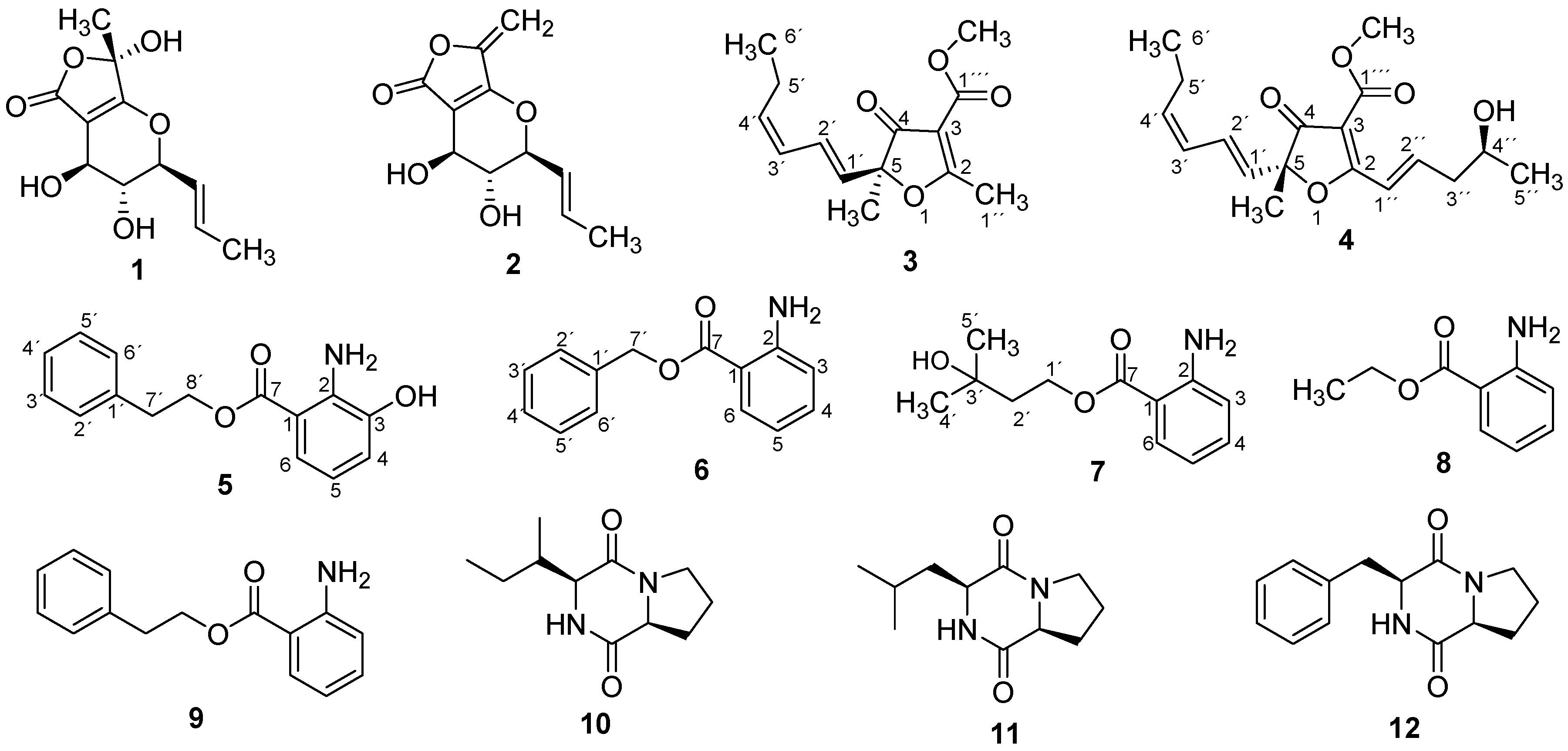
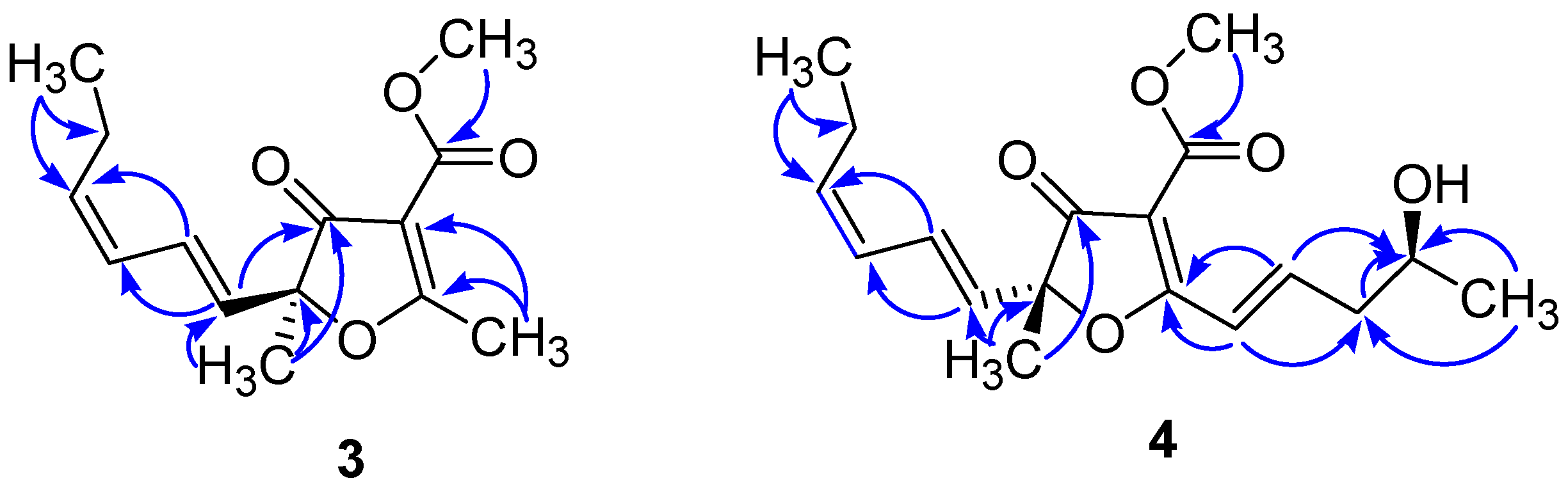
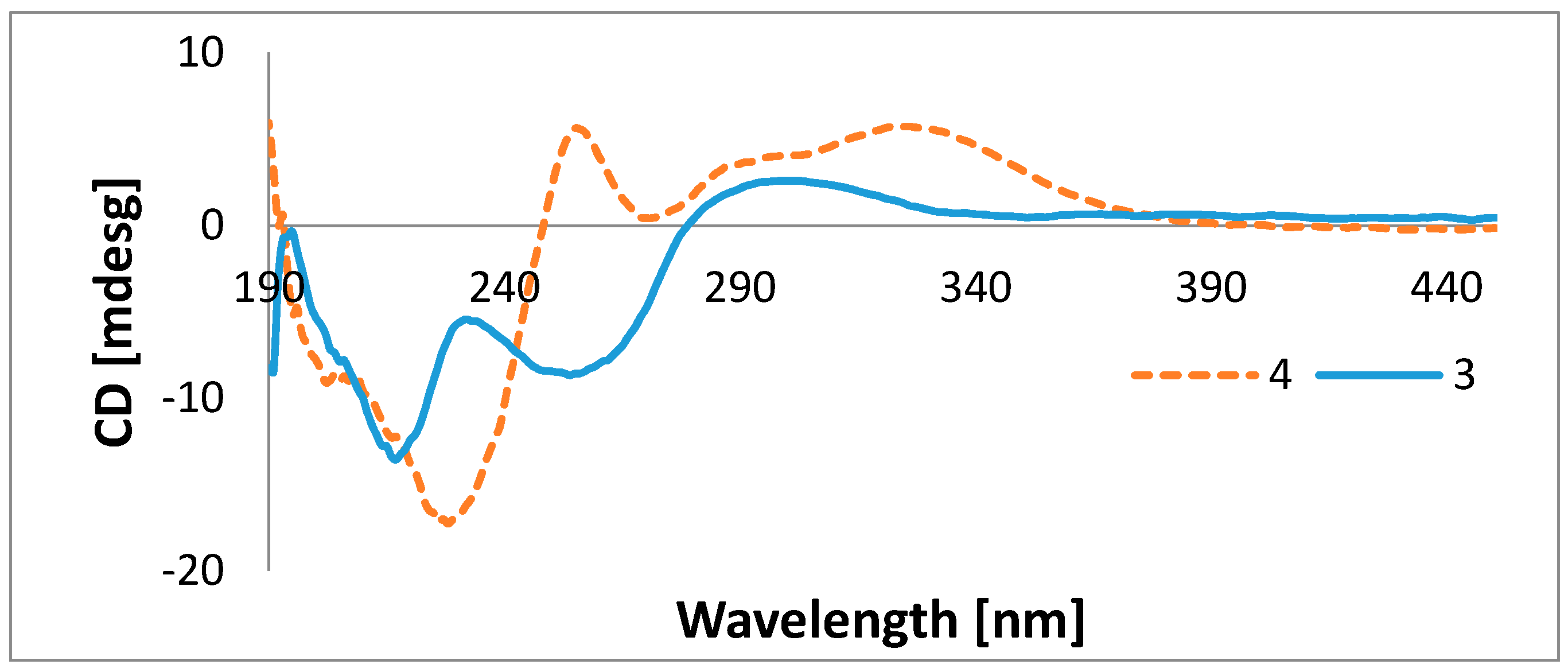
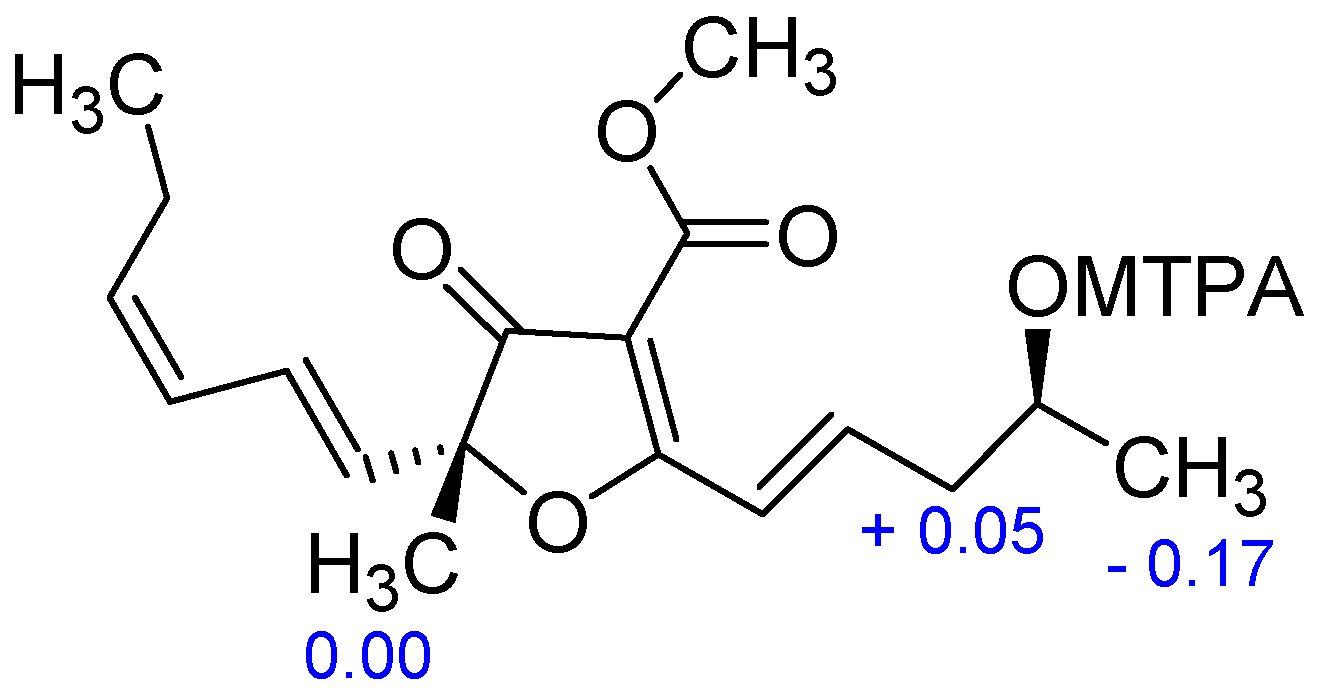
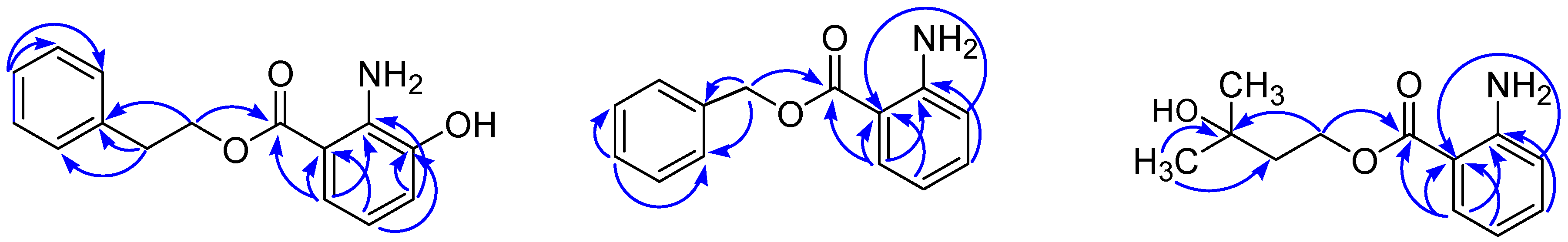
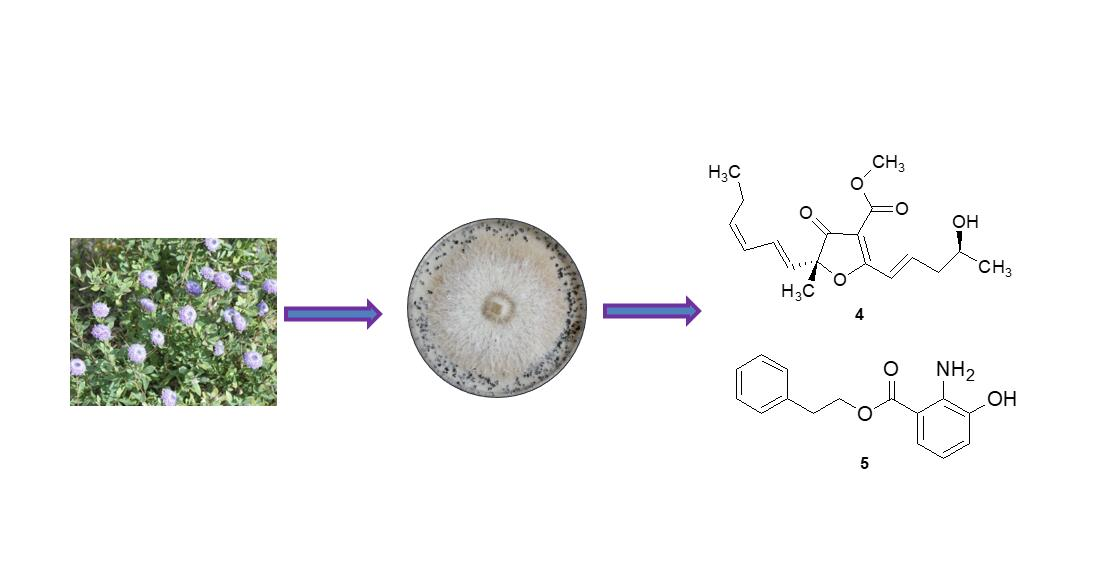
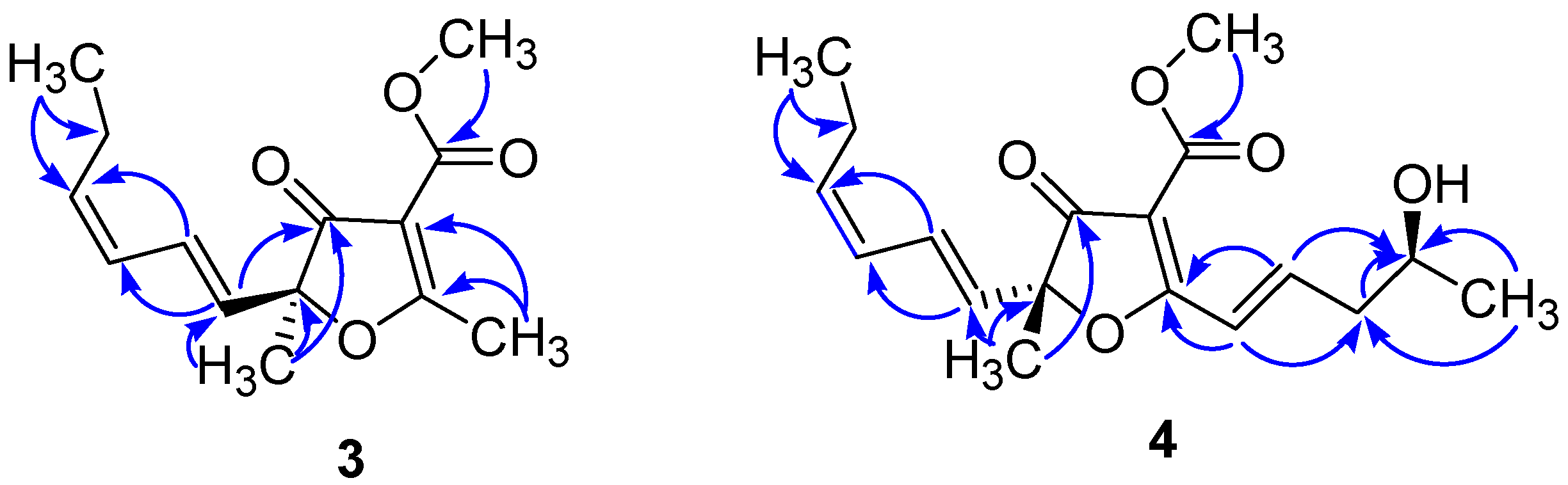
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation of Compounds 1–12
3.5. Preparation of the S- and R-MTPA Esters of 4
3.6. Biological Activities
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karwehl, S.; Stadler, M. Exploitation of fungal biodiversity for discovery of novel antibiotics. Curr. Top. Microbiol. Immunol. 2016, 398, 303–338. [Google Scholar] [PubMed]
- Noumeur, S.R.; Helaly, S.E.; Jansen, R.; Gereke, M.; Stradal, T.E.B.; Harzallah, D.; Stadler, M. Preussilides A−F, bicyclic polyketides from the endophytic fungus Preussia similis with antiproliferative activity. J. Nat. Prod. 2017, 80, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Verkley, G.J.M.; Dukik, K.; Renfurm, R.; Göker, M.; Stielow, J.B. Novel genera and species of the coniothyrium-like fungi in Montagnulaceae (Ascomycota). Persoonia 2014, 32, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Chepkirui, C.; Richter, C.; Matasyoh, J.C.; Stadler, M. Monochlorinated calocerins A–D and 9-oxostrobilurin derivatives from the basidiomycete Favolaschia calocera. Phytochemistry 2016, 132, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.; El Aouad, N.; Martín, J.; Perez-Victoria, I.; Gonzalez-Menendez, V.; Platas, G.; De la Cruz, M.; Monteiro, M.C.; De Pedro, N.; Bills, G.F.; et al. Graminin B, a furanone from the fungus Paraconiothyrium sp. J. Antibiot. 2014, 67, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, E.M.K.; Xu, Y.; Arnold, A.E.; Gunatilaka, A.A.L. Graminin C, and cis-Gregatin B–New natural furanones from Pulvinula sp. 11120, a fungal endophyte of Cupressus arizonica. Nat. Prod. Commun. 2015, 10, 107–111. [Google Scholar] [PubMed]
- Tang, H.-Y.; Zhang, Q.; Gao, Y.-Q.; Zhang, A.-L.; Gao, J.-M. Miniolins A–C, novel isomeric furanones induced by epigenetic manipulation of Penicillium minioluteum. RSC Adv. 2015, 5, 2185–2190. [Google Scholar] [CrossRef]
- Hoye, T. R.; Jeffrey, C. S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Burghart-Stoll, H.; Brückner, R. Total syntheses of the gregatins A–D and aspertetronin A: Structure revisions of these compounds and of aspertetronin B, together with plausible structure revisions of gregatin E, cyclogregatin, graminin A, the penicilliols A and B, and the huaspenones A and B. Eur. J. Org. Chem. 2012, 3978–4017. [Google Scholar]
- Li, C.-S.; Li, X.-M.; Gao, S.-S.; Lu, Y.-H.; Wang, B.-G. Cytotoxic anthranilic acid derivatives from deep sea sediment-derived fungus Penicillium paneum SD-44. Mar. Drugs 2013, 11, 3068–3076. [Google Scholar] [CrossRef] [PubMed]
- Miltojević, A.B.; Radulović, N.S. Complete assignment of 1H- and 13C-NMR spectra of anthranilic acid and its hydroxyl derivatives and salicylic acid and its amino derivatives. FU Phys. Chem. Technol. 2015, 13, 121–132. [Google Scholar] [CrossRef]
- Hirata, M.; Isoda, S.; Kanao, M.; Shimizu, H.; Inoue, S. Anesthetics for fish. Nippon Suisan Gakk. 1970, 36, 1127–1135. [Google Scholar] [CrossRef]
- Kock, I.; Krohn, K.; Egold, H.; Draeger, S.; Schulz, B.; Rheinheimer, J. New massarilactones, massarigenin E, and coniothyrenol, isolated from the endophytic fungus Coniothyrium sp. from Carpobrotus edulis. Eur. J. Org. Chem. 2007, 2186–2190. [Google Scholar]
- Zhang, G.F.; Han, W.B.; Cui, J.T.; Ng, S.W.; Guo, Z.K.; Tan, R.X.; Ge, H.M. Neuraminidase inhibitory polyketides from the marine-derived fungus Phoma herbarum. Planta Med. 2012, 78, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Qian, M.C. Quantification of selected aroma-active compounds in Pinot Noir wines from different grape maturities. J. Agric. Food Chem. 2006, 54, 8567–8573. [Google Scholar] [CrossRef] [PubMed]
- Raina, V.K.; Srivastava, S.K.; Aggarwal, K.K.; Ramesh, S.; Kumar, S. Essential oil composition of Cinnamomum zeylanicum Blume leaves from Little Andaman, India. Flavour Frag. J. 2001, 16, 374–376. [Google Scholar] [CrossRef]
- Ren, S.; Ma, W.; Xu, T.; Lin, X.; Yin, H.; Yang, B.; Zhou, X.-F.; Yang, X.-W.; Long, L.; Lee, K.J.; et al. Two novel alkaloids from the South China Sea marine sponge Dysidea sp. J. Antibiot. 2010, 63, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Sansinenea, E.; Salazar, F.; Jiménez, J.; Mendoza, A.; Ortiz, A. Diketopiperazines derivatives isolated from Bacillus thuringiensis and Bacillus endophyticus, establishment of their configuration by X-ray and their synthesis. Tetrahedron Lett. 2016, 57, 2604–2607. [Google Scholar] [CrossRef]
- Kusari, S.; Lamshöft, M.; Zühlke, S.; Spiteller, M. An endophytic fungus from Hypericum perforatum that produces hypericin. J. Nat. Prod. 2008, 71, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Helaly, S.E.; Thongbai, B.; Hyde, K.D.; Stadler, M. Pyristriatins A and B: Pyridino-cyathane antibiotics from the basidiomycete Cyathus cf. striatus. J. Nat. Prod. 2016, 79, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sudarman, E.; Kuhnert, E.; Hyde, K.D.; Esteban Benjamin Sir, E.B.; Surup, F.; Stadler, M. Truncatones A–D, benzo[j]fluoranthenes from Annulohypoxylon species (Xylariaceae, Ascomycota). Tetrahedron 2016, 72, 6450–6454. [Google Scholar] [CrossRef]
- Wittstein, K.; Rascher, M.; Rupcic, Z.; Löwen, E.; Winter, B.; Köster, R.W.; Stadler, M. Corallocins A-C, Nerve growth and brain-derived neurotrophic factor inducing metabolites from the Mushroom Hericium coralloides. J. Nat. Prod. 2016, 79, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1, 2, 10–12 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
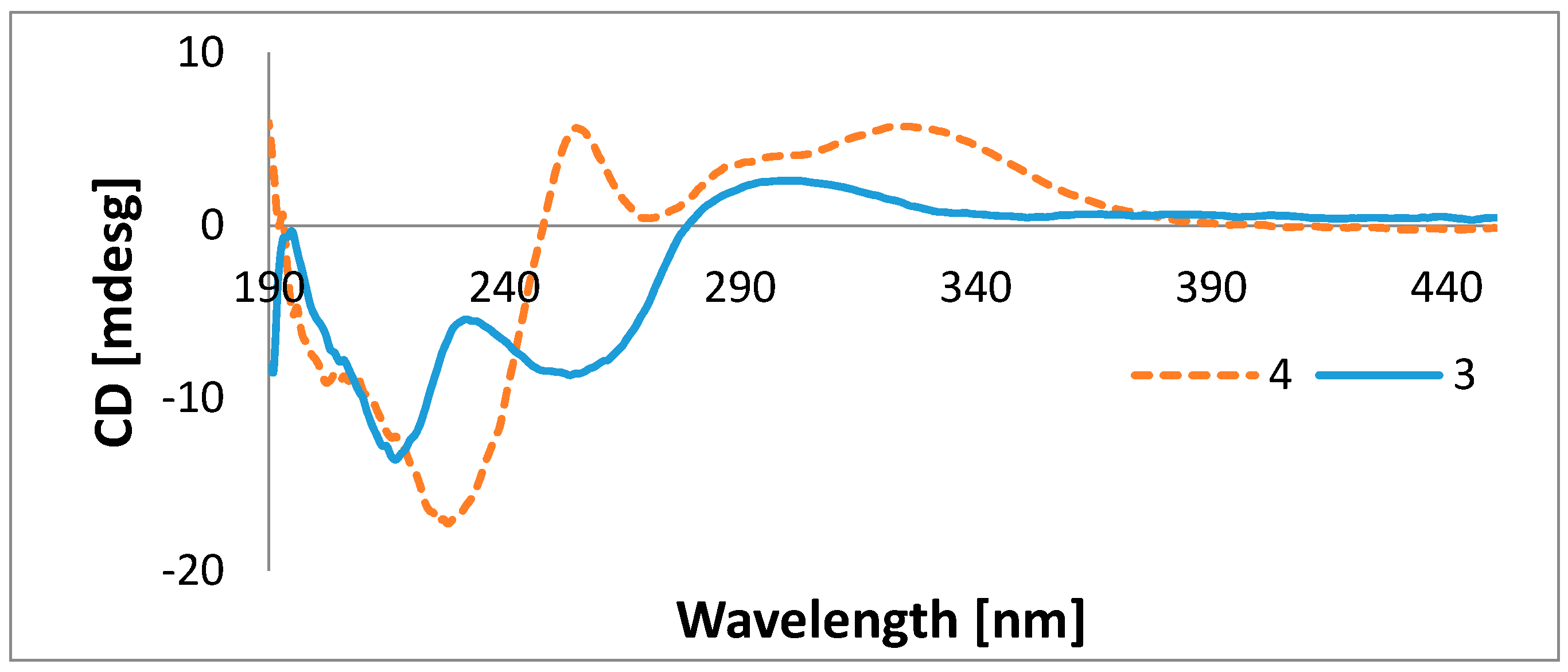{kind=link}
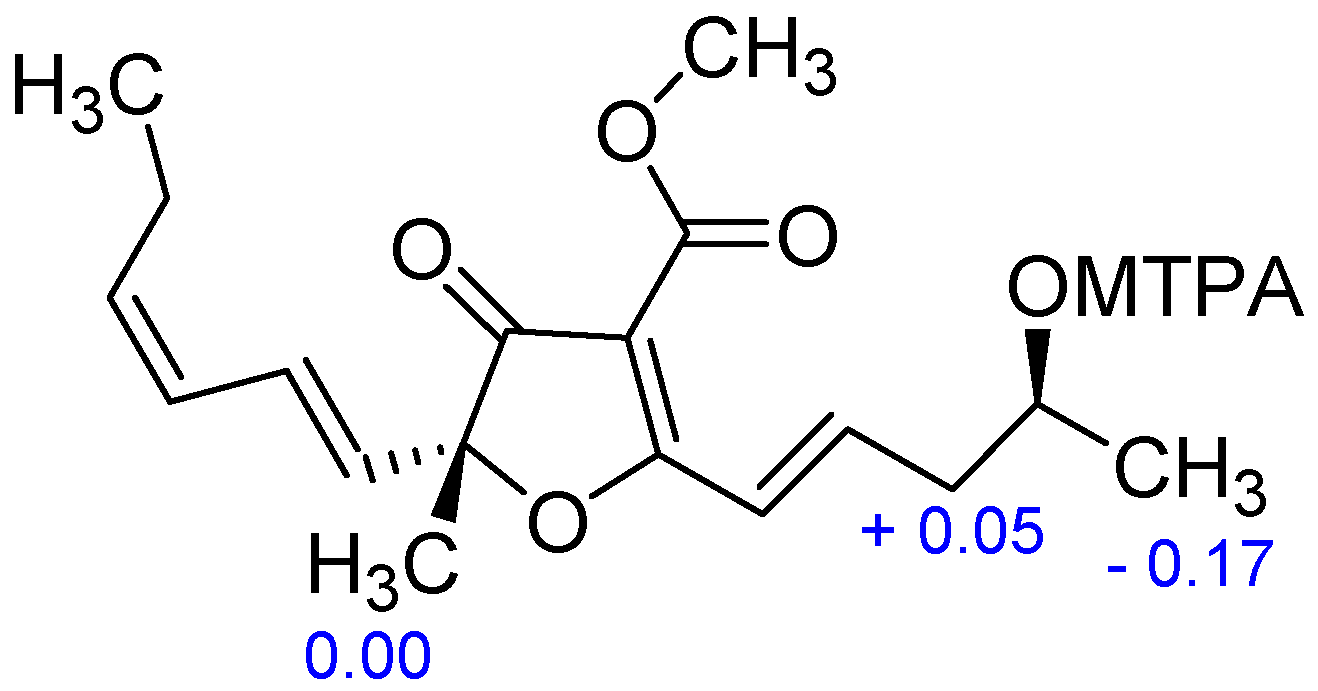{kind=link}
{kind=link}
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 195.6, C | / | 184.3, C | / |
| 3 | 106.8, C | / | 104.3, C | / |
| 4 | 197.8, C | / | 198.2, C | / |
| 5 | 91.4, C | / | 90.7, C | / |
| 1′ | 127.6, CH | 5.63, d (15.5) | 128.2, CH | 5.65, d (15.5) |
| 2′ | 126.5, CH | 6.59, ddd (15.5, 11.2, 1.3) | 126.5, CH | 6.59, ddd (15.5, 11.2, 1.3) |
| 3′ | 126.2, CH | 5.90, t (11.2) | 126.3, CH | 5.90, t (11.2) |
| 4′ | 136.9, CH | 5.53, d (10.8, 7.7) | 136.7, CH | 5.52, dt (10.8, 7.7) |
| 5′ | 21.2, CH2 | 2.21, m | 21.1, CH2 | 2.19, m |
| 6′ | 14.1, CH3 | 1.01, t (7.3) | 14.1, CH3 | 1.00, t (7.3) |
| 1′′ | 121.6, CH | 7.41, dt (15.9, 1.7) | ||
| 2′′ | 145.1, CH | 7.21, dt (15.9, 7.3) | ||
| 3′′ | 42.9, CH2 | 2.55, m | ||
| 4′′ | 67.0, CH | 4.08, m | ||
| 5′′ | 23.6, CH3 | 1.31, d (6.2) | ||
| 1′′′ | 163.4, C | / | 163.4, C | / |
| Me-5 | 22.5, CH3 | 1.56, s | 22.5, CH3 | 1.58, s |
| OMe | 51.6, CH3 | 3.83, s | 51.7, CH3 | 3.85, s |
| Position | 5 a | 6 a | 7 c | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 115.0, C | / | 111.7, C | / | 112.3, C | / |
| 2 | 137.5, C | / | 152.5, C | / | 151.9, C | / |
| 3 | 147.8, C | / | 118.2, CH | 6.77, dd (8.3, 1.0) | 118.4, CH | 6.78, dd (8.3, 1.0) |
| 4 | 119.5, CH | 6.90, dd (7.8, 1.4) | 135.4, CH | 7.25, ddd (8.5, 7.0, 1.6) | 135.2, CH | 7.26, ddd (8.5, 7.1, 1.6) |
| 5 | 119.0, CH | 6.66, t (7.9) | 117.2, CH | 6.60, ddd (8.1, 7.1, 1.1) | 117.5, CH | 6.62, ddd (8.2, 7.1, 1.1) |
| 6 | 122.8, CH | 7.35, dd (8.1, 1.4) | 132.2, CH | 7.83, dd ( 8.1, 1.6) | 132.2, CH | 7.81, dd ( 8.1, 1.6) |
| 7 | 169.2, C | / | 169.3, C | / | 169.6, C | / |
| 1′ | 139.7, C | / | 138.2, C | / | 62.4, CH2 | 4.41, t (7.0) |
| 2′ | 130.2, CH | 7.30, m b | 129.2, CH | 7.44, dd (8.0, 0.9) | 43.0, CH2 | 1.94, t (7.0) |
| 3′ | 129.7, CH | 7.30, m b | 129.7, CH | 7.38, td (7.1, 1.6) | 70.6, C | / |
| 4′ | 127.7, CH | 7.22, m | 129.3, CH | 7.31, m | 29.8, CH3 | 1.28, s |
| 5′ | 129.7, CH | 7.30, m b | 129.7, CH | 7.38, td (7.1, 1.6) | 29.8, CH3 | 1.28, s |
| 6′ | 130.2, CH | 7.30, m b | 129.2, CH | 7.44, dd (8.0, 0.9) | ||
| 7′ | 36.3, CH2 | 3.06, t (6.9) | 67.1, CH2 | 5.31, s | ||
| 8′ | 66.5, CH2 | 4.48, t (6.9) | ||||
| Test Organism | MIC (μg/mL) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | References | |
| Schizosaccharomyces pombe DSM 70572 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 16.66 n |
| Pichia anomala DSM 6766 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 8.33 n |
| Mucor hiemalis DSM 2656 | n.a. | n.a. | n.a. | 33.33 | n.a. | n.a. | n.a. | 16.66 | n.a. | n.a. | n.a. | 16.66 n |
| Candida albicans DSM 1665 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 16.66 n |
| Rhodoturula glutinis DSM 10134 | n.a. | n.a. | n.a. | 66.67 | n.a. | n.a. | n.a. | 33.33 | n.a. | n.a. | n.a. | 2.08 n |
| Micrococcus luteus DSM 1790 | n.a. | n.a. | n.a. | 16.66 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 0.40 o |
| Bacillus subtilis DSM 10 | n.a. | n.a. | n.a. | 8.33 | n.a. | n.a. | n.a. | 66.67 | n.a. | n.a. | n.a. | 4.16 o |
| Escherichia coli DSM 1116 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 3.33 o |
| Staphylococcus aureus DSM 346 | n.a. | n.a. | n.a. | 66.67 | n.a. | n.a. | n.a. | 66.67 | n.a. | n.a. | n.a. | 0.10 o |
| Mycobacterium smegmatis DSM ATCC 700084 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 2.08 k |
| Chromobacterium violaceum DSM 30191 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 0.40 o |
| Pseudomonas aeruginosa DSM PA14 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 0.52 g |
| IC50 (μg/mL) | IC50 (ng/mL) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Line | 1 | 2 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Epothilone B |
| KB3.1 | n.a. | n.a. | 18 | 18 | n.a. | n.a. | n.a. | 8.8 | n.a. | n.a. | n.a. | 0.052 |
| L929 | n.a. | n.a. | 19 | no | n.a. | n.a. | n.a. | n.o. | n.a. | n.a. | n.a. | 1.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teponno, R.B.; Noumeur, S.R.; Helaly, S.E.; Hüttel, S.; Harzallah, D.; Stadler, M. Furanones and Anthranilic Acid Derivatives from the Endophytic Fungus Dendrothyrium variisporum. Molecules 2017, 22, 1674. https://doi.org/10.3390/molecules22101674
Teponno RB, Noumeur SR, Helaly SE, Hüttel S, Harzallah D, Stadler M. Furanones and Anthranilic Acid Derivatives from the Endophytic Fungus Dendrothyrium variisporum. Molecules. 2017; 22(10):1674. https://doi.org/10.3390/molecules22101674
Chicago/Turabian StyleTeponno, Rémy B., Sara R. Noumeur, Soleiman E. Helaly, Stephan Hüttel, Daoud Harzallah, and Marc Stadler. 2017. "Furanones and Anthranilic Acid Derivatives from the Endophytic Fungus Dendrothyrium variisporum" Molecules 22, no. 10: 1674. https://doi.org/10.3390/molecules22101674