Solid-State Form Characterization of Riparin I

Abstract
:1. Introduction
2. Results and Discussion
2.1. Scanning Electron Microscopy (SEM)
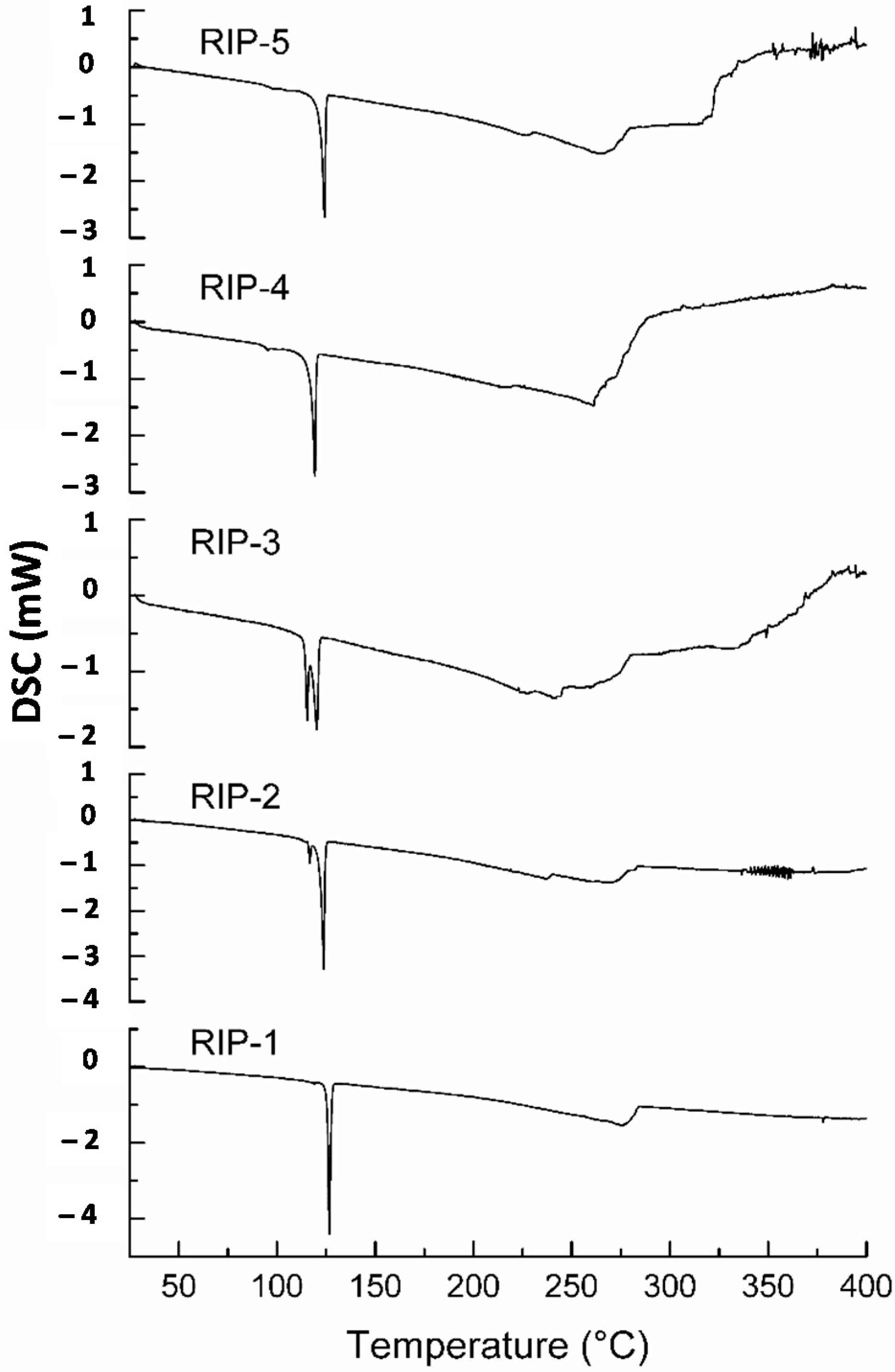
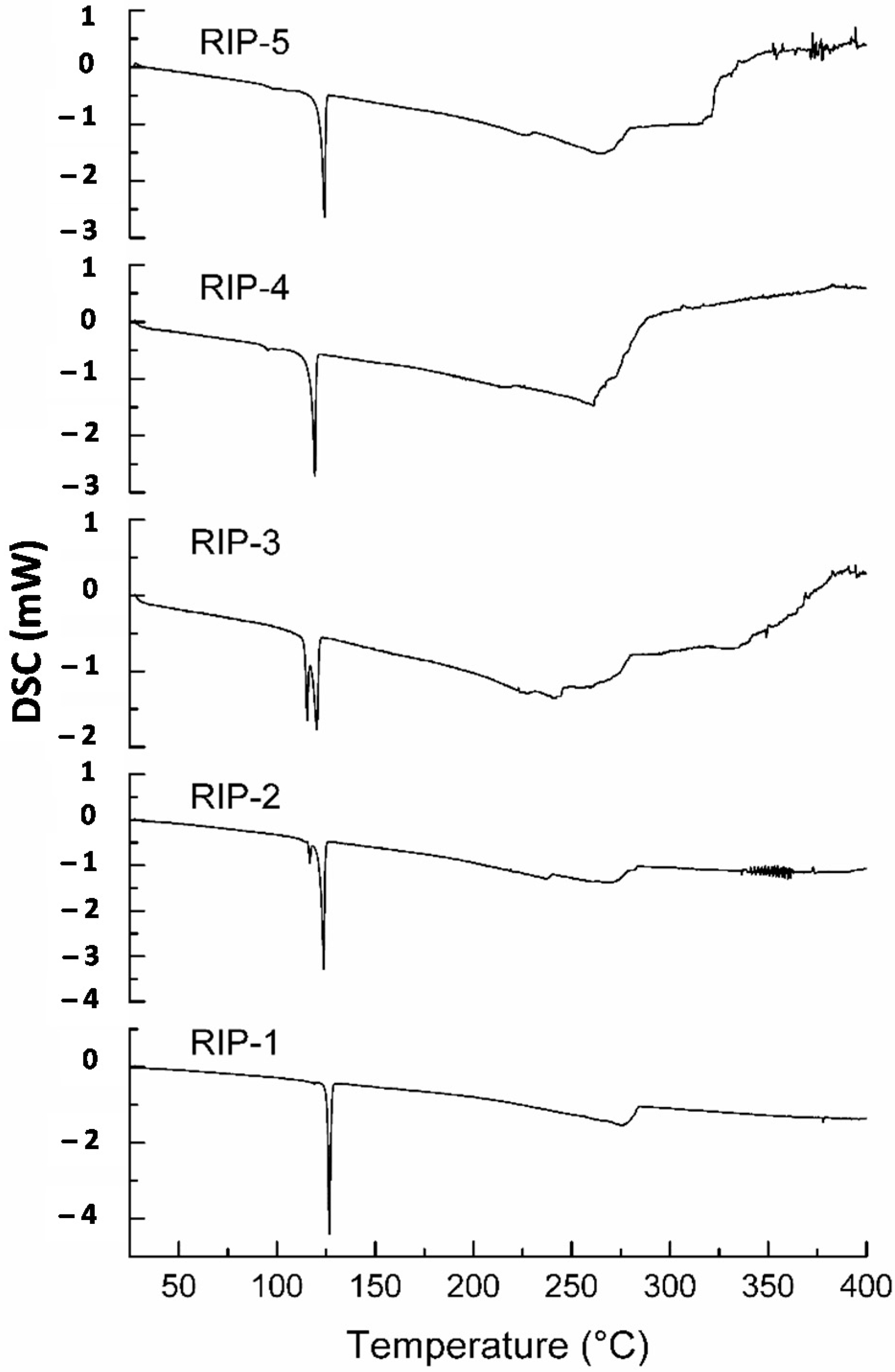
2.2. Differential Scanning Calorimetry (DSC)
2.3. DSC Coupled with the System Photovisual
2.4. DSC Coupled with the System Photovisual
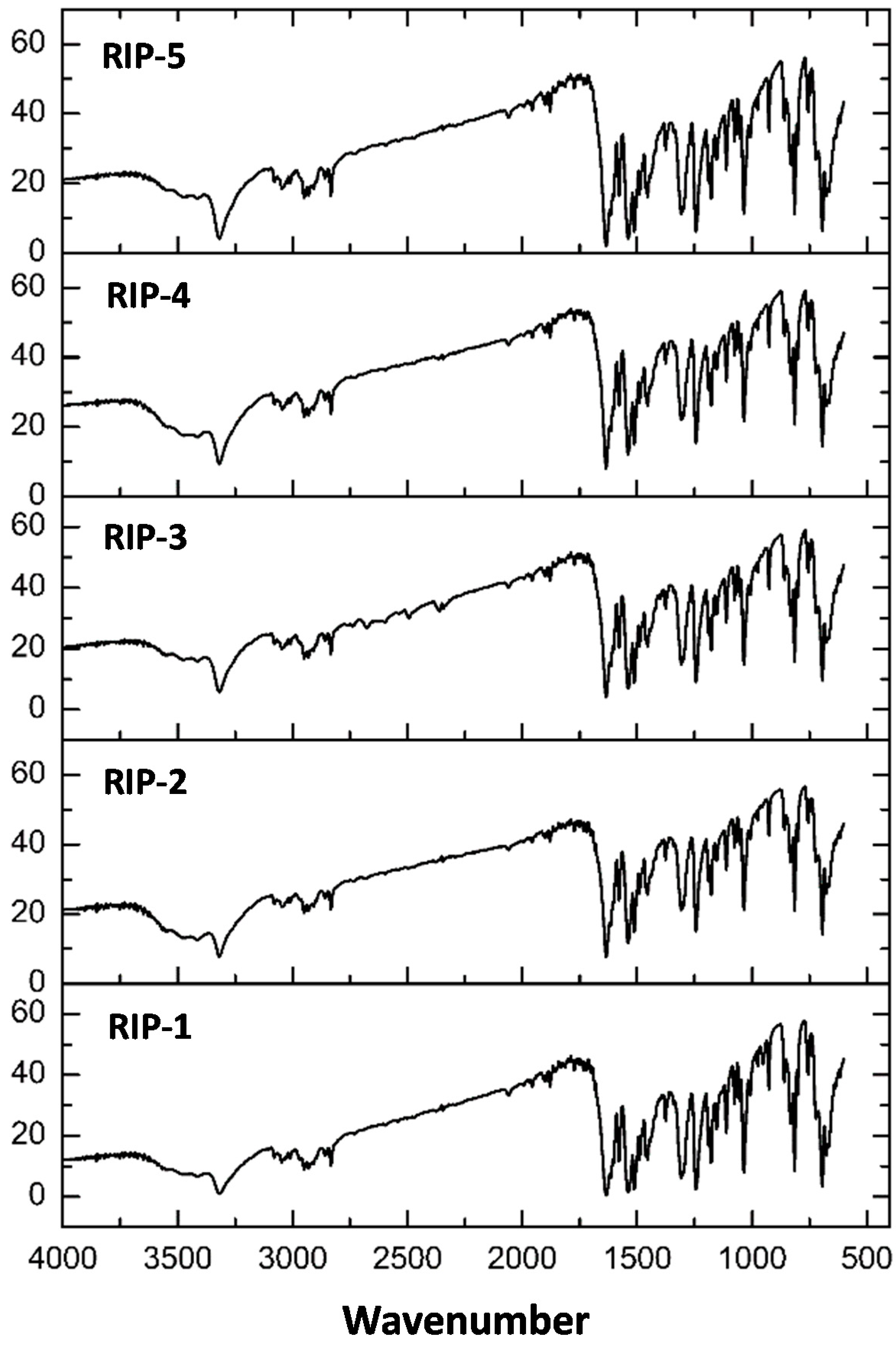
2.5. Fourier Transform Infrared (FTIR)
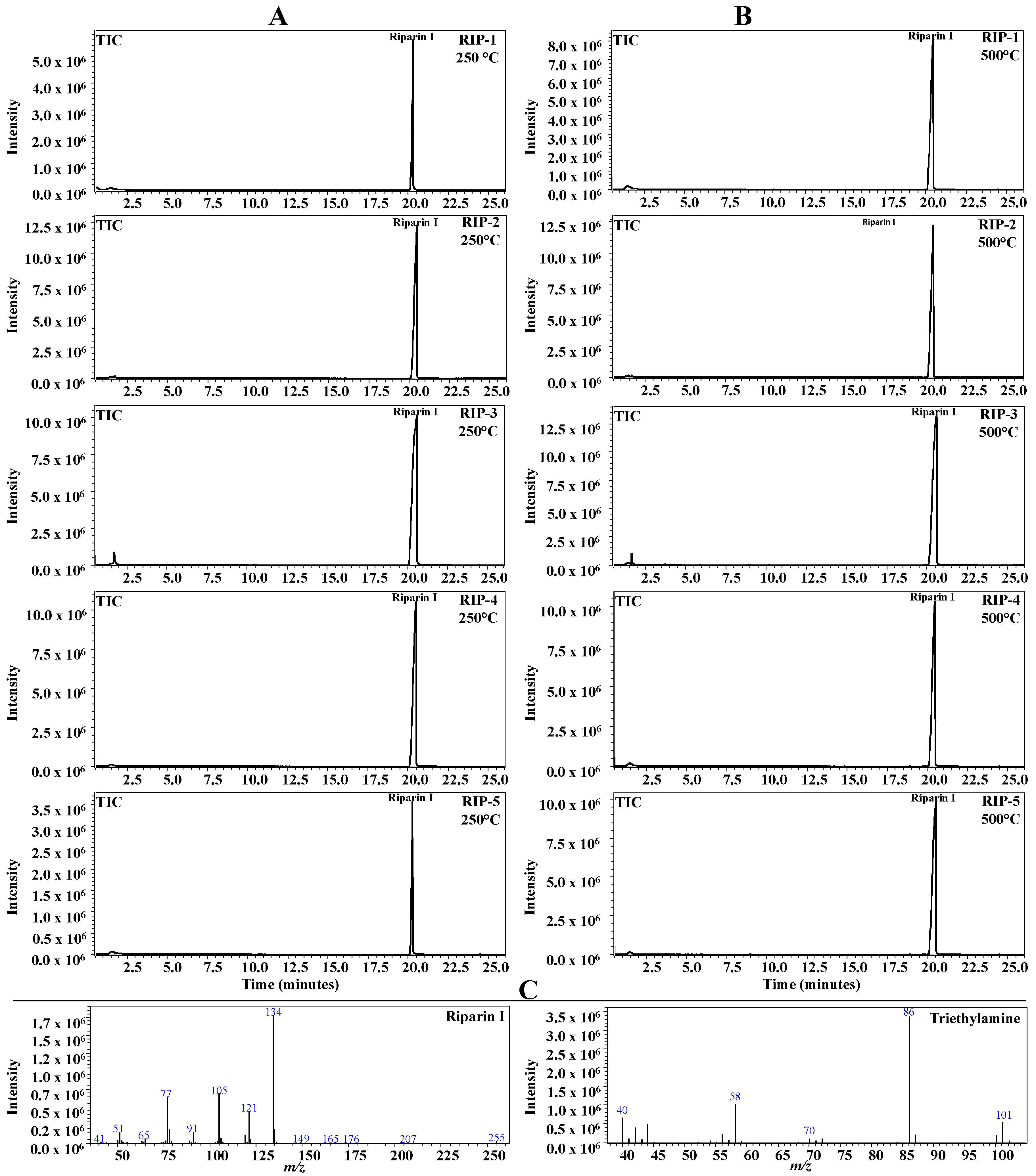
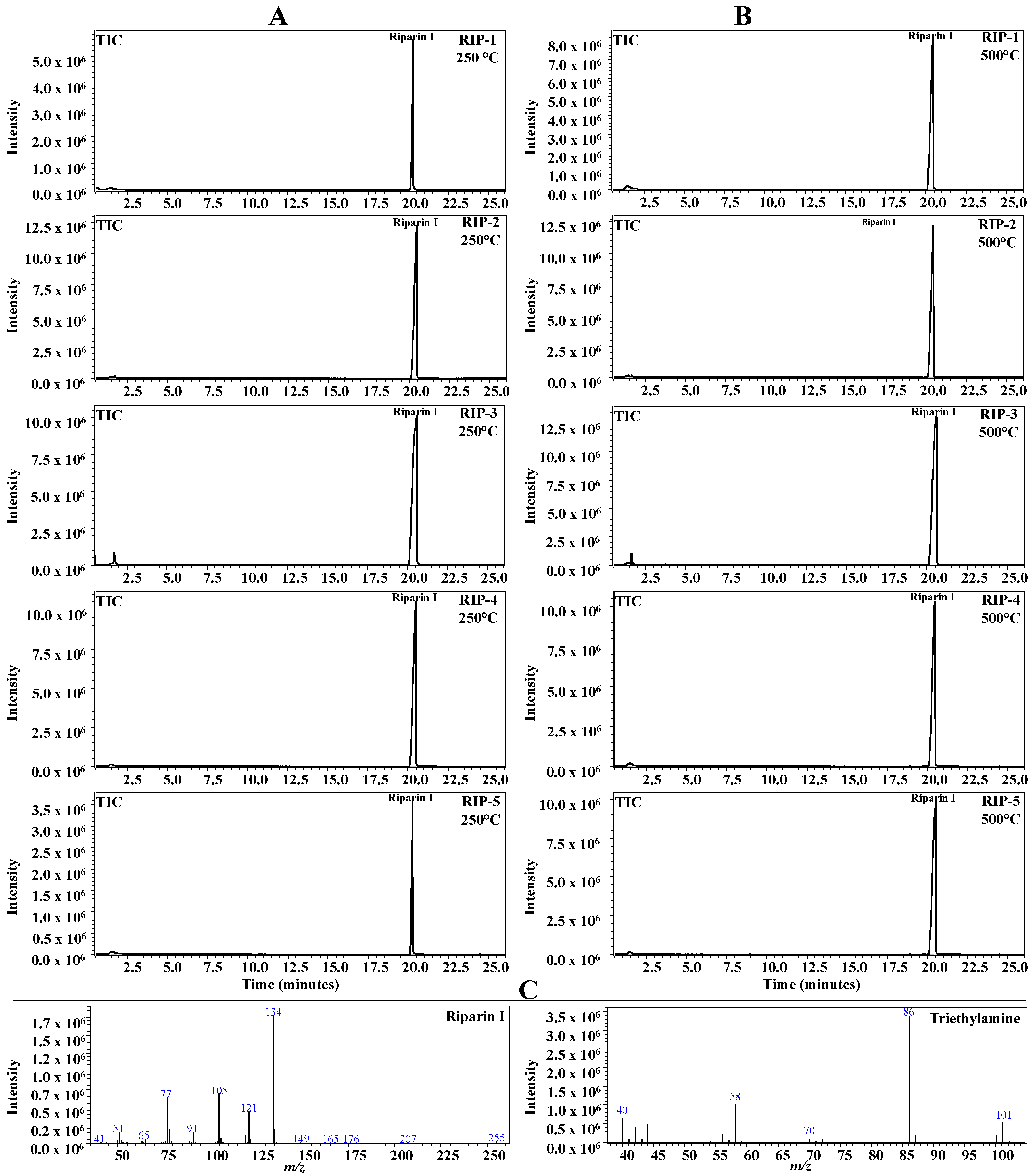
2.6. Pyrolysis Coupled with Gas Chromatography/Mass Spectrometry (Pyr-GC-MS)
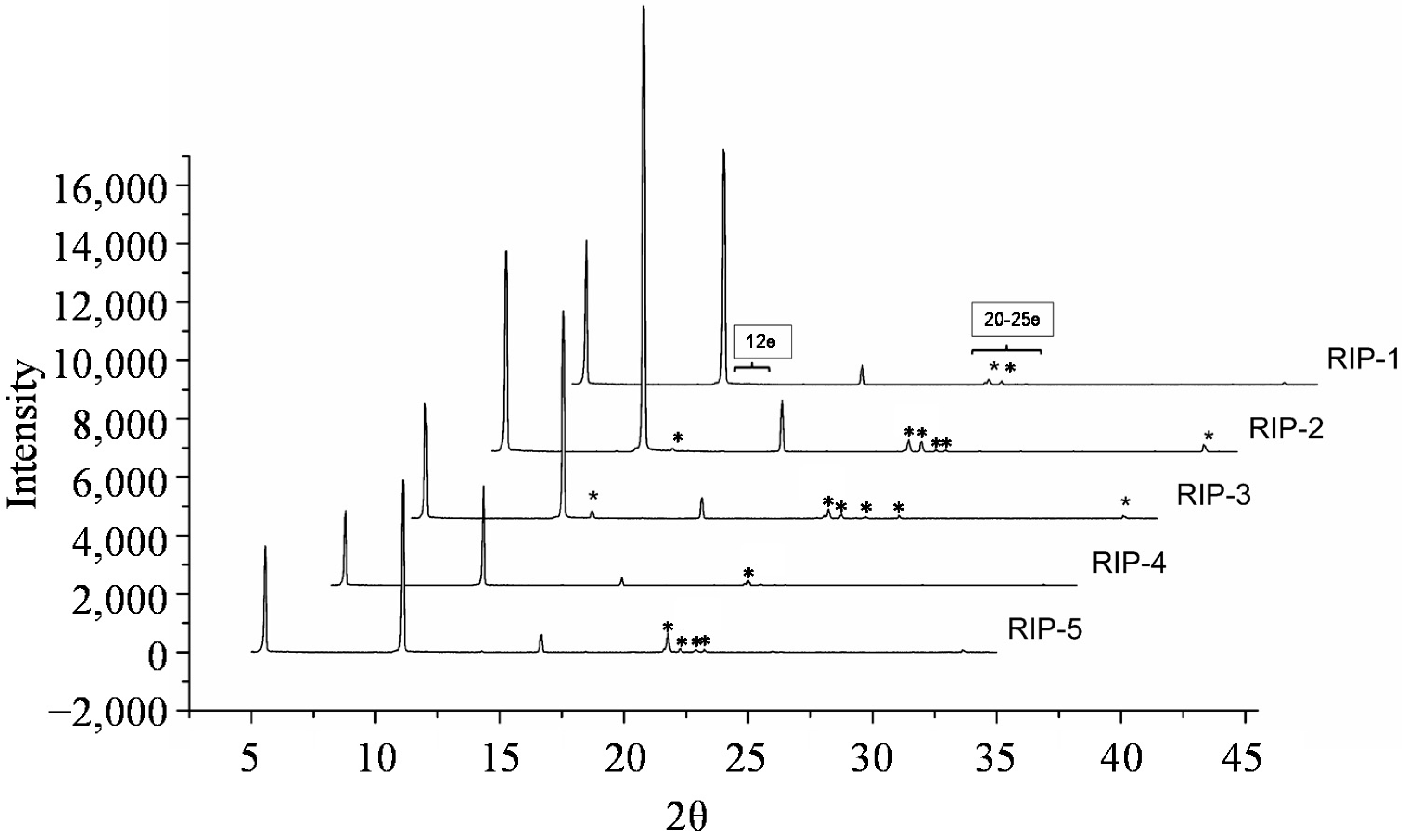
2.7. Powder X-Ray Diffractometry (PXRD)
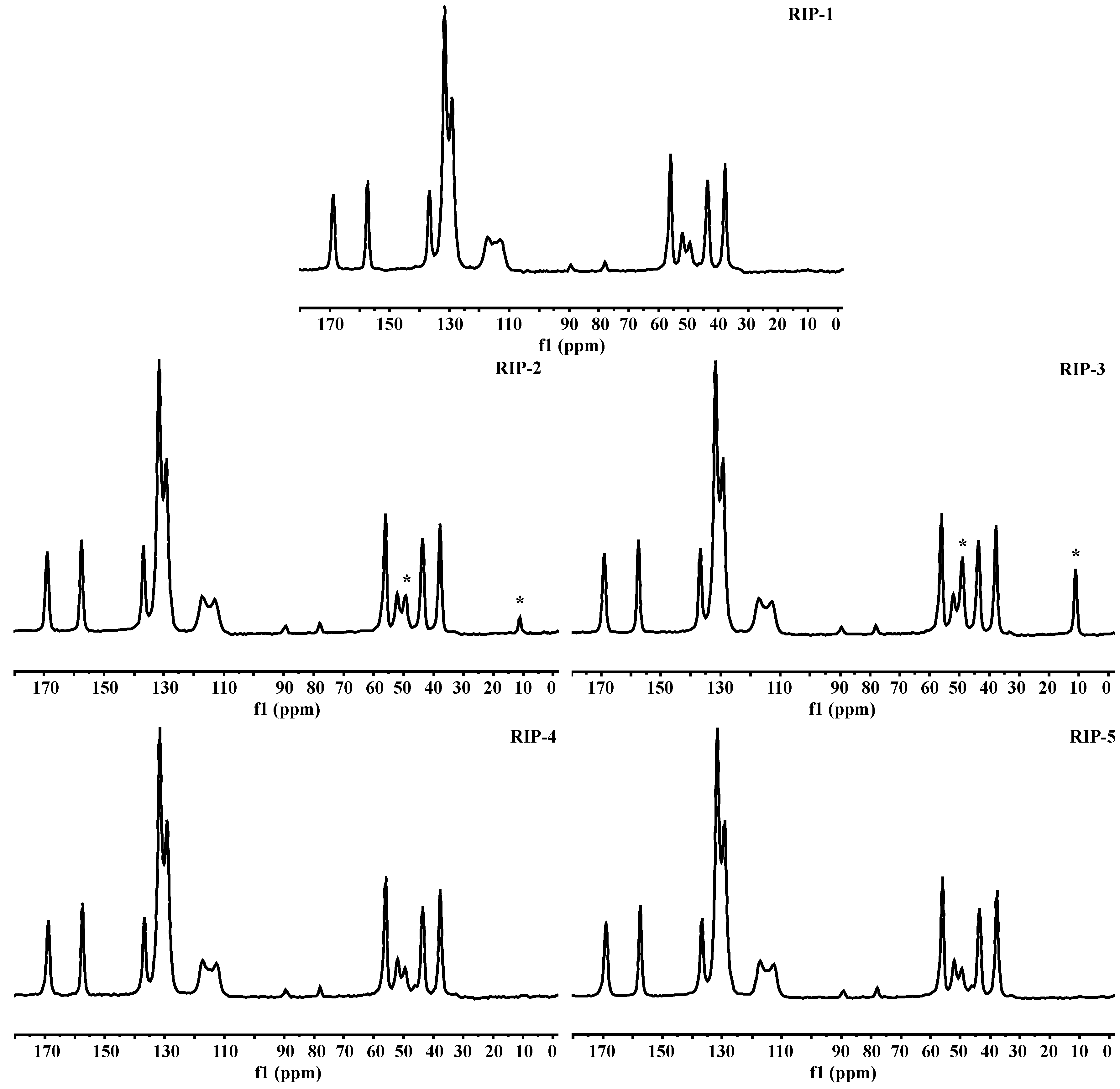
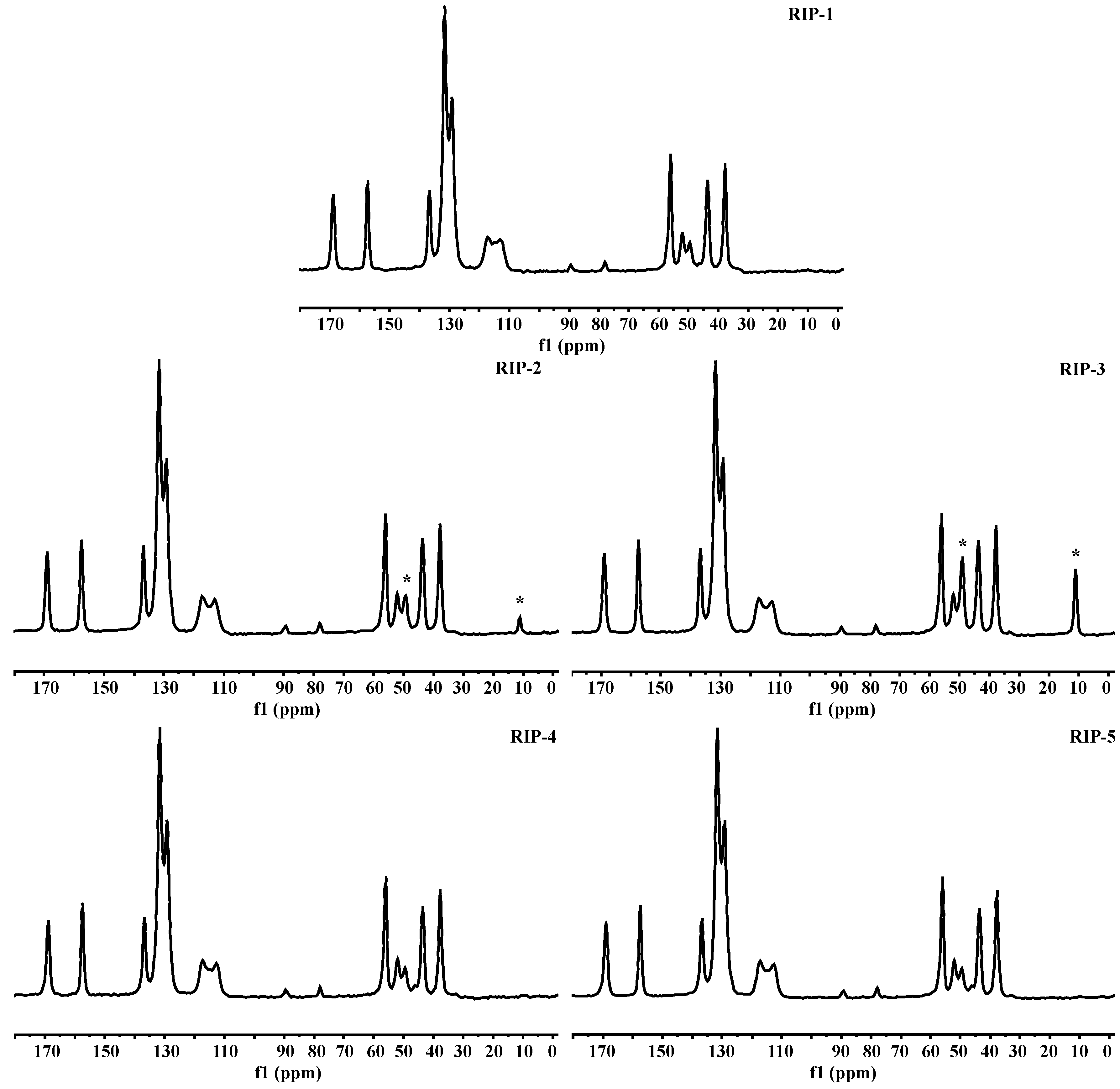
2.8. Solid-State Nuclear Magnetic Resonance (ssNMR)
3. Materials and Methods
3.1. Materials
3.2. Scanning Electron Microscopy (SEM)
3.3. Differential Scanning Calorimetry (DSC)
3.4. DSC Coupled with a Photovisual System (DSC-Photovisual)
3.5. Thermogravimetry (TG)
3.6. Fourier Transform Infrared (FTIR)
3.7. Pyrolysis Coupled with Gas Chromatography/Mass Spectrometry (Pyr-GC/MS)
3.8. Powder X-Ray Diffractometry (PXRD)
3.9. Solid-State 13C Nuclear Magnetic Resonance (ssNMR)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bernardia, L.S.; Ferreira, F.F.; Cuffini, S.L.; Campos, C.E.M.; Monti, G.A.; Kuminek, G.; Oliveira, P.R.; Cardoso, S.G. Solid-state evaluation and polymorphic quantification of venlafaxine hydrochloride raw materials using the Rietveld method. Talanta 2013, 117, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Garcia, L.C.; Checura, C.; DOnadio, L.G.; Fernandez, C.; Orgueira, H.; Comin, M.J. Synthesis and characterization of new related substances of the antiarrhythmic drug dronedarone hydrochloride. J. Pharm. Biomed. Anal. 2015, 114, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodriguez-Hornedo, N. General principles of pharmaceutical solid polymorphism: A supramolecular perspective. Adv. Drug Deliv. Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Gilles, F. New perspectives for the on-line monitoring of pahrmaceutical crystallization processes using in situ infrared spectroscopy. Int. J. Pharm. 2002, 241, 263–278. [Google Scholar] [CrossRef]
- Llinàs, A.; Goodman, J.M. Polymorph control: Past, present and future. Drug Discov. Today 2008, 13, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Vippagunta, S.R.; Brittain, H.G.; Grant, D.J.W. Crystalline solids. Adv. Drug Deliv. Rev. 2001, 48, 3–26. [Google Scholar] [CrossRef]
- Borghetti, G.S.; Carini, J.P.; Honorato, S.B.; Ayala, A.P.; Moreira, J.C.F.; Bassani, V.L. Physicochemical properties and thermal stability of quercetin hydrates in the solid state. Thermochim. Acta 2012, 539, 109–114. [Google Scholar] [CrossRef]
- Xu, K.; Xiong, X.; Zhai, Y.; Wang, L.; Li, S.; Yan, J.; Wu, D.; Ma, X.; Li, H. Effect of milling conditions on solid-stateamorphization of glibizide, and characterization and stability of solid forms. J. Pharm. Biomed. Anal. 2016, 129, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y. Molecular perspectives on solid-state phase transfomation and chemical reactivity of drugs: Metoclopramide as an example. Drug Discov. Today 2015, 20, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Rauber, G.S.; Riekes, M.K.; Campos, C.E.M.; Monti, G.A.; Bortoluzzi, A.J.; Cuffini, S.L.; Cardoso, S.G. Single Crystal structure, solid state characterization and dissolution rate of terbinafine hydrochloride. J. Pharm. Biomed. Anal. 2013, 78–79, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Shalaev, E.; Smith, S. Physical stability of pharmaceutical formulations: Solid-state characterization of amorphous dispersions. Trends Anal. Chem. 2013, 49, 137–144. [Google Scholar] [CrossRef]
- Santos, L.M.; Santos, O.M.M.; Mendes, P.F.; Rosa, I.M.L.; Silva, C.C.; Bonfilio, R.; Araujo, M.B.; Boralli, V.B.; Doriguetto, A.C.; Martins, F.T. Identification and proportion of the enantiomers of the antihypertensive drug chlortalidone in its form II by high quality sinlge-crystal X-ray diffraction data. J. Pharm. Biomed. Anal. 2016, 118, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Boer, T.M.; Procópio, J.V.V.; Nascimento, T.G.; Macêdo, R.O. Correlation of thermal analysis and pyrolysis couped to GC-MS in the characterization of tacrolimus. J. Pharm. Biomed. Anal. 2013, 73, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.D.S.; Zheng, C.; Lin, C.T.; Takahata, Y.; Barbosa-Filho, M.; Gutierrez, S.J.C. Electronic and structural effects in muscular relaxants: Riparin I and Riparin III. J. Mol. Struct. 2005, 753, 13–21. [Google Scholar] [CrossRef]
- Santos, S.G.; Queiroga, K.F.; Oliveira, A.M.F.; Tavares, J.F.; Gutierres, S.J.C.; Diniz, M.F.F.; Barbosa-Filho, M.J.M.; Silva, M.S. Blood matrix effects for male and female wistar rats, in simultaneous HPLC-UV determination of riparin I and III from Aniba riparia (Nees) Mez. (Laureceae). Talanta 2011, 86, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.T.V.; Monteiro, A.P.; Leite, C.P.; Araujo, F.L.O.; Lima, V.T.M.; Barbosa-Filho, J.M.; Fonteles, M.M.F.; Vasconcelos, S.M.M.; Viana, G.S.B.; Sousa., F.C.F. Anxiolytic-like effects of (O-methyl)-N-benzoyl-tyramine (riparin III) from Aniba riparia (Nees) Mez (Lauraceae) in mice. Biol. Pharm. Bull. 2006, 29, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Moura, E.A.; Correia, L.P.; Pinto, M.F.; Procópio, J.V.V.; Souza, F.S.; Macêdo, R.O. Thermal characterization of the solid state and raw material fluconazole by thermal analysis and pyrolysis coupled to GC/MS. J. Therm. Anal. Calorim. 2010, 100, 289–293. [Google Scholar] [CrossRef]
- Oliveira, A.H.; Moura, E.A.; Pinto, M.F.; Procópio, J.V.V.; Souza, V.G.; Souza, F.S.; Macêdo, R.O. Thermal characterization of raw material pentoxifylline using thermoanalytical techniques and Pyr-GC/MS. J. Therm. Anal. Calorim. 2011, 106, 763–766. [Google Scholar] [CrossRef]
- Sousa, F.C.F.; Melo, C.T.V.; Monteiro, A.P.; Lima, V.T.M.; Gutierrez, S.J.C.; Pereira, B.A.; Brabosa-Filho, J.M.; Vasconcelos, S.M.M.; Fonteles, M.F.; Viana, G.S.B. Antianxiety and antidepressant effects of riparin III from Aniba riparia (Ness) (Lauraceae) in mice. Pharmacol. Biochem. Behav. 2004, 78, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.C.J. Polymer: Additive analysis by flash pyrolysis techniques. J. Anal. Appl. Pyrolysis 2001, 58, 3–28. [Google Scholar] [CrossRef]
- Uvarov, V.; Popov, I. Metrological characterization of X-ray diffraction methods at different acquisition geometries for determination of crystallite size in nanoscale materials. Mater. Charact. 2013, 85, 111–123. [Google Scholar] [CrossRef]
- Procópio, J.V.V.; Souza, V.G.; Costa, R.A.; Correia, L.P.; Souza, F.S.; Macêdo, R.O. Application of thermal analysis and pyrolysis coupled to GC/MS in the qualification of simvastatin pharmaceutical raw material. J. Therm. Anal. Calorim. 2011, 106, 665–670. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds and riparin I batches are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
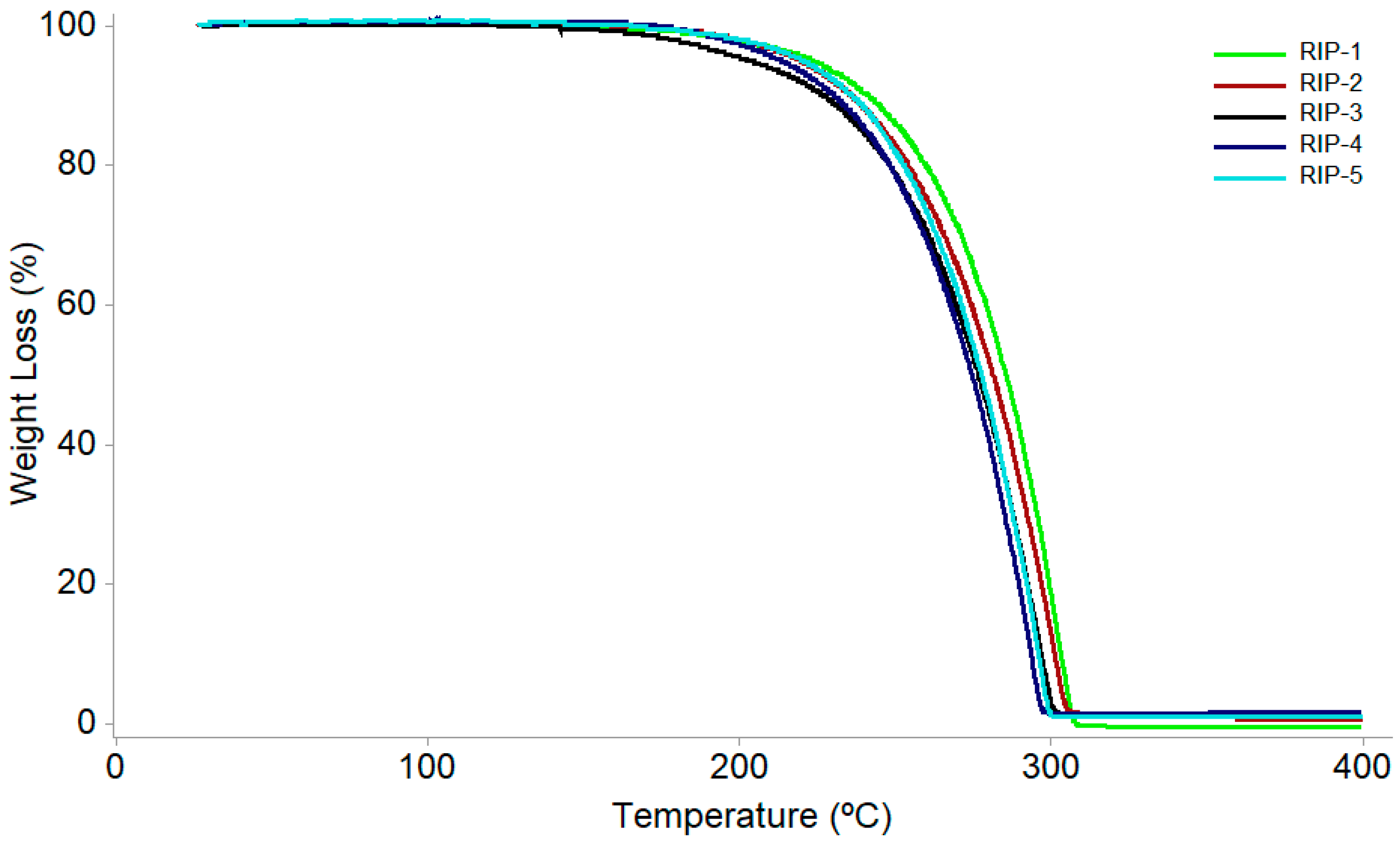
| Kinetic Parameter | Sample | ||||
|---|---|---|---|---|---|
| RIP-1 | RIP-2 | RIP-3 | RIP-4 | RIP-5 | |
| Activation energy (kJ·mol−1) | 156.85 | 138.45 | 98.87 | 106.50 | 113.45 |
| Frequency factor | 6.34 × 108 | 5.46 × 108 | 2.38 × 106 | 8.23 × 107 | 4.11 × 108 |
| Reaction order | 0.0 | 0.0 | 0.2 | 0.0 | 0.4 |
| Batches | Wavenumber (cm−1) | |||
|---|---|---|---|---|
| N–H | C=O | –C=C | ||
| Rip-1 | 3325.49 | 1637.71 | 1537.27 | 1512.19 |
| Rip-2 | 3323.56 | 1637.71 | 1539.21 | 1512.19 |
| Rip-3 | 3323.56 | 1635.64 | 1535.34 | 1512.19 |
| Rip-4 | 3325.49 | 1637.71 | 1539.21 | 1512.19 |
| Rip-5 | 3325.49 | 1637.71 | 1537.27 | 1512.19 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Moura, E.A.; Terto, M.V.C.; De Moura Mendonça, E.A.; Procópio, J.V.V.; De O. Costa, V.C.; Barbosa Filho, J.M.; Chavez Gutierrez, S.J.; Tavares, J.F.; Oliveira Macedo, R.; Da Silva, M.S. Solid-State Form Characterization of Riparin I. Molecules 2017, 22, 1615. https://doi.org/10.3390/molecules22101615
De Moura EA, Terto MVC, De Moura Mendonça EA, Procópio JVV, De O. Costa VC, Barbosa Filho JM, Chavez Gutierrez SJ, Tavares JF, Oliveira Macedo R, Da Silva MS. Solid-State Form Characterization of Riparin I. Molecules. 2017; 22(10):1615. https://doi.org/10.3390/molecules22101615
Chicago/Turabian StyleDe Moura, Elisana Afonso, Márcio Vinícius Cahino Terto, Elisângela Afonso De Moura Mendonça, José Valdilânio Virgulino Procópio, Vicente Carlos De O. Costa, José Maria Barbosa Filho, Stanley Juan Chavez Gutierrez, Josean Fechine Tavares, Rui Oliveira Macedo, and Marcelo Sobral Da Silva. 2017. "Solid-State Form Characterization of Riparin I" Molecules 22, no. 10: 1615. https://doi.org/10.3390/molecules22101615