Current Development Status of MEK Inhibitors


Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin 300192, China
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(10), 1551; https://doi.org/10.3390/molecules22101551
Submission received: 9 August 2017
/
Revised: 11 September 2017
/
Accepted: 12 September 2017
/
Published: 26 September 2017
(This article belongs to the Special Issue Kinase Inhibitors)
Abstract
:The current development status of mitogen-activated protein kinase kinase (MEK) inhibitors, including the preclinical data and clinical study progress, has been summarized in this review. Different MEK inhibitors, possessing specific physicochemical properties and bioactivity characteristics, may provide different options for patients seeking treatment for cancer. Moreover, the combination of the MEK inhibitors with other therapies—such as chemotherapy, targeted therapy, and immunotherapy—may be a promising approach for clinical use.
1. Introduction
The mitogen-activated protein kinase (MAPK) signaling pathway plays critical roles in the regulation of diverse cellular activities, including cell proliferation, survival, differentiation, and motility [1]. Dysregulation of the MAPK pathway occurs in more than one-third of all malignancies. The classical MAPK pathway consists of Ras (a family of related proteins which is expressed in all animal cell lineages and organs), Raf (a family of three serine/threonine-specific protein kinases that are related to retroviral oncogenes), MEK (mitogen-activated protein kinase kinase), and ERK (extracellular signal-regulated kinases), sequentially relaying proliferative signals generated at the cell surface receptors into the nucleus through cytoplasmic signaling. The MEK inhibitor targets the Ras/Raf/MEK/ERK signaling pathway, inhibiting cell proliferation and inducing apoptosis. It hence has potential in clinical use for cancer treatment, especially for those cancers induced by RAS/RAF dysfunction [2].
Owing to the widespread activation of this pathway in numerous neoplasms, MEK inhibitors have been in the process of development and study as a type of monotherapy or combination therapy with other targeted and cytotoxic drugs in a variety of clinical situations. More recently, the combination with the use of immune checkpoint inhibitors has emerged as an efficacious treatment for some cancers, expanding the efficacy of this class of agent [3].
This review summarized the recent progress of MEK inhibitors, complementary to an earlier review [4,5,6,7,8,9,10,11,12,13,14,15] but with a greater focus on those compounds that have been approved or are in clinical stages of development. We also give a brief summary of compounds in the preclinical phase.
2. Ras/Raf/MEK/ERK Pathway and MEK Inhibitors
Signal transduction occurs when an extracellular signaling molecule activates a specific receptor located on the cell surface. In turn, this receptor triggers a biochemical chain of events inside the cell and creates a response. The Ras/Raf/MEK/ERK pathway is one of the critical pathways involved in signal transduction, which results in the control of cell proliferation, survival, and differentiation [16,17] and plays a role in the development of multiple cancers including melanoma, non-small cell lung cancer (NSCLC), etc. MEK1 and MEK2 are closely related and participate in the Ras/Raf/MEK/ERK signal transduction cascade. Blockage of the pathway with MEK1/2 inhibitors could result in the clinical benefits for treatment of cancers with RAS/RAF dysfunction. Therefore, huge efforts have been made in past decades with respect to the development of MEK1/2 inhibitors. The first MEK inhibitor, PD098059, was reported in 1995 [18], since then a number of MEK inhibitors have been progressing into clinical stages of development. More recently, the combination of MEK with BRAF (a human gene that encodes a protein called B-Raf) inhibitors and/or other therapies has provided a new treatment option for multiple cancers.
3. MEK Inhibitors Approved by the US Food and Drug Administration (FDA)
Two MEK inhibitors, trametinib and cobimetinib, were approved by the FDA and the European Medicines Agency (EMA). The preclinical and clinical data for these two compounds are summarized in Table 1.
3.1. Trametinib
Trametinib (GSK1120212) was the first MEK inhibitor approved by the FDA (May, 2013) for treatment of melanoma. It is an allosteric, non-ATP-competitive inhibitor with sub-nanomolar activity against purified MEK1 and MEK2 kinases (half maximal inhibitory concentration (IC50) of 0.7 nM [19] and 0.9 nM, respectively) and its chemical structure is shown in Figure 1.
In a preclinical study, trametinib showed high efficacy in xenograft models. For the HT-29 xenograft model, oral administration of trametinib demonstrated efficacy in inhibiting tumor growth at a dose of either 0.3 mg/kg or 1 mg/kg once daily for 14 days, and could block tumor growth almost completely at a dose of 1 mg/kg [27]. Similar findings were observed in an A549 (KRAS (proto-oncogene corresponding to the oncogene first identified in Kirsten rat sarcoma virus) mutant cell line) xenograft model with tumor growth inhibition of 92% and 87% at 5.0 mg/kg and 2.5 mg/kg respectively, with inhibition to a lesser degree with a lower dose [28].
In clinical study as a single agent, treatment with trametinib resulted in a statistically significant and clinically meaningful improvement with respect to progression-free survival (PFS) compared to standard chemotherapy. In a global multi-center, randomized, open-label and controlled trial, 322 melanoma patients with the BRAF V600E or V600K-mutant were allocated with the ratio of 2:1 into either the trametinib group or conventional chemotherapy group (dacarbazine or paclitaxel), respectively. It was reported that trametinib resulted in an improved overall response rate (22% vs. 8%) and progression-free survival (4.8 vs. 1.5 months) when compared with the group receiving cytotoxic treatment [29]. Rash, diarrhea, and peripheral edema were the main adverse events, but those were manageable. A phase I/Ib study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced KRAS-mutant non-small cell lung cancer (NSCLC) showed that the primary endpoint of overall response rate (ORR) was met for both combinations [30] (ClinicalTrials.gov number, NCT01192165).
The FDA first approved the combination of trametinib and dabrafenib for patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations in January 2014, based on the results of median PFS extended from 8.8 months to 11.0 months when compared with dabrafenib monotherapy in a multi-center phase 3 trial [31,32]. The EMA approved this combination a year later for adults with unresectable or metastatic melanoma with the BRAF V600 mutation. MEK and BRAF inhibitors hence presented a new treatment method for metastatic melanoma [33].
For BRAF-mutant non-small cell lung cancer (NSCLC), the FDA gave the breakthrough designation for combination of trametinib and dabrafenib in 2015, and approved this combination in June 2017 [34] following the EMA’s approval in April [35]. The approval of this combination was based on the results from a three-cohort, multicenter, non-randomized, open-label study of patients with stage IV NSCLC. In this phase II trial, 36 treatment-naïve patients and 57 previously treated patients were assigned to the combination regimen with trametinib and dabrafenib, and 78 previously treated patients received the single agent dabrafenib. The results showed that there was an overall response rate (ORR) of more than 60%, with median PFS of longer than 9 months in the combination group, while there was an ORR of 27% with PFS of 5.5 months in the dabrafenib group [36]. The combination of MEK and BRAF inhibitors has provided a new treatment option [37].
Recently, the combination of MEK inhibitor with programmed cell death protein 1 (PD-1) inhibitors [38,39,40] has shown good prospects. Trametinib, in combination with dabrafenib and pembrolizumab, was developed by Merck Sharp & Dohme Corp. and Novartis for the treatment of melanoma in a phase 2 study. The phase 1 study has shown a manageable toxicity profile in patients with BRAF V600-mutant melanoma and the ongoing phase 2 study will further evaluate safety and efficacy of this triple combination as a first-line therapy for BRAF-mutant melanoma.
The development of triple regimens on the above approved combination with either Pembrolizumab or PDR 001 are ongoing by Merk Sharp & Dohme and Novartis, respectively, for the treatment of melanoma. A trial to assess the triple combination of trametinib, dabrafenib, and the anti-programmed death-ligand 1 (PD-L1) antibody durvalumab in 26 patients with BRAF-mutated advanced melanoma was conducted. There was an ORR of 69%, and 16 of 18 patients have ongoing responses [41].
Apart from melanoma and NSCLC, trametinib as a single agent or in combination with dabrafenib has also been tested in other types of cancers. As reported in American Society of Clinical Oncology (ASCO) recently, a two-arm phase II study with either trametinib or 5-fluororacil/capecitabine in refractory advanced biliary cancer was stopped due to the lack of efficacy in the trametinib arm. Meanwhile, a combination with dabrafenib was shown to be well tolerated in BRAF-mutated papillary thyroid carcinoma in one randomized phase II trial.
3.2. Cobimetinib
The second approved MEK inhibitor was cobimetinib (GDC-0973, XL518), which was developed by Exelixis and Genentech (Roche). The compound was awarded the status of orphan drug by the FDA in 2014 for malignant melanoma with the BRAFV600 mutation, and was then approved for combination treatment with vemurafenib for unresectable or metastatic melanoma with a BRAF V600E or V600K mutation in November 2015.
The chemical structure of cobimetinib is shown in Figure 2. Cobimetinib is a potent and highly selective MEK inhibitor, with a biochemical IC50 of 0.9 nM against MEK1 [20]. It also showed broad efficacy in vivo in xenograft models with BRAF- and KRAS-mutated cell lines [42]. Preclinical studies demonstrated that this agent was effective in inhibiting the growth of many tumor cells bearing a BRAF mutation. In preclinical studies, cobimetinib was associated with sustained ERK/MAPK inhibition in tumor tissues, with minimal drug exposure in the brain.
In a phase 3 trial, median PFS was 12.3 months for cobimetinib plus vemurafenib versus 7.2 months for vemurafenib alone, and these data supported the use of combination as a standard first-line approach to improve survival in patients with advanced BRAF V600-mutant melanoma [43].
Currently, there are many ongoing clinical studies for cobimetinib combined with other targeted therapies. For example, there is the combination of cobimetinib with GDC-0941 (a PI3K inhibitor) or GDC-0994 (a Erk1/2 inhibitor) for metastatic solid tumors (ClinicalTrials.gov number, NCT02457793) and the combination with idasanutlin (p53-MDM2 inhibitor) and venetoclax (BCL-2 inhibitor) for the treatment of leukemia.
The combination of cobimetinib with immunotherapy is also a highly investigated topic. A phase Ib dose-escalation and dose-expansion study (ClinicalTrials.gov number, NCT01988896) achieved longer PFS, with a median of 12.0 months in the combination treatment group of cobimetinib with atezolizumab when compared with atezolizumab or cobimetinib alone in melanoma [44]. An updated phase Ib study (ClinicalTrials.gov number, NCT01656642) showed that cobimetinib + atezolizumab + vemurafenib in BRAFV600-mutant metastatic melanoma had a manageable safety profile and promising antitumor activity [45]. Cobimetinib, in combination with atezolizumab and regorafenib, was used for the treatment of colorectal cancer in the phase III trial (ClinicalTrials.gov number, NCT02788279).
The clinical study results of clinical activity and safety of cobimetinib (cobi) and atezolizumab (atezo) in colorectal cancer (CRC) reported that the combination of cobi and atezo in CRC is well tolerated at the maximum administered doses. These results show that patients with MSS CRC can respond to the combination of cobi and atezo, and provide support for continued evaluation of the combination [46] (ClinicalTrials.gov number, NCT01988896).
4. MEK Inhibitors under Clinical Development
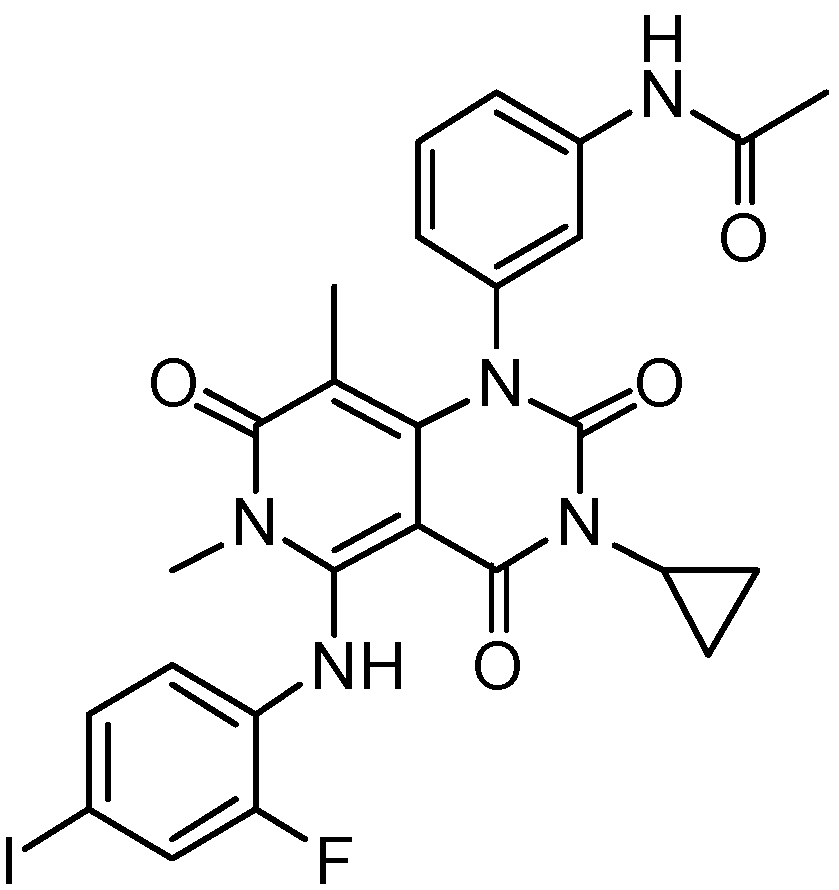
The MEK inhibitors in clinical trials—including their efficacy, therapeutic indications, sponsors, and status—are shown in Table 2. At the same time, the chemical structures of the compounds are presented in Figure 3.
4.1. CI-1040 (PD184352)
CI-1040 was the first MEK inhibitor run in the clinical stage by Pfizer/Warner-Lambert [47]. This compound presented a novel series of benzhydroxamate esters derived from their precursor anthranilic acids as MEK inhibitors. CI-1040 was developed in phase II for breast cancer, colorectal cancer, lung cancer, and pancreatic cancer, but it was found that there was poor exposure related to its poor solubility and rapid clearance [57], leading to insufficient antitumor activity in two phase II studies completed in 2003 (ClinicalTrials.gov number, NCT00034827, NCT00033384). Hence, to resolve the problems of this compound, the second compound PD-0325901 had been developed into clinical trial.
4.2. PD-0325901
PD-0325901 is a derivative of the MEK inhibitor CI-1040. The optimization of the hydroxamate side chain of CI-1040 for the improvement of solubility and exposure with oral doses resulted in the discovery of the clinical candidate PD-0325901 by Pfizer/Warner-Lambert. The administration of oral dose (PO) or intravenous injection (IV) of PD-0325901 induced in a dose-dependent decrease in phosphorylation of MAPK (pMAPK) in liver and lung caused by MEK inhibition. Inhibition of pMAPK in liver was generally comparable between the routes of administration, whereas inhibition of pMAPK in lung occurred for a longer duration following IV administration of PD-0325901, possibly due to higher PD-0325901 plasma maximum concentration (Cmax) [58,59].
PD-0325901 failed in its phase II clinical trials for the treatment of KRAS mutant non-small cell lung cancer by not meeting its primary efficacy end-point (ClinicalTrials.gov number, NCT00174369) [60]. A phase I/II study for the treatment of melanoma, colonic neoplasms and breast neoplasms had been terminated in 2007 due to musculoskeletal, neurological, and ocular toxicity (ClinicalTrials.gov number, NCT 00147550) [61]. Furthermore, the combination of PD-325901 with palbociclib for the same indication is still ongoing to date (ClinicalTrials.gov number, NCT 02022982). A phase 2 study in adolescents and adults with neurofibromatosis type-1 is ongoing and is expected to be completed at the end of 2018 (ClinicalTrials.gov number, NCT 02196471). There are another two phase I or I/II studies ongoing in colorectal cancer or KRAS mutated malignancies (ClinicalTrials.gov number, NCT02510001, NCT02039336); both are combinations with target agents or chemotherapy.
4.3. Selumetinib (ARRY-142886; AZD6244)
Selumetinib (AZD6244) is an effective, high selective, non-ATP-competitive MEK1 inhibitor which was developed by Array BioPharma and then licensed to AstraZeneca.
In 2004, Array BioPharma and AstraZeneca co-developed selumetinib for clinical study with several phase I and II clinical trials carried out for solid tumors as monotherapy [59,62,63,64,65]. Unfortunately, it was discontinued in phase II trials due to its failure to distinguish from temozolomide.
Randomized phase II studies that compared selumetinib with conventional cytotoxic chemotherapy in a wide array of malignancies have also been performed. Selumetinib showed no superiority when compared with temozolomide in patients with chemotherapy-naive melanoma, when compared with pemetrexed in patients with NSCLC who had not responded to first-line and second-line therapies, and when compared with capecitabine in patients with pancreatic cancer and colorectal cancer. Nevertheless, an antitumor activity of selumetinib was detected in each of these comparative studies.
Several clinical trials on selumetinib in combination with other cancer drugs have been conducted. A phase Ib study of combination of selumetinib and cyclosporine A (CsA) in patients with advanced solid tumors with an expansion cohort in metastatic colorectal cancer (mCRC) showed that the combination therapy appeared to be well tolerated, with evidence of activity in mCRC (ClinicalTrials.gov number, NCT02188264) [66]. Selumetinib in combination with sorafenib in advanced hepatocellular carcinoma (HCC) is also under study (ClinicalTrials.gov number, NCT01029418) [67]. A randomized Phase II study for KRAS-mutated NSCLC did show very promising results for selumetinib + docetaxel vs. docetaxel, with significant improvement in median PFS (5.3 vs. 2.1 months, p = 0.014) and objective response rate (37% vs. 0%, p < 0.0001) when compared with the docetaxel monotherapy cohort, and the overall survival benefit is 9.4 vs. 5.2 months (p = 0.21), with higher toxicity and the overall survival benefit (9.4 vs. 5.2 months, p = 0.21), with high toxicity. However, such positive efficacy could not repeat in the multinational phase III clinical trial. A total of 510 KRAS-mutated NSCLC patients were randomized 1:1 to receive selumetinib + docetaxel or placebo + docetaxel. The result showed that addition of selumetinib to docetaxel did not improve progression-free survival compared with docetaxel alone (ClinicalTrials.gov number, NCT01933932) [68]. Combination therapy with selumetinib plus dacarbazine has also been compared with placebo plus dacarbazine as first-line treatment in patients with BRAF mutated melanoma in a phase II study. While markedly improved PFS was observed with the addition of selumetinib to the therapeutic regimen, no overall survival benefit was demonstrated.
Recently, a phase I trial in pediatric patients with recurrent or refractory low-grade glioma has been reported. In this study, 25 subjects received a median of 13 cycles (range: 1–26). Fourteen (37%) completed all protocol treatment (26 cycles (n = 13), 13 cycles (n = 1)) with at least stable disease; 2-year progression-free survival at the RP2D was 69 ± SE (standard error) 9.8% [69]. The study showed that selumetinib had promising antitumor activity in children with low-grade glioma. In the early phase 2 result, selumetinib was effective in treating children with recurrent/refractory low-grade glioma (LGG), including those with neurofibromatosis type 1 (NF-1)-associated LGG and pilocytic astrocytomas (PA) harboring BRAF V600E mutation or BRAF-KIAA 1549 fusion. Larger prospective studies are necessary to determine the specific role of this agent in treating children with LGG harboring specific molecular aberrations in the future [70].
4.4. Binimetinib (MEK162, ARRY-438162)
Binimetinib (MEK162, ARRY-438162) is an orally bioavailable, highly selective, non-ATP-competitive MEK inhibitor that has the potential to treat a range of malignant diseases. Binimetinib, 5-((4-bromo-2-fluorophenyl)amino)-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzo[d]imidazole-6-carboxamide, was discovered by Array BioPharm., and was then co-developed by Array BioPharm/Novartis. In preclinical studies, binimetinib either alone or in combination with other agents showed significant antitumor activities in cell lines and animal models [71].
Binimetinib was investigated in phase III trials as a single agent or in combination with other anticancer agents. Common adverse events (AEs) included rash, nausea, vomiting, diarrhea, peripheral edema, and fatigue [72].
The open-label and non-randomized phase II study assessed the safety and efficacy of single-agent binimetinib in adult patients with locally advanced and unresectable or metastatic malignant cutaneous melanoma, harboring BRAF V600E or NRAS (a member of the RAS gene family) mutations (ClinicalTrials.gov number, NCT01320085). The resulting data supported clinical activity of binimetinib in patients with NRAS-mutated and BRAF-mutated metastatic melanoma. Binimetinib was the first targeted therapy to show activity in patients with NRAS-mutated melanoma and offered a potential option for kinds of cancer with few effective treatments [71,73].
Then, a randomized phase III, open label, multicenter, two-arm study was designed to compare the efficacy of binimetinib versus dacarbazine in patients with advanced unresectable or metastatic NRAS mutation-positive melanoma (NCT01763164). The data showed that the ORR was 15% vs. 7% and median PFS was 2.8 months (95% CI 2.8–3.6) vs. 1.5 months (1.5–1.7) in the binimetinib group and dacarbazine group, respectively (hazard ratio 0.62 (95% CI (confidence interval) 0.47–0.80); one-sided p < 0.001) [74]. PFS by binimetinib was statistically improved but not clinically significant (2.8 vs. 1.5 months). A new drug application (NDA) application for binimetinib as monotherapy for NRAS-mutated melanoma was withdrawn finally from the FDA. Another prospective, randomized, open label, multi-center, parallel group, three-arm phase III study is in progress to study the efficacy and safety of binimetinib with LGX818 (encorafenib) compared vemurafenib and LGX818 monotherapy in locally advanced unresectable or metastatic melanoma with BRAF V600 mutation (ClinicalTrials.gov number, NCT01909453).
Other plans for binimetinib combinations with immunotherapy (e.g., with pembrolizumab and encorafenib for the treatment of malignant melanoma, with nivolumab, LGX818, and ipilimumab for the treatment of metastatic melanoma, etc.) are also underway.
4.5. AZD-8330 (ARRY-424704)
AZD8330 is an orally active, selective MEK inhibitor with potential antineoplastic activity. The structure core for this compound was 6-oxo-1,6-dihydropyridazine, which presents a different class of MEK inhibitors [75]. AZD-8330 was being developed for non-ATP-competitive MEK1/2 inhibitors, with an IC50 of 7 nM [76].
AZD8330 was employed as a single agent, and the results from phase I clinical trial for the treatment of solid tumors (ClinicalTrials.gov number, NCT00454090) showed that common reported toxicities included acneiform dermatitis, fatigue, diarrhea, and vomiting. Four patients experienced dose-limiting toxicities: mental status changes (40 mg once daily; 2/9 patients and 60 mg once daily; 1/3) and rash (20 mg BID (twice daily); 1/9). The maximum tolerated dose was defined as 20 mg BID. The exposure of AZD8330 increased approximately proportionally with dosage across a dose range of 0.5–60 mg once-daily. Phosphorylated ERK levels in peripheral blood mononuclear cells were measured and target inhibition was confirmed. AZD8330 demonstrated a manageable toxicity profile with fewer class-effect AEs compared with other MEK inhibitors [77]. No more clinical studies have been reported recently.
4.6. TAK-733
TAK-733 is an orally bioavailable, non-ATP-competitive small-molecule inhibitor of MEK1/2 with potential antineoplastic activity. TAK-733 is highly potent and selective MEK allosteric site inhibitor with IC50 of 3.2 nM, and potent enzymatic and cell activity with an EC50 (concentration for 50% of maximal effect) of 1.9 nM against ERK phosphorylation in cells [51]. TAK-733 reveals broad antitumor activity in mouse xenograft models of human cancer including models of melanoma, colorectal, NSCLC, and pancreatic and breast cancer.
Phase I clinical study (ClinicalTrials.gov number, NCT00948467) of TAK-733 has been developed by Millennium Pharmaceuticals Inc., and the results from the dose-escalation phase I showed that the maximum tolerated dose was 16 mg. Common drug-related AEs included dermatitis acneiform, diarrhea, and increased blood creatine phosphokinase. TAK-733 demonstrated a generally manageable toxicity profile and limited antitumor activity [78]. No further investigations have been published recently.
4.7. GDC-0623
GDC-0623, with the chemical name of (1-(5-((2-fluoro-4-iodophenyl)amino)imidazo[1,5-a]pyridin-6-yl)-2-(2-hydroxyethoxy)ethan-1-one), is a potent, orally active, selective non-ATP-competitive MEK inhibitor (MEK1, Ki = 0.13 nM, + ATP). It was developed by Genentech, and has a novel structure of imidazopyridine [52]. GDC-0623 has broad potency in cell-based assays, particularly in both KRAS and BRAF mutant cancer cell lines, with corresponding efficacy in xenograft tumor models. It exhibited higher activity in KRAS mutant tumors than BRAF mutant tumors [79]. In preclinical pharmacokinetics and efficacy assessments, GDC-0623 showed low clearance and a low volume of distribution. The results of safety, tolerability, and pharmacokinetics from its phase I study were revealed in 2014 (ClinicalTrials.gov number, NCT01106599).
4.8. Refametinib (RDEA-119, BAY-869766)
Refametinib, discovered by Ardea Biosciences and developed by Bayer, is a potent and orally bioavailable, non-ATP-competitive inhibitor with a low ability to accumulate in brain and other neural tissues. The IC50 of refametinib against MEK1 was 19 nM [53]. It was selected for clinical development due to its potency and favorable pharmacokinetic profile.
Several phase I or I/II or phase II clinical trials had been conducted for single or combination therapy. One phase I/II study concluded that refametinib plus gemcitabine was well tolerated, with a promising objective response rate [80]. One phase II clinical study of the combination therapy with rafametinib and sorafenib for the treatment of RAS-mutated HCC has been carried out as the first line systemic treatment due to its high prevalence of constitutive activation of MAPK pathway in HCC. The results showed the efficacy benefit; among 70 enrolled patients, three had confirmed partial response and 25 had prolonged stable disease [81]. However, this combination was poorly tolerated, with several severe adverse events reported and almost all patients required dose modifications due to drug toxicity.
4.9. Pimasertib (AS703026)
Pimasertib, developed by Merck KGaA and known as AS703026 or MSC1936369B, is a selective, orally bioavailable, non-ATP-competitive MEK1/2 inhibitor with potent antitumor activity in cell lines and xenograft models with constitutive activation of the MAPK pathway. Its structure, including (2-fluoro-4-iodophenyl) amino, core structure pyridine, and side chain (S)-N-(2,3-dihydroxypropyl)acetamide, is different from other MEK inhibitors. The first-in-human trial reported pharmacokinetics (PK) and pharmacodynamics (PD) of pimasertib in patients with advanced solid tumors. Pimasertib exhibited a favorable PK profile in patients with solid tumors, and target engagement demonstrated by phospho-ERK (pERK) inhibition in peripheral blood mononuclear cell (PBMC) was observed [82,83]. Pimasertib showed clinical activity in dose-dependent manner associated with target inhibition. Sustained responses were observed mostly in BRAF or NRAS-mutated melanoma [84,85]. Currently, a few phase I/II studies are ongoing to evaluate pimasertib in the setting of advanced or metastatic solid tumors including ovarian cancer, NRAS-mutated cutaneous melanoma, ovarian cancer, breast cancer, NSCLC, hepatocellular carcinoma, metastatic colorectal cancer, and pancreatic adenocarcinoma.
4.10. RO4987655 (CH4987655)
RO4987655, possessing a unique 3-oxo-oxazinane ring structure at the 5-position of the benzamide core structure [54], was developed by Hoffman–La Roche. RO4987655 is an orally active small molecule, targeting MEK1 with potential antineoplastic activity. It was designed based on the X-ray crystal structure information of the target enzyme and then given multidimensional optimization including metabolic stability, physicochemical properties, and safety profiles.
RO4987655 exhibited slow dissociation from the MEK1 enzyme, remarkable antitumor efficacy both as monotherapy and combination therapy in vivo, desirable metabolic stability, and insufficient MEK inhibition in mouse brain, implying few central nervous system (CNS)-related side effects in humans.
An excellent PK profile and clear target inhibition in PBMC were demonstrated in one phase I study with healthy volunteers [86], and also showed manageable toxicity, a favorable PK/PD profile, and promising preliminary antitumor activity with heavily pretreated patients [76,87,88], which has been further investigated in specific populations of patients with RAS and/or RAF mutation driven tumors (ClinicalTrials.gov number, NCT00817518).
4.11. RO5126766
RO5126766, known as CH5126766, is a dual Raf/MEK inhibitor specifically for the kinase activities of Raf and MEK, resulting in the blockage of target gene transcription that promotes malignant transformation of cells. RO5126766 specifically inhibits the kinase activities of Raf and MEK, resulting in the inhibition of target gene transcription that promotes malignant transformation of cells. RO5126766, bearing a sulfamide moiety instead of aniline in coumarin, was identified as a clinical compound with enhanced inhibitory activity, satisfactory PK/PD profiles, and manageable toxicity [89]. More studies are needed to further clarify the safety and efficacy of this agent, as well as this novel class of MEK-RAF inhibitors in various cancers.
4.12. WX-554
WX-554 is a selective, noncompetitive MEK1/2 inhibitor entering preliminary human studies. Phase I pharmacokinetic and pharmacodynamic study showed that WX-554 was well tolerated, with the recommended phase 2 dose being 75 mg twice weekly [55]. Unfortunately, two dose-escalation phase I/II studies in patients with advanced solid tumors were terminated as reported (ClinicalTrials.gov number, NCT01859351, NCT01581060).
4.13. HL-085
HL-085 is an orally active, selective MEK inhibitor with significant inhibitory activity against MEK kinase and an IC50 value of 1.9–10 nM. In vitro, the IC50 values of tumor cell lines were 0.41–6.2 nM in A375, 0.1–7.8 nM in Colo205 and 0.88–2.9 nM in HT29, respectively. Oral dosing of HL-085 (1 mg/kg, QD, 21 days) in BRAF-mutant Colo 205 and A375 xenograft models showed a high tumor growth inhibition (TGI) value (70–76%, 60–70%). HL-085 is effective in inhibiting tumor proliferation in other tumor cells as well. Currently, HL-085 is under phase I clinical study.
5. MEK Inhibitors in Preclinical Development
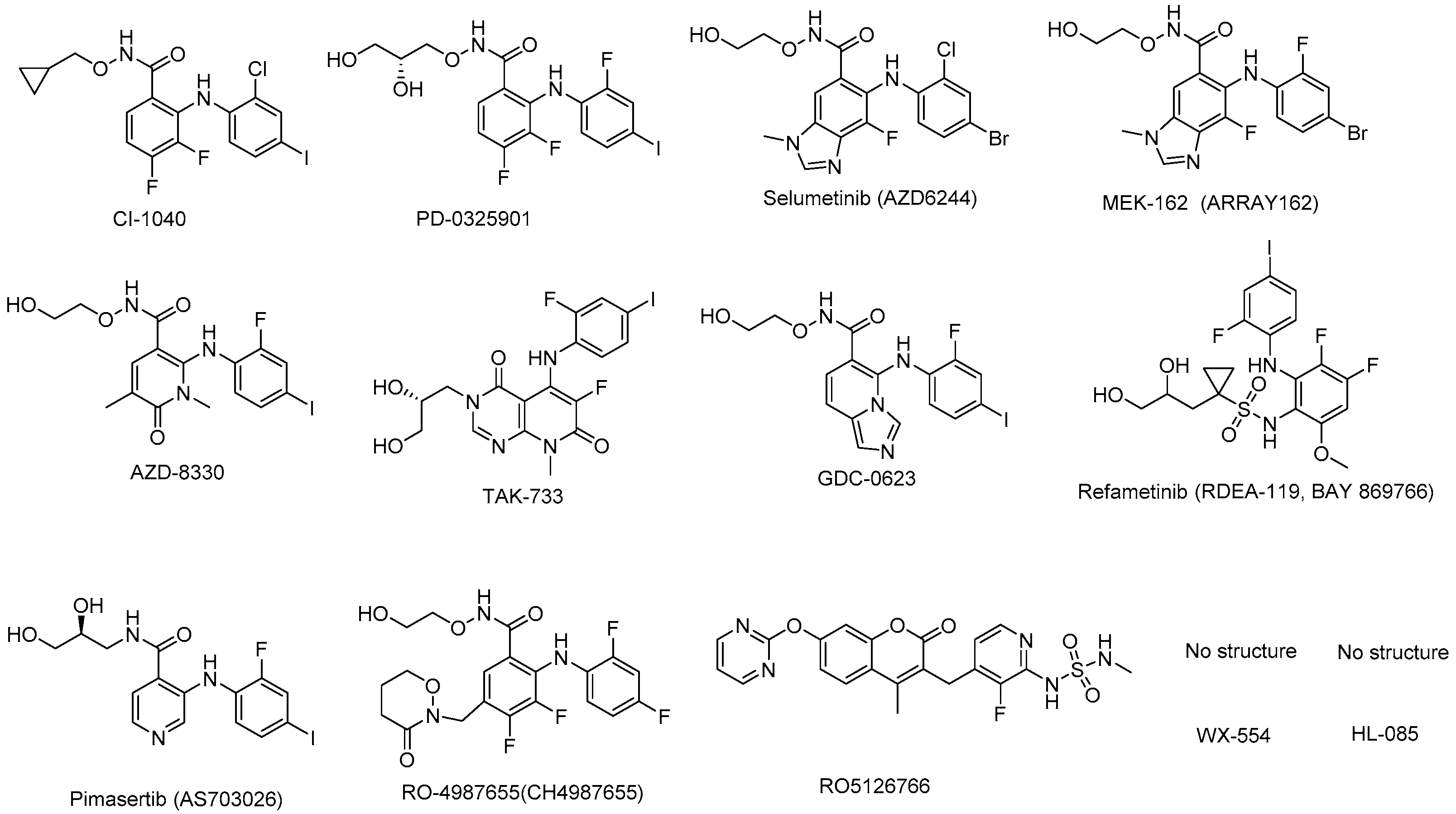
There are several MEK inhibitors in preclinical development (Table 3) and the structures are shown in Figure 4.
5.1. CInQ-03
CInQ-03 is a novel and specific MEK inhibitor both in vitro and in vivo. CInQ-03 has a distinct chemical structure compared with current MEK inhibitors, such as PD318088, PD184352, PD0325901, and selumetinib. The compound has similar binding affinity and displays almost the same binding mode with known inhibitors. CInQ-03 binds deeply into the binding pocket in a manner similar to the crystal ligand; the docking and experimental data suggest that CInQ-03 shows an ability to inhibit MEK1/2.
A xenograft mouse model indicated that administration of CInQ-03 at 1 or 5 mg/kg for 11 days could suppress colon cancer cell growth significantly without toxicity. Furthermore, CInQ-03 could decrease phosphorylation level of ERKs in a cell-based assay, which was highly consistent with the results in vivo. Based on these findings, CInQ-03 is considered and is being developed as a novel MEK inhibitor [90].
5.2. G-573
G-573 is a potent and selective allosteric inhibitor of MEK. Structural and functional analysis illustrates that G-573 could form a strong hydrogen bond with MEK kinase. The IC50 value for pERK inhibition in HCT116 tumor by G-573 was estimated to be 0.406 μM, and ED50 values in HCT116 and H2122 mouse xenograft models were estimated to be 4.6 and 1.9 mg/kg/day, respectively [91].
5.3. PD184161
PD184161 is an orally-active MEK inhibitor which could inhibit MEK activity more effectively (IC50 = 10–100 nM) in a time- and concentration-dependent manner than PD098059 or U0126. PD184161, a structurally related analog of CI-1040 but distinct from PD098059 and U0126, joins the mechanistic class of agents that inhibit the downstream phosphorylation of ERK through their direct effects on MEK. Unlike PD098059 and U0126, PD184161 has the obvious advantages of solubility and oral bioavailability. It exerts antitumor effects against HCC (hepatic cellular cancer) in vitro and in vivo which appears to correlate with suppression of MEK activity. PD184161 is unable to suppress MEK activity in HCC xenografts in the long term [92,93].
5.4. PD318088
PD318088, an analog of CI-1040 with a biarylamine structure, is a novel non-ATP-competitive MEK1 inhibitor. The compound-binding site of PD318088 is adjacent to the ATP binding site. Its potency in inhibiting MEK1 activation is not affected by ATP concentration, suggesting that PD318088 is not competitive with ATP [94].
5.5. PD98059
PD98059 is a potent and selective non-ATP-competitive MEK1 inhibitor. It mediates its inhibitory properties by binding to the ERK-specific MAP kinase MEK, therefore preventing phosphorylation of ERK1/2 (p44/p42 MAPK) by MEK1/2. PD98059 can interact with GST-MEK1 or partially activate MEK, but does not inhibit the MAPK homologues JNK and P38 (D.R. Alessi and P. Cohen, personal communication) [95]. In addition, PD98059 can highly inhibit MEK in a selective manner, but not the other kinases such as Raf kinase, cAMP (cyclic Adenosine monophosphate)-dependent kinase, protein kinase C, v-Src (a gene found in Rous sarcoma virus), epidermal growth factor (EGF) receptor kinase, and phosphatidyl inositol kinase 3 [96,97].
5.6. RO5068760
RO5068760, a substituted hydantoin, is a potent, highly selective, non-ATP-competitive MEK1/2 inhibitor. In vitro, RO5068760 could inhibit MEK1 kinase activity potently with an IC50 value of 0.025 ± 0.012 μmol/L in Raf/MEK/ERK cascade assay. It also shows significant efficacy in a broad spectrum of tumors via the activation of aberrant mitogen-activated protein kinase pathway. RO5068760 shows superior efficacy in tumors harboring the BRAF V600E mutation [98]. The assessment study of target suppression in healthy volunteers showed a peak time (tmax) of two hours and biphasic disposition with reaction half-time (T1/2) of 5~9 h. The inhibition of pERK was relatively modest, with mean maximal pERK suppression of 55% [99].
5.7. U0126
U0126 is a potent and selective non-competitive MEK inhibitor, inhibiting MEK1 and MEK2 (IC50 values of 70 nM and 60 nM, respectively). It could inhibit ERK phosphorylation up to 80% in astrocytes. Surprisingly, U0126 also causes profound depletion of ATP in glucose-deprived cells, leading to death by necrosis [100]. It was reported and demonstrated [101] that U0126 could inhibit the cellular target MEK, and have an antiviral potential not only in cell culture in vitro, but also in the mouse mode in vivo. A promising approach might need to be considered and evaluated for MEK inhibitors as new antivirals against influenza.
5.8. SL327
SL327 is a homolog of U0126, with a mixture of E and Z isomers. SL327 inhibits MEK1 and MEK2, with IC50 values of 0.18 μM and 0.22 μM, respectively. In vivo, the study showed that the combination of SL327 with sunitinib malate induced significant additive suppression of doxorubicin-resistant anaplastic thyroid carcinoma (ATC) tumor growth [102].
6. Conclusions
In recent years, MEK inhibitors have been discovered and developed very rapidly. Trametinib became the first approved drug among MEK inhibitors, and cobimetinib soon followed suit. MEK inhibitors as single agents or in combination with other therapies have shown to be efficacious in treating melanoma, lung cancer, and colorectal cancer.
According to available clinical data, MEK inhibitor plus a BRAF inhibitor could be a superior therapy. However, a proportion of patients being treated with the combination may not achieve optimal results.
Some preclinical studies show that a MEK inhibitor plus a BRAF inhibitor or other targeted agents may modulate the immunization and enhance immune activation. To further improve the efficacy, the combination of an MEK inhibitor plus a BRAF inhibitor or other targeted agents and immunotherapy could represent a promising form of cancer treatment.
However, there are still concerns regarding toxicity, which is one of the obstacles for the development of MEK inhibitors. MEK inhibitor treatment is still considered one of the most promising areas in cancer research. Novel small molecular inhibitors are expected to become the new breakthrough in cancer treatment. Moreover, dual inhibition of MEK and RAF kinase offers advantage in terms of both increased efficacy and minimized toxicity, and may be a prospective therapeutic strategy in targeting the MARK pathway in the near future.
Acknowledgments
This work was supported by the innovation team funding (1649) from the Institute of Radiation Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, and Fundamental Research Fund for CAMS&PUMC (2016ZX310199), CAMS Innovation Fund for Medical Sciences (CIFMS, 2017-I2M-3-019) from the Chinese Academy of Medical Sciences & Peking Union Medical College.
Author Contributions
Hongqi Tian designed the structure of the review; Ying Cheng wrote this paper; Hongqi Tian revised the paper.
Conflicts of Interest
The authors are the inventors of HL-085.
References
- Karin, L.C.M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar]
- Leonard, J.T.; Raess, P.; Dunlap, J.; Hayes-Lattin, B.; Tyner, J.W.; Traer, E. Functional and genetic screening of acute myeloid leukemia associated with mediastinal germ cell tumor identifies MEK inhibitor as an active clinical agent. J. Hematol. Oncol. 2016, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Lyons, J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr. Opin. Pharmacol. 2005, 5, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. MEK inhibitors in oncology: A patent review (2015–Present). Expert Opin. Ther. Pat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Templeton, I.E.; Musib, L. MEK inhibitors beyond monotherapy: Current and future development. Curr. Opin. Pharmacol. 2015, 23, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Martin-Liberal, J.; Lagares-Tena, L.; Larkin, J. Prospects for MEK inhibitors for treating cancer. Expert Opin. Drug Saf. 2014, 13, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alex, A.A. The clinical development of MEK inhibitors. Nat. Rev. 2014, 11, 385–400. [Google Scholar]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef] [PubMed]
- Antonio, M.G.; Ester, S.; Lucia, F.; Vito, V.; Martina, S.; Paolo, A.A. MEK Inhibitors in the treatment of metastatic melanoma and solid tumors. Am. J. Clin. Dermatol. 2017. [Google Scholar] [CrossRef]
- David, E.U.; Philip, A.H. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 19, 4047–4056. [Google Scholar]
- Steve, P. Putative allosteric MEK1 and MEK2 inhibitors. Expert Opin. Ther. Pat. 2008, 18, 603–627. [Google Scholar]
- Juliet, R.; Juan, M.L.; Stefan, D.; James, L. BRAF and MEK inhibition for the treatment of advanced BRAF mutant melanoma. Expert Opin. Pharmacother. 2015, 16, 1285–1297. [Google Scholar]
- Carolina, H.J.; Suzanne, E.B. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar]
- Carsten, H.; Jonathan, L.B. The ups and downs of MEK kinase interactions. Cell. Signal. 2001, 13, 863–875. [Google Scholar]
- Paola, Q.; Virginia, P.; Francesco, S. Combined BRAF and MEK inhibition for the treatment of BRAF-mutated metastatic melanoma. Cancer Treat. Rev. 2015, 41, 519–526. [Google Scholar]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharmacol. Ther. 2014, 141, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Boerner, S.A.; Winkler, J.D.; LoRusso, P.M. Clinical experience of MEK inhibitors in cancer therapy. Biochim. Biophys. Acta 2007, 1773, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-activated Protein Kinase Kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.D.; Aay, N.; Anand, N.K.; Blazey, C.M.; Bowles, O.J.; Bussenius, J.; Costanzo, S.; Curtis, J.K.; Defina, S.C.; Dubenko, L.; et al. Novel Carboxamide-Based Allosteric MEK Inhibitors: Discovery and Optimization Efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012, 3, 416–421. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000TOC.cfm (accessed on 29 May 2013).
- U.S. Food and Drug Administration (FDA). 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206192Orig1s000PharmR.pdf (accessed on 10 November 2015).
- U.S. Food and Drug Administration (FDA). 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000ClinPharmR.pdf (accessed on 29 May 2013).
- Takahashi, R.H.; Ma, S.; Yue, Q.; Kim-Kang, H.; Yi, Y.; Ly, J.; Boggs, J.W.; Fettes, A.; McClory, A.; Deng, Y.; et al. Absorption, metabolism and excretion of cobimetinib, an oral MEK inhibitor, in rats and dogs. Xenobiotica 2017, 47, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Rosen, L.S.; LoRusso, P.; Ma, W.W.; Goldman, J.W.; Weise, A.; Colevas, A.D.; Adjei, A.; Yazji, S.; Shen, A.; Johnston, S.; et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [PubMed]
- Abe, H.; Kikuchi, S.; Hayakawa, K.; Iida, T.; Nagahashi, N.; Maeda, K.; Sakamoto, J.; Matsumoto, N.; Miura, T.; Matsumura, K.; et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Leighl, N.; Delord, J.-P.; Barlesi, F.; Bennouna, J.; Zalcman, G.; Infante, J.R.; Reckamp, K.L.; Kelly, K.; Shepherd, F.A.; et al. A phase 1/1b study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204114s001lbl.pdf (accessed on 1 January 2014).
- Hsueh, E.C.; Gorantla, K.C. Novel melanoma therapy. Exp. Hematol. Oncol. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). FDA Grants Regular Approval to Dabrafenib and Trametinib Combination for Metastatic NSCLC with BRAF V600E Mutation; FDA: Silver Spring, MD, USA, 2017.
- EMA. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002643/WC500222159.pdf (accessed on 23 February 2017).
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Dholaria, B.; Hammond, W.; Shreders, A.; Lou, Y. Emerging therapeutic agents for lung cancer. J. Hematol. Oncol. 2016, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Daud, A. Nivolumab plus ipilimumab in the treatment of advanced melanoma. J. Hematol. Oncol. 2015, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Diggs, L.P.; Hsueh, E.C. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark. Res. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Simeone, E.; Grimaldi, A.M.; Festino, L.; Vanella, V.; Palla, M.; Ascierto, P.A. Combination treatment of patients with BRAF-mutant melanoma: A new standard of care. BioDrugs 2017, 31, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.F.; Ng, C.M.; Berry, L.; Belvin, M.; Lewin-Koh, N.; Merchant, M.; Salphati, L. PK-PD modeling of combination efficacy effect from administration of the MEK inhibitor GDC-0973 and PI3K inhibitor GDC-0941 in A2058 xenografts. Cancer Chemother. Pharmacol. 2013, 71, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Miller, W.H.; Kim, T.M.; Lee, C.B.; Flaherty, K.T.; Reddy, S.; Jamal, R.; Chow, L.Q.; Rooney, I.A.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + cobimetinib (C) in metastatic melanoma (mel): Updated safety and clinical activity. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Wallin, J.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + Cobimetinib (C) + Vemurafenib (V) in BRAFV600-Mutant Metastatic Melanoma (mel): Updated Safety and Clinical Activity. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 2–6 June 2017; p. 3063. [Google Scholar]
- Johanna, C.B.; Tae, W.K.; Boon, C.G.; Jeffrey, W.; Do-Youn, O.; Sae-Won, H.; Carrie, B.L.; Matthew, D.H.; Jayesh, D.; Jeremy, H.L.; et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Barrett, S.D.; Bridges, A.J.; Dudley, D.T.; Saltiel, A.R.; Fergus, J.H.; Flamme, C.M.; Delaney, A.M.; Kaufman, M.; LePage, S.; Leopold, W.R.; et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg. Med. Chem. Lett. 2008, 18, 6501–6504. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Duesbery, N.S. Highly selective MEK inhibitors. Curr. Enzym. Inhib. 2010, 6, 146–157. [Google Scholar] [CrossRef]
- Pheneger, J.; Wallace, E.; Marlow, A.; Hurley, B.; Lyssikatos, J.; Bendele, A.M.; Lee, P.A. Characterization of ARRY-438162, a Potent MEK Inhibitor in Combination with Methotrexate or Ibuprofen in In Vivo Models of Arthritis. In Proceedings of the 2006 Annual Scientific Meeting, Boston, MA, USA, 20–24 October 2006; p. 794. [Google Scholar]
- Wallace, E.; Lyssikatos, J.; Blake, J.; Marlow, A.; Greschuk, J.; Yeh, T.; Callejo, M.; Marsh, V.; Poch, G.; Otten, J.; et al. AZD8330 (ARRY-424704): Preclinical evaluation of a potent, selective MEK 1/2 inhibitor currently in phase I trials. In Proceedings of the AACR, Denver, CO, USA, 18–22 April 2009; Volume 69. [Google Scholar]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Iverson, C.; Larson, G.; Lai, C.; Yeh, L.T.; Dadson, C.; Weingarten, P.; Appleby, T.; Vo, T.; Maderna, A.; Vernier, J.M.; et al. RDEA119/BAY 869766: A potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009, 69, 6839–6847. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, Y.; Kohchi, Y.; Iikura, H.; Matsubara, Y.; Asoh, K.; Murata, T.; Kohchi, M.; Mizuguchi, E.; Tsujii, S.; Hattori, K.; et al. Design and synthesis of novel allosteric MEK inhibitor CH4987655 as an orally available anticancer agent. Bioorg. Med. Chem. Lett. 2011, 21, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, D.; Griffin, M.J.; Sludden, J.; Drew, Y.; Cresti, N.; Swales, K.; Merriman, M.; Allen, R.; Bevan, P.; Buerkle, M.; et al. A phase I pharmacokinetic and pharmacodynamic study of the oral mitogen-activated protein kinase kinase (MEK) inhibitor, WX-554, in patients with advanced solid tumours. Eur. J. Cancer 2016, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ji, C.; Liu, C.; Kong, L.; Cheng, Y.; Huang, G.C. Benzoheterocyclic Compounds and Use Thereof. Patent EP 2804855 A4, 25 July 2013. [Google Scholar]
- Wabnitz, P.A.; Mitchell, D.; Wabnitz, D.A. In vitro and in vivo metabolism of the anti-cancer agent CI-1040, a MEK inhibitor, in rat, monkey, and human. Pharm. Res. 2004, 21, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Carlson, T.C.; Loi, C.M.; Graziano, M.J. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother. Pharmacol. 2007, 59, 671–679. [Google Scholar] [CrossRef] [PubMed]
- El-Hoss, J.; Kolind, M.; Jackson, M.T.; Deo, N.; Mikulec, K.; McDonald, M.M.; Little, C.B.; Little, D.G.; Schindeler, A. Modulation of endochondral ossification by MEK inhibitors PD0325901 and AZD6244 (Selumetinib). Bone 2014, 59, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Ricart, A.D.; Larson, T.G.; Stella, P.J.; Bazhenova, L.; Miller, V.A.; Cohen, R.B.; Eisenberg, P.D.; Selaru, P.; Wilner, K.D.; et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Boasberg, P.D.; Redfern, C.H.; Daniels, G.A.; Bodkin, D.; Garrett, C.R.; Ricart, A.D. Pilot study of PD-0325901 in previously treated patients with advanced melanoma, breast cancer, and colon cancer. Cancer Chemother. Pharmacol. 2011, 68, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Lui, V.W.; Cheung, C.S.; Lau, C.P.; Ho, K.; Hui, E.P.; Tsui, S.K.; Ng, M.H.; Cheng, S.H.; Ng, P.K.; et al. Activity of the MEK inhibitor selumetinib (AZD6244; ARRY-142886) in nasopharyngeal cancer cell lines. Investig. New Drugs 2013, 31, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Beloueche-Babari, M.; Jamin, Y.; Arunan, V.; Walker-Samuel, S.; Revill, M.; Smith, P.D.; Halliday, J.; Waterton, J.C.; Barjat, H.; Workman, P.; et al. Acute tumour response to the MEK1/2 inhibitor selumetinib (AZD6244, ARRY-142886) evaluated by non-invasive diffusion-weighted MRI. Br. J. Cancer 2013, 109, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.; Van, C.N.; Heng, N.K.; Donald, P.; Su, P.C.; Han, C.T.; Choon, H.T.; Pierce, C.; Hock, S.O.; Alexander, C.; et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J. Hepatol. 2010, 52, 79–87. [Google Scholar]
- Krishnamurthy, A.; Dasari, A.; Noonan, A.M.; Mehnert, J.M.; Lockhart, A.C.; Stein, M.N.; Sanoff, H.K.; Lee, J.J.; Hansen, A.R.; Malhotra, U.; et al. A phase IB study of the combination of selumetinib (AZD6244, ARRY-142886) and cyclosporin A (CsA) in patients with advanced solid tumors with an expansion cohort in metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Tai, W.M.; Yong, W.P.; Lim, C.; Low, L.S.; Tham, C.K.; Koh, T.S.; Ng, Q.S.; Wang, W.W.; Wang, L.Z.; Hartano, S.; et al. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC). Ann. Oncol. 2016, 27, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non–small cell lung cancer. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A pediatric brain tumor consortium (PBTC) study. Neuro-Oncology 2017, 19, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.R.; Onar-Thomas, A.; Young-Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.I.; Banerjee, A.; Packer, R.; Kilburn, L.B.; Pollack, I.; et al. A phase II prospective study of selumetinib in children with recurrent or refractory low-grade glioma (LGG): A Pediatric Brain Tumor Consortium (PBTC) study. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Chappell, W.H.; Russo, S.; Ove, R.; Milella, M.; Tafuri, A.; Lunghi, P.; Bonati, A.; et al. Emerging MEK inhibitors. Expert Opin. Emerg. Drugs 2010, 15, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D.; Tan, B.R.; Khan, G.N.; Zalupski, M.M.; et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer 2017, 116, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Marlow, A.L.; Wallace, E.; Seo, J.; Lyssikatos, J.P.; Yang, H.W.; Blake, J.; Storey, R.A.; Booth, R.J.; Pittam, J.D. Heterocyclic Inhibitors of Mek and Methods of Use Thereof. Patent WO2007044084 A3, 19 April 2007. [Google Scholar]
- Haasbach, E.; Hartmayer, C.; Planz, O. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro. Antivir. Res. 2013, 98, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B.; Aamdal, S.; Nyakas, M.; Cavallin, M.; Green, D.; Learoyd, M.; Smith, I.; Kurzrock, R. A phase I dose-finding, safety and tolerability study of AZD8330 in patients with advanced malignancies. Eur. J. Cancer 2013, 49, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S.; et al. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.; Belvin, M.; Merchant, M.; Chan, E.; Hollingshead, P.; Orr, C.; Boggs, J.; Plise, E.; Robarge, K.; Zak, M. Preclinical Pharmacokinetics and efficacy assessment of a potent and selective MEK inhibitor, GDC-0623. Eur. J. Cancer 2012, 48, 155. [Google Scholar] [CrossRef]
- Van Laethem, J.L.; Riess, H.; Jassem, J.; Haas, M.; Martens, U.M.; Weekes, C.; Peeters, M.; Ross, P.; Bridgewater, J.; Melichar, B.; et al. Phase I/II study of refametinib (BAY 86-9766) in combination with gemcitabine in advanced pancreatic cancer. Target. Oncol. 2017, 12, 97–109. [Google Scholar] [PubMed]
- Lim, H.Y.; Heo, J.; Choi, H.J.; Lin, C.Y.; Yoon, J.H.; Hsu, C.; Rau, K.M.; Poon, R.T.; Yeo, W.; Park, J.W.; et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5976–5985. [Google Scholar] [CrossRef] [PubMed]
- Houédé, N.; Delord, J.P.; Awada, A.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; von Richter, O.; Raymond, E. Pharmacokinetics and pharmacodynamics of a selective oral MEK1/2 inhibitor, pimasertib (MSC1936369B/AS703026), in patients with advanced solid tumors. Eur. J. Cancer 2012, 48, 184. [Google Scholar] [CrossRef]
- Naing, A.; Mita, M.; Komarnitsky, P.; Milner, A.; von Richter, O.; Ogden, J.; Piha-Paul, S.; Fu, S.; Asatiani, E.; Kurzrock, R. Phase I dose-escalation trial of a selective oral MEK1/2 inhibitor, pimasertib (MSC1936369B), combined with an mTOR inhibitor, temsirolimus, in patients with advanced solid tumors. Eur. J. Cancer 2012, 48, 187. [Google Scholar] [CrossRef]
- Awada, A.; Delord, J.P.; Houédé, N.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; Rejeb, N.; Raymond, E. Safety and recommended phase II Dose (RP2D) of the selective oral MEK1/2 inhibitor pimasertib (MSC1936369B/AS703026): Results of a phase I trial. Eur. J. Cancer 2012, 48, 185–186. [Google Scholar] [CrossRef]
- Delord, J.P.; Houédé, N.; Awada, A.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; Rejeb, N.; Raymond, E. 616 pimasertib (MSC1936369B/AS703026), a selective oral MEK1/2 inhibitor, shows clinical activity in melanoma. Eur. J. Cancer 2012, 48, 190. [Google Scholar] [CrossRef]
- Kraeber-Bodere, F.; Carlier, T.; Naegelen, V.M.; Shochat, E.; Lumbroso, J.; Trampal, C.; Nagarajah, J.; Chua, S.; Hugonnet, F.; Stokkel, M.; et al. Differences in the biologic activity of 2 novel MEK inhibitors revealed by 18F-FDG PET: Analysis of imaging data from 2 phase I trials. J. Nucl. Med. 2012, 53, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Middleton, M.R.; Tresca, P.; Kraeber-Bodere, F.; Dieras, V.; Scheulen, M.E.; Gupta, A.; Lopez-Valverde, V.; Xu, Z.X.; Rueger, R.; et al. Phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of the MEK inhibitor RO4987655 (CH4987655) in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 4794–4805. [Google Scholar] [CrossRef] [PubMed]
- Shinji, N.; Hiroshi, N.; Noboru, Y.; Yasuhide, Y.; Yutaka, F.; Yosuke, T.; Hiroshi, W.; Kazunori, H.; Hidenori, M.; Satoru, K.; et al. Phase I and pharmacokinetics/pharmacodynamics study of the MEK inhibitor RO4987655 in Japanese patients with advanced solid tumors. Investig. New Drugs 2015, 41, 641–651. [Google Scholar]
- Martinez-Garcia, M.; Banerji, U.; Albanell, J.; Bahleda, R.; Dolly, S.; Kraeber-Bodere, F.; Rojo, F.; Routier, E.; Guarin, E.; Xu, Z.X.; et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin. Cancer Res. 2012, 18, 4806–4819. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, M.H.; Reddy, K.; Li, Y.; Lim, D.Y.; Xie, H.; Lee, S.Y.; Yeom, Y.I.; Bode, A.M.; Dong, Z. CInQ-03, a novel allosteric MEK inhibitor, suppresses cancer growth in vitro and in vivo. Carcinogenesis 2013, 34, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.F.; Belvin, M.; Chan, J.; Hoeflich, K.; Orr, C.; Robarge, K.; Yang, X.; Zak, M.; Boggs, J. Preclinical disposition and pharmacokinetics-pharmacodynamic modeling of biomarker response and tumour growth inhibition in xenograft mouse models of G-573, a MEK inhibitor. Xenobiotica 2010, 40, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.J.; Schmidt, C.M.; Wiesenauer, C.A.; Choi, J.N.; Gage, E.A.; Yip-Schneider, M.T.; Wiebke, E.A.; Wang, Y.; Omer, C.; Sebolt-Leopold, J.S. The effects of a novel MEK inhibitor PD184161 on MEK-ERK signaling and growth in human liver cancer. Neoplasia 2006, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Wyttenbach, A.; Tolkovsky, A.M. Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem. Pharmacol. 2004, 68, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhou, V.; Pan, S.; Liu, Y.; Hornsby, M.; McMullan, D.; Klock, H.E.; Haugen, J.; Lesley, S.A.; Gray, N.; et al. Identification of coumarin derivatives as a novel class of allosteric MEK1 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5467–5473. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.T.; Pang, L.; Decker, S.J.; Bridges, A.J.; Saltiel, A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Galuppo, M.; Mazzon, E.; Paterniti, I.; Bramanti, P.; Cuzzocrea, S. PD98059, a specific MAP kinase inhibitor, attenuates multiple organ dysfunction syndrome/failure (MODS) induced by zymosan in mice. Pharmacol. Res. 2010, 61, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Cerioni, L.; Palomba, L.; Cantoni, O. The Raf/MEK inhibitor PD98059 enhances ERK1/2 phosphorylation mediated by peroxynitrite via enforced mitochondrial formation of reactive oxygen species. FEBS Lett. 2003, 547, 92–96. [Google Scholar] [CrossRef]
- Daouti, S.; Higgins, B.; Kolinsky, K.; Packman, K.; Wang, H.; Rizzo, C.; Moliterni, J.; Huby, N.; Fotouhi, N.; Liu, M.; et al. Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Mol. Cancer Ther. 2010, 9, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Niu, H.; Goelzer, P.; Rueger, R.; Deutsch, J.; Busse-Reid, R.; DeSchepper, S.; Blotner, S.; Barrett, J.; Weissgerber, G.; et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of RO5068760, an MEK inhibitor, in healthy volunteers: Assessment of target suppression. J. Clin. Pharmacol. 2010, 50, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.; Houpt, T.A. Mitogen-activated protein kinase in the amygdala plays a critical role in lithium chloride-induced taste aversion learning. Neurobiol. Learn. Mem. 2012, 97, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Dokladda, K.; Green, K.A.; Pan, D.A.; Hardie, D.G. PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP: ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett. 2005, 579, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Zhao, L.; Chen, S. Combination of SL327 and Sunitinib Malate leads to an additive anti-cancer effect in doxorubicin resistant thyroid carcinoma cells. Biomed. Pharmacother. 2017, 88, 985–990. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical formula of trametinib.
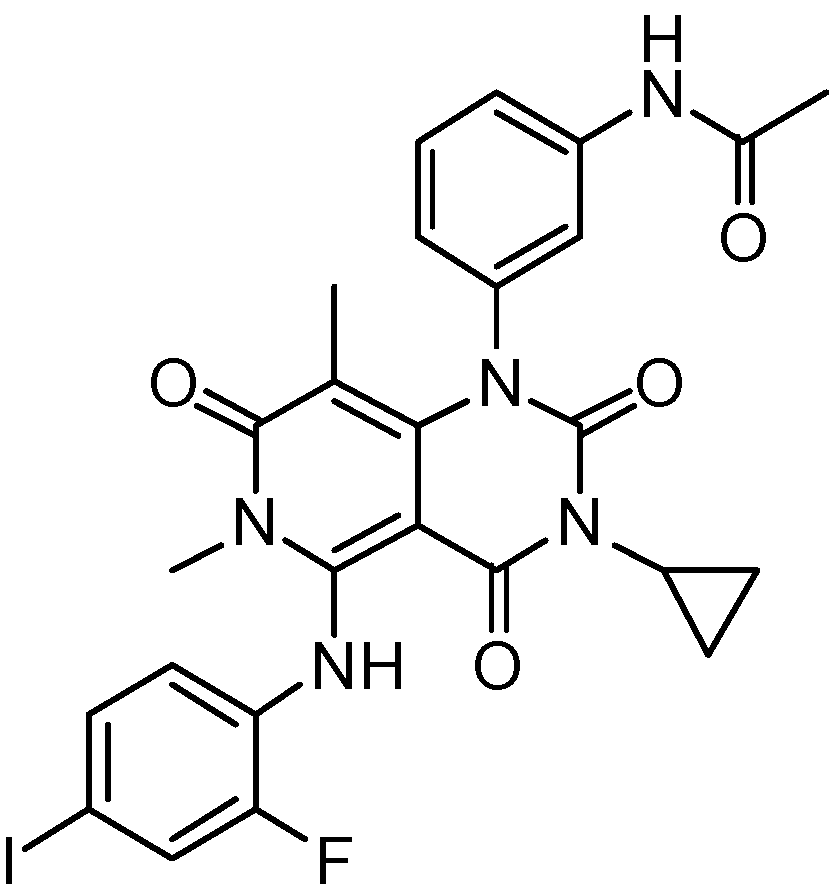
Figure 2.
Chemical formula of cobimetinib.
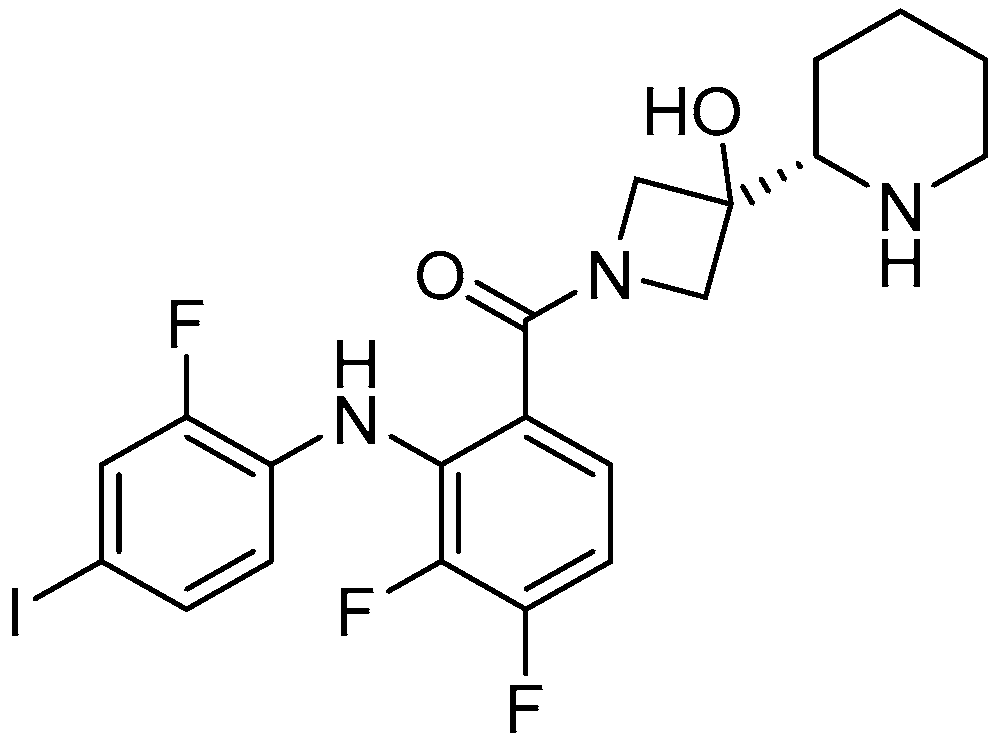
Figure 3.
MEK inhibitors in clinical study.
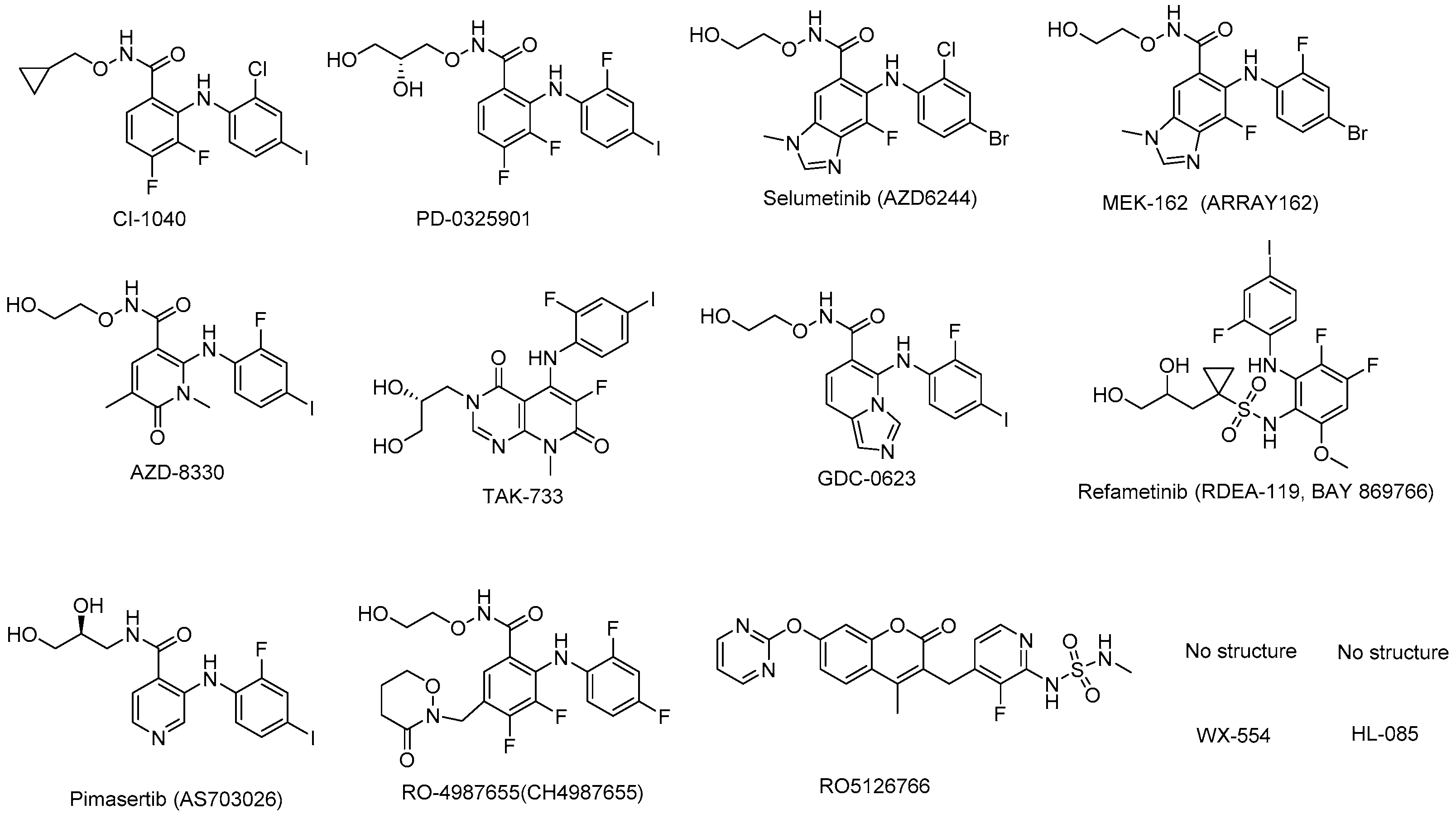
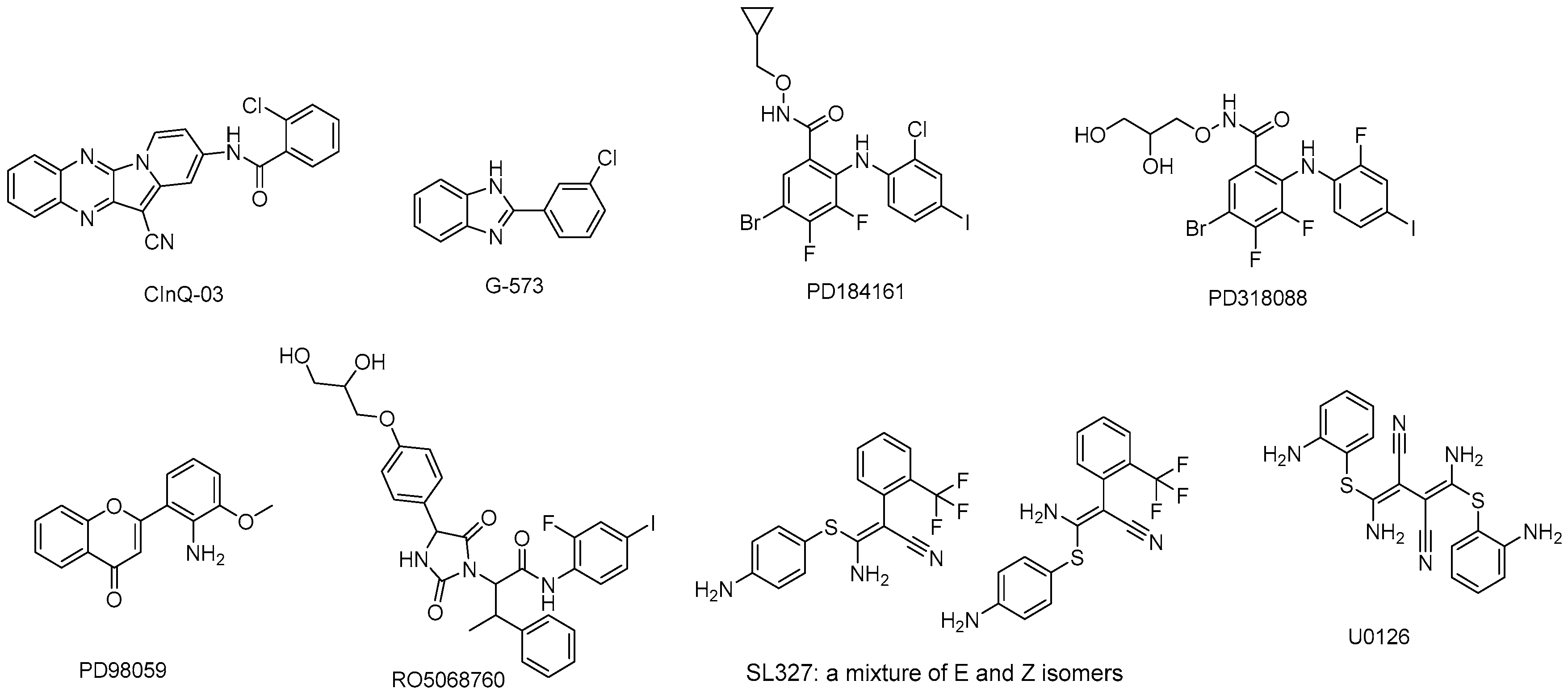
Figure 4.
MEK inhibitors in preclinical study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Preclinical and clinical data for trametinib and cobimetinib.
| Item | Trametinib | Cobimetinib | |
|---|---|---|---|
| Structure | 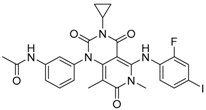 | 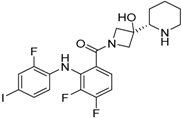 | |
| BCS 1 class | BCS II (high permeability, low solubility) | BCS I (high permeability, high solubility) | |
| Salt form | dimethyl sulfoxide solvate (1:1) | hemifumarate | |
| Molecule weight (free base) | 615.4 | 531.32 | |
| In vitro (enzyme) [19,20] | MEK 2 kinase | MEK1 IC50 3 = 0.7 nM; | MEK1 IC50 = 0.95 nM; |
| MEK2 IC50 = 0.9 nM | MEK2 IC50 = 199 nM | ||
| In vitro (cell potency) [21,22] | A375 | 0.74 nM | 5 nM |
| In vivo efficacy (xenograft) [19,20] | A375 | TGI 4 = 60% @ 0.1 mpk; 14 days; | TGI = 87% @ 3 mg/kg, 21 days, QD 5; |
| TGI = 102% @ 0.3 mpk; 14 days; TGI = 118% @ 3 mpk; 14 days | TGI = 106% @ 5 mg/kg, 21 days, QD | ||
| Pharmacokinetics (rats) [23,24] | Cmax 6 | 2.7 μM (3 mg/kg, mice); | 0.997 μM (30 mg/kg, male rats) |
| 0.47 μM (3 mg/kg, rats) | |||
| Tmax 7 | 4 h (mice, 3 mg/kg, 14 days, repeat); | 2 h (30 mg/kg, male rats) | |
| 4 h (rats, 1 mg/kg, 21days, repeat) | |||
| T1/2 8 | 3.8 h (mice); | 5.56 h (30 mg/kg, male rats) | |
| 5.5 h (rats) | |||
| protein binding rate | 97.40% (human) | 98.8% (5 μM, dog); | |
| 93.5% (5 μM, human) | |||
| Toxicokinetics (rats) [21,22] | Tmax | 4 h (male rats, 0.1667 mg/kg, week 4) | N/A |
| Cmax | 10.1 ng/mL (male rats, 0.1667 mg/kg, week 4) | 39.9 ng/mL (male rats, 3 mg/kg, day 22) | |
| AUC0-t 9 | 188 ng·h/mL (male rats, 0.1667 mg/kg, week 4) | 244 ng·h/mL (male rats, 3 mg/kg, day 22) | |
| Clinical PK [25,26] | MTD 10 | 3 mg/day (QD) | 60 mg/day, QD, 21/7; |
| 100 mg/day, QD, 14/14 | |||
| Tmax | 1.5 h (2 mg, QD) | 2.4 h (60 mg, QD) | |
| Cmax | 22.2 ng/mL (2 mg, QD) | 273 ng/mL (60 mg, QD) | |
| T1/2 | 4–5 d (2 mg, QD) | 43.6 h (60 mg, QD) | |
| CL/F 11 | 5.4 L/h (2 mg, QD) | 13.8 L/h (60 mg, QD) | |
| AUC | 370 ng·h/mL (0-t, day 15, 2 mg, QD) | 4340 ng·h/mL (60 mg, QD) | |
| period/cycle | 21 days/7 days | 21 days/7 days | |
| absolute bioavailability | 72% (2 mg, QD) | 46% (20 mg, QD) | |
| recommended dose | 2 mg, QD | 60 mg, QD | |
| Adverse reactions [25,26] | rash, diarrhea, fatigue, peripheral edema, nausea, and dermatitis acneiform | gastrointestinal disorders, rash, pyrexia, increased blood CPK 12, chorioretinopathy | |
1 BSC: biopharmaceutics classification system. 2 MEK: mitogen-activated protein kinase kinase. 3 IC50: half maximal inhibitory concentration. 4 TGI: tumor growth inhibition. 5 QD: once a day. 6 Cmax: peak concentration. 7 Tmax: peak time. 8 T1/2: elimination half-life. 9 AUC0-t: Area under the plasma drug concentration-time curve. 10 MTD: maximum tolerated dose. 11 CL/F: apparent plasma clearance. 12 CPK: creatine phosphokinase.
Table 2.
MEK inhibitors in clinical trials.
| MEK Inhibitor | Target | IC50 | Indications | Company | Clinical Phase |
|---|---|---|---|---|---|
| CI-1040 (PD184352) [47] | MEK1/2 | 2.3 nM | breast cancer, colorectal cancer, lung cancer, and pancreatic cancer | Pfizer | Phase II |
| PD0325901 [47] | MEK1/2 | 0.33 nM | melanoma, colonic neoplasms, breast neoplasms, carcinoma, NSCLC 1 | Pfizer | Phase II |
| Selumetinib (AZD6244) [48] | MEK1 | 14 nM | melanoma, NSCLC | Array BioPharma and AstraZeneca | Phase III |
| MEK162 [49] | MEK1/2 | 12 nM | BRAF 2 or NRAS 3 mutant melanoma | Array Biopharma/Novartis | Phase III |
| AZD8330 [50] | MEK1/2 | 7 nM | advanced solid tumors | AstraZeneca | Phase I |
| TAK-733 [51] | MEK1/2 | 3.2 nM | advanced non-hematologic malignancies, advanced metastatic melanoma | Millennium Pharmaceutical, Inc./Takeda Pharmaceutical Company Limited | Phase I |
| GDC-0623 [52] | MEK1/2 | 0.13 nM | metastatic solid tumors | Genentech | Phase I |
| Refametinib (RDEA119; BAY 869766) [53] | MEK1/2 | 19 nM/47 nM | hepatocellular cancer, melanoma, colorectal cancer | Ardea Biosciences/Bayer | Phase II |
| Pimasertib (AS703026) | MEK1/2 | 5–11 nM | colorectal cancer, multiple myeloma | Merck and Co. | Phase II |
| RO4987655 (CH4987655) [54] | MEK1 | 42 nM | neoplasms | Hoffman-La Roche | Phase I |
| RO5126766 [54] | RAF/MEK1/2 | 160 nM | neoplasms | Hoffmann-La Roche | Phase I |
| WX-554 [55] | MEK1/2 | 4.7 nM/10.7 nM | advanced solid tumors | Wilex, AG. Germany | Phase I/II (terminated) |
| HL-085 [56] | MEK1 | 1.9–10 nM | no data | Binjiang Pharma | Phase I |
1 NSCLC: non-small-cell lung cancer. 2 BRAF: a human gene that encodes a protein called B-Raf. 3 NRAS: a member of the RAS gene family.
Table 3.
MEK inhibitors in preclinical development
| MEK Inhibitor | Target | IC50 | Current Sponsor | Research Progress |
|---|---|---|---|---|
| CInQ-03 | MEK1/2 | 5/10 μM | No data | in preclinical study |
| G-573 | MEK | No data | Genentech | in preclinical study |
| PD184161 | MEK | 10–100 nM | Pfizer | in preclinical study |
| PD318088 | MEK1 | No data | Pfizer Global Research & Development | in preclinical study |
| PD98059 | MEK1 | 2 μM | No data | in preclinical study |
| RO5068760 | MEK1 | 0.025 ± 0.012 μM | Hoffmann-La Roche, Inc. | in preclinical study |
| U0126 | MEK1/2 | 0.07 μM/0.06 μM | No data | in preclinical study |
| SL327 | MEK1/2 | 0.18 μM/0.22 μM | No data | in preclinical study |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. https://doi.org/10.3390/molecules22101551
AMA Style
Cheng Y, Tian H. Current Development Status of MEK Inhibitors. Molecules. 2017; 22(10):1551. https://doi.org/10.3390/molecules22101551
Chicago/Turabian StyleCheng, Ying, and Hongqi Tian. 2017. "Current Development Status of MEK Inhibitors" Molecules 22, no. 10: 1551. https://doi.org/10.3390/molecules22101551