
Oligonucleotide Therapy for Obstructive and Restrictive Respiratory Diseases

Abstract
:1. Major Respiratory Diseases and the Current Therapies
1.1. Asthma
1.2. Chronic Obstructive Pulmonary Disease (COPD)
1.3. Pulmonary Fibrosis
2. Oligonucleotides as Therapeutics for Respiratory Diseases
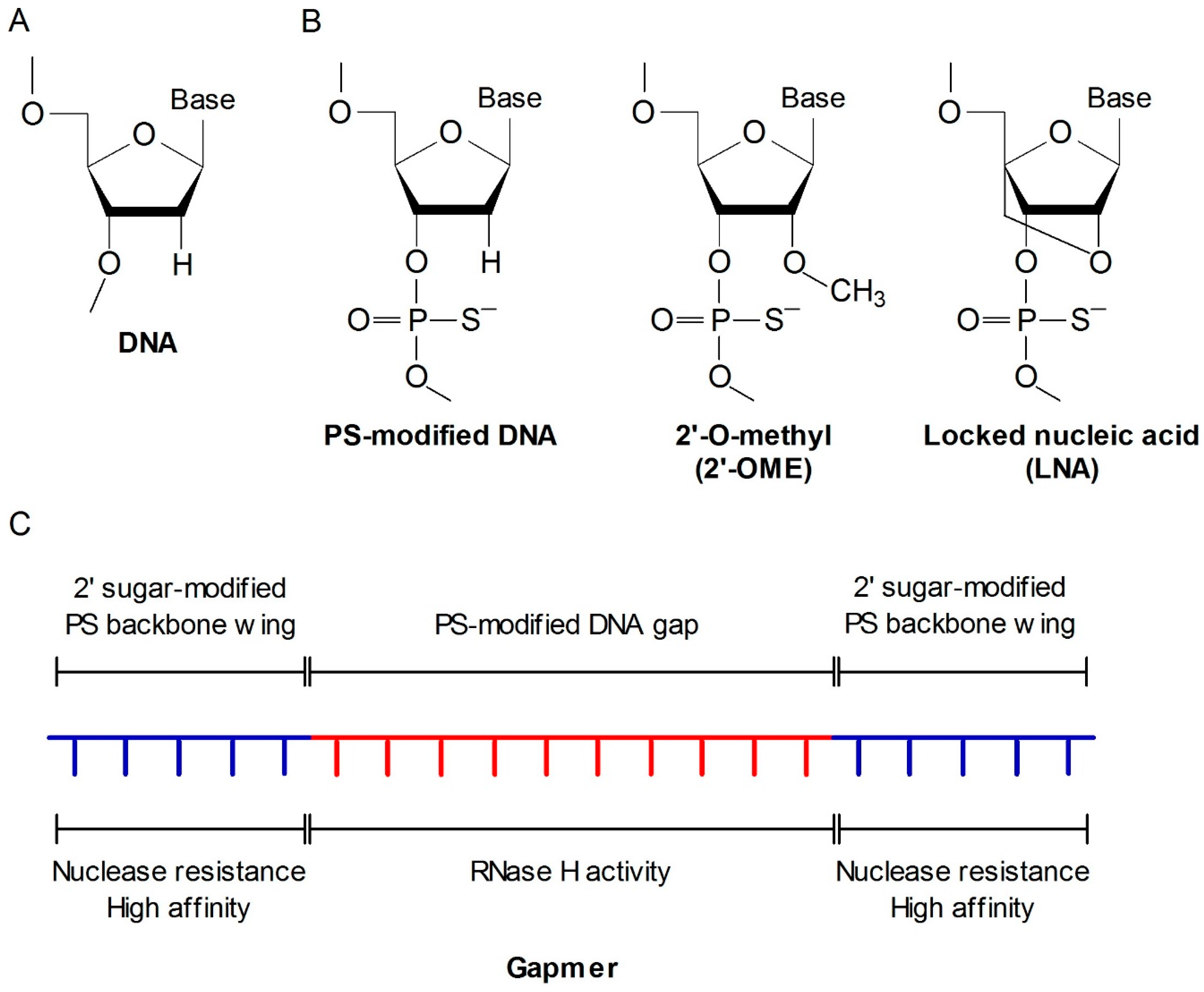
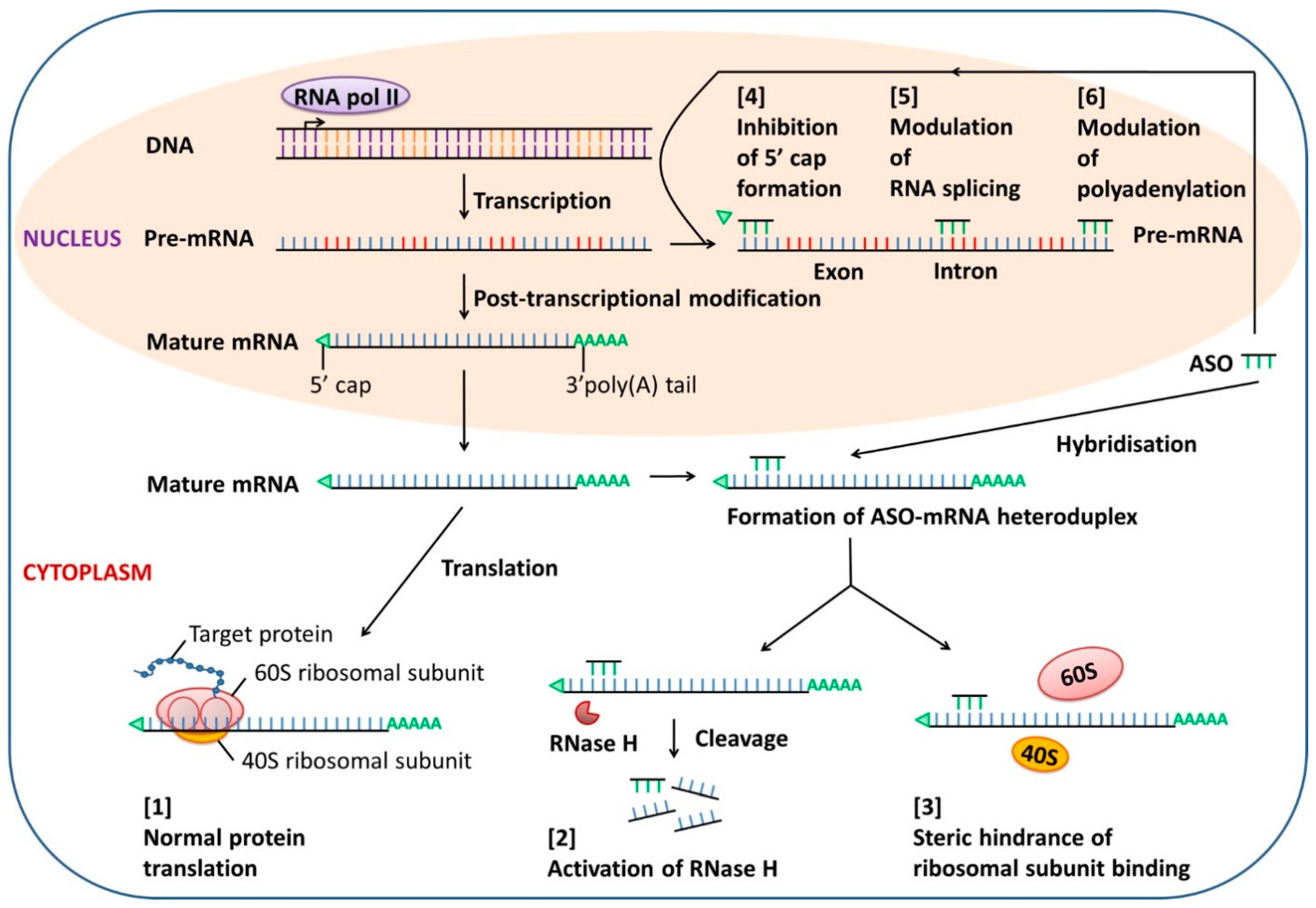
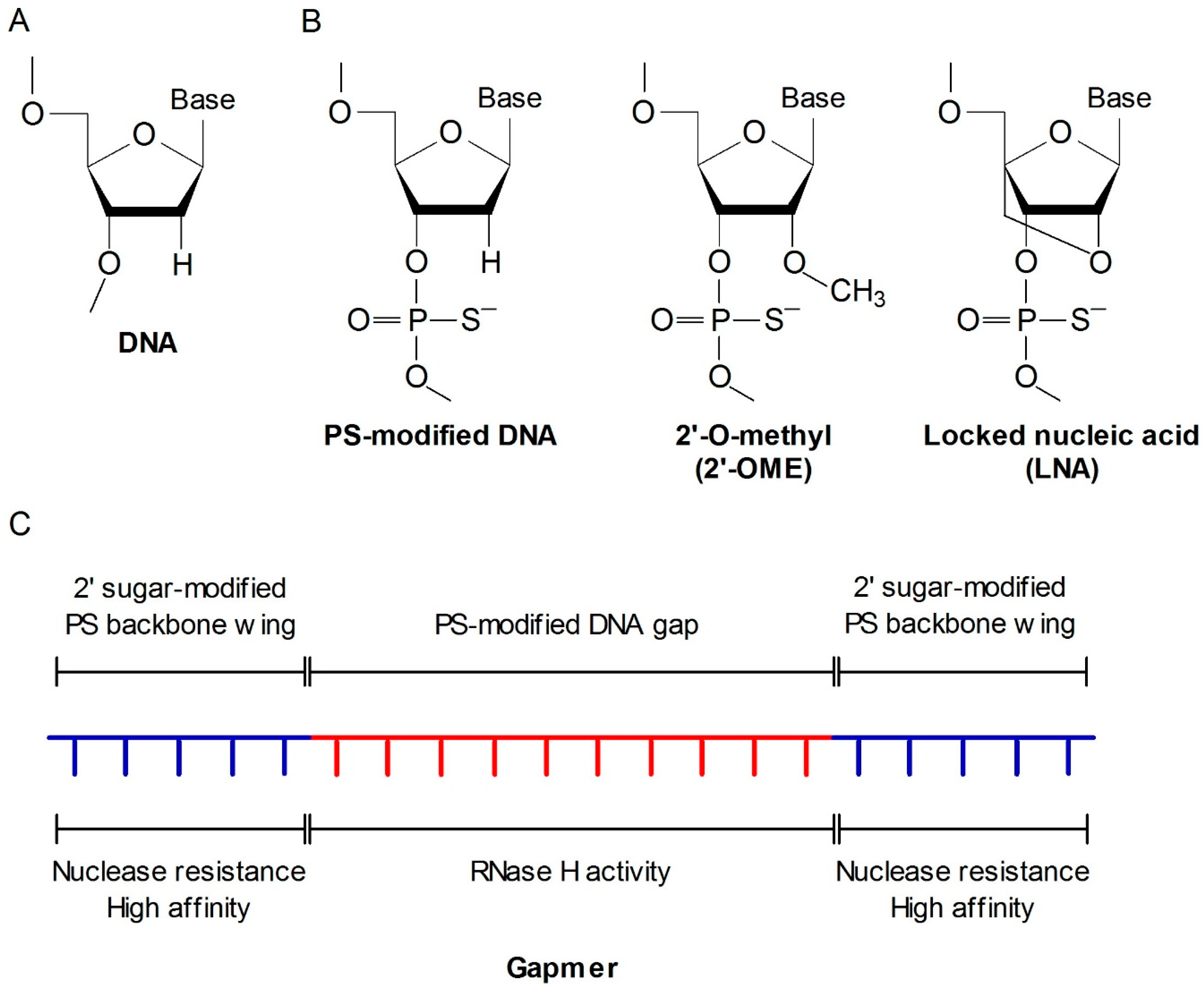
2.1. Antisense Oligonucleotide (ASO)
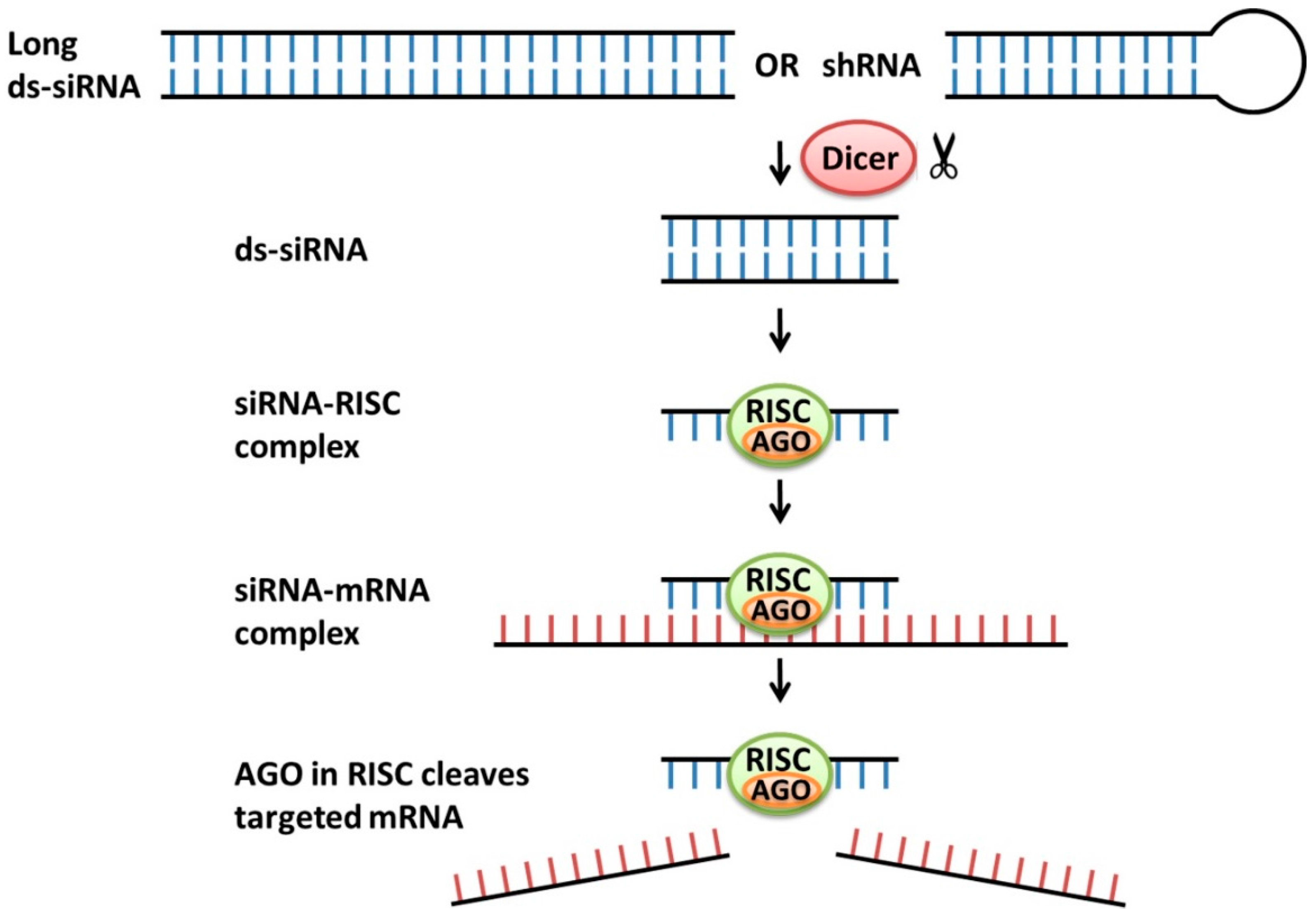
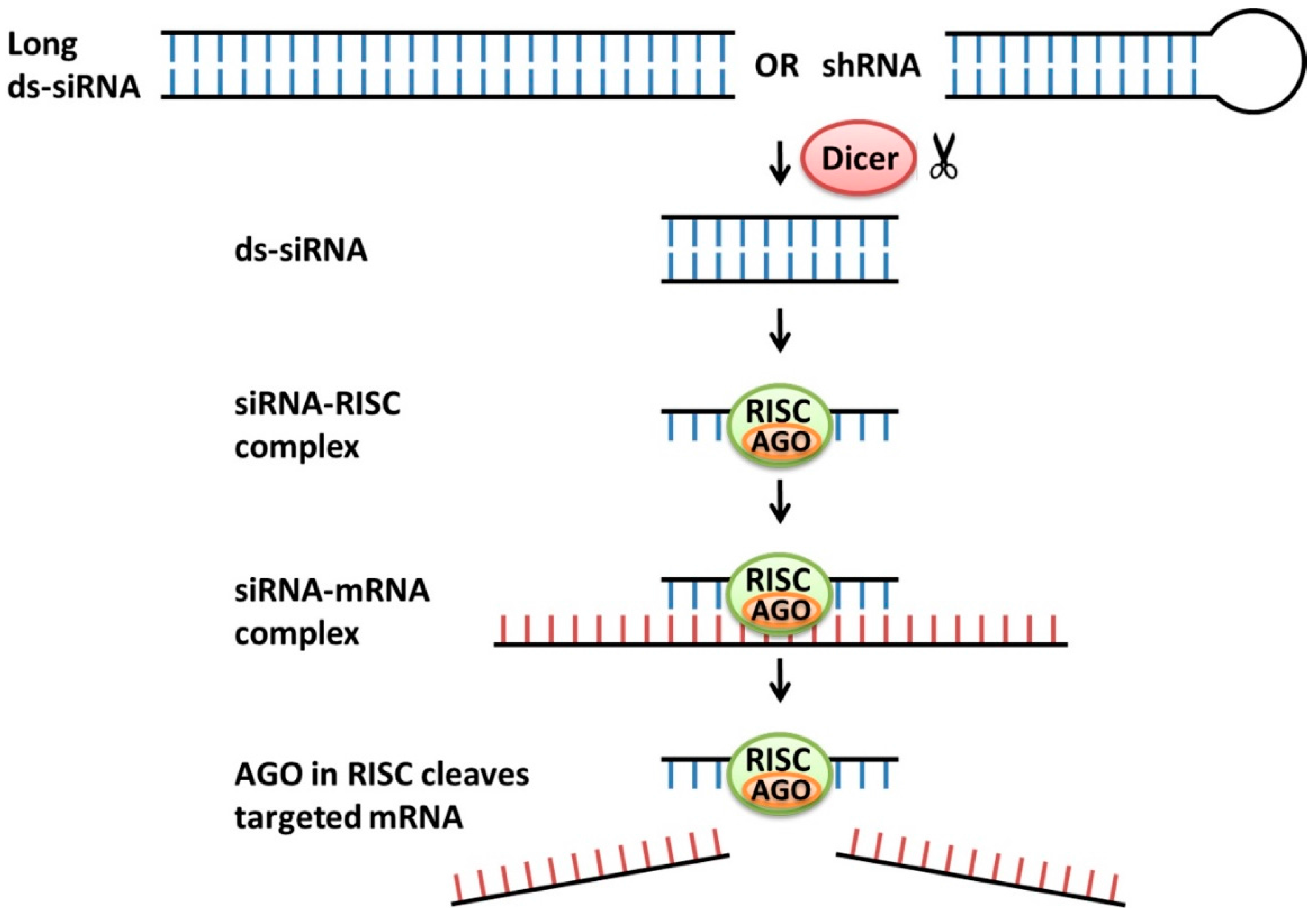
2.2. siRNA
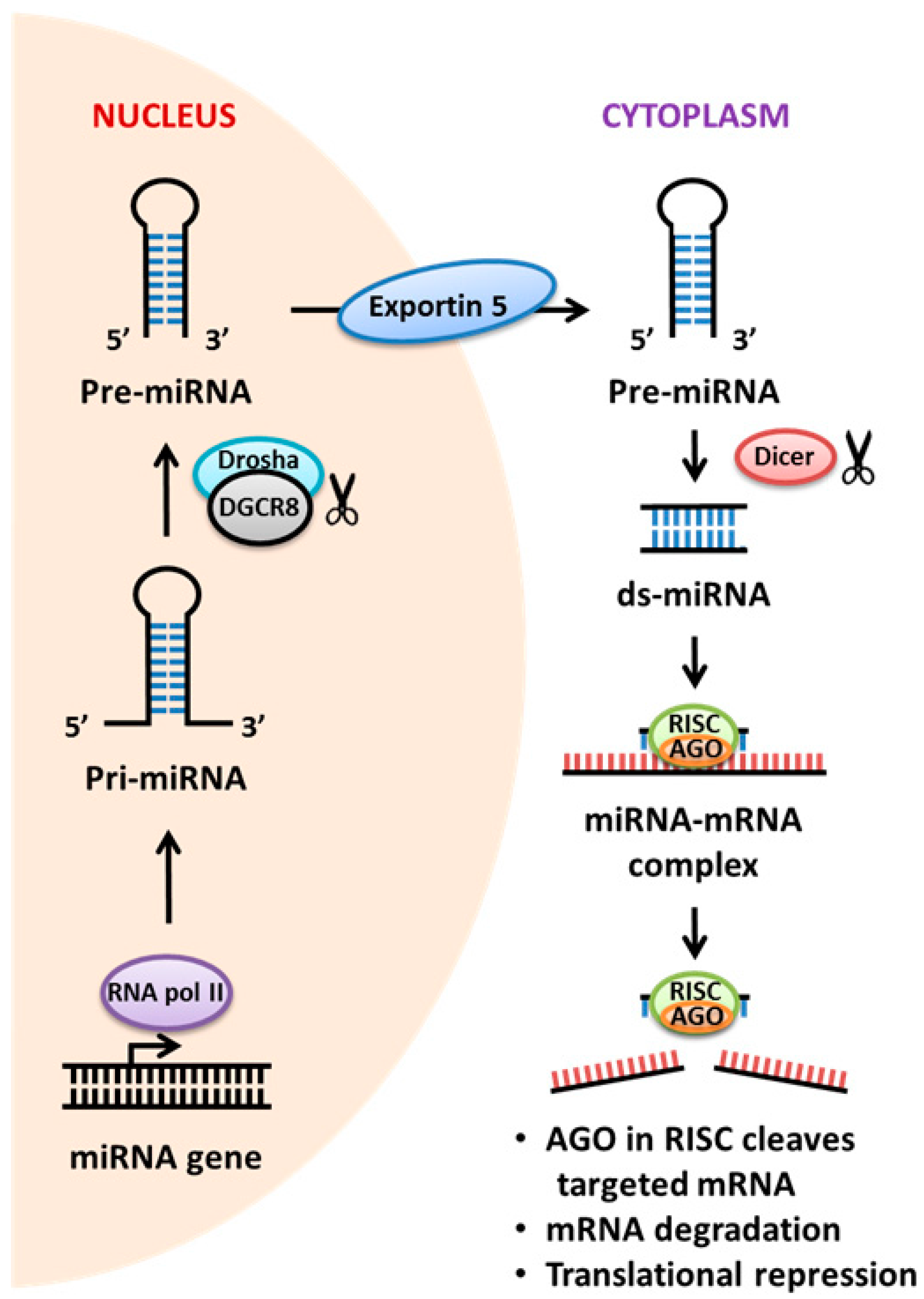
2.3. miRNA
3. Future Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AHR | airway hyperresponsiveness |
| Ago | Argonaute 2 |
| ASO | antisense oligonucleotide |
| BAL | bronchoalveolar lavage |
| CCR | CC chemokine receptor |
| CHST | carbohydrate sulfotransferase |
| COPD | chronic obstructive pulmonary disease |
| CTGF | connective tissue growth factor |
| DGCR | DiGeorge syndrome critical region gene |
| ds | double-stranded |
| FGF | fibroblast growth factor |
| ICS | inhaled corticosteroid |
| IPF | idiopathic pulmonary fibrosis |
| LNA | locked nucleic acid |
| lncRNA | long non-coding RNA |
| LT | leukotriene |
| MAPK | mitogen-activated protein kinase |
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| NPRA | natriuretic peptide receptor A |
| OVA | ovalbumin |
| PAI | plasminogen activator |
| PDE | phosphodiesterase |
| PMO | phosphoroamide morpholino oligomer |
| PNA | peptide nucleic acid |
| PS | phosphorothioate |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| RSV | respiratory syncytial virus |
| SCF | stem cell factor |
| sh | short hairpin |
| siRNA | small interfering RNA |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducer and activator of transcription |
| TGF | transforming growth factor |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| UTR | untranslated region |
| VLA | very late antigen |
References
- Bush, A.; Zar, H.J. WHO universal definition of severe asthma. Curr. Opin. Allergy Clin. Immunol. 2011, 11, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.D.; Vercelli, D. Asthma. Lancet 2013, 382, 1360–1372. [Google Scholar] [CrossRef]
- Braman, S.S. The global burden of asthma. Chest 2006, 130, 4S–12S. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S. Centers for Disease Control and Prevention. 2013. Vital signs: Binge drinking among women and high school girls—United States, 2011. Adv. Nutr. 2013, 4, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Fanta, C.H. Asthma. N. Engl. J. Med. 2009, 360, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Eosinophilic and neutrophilic inflammation in asthma: Insights from clinical studies. Proc. Am. Thorac. Soc. 2009, 6, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Chung, K.F.; Adcock, I.M. Update on glucocorticoid action and resistance. J. Allergy Clin. Immunol. 2006, 117, 522–543. [Google Scholar] [CrossRef] [PubMed]
- WTO. Burden of COPD. Available online: http://www.who.int/respiratory/copd/burden/en/ (accessed on 30 May 2016).
- Lopez, A.D.; Shibuya, K.; Rao, C.; Mathers, C.D.; Hansell, A.L.; Held, L.S.; Schmid, V.; Buist, S. Chronic obstructive pulmonary disease: Current burden and future projections. Eur. Respir. J. 2006, 27, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, E.S.; Murphy, L.B.; Khavjou, O.; Giles, W.H.; Holt, J.B.; Croft, J.B. Total and state-specific medical and absenteeism costs of COPD among adults aged ≥ 18 years in the United States for 2010 and projections through 2020. Chest 2015, 147, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Lomborg, B. Global Problems, Local Solutions: Costs and Benefits; Cambridge University Press: Cambridge, UK, 2013; p. 143. [Google Scholar]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Zervas, E.; Samitas, K.; Gaga, M.; Beghe, B.; Fabbri, L.M. Inhaled corticosteroids in COPD: Pros and cons. Curr. Drug Targets 2013, 14, 192–224. [Google Scholar] [CrossRef] [PubMed]
- Wedzicha, J.A.; Calverley, P.M.; Rabe, K.F. Roflumilast: A review of its use in the treatment of COPD. Int. J. Chron. Obstr. Pulm. Dis. 2016, 11, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Harari, S.; Caminati, A. IPF: New insight on pathogenesis and treatment. Allergy 2010, 65, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Loomis-King, H.; Flaherty, K.R.; Moore, B.B. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr. Opin. Pharmacol. 2013, 13, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Marwick, C. First “antisense” drug will treat CMV retinitis. JAMA 1998, 280. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 5, 347–355. [Google Scholar]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nyce, J.W. Respirable antisense oligonucleotides: A new drug class for respiratory disease. Respir. Res. 2001, 2, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T. Progress in antisense technology. Annu. Rev. Med. 2004, 55, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.S. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.H.; Fabbro, D.; Dean, N.M.; Geiger, T.; Monia, B.P.; Müller, M.; Nicklin, P. Second-generation antisense oligonucleotides: Structure-activity relationships and the design of improved signal-transduction inhibitors. Biochem. Soc. Trans. 1996, 24, 630–637. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.A.; Miraglia, L.J.; Cummins, L.L.; Owens, S.R.; Sasmor, H.; Dean, N.M. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J. Biol. Chem. 1999, 274, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Vaishnaw, A.; Fitzgerald, K. Liver as a target for oligonucleotide therapeutics. J. Hepatol. 2013, 59, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Aher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 9, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Nyce, J.W.; Metzger, W.J. DNA antisense therapy for asthma in an animal model. Nature 1997, 385, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.A.; Van Scott, M.R.; Robinson, C.B. Sense and antisense: Therapeutic potential of oligonucleotides and interference RNA in asthma and allergic disorders. Clin. Rev. Allergy Immunol. 2004, 27, 207–217. [Google Scholar] [CrossRef]
- Seong, J.H.; Lee, K.M.; Kim, S.T.; Jin, S.E.; Kim, C.K. Polyethylenimine-based antisense oligodeoxynucleotides of IL-4 suppress the production of IL-4 in a murine model of airway inflammation. J. Gene Med. 2006, 8, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xiong, W.N.; Xu, Y.J.; Zhang, Z.X.; Gao, B.A.; Du, C.L.; Lu, J.Y.; Ye, T. Recombinant adeno-associated virus-mediated inhibiting of interleukin-4 expression in rat model of asthma. Chin. Med. J. 2006, 119, 223–225. [Google Scholar] [PubMed]
- Cao, Y.; Zeng, D.; Song, Q.; Cao, C.; Xie, M.; Liu, X.; Xiong, S.; Xu, Y.; Xiong, W. The effects of antisense interleukin-4 gene transferred by recombinant adeno-associated virus vector on the airway remodeling in allergic rats. J. Asthma 2010, 47, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Karras, J.G.; McGraw, K.; McKay, R.A.; Cooper, S.R.; Lerner, D.; Lu, T.; Walker, C.; Dean, N.M.; Monia, B.P. Inhibition of antigen-induced eosinophilia and late phase airway hyperresponsiveness by an IL-5 antisense oligonucleotide in mouse models of asthma. J. Immunol. 2000, 164, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Cao, Y.; Song, Q.; Cao, C.; Liu, X.; Xu, Y.; Xiong, W. Effects of antisense interleukin-5 gene transferred by recombinant adeno-associated virus to allergic rats. Respirology 2010, 15, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Cao, Y.; Song, Q.; Cao, C.; Xie, M.; Liu, X.; Xiong, S.; Xu, Y.; Xiong, W. Recombinant adeno-associated virus vector-mediated delivery of antisense interleukin-5 gene attenuates airway remodeling in allergic rats. Int. Arch. Allergy Immunol. 2011, 154, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Karras, J.G.; Crosby, J.R.; Guha, M.; Tung, D.; Miller, D.A.; Gaarde, W.A.; Geary, R.S.; Monia, B.P.; Gregory, S.A. Anti-inflammatory activity of inhaled IL-4 receptor-alpha antisense oligonucleotide in mice. Am. J. Respir. Cell Mol. Biol. 2007, 36, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Fey, R.A.; Templin, M.V.; McDonald, J.D.; Yu, R.Z.; Hutt, J.A.; Gigliotti, A.P.; Reed, M.D. Local and systemic tolerability of a 2′O-methoxyethyl antisense oligonucleotide targeting interleukin-4 receptor-α delivery by inhalation in mouse and monkey. Inhal. Toxicol. 2014, 26, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Allam, M.; Renzi, P.M. Inhibition of allergen-induced eosinophilia and airway hyperresponsiveness by antisense oligonucleotides directed against the common beta chain of IL-3, IL-5, GM-CSF receptors in a rat model of allergic asthma. Am. J. Respir. Crit. Care Med. 2002, 165, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Fortin, M.; Ferrari, N.; Higgins, M.E.; Séguin, S.; Allam, M.; Allakhverdi, Z.; Piaget-Rodriguez, C.; Paquet, L.; Renzi, P.M. Effects of antisense oligonucleotides targeting CCR3 on the airway response to antigen in rats. Oligonucleotides 2006, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Allam, M.; Guimond, A.; Ferrari, N.; Zemzoumi, K.; Séguin, R.; Paquet, L.; Renzi, P.M. Multi-targeted approach using antisenseoligonucleotides for the treatment of asthma. Ann. N. Y. Acad. Sci. 2006, 1082, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Pang, Z.; Zhu, Q.; Cai, X.; Yin, Y.; Wang, M.; Zhu, J.; Chen, J.; Zeng, K.; Zhang, C.; et al. Locally instilled tumor necrosis factor-α antisense oligonucleotide inhibits allergic inflammation via the induction of Tregs. J. Gene Med. 2012, 14, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Yamazumi, Y.; Sasaki, O.; Imamura, M.; Oda, T.; Ohno, Y.; Shiozaki-Sato, Y.; Nagai, S.; Suyama, S.; Kamoshida, Y.; Funato, K.; et al. The RNA Binding Protein Mex-3B Is Required for IL-33 Induction in the Development of Allergic Airway Inflammation. Cell Rep. 2016, 16, 2456–2471. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.R.; Guha, M.; Tung, D.; Miller, D.A.; Bender, B.; Condon, T.P.; York-DeFalco, C.; Geary, R.S.; Monia, B.P.; Karras, J.G.; et al. Inhaled CD86 antisense oligonucleotide suppresses pulmonary inflammation and airway hyper-responsiveness in allergic mice. J. Pharmacol. Exp. Ther. 2007, 321, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Finotto, S.; De Sanctis, G.T.; Lehr, H.A.; Herz, U.; Buerke, M.; Schipp, M.; Bartsch, B.; Atreya, R.; Schmitt, E.; Galle, P.R.; et al. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J. Exp. Med. 2001, 193, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.R.; Tian, X.L.; Bo, J.P.; Li, S.G.; Liu, Z.L.; Niu, B. Inhibition of allergic airway inflammation by antisense-induced blockade of STAT6 expression. Chin. Med. J. 2011, 124, 26–31. [Google Scholar] [PubMed]
- Choi, I.W.; Kim, D.K.; Ko, H.M.; Lee, H.K. Administration of antisense phosphorothioate oligonucleotide to the p65 subunit of NF-κB inhibits established asthmatic reaction in mice. Int. Immunopharmacol. 2004, 4, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Stenton, G.R.; Ulanova, M.; Déry, R.E.; Merani, S.; Kim, M.K.; Gilchrist, M.; Puttagunta, L.; Musat-Marcu, S.; James, D.; Schreiber, A.D.; et al. Inhibition of allergic inflammation in the airways using aerosolized antisense to Syk kinase. J. Immunol. 2002, 169, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Chan, J.H.; McKay, K.; Crosby, J.R.; Choo, H.H.; Leung, B.P.; Karras, J.G.; Wong, W.S. Inhaled p38α mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am. J. Respir. Crit. Care Med. 2005, 171, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Seo, J.M.; Shin, Y.; Yoo, M.H.; Park, C.S.; Lee, S.H.; Chang, Y.S.; Cho, S.H.; Kim, J.H. Blockade of airway inflammation and hyperresponsiveness by inhibition of BLT2, a low-affinity leukotriene B4 receptor. Am. J. Respir. Cell Mol. Biol. 2010, 42, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Cabral, M.D.; Paulet, P.E.; Robert, V.; Gomes, B.; Renoud, M.L.; Savignac, M.; Leclerc, C.; Moreau, M.; Lair, D.; Langelot, M.; et al. Knocking down Cav1 calcium channels implicated in Th2 cell activation prevents experimental asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Lofthouse, S.A.; Crosby, J.R.; Tung, D.; Guha, M.; Kowalski, D.; Luther, D.; Arberg, C.C. Aerosol delivery of VLA-4 specific antisense oligonucleotides inhibit airway inflammation and hyperresponsiveness in mice. Respirology 2005, 10, A11. [Google Scholar]
- Limmroth, V.; Barkhof, F.; Desem, N.; Diamond, M.P.; Tachas, G.; ATL1102 Study Group. CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS. Neurology 2014, 83, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Cieslinski, L.B.; Newton, R.; Donnelly, L.E.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a Selective Inhibitor of Phosphodiesterase 7: In Vitro Studies in Human Monocytes, Lung Macrophages, and CD8+ T-Lymphocytes. Mol. Pharmacol. 2004, 66, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishnan, L.; Rudra, S.; Eapen, M.S.; Dastidar, S.; Ray, A. Small-molecule inhibitors of PDE-IV and -VII in the treatment of respiratory diseases and chronic inflammation. Expert Opin. Investig. Drugs 2007, 16, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- Fortin, M.; D’Anjou, H.; Higgins, M.E.; Gougeon, J.; Aubé, P.; Moktefi, K.; Mouissi, S.; Séguin, S.; Séguin, R.; Renzi, P.M.; et al. A multi-target antisense approach against PDE4 and PDE7 reduces smoke-induced lung inflammation in mice. Respir. Res. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tan, W.; Zhang, L.; Tan, Q.; Yang, J. Beneficial impact of bFGF antisense therapy in a rat model of pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32, 22–31. [Google Scholar]
- Luo, Y.; Wang, M.; Pang, Z.; Jiang, F.; Chen, J.; Zhang, J. Locally instilled tumor necrosis factor α antisense oligonucleotide contributes to inhibition of TH2-driven pulmonary fibrosis via induced CD4+CD25+Foxp3+ regulatory T cells. J. Gene Med. 2013, 15, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.Y.; Zhang, X.Y.; Tan, M.Q.; Zheng, R.; Zhao, L. Intervention of transforming pulmonary fibrosis with NF-κB p65 antisense oligonucleotide. Int. J. Clin. Exp. Med. 2014, 7, 5252–5259. [Google Scholar] [PubMed]
- Ball, H.A.; Sandrasagra, A.; Tang, L.; Van Scott, M.; Wild, J.; Nyce, J.W. Clinical potential of respirable antisense oligonucleotides (RASONs) in asthma. Am. J. Pharmacogenom. 2003, 3, 97–106. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Boulet, L.P.; Cockcroft, D.W.; Baatjes, A.; Cote, J.; Deschesnes, F.; Davis, B.; Strinich, T.; Howie, K.; Duong, M.; et al. Antisense therapy against CCR3 and the common beta chain attenuates allergen-induced eosinophilic responses. Am. J. Respir. Crit. Care Med. 2008, 177, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Gauvreau, G.M.; Pageau, R.; Séguin, R.; Carballo, D.; Gauthier, J.; D’Anjou, H.; Campbell, H.; Watson, R.; Mistry, M.; Parry-Billings, M.; et al. Dose-response effects of TPI ASM8 in asthmatics after allergen. Allergy 2011, 66, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, H.; Campbell, H.; Babirad, I.; Watson, R.M.; Mistry, M.; Sehmi, R.; Gauvreau, G.M. TPI ASM8 reduces eosinophil progenitors in sputum after allergen challenge. Clin. Exp. Allergy 2011, 41, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Hodges, M.R.; Castelloe, E.; Chen, A.; Geary, R.S.; Karras, J.G.; Shapiro, D.; Yeung, B.; Yu, R.; Gregory, S.A. Randomized, Double-Blind, Placebo Controlled First in Human Study of Inhaled AIR645, an IL-4Rα Oligonucleotide, in Healthy Volunteers. In Proceedings of the American Thoracic Society 2009 International Conference, San Diego, CA, USA, 15–20 May 2009; p. A3640.
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 2, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Lowe, S.W. Stable RNA interference rules for silencing. Nat. Cell Biol. 2014, 16, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Boil. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-Berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Durcan, N.; Murphy, C.; Cryan, S.A. Inhalable siRNA: Potential as a therapeutic agent in the lungs. Mol. Pharm. 2008, 5, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Khaitov, M.R.; Shilovskiy, I.P.; Nikonova, A.A.; Shershakova, N.N.; Kamyshnikov, O.Y.; Babakhin, A.A.; Zverev, V.V.; Johnston, S.L.; Khaitov, R.M. Small interfering RNAs targeted to interleukin-4 and respiratory syncytial virus reduce airway inflammation in a mouse model of virus-induced asthma exacerbation. Hum. Gene Ther. 2014, 7, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lee, C.C.; Chiang, B.L. Small interfering RNA against interleukin-5 decreases airway eosinophilia and hyper-responsiveness. Gene Ther. 2008, 15, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S.; Lockey, R.F.; Vesely, D.L.; Gower, W.R., Jr. Natriuretic peptides and genesis of asthma: An emerging paradigm? J. Allergy Clin. Immunol. 2004, 114, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, W.; Mohapatra, S.; Kong, X.; Li, X.; Lockey, R.F.; Mohapatra, S.S. Prevention of airway inflammation with topical cream containing imiquimod and small interfering RNA for natriuretic peptide receptor. Genet. Vaccines Ther. 2008, 6. [Google Scholar] [CrossRef] [PubMed]
- Darcan-Nicolaisen, Y.; Meinicke, H.; Fels, G.; Hegend, O.; Haberland, A.; Kühl, A.; Loddenkemper, C.; Witzenrath, M.; Kube, S.; Henke, W.; et al. Small interfering RNA against transcription factor STAT6 inhibits allergic airway inflammation and hyperreactivity in mice. J. Immunol. 2009, 182, 7501–7508. [Google Scholar] [CrossRef] [PubMed]
- Asai-Tajiri, Y.; Matsumoto, K.; Fukuyama, S.; Kan-o, K.; Nakano, T.; Tonai, K.; Ohno, T.; Azuma, M.; Inoue, H.; Nakanishi, Y. Small interfering RNA against CD86 during allergen challenge blocks experimental allergic asthma. Respir. Res. 2014, 1, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Inoue, H.; Nagata, N.; Hayashi, K.; Fukuyama, S.; Matsumoto, K.; Komine, O.; Hamano, S.; Himeno, K.; Inagaki-Ohara, K.; et al. SOCS-3 regulates onset and maintenance of TH2-mediated allergic responses. Nat. Med. 2003, 9, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Zafra, M.P.; Mazzeo, C.; Gámez, C.; Marco, A.R.; de Zulueta, A.; Sanz, V.; Bilbao, I.; Ruiz-Cabello, J.; Zubeldia, J.M.; del Pozo, V. Gene silencing of SOCS3 by siRNA intranasal delivery inhibits asthma phenotype in mice. PLoS ONE 2014, 3, e91996. [Google Scholar]
- Binia, A.; Van Stiphout, N.; Liang, L.; Michel, S.; Bhavsar, P.K.; Fan Chung, K.; Brightling, C.E.; Barnes, P.J.; Kabesch, M.; Bush, A.; et al. A polymorphism affecting MYB binding within the promoter of the PDCD4 gene is associated with severe asthma in children. Hum. Mutat. 2013, 8, 1131–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Yang, X.; Sun, Q.; Liu, L.; Lan, X.; Tian, J.; He, Q.; Hou, W.; Liu, L.; Jiang, C.; et al. Pdcd4 modulates markers of macrophage alternative activation and airway remodeling in antigen-induced pulmonary inflammation. J. Leukoc. Biol. 2014, 6, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.; Da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol. 2006, 1, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, H.; Li, Y.M.; Wang, S.Y.; Diao, X.; Liu, K.G. Intranasal sirna targeting c-kit reduces airway inflammation in experimental allergic asthma. Int. J. Clin. Exp. Pathol. 2014, 7, 5505–5514. [Google Scholar] [PubMed]
- Goh, F.Y.; Cook, K.L.; Upton, N.; Tao, L.; Lah, L.C.; Leung, B.P.; Wong, W.S.F. Receptor-interacting protein 2 gene silencing attenuates allergic airway inflammation. J. Immunol. 2013, 191, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Tomoda, K.; Yoneyama, H.; Yoshikawa, M.; Kimura, H. RNA interference targeting carbohydrate sulfotransferase 3 diminishes macrophage accumulation, inhibits MMP-9 expression and promotes lung recovery in murine pulmonary emphysema. Respir. Res. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Boveda-Ruiz, D.; Takagi, T.; Toda, M.; Gil-Bernabe, P.; Miyake, Y.; Yasukawa, A.; Matsuda, Y.; Suzuki, N.; et al. Development and preclinical efficacy of novel transforming growth factor-β1 short interfering RNAs for pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Senoo, T.; Hattori, N.; Tanimoto, T.; Furonaka, M.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Yokoyama, A.; Kohno, N. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax 2010, 65, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Li, W.B.; Wang, W.L.; Liu, J.; Song, S.X.; Bai, L.L.; Hu, Y.Y.; Yuan, Y.D.; Zhang, M. siRNA against plasminogen activator inhibitor-1 ameliorates bleomycin-induced lung fibrosis in rats. Acta Pharmacol. Sin. 2012, 7, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Sung, D.K.; Kong, W.H.; Park, K.; Kim, J.H.; Kim, M.Y.; Kim, H.; Hahn, S.K. Noncovalenly PEGylated CTGF siRNA/PDMAEMA complex for pulmonary treatment of bleomycin-induced lung fibrosis. Biomaterials 2013, 4, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Ulanova, M.; Puttagunta, L.; Marcet-Palacios, M.; Duszyk, M.; Steinhoff, U.; Duta, F.; Kim, M.K.; Indik, Z.K.; Schreiber, A.D.; Befus, A.D. Syk tyrosine kinase participates in beta1-integrin signaling and inflammatory responses in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 3, L497–L507. [Google Scholar] [CrossRef] [PubMed]
- Ulanova, M.; Marcet-Palacios, M.; Muñoz, S.; Asfaha, S.; Kim, M.K.; Schreiber, A.D.; Befus, A.D. Involvement of Syk kinase in TNF-induced nitric oxide production by airway epithelial cells. Biochem. Biophys. Res. Commun. 2006, 2, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ulanova, M.; Asfaha, S.; Stenton, G.; Lint, A.; Gilbertson, D.; Schreiber, A.; Befus, D. Involvement of Syk protein tyrosine kinase in LPS-induced responses in macrophages. J. Endotoxin Res. 2007, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.W.; Bruford, E.A. Naming “junk”: Human non-protein coding RNA (ncRNA) gene nomenclature. Hum. Genom. 2011, 5, 90–98. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Cir. 2013, 3, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Rahman, M.; Nana-Sinkam, S.P. MicroRNAs in respiratory disease. A clinician’s overview. Ann. Am. Thorac. Soc. 2014, 11, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Borucka, J.; Petrkova, J.; Petrek, M. Novel Insights into miRNA in Lung and Heart Inflammatory Diseases. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- miRBase: The microRNA Database v21 (Released in 2014 on Human Genome Version GRCh38). Available online: http://mirbase.org/ (accessed on 12 December 2016).
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Reinhart, B.J.; Jones-Rhoades, M.W.; Tang, G.; Zamore, P.D.; Barton, M.K.; Bartel, D.P. MicroRNA control of PHABULOSA in leaf development: Importance of pairing to the microRNA 5′ region. EMBO J. 2004, 23, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; McElvaney, N.G.; Greene, C.M. MicroRNAs in inflammatory lung disease-master regulators or target practice? Respir. Res. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Rupani, H.; Sanchez-Elsner, T.; Howarth, P. MicroRNAs and respiratory diseases. Eur. Respir. J. 2013, 41, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.J.; Legesse-Miller, A.; Coller, H.A. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl. Acad. Sci. USA 2008, 105, 14879–14884. [Google Scholar] [CrossRef] [PubMed]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiang, X.; Zhuang, X.; Zhang, S.; Liu, C.; Cheng, Z.; Michalek, S.; Grizzle, W.; Zhang, H.G. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am. J. Pathol. 2010, 176, 2490–2499. [Google Scholar] [CrossRef] [PubMed]
- Redis, R.S.; Calin, S.; Yang, Y.; You, M.J.; Calin, G.A. Cell-to-cell miRNA transfer: From body homeostasis to therapy. Pharmacol. Ther. 2012, 136, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Liston, A. MicroRNA in the immune system, microRNA as an immune system. Immunology 2009, 127, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.J.; Patel, S.; Bhakta, N.R.; Choy, D.F.; Brightbill, H.D.; Ren, X.; Wang, Y.; Pua, H.H.; Baumjohann, D.; Montoya, M.M.; et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat. Immunol. 2014, 15, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Tay, H.L.; Maltby, S.; Xiang, Y.; Eyers, F.; Hatchwell, L.; Zhou, H.; Toop, H.D.; Morris, J.C.; Nair, P.; et al. MicroRNA-9 regulates steroid-resistant airway hyperresponsiveness by reducing protein phosphatase 2A activity. J. Allergy Clin. Immunol. 2015, 136, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, M.; Ahmad, T.; Mabalirajan, U.; Aich, J.; Agrawal, A.; Ghosh, B. Antagonism of mmu-mir-106a attenuates asthma features in allergic murine model. J. Appl. Physiol. 2012, 113, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, M.; Aich, J.; Hariharan, M.; Brahmachari, S.K.; Agrawal, A.; Ghosh, B. Posttranscriptional regulation of interleukin-10 expression by hsa-miR-106a. Proc. Natl. Acad. Sci. USA 2009, 106, 5761–5766. [Google Scholar] [CrossRef] [PubMed]
- Malmhäll, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Rådinger, M. MicroRNA-155 is essential for TH2-mediated allergen-induced eosinophilic inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Czieso, S.; Ktistaki, E.; Roderick, K.; Coomes, S.M.; Pelly, V.S.; Kannan, Y.; Perez-Lloret, J.; Zhao, J.L.; Baltimore, D.; et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc. Natl. Acad. Sci. USA 2014, 111, E3081–E3090. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.B.; Xu, B.; Mei, J.J.; Li, D.; Liu, J.J.; Zhao, D.Y.; Liu, F. Inhibition of miRNA-221 suppresses the airway inflammation in asthma. Inflammation 2012, 35, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.M.; Moschos, S.A.; Williams, A.E.; Shepherd, N.J.; Larner-Svensson, H.M.; Lindsay, M.A. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J. Immunol. 2008, 15, 5689–5698. [Google Scholar] [CrossRef]
- Perry, M.M.; Williams, A.E.; Tsitsiou, E.; Larner-Svensson, H.M.; Lindsay, M.A. Divergent intracellular pathways regulate interleukin-1β-induced miR-146a and miR-146b expression and chemokine release in human alveolar epithelial cells. FEBS Lett. 2009, 583, 3349–3355. [Google Scholar] [CrossRef] [PubMed]
- Comer, B.S.; Camoretti-Mercado, B.; Kogut, P.C.; Halayko, A.J.; Solway, J.; Gerthoffer, W.T. MicroRNA-146a and microRNA-146b expression and anti-inflammatory function in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L727–L734. [Google Scholar] [CrossRef] [PubMed]
- Akbas, F.; Coskunpinar, E.; Aynaci, E.; Oltulu, Y.M.; Yildiz, P. Analysis of serum micro-RNAs as potential biomarker in chronic obstructive pulmonary disease. Exp. Lung Res. 2012, 38, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wu, M.; Lin, H.; Liu, C.; Yang, H.; Zhan, J.; Sun, S. An increased ratio of serum miR-21 to miR-181a levels is associated with the early pathogenic process of chronic obstructive pulmonary disease in asymptomatic heavy smokers. Mol. Biosyst. 2014, 10, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Hassan, T.; Carroll, T.P.; Buckley, P.G.; Cummins, R.; O’Neill, S.J.; McElvaney, N.G.; Geene, C.M. miR-199a-5p silencing regulates the unfolded protein response in chronic obstructive pulmonary disease and alpha1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2014, 189, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chatila, W.M.; Criner, G.J.; Hancock, W.W.; Akimova, T.; Moldover, B.; Chang, J.K.; Cornwell, W.; Santerre, M.; Rogers, T.J. Blunted expression of miR-199a-5p in regulatory T cells of patients with chronic obstructive pulmonary disease compared to unaffected smokers. Clin. Exp. Immunol. 2014, 177, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Halappanavar, S.; Nikota, J.; Wu, D.; Williams, A.; Yauk, C.L.; Stampfli, M. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J. Immunol. 2013, 190, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Xu, C.; Pan, Z.; Zhang, Y.; Xu, Z.; Chen, Y.; Li, T.; Li, X.; Liu, Y.; Huangfu, L.; et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol. Ther. 2014, 22, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Banerjee, S.; de Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in pulmonary fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kumar, M.; Negi, V.; Pattnaik, B.; Prakash, Y.S.; Agrawal, A.; Ghosh, B. MicroRNA-326 regulates profibrotic functions of transforming growth factor-beta in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wu, B.; Fan, J.; Han, R.; Luo, C.; Wang, T.; Yang, J.; Han, L.; Zhu, B.; Wei, D.; et al. The Anti-fibrotic effects and mechanisms of MicroRNA-486–5p in pulmonary fibrosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2015, 31. [Google Scholar] [CrossRef] [PubMed]
- Maltby, S.; Plank, M.; Tay, H.L.; Collison, A.; Foster, P.S. Targeting MicroRNA Function in Respiratory Diseases: Mini-Review. Front Physiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. RNA interference: Homing in on delivery. Nature 2010, 464, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Popescu, F.D. Antisense- and RNA interference-based therapeutic strategies in allergy. J. Cell. Mol. Med. 2005, 9, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, Y.; Liu, L. Interaction of long noncoding RNAs and microRNAs in the pathogenesis of idiopathic pulmonary fibrosis. Physiol. Genom. 2015, 47, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhou, J.; Wu, D.; Gao, W.; Li, L.; Yu, L.; Liu, F.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Microarray analysis of long non-coding RNAs in COPD lung tissue. Inflamm. Res. 2015, 64, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Austin, P.J.; Tsitsiou, E.; Boardman, C.; Jones, S.W.; Lindsay, M.A.; Adcock, I.M.; Chung, K.F.; Perry, M.M. Transcriptional profiling identifies the long noncoding RNA plasmacytoma variant translocation (PVT1) as a novel regulator of the asthmatic phenotype in human airway smooth muscle. J. Allergy Clin. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Booton, R.; Lindsay, M.A. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest 2014, 146, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shan, P.; Jiang, D.; Noble, P.W.; Abraham, N.G.; Kappas, A.; Lee, P.J. Small interfering RNA targeting heme oxygenase-1 enhances ischemia-reperfusion-induced lung apoptosis. J. Biol. Chem. 2004, 279, 10677–10684. [Google Scholar] [CrossRef] [PubMed]
- Griesenbach, U.; Kitson, C.; Escudero Garcia, S.; Farley, R.; Singh, C.; Somerton, L.; Painter, H.; Smith, R.L.; Gill, D.R.; Hyde, S.C.; et al. Inefficient cationic lipid-mediated siRNA and antisense oligonucleotide transfer to airway epithelial cells in vivo. Respir. Res. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.L.; Bayley, D.; Giddings, J.; Craig, S.J.; Cummins, L.L.; Hastewell, J.G.; Phillips, J.A. Pulmonary bioavailability of a phosphorothioate oligonucleotide (CGP 64128A): Comparison with other delivery routes. Pharm. Res. 1998, 15, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Frick, M.; Taylor, B.; Turnpenny, P.; Graves, H.; Spink, K.G.; Brady, K.; Lamb, D.; Collins, D.; Rockel, T.D.; et al. Uptake, efficacy, and systemic distribution of naked, inhaled short interfering RNA (siRNA) and locked nucleic acid (LNA) antisense. Mol. Ther. 2011, 19, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
- Kuruba, R.; Wilson, A.; Gao, X.; Li, S. Targeted delivery of nucleic-acid-based therapeutics to the pulmonary circulation. AAPS J. 2009, 11, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Séguin, R.M.; Ferrari, N. Emerging oligonucleotide therapies for asthma and chronic obstructive pulmonary disease. Expert Opin. Investig. Drugs 2009, 18, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lopes, C.M.; Sousa Lobo, J.M.; Amaral, M.H. Nucleic Acids Delivery Systems: A Challenge for Pharmaceutical Technologists. Curr. Drug Metab. 2015, 16, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Busse, W.; Corren, J.; Lanier, B.Q.; McAlary, M.; Fowler-Taylor, A.; Cioppa, G.D.; van As, A.; Gupta, N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J. Allergy Clin. Immunol. 2001, 108, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Spergel, J.M.; Rothenberg, M.E.; Collins, M.H.; Furuta, G.T.; Markowitz, J.E.; Fuchs, G., 3rd; O’Gorman, M.A.; Abonia, J.P.; Young, J.; Henkel, T.; et al. Reslizumab in children and adolescents with eosinophilic esophagitis: Results of a double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2012, 129, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.P.; Saif, M.W. Infusion-related and hypersensitivity reactions of monoclonal antibodies used to treat colorectal cancer—Identification, prevention, and management. J. Support Oncol. 2007, 5, 451–457. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Type | Target | Disease | Company | Status |
|---|---|---|---|---|---|
| EPI-2010 | ASO | Adenosine A1 Receptor | Asthma | Epigenesis | Phase II/Discontinued |
| TPI ASM8 | ASO | Combined βc & CCR3 | Asthma | Topigen | Phase II |
| AIR645 | ASO | IL-4/IL-13 Receptor α chain | Asthma | Altair/ISIS | Phase II/Discontinued |
| TPI 1100 | ASO | Combined PDE4B/4D/7A | COPD | Topigen | Phase I/Discontinued |
| Excellair | siRNA | Syk Kinase | Asthma | ZaBeCor | Phase II/Discontinued |
| Therapeutic Targets or Biomarkers | ||||||
|---|---|---|---|---|---|---|
| Disease | ASO | Ref. | siRNA | Ref. | miRNA | Ref. |
| Asthma | Adenosine A1 Receptor | [39,40] | IL-4 | [84] | miR-19a # | [126] |
| IL-4 | [41,42,43] | IL-5 | [85] | miR-9 | [127] | |
| IL-5 | [44,45,46] | NPRA | [87] | miR-106a | [128,129] | |
| IL-4α | [47,48] | STAT6 | [88] | miR-155 | [130,131] | |
| βc & CCR3 | [49,50,51] | CD86 | [89] | miR-221 | [132] | |
| TNF-α | [52] | SOCS3 | [91] | miR-146 | [133,134,135] | |
| Mex-3B | [53] | Pdcd4 | [93] | |||
| CD86 | [54] | c-kit | [95] | |||
| GATA-3 | [55] | RIP-2 | [96] | |||
| STAT6 | [56] | |||||
| NF-κB p65 Subunit | [57] | |||||
| Syk Kinase | [58] | |||||
| p38α MAPK | [59] | |||||
| BLT2 | [60] | |||||
| Ca(v)1 | [61] | |||||
| VLA-4 | [62] | |||||
| COPD | PDE4B/4D/7A | [66] | CHST3 | [97] | miR-20a #, -28-3p # | [136] |
| miR-34c-5p #, -100 #, -7 # | [136] | |||||
| miR-21 #, -181a # | [137] | |||||
| miR-199a-5p # | [138,139] | |||||
| miR-135b | [140] | |||||
| IPF | bFGF | [67] | TGF-β1 | [98] | miR-26a # | [141] |
| TNF-α | [68] | PAI-1 | [99,100] | miR-200 | [142] | |
| NF-κB p65 Subunit | [69] | CTGF | [101] | miR-326 # | [143] | |
| miR-485-5p | [144] | |||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.; Dong, J.; Peh, H.Y.; Tan, L.H.; Lim, K.S.; Li, L.; Wong, W.-S.F. Oligonucleotide Therapy for Obstructive and Restrictive Respiratory Diseases. Molecules 2017, 22, 139. https://doi.org/10.3390/molecules22010139
Liao W, Dong J, Peh HY, Tan LH, Lim KS, Li L, Wong W-SF. Oligonucleotide Therapy for Obstructive and Restrictive Respiratory Diseases. Molecules. 2017; 22(1):139. https://doi.org/10.3390/molecules22010139
Chicago/Turabian StyleLiao, Wupeng, Jinrui Dong, Hong Yong Peh, Lay Hong Tan, Kah Suan Lim, Li Li, and Wai-Shiu Fred Wong. 2017. "Oligonucleotide Therapy for Obstructive and Restrictive Respiratory Diseases" Molecules 22, no. 1: 139. https://doi.org/10.3390/molecules22010139