
Chemical Constituents from the Roots and Rhizomes of Asarum heterotropoides var. mandshuricum and the In Vitro Anti-Inflammatory Activity

 ,
,
Abstract
:
1. Introduction
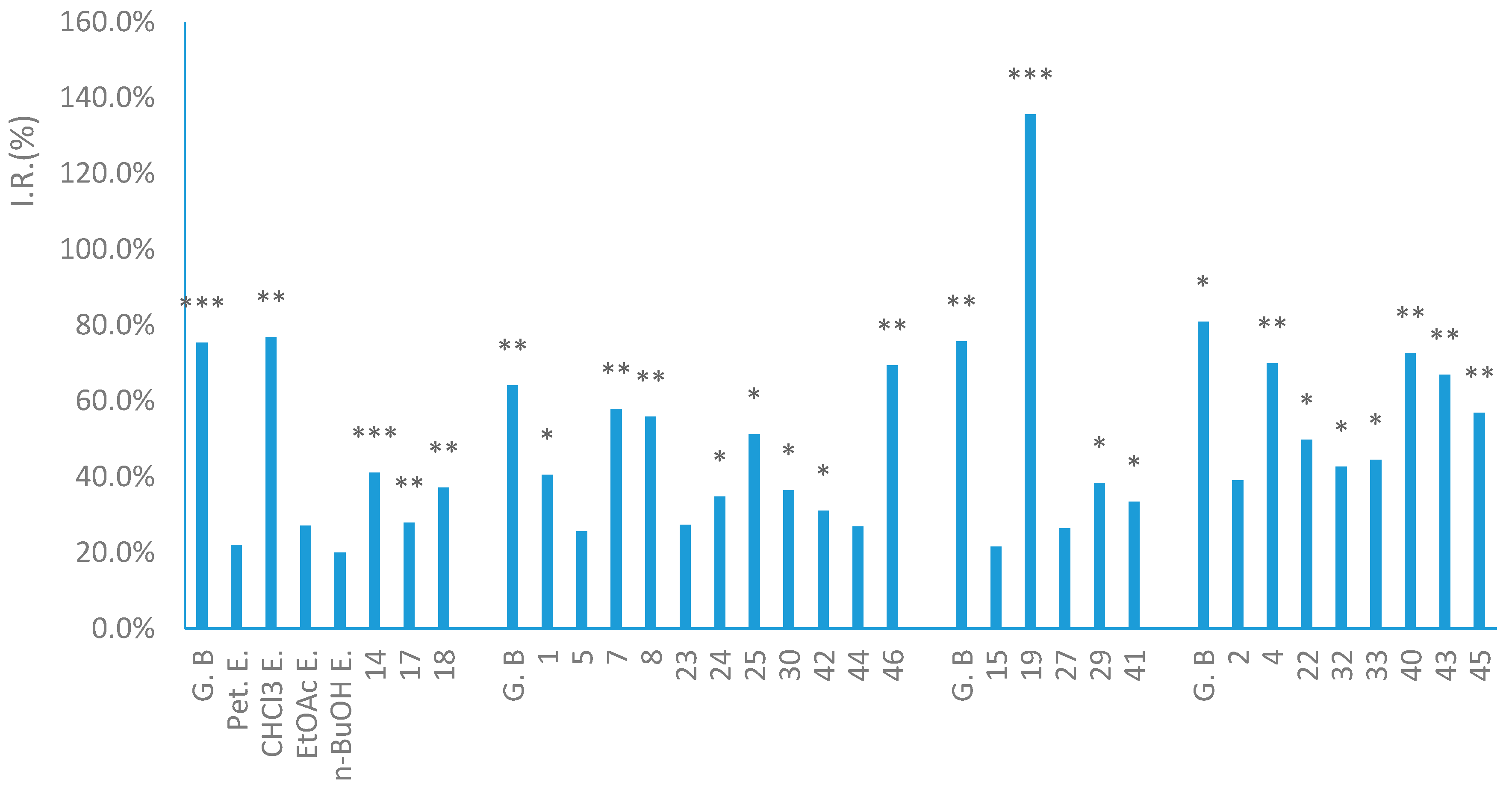
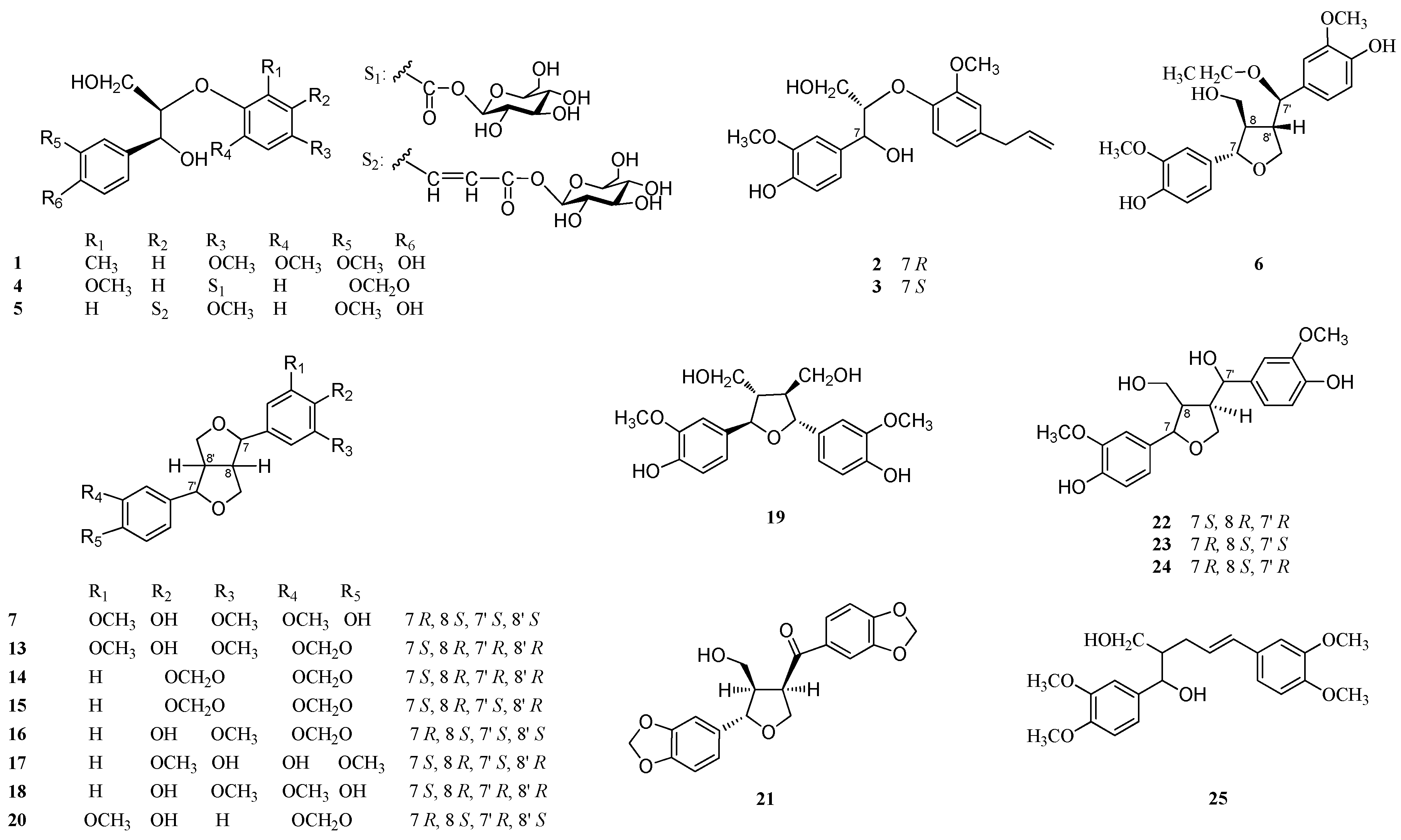
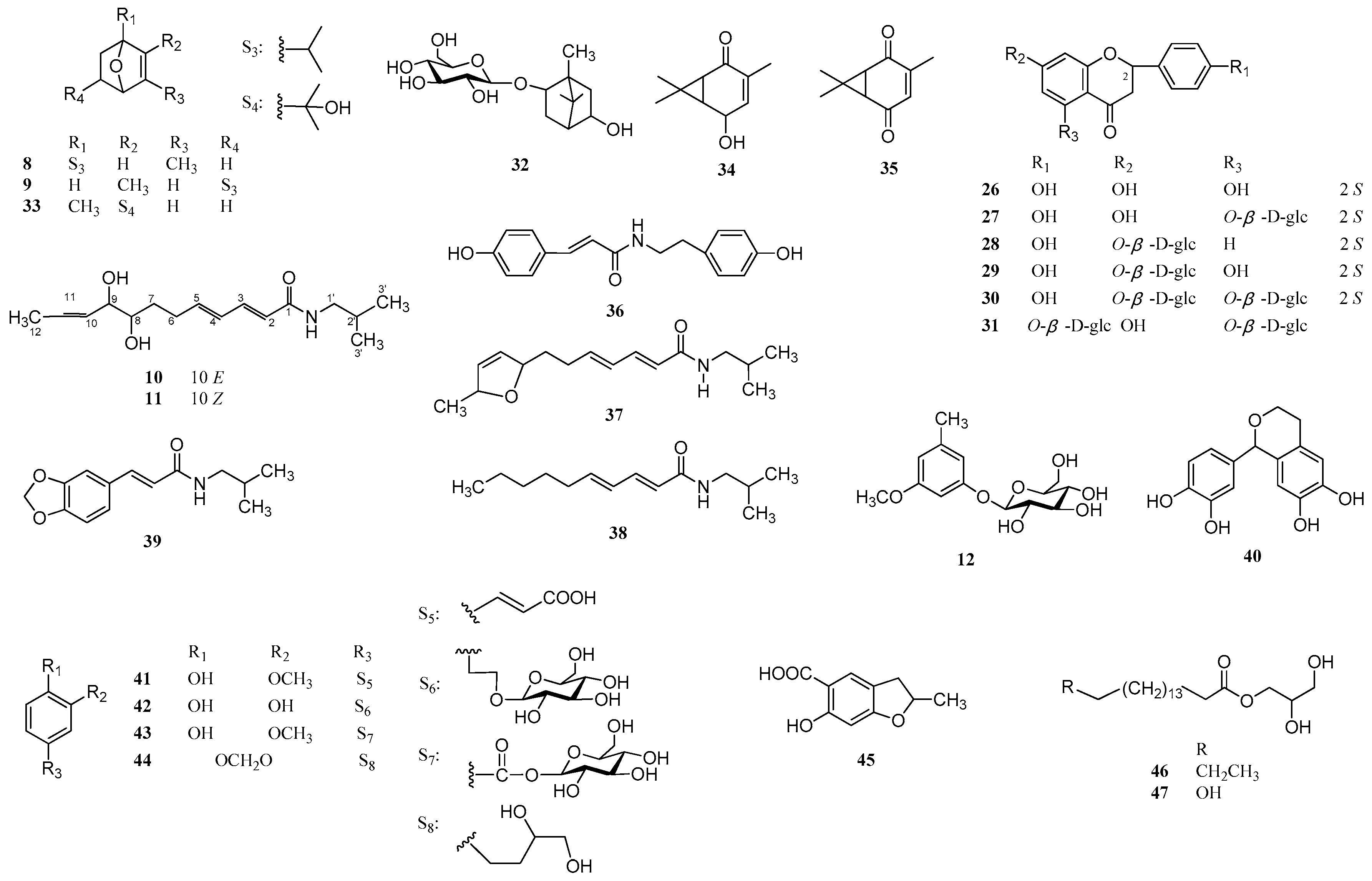
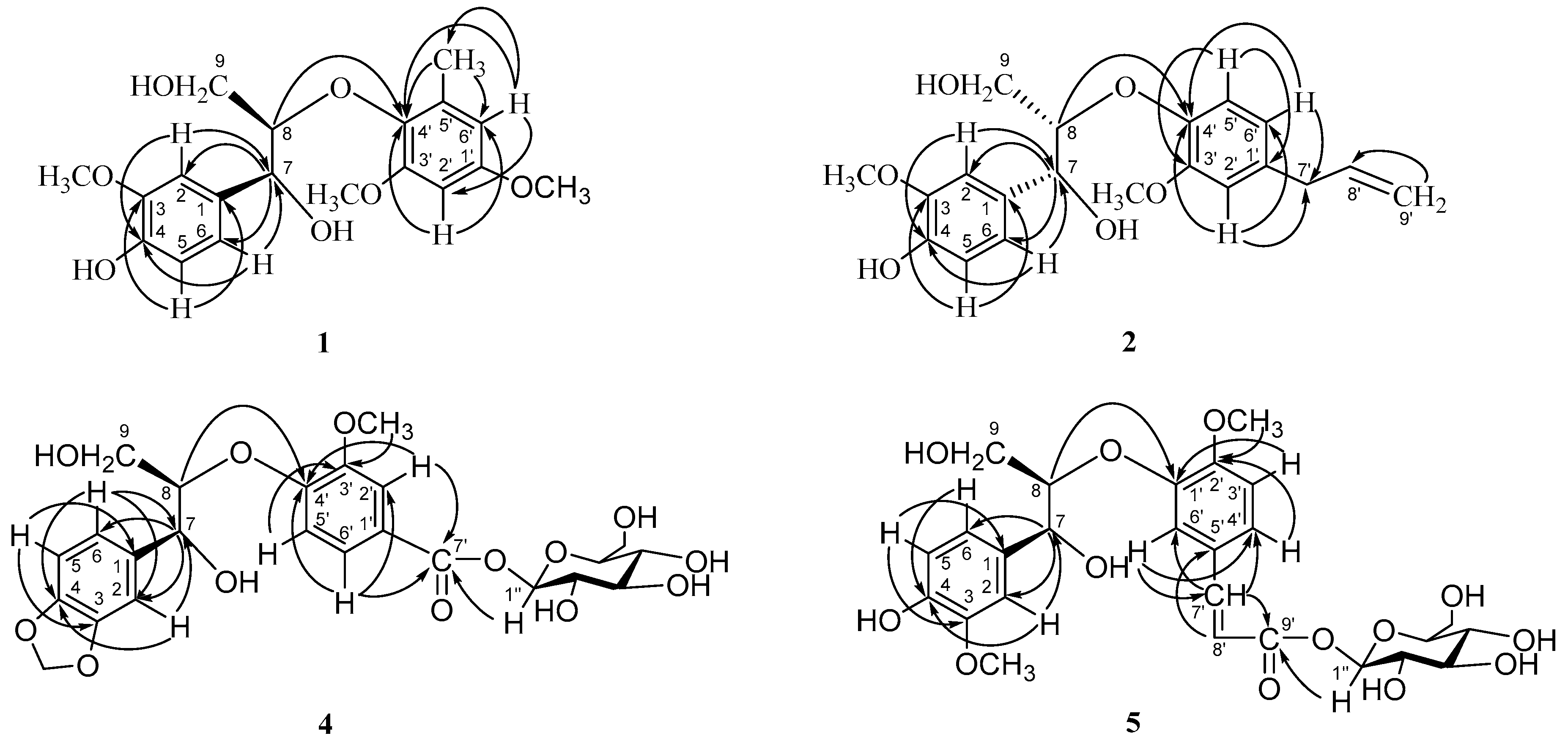
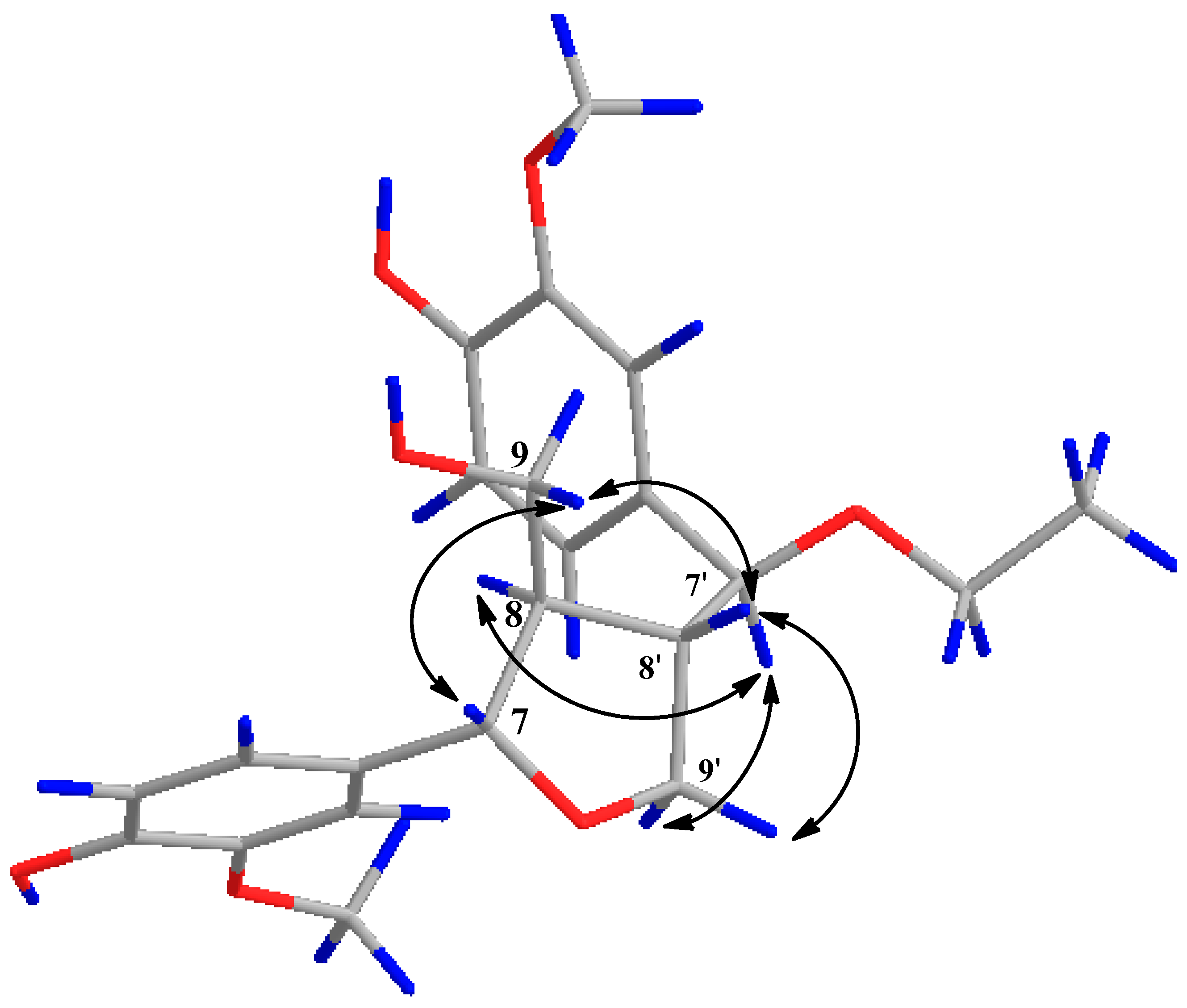
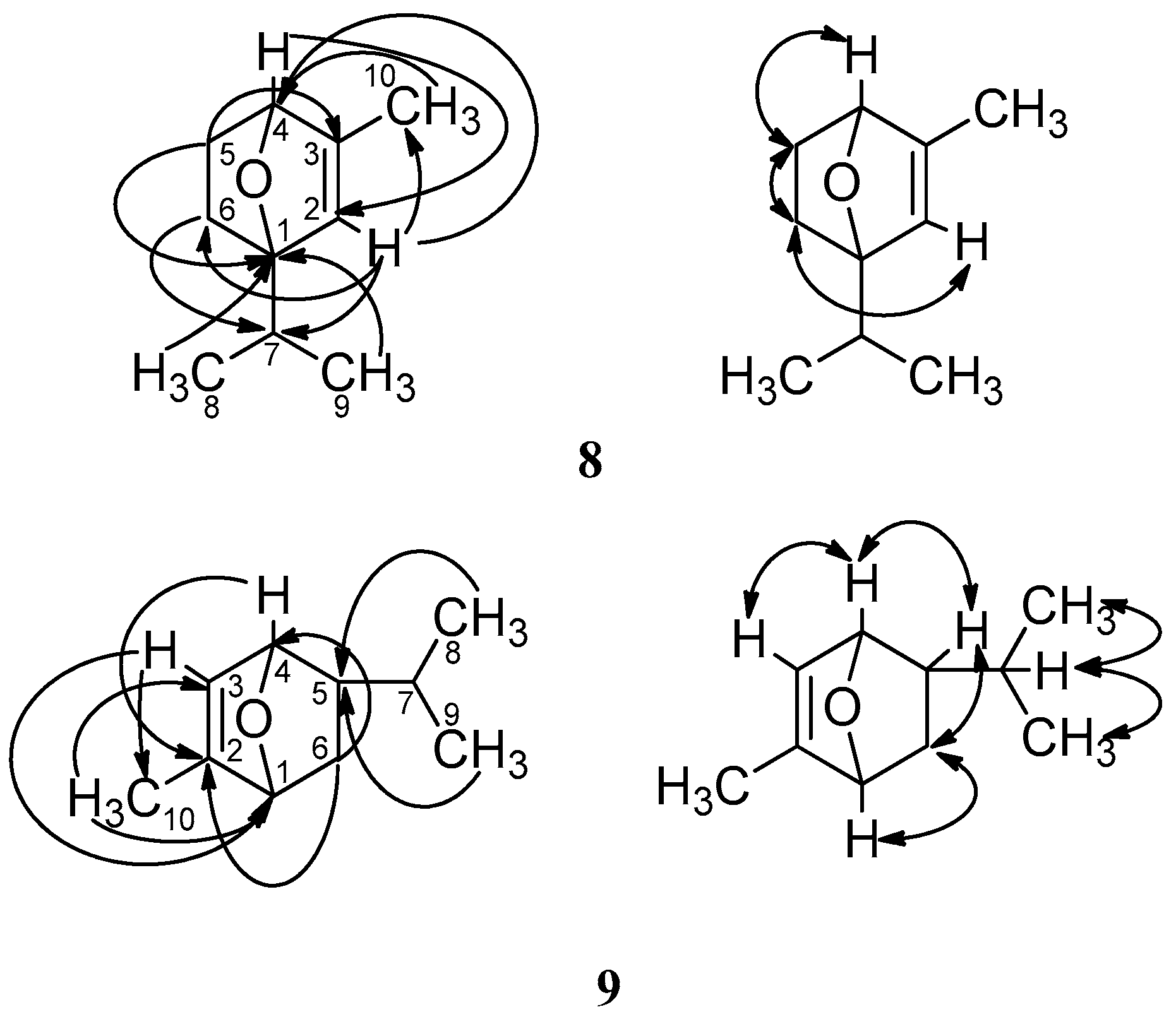
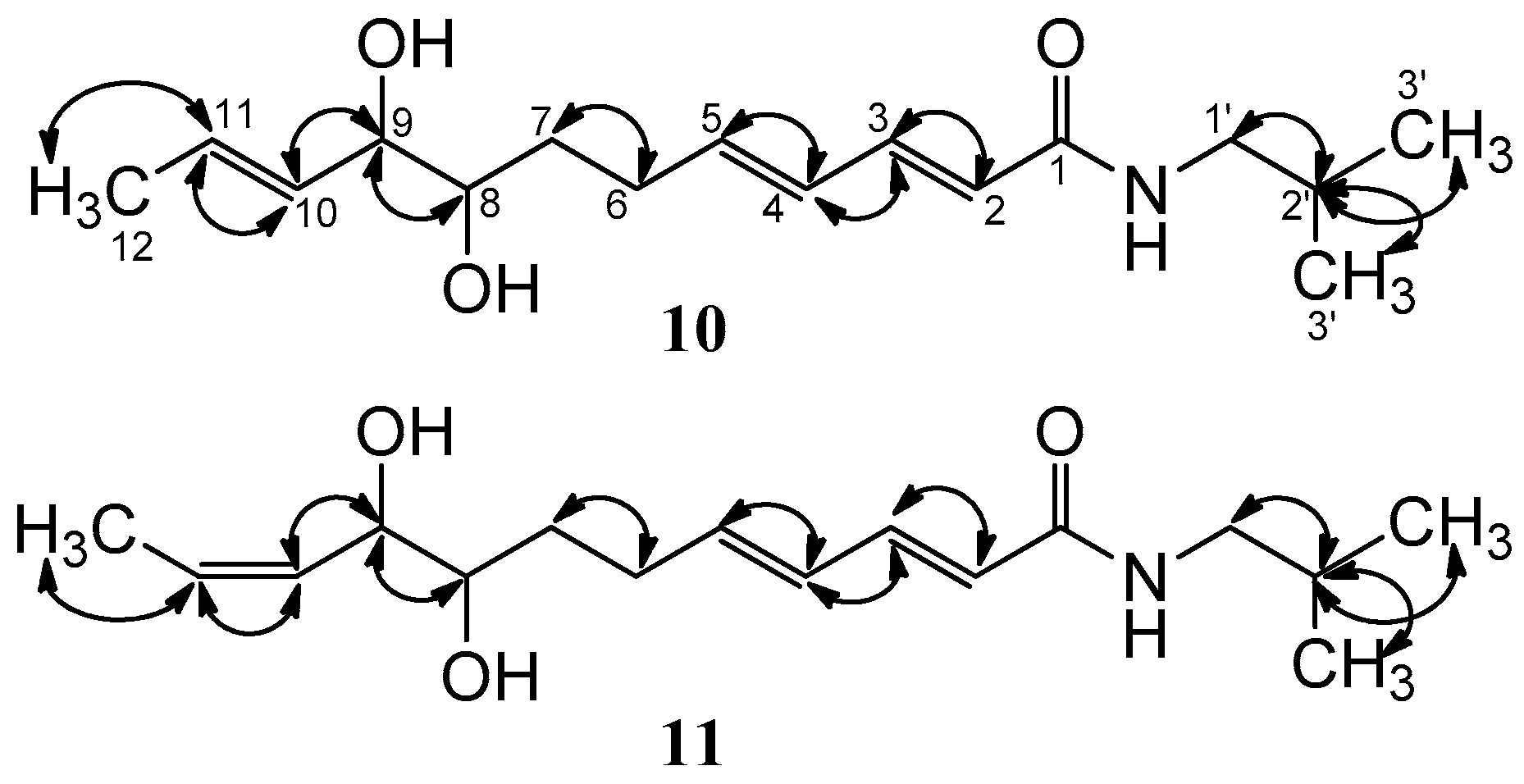
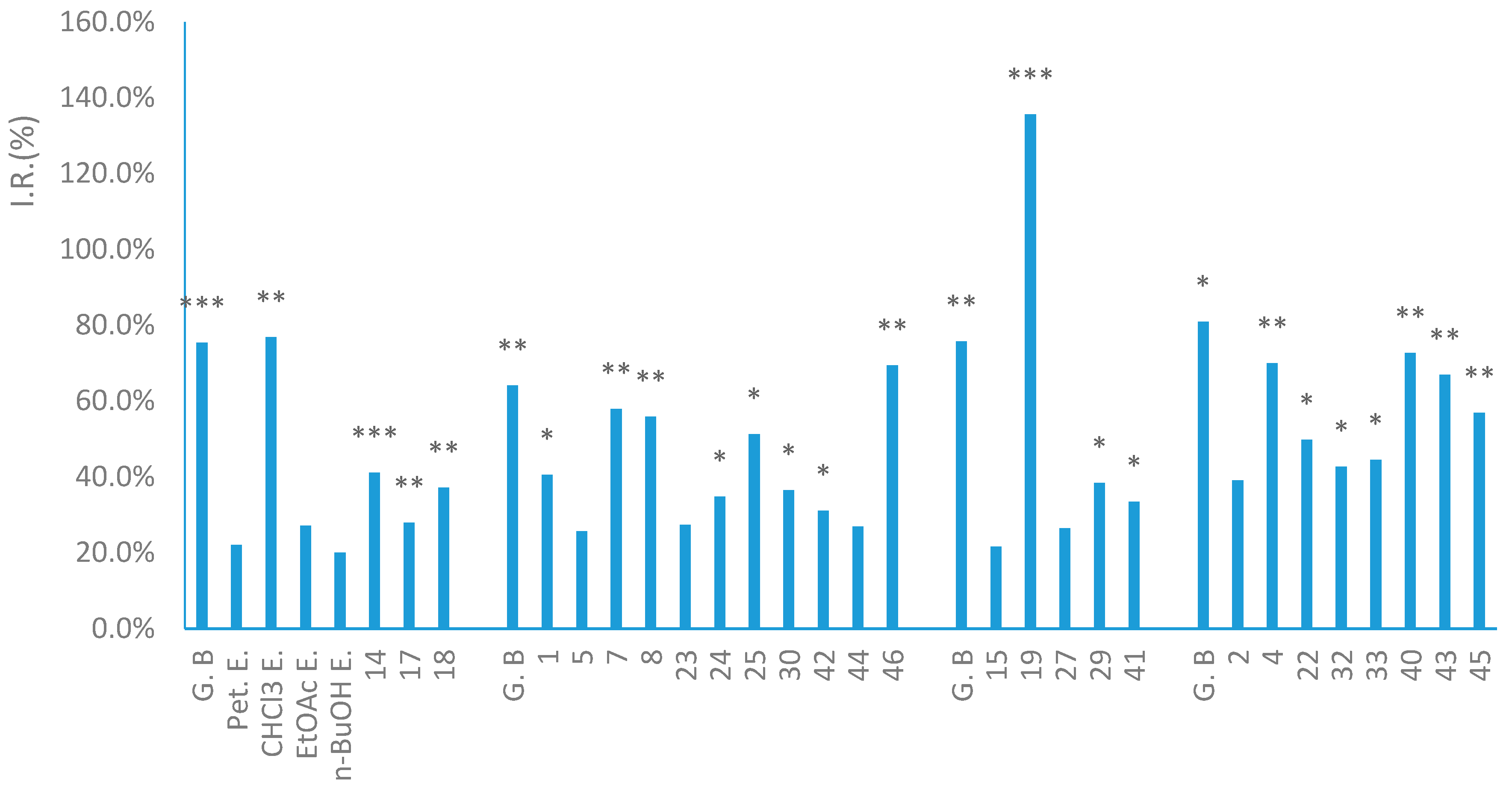
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compounds 4 and 5, and GC Analysis
3.5. Anti-Inflammatory Activity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, R.H. Resource Science of Chinese Medicinal Materials; China Medical & Pharmaceutical Sciences Press: Beijing, China, 1993; pp. 202–211. [Google Scholar]
- Chen, Y.L.; Liu, Q.Q.; Wang, J.Z.; Chen, C.P.; Wang, J.Y. Composition analysis of volatile oil of Asarum heterotropoides var. mandshuricum extracted by three different methods. J. Chin. Med. Mater. 2013, 36, 1447–1451. [Google Scholar]
- Cai, S.Q.; Wang, H.; Chen, S.Z.; Lou, Z.C. Studies on the non-volatile chemical constituents of Asarum heterotropoides var. mandshuricum. J. Beijing Med. Univ. 1996, 28, 228–230. [Google Scholar]
- Lu, S.; Xu, L.; Huang, J.; Gao, H.Y.; Wu, L.J. Chemical contituents of the roots of Asarum heterotropoides Fr. Schmidt var. mandshuricum. J. Shenyang Pharm. Univ. 2008, 25, 702–704. [Google Scholar]
- Huang, J.; Wang, H.Q.; Zhang, C.; Li, G.Y.; Lin, R.C.; Wang, J.H. A new tetrahydrofuran-type lignan with anti-inflammatory activity from Asarum heterotropoides Fr. Schmidt var. mandshuricum. J. Asian Nat. Prod. Res. 2014, 16, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, Y.J.; Oh, S.M.; Yi, J.M.; Kim, N.S.; Bang, O.S. Bioactive compounds from the roots of Asiasarum heterotropoides. Molecules 2014, 19, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, I.; Takeya, K.; Itokawa, H. Structures of amides from Asiasarum heterotropoides. Chem. Pharm. Bull. 1981, 29, 564–566. [Google Scholar] [CrossRef]
- Hashimoto, K.; Okui, Y.; Hiroyoki, I. Monoterpenes from Asiasari Radix from Asarum sp. Phytochemistry 1990, 29, 3571–3574. [Google Scholar] [CrossRef]
- Wang, D.; Xia, X.H. Chemical constituents of aerial parts of Asarum heterotropides var. mandshuricum. Chin. Tradit. Herbal Drugs 1998, 29, 83–84. [Google Scholar]
- Xu, L.; Lu, S.; Sun, B.H.; Gao, H.Y.; Wu, L.J. Chemical contituents of aerial parts of Asarum heterotropoides Fr. Schmidt var. mandshuricum. J. Shenyang Pharm. Univ. 2008, 25, 699–701. [Google Scholar]
- Xu, L.; Wu, D.; Wu, Z.H.; Lu, S.; Gao, H.Y.; Wu, L.J. Chemical Contituents of aerial parts of Asarum heterotropoides Fr. Schmidt var. mandshuricum (2). J. Shenyang Pharm. Univ. 2009, 26, 964–967. [Google Scholar]
- Lu, S.; Wu, D.; Wu, Z.H.; Gao, H.Y.; Sun, B.H.; Wu, L.J. Isolation and identification of chemical constituents from roots of Asarum heterotropides Fr Schmidt. var. mandshuricum (Maxim.) Kitag. (II). J. Shenyang Pharm. Univ. 2010, 27, 707–710. [Google Scholar]
- Miyazawa, M.; Ishikawa, Y.; Toshikura, M. Insecticidal Compounds from Asarum heterotropoides var. mandshuricum. Chem. Express 1991, 6, 703–706. [Google Scholar]
- Miyazawa, M.; Ishikawa, Y.; Toshikura, M. Insecticidal allylbenzenes from Asarum heterotropoides var. mandshuricum. Chem. Express 1992, 7, 69–72. [Google Scholar]
- Hu, Y.J.; Zhou, H.; Wang, J.G.; Zhang, Y.; Li, Y.K. The pharmacological effect of “Xi Xin” oil. Chin. Pharm. Bull. 1986, 2, 41–44. [Google Scholar]
- Qu, S.Y.; Wu, Y.J. The anti-inflammatory effect of Asarum heterotropoides var. mandshuricum oil. Yao Xue Xue Bao 1982, 17, 12–16. [Google Scholar] [PubMed]
- Gao, J.J.; Zhu, L.L.; Wang, H.M. Comparative study between heavy dosage and conventional dosage herba Asari for treatment of late stage severe rheumatoid arthitis. J. Tradit. Chin. Med. 1997, 38, 283–285. [Google Scholar]
- Zhu, X.H.; Tian, H.M. Impact on the effect of decoction of Xixin due to dosage, decoction time and species. Tianjin Pharm. 1999, 11, 35–36. [Google Scholar]
- Cai, S.Q.; Li, J. Species Systematization and Quality Evaluation of Commonly Used Chinese Traditional Drugs, North, 5th ed.; Beijing Medical University Press: Beijing, China, 2001; p. 91. [Google Scholar]
- Liu, J. Study the clinical application of Xixin through determination volatile oil of different parts. Res. Tradit. Chin. Med. 1996, 6, 54–55. [Google Scholar]
- Xiong, Y.L.; Jing, Y.; Shang, M.Y.; Li, C.L.; Ye, J.; Wang, X.; Cai, S.Q. Anti-inflammatory and anti-nociceptive effects in mice of water and ethanol extracts of root and rhizomes of Asarum heterotropoides var. mandshuricum. Chin. J. Chin. Mater. Med. 2009, 34, 2252–2257. [Google Scholar]
- Kosuge, T.; Yokota, M.; Nukaya, H. Studies on antitussive principles of Asiasari Radix. Chem. Pharm. Bull. 1978, 26, 2284–2285. [Google Scholar] [CrossRef]
- Takasaki, M.; Konoshima, T.; Yasuda, I. Inhibitory Effects of Shouseiryu-to on Two-Stage Carcinogenesis II Anti-tumor-promoting activities of lignans from Asiasarum hetertropoides var. mandshuricum. Biol. Pharm. Bull. 1997, 20, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Yanagisawa, T.; Okui, Y. Study on anti-allergic components in the roots of Asiasarum sieboldi. Planta Med. 1994, 60, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Han, A.R.; Kim, H.J.; Shin, M.; Hong, M.; Kim, Y.S.; Bae, H. Constituents of Asarum sieboldii with inhibitory activity on lipopolysaccharide (LPS)-induced NO production in BV-2 microglial cells. Chem. Biodivers. 2008, 5, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Quang, T.H.; Ngan, N.T.T.; Minh, C.V.; Kiem, P.V.; Tai, B.H.; Thao, N.P.; Song, S.B.; Kim, Y.H. Anti-inflammatory and PPAR transactivational effects of secondary metabolites from the roots of Asarum sieboldii. Bioorg. Med. Chem. Lett. 2012, 22, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.; Dewald, B.; Baggiolini, M. Neutrophil signal transduction and activation of the respiratory burst. Physiol. Rev. 1993, 73, 797–821. [Google Scholar] [PubMed]
- Sergio, H.C.; Massayoshi, Y.; Otto, R.G. Neolignans from Virola carinata fruit. Phytochemistry 1985, 24, 1051–1055. [Google Scholar]
- Takeshi, D.; Takako, I.; Shizuke, K. The constituents of Eucommia ulmoides Oliv. VI. Isolation of a new sesquilignan and neolignan glycosides. Chem. Pharm. Bull. 1987, 35, 1803–1807. [Google Scholar]
- Yuan, Z.; Li, X. NMR Methods for determining the configuration of 8-O-4′ neolignans. Chin. J. Magnet. Reson. 2003, 20, 307–314. [Google Scholar]
- Wang, C.Z.; Jia, Z.J. Neolignan glycosides from pedicularis longiflora. Planta Med. 1997, 63, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Greca, M.D.; Molinaro, A.; Monaco, P. Neolignans from arum italicum. Phytochemistry 1994, 35, 777–779. [Google Scholar] [CrossRef]
- Sakushima, A.; Coskun, M.; Maoka, T. Hydroxybenzoic acids from Boreava orientalis. Phytochemistry 1995, 40, 257–261. [Google Scholar] [CrossRef]
- Ichikawa, M.; Ryu, K.; Yoshida, J. Identification of six phenylpropanoids from Garlic Skin as major antioxidants. J. Agric. Food Chem. 2003, 51, 7313–7317. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Z.; Zhang, W.D.; Gu, Z.B.; Liu, W.Y.; Zhou, J.; Chen, W.S. Studies on the lignans from Patrinia scabra. Yao Xue Xue Bao 2003, 38, 520–522. [Google Scholar] [PubMed]
- Macias, F.A.; Lopez, A.; Varela, R.M.; Torres, A.; Molinillo, J.M. Bioactive lignans from a cultivar of Helianthus annuus. J. Agric. Food Chem. 2004, 52, 6443–6447. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, D.H.; Jang, D.S. Two new stereoisomers of tetrahydrofuranoid lignans from the flower buds of Magnolia fargesii. Chem. Pharm. Bull. 2007, 55, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Miyase, T.; Ueno, A. Lignan and terpene glycosides from Epimedium sagittarium. Phytochemistry 1991, 30, 2025–2027. [Google Scholar] [CrossRef]
- Miyazawa, M.; Kasahara, H.; Kameoka, H. Microbial oxidation of (+)-epimagnolin A by Aspergillus niger. Phytochemistry 1994, 35, 1191–1193. [Google Scholar] [CrossRef]
- Li, N.; Wu, J.L.; Hasegawa, T.; Sakai, J.I.; Bai, L.M.; Wang, L.Y.; Kakuta, S.; Furuya, Y.; Ogura, H.; Kataoka, T.; et al. Bioactive lignans from Peperomia duclouxii. J. Nat. Prod. 2007, 70, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Blumann, A.; Ryder, L. Autoxidation of α-phellandrene. J. Chem. Soc. 1949, 2040–2043. [Google Scholar] [CrossRef]
- Opitz, M.; Pachaly, P.; Sin, K.S. New polar ingredients from Asarum sieboldii. Pharmazie 1999, 54, 218–223. [Google Scholar]
- Chen, I.S.; Chen, T.L.; Lin, W.Y. Isobutylamides from the fruit of Zanthoxylum integrifoliolum. Phytochemistry 1999, 52, 357–360. [Google Scholar] [CrossRef]
- Xiong, Q.B.; Shi, D.W.; Hirofumi, Y. Alkylamides from pericarps of Zanthoxylum bungeanum. Phytochemistry 1997, 46, 1123–1126. [Google Scholar] [CrossRef]
- Takaku, N.; Choi, D.H.; Mikame, K.; Okunishi, T.; Suzuki, S.; Ohashi, H.; Umezawa, T.; Shimada, M. Lignans of Chamaecyparis obtusa. J. Wood Sci. 2001, 47, 476–482. [Google Scholar] [CrossRef]
- He, M.; Zhang, J.H.; Hu, C.Q. Studies on the chemical components of Clematic chineniss. Yao Xue Xue Bao 2001, 36, 278–280. [Google Scholar] [PubMed]
- Okuyama, E.; Suzumura, K.; Yamazaki, M. Pharmacologically active components of Todopon Puok (Fagraea racemosa), a medicinal plant from Borneo. Chem. Pharm. Bull. 1995, 43, 2200–2204. [Google Scholar] [CrossRef] [PubMed]
- Schottner, M.; Reiner, J.; Tayman, F.S.K. (+)-Neo-olivil from roots of Urtica dioica. Phylochemistry 1997, 46, 1107–1109. [Google Scholar] [CrossRef]
- Kato, M.J.; Chu, A.; Davin, L.B.; Lewis, N.G. Biosynthesis of antioxidant lignans in Sesamum indicum seeds. Phytochemistry 1998, 47, 583–591. [Google Scholar] [CrossRef]
- Patrice, A.M.; Massuo, J.K.; Norman, G.L. Episesaminone, a Sesamum indicum furofuran lignin, isolation and hemisynthesis. J. Nat. Prod. 1997, 60, 1189–1192. [Google Scholar]
- Barrero, A.F.; Haidour, A.; Dorado, M.M. Lignans from the wood of Abzes pznsapo. J. Nat. Prod. 1994, 57, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Cutillo, F.; D’Abrosca, B.; DellaGreca, M.; Fiorentino, A.; Zarrelli, A. Lignans and neolignans from Brassica fruticulosa: Effects on seed germination and plant growth. J. Agric. Food Chem. 2003, 51, 6165–6172. [Google Scholar] [CrossRef] [PubMed]
- Su, B.N.; Takaishi, Y.; Kusumi, T.; Morinols, A.L. Twelve novel sesquineolignans and neolignans with a new carbon skeleton from Marina chinensis. Tetrahedron 1999, 55, 14571–14586. [Google Scholar] [CrossRef]
- Li, J.; Li, F.; Lu, Y.Y.; Wen, G.Q. Study on the an tiphlogistic constituents in seed of Schisandra propinqua (Wall) Hook. F. et Thoms. Chin. Pharm. J. 2007, 142, 255–257. [Google Scholar]
- Long, C.F.; Wang, X.; Yang, Y.X.; Cai, S.Q. Studies on flavonoids from the roots of Asarum maximum. J. Beijing Med. Univ. 2000, 32, 229–232. [Google Scholar]
- Dong, C.X.; Wu, K.S.; Shi, S.P. Flavanoids from Clematis hexapetala. J. Chin. Pharm. Sci. 2006, 15, 15–20. [Google Scholar]
- Zhang, S.X.; Tani, T.; Yamaji, S.; Ma, C.M.; Wang, M.C.; Cai, S.Q.; Zhao, Y.Y. Flavonoids from the roots and rhizomes of Asarum longerhizomatosum. J. Asian Nat. Prod. Res. 2003, 5, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Katsuhara, T.; Niitsu, K.; Ikeya, Y.; Okada, M.; Mitsuhashi, H. Two glycosides from roots of Asarum sieboldii. Phytochemistry 1992, 31, 2477–2480. [Google Scholar] [CrossRef]
- Morikawa, T.; Wang, L.B.; Nakamura, S.; Ninomiya, K.; Yokayama, E.; Matsuda, H.; Muraoka, O.; Wu, L.J.; Yoshikawa, M. Medicinal flowers. XXVII.1 new flavanone and chalcone glycosides, Arenariumosides I, II, III, and IV, and tumor necrosis factor-alpha inhibitors from everlasting, flowers of Helichrysum arenarium. Chem. Pharm. Bull. 2009, 57, 361–367. [Google Scholar] [PubMed]
- Atta-ur-Rahman; Bhatti, M.K.; Akhtar, F.; Choudhary, M.I. Alkaloids of Fumaria indica. Phytochemistry 1992, 31, 2869–2872. [Google Scholar] [CrossRef]
- Zhang, S.X.; Cai, S.Q.; Zhao, Y.Y. Studies on chemical constituents from radix and rhizome of Asarum longerhizomatosum. Chin. Tradit. Herbal Drugs 2002, 4, 297–299. [Google Scholar]
- Wang, Z.H.; Huang, J.; Ma, X.C.; Li, G.Y.; Ma, Y.P.; Li, N.; Wang, J.H. Phenolic glycosides from Curculigo orchioides Gaertn. Fitoterapia 2013, 86, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Dai, H.Q.; Du, Y.H.; Schneider, B.; Guo, H.; Li, D.P.; Zhang, L.X.; Fu, H.A.; Dong, X.P.; Cheng, Y.X. Identification of blapsins A and B as potent small-molecule 14-3-3 inhibitors from the insect Blaps japanensis. Bioorg. Med. Chem. Lett. 2012, 22, 4179–4181. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.Z.; Piao, H.S. Studies on chemical constituents from roots of Caragana microphylla. Chin. J. Chin. Mater. Med. 2007, 32, 698–700. [Google Scholar]
- Greca, M.D.; Ferrara, M.; Fiorentino, A. Antialgal compounds from Zantedeschia aethiopica. Phytochemistry 1998, 49, 1299–1304. [Google Scholar] [CrossRef]
- Klick, S.; Herrmann, K. Glucosides and glucose esters of hydroxybenzoic acids in plants. Phytochemistry 1988, 27, 2177–2180. [Google Scholar] [CrossRef]
- Rukachaisirikul, T.; Intaraudom, J.; Chawanasak, S. Phenylpropanoids from Cinnamomum parthenoxylon. Sci. Asia 2000, 26, 159–161. [Google Scholar]
- Banerjee, S.K.; Singh, K. A novel acid from Apium leptophyllum. J. Indian Council Chem. 1995, 11, 39–41. [Google Scholar]
- Cai, L.N.; Wang, H.S.; Cao, H.X. The constituents from the rhizome of Alisma orientalis (SAM) Juzep. Tianran Chanwu Yanjiu Yu Kaifa 1996, 8, 5–9. [Google Scholar]
- Kuo, Y.H.; Yeh, M.H.; Huang, S.L. A novel 6-butyl-3-hydroxyflavanone from heartwood of Bauhinia purpurea. Phytochemistry 1998, 49, 2529–2530. [Google Scholar] [CrossRef]
- Dai, S.J.; Mi, Z.M.; Ma, Z.B.; Li, S.; Chen, R.Y.; Yu, D.Q. Bioactive Diels-Alder type adducts from the stem bark of Morus macroura. Planta Med. 2004, 70, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Iwashina, T.; Kitajima, J. Chalcone and flavonol glycosides from Asarum canadense (Aristolochiaceae). Phytochemistry 2000, 55, 971–974. [Google Scholar] [CrossRef]
- Xie, B.B.; Shang, M.Y.; Lee, K.H.; Wang, X.; Komatsu, K.; Cai, S.Q. 5-methoxyaristololactam I, the first natural 5-substituted aristololactam from Asarum ichangense. Nat. Prod. Commun. 2011, 6, 11–14. [Google Scholar] [PubMed]
- Bai, X.; Zhang, X.; Chen, L.; Zhang, J.; Zhang, L.; Zhao, X.; Zhao, T.; Zhao, Y. Protective effect of naringenin in experimental ischemic stroke: Down-regulated NOD2, RIP2, NF-κB, MMP-9 and up-regulated claudin-5 expression. Neurochem. Res. 2014, 39, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, W.; Wang, Y. Anti-inflammation of flavonoid compounds from Dalbergia odorifera T. Chen in lipopolysaccharide stimulated RAW264.7 macrophage. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2013, 29, 681–684. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
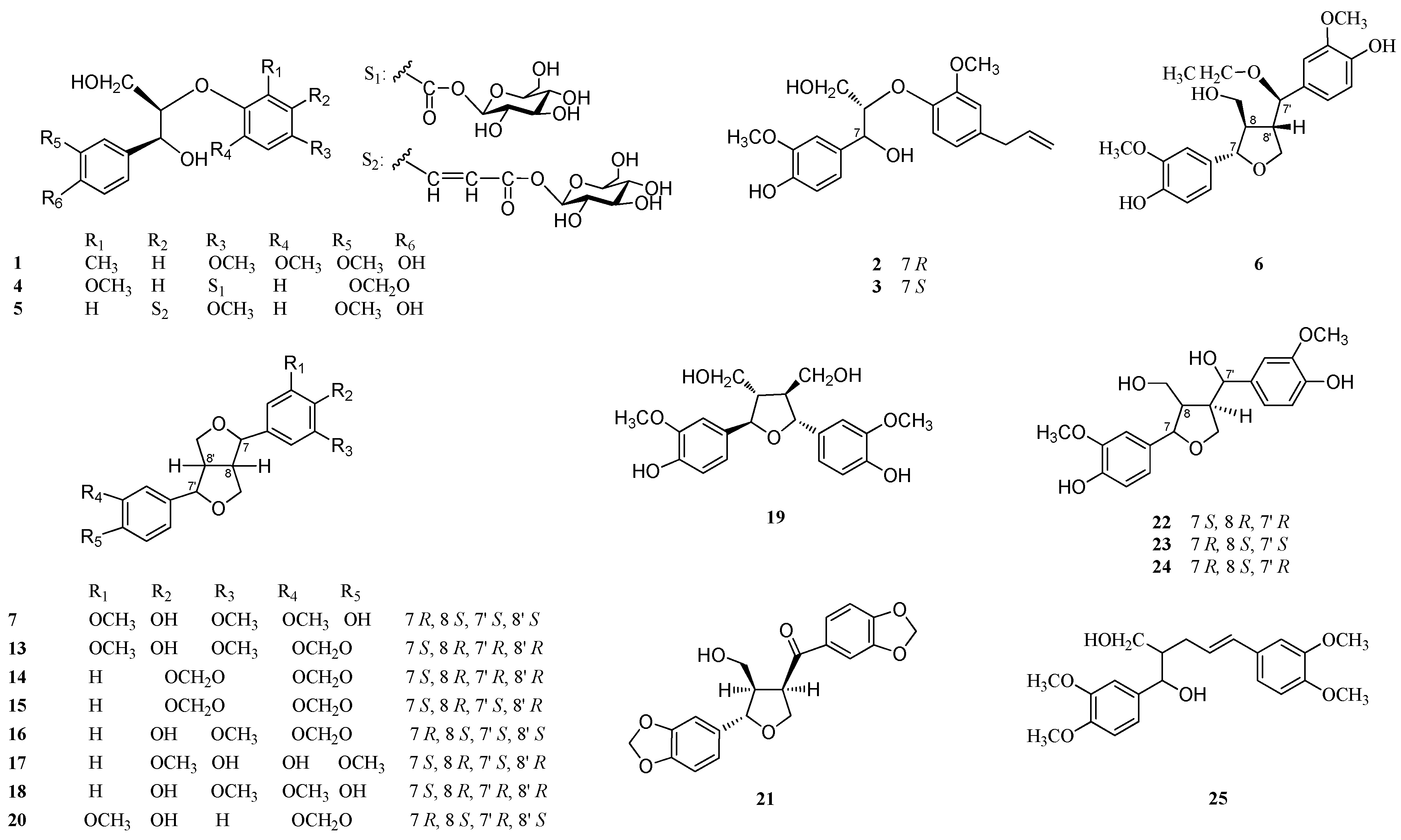

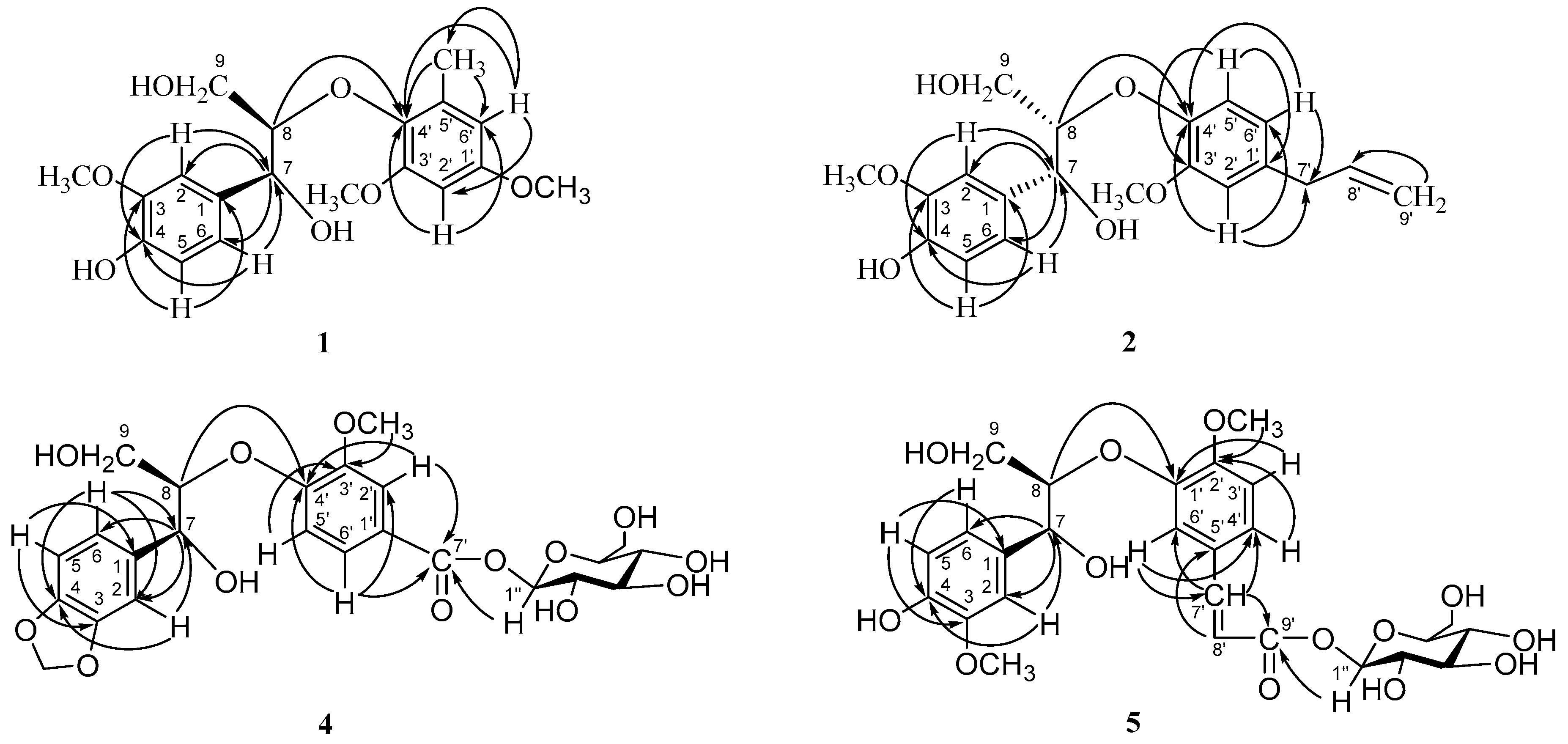
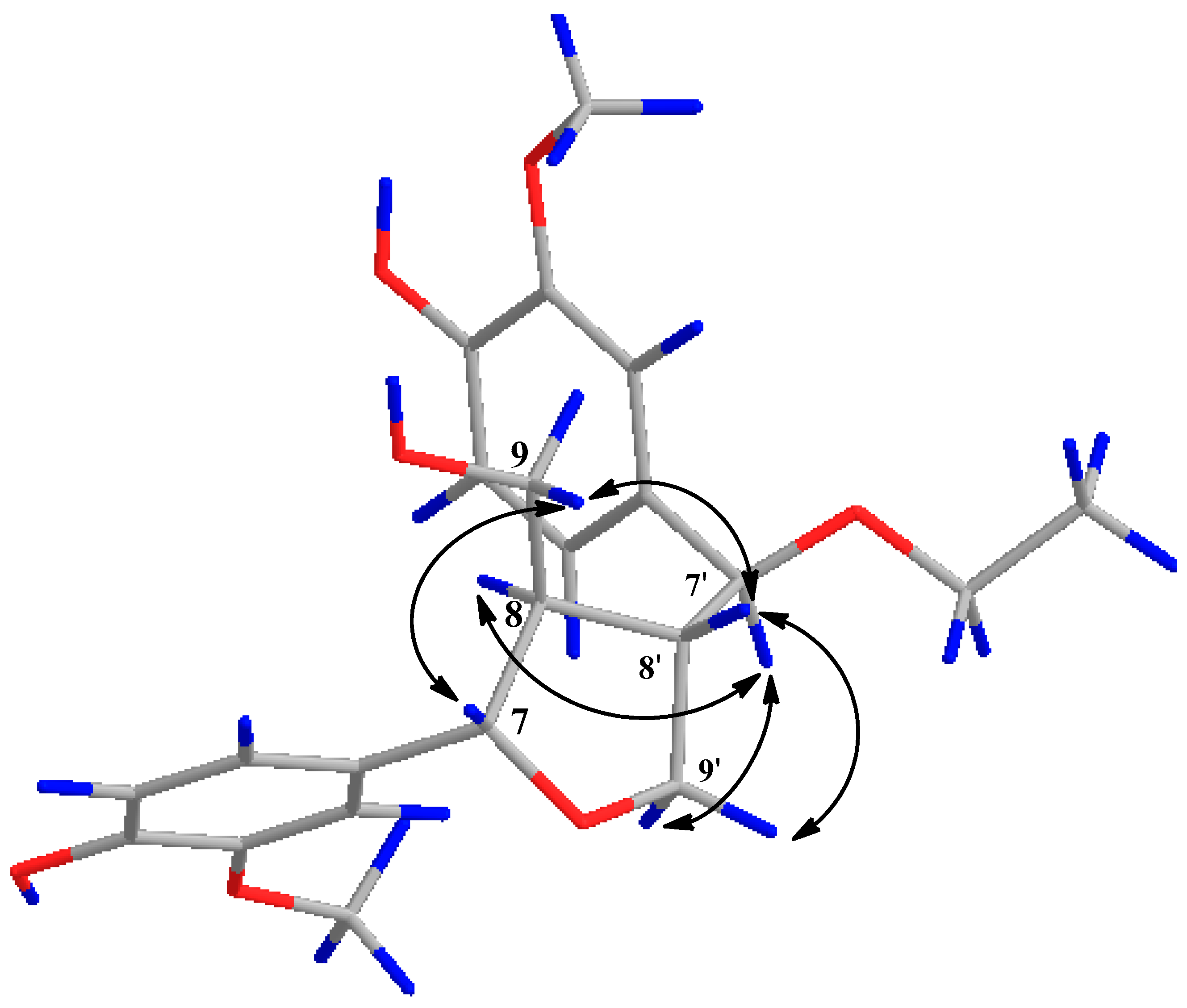
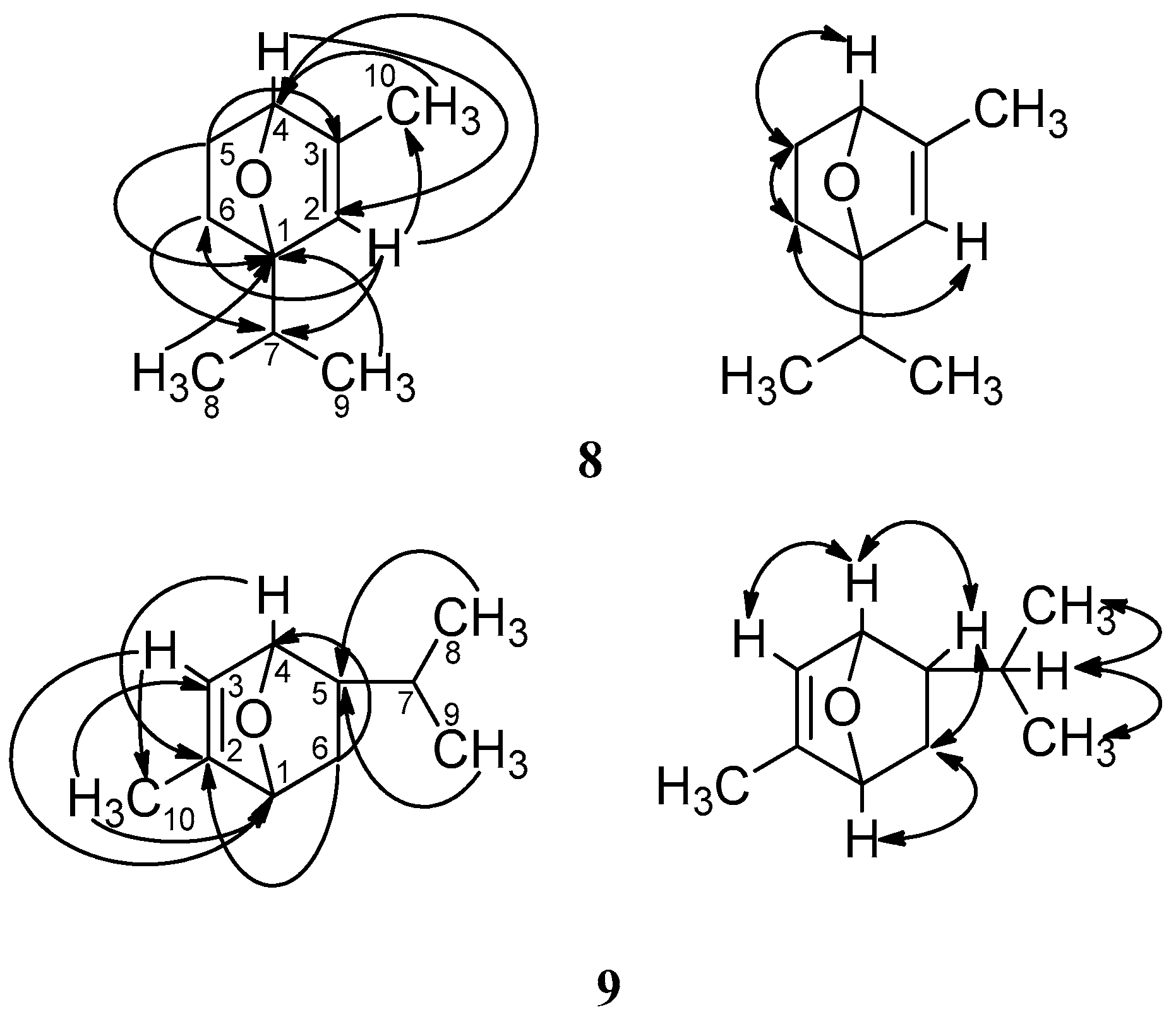

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 133.1 | — | 132.8 | — | 132.2 | — |
| 2 | 110.1 | 6.96, d (2.4) | 110.5 | 7.00, d (2.4) | 110.4 | 7.01, d (1.8) |
| 3 | 147.4 | — | 147.4 | — | 147.5 | — |
| 4 | 145.5 | — | 145.7 | — | 145.9 | — |
| 5 | 114.4 | 6.73, d (8.4) | 114.3 a | 6.73, d (8.4) | 114.5 b | 6.75, d (8.4) |
| 6 | 119.2 | 6.77, dd (8.4, 2.4) | 119.6 | 6.82, dd (8.4, 2.4) | 119.4 | 6.85, dd (8.4, 1.8) |
| 7 | 73.0 | 4.87, d (4.2) | 72.8 | 4.82, d (5.4) | 72.8 | 4.88, d (6.0) |
| 8 | 85.1 | 4.18, m | 85.3 | 4.28, m | 86.3 | 4.20, m |
| 9 | 60.2 | 3.83, m, H-9a | 60.8 | 3.75–3.83, m | 60.5 | 3.70, dd (12.0, 3.6), H-9a |
| 3.62, dd (12.6, 3.6), H-9b | 3.41, m, H-9b | |||||
| 1′ | 152.9 | — | 134.6 | — | 134.9 | — |
| 2′ | 97.4 | 6.41, d (3.0) | 112.8 | 6.75, d (1.8) | 112.7 | 6.81, d (1.8) |
| 3′ | 156.0 | — | 150.6 | — | 150.4 | — |
| 4′ | 138.2 | — | 146.1 | — | 146.5 | — |
| 5′ | 132.6 | — | 118.3 | 6.83, d (7.2) | 118.2 | 6.98, d (7.8) |
| 6′ | 106.6 | 6.29, d (3.0) | 120.7 | 6.64, dd (7.2, 1.8) | 120.8 | 6.69, dd (7.8, 1.8) |
| 7′ | — | — | 39.4 | 3.28, d (5.4) | 39.5 | 3.33, m |
| 8′ | — | — | 137.7 | 5.92, m | 137.7 | 5.93, m |
| 9′ | — | — | 114.4 a | 5.03, m | 114.5 b | 5.03, m |
| 3-OCH3 | 54.9 | 3.71, s | 55.4 | 3.79, s | 55.0 | 3.82, s |
| 1′-OCH3 | 54.5 | 3.80, s | ||||
| 3′-OCH3 | 55.0 | 3.81, s | 55.3 | 3.76, s | 55.2 | 3.84, s |
| 5′-CH3 | 15.7 | 2.12, s | ||||
| Position | 4 | 5 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 135.1 | — | 136.2 | — |
| 2 | 107.4 a | 6.87, d (1.8) | 111.0 | 7.09, d (1.2) |
| 3 | 147.5 | — | 149.1 | — |
| 4 | 147.1 | — | 146.1 | — |
| 5 | 107.1 a | 6.59, d (7.8) | 116.21 d | 7.07, m |
| 6 | 120.4 | 6.78, m | 119.8 | 6.94, dd (7.8, 1.2) |
| 7 | 72.5 | 4.71, d (4.8) | 72.4 | 4.84, d (4.8) |
| 8 | 83.8 | 4.44, m | 84.1 | 4.47, m |
| 9 | 61.0 b | 3.77–3.74 c, m | 60.8 | 3.85–3.76 e, m, H-9a 3.65, dd (12.0, 4.8), H-9b |
| 1′ | 122.1 | — | 150.4 | — |
| 2′ | 113.0 | 7.46, d (1.2) | 150.2 | — |
| 3′ | 149.7 | — | 116.15 d | 6.93, d (7.8) |
| 4′ | 152.9 | — | 122.0 | 7.04, dd (7.8, 1.8) |
| 5′ | 114.7 | 6.92, d (8.0) | 128.4 | — |
| 6′ | 123.8 | 7.52, dd (7.8, 1.2) | 110.8 | 7.12, m |
| 7′ | 165.1 | — | 144.8 | 7.54, d (15.6) |
| 8′ | — | — | 116.11 d | 6.31, d (15.6) |
| 9′ | — | — | 169.5 | — |
| 1″ | 94.8 | 5.58, d (7.8) | 101.6 | 4.80, d (7.2) |
| 2″ | 72.7 | 3.62–3.32, m | 73.6 | 3.45–3.36 , m |
| 3″ | 76.7 | 76.5 | ||
| 4″ | 69.7 | 69.9 | ||
| 5″ | 77.5 | 76.8 | ||
| 6″ | 61.0 b | 3.77–3.74 c, m | 61.2 | 3.85–3.76 e, m |
| -OCH2O- | 100.9 | 5.77, s | — | — |
| 3-OCH3 | — | — | 55.3 | 3.79, s |
| 2′-OCH3 | — | — | 55.3 | 3.79, s |
| 3′-OCH3 | 55.2 | 3.75, s | — | — |
| Position | 7 | |
|---|---|---|
| δC | δH (J in Hz) | |
| 1 | 133.1 | — |
| 2 | 102.4 | 6.60, s |
| 3 | 147.1 | — |
| 4 | 133.7 | — |
| 5 | 147.1 | — |
| 6 | 102.4 | 6.60, s |
| 7 | 87.8 | 4.44, d (6.6) |
| 8 | 54.5 | 2.90, dd (7.2, 13.8) |
| 9 | 71.1 | 4.13, d (9.6) 3.87–3.83, m, H-9b |
| 1′ | 129.6 | — |
| 2′ | 108.6 | 6.91, d (1.2) |
| 3′ | 146.8 | — |
| 4′ | 145.4 | — |
| 5′ | 114.3 | 6.89, d (7.8) |
| 6′ | 119.2 | 6.83, dd (7.8, 1.2) |
| 7′ | 82.3 | 4.85, d ( 6.0) |
| 8′ | 50.2 | 3.34–3.31, m, H-8′ |
| 9′ | 69.7 | 3.87–3.83, m, H-9′a 3.34–3.31, m, H-9′b |
| 7′-OCH2- | — | — |
| -CH3 | — | — |
| 3-OCH3 | 56.4 | 3.90, s |
| 5-OCH3 | 56.4 | 3.90, s |
| 3′-OCH3 | 56.0 | 3.91, s |
| 4-OH | — | 5.60 a, brs |
| 4′-OH | — | 5.48 a, brs |
| Position | 8 | 9 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 71.4 | — | 67.0 | 3.90, d (3.0) |
| 2 | 124.8 | 5.54, t (4.2, 1.2) | 136.0 | — |
| 3 | 134.0 | — | 129.6 | 5.46, d (1.2) |
| 4 | 67.9 | 3.94, t (3.0, 2.4) | 68.6 | 3.85, dd (3.0, 1.2) |
| 5 | 32.8 | 1.94, dt (13.8, 2.4), H-5a 1.35, ddd (13.8, 3.5), H-5b | 41.5 | 1.58, m |
| 6 | 26.7 | 2.09, m, H-6a 1.77, m, H-6b | 29.5 | 1.71, dt (13.2, 3.0), H-6a 1.38, ddd (13.2, 4.2), H-6b |
| 7 | 38.4 | 1.77, m, | 25.6 | 2.10, m |
| 8 | 25.7 | 1.15, brs | 20.0 | 0.96, d (6.6) |
| 9 | 25.6 | 1.15, brs | 15.7 | 0.81, d (7.2) |
| 10 | 19.8 | 1.75, s | 19.3 | 1.76, s |
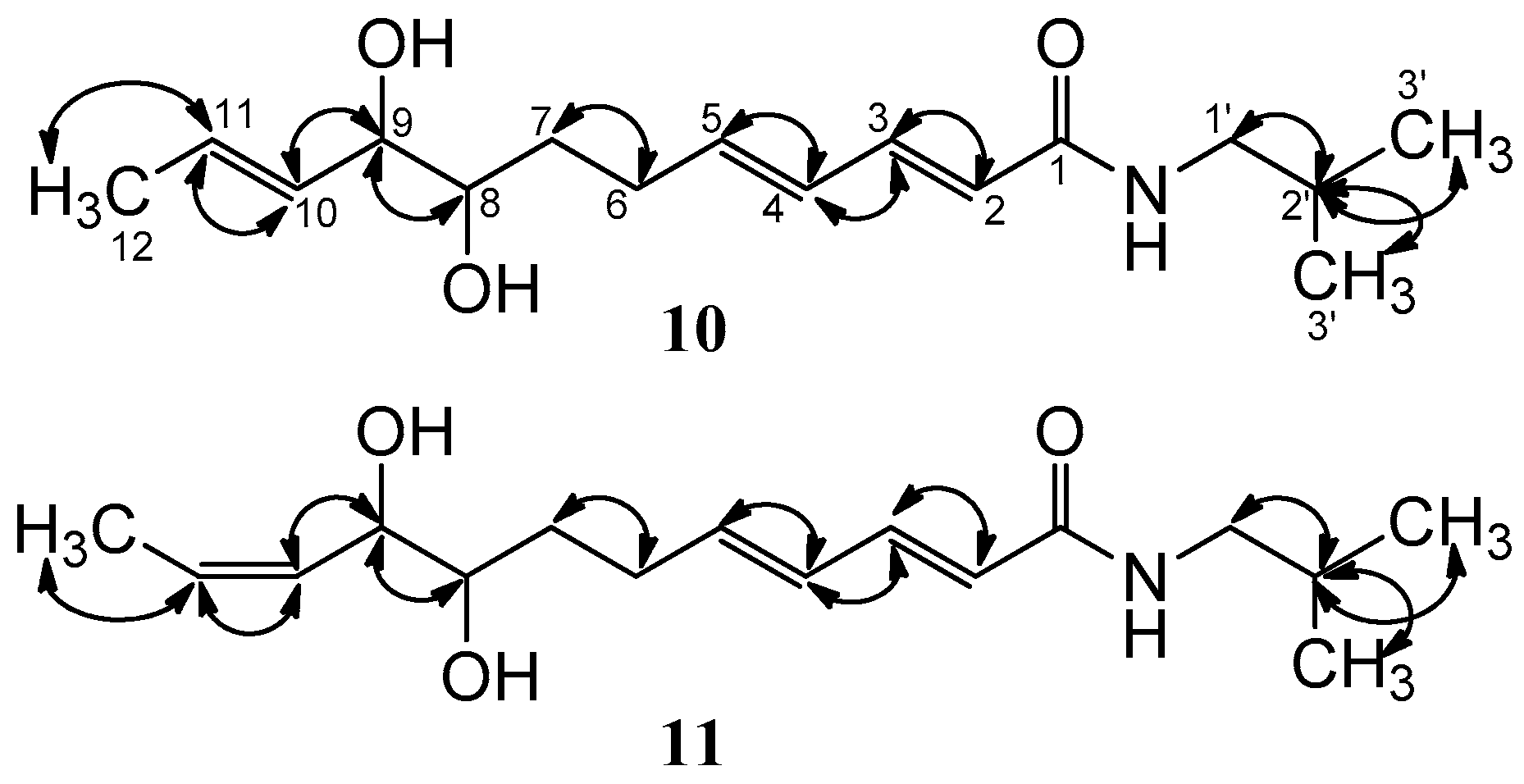
| Position | 10 | 11 | ||
|---|---|---|---|---|
| δC, Mult. | δH (J in Hz) | δC, Mult. | δH (J in Hz) | |
| 1 | 167.7, C | — | 167.6, C | — |
| 2 | 121.8, CH | 5.94, d (15.0) | 121.8, CH | 5.92, d (15.0) |
| 3 | 140.7, CH | 7.10, dd (15.0, 10.8) | 140.7, CH | 7.10, dd (15.0, 10.8) |
| 4 | 128.0, CH | 6.21, m | 127.1, CH | 6.22, m |
| 5 | 142.2, CH | 6.10, m | 142.2, CH | 6.11, m |
| 6 | 28.9, CH2 | 2.35, m | 28.9, CH2 | 2.38, m |
| 7 | 31.6, CH2 | 2.23, m | 31.3, CH2 | 2.22, m |
| 8 | 73.6, CH | 3.38, m | 73.6, CH | 3.49, m |
| 9 | 75.8, CH | 3.81, m | 69.9, CH | 4.29, m |
| 10 | 130.6, CH | 5.48, dd (15.0, 7.2) | 129.7, CH | 5.46, dd (9.0, 7.2) |
| 11 | 128.6, CH | 5.71, m | 128.7, CH | 5.65, m |
| 12 | 16.7, CH3 | 1.70, d (6.0) | 12.3, CH3 | 1.67, d (7.2) |
| 1′ | 46.5, CH2 | 3.05, d (7.2) | 46.7, CH2 | 3.05, d (7.2) |
| 2′ | 28.3, CH | 1.78, m | 28.4, CH | 1.78, m |
| 3′, 4′ | 19.1, CH3 | 0.91, m | 19.1, CH3 | 0.91, d (6.6) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, Y.; Zhang, Y.-F.; Shang, M.-Y.; Liu, G.-X.; Li, Y.-L.; Wang, X.; Cai, S.-Q. Chemical Constituents from the Roots and Rhizomes of Asarum heterotropoides var. mandshuricum and the In Vitro Anti-Inflammatory Activity. Molecules 2017, 22, 125. https://doi.org/10.3390/molecules22010125
Jing Y, Zhang Y-F, Shang M-Y, Liu G-X, Li Y-L, Wang X, Cai S-Q. Chemical Constituents from the Roots and Rhizomes of Asarum heterotropoides var. mandshuricum and the In Vitro Anti-Inflammatory Activity. Molecules. 2017; 22(1):125. https://doi.org/10.3390/molecules22010125
Chicago/Turabian StyleJing, Yu, Yi-Fan Zhang, Ming-Ying Shang, Guang-Xue Liu, Yao-Li Li, Xuan Wang, and Shao-Qing Cai. 2017. "Chemical Constituents from the Roots and Rhizomes of Asarum heterotropoides var. mandshuricum and the In Vitro Anti-Inflammatory Activity" Molecules 22, no. 1: 125. https://doi.org/10.3390/molecules22010125