
Keratin Protein-Catalyzed Nitroaldol (Henry) Reaction and Comparison with Other Biopolymers


 and
and
Abstract
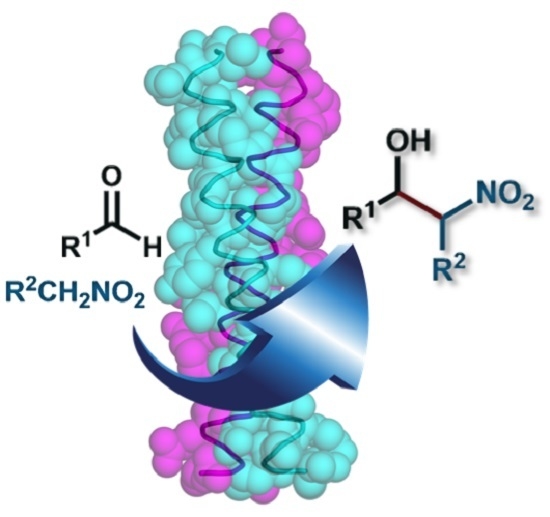
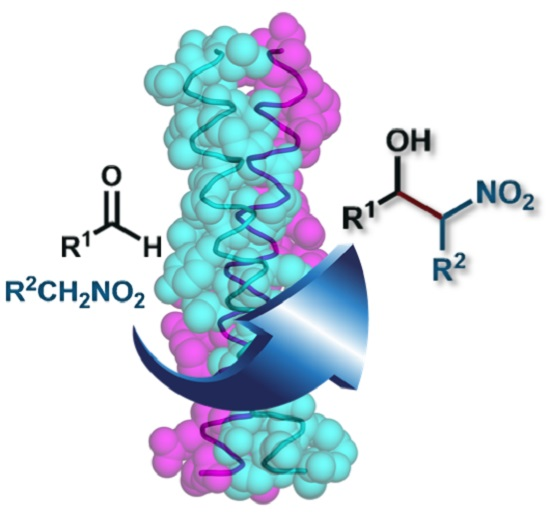
:
1. Introduction
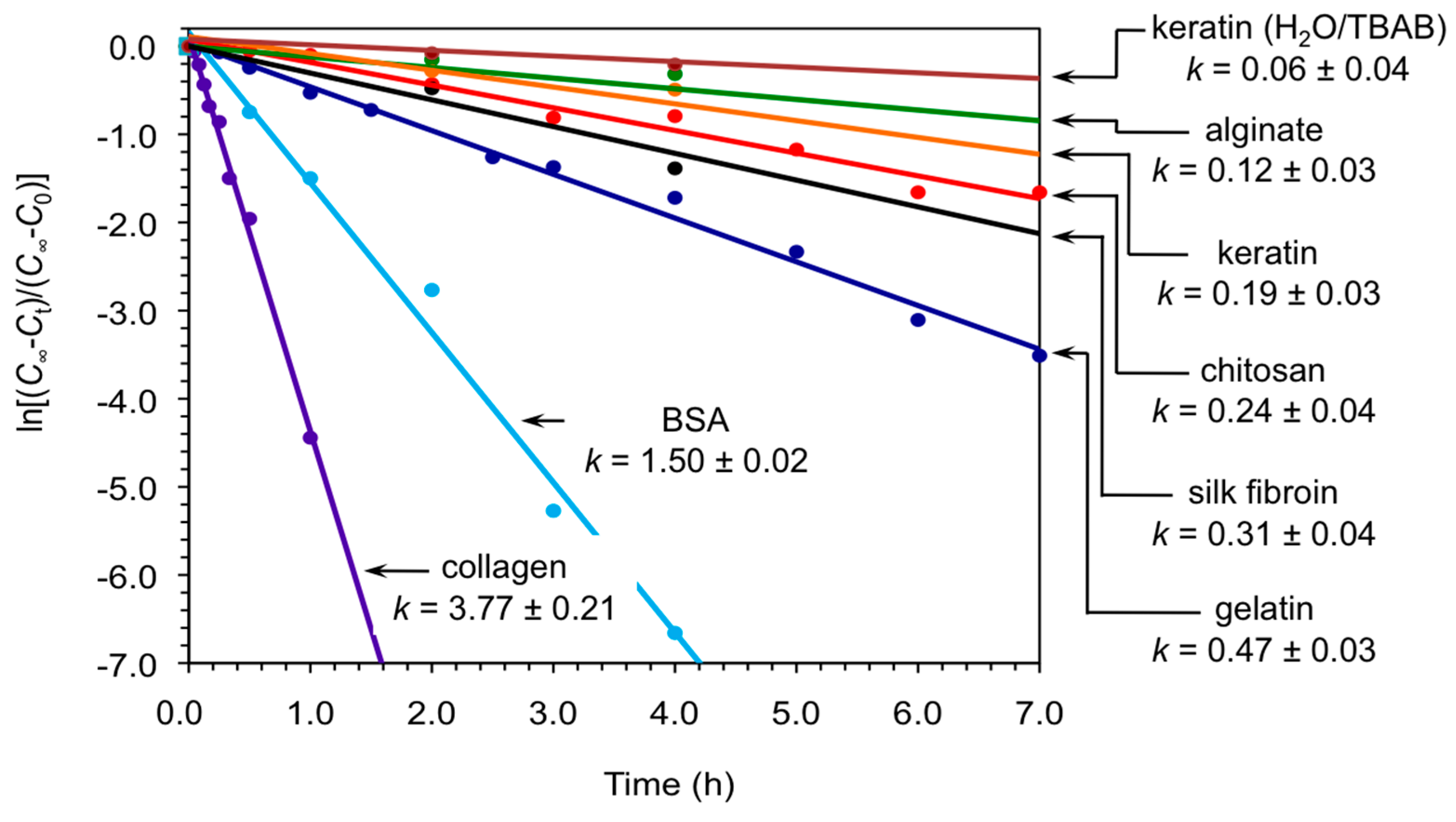
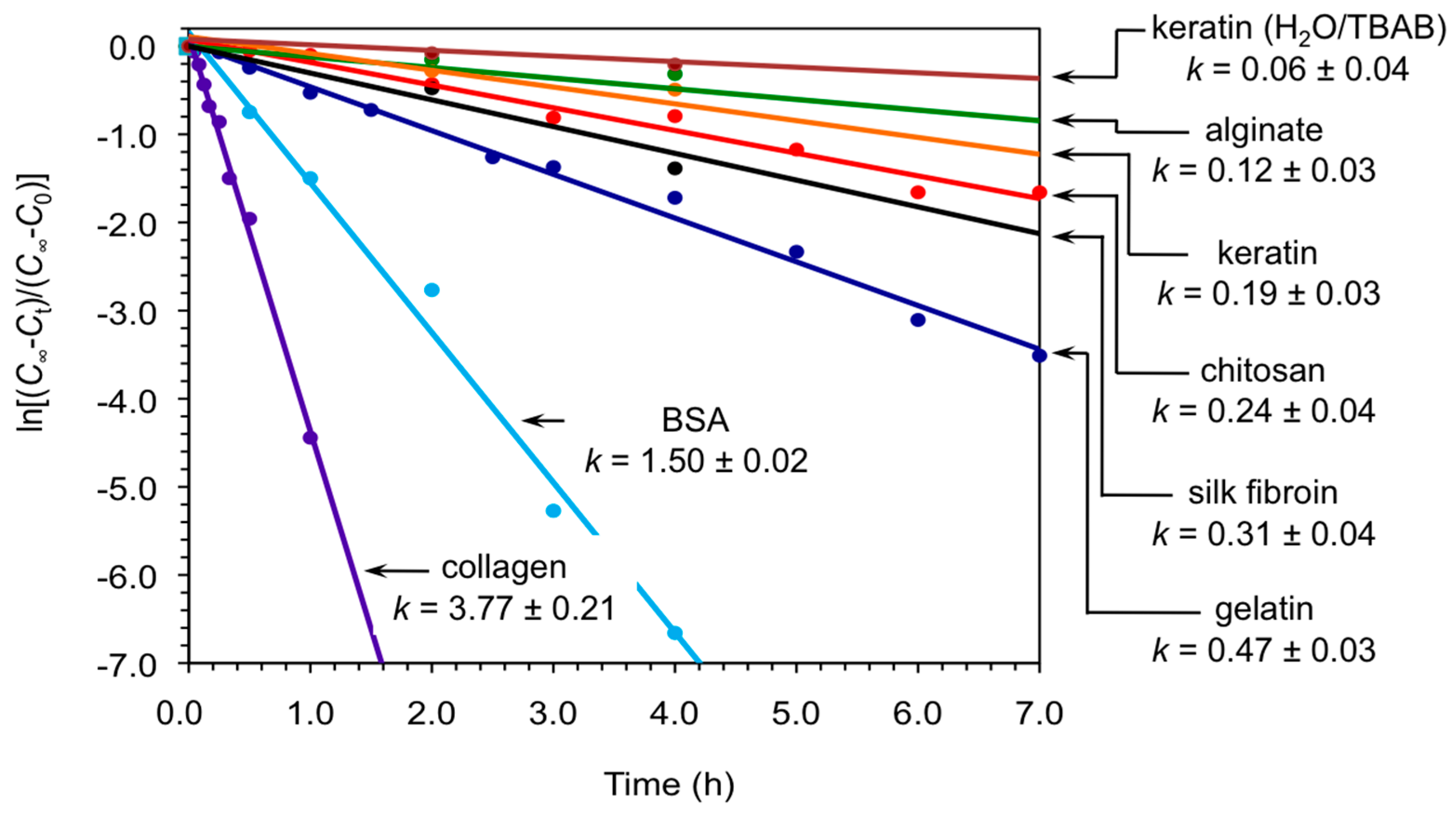
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. General Procedure for Keratin-Catalyzed Nitroaldol (Henry) Reaction
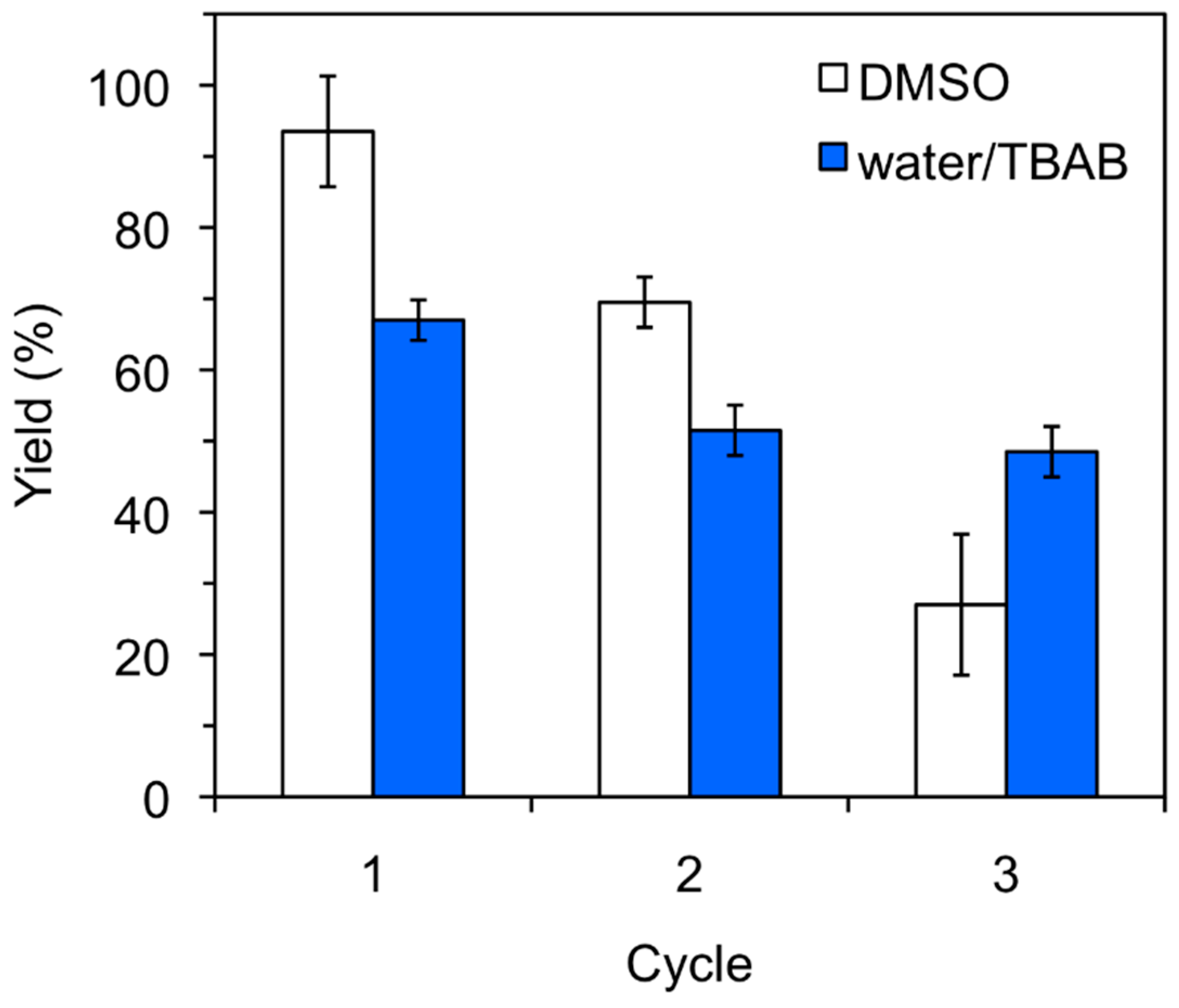
3.4. Typical Recycling Procedure
3.5. Kinetic Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rouse, J.G.; Van Dyke, M.E. A review of keratin-based biomaterials for biomedical applications. Materials 2010, 3, 999–1014. [Google Scholar] [CrossRef]
- Kornillowics-Kowalska, T.; Bohacz, J. Biodegradation of keratin waste: Theory and practical aspects. Waste Manag. 2011, 31, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, M.S.; Chung, B.M.; Leahy, D.J.; Coulombe, P.A. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 2012, 19, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Idler, W.W.; Steven, A.C.; Roop, D.R.; Steinert, P.M. The complete sequence of the human intermediate filament chain keratin 10. Subdomainal divisions and model for folding of end domain sequences. J. Biol. Chem. 1988, 263, 15584–15589. [Google Scholar] [PubMed]
- O′Donnell, J. The complete amino acid sequence of a feather keratin from Emu (Dromaius Novae-Hollandiae). Aust. J. Bioi. Sci. 1973, 26, 415–437. [Google Scholar] [CrossRef]
- Arai, K.M.; Takahashi, R.; Yokote, Y.; Akahane, K. Amino-acid sequence of feather keratin from fowl. Eur. J. Biochem. 1983, 132, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Rice, R.H.; Roop, D.R.; Trus, B.L.; Steven, A.C. Complete amino acid sequence of a mouse epidermal keratin subunit and implications for the structure of intermediate filaments. Nature 1983, 302, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Tonin, C.; Aluigi, A.; Zoccola, M. Characterisation of keratin biomass from butchery and wool industry wastes. J. Mol. Struct. 2009, 938, 35–40. [Google Scholar]
- Sierpinski, P.; Garrett, J.; Ma, J.; Apel, P.; Klorig, D.; Smith, T.; Koman, L.A.; Atala, A.; Van Dyke, M. The use of keratin biomaterials derived from human hair for the promotion of rapid regeneration of peripheral nerves. Biomaterials 2008, 29, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Saul, J.M.; Ellenburg, M.D.; de Guzman, R.C.; van Dyke, M. Keratin hydrogels support the sustained release of bioactive ciprofloxacin. J. Biomed. Mater. Res. A 2011, 98A, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Hengchang, M.; Zhikang, B.; Guobin, H.; Ningning, Y.; Yufei, X.; Zengming, Y.; Wei, C.; Yuan, M. Nanoparticulate palladium catalyst stabilized by supported on feather keratin for Suzuki coupling reaction. Chin. J. Catal. 2013, 52, 578–584. [Google Scholar]
- Karthikeyan, R.; Balaji, S.; Sehgal, P.K. Industrial applications of keratins—A review. J. Sci. Ind. Res. 2007, 52, 710–715. [Google Scholar]
- Luzzio, F.A. The Henry reaction: Recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Laso, A. Recent advances in the catalytic asymmetric nitroaldol (Henry) reaction. Eur. J. Org. Chem. 2007, 2007, 2561–2574. [Google Scholar] [CrossRef]
- Boruwa, J.; Gogoi, N.; Saikia, P.P.; Barua, N.C. Catalytic asymmetric Henry reaction. Tetrahedron Asymmetry 2006, 17, 3315–3326. [Google Scholar] [CrossRef]
- Sharma, K.K.; Biradar, A.V.; Asefa, T. Substituent- and catalyst-dependent selectivity to aldol or nitrostyrene products in a heterogeneous base-catalyzed Henry reaction. ChemCatChem 2010, 2, 61–66. [Google Scholar] [CrossRef]
- Ballini, R.; Gabrielli, S.; Palmieri, A.; Petrini, M. Nitroalkanes as key compounds for the synthesis of amino derivatives. Curr. Org. Chem. 2011, 15, 1482–1506. [Google Scholar] [CrossRef]
- Kühbeck, D.; Saidulu, G.; Reddy, K.R.; Díaz, D.D. Critical assessment of the efficiency of chitosan biohydrogel beads as recyclable and heterogenous organocatalyst for C-C bond formation. Green Chem. 2012, 14, 378–392. [Google Scholar] [CrossRef]
- Kühbeck, D.; Dhar, B.B.; Schön, E.-M.; Cativiela, C.; Gotor-Fernández, V.; Díaz, D.D. C-C bond formation catalyzed by natural gelatin and collagen proteins. Beilstein J. Org. Chem. 2013, 9, 1111–1118. [Google Scholar]
- Kühbeck, D.; Ghosh, M.; Gupta, S.S.; Díaz, D.D. Investigation of C-C Bond formation mediated by Bombyx Mori silk fibroin materials. ACS Sustain. Chem. Eng. 2014, 2, 1510–1517. [Google Scholar] [CrossRef]
- Kühbeck, D.; Mayr, J.; Häring, M.; Hofmann, M.; Quignard, F.; Díaz, D.D. Evaluation of the nitroaldol reaction in the presence of metal ion-crosslinked alginates. New J. Chem. 2015, 39, 2306–2315. [Google Scholar]
- Akutu, K.; Kabashima, H.; Seki, T.; Hattori, H. Nitroaldol reaction over solid base catalysts. Appl. Catal. A 2003, 247, 65–74. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Protein-mediated nitroaldol addition in aqueous media. Catalytic promiscuity or unspecific catalysis? Org. Process Res. Dev. 2011, 15, 236–240. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.


| Entry | Solvent | Yield c of 3a (%) |
|---|---|---|
| 1 | Toluene | 0 |
| 2 | THF | 0 |
| 3 | CH3CN | 0 |
| 4 | CH2Cl2 | 11 |
| 5 | CHCl3 | 10 |
| 6 | DMSO | 66 (0) d |
| 7 | H2O | 11 |
| 8 | H2O/TBAB b | 57 (3) d |

| Entry | R (1) | R′ (2) | Yield b of 3 (%) | 3 | dr d (anti/syn) |
|---|---|---|---|---|---|
| 1 | (4-NO2)-C6H4 | H | 84 | 3a | - |
| 2 | (4-NO2)-C6H4 | CH3 | 96 | 3b | 1:1 |
| 3 | (3-NO2)-C6H4 | H | 93 | 3c | - |
| 4 | (3-NO2)-C6H4 | CH3 | 98 | 3d | 1:0.8 |
| 5 | (2-NO2)-C6H4 | H | 94 | 3e | - |
| 6 | (2-NO2)-C6H4 | CH3 | 96 | 3f | 1:1.3 |
| 7 | (4-F)-C6H4 | H | 10 | 3g | - |
| 8 | (4-Cl)-C6H4 | H | 25 | 3h | - |
| 9 | (3-Cl)-C6H4 | H | 48 | 3i | - |
| 10 | (4-Br)-C6H4 | H | 25 | 3j | - |
| 11 | (4-OH)-C6H4 | H | 5 | 3k | - |
| 12 | (4-CN)-C6H4 | H | 30 | 3l | - |
| 13 | (4-Me)-C6H4 | H | 7 | 3m | - |
| 14 | C6H4 | H | 9 | 3n | - |
| 15 | (4-CHO)-C6H4 | H | 65 c | 3o | - |
| 16 | Pyrid-2-yl | H | 90 | 3p | - |
| 17 | Pyrid-2-yl | CH3 | 96 | 3q | 1:1.7 |
| 18 | Furfur-2-yl | H | 3 | 3r | - |
| 19 | Naphtha-1-yl | H | 13 | 3s | - |

| Entry | R (1) | R’ (2) | Yield b of 3 (%) | 3 | dr e (anti/syn) |
|---|---|---|---|---|---|
| 1 | (4-NO2)-C6H4 | H | 73 (50) c | 3a | - |
| 2 | (4-NO2)-C6H4 | CH3 | 94 | 3b | n.d. |
| 3 | (3-NO2)-C6H4 | H | 21 | 3c | - |
| 5 | (2-NO2)-C6H4 | H | 70 | 3e | - |
| 6 | (2-NO2)-C6H4 | CH3 | 98 | 3f | 1:0.8 |
| 7 | (4-F)-C6H4 | H | 9 | 3g | - |
| 8 | (4-Cl)-C6H4 | H | 18 | 3h | - |
| 9 | (3-Cl)-C6H4 | H | 34 | 3i | - |
| 10 | (4-Br)-C6H4 | H | 14 | 3j | - |
| 11 | (4-OH)-C6H4 | H | 1 | 3k | - |
| 12 | (4-CN)-C6H4 | H | 66 | 3l | - |
| 13 | (4-CN)-C6H4 | CH3 | 89 | 3m | n.d. |
| 14 | (4-Me)-C6H4 | H | 4 | 3n | - |
| 15 | C6H4 | H | 8 | 3o | - |
| 16 | (4-CHO)-C6H4 | H | 19 d | 3p | - |
| 17 | Pyrid-2-yl | H | 72 | 3q | - |
| 18 | Pyrid-2-yl | CH3 | 85 | 3r | 1:0.9 |
| 19 | Furfur-2-yl | H | 11 | 3s | - |
| 20 | Naphtha-1-yl | H | 0 | 3t | - |

{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häring, M.; Pettignano, A.; Quignard, F.; Tanchoux, N.; Díaz Díaz, D. Keratin Protein-Catalyzed Nitroaldol (Henry) Reaction and Comparison with Other Biopolymers. Molecules 2016, 21, 1122. https://doi.org/10.3390/molecules21091122
Häring M, Pettignano A, Quignard F, Tanchoux N, Díaz Díaz D. Keratin Protein-Catalyzed Nitroaldol (Henry) Reaction and Comparison with Other Biopolymers. Molecules. 2016; 21(9):1122. https://doi.org/10.3390/molecules21091122
Chicago/Turabian StyleHäring, Marleen, Asja Pettignano, Françoise Quignard, Nathalie Tanchoux, and David Díaz Díaz. 2016. "Keratin Protein-Catalyzed Nitroaldol (Henry) Reaction and Comparison with Other Biopolymers" Molecules 21, no. 9: 1122. https://doi.org/10.3390/molecules21091122